

Abstract
Serine/threonine phosphatase (Stp1) is a member of the bacterial Mg2+- or Mn2+- dependent protein phosphatase/protein phosphatase 2C family, which is involved in the regulation of Staphylococcus aureus virulence. Aurintricarboxylic acid (ATA) is a known Stp1 inhibitor with an IC50 of 1.03 μM, but its inhibitory mechanism has not been elucidated in detail because the Stp1–ATA cocrystal structure has not been determined thus far. In this study, we performed 400 ns molecular dynamics (MD) simulations of the apo–Stp1 and Stp1–ATA complex models. During MD simulations, the flap subdomain of the Stp1–ATA complex experienced a clear conformational transition from an open state to a closed state, whereas the flap domain of apo–Stp1 changed from an open state to a semi-open state. In the Stp1–ATA complex model, the hydrogen bond (H-bond) between D137 and N142 disappeared, whereas critical H-bond interactions were formed between Q160 and H13, Q160/R161 and ATA, as well as N162 and D198. Finally, four residues (D137, N142, Q160, and R161) in Stp1 were mutated to alanine and the mutant enzymes were assessed using phosphate enzyme activity assays, which confirmed their important roles in maintaining Stp1 activity. This study indicated the inhibitory mechanism of ATA targeting Stp1 using MD simulations and sheds light on the future design of allosteric Stp1 inhibitors.
Keywords: antimicrobial resistance, Staphylococcus aureus, serine/threonine phosphatase, aurintricarboxylic acid, molecular dynamics simulations, flap subdomain
Introduction
The increasing problem of antimicrobial resistance has attracted the public’s attention for more than a decade [1]. Serious health complications can be caused by infections with antimicrobial-resistant bacteria [2–4], whose rapid evolution has been accompanied by the slowed development of antibiotics [5]. Antibiotics are widely used and overused in human medicine and livestock production [6], which has led to the spread of antimicrobial resistance. Bacterial resistance mechanisms can be divided into three categories based on the defense mechanism or the site of action of the antibiotic [7]. Bacterial biofilm formation is the first defense mechanism to protect bacterial cells and increase their resistance to antibiotics. The second line of defense includes the cytoderm, cytomembrane, and encased efflux pumps. Through these mechanisms, mutations result in alterations in the structural organization of the cell wall that prevent antibiotic molecules, such as vancomycin, from accessing the biosynthesis site [8]. Multiple adaptations provide resistance to antimicrobial agents that target the cell membrane, including reduced permeability of the outer membrane and reduced negative charges on the membrane resulting from modifications to phosphatidylglycerol headgroups [9]. The membrane also regulates the resistance to fosfomycin by decreasing uptake of the drug [10], and the activation of efflux pumps has also been shown to lead to resistance to many other antibiotics [11]. Intracellular alterations, which include gene mutations and alterations of antibiotic target sites, are the third line of defense [12]. As the problem of antimicrobial resistance becomes increasingly severe, the development of new antibiotic mechanisms and targets is extremely urgent.
In response to the problem of antimicrobial resistance, the development of treatments for bacterial infections in recent years has been focused on anti-virulence agents, which target pathways and proteins related to virulence [12]. Multiple strategies have been applied to the development of anti-virulence agents, including regulating virulence expression, inhibiting toxin function and delivery, and interfering with bacterial adhesion [12–14]. Staphylococcus aureus is a major medical pathogen that causes various infectious diseases, which range from mild skin infections to endocarditis and bacteremia [15–17]. Research has identified several S. aureus anti-virulence agents, including MAE4, which has been reported to block virulence [18], and the serine/threonine phosphatase (Stp1) and kinase Stk1, which have been suggested to be involved in regulating virulence [19–22]. Stp1 and Stk1 can regulate the phosphorylation level of the cysteine that is highly conserved in the virulence regulatory proteins, including SarA, MgrA, and SarZ [21]. The absence or mutation of the stp1 gene results in elevated cysteine phosphorylation of MgrA/SarA family proteins and significantly reduces S. aureus virulence [21]. In addition, it has been reported that Stp1 plays a role in decreasing both the virulence and susceptibility to vancomycin of S. aureus [22]. These studies suggest that Stp1 is a promising target for anti-virulence agents.
Stp1 is a member of the Mg2+- or Mn2+- dependent protein phosphatases/protein phosphatase 2C (PPM/PP2C) family, [23, 24] which is a large family of Phospho-Ser/Thr protein phosphatases [25]. Structural analysis of PPM/PP2C protein phosphatases suggests that three or four metal ions are embedded in the catalytic site and that a flap subdomain that contains helices and flexible loops is located next to the metal 3 (M3, the third manganese ion or magnesium ion in the PPM/PP2C family) binding site [26–32]. In addition, studies of the PP2C phosphatase tPphA from Thermosynechococcus elongatus report that the flexible flap subdomain is involved in the regulation of enzyme activity [27] and that it plays an important role in substrate specificity [28].
To date, 5,5’-methylenedisalicylic acid (MDSA), aurintricarboxylic acid (ATA), and aurin (a derivative of ATA) (Table 1) are the only known inhibitors that target S. aureus Stp1, with half maximal inhibitory concentration (IC50) values of 9.68 μM, 1.03 μM, and 19.42 μM, respectively [33, 34]. A structure–activity relationship study and surface plasmon resonance experiments showed that ATA directly binds with Stp1 [34]. These experiments also showed that the Stp1 variants N162A and D198A both exhibited attenuated ATA inhibition ratios and weakened stabilization between ATA and Stp1, thus confirming that N162 and D198 play important roles in ATA binding [34]. In addition, ATA was found to inhibit Stp1 mainly via noncompetitive mechanisms, as indicated by enzymatic-kinetic assays [34]. However, the mechanism of ATA inhibition of the activity and biological function of Stp1 has not previously been determined in detail due to the lack of a crystal structure of the Stp1–ATA complex.
Table 1.
Inhibitors targeting Stp1 of S. aureus
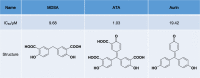
|
Stp1 serine/threonine phosphatase, MDSA 5,5’-methylenedisalicylic acid, ATA aurintricarboxylic acid
Because of the dynamic nature of biomolecules, a single-crystal structure is insufficient for predicting putative mechanisms or binding modes [35]. MD simulation is a powerful research approach in drug discovery that can be used to calculate the dynamics and time-dependent behavior of macromolecular models [36–38]. Multiple molecular conformations obtained by MD simulations can be used to explain the dynamics of molecular structures [35]. In addition, the combination of docking and MD simulations has been used to investigate the mechanisms of molecules that target proteins, [39, 40] prediction of protein–ligand binding [41], and drug design [42, 43].
