

Abstract
Inorganic arsenic is an environmental carcinogen that poses a major global public health risk. A high percentage of drinking water from wells in the U.S. contains higher-than-normal levels of arsenic, suggesting an increased risk of arsenic-induced deleterious effects. In addition to primary preventive measures, therapeutic strategies need to effectively address and integrate multiple molecular mechanisms underlying arsenic-induced carcinogenesis. We previously showed that the loss of miR-199a-5p in arsenic-transformed cells is pivotal to promote arsenic-induced angiogenesis and tumor growth in lung epithelial cells. In this study, we further showed that subacute or chronic exposure to arsenic diminished miR-199a-5p levels largely due to DNA methylation, which was achieved by increased DNA methyltransferase-1 (DNMT1) activity, mediated by the formation of specific protein 1 (Sp1)/DNMT1 complex. In addition to the DNA hypermethylation, arsenic exposure also repressed miR-199a transcription through a transcriptional repressor Sp1. We further identified an association between miR-199a-5p repression and the arsenic-mediated energy metabolic shift, as reflected by mitochondria defects and a switch to glycolysis, in which a glycolytic enzyme pyruvate kinase 2 (PKM2) was a functional target of miR-199a-5p. Taken together, the repression of miR-199a-5p through both Sp1-dependent DNA methylation and Sp1 transcriptional repression promotes an arsenic-mediated metabolic shift from mitochondria respiration to aerobic glycolysis via PKM2.
Keywords: arsenic, miR-199a-5p, DNA methylation, Sp1, glycolysis, PKM2
Introduction
Inorganic arsenic and arsenic compounds were identified as human carcinogens associated with the increased risks of skin, lung, and bladder cancers (1980). Exposure to arsenic remains a major public concern in the U.S. and around the world. Arsenic occurs naturally in the form of various compounds in the earth’s crust that can be released to the environment. A recent investigation by U.S. Geological Survey and Centers for Diseases and Prevention (CDC) estimated 2.1 million people in US drinking water from wells that contain high levels of arsenic (greater than 10 μg/L) (Ayotte et al. 2017).
The molecular etiology involved in arsenic-induced diverse toxic effects and cell malignant transforming activity remains to be elucidated. Unlike many other typical chemical carcinogens, arsenic does not form DNA adducts nor induce point mutations, and fail to initiate carcinogenesis in experimental animal models (Jacobson-Kram and Montalbano 1985; Ren et al. 2011). One of the common routes of arsenic exposure to humans is through drinking water contaminated with inorganic pentavalent arsenic (iAs5+) which can rapidly convert to trivalent arsenic (iAs3+). The latter then metabolizes into monomethylated and dimethylated arsenicals (MMA, DMA) in human bodies (Drobna et al. 2009). Given the arsenic biotransformation and DNA methylation use the same methyl group donor S-adenosyl methionine (SAM), it is plausible to speculate that epigenetic regulations, in particular DNA methylation, plays a role in activating oncogenic signaling pathways, resulting in arsenic carcinogenicity. To date, arsenic exposure related global DNA hypomethlaytion and/or gene-specific DNA hypermethylation have been extensively reported based on population-based investigations and in vitro studies (Bailey and Fry 2014; Brocato and Costa 2013; Roy et al. 2015). For example, the earliest findings in the area suggested positive associations between arsenic exposure and hypermethylation of tumor suppressor genes p53 and p16 (Hossain et al. 2012; Intarasunanont et al. 2012). More importantly, studies involving human subjects have observed that low to moderate levels of arsenic exposure sufficiently alter DNA methylation signatures (Bailey and Fry 2014). However, the functional consequences of epigenetic changes are not well-established.
In addition to DNA methylation, other epigenetic changes such as histone modification and dysregulation of miRNAs have also been linked with arsenic-induced cell malignant transformation (Cardoso et al. 2018; Chervona et al. 2012; Salnikow and Zhitkovich 2008; Suzuki and Nohara 2013), indicating a possible interplay between different layers of epigenetic control. We previously conducted a miRNA microarray analysis using a well-characterized cell model, arsenic-transform bronchial epithelial cell Beas2B (desigated as AsT) and its passage-matched parental control Beas-2B cells (designated as B2B) (Carpenter et al. 2011; Stueckle et al. 2012). With the microarray result and subsequent RT-PCR assays, we identified that the expression level of miR-199a-5p was dramatically downregulated in transformed AsT cells. We further showed that the negative regulation of miR-199a-5p on proangiogenic genes hypoxia inducible factor 1 α (HIF-1 α) and cyclooxygenase-2 (COX-2) was released in AsT cells resulting in enhanced tumor angiogenesis and tumor growth (He et al. 2014). However, the question of how arsenic regulates miR-199a-5p expression has yet to be answered.
In this study, we utilized the cells that had been exposed to arsenic for weeks and were in the intermediate stages of malignant transformation, and earlier established AsT cells to investigate: 1) the miR-199a-5p expression levels in the intermediate stages of arsenic-induced transformation; 2) the mechanism of arsenic regulation of miR-199a-5p; and 3) the function of miR-199a-5p DNA methylation upon arsenic exposure.
Materials and Methods
Cell Culture, reagents, and antibodies
Human bronchial epithelial Beas2B were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS). Human lung fibroblast cells (HFL1) were purchased from ATCC and were cultured in F-12K medium (ATCC) with 10% FBS. Sodium arsenic (NaAsO2) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Inhibitors RG-108 and 5-azacytidine were purchased from Selleckchem (Boston, MA, USA). DNMT1 and SP1 antibodies using for Western-blots and/or Immunoprecipitation assay were purchased from Cell Signaling Technology (Danvers, MA) and PKM2 antibody from Santa Cruz Biotechology (Dallas, TX). Primary antibodies used for Western analysis were diluted 1:1000 in 5% bovine serum albumin as a working concentration.
RT-qPCR:
Total RNAs were extracted by Trizol (ThermoFisher, Waltham, MA, USA). Two-step Taqman RT-qPCR assay was conducted to assess miRNA levels using Taqman miRNA reverse transcription kit and Taqman universal PCR master mix (Applied Biosystem, Austin, TX, USA) in accordance with the manufacturer’s instructions. Normalization was performed using U6 RNA level.
DNA methylation analysis:
Genomic DNA was extracted using a Quick-gDNA kit (Zymo Research, Irvine, CA, USA). Total 1 mg genomic DNA was treated with sodium bisulphite using the EpiTect Kit (Qiagen). Bisulphite-treated DNA was then amplified with methylation specific primers for CpG islands surrounding miR-199a-1 genomic locations. Methylation-specific-PCR was performed to determine the methylation status of miR-199a-1 gene with arsenic treatments. The methylation-specific PCR primers used for miR-199a-1 amplification were designed by MethPrimer (Li and Dahiya 2002) as follows:
Methylated F: 5’- ATTTCGTCGAGAAATTAGTGGTC −3’
Methylated R: 5’- AATCCTAAAAAATAAAAAAAACGAA −3’
Unmethylated F: 5’-ATTTTGTTGAGAAATTAGTGGTTGT −3’
Unmethylated R: 5’- AATCCTAAAAAATAAAAAAAACAAA −3’
Methylated DNA immunoprecipitation (MeDIP assay) was performed using a MeDIP ChIP kit (Abeam, Cambridge, MA, USA) following the manufacturer’s instruction. Briefly, Genomic DNA was extracted in Beas2B cells treated with or without arsenic for four weeks. The genomic DNA were sonicated to shear to 500bp fragments and quantified by a spectrophotometer. The sheared genomic DNA fragments were incubated with the 5-Methylcytosine (5-mC) (D3S2Z) Rabbit mAb overnight at 4°C with rotation. Then, the complex was captured by ChIP-Grade Protein G Magnetic Beads with 2 hour-incubation at room temperature. The DNA fragments were eluted from the beads. After purification, the immunoprecipitated DNA was quantified using a SYBR Green qPCR kit (Applied Biosystem, Foster City, CA, USA). The primers used were:
Forward: 5’-GAGCCCAGAAGCCACGAT-3’
Reverse: 5’-CAGCCAAGGAAACCACCAC-3’
The enrichment of miR-199a-1 was presented as the percent of input with the formula: percent of INPUT = 10% × 2(C[T] 10%Input Sample − C[T] IP Sample). DNMT1 activity assay was analyzed using a DNMT1 activity screening assay core kit (EpiGentek, Farmingdale,NY, USA). Briefly, nuclear extracts from cells were prepared and added into the wells coated with cytocine-rich DNA substrate for two hours. The amount of methylated DNA can be colorimetrically detected by a spectrophotometer using an antibody against 5-methylcytosine.
