

Abstract
Innate immune system quickly responds to invasion of microbes and foreign substances through the extracellular and intracellular sensing receptors, which recognize distinctive molecular and structural patterns. The recognition of innate immune receptors leads to the induction of inflammatory and adaptive immune responses by activating downstream signaling pathways. Allergy is an immune‐related disease and results from a hypersensitive immune response to harmless substances in the environment. However, less is known about the activation of innate immunity during exposure to allergens. New insights into the innate immune system by sensors and their signaling cascades provide us with more important clues and a framework for understanding allergy disorders. In this review, we will focus on recent advances in the innate immune sensing system.
Keywords: inflammasome, pattern recognition receptors, pyroptosis
1. INTRODUCTION
Our body is endlessly exposed to microbial agents and environmental noxious substances. These may cause serious illness, or toxicity to the body; therefore, they must be eliminated. This is mediated by the innate immune system, which is the first line of host defense against foreign invasion. Any disruption in the physical barriers that prevent pathogens from entering the body triggers pro‐inflammatory responses by activating myeloid cells and dendritic cells (DCs) that are central players of the innate immune defense. Furthermore, pro‐inflammatory responses induce antigen presentation shifting from an innate immune response to an acquired immune response. B cells and T cells, in which antigen receptors are individually specialized by DNA rearrangement, mainly mediate acquired immune responses. One of the main features making the innate immune system highly specialized is the germline‐encoded receptors distinguishing between self and nonself. This discrimination is mediated by membrane‐bound or cytoplasmic pattern recognition receptors (PRRs).1 The membrane‐bound receptors are Toll‐like receptors (TLRs) and C‐type lectin receptors (CLRs). The cytoplasmic‐type receptors are retinoic acid‐inducible gene I (RIG‐I)‐like receptors (RLRs) and nucleotide‐binding and oligomerization domain (NOD)‐like receptors (NLRs). These receptors directly sense various components from pathogens and distinguish conserved microbial structural features, called pathogen‐associated molecular patterns (PAMPs).2 The recognition of PAMPs leads to robust innate immune responses through the activation of these downstream signaling pathways. PRRs also recognize self‐components released from the damaged cells, called damage/danger‐associated molecular patterns (DAMPs), and can thus be associated with the pathogenesis of many diseases.
Allergy is an increasing problem in the health sector, with a soaring number of patients in recent years. Accumulating evidence suggests the importance of the innate immune system in the development of allergy susceptibility. Therefore, in this review, we will summarize recent advances in the involvement of PRRs in allergic diseases, and improvements to current allergy treatment modalities.
2. T‐HELPER 2 RESPONSE AND ALLERGY
In recent years, although highly criticized, the hygiene hypothesis has been proposed to explain the increase in frequency of patients with allergy worldwide.3 It suggests that a cleaner environment may lead to development of allergic diseases, highlighting that early exposure to microbes and parasites during childhood is essential to reduce development of susceptibility.4 As evidence in favor of the hygiene hypothesis, it is demonstrated that maternal intranasal exposure to the nonpathogenic microbe Acinetobacter protected against the development of experimental asthma in the progeny.5
Allergy is characterized by a T‐helper 2 (Th2) hypersensitivity response with a significant increase in immunoglobulin (Ig) E. Type 2 immune responses are characterized by the expression of type 2 cytokines, such as interleukin (IL)‐4, IL‐5, IL‐9, and IL‐13. Contact with bacteria during early development may be protective by inducing T‐helper 1 (Th1) cell differentiation. Intestinal microbiota also plays an important role in the prevention of allergies.6, 7 Although oral administration of ovalbumin (OVA) abrogates both Th1 and Th2 responses in specific pathogen‐free mice, only Th1 responses are reduced in germ‐free mice.8 The reconstitution of the intestinal microbiota of germ‐free mice suppressed the susceptibility of the Th2 responses in neonates,9 suggesting the importance of exposure to intestinal microbiota at the neonatal stage for prevention of allergies.
A high dose of lipopolysaccharide (LPS) promotes Th1 immune responses and prevents allergic disease in an OVA‐induced allergic asthma model.10 Conversely, a low amount of LPS skews the immune response to Th2 type and induces allergic airway inflammation in a thymic stromal lymphopoietin (TSLP)‐dependent manner.11 TSLP is an epithelial cell‐derived cytokine expressed in the thymus, lung, skin, and gut. The release of TSLP,12, 13 IL‐25,14, 15 IL‐33,16 and granulocyte‐macrophage colony‐stimulating factor (GM‐CSF)17 from nonlymphoid cells is important for the initiation of Th2 immune responses. Furthermore, epithelial cells in the damaged barriers may also cooperate in the initiation of Th2 immune responses to repair the tissue injury. Activation of group 2 innate lymphoid cells (ILC2s) by epithelium‐derived cytokines such as TSLP, IL‐7, IL‐25, IL‐33, and also IL‐4 induces the production of type 2 cytokines including IL‐5, IL‐9, IL‐13, and epidermal growth factor receptor (EGFR) ligand amphiregulin (AREG) for leading innate type 2 immunity.18, 19, 20 In this context, the intensity of activation of downstream signaling molecules and pathways may be strongly associated with the development of allergy.
3. TOLL‐LIKE RECEPTORS
The TLRs were the first discovered PRR and are mammalian homologues of Drosophila Toll protein involved in innate immune response.21, 22 Studies over the past two decades have revealed important roles of TLRs in a variety of biological phenomena including inflammation, the bridging between innate and acquired immune responses, and cancer cell proliferation and survival.