Herein, we have run 400 ns molecular dynamics (MD) simulations for the apo–Stp1 model and Stp1–ATA complex model, with the aim to determine the inhibitory mechanism of ATA. These MD simulations enable us to observe conformational changes in the flap subdomain of Stp1–ATA complex models and clearly provide insights into the inhibitory mechanism of ATA targeting Stp1. In addition, we have designed mutation studies, which verified the critical roles of several residues for the first time. These results will contribute to the development of new anti-virulence agents against severe S. aureus infections.
Materials and methods
Structural comparisons and sequence alignments
Crystal structures were obtained from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB; https://www.rcsb.org/) and superimposed using PyMOL [44] (Schrödinger Inc., LLC, New York, NY, USA, https://pymol.org/). Multiple sequence alignment was performed using Clustal Omega [45, 46] (https://www.ebi.ac.uk/Tools/msa/clustalo/) and prepared using ESPript 3.0 [47] (http://espript.ibcp.fr).
MD simulations of apo–Stp1 and Stp1–ATA complex models
Model construction and preparation
In this study, two molecular models were constructed: an apo–Stp1 model and a Stp1–ATA complex model that combined Stp1 with an ATA molecule. For the apo–Stp1 model, the crystal coordinate of 5F1M was obtained from the RCSB PDB [48]. The Stp1–ATA complex model was built with molecular docking simulations using Glide [49] in the Maestro package (Schrödinger Inc., LLC, New York, NY, USA, 2015) [50]. First, the Stp1 crystal structure 5F1M obtained from the RCSB PDB [48] was prepared using the Protein Preparation Wizard panel in Maestro. The initial Stp1 structure was prepared by assigning the bond order, adding missing hydrogen and disulfide bonds, and deleting water molecules but retaining the metal ions, followed by impref minimization. Several residues, such as Asp37, Asn162, and Asp198, which play critical roles in molecule binding, located at the catalytic site were then selected to conform to the docking site, followed by grid generation with enclosing box of 16 Å by using Maestro. The ATA molecule was prepared using the LigPrep module in Maestro with the OPLS_2005 force field and a target pH 7.0 ± 2.0. The protonation state of ATA is shown in Supplementary Figure S1. Finally, a molecular docking simulation was performed using Glide in extra precision mode (GlideXP) [51]. The GlideXP mode enables dockings of flexible ligand with rigid receptor side chains. As a result, the GlideXP docking produced five binding poses, from which the proper pose with a docking score value of –7.143 was selected as the initial binding mode of Stp1 and ATA. Moreover, the proper pose was similar to the binding pose proposed by Zheng et al. [34]. The other four poses were bad with poor docking greater than –5. PyMOL viewer (http://www.pymol.org/) was used to visualize structures and generate molecular images. In addition, the protonated states of the titratable amino acids in the two previously described models were set according to the pKa values predicted by Propka [52, 53] at a neutral bulk pH.
The structure and coordinates of ATA were obtained from the initial binding mode. The parameters for ATA were subsequently produced by antechamber [54]. Epik module [55] in the Schrodinger suite was used to predict the protonated state of ATA. Optimal quantization of ATA was performed with Gaussian 09 software (Gaussian, Inc.) at the HF/6-31 (d, p) level of theory. The Restrained Electrostatic Potential (RESP) charges of ATA were obtained using antechamber. The two simulation systems were subsequently constructed with tleap in AmberTools 13. Both the apo–Stp1 model and the Stp1–ATA complex model were solvated in a cubic water box of TIP3P water molecules [56], maintaining a distance of 10 Å from any protein atom. A suitable number of counterions (Na+ and Cl−) was added to neutralize the charge of the entire simulation model. The ff14SB force field [57, 58] was used for each protein, and the general Amber force field [59] was used for the ATA molecule. Finally, ACPYPE (AnteChamber PYthon Parser interfacE) [60] was used to convert the amber topology of both systems into Gromacs format.
Conventional MD simulations
All conventional MD simulations were carried out using Gromacs version 5.0.2 [61] (http://www.gromacs.org/) with standard periodic boundary conditions, which were used to avoid problems with boundary effects caused by finite size. During the first process of energy minimization, a conjugate gradient algorithm was used. A 1 ns MD simulation using a canonical (NVT) ensemble [62] was then performed to equilibrate the structures by gradually heating the system to 300 K. Another 1 ns MD simulation with an isothermal–isobaric (NPT) ensemble [62] at 1 bar was then carried out to equilibrate the system density. Finally, 400 ns MD simulations were performed. During the production run, which had a coupling time (τT) of 1.0 ps, a V-rescale thermostat [63] was used to maintain the system temperature at 300 K, while the Parrinello–Rahman barostat method [64] was used to maintain a system pressure of 1 bar. For each production run and each model, coordinates or conformations were extracted every 10 ps.
Analysis of MD trajectory
The trajectories were initially processed to correct for periodicity with the Trjconv tool in Gromacs 5.0.2. The atomic characteristics of the apo–Stp1 model and Stp1–ATA complex were subsequently compared using a series of analysis tools in Gromacs 5.0.2 software. The root mean square deviation (RMSD), radius of gyration (Rg), and root mean square fluctuations (RMSF) are critical indicators of the stability of trajectories. The RMSD, Rg, and RMSF values of the Cα backbone of the entire models and the flap subdomain were, respectively, calculated using the initial structure as references. Furthermore, the trajectories of the apo–Stp1 model and Stp1–ATA complex model were clustered into 10 classes using the Class tool of Gromacs 5.0.2 with cut-offs of 0.15 nm and 0.18 nm, respectively. The central conformation of each cluster was extracted for superimposition. In addition, accurate hydrogen bond (H-bond) analysis was performed using the H-bond tool in Gromacs 5.0.2.
Mutation studies
Expression and purification of Stp1 and variants
The materials used in the mutation studies were purchased from Sigma-Aldrich. The wild-type (WT) stp1 gene was cloned into the vector Pet28a. The primers used to produce the four variants (D137A, N142A, Q160A, and R161A) are shown in Supplementary Table S1. The expression of WT-Stp1 and N-His-tagged variant proteins in Escherichia coli BL21-Gold (DE3) was induced with 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). One liter of cells was diluted 1:100 in Luria-Bertani (LB) medium and cultured at 37 °C. Following overnight culture, 0.5 mM IPTG was added to the liquid medium to induce cells until the optical density at 600 nm reached 0.6–0.8. After overnight growth at 16 °C with shaking, cells were collected by centrifugation and resuspended in 30 mL Ni-NTA buffer A (50 mM Tris-HCl [pH 8.0], 100 mM NaCl, 50 mM imidazole, 2 mM MgCl2, and 1 mM dithiothreitol [DTT]) that contained the protease inhibitor phenylmethanesulfonyl fluoride. The samples were then purified using Ni-NTA (GE Healthcare) with gradient washing using buffer B (50 mM Tris-HCl [pH 8.0], 100 mM NaCl, 400 mM imidazole, 2 mM MgCl2, and 1 mM DTT). Eluted target fractions were further purified using Superose 75 gel filtration chromatography (GE Healthcare). The fractions were collected, concentrated, and further purified using a gel filtration column and desalting buffer (50 mM Tris-HCl [pH 8.0], 100 mM NaCl, 2 mM MgCl2, and 1 mM DTT). The purity of the final proteins was assessed using sodium dodecyl sulfate polyacrylamide gel electrophoresis on a 12% (wt/vol) gel and was determined to be > 90%.