Western-blots and immunoprecipitation assay
Cells were lysed in M-PER mammalian protein extract buffer supplemented with protease and phosphatase inihibitors (ThermoFisher Scientific) for 30 min, and followed by centrifugation at 12,000 rpm for 10 min. The protein concentrations were assayed and subjected to Western-blots analysis. The proteins of interest were visualized by the Pierce™ ECL Western Blotting Substrate (ThermoFisher Scientific) with the ImageQuant LAS 4000 (GE Healthcare, Chicago, IL, USA). Nuclear fraction of cells was prepared using a kit NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (ThermoFisher Scientific). Immunoprecipitation assay was conducted using the Pierce Classic IP Kit (ThermoFisher Scientific) following the manufacture’s instruction. The Sp1 antibody was used to incubate with total proteins overnight. Rabbit IgG was used as a negative control. The immune complex was captured by Protein A/G agarose beads. The elution of immune complex was subject to Western-blotting.
Sp1 luciferase reporter assay and PKM2 3’UTR reporter assay
MiR-199a-1 or miR-199a-2 promoter region containing Sp1 binding sequences was inserted into a pGL3-basic vector (Promega, Madison, WI, USA) between the restriction enzyme sites Sac-I and Xho-I (for miR-199a-1), or Kpn-1 and Xho-I (for miR-199a-2). The primers used for the generation of the linear promoter regions were: miR-199a-1 promoter Forward: 5’-CGAGCTCTTATCTGGGCTCCATCTTTG-3’ miR-199a-1 promoter Reverse: 5’-CCGCTCGAGGCTGGCATCACCTCAACC-3’ miR-199a-2 promoter Forward: 5’-GGGGTACCTTGGTATCCACATATAATACAAGTA-3’ miR-199a-2 promoter Reversed-CGCTCGAGGGTAGATGAAATAGTAAACAGAAA-3’ The PKM2 3’UTR luciferase reporter constructs containing the wild type and mutant binding sites of miR-199a-5p were amplified using PCR method. The PCR products were cloned into the pMiR-Report luciferase vector (Ambion), immediately downstream of the luciferase gene. The mutant 3’UTR constructs were made by introducing 4 mismatch mutations into the putative seed regions of PKM2. Cells were co-transfected with the reporter constructs and miR-199a-5p mimic for 48 h and then lysed with the reporter lysis buffer (Promega, WI, USA). Firefly and Renilla luciferase activities were measured 24h after transfection using a dual luciferase assay kit (Promega, WI, USA).
JC-1 staining for mitochondria membrane potential
JC-1 dye (ThermoFisher Scientific) was used to evaluate the mitochondrial membrane potential. JC-1 staining solution (solved in DMSO) was added to each well at the final concentration of 5 μg/ml, and incubated in a 5 % CO2 incubator at 37°C for 20 min protected from the light. After washing twice with a buffer solution, cells were observed and photographed by a fluorescence microscope (Zeiss, Germany).
Glycolysis analysis:
For the L-lactate measurement, 5 million cells were seeded in 6-well plates. After the arsenic treatments, the cell media was collected for the determination of extracellular L-lactate production while the cells were homogenized in the assay buffer and centrifuged. The insoluble fraction was used to measure intracellular L-lactate using a lactate colorimetric assay kit (Biovision incorporated, Milpitas, CA, USA) according to the manufacture’s instruction. The extracellular acidification rate (ECAR) was measured using a Seahorse XFp analyzer (Agilent Technology, Santa Clara, CA, USA). In brief, the sensor cartridge was hydrated with the XF calibrant and incubated at 37°C without CO2 overnight the day prior to the assay. The cells were seeded in XFp cell culture miniplates at the density of 20000 cells /well. The outer wells weren’t seeded with cells but only growth medium to minimize the edge effect. On the assay day, the assay medium was prepared and adjusted pH to 7.4. Cells were washed with the warm assay medium and then put in a non-CO2 incubator at 37°C for 1 h. The sensor cartridge injection ports were loaded with glucose (final concentration 10 mM), oligomycin (final concentration 1 μM), and 2-DG (final concentration 50 mM), and then the cartridge was placed onto the XFp Analyzer for calibration. The XFp cell plate was then put into the analyzer and assayed. The values were normalized by the protein concentration for each well. The XF Report Generator was used for the data analysis.
Statistical Analysis
All data points represent means and standard deviations (error bar) were obtained from at least three independent experiments. Student’s t test or one-way ANOVA was used for comparison between groups. All results were analyzed by SPSS for Windows, version 23. P <0.05 was considered statistically different.
Results
miR-199a-5p repression is due to arsenic-induced DNA hypermethylation
We previously reported that miR-199a-5p expression was 100-fold lower in arsenic-transformed cell AsT than its parental control B2B (He et al. 2014). In this study, BEAS-2B cells were continuously cultured in the presence of sodium arsenic for up to 16 weeks. We observed that arsenic decreased the miR-199a-5p level starting at 2 weeks’ exposure (Fig.1A). We also treated lung fibroblast cells HFL with arsenic for 2 weeks and obtained similar results (Fig.1B), suggesting that subacute (24 hours to a month) and chronic exposure (more than a month) to arsenic are capable of reducing miR-199a-5p levels.
Fig.1. miR-199a-5p is downregulated in lung cells with chronic exposure to arsenic.
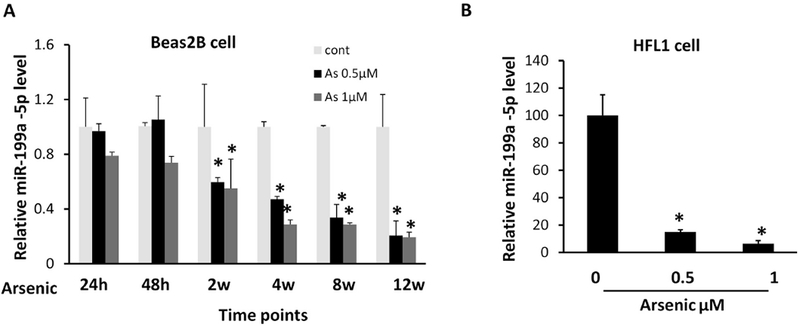
(A) BEAS-2B cells were treated with sodium arsenic for 24 h, 48 h, 2 weeks, 4 weeks, 8 weeks, and 12 weeks at indicated doses. Taqman-qRT assay was used to determine miR-199a-5p expression level. (B) HFL cells were treated with arsenic for 2 weeks followed by miR-199a-5p determination. Data are presented as mean ± SD, *P < 0.05, compared with control cells.
Our earlier study indicated that endogenous reactive oxygen species (ROS) inhibit miR-199a-5p expression by DNMT1-mediated DNA hypermethylation in ovarian cancer cells (He et al. 2012). We speculate whether the loss of miR-199a-5p resulted from arsenic-mediated the gene hypermethylation. The mature miR-199a-5p derives from two genes, miR-199a-1 and miR-199a-2, located in chromosome 19 and 1, respectively. Using UCSC genome browser, we found CpG islands only in the miR-199a-1 gene promoter region but not in the miR-199a-2 gene. We then performed methylation-specific PCR (MSP) assay to analyze the methylation status of the miR-199a-1 gene in arsenic-treated cells and control cells. The MSP result showed that arsenic treatment for 4 weeks significantly increased the hypermethylation level of the miR-199a-1 gene (Fig.2A, left panel). Similarly, the transformed AsT cells displayed a high level of methylated miR-199a-1 relative to that in parental control B2B cells (Fig.2A, right panel).