3.1. TLR family
Toll‐like receptors include 10 and 13 family member proteins in humans and mice, respectively. The TLR family is evolutionally conserved and contains the ligand‐binding domains via leucine‐rich repeat (LRR) motifs at the N‐terminus and intracellular Toll/IL‐1 receptor (TIR) domain at the C‐terminus (Figure 1). TLRs act as the gatekeepers of host defense to various pathogens through structure‐ and sequence‐dependent immune recognition (Figure 1).23, 24, 25, 26 Most TLRs in humans and mice recognize similar PAMPs with some exceptions. TLR11, TLR12, and TLR13 have been lost in the human genome, and the Tlr10 gene is disrupted in the mouse genome. TLR1 through to TLR9 are conserved in both species. TLR4 recognizes bacterial LPS. Triacyl and diacyl lipopeptides are recognized by dimerization of TLR2 with TLR1 and TLR6, respectively. TLR5 recognizes bacterial flagellin derived from flagella. TLR9 is a deoxyribonucleic acid (DNA) sensor and recognizes nonmethylated cytosine‐phosphate‐guanine (CpG) DNA. TLR3 recognizes double‐stranded RNAs (dsRNAs), and both TLR7 and TLR8 detect unmodified uridine‐rich single‐stranded RNAs (ssRNAs). TLRs localize either on the cell surface (TLR1, TLR2, TLR4, TLR5, and TLR6) or in endosomes (TLR3, TLR7, TLR8, and TLR9) through the transmembrane (TM) domain (Figure 1).
Figure 1.
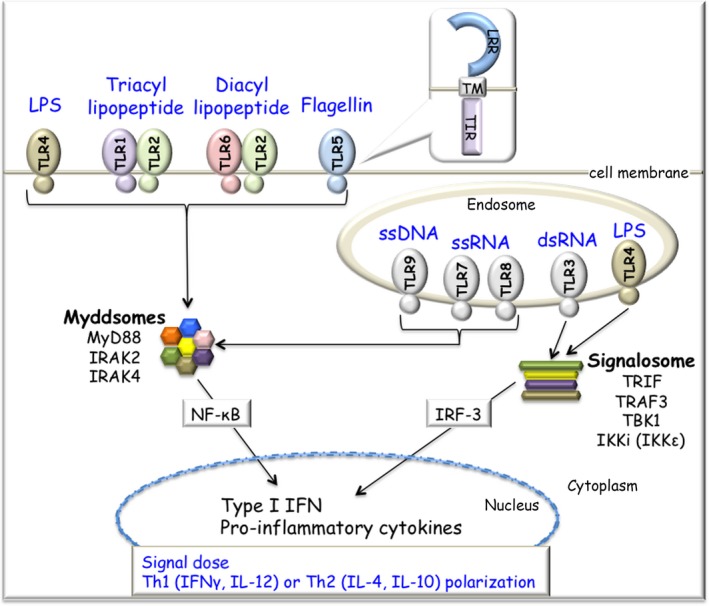
TLR‐mediated signaling pathway. All TLR proteins have LRR and TIR domains (in the balloon). Individual TLRs recognize different ligands, such as LPS, triacyl lipopeptide, diacyl lipopeptides, bacterial flagellin, DNA, and RNA. TLRs localize at the cell surface or in endosomes. TLRs recruit two adaptor proteins, MyD88 and TRIF. The ligand engagement of TLRs induces the formation of the Myddosome (MyD88 and IRAKs) and activates the NF‐κB pathway. TLR3 and TLR4 also induce the formation of a signalosome (TRIF, TRAF3, TBK1, and IKKi [IKKε]). Activated IRF‐3 induces type I IFN production. Th1/Th2 polarization into either a Th1 immune response or Th2 immune response is dependent on the signal dose through TLRs. IKKi, inducible inhibitor of NF‐κB (IκB) kinase; IRAK, IL‐1 receptor‐associated kinase; IRF‐3, IFN regulatory factor 3; LPS, lipopolysaccharide; LRR, leucine‐rich repeat; MyD88, myeloid differentiation primary response 88; NF‐κB, nuclear factor‐kappa B; TBK1, TRAF‐associated NF‐κB activator (TANK)‐binding kinase 1; Th1, T‐helper 1; Th2, T‐helper 2; TIR, Toll/interleukin‐1 receptor; TLR, Toll‐like receptor; TM, transmembrane; TRAF3, TNF receptor‐associated factor 3; TRIF, TIR‐domain‐containing adaptor inducing interferon (IFN)‐β
3.2. TLR signaling
Once activated by their ligands, individual TLRs recruit two major TIR‐containing adaptor molecules, myeloid differentiation primary response 88 (MyD88) and TIR‐domain‐containing adaptor inducing interferon‐β (IFN‐β) (TRIF) (Figure 1). The engagement of all TLRs except for TLR3 induces the signaling complex, named the Myddosome, which consists of MyD88 and IL‐1 receptor‐associated kinases (IRAKs), leading to activation of NF‐κB.27 The ligand engagement to TLR3 or TLR4 recruits TRIF, which results in activation of IFN regulatory factor 3 (IRF‐3) through a signalosome complex (TNF receptor‐associated factor 3 [TRAF3], TRAF‐associated NF‐κB activator [TANK]‐binding kinase 1 [TBK1], and inducible inhibitor of nuclear factor [NF]‐κB [IκB] kinase [IKKi], also known as IKKε, IKBKE). Phosphorylated IRF‐3 translocates into the nucleus and eventually induces the production of type I IFN.
3.3. TLR and allergy
Allergic development is believed to result from genetic backgrounds and environmental factors. Mutations in TLR family genes have been investigated using single nucleotide polymorphism (SNP) analysis and meta‐genome‐wide association studies (GWAS). Indeed, SNPs in the TLR4 gene are a risk factor for asthma,28, 29 indicating that genetic variations of TLR family genes are related to susceptibility to allergic diseases.
Toll‐like receptor family proteins are differentially expressed in all cells types including macrophages, DCs, B cells, regulatory T (Treg) cells, and epithelial cells. They are directly capable of interacting with pathogens or foreign particles in the epithelial barrier and influence host immune cell responses with environmental factors. Barrier epithelial cells function as an origin of allergic response to external signals from the mucous membranes of the respiratory tract, intestinal tract, or skin. Tight junction barriers are extremely sensitive to detergents.30 LPS also increases tight junction permeability in a TLR4‐dependent manner.31 The dysregulation of the epithelial barrier may increase uptake of allergens in the pathogenesis of allergy.