Phosphatase activity assay
Phosphatase activity assays were performed at room temperature and were based on the formation of p-nitrophenol from para-nitrophenylphosphate (pNPP). Each reaction was performed in a 200 μL mixture that contained 0.3 μM WT-Stp1 or Stp1 variant in buffer (10 mM Tris-HCl [pH 7.5], 1 mM EDTA, 5 mM MnCl2, and 0.02% β-mercaptoethanol) and 1 mM pNPP and was incubated for 15 min. In the control experiments, pNPP was replaced with dimethyl sulfoxide. Finally, the reactions were quenched by the addition of 20 μL of 5 M NaOH, and the absorbance at 405 nm was measured.
Experimental data were analyzed using GraphPad Prism software (GraphPad Software Inc.). All reactions were carried out in triplicate.
Results and discussion
Structural comparisons and sequence alignments
The Stp1 crystal structure (PDB code: 5F1M) has previously been published with a resolution of 2.32 Å (Fig. 1a) [34]. To observe structural differences, particularly those in the flap subdomain, the crystal structures of the PP2Cs that include Stp1, tPphA (PDB code: 5ITI), PstP (PDB code: 1TXO), and human PPM1A (PDB code: 1A6Q) were aligned. The results showed that the flap subdomain conformations were similar among the different bacteria strains; however, the conformation of the flap subdomain of the human protein differed from that of the bacterial proteins (Fig. 1b). In contrast, multiple sequence alignment indicated that the flap subdomains of the bacterial PPMs have a low sequence identity with a value of 26.92% (Fig. 1c). Based on sequence alignment with the tPphA flap domain (T138–L163) [65], the S. aureus flap subdomain is located between residues S139 and I164. Given the flexibility of the flap subdomain, its conformation should be variable. However, no conformational changes have previously been reported, and the biological function of the S. aureus flap subdomain has not previously been elucidated.
Fig. 1.

Structural comparisons and sequence alignments of multiple PPM/PP2C protein phosphatases. a Representation of serine/threonine phosphatase (Stp1) structure (Protein Data Bank (PDB) code: 5F1M) showing two main functional domains: the active site (surface) and the flap subdomain (dark blue). b Three-dimensional (3D) structural comparisons between PPM/PP2C structures, including Stp1 (dark blue), tPphA (PDB code: 5ITI; pink), PstP (PDB code: 1TXO; yellow), and human PPM1A (PDB code: 1A6Q; gray). c Multiple sequence alignment of flap domains of diverse bacterial PPM, including Stp1, tPphA, PstP, and SaSTP (PDB code: 2PK0)
As previously discussed, N162/D198 might participate in the binding of ATA and Stp1 [34]. ATA inhibits Stp1 in a mainly noncompetitive manner [34]. Furthermore, the flexible flap subdomain located next to the M3 site plays an important role in enzyme activity and substrate binding [26–28]. Therefore, we speculated that the introduction of ATA causes conformational changes in the flexible flap subdomain, which result in the loss of Stp1 function. To investigate the conformational changes in the flap domain and the ATA inhibitory mechanism, this study ran 400 ns all-atom MD simulations of the apo–Stp1 and Stp1–ATA complex models.
Conformational changes in the flap subdomain
To explore the stability and dynamics of the two models, the RMSD, Rg, and RMSF values of each model were calculated using Gromacs tools. Figure 2 shows that the flap subdomain of the apo–Stp1 model remained stable, whereas that of the Stp1–ATA complex model was flexible and unstable. The RMSD values (Fig. 2a) showed that the apo–Stp1 model remained stable throughout the entire MD simulation, whereas the Stp1–ATA complex model experienced conformational changes in the first 200 ns simulation and then remained stable during the 200–400 ns simulations. In particular, the RMSD values of the complex model exhibited apparent fluctuations during 0–100 ns and less obvious fluctuations during 150–200 ns. The Rg values (Fig. 2b) suggested that the Stp1–ATA complex model expanded during the 0–100 ns simulations and contracted during the 150–200 ns simulations, thus indicating that the complex model is relatively unstable at 0–200 ns. In addition, the RMSF values shown in Fig. 2c demonstrated that the flap subdomain (residue 139–164) of the Stp1–ATA complex model rather than the other domains underwent fluctuations in the range from 0.3 nm to 0.9 nm. Thus, the RMSD and Rg values of the flap subdomain in both models were calculated (Figs. 2d, e). The results indicated that the flap subdomain in the Stp1–ATA complex model was changeable, whereas it was stable in apo–Stp1. Furthermore, the comparison of the RMSF values of the Stp1–ATA complex model during the first 200 ns and the subsequent 200 ns was performed, which suggested that the flap subdomain was stable during 200–400 ns (Fig. 2f). This analysis confirmed that the flap subdomain was stable in the apo–Stp1 model during entire MD simulations and was unstable in the Stp1–ATA complex model during 0–200 ns simulations.
Fig. 2.