Fig.2. miR-199a-5p repression is due to arsenic-induced DNA hypermethylation.
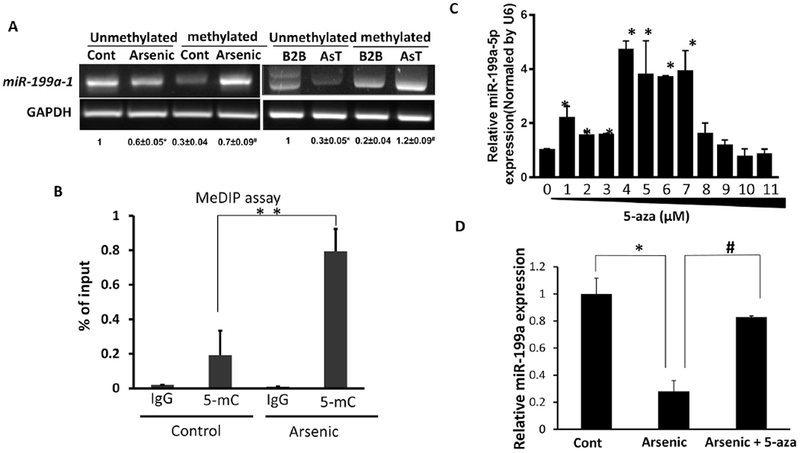
(A) Total DNA was extracted and then treated with sodium bisulphite. The hypermethylation level of miR-199a-1 gene promoter was determined by methylation-specific PCR assay. Left: BEAS-2B cells were treated with or without sodium arsenic (0.5 μM) for 4 weeks. Right: B2B cells and AsT cells. The quantitative densitometry was done by ImageJ. All of values were normalized with the unmethylated control Beas2B and unmethylated B2B, respectively. *P<0.05 compared with the unmethylated control or B2B; #P<0.05, compared with the methylated control or B2B. (B) Genomic DNA was extracted from Beas2B cells treated with arsenic (4 weeks, 0.5 μM) and control cells. MeDIP assay was conducted as described in Materials. The amount of DNA pulled down by the 5-mC antibody or negative control IgG antibody was determined by qPCR using specific primers for determination of miR-199a-1 levels. (C) AsT cells were treated with 5-aza for 5 d at the indicated doses. miR-199a-5p level was determined by Taqman RT-PCR assay. (D) Arsenic-treated Beas2B cells (four weeks, 0.5 μM) were treated with or without 5-aza (5 μM) for consecutive 5 d. miR-199a-5p level was determined by Taqman RT-PCR assay. Data are presented as mean ± SD, * P<0.05, compared with control cells.
To determine whether the arsenic treatment suppresses miR-199a-5p expression through DNA methylation, we conducted methylated DNA immunoprecipation (MeDIP) assay. The amount of methylated DNA pulled down by an antibody specific to 5-methylcytosine (5-mC) dramatically boosted in arsenic-treated cells (4 weeks), which was in line with MSP results (Fig.2B). The inhibition of methylation by an inhibitor 5-azacytidine effectively restored miR-199a-5p levels in AsT cells at doses ranging between 1 μM to 7 μM (Fig.2C). Demethylation treatment by 5-aza partially restored the miR-199a-5p level in BEAS-2B cells exposed to arsenic for 4 weeks (Fig.2D). In addition to DNA methylation, other epigenetic modifications induced by heavy metals have been reported (Arita and Costa 2009; Roy et al. 2015). We examined whether histone modification contributes to the loss of miR-199a-5p using a histone deacetylase inhibitor trichostatin A. However, the inhibitor could not rescue miR-199a-5p repression in arsenic-treated cells (Suppl.Fig.1). Collectively, these results suggest that DNA methylation is the underlying cause of arsenic-mediated miR-199a-5p repression.
Chronic exposure to arsenic increases DNMT1 activity
DNA methyltransferases (DNMTs) are the major enzymes to catalyze the transfer of methyl groups to the carbon atom at the position of carbon 5 of cytosines. We found that chronic arsenic exposure did not affect total DNMT1, the most abundant form of DNMTs in cells (Fig.3A), nor did it affect DNMT3a and DNMT3b. Notably, the arsenic treatments did markedly increase the expression levels of nuclear DNMT1 (Fig.3B). This result was supported by the observation of increased DNMT1 activities with chronic exposure to arsenic (Fig.3C). We then examined whether DNMT1 inhibition could reverse arsenic-mediated miR-199a repression. The cells with the 4-week arsenic exposure were treated with a DNMT1 inhibitor RG-108 for 24 h. DNMT1 inhibition was able to rescue miR-199a-5p levels, suggesting the involvement of DNMT1 in the loss of the miRNA (Fig.3D).
Fig.3. Increased DNMT1 activity with chronic exposure to arsenic.
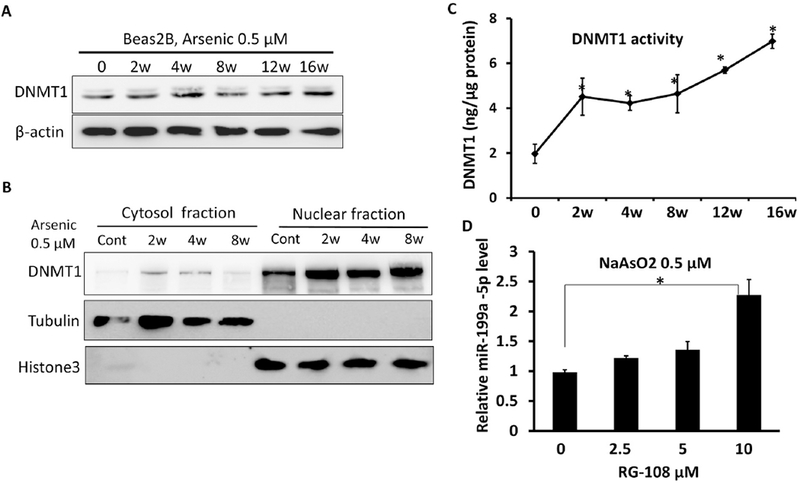
BEAS-2B cells were continuously treated with sodium arsenic (0.5 μM) for different time points as indicated: (A) Total DNMT1 protein levels were determined by Western-blots. (B) DNMT1 protein levels were determined in cytosol and nuclear fractions, respectively. (C) DNMT1 enzyme activities in arsenic-treated Beas2B cells (0.5 μM) were determined by a colorimetric kit. (D) BEAS-2B cells exposed to arsenic for 4 weeks were treated with RG-108 at the indicated doses for 24 h. miR-199a-5p was measured by Taqman RT-qPCR. Data are presented as mean ± SD, * P<0.05, compared with control cells.
Both Spl-dependent DNA methylation and Sp1 transcriptional repression contribute to the loss of miR-199a-5p
It is unclear how arsenic exposure induces DNMT1 activation and silences the miR-199 gene. Given DNMT1 functions globally for both de novo and maintenance of DNA methylation, we hypothesized that DNMT1 may form a complex with specific transcription factors and the promoter region of miR-199a for gene-specific methylation. Kishikawa et al. reported that the Specific protein (Sp) family of transcription factors Sp1 and Sp3 directly bind to a cis-element in the promoter region of DNMT1 gene that is essential for the gene expression (Kishikawa et al. 2002; Liu et al. 2008). We showed that nuclear Sp1 protein levels accumulated in arsenic-exposed cells and arsenic-transformed AsT cells (Fig.4A). Immunoprecipitation assay revealed a direct interaction between DNMT1 Sp1 in BEAS-2B cells (Fig.4B). Overexpression of Sp1 raised DNMT1 activity and augmented the miR-199a-1 gene methylation (Suppl.Fig.2, Fig.4C, 4D), suggesting that arsenic-induced miR-199a-1 gene methylation is mediated by Sp1/DNMT1. Notably, we found that both miR-199a-1 and miR-199a-2 genes harbor multiple Sp1 binding sites (Fig.4E).