Microbial‐treated TLR2/3/4/7/9 knockout mother mice are no longer protected from the development of asthma in their offspring,5 suggesting that maternal TLR signaling plays a pivotal role in the transfer of protective effects. However, the precise roles of TLRs in the development of allergic diseases are greatly influenced by many factors, such as cell types, expression level, and the nature of antigens. Indeed, TLR4 signaling leads to allergic responses.32, 33, 34 This TLR4‐mediated allergic reaction develops only by intranasal sensitization but not by subcutaneous or intraperitoneal sensitization, suggesting that TLR‐mediated reactions influence the dose of immune‐stimulatory components, as well as the route of administration and the timing of exposure. Treg cells have also been implicated in allergy development.35 Manipulation of Th1/Th2 balance or Treg cell function by administrating TLR agonists may be promising for the treatment of allergic diseases.36
3.4. Allergen‐specific immunotherapy for TLRs
Different TLR agonists have been assayed in clinical trials as adjuvants.37 These were further developed in the context of allergen‐specific immunotherapy (AIT) with different outcomes. Oral administration of TLR9 agonists displayed a significant benefit in the treatment of asthma and food allergy in mice.38 Several kinds of TLR agonists have now been applied to asthmatic or allergic patients in clinical trials.37, 39 In the case of TLR9 agonists, CpG‐containing nucleotides (Amb a 1‐CpG vaccine)40, 41 and bacteriophage‐derived virion‐like particles (VLPs) packaging A‐type CpG motif42 have been shown to be effective in treating rhinitis and allergic asthma. Intranasal administration of a TLR7 agonist (AZD8848) and TLR8 (VTX‐1463) has also reduced nasal symptoms in patients with allergic rhinitis.43 TLR4 agonist monophosphoryl A (MPL) promotes Th1 and Treg cell responses in cooperation with switching from IgE to IgG blocking antibody production.39
4. C‐TYPE LECTIN RECEPTORS
CLRs recognize a diverse range of nonself PAMPs derived from microbes, especially fungi and house dust mites.44, 45, 46 Most cells, including DCs and macrophages, express CLRs. CLRs belong to the C‐type lectin‐like domain (CTLD) superfamily, which carries the carbohydrate recognition domain (CRD). CLRs contain one or more conserved CTLD.47 Type II transmembrane CLRs, which possess a single CRD, have been most extensively studied among CLRs (Figure 2). This subfamily includes DC‐associated C‐type lectin‐1 (Dectin‐1, CLEX7A), Dectin‐2 (CLEC6A), macrophage‐inducible C‐type lectin (Mincle, CLEC4E), DC‐specific intracellular adhesion molecule 3 (ICAM3)‐grabbing nonintegrin (DC‐SIGN, CD209), and DC NK lectin group receptor‐1 (DNGR‐1, CLEC9A).
Figure 2.
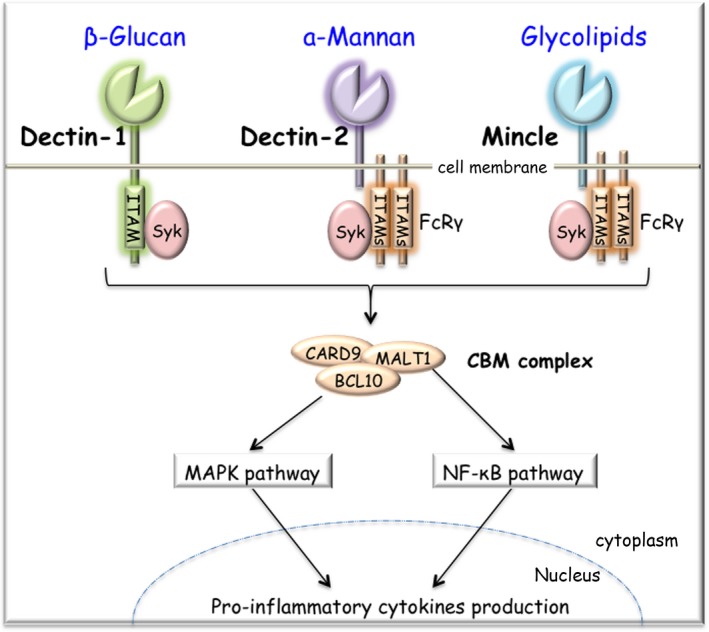
Type II transmembrane CLR. Type II transmembrane CLR proteins possess a single CRD. Dectin‐1 (CLEC7A) recognizes fungal wall‐derived β‐glucans. Dectin‐2 (CLEC6A) recognizes the structure of α‐mannans. Mincle (CLEC4E) recognizes diverse glycolipids including TDM, Glc‐DAG, and MGDG. Dectin‐1 transduces the signal through its ITAM‐like motif. Both Dectin‐2 and Mincle associate with FcRγ for signaling. Ligand‐bound CLRs result in the Syk‐dependent formation of the CBM (CARD9‐BCL10‐MALT1) complex. The CBM complex activates the pathways of MAPK and NF‐κB, leading to pro‐inflammatory cytokine production. BCL10, B‐cell CLL/lymphoma 10; CARD9, caspase‐recruitment domain 9; CLR, C‐type lectin receptor; CRD, carbohydrate recognition domain; Dectin‐1, dendritic cell (DC)‐associated C‐type lectin‐1; Dectin‐2, DC‐associated C‐type lectin‐2; FcRγ, Fc receptor common gamma chain; Glc‐DAG, glucosyl diacylglycerol; ITAM, immunoreceptor tyrosine‐based activation motif; MALT1, mucosa‐associated lymphoid tissue protein 1; MAPK, mitogen‐activated protein kinase; MGDG, monoglucosyldiacylglycerol; Mincle, macrophage‐inducible C‐type lectin; NF‐κB, nuclear factor‐kappa B; Syk, spleen tyrosine kinase; TDM, trehalose‐6,6′‐dimycolate
4.1. Dectin‐1, Dectin‐2, and Mincle
Dectin‐1 and Dectin‐2 recognize fungal wall‐derived β‐glucan and α‐mannan structure, respectively48 (Figure 2). Both are organized in the gene cluster in the human and mouse genomes.49, 50 Mincle, a member of the Dectin‐2 family, recognizes various glycolipids (Figure 2), such as trehalose‐6,6′‐dimycolate (TDM) in the cell wall of Mycobacterium tuberculosis,51 glucosyl diacylglycerol (Glc‐DAG) of Streptococcus pneumoniae,52 monoglucosyldiacylglycerol (MGDG) produced by Group A Streptococcus,53 and others derived from self and nonself.54
4.2. CLR signaling
Dectin‐1 directly transduces the signal through its immunoreceptor tyrosine‐based activation motif (ITAM)‐like motif in the cytoplasmic domain. Dectin‐2 and Mincle are required for the ITAM‐containing adaptor protein Fc receptor common gamma chain (FcRγ, FCER1G) (Figure 2). Once ligands are bound to CLRs in a calcium‐dependent manner, spleen tyrosine kinase (Syk) is recruited to phosphorylated ITAM motifs, leading to cellular activation. In this signaling cascade, caspase‐recruitment domain (CARD)‐containing adaptor proteins, CARD9 and B‐cell CLL/lymphoma 10 (BCL10), form a complex with caspase‐like cysteine protease mucosa‐associated lymphoid tissue protein 1 (MALT1). CARD9‐BCL10‐MALT1 (CBM) complex activates NF‐κB and mitogen‐activated protein kinase (MAPK) pathways, resulting in the production of pro‐inflammatory cytokines (Figure 2).