Values for the apo–serine/threonine phosphatase (Stp1) model (blue) and Stp1–aurintricarboxylic acid (ATA) complex (red) model were calculated using Gromacs version 5.0.2. a Root mean square deviation (RMSD) values of Cα backbone in both models. b ATA complex Radius of gyration (Rg) values of Cα backbone in both models. c Root mean square fluctuation (RMSF) values of every residue in two models during entire molecular dynamics (MD) simulations. d RMSD values of the Cα of flap subdomains in both models. e Rg values of the Cα of flap subdomains in both models. f RMSF values of every residue in Stp1–ATA complex model during 0–200 ns and 200–400 ns
To investigate conformational changes in the structure of the flap subdomain during the first 200 ns of MD simulations, five frames of conformations were extracted for each model (Fig. 3). The flap subdomain in the apo–Stp1 model retained its stability (Fig. 3a), whereas the flap subdomain of the Stp1–ATA complex fluctuated between a horizontal and a nearly vertical position (Fig. 3b). Based on the influence of the flap subdomain on the active site, the horizontal position of the flap subdomain is considered as the open state because the active pocket is open, whereas the vertical conformation is considered as the closed state. These conformational changes in the flap subdomain (Fig. 3) are in line with the value changes shown in Fig. 2. Overall, the conformations of each model throughout the MD simulations were clustered into 10 classes. For each model, the central conformations of every cluster were superimposed, as depicted in Supplementary Figure S2a and S2b. Moreover, their fluctuation extents were quantified (Supplementary Figure S2c). The distance between residue Pro152 in orange ribbon (open state) and residue Pro152 in cyan ribbon was 16.16 Å (Supplementary Figure S2c). However, compared with the Stp1–ATA complex, the conformations of the flap domain of the apo–Stp1 model fluctuated limited fluctuations, changing from a yellow conformation to cyan conformation (Supplementary Figure S2a). The distance of residue Pro152 between the two conformations was 7.48 Å (Supplementary Figure S2c), half as long as the distance in the Stp1–ATA complex model. Therefore, the transformation of the apo–Stp1 model was termed as changing from an open state (Supplementary Figure S2a, yellow) to a semi-open state (Supplementary Figure S2a, cyan). One potential explanation for these distinct differences in the flap subdomain conformation between the two models is that the introduction of the ATA molecule causes changes in the interaction mode.
Fig. 3.

Conformational changes in the flap subdomains of the apo–serine/threonine phosphatase (Stp1) and Stp1–aurintricarboxylic acid (ATA) complex models in 200 ns molecular dynamics (MD) simulations. a Flap domain of apo–Stp1 model. Conformations remained horizontal during the former 200 ns simulation. b Flap domain of the Stp1–ATA complex model. Conformational changes from the horizontal position to the nearly vertical position occurred during the first 200 ns simulation
Changes in the interaction mode of the flap subdomain in the Stp1–ATA complex model
The initial conformation (0 ns) and stable conformation (400 ns) of the Stp1–ATA complex model were analyzed to investigate changes in the interaction mode. The ions M1/M2/M3 located in the catalytic site were clearly involved in ATA binding, similar to the binding mode of the simulation models in the study of Zheng et al. [34]. The observations were in accordance with the roles of metal ions in the published PPM/PP2C. There was little difference in the metal ions binding after MD simulations. Therefore, the binding poses of ATA with the flap subdomain were mainly investigated.