Fig.4. Both Sp1 -dependent DNA methylation and SP1 transcriptional repression contribute to the loss of miR-199a-5p.
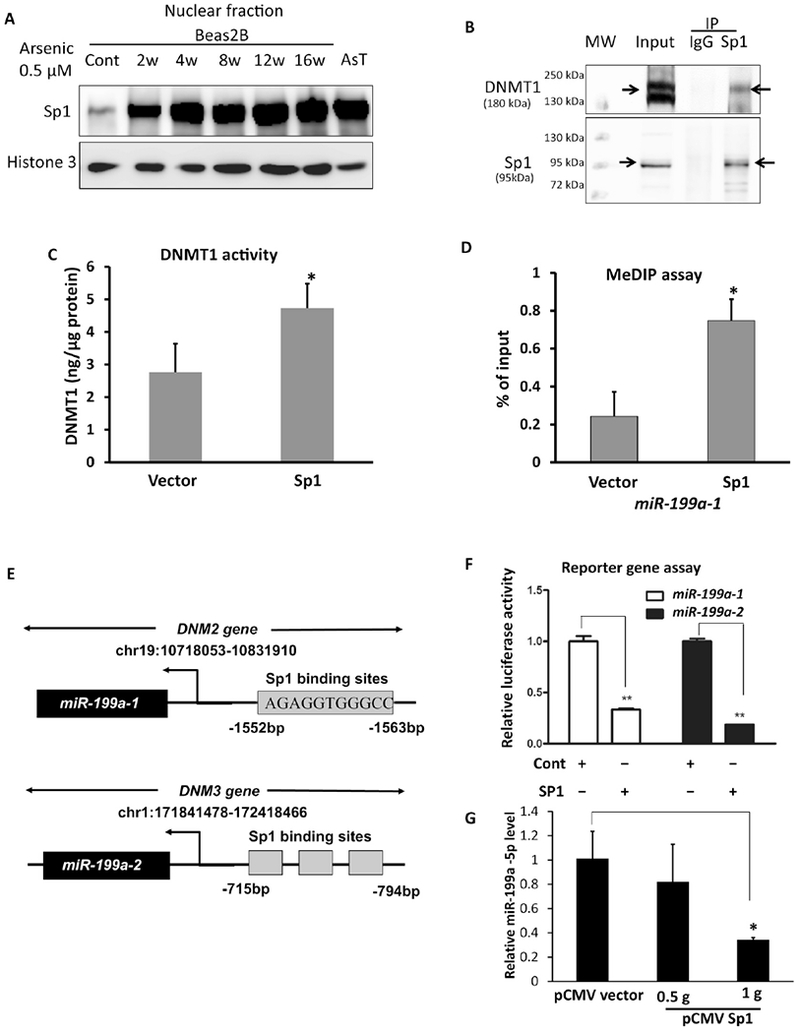
(A) Nuclear Sp1 protein levels in BEAS-2B cells exposed to arsenic were determined by Western-blots. (B) Beas2B cell lysate was prepared for IP assay. Sp1 antibody was used to pull down the immune complex and IgG antibody was used as a negative control. The elution of immune complex and the whole cell lysate (input) were subject to the Western-blots using antibodies against DNMT1 and Sp1. (C) DNMT1 enzyme activities were measured in BEAS-2B cells transiently transfected with a Sp1 plasmid or a vector control. (D) MeDIP assay was performed in BEAS-2B cells transfected with Sp1 plasmid or vector control. miR-199a-1 level was determined by PCR. (E) Sequence alignments of miR-199a-1 within the host gene DNM2 and miR-199a-2 within the host gene DNM3 with Sp1 binding sites for the generation of luciferase reporter constructs. (F) miR-199a genes luciferase activities were determined in BEAS-2B cells cotransfected with Sp1 plasmid, luciferase reporter constructs and a β-gal plasmid for 48 h. (G) BEAS-2B cells were transfected with a pCMV-SP1 plasmid or a pCMV vector, respectively. Total RNA was collected 72 h after the transfection. miR-199a-5p level was determined by RT-qPCR. Data are presented as mean ± SD, * P<.05, compared with control cells.
To determine whether Sp1 can directly regulate miR-199a transcription, we made luciferase reporter constructs containing the potential binding sites of Sp1 in the upstream promoters of miR-199-1 gene or miR-199a-2 gene. We showed that forced expression of Sp1 lowered the luciferase activity, an indicator of miR-199a gene transcription activity (Fig.4F) and of mature miR-199a-5p expression levels (Fig.4G). These data provide evidence that Sp1 is a transcriptional repressor of miR-199a genes. Taken together, both Sp1-dependent DNMT1 activation and Sp1-repressive transcriptional activity to miR-199a genes contribute to the loss of miR-199a-5p levels with arsenic exposure.
Loss of miR-199a-5p associates with an arsenic-mediated energy metabolic shift
Studies have shown that arsenic exposure leads to a cell metabolic shift from mitochondria respiration to aerobic glycolysis (Li et al. 2015; Luo et al. 2016; Zhao et al. 2013), and that miR-199a-5p regulates glycolysis and lactate production in cancer cells by targeting HuR, HIF, or PKM2 (Zhang et al. 2015). We wanted to find out whether the loss of miR-199a-5p contributes to the arsenic-mediated mitochondria dysfunction and aerobic glycolysis. The maintenance of mitochondria membrane potential (ΔΨm) is very important for the respiratory chain to generate ATP (Joshi and Bakowska 2011). We stained cells with a JC-1 dye that fluoresces red when the membrane potential is high and green when it is low. A decreased ratio of red/green fluorescence intensity through JC-1 dye staining indicated mitochondrial depolarization in cells exposed to arsenic for 4 weeks (Fig.5A), suggesting arsenic-mediated mitochondrial dysfunction. Zhao et al. observed the accumulation of lactate and elevated extracellular acidification rate (ECAR) upon chronic arsenic exposure (1 week to 4 weeks) in several cell types (Zhao et al. 2013). We obtained similar results in BEAS-2B cells exposed to arsenic for relatively longer (up to 8 weeks) (Suppl. Fig.3). We also observed a significant increase in extracellular and intracellular lactate production in arsenic-transformed AsT cells relative to their parental control B2B cells (Fig.5B).
Fig.5. Loss of miR-199a associates with arsenic-mediated energy metabolic shift.