4.3. CLRs and allergy
Dectin‐1 is involved in fungal‐mediated allergic inflammation mediating T‐helper 17 (Th17) cell differentiation.55, 56 Genetic polymorphisms of Dectin‐1, TLR3, and TLR9 are significantly associated with susceptibility to severe asthma with fungal sensitization.57 Dectin‐2 has been implicated in allergic inflammation to house dust mites with Th2 polarization.58, 59, 60 A recent study shows that Mincle recognizes not only glycolipids but also self‐derived cholesterol sulfate in skin epithelial cells and is involved in the induction of allergic skin inflammatory response.61
4.4. Allergen‐specific immunotherapy for CLRs
Recent findings have shown that allergoids conjugated to nonoxidized mannan from Saccharomyces cerevisiae are next‐generation vaccines targeting DCs through CLRs. These vaccines are candidates for AIT of allergic diseases as they promote the generation of Treg cells by mechanisms partially depending on programmed death‐ligand 1 (PD‐1) and IL‐10 in both humans and mice.62, 63 Phase II clinical trials for grass‐pollen and house dust mite allergens are currently ongoing.64
5. RIG‐I‐LIKE RECEPTORS
5.1. RLR family
Tlr3 ‐deficient cells showed normal type I IFN production toward viral infection,65 suggesting that additional mechanisms were hidden in the RNA sensing. As the cytosolic dsRNA sensor sensing both RNA helicases, RIG‐I (DDX58)66 and melanoma differentiation‐associated gene 5 (MDA5, IFIH1) 67 were identified.68 The RLR family proteins possess the DExHD motif containing the helicase domain for dsRNA recognition and have the two CARD domains at the N‐terminus (Figure 3).69 Laboratory of genetics and physiology 2 (LGP2, DHX58) lacks a CARD domain and, therefore, has no intrinsic signaling activity.
Figure 3.
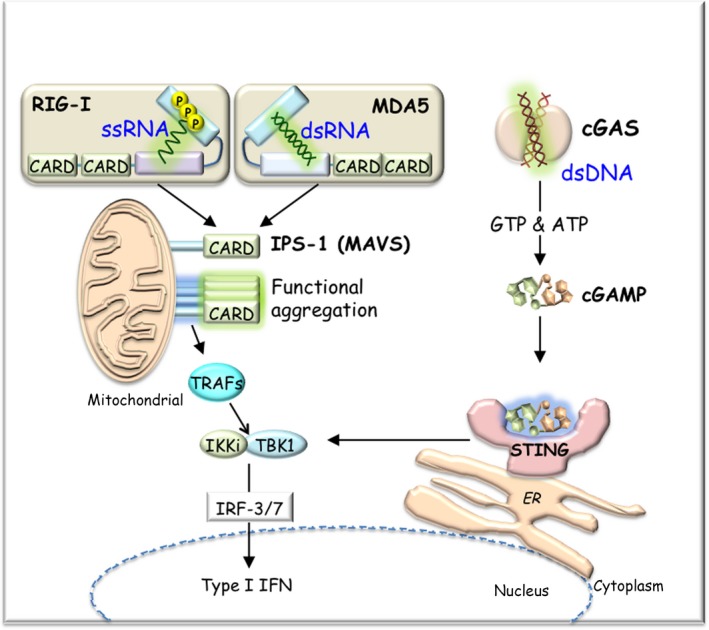
Nucleic acid sensors. RIG‐I and MDA5 have two CARD domains at the N‐terminus and a helicase domain at the center. Both RIG‐I and MDA5 bind viral RNAs bearing 5’‐triphosphate or 5’‐diphosphate distinct from the mammalian RNA with 5’ cap structure. After viral RNA recognition, RIG‐I and MDA5 interact with IPS‐1 (MAVS) through CARD‐CARD interactions. IPS‐1 (MAVS) is localized on the mitochondrial outer membrane. Aggregated IPS‐1 (MAVS) activates TBK1 and IKKi (IKKε) through TRAFs, leading to induction of type I IFN via phosphorylation of IRF‐3/7. cGAS binds dsDNA and produces a second messenger, cGAMP, from ATP and GTP. cGAMP binds to STING localized on the ER membrane and induces type I IFN production via the TBK1/IRF‐3 pathway. CARD, caspase‐recruitment domain; cGAMP, cyclic GMP‐AMP; cGAS, cyclic GMP‐AMP synthase; ER, endoplasmic reticulum; IKKi, inducible inhibitor of NF‐κB (IκB) kinase; IPS‐1, IFN‐β promoter stimulator 1; IRF, IFN regulatory factor; MDA5, melanoma differentiation‐associated gene 5; RIG‐I, retinoic acid‐inducible gene I; STING, stimulator of IFN genes; TBK1, TRAF‐associated NF‐κB activator (TANK)‐binding kinase 1; TRAF, TNF receptor‐associated factor
5.2. RLR recognition and signaling
Retinoic acid‐inducible gene I and MDA5 have different roles in the recognition of RNA viruses.70 RIG‐I recognizes relatively short viral RNA blunt ends bearing 5′‐di‐/triphosphate distinct from the host cellular RNA with 5′ cap structure.71, 72, 73 RIG‐I detects many RNA viruses, such as rhinovirus, Sendai virus, vesicular stomatitis virus, and influenza virus. RIG‐I‐mediated RNA recognition mechanisms are viral replication‐independent.71 MDA5 responds to longer (over 1k bp) dsRNA,74 such as polyinosinic‐polycytidylic acid [poly(I:C)], as well as dsRNA generated after infection of picornaviruses including encephalomyocarditis virus (EMCV), Mengo virus, and Theiler's virus. After viral RNA recognition, RIG‐I and MDA5 interact with the downstream CARD‐containing adaptor protein, IFN‐β promoter stimulator 1 (IPS‐1, MAVS), through CARD‐CARD interactions75, 76, 77, 78 (Figure 3). IPS‐1 localizes on the mitochondrial outer membrane, which triggers prion‐like aggregation of IPS‐179 (Figure 3). The aggregated IPS‐1 recruits IRF‐3 kinase and activates the IRF‐3‐TBK1‐IKKi (IKKε)‐IRF‐3/7‐IFN‐dependent signaling pathway through TRAFs.