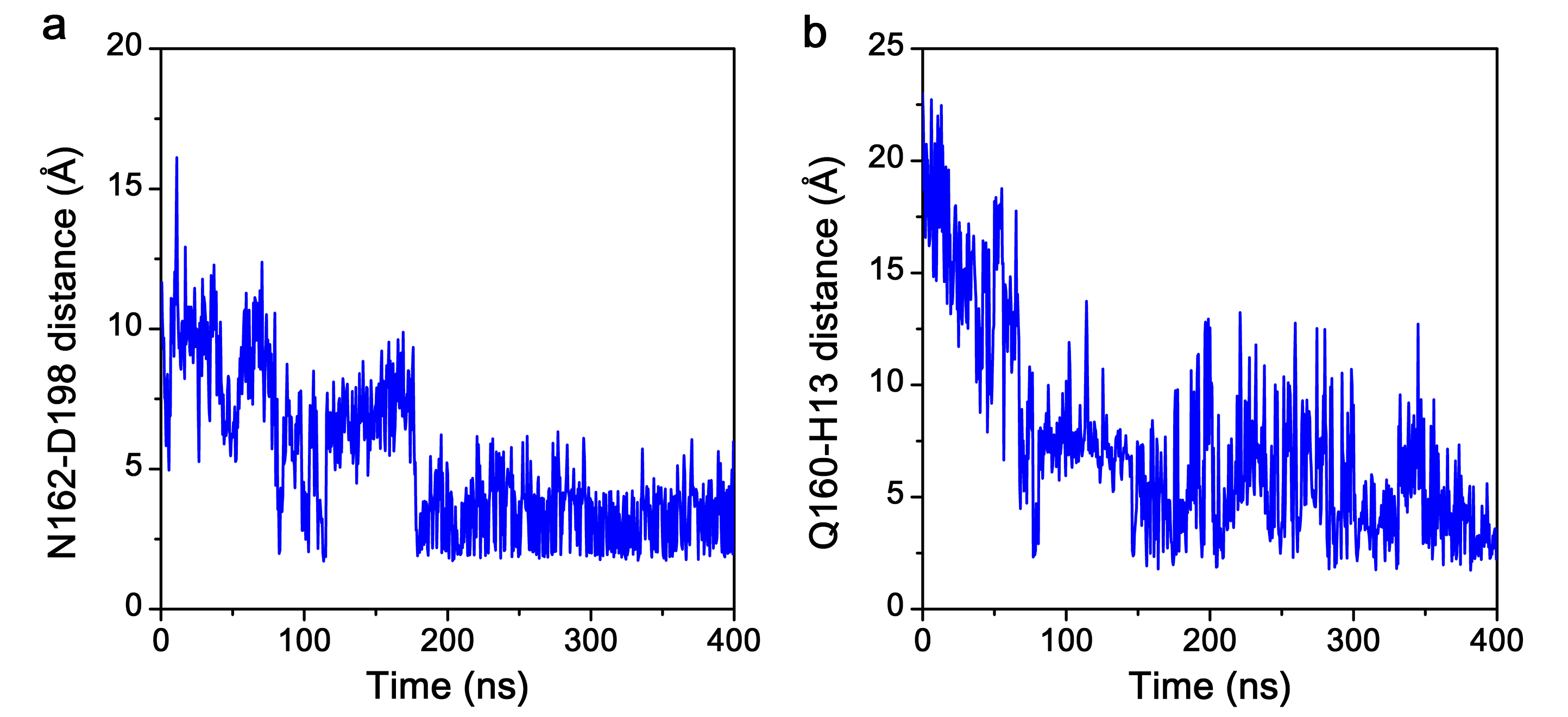
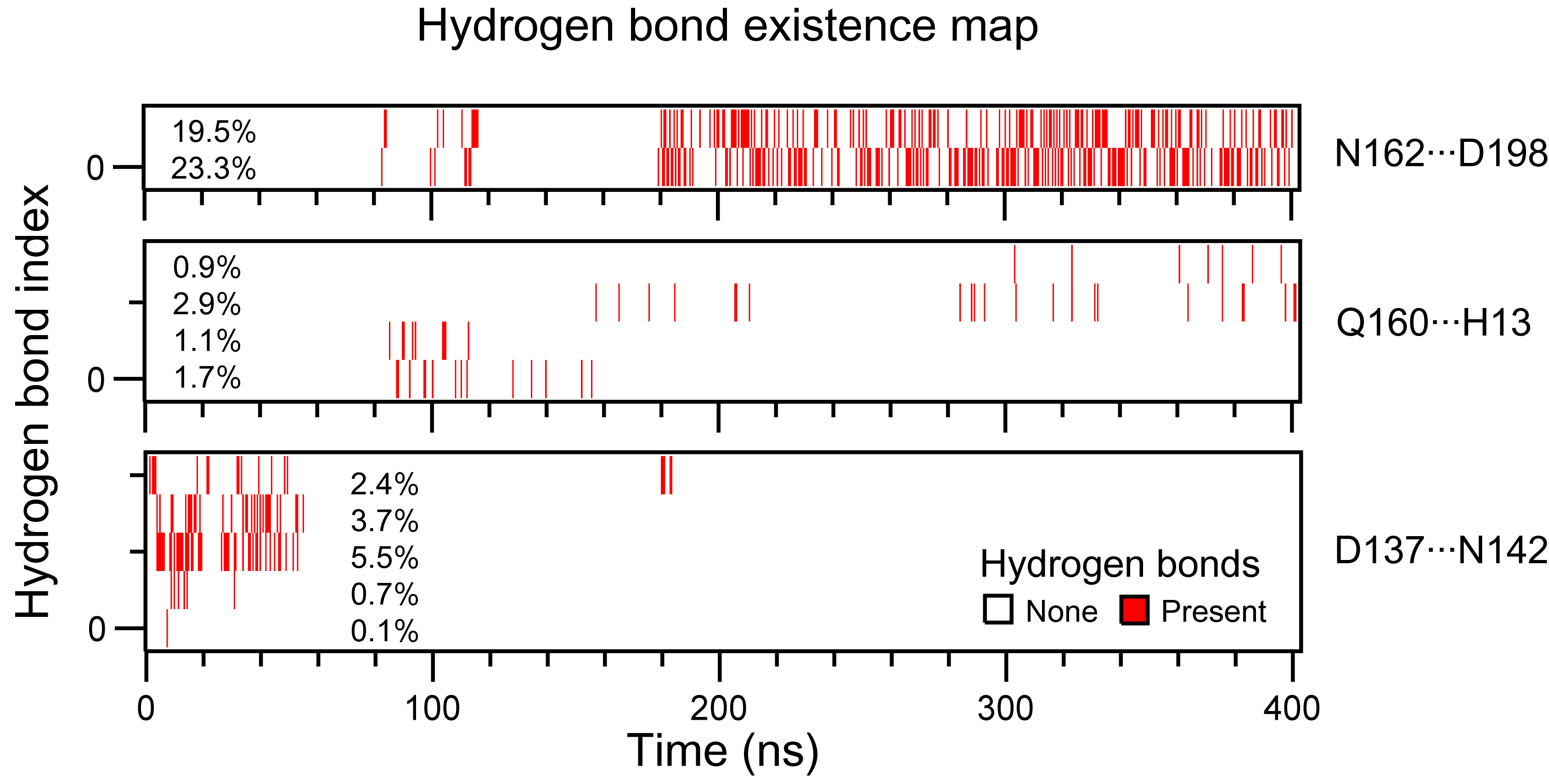
Figures 4a, b show the binding mode of ATA with the flap subdomain in the Stp1–ATA complex at 0 ns and 400 ns, respectively. The surface of the complex model is also shown at 0 ns (Fig. 4c) and 400 ns (Fig. 4d). First, N162···D198 formed at 400 ns (Fig. 4b) but not at 0 ns (Fig. 4a). Moreover, the atomic distances of the residue pair (N162 and D198) (Supplementary Figure S3a) became shorter as the simulation time increased. More obviously, the H-bond analysis (Supplementary Figure S4) shows that two types of H-bonds between N162 and D198 were formed in the later part simulations. Their occupancies were 19.5% and 23.3%, respectively, during the 400 ns simulations. These observations are consistent with previous mutation studies of N162A and D198A [34], which have been shown to weaken the stabilizing effect of ATA on enzymes and attenuate the inhibition of ATA.
Fig. 4.

The initial conformation (0 ns) and stable conformation (400 ns) of the serine/threonine phosphatass (Stp1)–aurintricarboxylic acid (ATA) complex model. The flap subdomain is shown in dark blue. Residues located at the active site are shown in light cyan. a The initial interaction mode (0 ns) of Stp1 with ATA. b The interaction mode of Stp1–ATA complex is shown at 400 ns. c The conformation of ATA in the active pocket is shown at 0 ns. d At 400 ns, ATA was surrounded by Q160 and H13
Second, the flap residues R161 and N162 interact with ATA at 0 ns (Fig. 4a), whereas Q160 and R161 interact with ATA at 400 ns (Fig. 4b). As the conformation of the flap subdomain changes, the Q160 residue stretches over the catalytic site and interacts with H13, surrounding the ATA molecule within the Stp1 catalytic site (Figs. 4b, d). Although four pairs of Q160···H13 are present with low occurrences (Supplementary Figure S4), the atomic distance between them becomes shortened with simulations (Supplementary Figure S3). These results indicate that residues Q160 and R161 may play important roles in Stp1 enzyme activity. These changes in H-bond interactions support that residues Q160, N162, and D198 exert critical roles in the catalytic activity of Stp1 and ATA inhibition.
The changes in the interaction between D137 and N142, which are located at the back of the flap subdomain, were also assessed. In the Stp1–ATA complex model, an H-bond D137···N142 was present in the initial conformation (0 ns; Fig. 5a); however, it disappeared at 400 ns (Fig. 5b). Figure 5c suggests that the distance between residues D137 and N142 also changed with the MD simulation time. In the initial conformation, the distance was 3 Å, which is consistent with the presence of an H-bond interaction (Fig. 5a). However, this space flattened as the conformation stabilized, which resulted in a distance of 7 Å and the absence of an H-bond (Figs. 5b, c). The changes in D137···N142 are more clearly shown in the H-bond existence map (Supplementary Figure S4). The map indicated that D137···N142 pairs were only presented in the first part simulations. In contrast, the flap subdomain of apo–Stp1 was relatively stable, with D137···N142 H-bonds that were similar to those observed in the initial conformation of the Stp1–ATA complex. Therefore, residues D137 and N142 may participate in maintaining the stability of Stp1, and the interaction between D137 and N142 may explain the stability of the flap subdomain in the apo–Stp1 model.
Fig. 5.