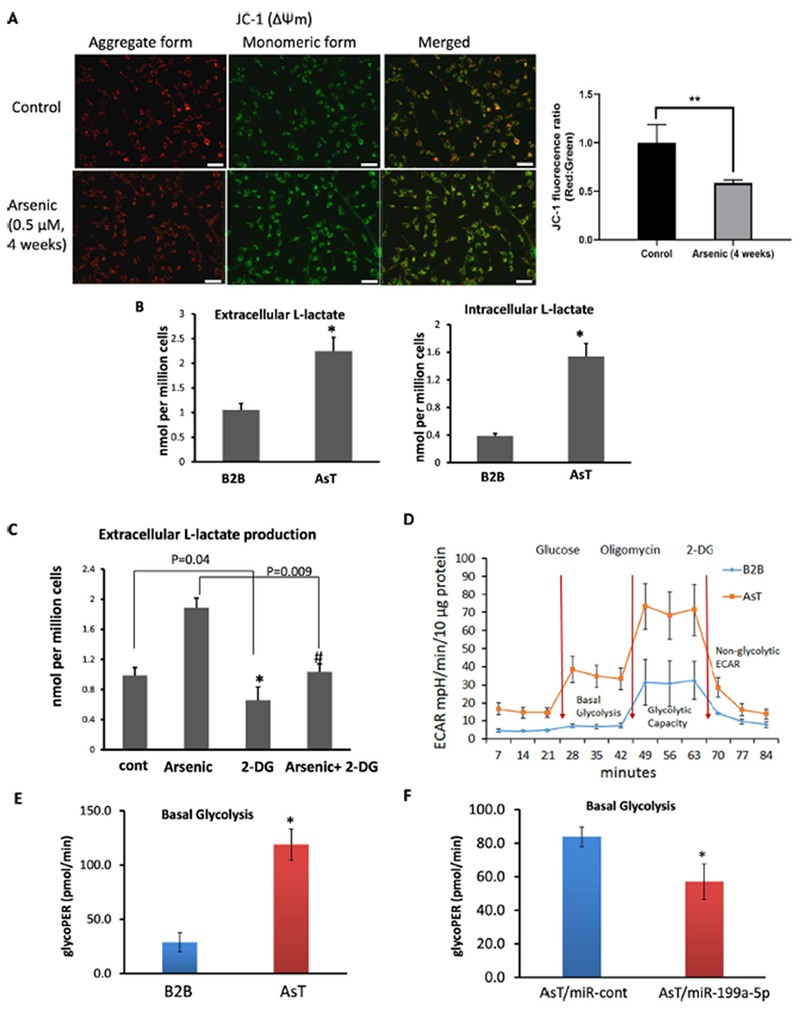
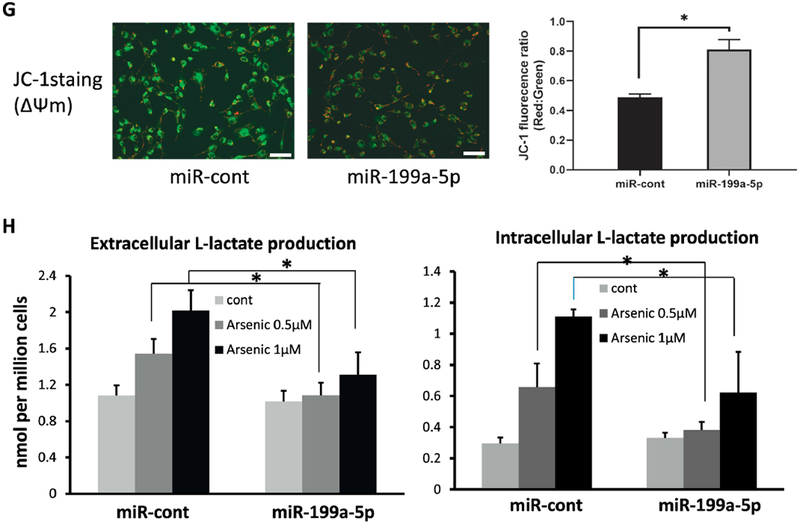
(A) Mitochondria membrane potential was evaluated by JC-1 dye in BEAS-2B cells exposed to sodium arsenic for 4 weeks (0.5 μM) and control cells. Representative images by fluorescence microscope were shown (left). Red fluorescence represents J-aggregates, indicating high mitochondrial membrane potential (ΔΨm). Green fluorescence represents monomers, indicating impaired mitochondrial membrane potential (ΔΨm). Scale bar= 100 μm. Quantification analysis for fluorescence was shown (right) (B) Extracelluar and intracellular Lactate productions were measured in culture medium or cells, respectively in AsT cells and B2B cells. (C) BEAS-2B cells exposed to arsenic for 4 weeks (0.5 μM) were treated with 2-DG for 24 h at 2 mM. Extracellular L-Lactate level was measured. (D, E) Extracellular acidification rate (ECAR) was determined by Seahorse assay in AsT cells and B2B cells. The basal glycolysis results were generated by the XF Report Generator based on the Seahorse glycolysis stress assay. (F) ECAR was determined by Seahorse assay in AsT cells overexpressing miR-199a-5p or miR-control. (G) AsT cells were transfected with miR-199a-5p mimic or scrambled control miR-cont for 96 h. Cells were stained with JC-1 dye. Representative images showed the merged red fluorescence (aggregate form) and green fluorescence (monomeric form). Scale bar: 100 μm. (H) BEAS-2B cells exposed to arsenic at the doses of 0.5 or 1μM for 4 weeks were transfected with miR-199a-5p mimic or miR-cont for 72 h. Extracellular and intracellular L-Lactate level was measured in culture medium or cell lysates. Data are presented as mean±SD, * P<0.05, compared with control cells.
To determine whether the increased lactate production is due to glycolysis, the arsenic-treated BEAS-2B cells were given a glycolysis inhibitor 2-DG for 24 h. The 2-DG reversed the elevation of lactate production, an indicator of glycolysis-dependent process (Fig.5C). Next, the ECAR associated with the conversion of glucose to pyruvate and lactate, was measured in cells stimulated with glucose, oligomycin, and 2-DG sequentially by a Seahorse glycolysis analyzer. AsT cells had much higher levels of basal glycolysis and glycolytic capacity (Fig. 5D, E). To determine the role of miR-199a-5p in the arsenic-induced metabolic shift, we transfected miR-199a-5p or a scrambled control miR-cont in AsT cells (Suppl. Fig.4). We then examined the ECAR, mitochondria membrane potential, and lactate production as well. Overexpression of miR-199a-5p in AsT cells decreased the basal glycolysis (Fig. 5F), raised the ratio of red/green fluorescence in arsenic-exposed cells (Fig.5G), and reduced arsenic-induced lactate accumulation (Fig.5H). All of these findings indicate that miR-199a-5p is highly involved in arsenic-induced energy metabolic shift.
PKM2 is the functional target of miR-199a in arsenic-induced energy metabolic shift
Pyruvate kinase M2 (PKM2) is a key enzyme for pyruvate and ATP production in the glycolysis pathway (Jurica et al. 1998). Zhang et a I. showed that the downregulation of miR-199a enhanced glycolysis by directly binding PKM2 in hepatocellular carcinomas (Zhang et al. 2015) (Fig.6A). Indeed, overexpression of miR-199a-5p in AsT cells markedly reduced PKM2 protein levels whereas the inhibition of miR-199a in BEAS-2B cells rescued PKM2 expression (Fig.6B). As shown in Fig.6C, cotransfection of the miR-199a-5p mimic with the wild-type PKM2 3’ UTR reporter construct significantly decreased the luciferase activities in BEAS-2B cells, whereas cotransfection with a PKM2 3’ UTR reporter containing point mutations at putative miR-199a-5p binding sites did not affect the luciferase activities, suggesting direct interaction between miR-199a-5p and PKM2 mRNA. Chronic exposure to arsenic increased PKM2 levels starting at 2-week, as well as in AsT cells compared with the control (Fig.6D). PKM2 enzyme activities also went up in arsenic-exposed cells at different time points (Fig.6E). To determine whether PKM2 participates in arsenic-induced glycolysis, we established a stable AsT cell line with PKM2 knockout using a CRISPR/Cas9 knockout plasmid (Fig.6F). The ECAR rate dropped in AsT PKM2/KO cells compared with AsT cells (Fig. 6G, H). Moreover, overexpressing PKM2 in AsT/miR-199a-5p cells reversed the inhibitory effect of miR-199a-5p on ECAR rates (Fig.6I), suggesting that PKM2 is a downstream effector of miR-199a-5p on arsenic-induced glycolysis.
Fig.6. PKM2is the functional target of miR-199a-5p in arsenic-induced energy metabolic shift.