5.3. RLRs and allergy
Early viral infections in children are associated with further allergic sensitization and asthma persistence. Similarly, viral infections in asthma patients (both allergic and nonallergic) are also associated with asthma exacerbations. In the development of respiratory disease, innate immune mechanisms are involved in virus‐infected airway epithelial cells.80 Loss‐of‐function mutations in the IFIH1 gene increase susceptibility to severe respiratory infection caused by human rhinovirus in children.81, 82 A meta‐phenome‐wide association study also revealed a novel association of an IFIH1 allele mutation to increased risk for asthma.83 In an experimental model, poly(I:C) and rhinovirus‐derived dsRNA exacerbated asthma.84, 85, 86 Taken together, RLRs play a nonredundant and critical role in the development and progression of asthma.
5.4. cGAS—DNA sensor
Recent studies have revealed the existence of a new intracellular DNA sensing system. Cyclic GMP‐AMP synthase (cGAS), a member of nucleotidyltransferase family, binds dsDNA in a sequence‐independent manner but is activated in a length‐dependent manner (longer than 94‐bp DNA).87 cGAS undergoes a conformational change of its catalytic center and then produces the cyclic GMP‐AMP (cGAMP) from ATP and GTP (Figure 3).88, 89, 90 cGAMP acts as second messenger,91, 92 binds to the stimulator of IFN genes (STING),93, 94, 95 localizes on the endoplasmic reticulum (ER) membrane, and induces type I IFN production via the TBK1/IRF‐3 pathway (Figure 3).96, 97 cGAMP senses viral‐derived DNA as well as host‐derived DNA.98, 99 cGAS is involved in DNA damage‐induced inflammatory signaling in cancer cells.100, 101 The cGAS‐STING‐mediated DNA sensing system also contributes to the induction of apoptosis, control of ER stress response, and autophagy.
6. NOD‐LIKE RECEPTORS
A third group of PRRs are NLRs.102 NLRs are localized in the cytosol and recognize PAMPs and DAMPs. NLRs carry three unique structural properties (Figure 4); the central region is named NOD or nucleotide‐binding domain (NACHT), which consists of conserved motifs including ATP/GTPase‐specific phosphate‐binding loop and magnesium‐binding site; the C‐terminal region contains LRRs, which respond to ligand specificity similar to TLRs; the N‐terminal region is different among NLRs (Figure 4). Based on the name, NLRs are divided into five major subgroups (NLRA, NLRB, NLRC, NLRP, and NLRX) (Figure 4). So far, twenty‐five NLR family genes have been identified in the human genome.
Figure 4.
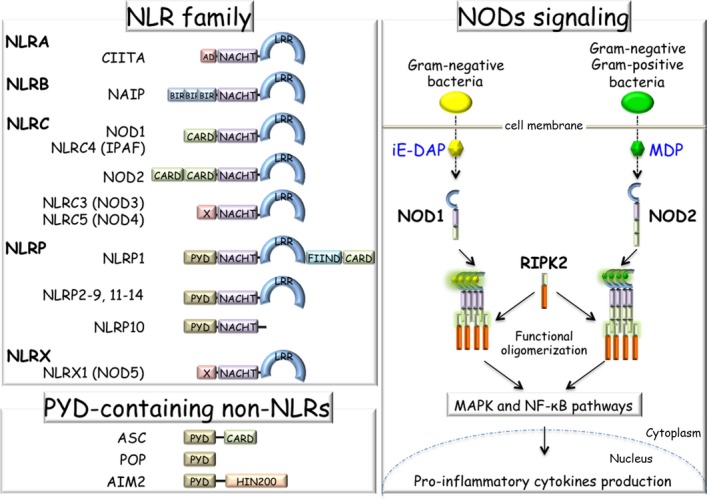
NLR family, PYD‐containing non‐NLR proteins, and NOD signaling. In the NLR family, there are five major subfamilies based on the unique N‐terminal domain structure: NLRA (AD‐type), NLRB (BIR‐type), NLRC (CARD‐type), NLRP (PYD‐type) and NLRX (X‐type). All family proteins have a NACHT domain in the central region. NOD2 senses bacterial cell wall‐derived peptidoglycan derivative MDP; however, NOD1 only senses Gram‐negative bacteria‐derived iE‐DAP. After sensing ligands, NODs oligomerize and interact with RIPK2 through the CARD domain. RIPK2 further activates downstream cascades and MAPK and NF‐κB pathways, leading to cytokine production. AD, transactivation domain; BIR, baculoviral inhibitor of apoptosis repeat; CARD, caspase‐recruitment domain; FIIND, function‐to‐find domain; HIN200, hematopoietic interferon‐inducible nuclear antigens with 200 amino acid repeats; iE‐DAP, γ‐D‐glutamyl‐meso‐diaminopimelic acid; LRR, leucine‐rich repeat; MAPK, mitogen‐activated protein kinase; MDP, muramyl dipeptide; NACHT, NAIP, CIIA, HeT‐E, and TEP1; NF‐κB, nuclear factor‐kappa B; NOD1, nucleotide‐binding oligomerization domain‐containing protein 1; NOD2, nucleotide‐binding oligomerization domain‐containing protein 2; PYD, Pyrin domain; RIPK2, receptor‐interacting serine/threonine‐protein kinase 2
6.1. NOD1 and NOD2
NOD1 (CARD4) and NOD2 (CARD15) are founding members of NLR family103 and belong to the NLRC subfamily. NOD1 has a single CARD domain and NOD2 has two CARD domains at the N‐terminus (Figure 4). Both NOD1 and NOD2 sense bacterial cell wall‐derived peptidoglycan derivative γ‐D‐glutamyl‐meso‐diaminopimelic acid (iE‐DAP) and muramyl dipeptide (MDP) structures, respectively (Figure 4). iE‐DAP is derived from Gram‐negative bacteria, whereas MDP is derived both from Gram‐positive and Gram‐negative strains.