The interactions and conformations of several residues located at the back of the flap subdomain of the serine/threonine phosphatase (Stp1)–aurintricarboxylic acid (ATA) complex model. a The initial interaction mode (0 ns) of several residues. b The interaction mode of residues at 400 ns. c The distance between residues D137 and N142 changed over time. The distance was 3 Å in the initial conformation and 7 Å in the final conformation
Mechanism of ATA inhibition
Based on the previously described analyses, an inhibitory mechanism of ATA targeting Stp1 was proposed. As a mixed-type inhibitor, the ATA molecule occupies the catalytic site of Stp1 [34], first in a competitive role, interacting with R161 in the flap subdomain, and second in a noncompetitive manner, with ATA causing the Q160 residue to stretch over the catalytic site and interact with H13 and the ATA molecule. The H-bond interaction between D137 and N142 subsequently disappears. At the same time, the distance between N162 and D198 becomes shorter, which leads to the formation of an H-bond. Finally, the flap domain transitions from the initial open state to a stable closed state. As a result, the ATA molecule becomes tightly wrapped in the active pocket, inhibiting the biological function of Stp1.
The important roles of D137, N142, Q160, and R161
To confirm the key roles of residues D137, N142, Q160, and R161, four variants (D137A, N142A, Q160A, and R161A) and WT-Stp1 were constructed, cloned, and purified, followed by phosphate enzyme activity assays. As shown in Fig. 6, compared with the enzyme activity of WT-Stp1, the D137A and R161A variants lost their enzyme activity almost completely, whereas the N142A and Q160A mutations reduced their enzyme activity by 40 and 90%, respectively. These mutation studies indicated that these four residues play extremely important roles in maintaining the dephosphorylation activity of Stp1. Moreover, D137 and N142 are related to the stability of the Stp1 structure. The results for these four variants are in accordance with the interactional changes observed in MD simulations (Figs. 4 and 5), further supporting our proposed mechanism.
Fig. 6.
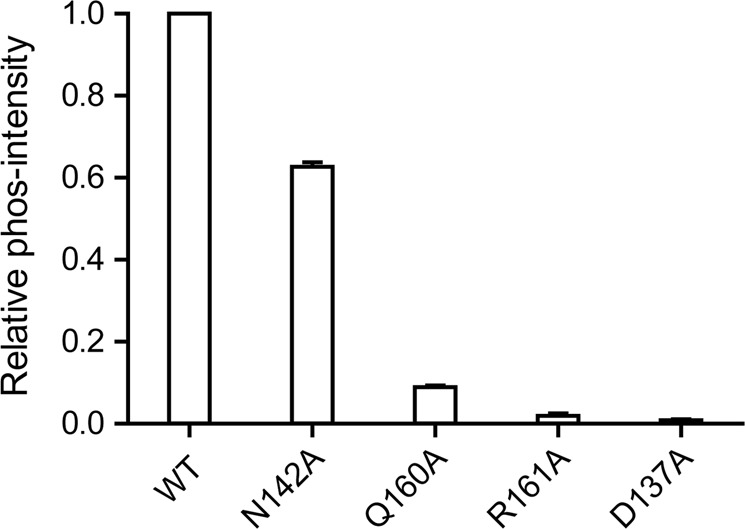
Enzyme activity of variants D137A, N142A, Q160A, and R161A relative to wild-type (WT) Stp1. D137A and R161A variants lost almost all enzyme activity. Enzyme activity of the Q160A variant was reduced by 90%. The N142A variant retained 60% enzyme activity
Conclusions
In this study, conventional MD simulations of the apo–Stp1 and Stp1–ATA complex models were run to explore the mechanism of ATA inhibition of the activity and biological function of Stp1. The RMSD, Rg, and RMSF values of each model indicated that the flap subdomain of the Stp1–ATA complex was substantially more flexible than that of the apo–Stp1 model. Conformational changes during 0–200 ns MD simulations suggested that the flap domain of the Stp1–ATA complex fluctuated between horizontal (open state) and vertical (closed state) positions. The analysis of the interaction modes and atomic distances subsequently showed that two H-bonds (N162···D198 and Q160···H13) formed near the active site, and the H-bond between D137 and N142 disappeared in the 400 ns conformation. Therefore, a mechanism of inhibition of Stp1 by ATA was proposed from the MD simulation perspective. Variants and phosphate enzyme activity assays verified that four residues (D137, N142, Q160, and R161) play critical roles in maintaining the dephosphorylation activity of Stp1 and that D137 and N142 also contribute to flap subdomain stabilization. Furthermore, the mutation studies confirmed the proposed ATA inhibitory mechanism. Initially, ATA acts as a competitive inhibitor, interacting with not only the residues in the Stp1 active site but also the R161 residue located in the flap subdomain. ATA subsequently caused a series of changes, including the stretching of Q160 over the active site, the interaction of N162 with D198, and the disappearance of the H-bond between D137 and N142. As a result, the conformation of the flap subdomain changed to the closed state, tightly wrapping ATA into the active site and making it a noncompetitive inhibitor.
In summary, this study indicated the inhibitory mechanism of ATA targeting Stp1 and provided a model for the binding interactions between ATA and Stp1 using MD simulations. Four residues were shown to play important roles in maintaining Stp1 activity. The present study provided useful information for the future discovery and design of allosteric inhibitors targeting flap subdomains.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by the National Key R & D Program of China (2016YFB0201700), the National Science and Technology Major Projects for “Major New Drugs Innovation and Development” (2018ZX09711003-003-005), and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDC01040100). The MD simulations were run on the TianHe-2 supercomputer at the National Supercomputing Center, Guangzhou, China.
Author contributions
K-qY conceived the project; K-qY and T-tL designed the research; T-tL performed MD simulations; TY performed mutation studies; M-nG performed sequence alignments; T-tL analyzed data; T-tL wrote the paper with help from TY and M-nG; K-qY, SY, K-xC, and H-lJ provided reagents and/or technical assistance. All authors reviewed the results and approved the manuscript.
Competing interests
The authors declare no competing financial interest.
Contributor Information
Song Yang, Email: jhzx.msm@gmail.com.
Kun-qian Yu, Email: yukunqian@simm.ac.cn.
Supplementary information
The online version of this article (10.1038/s41401-019-0216-x) contains supplementary material, which is available to authorized users.