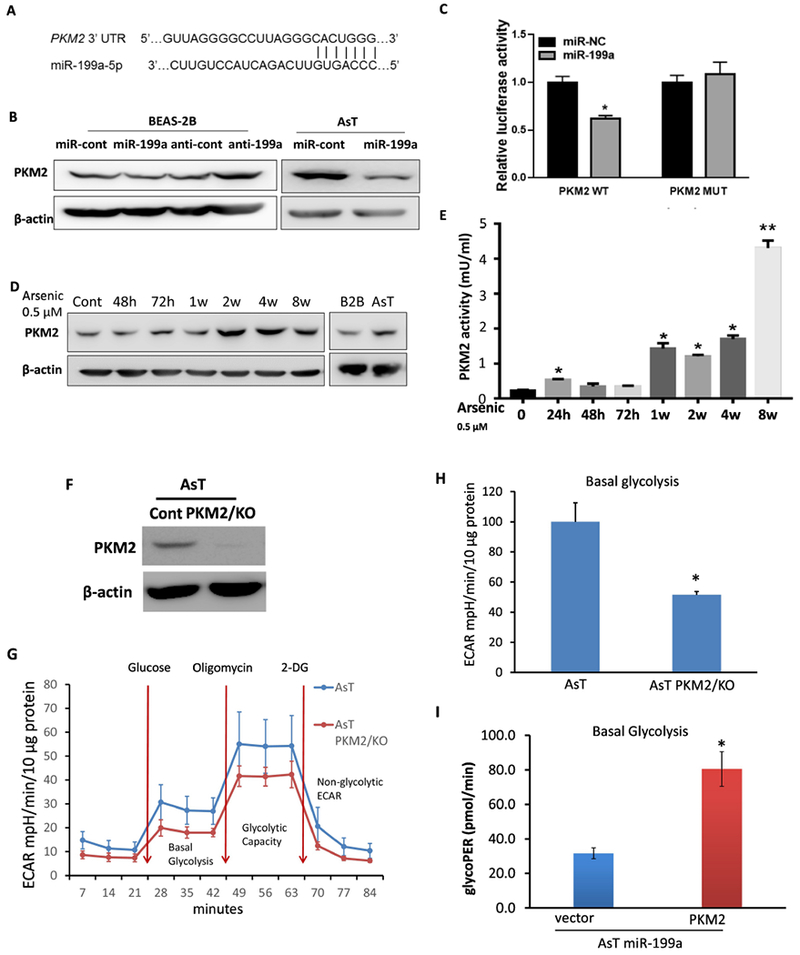
(A) Sequence alignment of 3’UTR of PKM2 mRNA and human miR-199a-5p with potential binding sites. (B) Cells were transiently transfected with 50 μM miR-199a-5p mimic or anti-miR-199a, and their corresponding control oligos respectively. Total proteins were prepared 72 h after the transfection and used to determine PKM2 level by Western-blotting. (C) Beas2B cells were cotransfected with PKM2 3’UTR wild-type or mutant luciferase reporter, miR-199a-5p mimic or negative control miR-cont for 48 h, then harvested for the dual luciferase activity assay. The luciferase activities were presented as relative luciferase activities normalized to those of the cells cotransfected with wild-type 3’UTR reporter and miR-control. (D) PKM2 protein levels were determined in BEAS-2B cells chronically exposure to arsenic at different time points. (E) PKM2 enzyme activity was determined in BEAS-2B cells chronically exposure to arsenic (0.5 μM) at different time points. (F) AsT PKM2 knockout stable cell was established using CRISPR/Cas9 KO plasmid. (G, H) Extracellular acidification rate (ECAR) was determined in AsT cont cells vs AsT PKM2/KO cells. (I) AsT PKM2/KO cell and vector control cells were transfected with miR-199a-5p. Basal glycolysis results were generated by the XF Report Generator based on the Seahorse glycolysis stress assay. All tests were performed in triplicate and presented as mean ± SD. *lndicates significant difference compared with control (P < 0.05).
Discussion
The toxic effects of arsenic-exposure have been widely investigated with multiple mechanisms being proposed. Given the diverse of arsenic-associated cancers and other diseases, it is reasonable to speculate that several key mechanisms may be intertwined synergistically or sequentially (Bailey and Fry 2014). Epigenetic changes such as DNA methylation, histone modification, and small non-coding RNAs (miRNAs) regulation alter driver genes expression levels in ways that impact cancer initiation and progression, and emerges as one of hallmarks of cancer (Esteller 2008). Compelling evidence of epigenetic modifications in cancers include the global hypomethylation of oncogenes and hypermethylation of tumor suppressor genes found in tumors (Baylin and Ohm 2006; Ganapathy et al. 2014; Jones and Baylin 2007). Specifically, genomewide approaches have repeatedly documented DNA global hypomethylation and gene-specific hypermethylation associated with arsenic (Bailey and Fry 2014; Pelch et al. 2015; Ren et al. 2011). However, researchers had not previously been able to determine how arsenic-induced DNA methylation occurs in the promoter regions of specific genes. It was generally believed that DNA-binding transcription factors may mediate site-specific DNA methylation by interacting with specific promoter regions (Blattler and Farnham 2013). In this study, we found that the miR-199a-1 gene had undergone hypermethylation after the chronic arsenic treatments. We then tried to identify how miR-199a-5p is silenced through DNA methylation by arsenic. DNA (cytosine-5-)-methyltransferase 1 (DNMT1) is the most abundant DNMT in cells, a principal enzyme responsible for both de novo and maintenance CpG methylation in the promoter regions of genes; which is required for early embryo development and maintaining cell phenotypes (Dean 2008; Dhe-Paganon et al. 2011). The overexpression of DNMT1 causes the aberrant epigenetic signatures found in lung, colorectal, liver, and breast cancers (De Marzo et al. 1999; Girault et al. 2003; Lin et al. 2007; Saito et al. 2003). In particular, studies revealed that DNMT1 is overexpressed in patients who are smokers and have lung and bladder cancer, and a key component of tobacco smoke carcinogen NNK was found to induce lung tumors in rats and mice through DNMT1 accumulation (Jin et al. 2017; Lin et al. 2010a). These findings indicate DNMT1 can be induced by environmental carcinogens. Indeed, our results showed DNMT1 enzyme was activated by chronic exposure to arsenic. Some earlier studies showed that arsenic exposure leads to a reduction of DNMT1 mRNA levels and activity resulting hypomethylation of genes ((Cui et al. 2006; Reichard et al. 2007), which seems not be in line with our results. We did not see any change of mRNA and protein levels of total DNMT1 upon arsenic treatments. Instead, we observed increased nuclear DNMT1 expression levels. As it remains enigmatic how stable an epigenetic change is, the doses and duration of treatments are very likely to affect the final measurements. In addition, the epigenetic patterns related with environmental carcinogens vary greatly between tissues and cell types. We then to determine how DNMT1 regulates specific miR-199a-1 gene methylation. According to studies about its structure and enzymatic nature, DNMT1 has physical and/or functional interaction with more than 30 different molecules (Svedruzic 2011). It often functions in a way of DNMT1/transcription factors-including complexes as a transcriptional repressor (Damelin and Bestor 2007). Lin et al. reported that dysregulation of DNMT1 is associated with gain of Sp1 and/or loss of negative regulation of p53 (Lin et al. 2010b). Herein, we report that Sp1 induced by arsenic interacts with DNMT1 to promote the miR-199a-1 methylation. Notably, by using the promoter luciferase reporter assay, we found that Sp1 also acts as a transcriptional repressor of miR-199a-1 and miR-199a-2; this finding suggests that Sp1 not only activates and recruits DNMT1 to the promoter region of miR-199a-1 gene, but also negatively regulates the gene transcription directly.