6.2. NOD signaling
Upon recognition, NODs oligomerize and interact with CARD‐containing receptor‐interacting serine/threonine‐protein kinase 2 (RIPK2) through the CARD domain, and then activate transforming growth factor (TGF)‐β‐activated kinase 1 (TAK1, MAP3K7) and IKK complex for leading to MAPK and NF‐κB activation104 (Figure 4). NOD1 is widely expressed by a variety of cell types, whereas NOD2 expression is limited to certain cell types such as hematopoietic cells105 and Paneth cells in the intestine.106
6.3. NODs and allergy
NOD1 and NOD2 promote Th1 and Th17 adaptive immunity by inducing the secretion of TNF and IL‐1107, 108 in addition to Th2 immune response,109 suggesting that signaling through these receptors may be central to susceptibility and exacerbation of allergies. Although inhalation of NOD1 and NOD2 ligands induces Th2 response, NOD2 appears to have more potent activity than NOD1 in Th2‐driven allergic inflammation. It is shown that NOD2 displays this function by promoting the expression of TSLP, OX40 ligand (OX40L/CD252, TNFSF4), and IL‐25.110 NOD2‐induced suppression of Treg cell development and induction of early IL‐4‐secreting cells are completely dependent on TSLP. NOD1 cannot induce strong TSLP expression. However, intranasal infusion of high doses of NOD2 ligands did not break tolerance nor lead to asthma susceptibility, indicating a dose‐dependent effect of NOD2 in allergy development.
Polymorphisms of NOD1 and NOD2 are highly associated with inflammation development in the respiratory airways,111 childhood asthma,112 and atopic diseases.113, 114, 115 It has been thought that these phenotypes arise from a defect in NOD sensing fragments of bacterial peptidoglycan. However, a recent study shows that NODs also participate in sensing infection with viruses and parasites by inducing ER stress‐induced inflammation,116 and further research is needed to elucidate the role of NODs in allergic diseases.
6.4. PYD‐containing non‐NLRs
The NLRP family has the Pyrin domain (PYD) at the N‐terminal region. Similarly to the CARD domain, PYD prefers to assemble together; thus, PYD‐containing proteins give rise to a complex. The complex formation of NLRs is important for signaling and inflammation. The overall composition of NLRs is not well characterized yet, but it is important to uncover the role of NLRs, especially in the inflammasome (Section 7). Studies on additional small molecules, such as PYD‐only proteins (POPs) family (Figure 4) and CARD‐only proteins (COPs) family, may provide a more detailed mechanism.117, 118, 119
7. INFLAMMASOME
NLRP family proteins, a subgroup of NLR family, and absent in melanoma 2 (AIM2) form a complex with apoptosis‐associated speck‐like protein containing a CARD (ASC, PYCARD) through their PYD domains and recruit the CARD domain of pro‐caspase‐1, forming the inflammasome120 (Figure 5). Inflammasomes are cytosolic multimeric protein complexes sensing and responding to pathogenic microbes and cellular damage.121 To date, various inflammasomes including NLR subfamily proteins (NLRP1 [CARD7], NLRP2, NLRP3, NLRP4, NLRP6, and NLRC4 [CARD12]) and PYD‐containing non‐NLR proteins (AIM2, pyrin (MEFV), and IFN‐γ‐inducible protein (IFI) 16) have been identified. Based on the activation mechanism, the inflammasomes are categorized as canonical or noncanonical.
Figure 5.
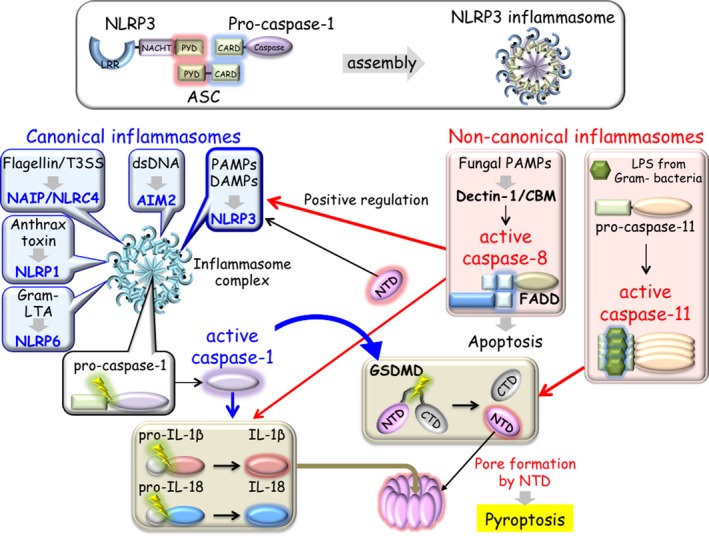
Inflammasome. NLRP3 forms a complex with ASC through their PYD binding each other and recruiting the CARD domain of pro‐caspase‐1. After assembly, this multimeric complex is called the inflammasome (upper box). In the canonical inflammasome assembly, ligand‐sensing NLRs (NLRP1, NLRP3, NLRP6, NAIP/NLRC4, and AIM2) (blue balloons) form multisubunit disk‐like structures comprising an inner ring and outer ring. Pro‐caspase‐1 is located in the central portion (black balloon). Activated caspase‐1 processes IL‐1β/IL‐18 and triggers proteolytic cleavage of GSDMD (brown boxes). In the noncanonical inflammasomes, caspase‐11 and caspase‐8 participate in the cytoplasmic LPS sensing pathway and Dectin‐1‐CBM signaling pathway, respectively (pink boxes). Activated caspase‐11 cleaves GSDMD similarly to caspase‐1 (red arrow). GSDMD‐derived NTD induces pore formation for pyroptosis and stimulates the NLRP3 inflammasome pathway (black arrows). Activated caspase‐8 with FADD is capable of cleaving pro‐IL‐1β/pro‐IL‐18 (red arrow). Caspase‐8 may positively regulate NLRP3 inflammasome pathway (red arrow). AIM2, absent in melanoma 2; ASC, apoptosis‐associated speck‐like protein containing a CARD; CARD, caspase‐recruitment domain; CBM, CARD9‐BCL10‐MALT1; Dectin‐1, lipopolysaccharide; GSDMD, gasdermin D; LPS, lipopolysaccharide; LTA, lipoteichoic acid; NAIP, NLR family apoptosis inhibitory protein; NLRC4, NLR family CARD domain containing 4; NLRP1, NLR family Pyrin domain containing 3; NLRP3, NLR family Pyrin domain containing 3; NLRP6, NLR family Pyrin domain containing 6; NTD, N‐terminal domain; PYD, Pyrin domain
7.1. Canonical inflammasome