References
- 1.Williams KJ, Bax RP. Challenges in developing new antibacterial drugs. Curr Opin Investig Drugs. 2009;10:157–63. [PubMed] [Google Scholar]
- 2.Thangamani S, Mohammad H, Abushahba MF, Sobreira TJ, Hedrick VE, Paul LN, et al. Antibacterial activity and mechanism of action of auranofin against multi-drug resistant bacterial pathogens. Sci Rep. 2016;6:22571. doi: 10.1038/srep22571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun D, Xu D, Yang C, Shahzad MB, Sun Z, Xia J, et al. An investigation of the antibacterial ability and cytotoxicity of a novel cu-bearing 317L stainless steel. Sci Rep. 2016;6:29244. doi: 10.1038/srep29244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker S. A return to the pre-antimicrobial era? Science. 2015;347:1064–66. doi: 10.1126/science.aaa2868. [DOI] [PubMed] [Google Scholar]
- 5.Xie R, Zhang XD, Zhao Q, Peng B, Zheng J. Analysis of global prevalence of antibiotic resistance in Acinetobacter baumannii infections disclosed a faster increase in OECD countries. Emerg Microbes Infect. 2018;7:31. doi: 10.1038/s41426-018-0038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woolhouse MEJ, Ward MJ. Sources of antimicrobial resistance. Science. 2013;341:1460–61. doi: 10.1126/science.1243444. [DOI] [PubMed] [Google Scholar]
- 7.Zhou G, Shi QS, Huang XM, Xie XB. The three bacterial lines of defense against antimicrobial agents. Int J Mol Sci. 2015;16:21711–33. doi: 10.3390/ijms160921711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di Gregorio S, Fernandez S, Cuirolo A, Verlaine O, Amoroso A, Mengin-Lecreulx D, et al. Different vancomycin-intermediate Staphylococcus aureus phenotypes selected from the same ST100-hVISA parental strain. Microb Drug Resist. 2017;23:44–50. doi: 10.1089/mdr.2016.0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Epand RM, Walker C, Epand RF, Magarvey NA. Molecular mechanisms of membrane targeting antibiotics. Biochim Biophys Acta. 2016;1858:980–7. doi: 10.1016/j.bbamem.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 10.Karageorgopoulos DE, Wang R, Yu XH, Falagas ME. Fosfomycin: evaluation of the published evidence on the emergence of antimicrobial resistance in Gram-negative pathogens. J Antimicrob Chemother. 2012;67:255–68. doi: 10.1093/jac/dkr466. [DOI] [PubMed] [Google Scholar]
- 11.Jayaraman R. Antibiotic resistance: an overview of mechanisms and a paradigm shift. Curr Sci. 2009;96:1475–84. [Google Scholar]
- 12.Clatworthy AE, Pierson E, Hung DT. Targeting virulence: a new paradigm for antimicrobial therapy. Nat Chem Biol. 2007;3:541–8. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 13.Garland M, Loscher S, Bogyo M. Chemical strategies to target bacterial virulence. Chem Rev. 2017;117:4422–61. doi: 10.1021/acs.chemrev.6b00676. [DOI] [PubMed] [Google Scholar]
- 14.Spaulding CN, Klein RD, Schreiber HLt, Janetka JW, Hultgren SJ. Precision antimicrobial therapeutics: the path of least resistance? NPJ Biofilms Micro. 2018;4:4. doi: 10.1038/s41522-018-0048-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fialova S, Slobodnikova L, Veizerova L, GranCai D. Lycopus europaeus: phenolic fingerprint, antioxidant activity and antimicrobial effect on clinical Staphylococcus aureus strains. Nat Prod Res. 2015;29:2271–4. doi: 10.1080/14786419.2015.1010086. [DOI] [PubMed] [Google Scholar]
- 16.Lehar SM, Pillow T, Xu M, Staben L, Kajihara KK, Vandlen R, et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature. 2015;527:323–28. doi: 10.1038/nature16057. [DOI] [PubMed] [Google Scholar]
- 17.Bahcall OG. Pathogen genetics: S. aureus multi-host tropism. Nat Rev Genet. 2015;16:194–94. [Google Scholar]
- 18.Liu Y, Feng J, Lu Q, Zhang X, Gao Y, Yan J, et al. MAE4, an eLtaS monoclonal antibody, blocks Staphylococcus aureus virulence. Sci Rep. 2015;5:17215. doi: 10.1038/srep17215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun F, Zhou L, Zhao BC, Deng X, Cho H, Yi C, et al. Targeting MgrA-mediated virulence regulation in Staphylococcus aureus. Chem Biol. 2011;18:1032–41. doi: 10.1016/j.chembiol.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sajid A, Arora G, Singhal A, Kalia VC, Singh Y. Protein phosphatases of pathogenic bacteria: role in physiology and virulence. Annu Rev Microbiol. 2015;69:527–47. doi: 10.1146/annurev-micro-020415-111342. [DOI] [PubMed] [Google Scholar]
- 21.Sun F, Ding Y, Ji Q, Liang Z, Deng X, Wong CC, et al. Protein cysteine phosphorylation of SarA/MgrA family transcriptional regulators mediates bacterial virulence and antibiotic resistance. Proc Natl Acad Sci USA. 2012;109:15461–6. doi: 10.1073/pnas.1205952109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cameron DR, Ward DV, Kostoulias X, Howden BP, Moellering RC, Jr, et al. Serine/threonine phosphatase Stp1 contributes to reduced susceptibility to vancomycin and virulence in Staphylococcus aureus. J Infect Dis. 2012;205:1677–87. doi: 10.1093/infdis/jis252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hao Q, Yin P, Li W, Wang L, Yan C, Lin Z, et al. The molecular basis of ABA-independent inhibition of PP2Cs by a subclass of PYL proteins. Mol Cell. 2011;42:662–72. doi: 10.1016/j.molcel.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 24.Almo SC, Bonanno JB, Sauder JM, Emtage S, Dilorenzo TP, Malashkevich V, et al. Structural genomics of protein phosphatases. J Struct Funct Genom. 2007;8:121–40. doi: 10.1007/s10969-007-9036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bellinzoni M, Wehenkel A, Shepard W, Alzari PM. Insights into the catalytic mechanism of PPM Ser/Thr phosphatases from the atomic resolution. Struct a Mycobact Enzym Struct. 2007;15:863–72. doi: 10.1016/j.str.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 26.Su J, Schlicker C, Forchhammer K. A third metal is required for catalytic activity of the signal-transducing protein phosphatase M tPphA. J Biol Chem. 2011;286:13481–88. doi: 10.1074/jbc.M109.036467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schlicker C, Fokina O, Kloft N, Grune T, Becker S, Sheldrick GM, et al. Structural analysis of the PP2C phosphatase tPphA from Thermosynechococcus elongatus: a flexible flap subdomain controls access to the catalytic site. J Mol Biol. 2008;376:570–81. doi: 10.1016/j.jmb.2007.11.097. [DOI] [PubMed] [Google Scholar]
- 28.Su J, Forchhammer K. Determinants for substrate specificity of the bacterial PP2C protein phosphatase tPphA from Thermosynechococcus elongatus. FEBS J. 2013;280:694–707. doi: 10.1111/j.1742-4658.2011.08466.x. [DOI] [PubMed] [Google Scholar]
- 29.Wehenkel A, Bellinzoni M, Schaeffer F, Villarino A, Alzari PM. Structural and binding studies of the three-metal center in two mycobacterial PPM Ser/Thr protein phosphatases. J Mol Biol. 2007;374:890–98. doi: 10.1016/j.jmb.2007.09.076. [DOI] [PubMed] [Google Scholar]