Little is known about the functional consequence of epigenetic perturbations by environmental toxicants. Metabolic reprogramming in cancer cells, called the Warburg effect, is a hallmark of cancer, in which cancer cells produce energy mainly via glycolysis even in the presence of oxygen. Accumulating evidence suggests that the disruption of homeostasis of microenvironment is the driving factor of environmental carcinogenesis (Sun et al. 2016). Environmental carcinogens create an acidic tissue microenvironment in which aerobic glycolysis becomes a dominant source of energy production. Several studies have showed that chronic exposure to arsenic causes metabolic shift from mitochondrial respiration to aerobic glycolysis via activation or stabilization of HIF-1α, NF-κB, or polo-like kinase 1 (Plk1), or via inactivation of p53 (Ganapathy et al. 2014; Li et al. 2015; Luo et al. 2016). Pyruvate kinase M2 (PKM2) is a rate limiting glycolytic enzyme that converts phosphoenopyruvate (PEP) and ADP to pyruvate and ATP (Jurica et al. 1998). PKM2 promotes the Warburg effect by activating HIF-1α target genes SLC2A1, LDHA, and PDK1 that facilitate the shift from oxidative phosphorylation to glycolytic metabolism (Luo and Semenza 2012). Studies show PKM2 can be regulated at multiple levels, including DNA hypomethylation, transcription factors, miRNAs, and post-translational modifications (Yang and Lu 2015). Zhang etal. reported that the suppression of miR-199a maturation by HuR led to glucose metabolic reprogramming in hepatocellular carcinoma by targeting PKM2 and hexokinase-2 (Hk2) (Zhang et al. 2015). In this study, we showed that elevated PKM2 expression contributes to the arsenic-induced metabolic shift by repressing miR-199a-5p. Notably, HIF-1α is also a validated target of miR-199a-5p (He et al. 2014; Rane et al. 2009) (Suppl. Fig.5). Moreover, in addition to promoting DNMT1 transcription, Sp1 also constitutively activates PKM2 gene transcription (Luo and Semenza 2012; Yang and Lu 2015). Taken together, we conclude that arsenic-induced Sp1 overexpression recruits DNMT1 to facilitate miR-199a genes methylation resulting in the loss of mature miR-199a-5p, which releases its post-transcriptional regression of PKM2 and HIF-1α. In addition, Sp1 directly deactivates miR-199a genes and activates PKM2 at the transcriptional level (Fig.7). The overexpression of PKM2 by the aforementioned multiple pathways in turn leads to the energy metabolic dysfunction, which eventually contributes to arsenic transformation. Our current study links the aberrant epigenetic changes to metabolic dysfunction in arsenic-mediated malignant transformation. Further investigation is warranted about whether the proposed mechanism is essential in arsenic carcinogenesis and tumor growth using animal models.
Fig.7. Schematic graph of proposed pathway.

Arsenic exposure induces Sp1 that either interacts with DNMT1 to facilitate miR-199a genes methylation or repress miR-199a genes at the transcriptional level. The miR-199a-5p suppression releases its inhibitory effect on target genes PKM2 and HIF-1α. Additionally, Sp1 can also directly activate PKM2 transcription. PKM2 upregulation by all these pathways results in energy metabolic dysfunction.
Supplementary Material
Highlights.
Sp-1-dependent DNA methylation contributes to arsenic-induced miR-199a-5p repression
Loss of miR-199a-5p associates with an arsenic-mediated energy metabolic shift
PKM2 is the key effector of miR-199a-5p to promote arsenic-induced glycolysis
The study links the aberrant epigenetic changes to metabolic dysfunction in arsenic-mediated malignant transformation
Acknowledgements
Jun He’s K99-funded research is supported by the mentorship of Dr. Andrew E. Aplin, Sidney Kimmel Cancer Center.
Funding information
This work was supported by National Cancer Institute (K99/R00 CA 215316-01) and National Institutes of Environmental Health Science (R01-ES020868, R01-ES024151, and K02ES029119).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Conflicting interests
The authors declare no potential conflicts of interest.
Conflicts of Interest Statement
All authors declare no potential conflicts of interest.
Reference List
- 1.(1980). Arsenic and arsenic compounds. IARC Monogr Eval. Carcinog. Risk Chem. Hum 23, 39–141. [PubMed] [Google Scholar]
- 2.Arita A, and Costa M (2009). Epigenetics in metal carcinogenesis: nickel, arsenic, chromium and cadmium. Metallomics. 1(3), 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ayotte JD, Medalie L, Qi SL, Backer LC, and Nolan BT (2017). Estimating the High-Arsenic Domestic-Well Population in the Conterminous United States. Environ. Sci. Technol 51(21), 12443–12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailey KA, and Fry RC (2014). Arsenic-Associated Changes to the Epigenome: What Are the Functional Consequences? Curr. Environ. Health Rep 1,22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baylin SB, and Ohm JE (2006). Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer 6(2), 107–116. [DOI] [PubMed] [Google Scholar]
- 6.Blattler A, and Farnham PJ (2013). Cross-talk between site-specific transcription factors and DNA methylation states. J. Biol. Chem 288(48), 34287–34294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brocato J, and Costa M (2013). Basic mechanics of DNA methylation and the unique landscape of the DNA methylome in metal-induced carcinogenesis. Crit Rev. Toxicol 43(6), 493–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cardoso APF, Al-Eryani L, and States JC (2018). Arsenic-Induced Carcinogenesis: The Impact of miRNA Dysregulation. Toxicol. Sci 165(2), 284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carpenter RL, Jiang Y, Jing Y, He J, Rojanasakul Y, Liu LZ, and Jiang BH (2011). Arsenite induces cell transformation by reactive oxygen species, AKT, ERK1/2, and p70S6K1. Biochem. Biophys. Res. Commun 414(3), 533–538. [DOI] [PubMed] [Google Scholar]
- 10.Chervona Y, Hall MN, Arita A, Wu F, Sun H, Tseng HC, Ali E, Uddin MN, Liu X, Zoroddu MA, Gamble MV, and Costa M (2012). Associations between arsenic exposure and global posttranslational histone modifications among adults in Bangladesh. Cancer Epidemiol. Biomarkers Prev 21(12), 2252–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui X, Wakai T, Shirai Y, Yokoyama N, Hatakeyama K, and Hirano S (2006). Arsenic trioxide inhibits DNA methyltransferase and restores methylation-silenced genes in human liver cancer cells. Hum. Pathol 37(3), 298–311. [DOI] [PubMed] [Google Scholar]
- 12.Damelin M, and Bestor TH (2007). Biological functions of DNA methyltransferase 1 require its methyltransferase activity. Mol. Cell Biol 27(11), 3891–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Marzo AM, Marchi VL, Yang ES, Veeraswamy R, Lin X, and Nelson WG (1999). Abnormal regulation of DNA methyltransferase expression during colorectal carcinogenesis. Cancer Res 59(16), 3855–3860. [PubMed] [Google Scholar]
- 14.Dean W (2008). The elusive Dnmt1 and its role during early development. Epigenetics. 3(4), 175–178. [DOI] [PubMed] [Google Scholar]
- 15.Dhe-Paganon S, Syeda F, and Park L (2011). DNA methyl transferase 1: regulatory mechanisms and implications in health and disease. Int. J. Biochem. Mol. Biol 2(1), 58–66. [PMC free article] [PubMed] [Google Scholar]
- 16.Drobna Z, Naranmandura H, Kubachka KM, Edwards BC, Herbin-Davis K, Styblo M, Le XC, Creed JT, Maeda N, Hughes MF, and Thomas DJ (2009). Disruption of the arsenic (+3 oxidation state) methyltransferase gene in the mouse alters the phenotype for methylation of arsenic and affects distribution and retention of orally administered arsenate. Chem. Res. Toxicol 22(10), 1713–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esteller M (2008). Epigenetics in cancer. N. Engl. J. Med 358(11), 1148–1159. [DOI] [PubMed] [Google Scholar]
- 18.Ganapathy S, Xiao S, Yang M, Qi M, Choi DE, Ha CS, Little JB, and Yuan ZM (2014). A low-dose arsenic-induced p53 protein-mediated metabolic mechanism of radiotherapy protection. J. Biol. Chem 289(8), 5340–5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Girault I, Tozlu S, Lidereau R, and Bieche I (2003). Expression analysis of DNA methyltransferases 1, 3A, and 3B in sporadic breast carcinomas. Clin. Cancer Res 9(12), 4415–4422. [PubMed] [Google Scholar]