Caspases are a group of cysteine‐aspartic acid proteases, of which 12 and 10 family members have been identified in humans and mice, respectively. Caspases play an essential role in programmed cell death and are involved in either apoptosis or inflammation along with pyroptosis122 (Section 8). In the steady state, the caspase family is present in inactive forms called zymogens, which require activation. Inflammasome mediates the activation of caspase‐1 through CARD‐CARD interactions. Caspase‐1, caspase‐4 (human only), caspase‐5 (human only), caspase‐11 (mouse only), and caspase‐12 are known to be involved in the inflammatory pathway, and those inflammatory caspase genes are clustered in the human and mouse genome.123 Inflammasomes activate caspase‐1, resulting in production of pro‐inflammatory cytokines IL‐1β and IL‐18 upon activation by various signals124 (Figure 5). Thus, “canonical” inflammasome relies on the activation of caspase‐1.
7.1.1. NLRP3 inflammasome
NLRP3 inflammasomes can respond to a variety of substances including crystals such as urate, cholesterol, asbestos, silica, aggregated amyloid‐β, and islet amyloid polypeptide.125 As a direct interaction of NLRP3 with these substances has not been detected, NLRP3 inflammasome activation is considered to occur via mitochondrial and lysosomal damage. Reactive oxygen species (ROS) also trigger NLRP3 inflammasomes priming.126 Extracellular ATPs released from activated or necrotic cells activate P2X purinoceptor 7 (P2X7, P2RX7) receptor127 and induce caspase‐1 activation, leading to IL‐1β production via formation of an NLRP3 inflammasome.128 IL‐1β is critically involved in Th17 cell differentiation, production of Th17‐associated cytokines, and neutrophilic inflammation.129 In addition to the Th2 response, a Th17 response is associated with asthma, notably severe glucocorticoid‐resistant asthma.130 From various studies of human diseases and mouse models, the elevation of IL‐1β and IL‐18 is associated with the development of allergic diseases, such as asthma, dermatitis, rhinitis, and conjunctivitis.131 All these events are connected with inflammasome activation and suggest participation of the inflammasome in the development and progression of asthma. Th2 and Th17 inflammatory pathways are mutually regulated in asthmatic patient samples.132 Type II cytokine suppression promotes Th17 responses, indicating that combined therapy targeting both Th2 and Th17 responses may benefit asthmatic patients.
7.1.2. AIM2 inflammasome
Cytoplasmic dsDNA sensor AIM2 is a member of IFI20X/IFI16 family.133 In normal conditions, the N‐terminal PYD of AIM2 maintains an autoinhibitory state through interaction with C‐terminal hematopoietic IFN‐inducible nuclear proteins with a 200‐amino acid repeat (HIN‐200) domain, which directly binds to the sugar‐phosphate backbone of dsDNA in a sequence‐independent manner.134, 135 The ligand binding to the HIN‐200 domain triggers the activation of the AIM2 inflammasomes, leading to pyroptosis and the production of IL‐1β and IL‐18.136, 137, 138, 139 The AIM2 inflammasomes play an important role in infection, cancer, and autoimmunity 140 by sensing of virus/bacteria‐derived dsDNA and tumor‐derived DNA.141, 142 A recent study shows that AIM2 is dispensable for inflammasome activation in some primary human myeloid cells, where STING‐dependent cell death can trigger NLRP3 inflammasome activation by inducing potassium efflux.143 This result implicates the presence of a cell‐type‐specific alternative pathway in the dsDNA sensing system. Inflammasome‐derived caspase‐1 binds to cGAS and restricts its downstream STING‐TBK1‐IRF‐3/7‐IFN‐dependent signaling pathway by cutting out cGAS,142 implicating a crosstalk between type I IFN production and the inflammasome. cGAMP also functions in the priming and activation of AIM2 and NLRP3 inflammasomes.144
7.1.3. Other inflammasomes
NLRC4 inflammasomes are activated by NLR family apoptosis inhibitory protein (NAIP) harboring three baculoviral inhibitor of apoptosis repeat (BIR) domains at N‐terminus.145, 146 NAIP family proteins detect monomeric flagellin or the needle/rod regions of type III secretion system (T3SS),147, 148, 149, 150 leading to the activation of downstream NAIP/NLRC4 inflammasome responses. The transcription of Naips is regulated by the transcription factor IRF‐8.151 NLRC4 is implicated in the exacerbation of psoriatic lesions,152 and NLRP4 mutation is associated with exacerbation of asthma in smoking adults.153 Polymorphisms of NACHT‐LRR in NLRP12 and the promoter regions of NLRC4 and NLRP1 are associated with atopic dermatitis.113 The elusive other inflammasomes remain of outstanding interest.
7.2. Noncanonical inflammasome
Caspase‐11 senses cytosolic LPS with noncanonical function154, 155, 156, 157, 158 (Figure 5). This intracellular LPS sensing can trigger caspase‐11‐mediated pyroptosis under a TLR4‐independent mechanism.159, 160, 161 In this caspase‐1‐independent pathway, murine caspase‐11 (caspase‐4 and caspase‐5 in humans) directly senses LPS through its CARD domain.162 Thus, “noncanonical” inflammasomes can lead to the activation of caspase‐11. The NLRP3 inflammasome is involved in both canonical and noncanonical activation. Interestingly, the NLRP6 inflammasome senses Gram‐positive bacteria‐derived lipoteichoic acid (LTA) (Figure 5).163 In this activation mechanism, NLRP6 recruits both caspase‐1 and caspase‐11. The processed caspase‐11 induces caspase‐1 activation, resulting in the production of IL‐1β and IL‐18.