- 30.Pullen KE, Ng HL, Sung PY, Good MC, Smith SM, Alber T. An alternate conformation and a third metal in PstP/Ppp, the M. tuberculosis PP2C-family Ser/Thr protein phosphatase. Structure. 2004;12:1947–54. doi: 10.1016/j.str.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 31.Levdikov VM, Blagova EV, Rawlings AE, Jameson K, Tunaley J, Hart DJ, et al. Structure of the phosphatase domain of the cell fate determinant SpoIIE from Bacillus subtilis. J Mol Biol. 2012;415:343–58. doi: 10.1016/j.jmb.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dupeux F, Antoni R, Betz K, Santiago J, Gonzalez-Guzman M, Rodriguez L, et al. Modulation of abscisic acid signaling in vivo by an engineered receptor-insensitive protein phosphatase type 2C allele. Plant Physiol. 2011;156:106–16. doi: 10.1104/pp.110.170894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng W, Liang Y, Zhao H, Zhang J, Li Z. 5, 5 ′-Methylenedisalicylic acid (MDSA) modulates SarA/MgrA phosphorylation by targeting Ser/Thr phosphatase Stp1. Chembiochem. 2015;16:1035–40. doi: 10.1002/cbic.201500003. [DOI] [PubMed] [Google Scholar]
- 34.Zheng Weihao, Cai Xiaodan, Xie Mingsheng, Liang Yujie, Wang Tao, Li Zigang. Structure-Based Identification of a Potent Inhibitor Targeting Stp1-Mediated Virulence Regulation in Staphylococcus aureus. Cell Chemical Biology. 2016;23(8):1002–1013. doi: 10.1016/j.chembiol.2016.06.014. [DOI] [PubMed] [Google Scholar]
- 35.Sliwoski G, Kothiwale S, Meiler J, Lowe EW., Jr. Computational methods in drug discovery. Pharmacol Rev. 2014;66:334–95. doi: 10.1124/pr.112.007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dalkas GA, Vlachakis D, Tsagkrasoulis D, Kastania A, Kossida S. State-of-the-art technology in modern computer-aided drug design. Brief Bioinform. 2013;14:745–52. doi: 10.1093/bib/bbs063. [DOI] [PubMed] [Google Scholar]
- 37.Peréz-Sánchez Horacio, Fassihi Afshin, Cecilia José M., Ali Hesham H., Cannataro Mario. Bioinformatics and Biomedical Engineering. Cham: Springer International Publishing; 2015. Applications of High Performance Computing in Bioinformatics, Computational Biology and Computational Chemistry; pp. 527–541. [Google Scholar]
- 38.Liu T, Lu D, Zhang H, Zheng M, Yang H, Xu Y, et al. Applying high-performance computing in drug discovery and molecular simulation. Nat Sci Rev. 2016;3:49–63. doi: 10.1093/nsr/nww003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minini L, Alvarez G, Gonzalez M, Cerecetto H, Merlino A. Molecular docking and molecular dynamics simulation studies of Trypanosoma cruzi triosephosphate isomerase inhibitors. Insights into the inhibition mechanism and selectivity. J Mol Graph Model. 2015;58:40–9. doi: 10.1016/j.jmgm.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 40.Yadava U, Gupta H, Roychoudhury M. Stabilization of microtubules by taxane diterpenoids: insight from docking and MD simulations. J Biol Phys. 2015;41:117–33. doi: 10.1007/s10867-014-9369-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou S, Fang D, Tan S, Lin W, Wu W, Zheng K. Investigating the binding mechanism of novel 6-aminonicotinate-based antagonists with P2Y12 by 3D-QSAR, docking and molecular dynamics simulations. J Biomol Struct Dyn. 2017;35:2938–65. doi: 10.1080/07391102.2016.1237381. [DOI] [PubMed] [Google Scholar]
- 42.Sledz P, Caflisch A. Protein structure-based drug design: from docking to molecular dynamics. Curr Opin Struct Biol. 2018;48:93–102. doi: 10.1016/j.sbi.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 43.Cheng P, Li J, Wang J, Zhang X, Zhai H. Investigations of FAK inhibitors: a combination of 3D-QSAR, docking, and molecular dynamics simulations studies. J Biomol Struct Dyn. 2018;36:1529–49. doi: 10.1080/07391102.2017.1329095. [DOI] [PubMed] [Google Scholar]
- 44.Yuan S, Chan HCS, Hu Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip Rev: Comput Mol Sci. 2017;7:e1298. [Google Scholar]
- 45.Sievers F, Higgins DG. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018;27:135–45. doi: 10.1002/pro.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–W24. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. 2000;28:235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–59. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 50.Release S. 1: Maestro, version 10.1. New York, NY: Schrödinger, LLC; 2015.
- 51.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J Med Chem. 2006;49:6177–96. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 52.Søndergaard CR, Olsson MHM, Rostkowski M, Jensen JH. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J Chem Theory Comput. 2011;7:2284–95. doi: 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- 53.Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput. 2011;7:525–37. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 54.Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model. 2006;25:247–60. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 55.Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. Epik: a software program for pK(a) prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des. 2007;21:681–91. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- 56.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–35. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 57.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Case D, Babin V, Berryman J, Betz R, Cai Q, Cerutti D, et al. The FF14SB force field. Amber. 2014;14:29–31. [Google Scholar]
- 59.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–74. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 60.Sousa da Silva AW, Vranken WF. ACPYPE - AnteChamber PYthon Parser interfacE. BMC Res Notes. 2012;5:367. doi: 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 62.Panagiotopoulos AZ. Direct determination of phase coexistence properties of fluids by Monte Carlo simulation in a new ensemble. Mol Phys. 1987;61:813–26. doi: 10.1080/00268978700101491. [DOI] [Google Scholar]
- 63.Berendsen HJ, Jv Postma, van Gunsteren WF, DiNola A, Haak J. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–90. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 64.Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys. 1981;52:7182–90. doi: 10.1063/1.328693. [DOI] [Google Scholar]
- 65.Si Y, Yuan Y, Wang Y, Gao J, Hu Y, Feng S, et al. Structural and biochemical characterization of a cyanobacterial PP2C phosphatase reveals insights into catalytic mechanism and substrate recognition. Catalysts. 2016;6:60. doi: 10.3390/catal6050060. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.