- 20.He J, Wang M, Jiang Y, Chen Q, Xu S, Xu Q, Jiang BH, and Liu LZ (2014). Chronic arsenic exposure and angiogenesis in human bronchial epithelial cells via the ROS/miR-199a-5p/HIF-1alpha/COX-2 pathway. Environ. Health Perspect 122(3), 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He J, Xu Q, Jing Y, Agani F, Qian X, Carpenter R, Li Q, Wang XR, Peiper SS, Lu Z, Liu LZ, and Jiang BH (2012). Reactive oxygen species regulate ERBB2 and ERBB3 expression via miR-199a/125b and DNA methylation. EMBO Rep 13(12), 1116–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hossain MB, Vahter M, Concha G, and Broberg K (2012). Environmental arsenic exposure and DNA methylation of the tumor suppressor gene p16 and the DNA repair gene MLH1: effect of arsenic metabolism and genotype. Metallomics. 4(11), 1167–1175. [DOI] [PubMed] [Google Scholar]
- 23.Intarasunanont P, Navasumrit P, Waraprasit S, Chaisatra K, Suk WA, Mahidol C, and Ruchirawat M (2012). Effects of arsenic exposure on DNA methylation in cord blood samples from newborn babies and in a human lymphoblast cell line. Environ. Health 11, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacobson-Kram D, and Montalbano D (1985). The reproductive effects assessment group’s report on the mutagenicity of inorganic arsenic. Environ. Mutagen 7(5), 787–804. [DOI] [PubMed] [Google Scholar]
- 25.Jin F, Thaiparambil J, Donepudi SR, Vantaku V, Piyarathna DWB, Maity S, Krishnapuram R, Putluri V, Gu F, Purwaha P, Bhowmik SK, Ambati CR, von Rundstedt FC, Roghmann F, Berg S, Noldus J, Rajapakshe K, Godde D, Roth S, Storkel S, Degener S, Michailidis G, Kaipparettu BA, Karanam B, Terris MK, Kavuri SM, Lerner SP, Kheradmand F, Coarfa C, Sreekumar A, Lotan Y, El-Zein R, and Putluri N (2017). Tobacco-Specific Carcinogens Induce Hypermethylation, DNA Adducts, and DNA Damage in Bladder Cancer. Cancer Prev. Res. (Phila) 10(10), 588–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones PA, and Baylin SB (2007). The epigenomics of cancer. Cell 128(4), 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joshi DC, and Bakowska JC (2011). Determination of mitochondrial membrane potential and reactive oxygen species in live rat cortical neurons. J. Vis. Exp(51). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jurica MS, Mesecar A, Heath PJ, Shi W, Nowak T, and Stoddard BL (1998). The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure. 6(2), 195–210. [DOI] [PubMed] [Google Scholar]
- 29.Kishikawa S, Murata T, Kimura H, Shiota K, and Yokoyama KK (2002). Regulation of transcription of the Dnmt1 gene by Sp1 and Sp3 zinc finger proteins. Eur. J. Biochem 269(12), 2961–2970. [DOI] [PubMed] [Google Scholar]
- 30.Li LC, and Dahiya R (2002). MethPrimer: designing primers for methylation PCRs. Bioinformatics. 18(11), 1427–1431. [DOI] [PubMed] [Google Scholar]
- 31.Li Z, Lu Y, Ahmad N, Strebhardt K, and Liu X (2015). Low-dose arsenic-mediated metabolic shift is associated with activation of Polo-like kinase 1 (Plk1). Cell Cycle 14(19), 3030–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin RK, Hsieh YS, Lin P, Hsu HS, Chen CY, Tang YA, Lee CF, and Wang YC (2010a). The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J. Clin. Invest 120(2), 521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin RK, Hsu HS, Chang JW, Chen CY, Chen JT, and Wang YC (2007). Alteration of DNA methyltransferases contributes to 5’CpG methylation and poor prognosis in lung cancer. Lung Cancer 55(2), 205–213. [DOI] [PubMed] [Google Scholar]
- 34.Lin RK, Wu CY, Chang JW, Juan LJ, Hsu HS, Chen CY, Lu YY, Tang YA, Yang YC, Yang PC, and Wang YC (2010b). Dysregulation of p53/Sp1 control leads to DNA methyltransferase-1 overexpression in lung cancer. Cancer Res 70(14), 5807–5817. [DOI] [PubMed] [Google Scholar]
- 35.Liu S, Liu Z, Xie Z, Pang J, Yu J, Lehmann E, Huynh L, Vukosavljevic T, Takeki M, Klisovic RB, Baiocchi RA, Blum W, Porcu P, Garzon R, Byrd JC, Perrotti D, Caligiuri MA, Chan KK, Wu LC, and Marcucci G (2008). Bortezomib induces DNA hypomethylation and silenced gene transcription by interfering with Sp1/NF-kappaB-dependent DNA methyltransferase activity in acute myeloid leukemia. Blood 111(4), 2364–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo F, Liu X, Ling M, Lu L, Shi L, Lu X, Li J, Zhang A, and Liu Q (2016). The lncRNA MALAT1, acting through HIF-1alpha stabilization, enhances arsenite-induced glycolysis in human hepatic L-02 cells. Biochim. Biophys. Acta 1862(9), 1685–1695. [DOI] [PubMed] [Google Scholar]
- 37.Luo W, and Semenza GL (2012). Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol. Metab 23(11), 560–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pelch KE, Tokar EJ, Merrick BA, and Waalkes MP (2015). Differential DNA methylation profile of key genes in malignant prostate epithelial cells transformed by inorganic arsenic or cadmium. Toxicol. Appl. Pharmacol 286(3), 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, Vatner DE, Vatner SF, and Abdellatif M (2009). Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ. Res 104(7), 879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reichard JF, Schnekenburger M, and Puga A (2007). Long term low-dose arsenic exposure induces loss of DNA methylation. Biochem. Biophys. Res. Commun 352(1), 188–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ren X, McHale CM, Skibola CF, Smith AH, Smith MT, and Zhang L (2011). An emerging role for epigenetic dysregulation in arsenic toxicity and carcinogenesis. Environ. Health Perspect 119(1), 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roy RV, Son YO, Pratheeshkumar P, Wang L, Hitron JA, Divya SP, D R, Kim D, Yin Y, Zhang Z, and Shi X (2015). Epigenetic targets of arsenic: emphasis on epigenetic modifications during carcinogenesis. J. Environ. Pathol. Toxicol. Oncol 34(1), 63–84. [DOI] [PubMed] [Google Scholar]
- 43.Saito Y, Kanai Y, Nakagawa T, Sakamoto M, Saito H, Ishii H, and Hirohashi S (2003). Increased protein expression of DNA methyltransferase (DNMT) 1 is significantly correlated with the malignant potential and poor prognosis of human hepatocellular carcinomas. Int. J. Cancer 105(4), 527–532. [DOI] [PubMed] [Google Scholar]
- 44.Salnikow K, and Zhitkovich A (2008). Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic, and chromium. Chem. Res. Toxicol 21(1), 28–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stueckle TA, Lu Y, Davis ME, Wang L, Jiang BH, Holaskova I, Schafer R, Barnett JB, and Rojanasakul Y (2012). Chronic occupational exposure to arsenic induces carcinogenic gene signaling networks and neoplastic transformation in human lung epithelial cells. Toxicol. Appl. Pharmacol 261(2), 204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun Y, Shi Z, Lian H, and Cai P (2016). Energy metabolic dysfunction as a carcinogenic factor in cancer cells. Clin. Transl. Med 5(1), 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suzuki T, and Nohara K (2013). Long-term arsenic exposure induces histone H3 Lys9 dimethylation without altering DNA methylation in the promoter region of p16(INK4a) and down-regulates its expression in the liver of mice. J. Appl. Toxicol 33(9), 951–958. [DOI] [PubMed] [Google Scholar]
- 48.Svedruzic ZM (2011). Dnmt1 structure and function. Prog. Mol. Biol. Transl. Sci 101,221–254. [DOI] [PubMed] [Google Scholar]
- 49.Yang W, and Lu Z (2015). Pyruvate kinase M2 at a glance. J. Cell Sci 128(9), 1655–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang LF, Lou JT, Lu MH, Gao C, Zhao S, Li B, Liang S, Li Y, Li D, and Liu MF (2015). Suppression of miR-199a maturation by HuR is crucial for hypoxia-induced glycolytic switch in hepatocellular carcinoma. EMBO J 34(21), 2671–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao F, Severson P, Pacheco S, Futscher BW, and Klimecki WT (2013). Arsenic exposure induces the Warburg effect in cultured human cells. Toxicol. Appl. Pharmacol 271(1), 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.