7.2.1. Caspase‐8
Caspase‐8 is known to play a central role in apoptosis as initiator and apical activator. Caspase‐8 has two death effecter domains (DEDs) in its N‐terminus and is structurally different to CARD‐containing inflammatory caspases. Interestingly, caspase‐8 has been shown to form a noncanonical inflammasome in response to fungal and mycobacterial infection by Dectin‐1.164 Fungal PAMPs activate Dectin‐1 signaling to induce a noncanonical caspase‐8‐ASC with CBM complex in a caspase‐1‐independent manner.165 Caspase‐8 assumes its inflammatory roles by inducing IL‐1β activation165, 166 (Figure 5). Furthermore, caspase‐8 mediates both canonical and noncanonical NLRP3 inflammasome priming and activation with a death domain (DD)‐containing adaptor protein Fas‐associated protein with DD (FADD).167 Caspase‐8‐mediated IL‐1 signaling promotes Th2 responses in allergic airway inflammation,168 implicating its therapeutic potential for asthma.
7.3. Inhibition of inflammasome activation
Inflammasomes are multiple protein complexes; therefore, abnormal assembly causes hyperinflammatory conditions, as in the case of skin inflammation in NLRP1 germline mutation.169 Some pathogens are able to selectively inhibit the activation of the caspase‐11‐mediated noncanonical NLRP3 inflammasome.170 However, the regulatory mechanisms of inflammasomes are not fully understood and need further characterization. Interestingly, a recent study shows that TAK1 restricts the NLRP3 inflammasome to regulate cell homeostasis and death in myeloid cells.171 TAK deficiency promotes spontaneous NLRP3 inflammasome activation. TAK1 inhibits the activation of DD‐containing receptor‐interacting serine/threonine‐protein kinase 1 (RIPK1). Activated RIPK1 induces the caspase‐8‐FADD pathway. TAK1 inactivation induces RIPK1 activation, leading to the caspase‐8‐dependent pathway,172 indicating that RIPK1 plays a role upstream of caspase‐8. This machinery is also associated with neuroinflammation, aging, and infection.173 Hence, the mechanism requires further investigation to understand the implications of inhibiting inflammasome activation.
8. PYROPTOSIS
8.1. Pyroptosis
Pyroptosis is one form of cell death and is morphologically different to apoptosis and necrosis. Apoptosis is an immunologically silent death mode while necrosis and pyroptosis are pro‐inflammatory death modes tightly associated with inflammation.174 Caspase‐1 is a key player of pyroptosis in cell death (canonical inflammasome). As described above, caspase‐11 also contributes to the central mechanism of pyroptosis (Figure 5).155 Pyroptotic cells release their entire cellular contents including nuclear and mitochondrial DNA. Pyroptosis preferentially occurs in macrophages, monocytes, and DCs. Neutrophil cell death is called NETosis (neutrophil extracellular traps) and releases chromatin components to the extracellular space.175, 176 It is found that NETosis‐derived dsDNA mediates allergic asthma exacerbations during rhinovirus infection,177, 178 suggesting dsDNA acts as an adjuvant to boost type II‐mediated allergic inflammation.
8.2. Gasdermin
The gasdermin (GSDM) family consists of six member proteins (GSDMA, GSDMB, GSDMC, GSDMD, GSDME, and Pejvakin).179 The molecular mechanisms underlying pyroptosis and GSDM family functions have been elucidated.161, 180, 181, 182, 183, 184, 185, 186 Activated caspases directly cleave GSDMD into two fragments, the N‐terminal domain (NTD) and C‐terminal domain (CTD). The NTD of GSDMD oligomerizes to form a pore on the cell membrane. This formation perforates the plasma membrane and initiates pyroptosis, leading to inflammasome‐mediated secretion of mature IL‐1β and IL‐18 (Figure 5). Recent studies show that GSDMD is involved not only in pyroptosis but also in NETosis.187, 188, 189
8.3. GSDMB and allergy
Importantly, the genome locus of GSDMB and orosomucoid 1‐like 3 (ORMDL3) on chromosome 17q21 is strongly associated with childhood‐onset asthma.190, 191 GSDMB is highly expressed in airway epithelial cells.192 GSDMB‐mediated pyroptosis in epithelial cells may be involved in the pathogenesis of asthma. Furthermore, GSDMB transgenic mice assume asthma symptoms in the absence of airway inflammation,192 implicating that the induction of GSDMB triggers asthma.
9. ALPK1‐TIFA‐NF‐κB AXIS
Besides LPS itself, ADP‐β‐D‐manno‐heptose (ADP‐Hep) and D‐glycero‐β‐D‐manno‐heptose 1,7‐bisphosphate (HBP),193, 194, 195 intermediate products of the LPS biosynthetic pathway, activate the NF‐κB signaling pathway (Figure 6). ADP‐Hep is more potent than HBP. TRAF‐interacting protein with forkhead‐associated domain (TIFA) was originally identified as a TRAF2‐binding protein that is involved in the NF‐κB pathway.196 Both ADP‐Hep and HBP sensing trigger TIFA oligomerization.193 Recent studies have shown that TIFA oligomerization can be induced by ADP‐Hep or ADP‐heptose 7‐P that is converted from HBP by host adenylyltransferase enzymes of the nicotinamide mononucleotide adenylyltransferase (NMNAT) family (Figure 6).197 Alpha‐kinase 1 (ALPK1), a member of the atypical kinase family alpha kinases, is necessary for phosphorylation‐dependent formation of TIFA oligomerization.194, 195 These sugar molecules directly bind the N‐terminal domain of ALPK1, stimulating its kinase domain to phosphorylate and activate TIFA. The role of ADP‐Hep as a PAMP was further confirmed on comparison with synthetic HBP.198 This ADP‐heptose sensing system stimulates host innate immune responses.
Figure 6.
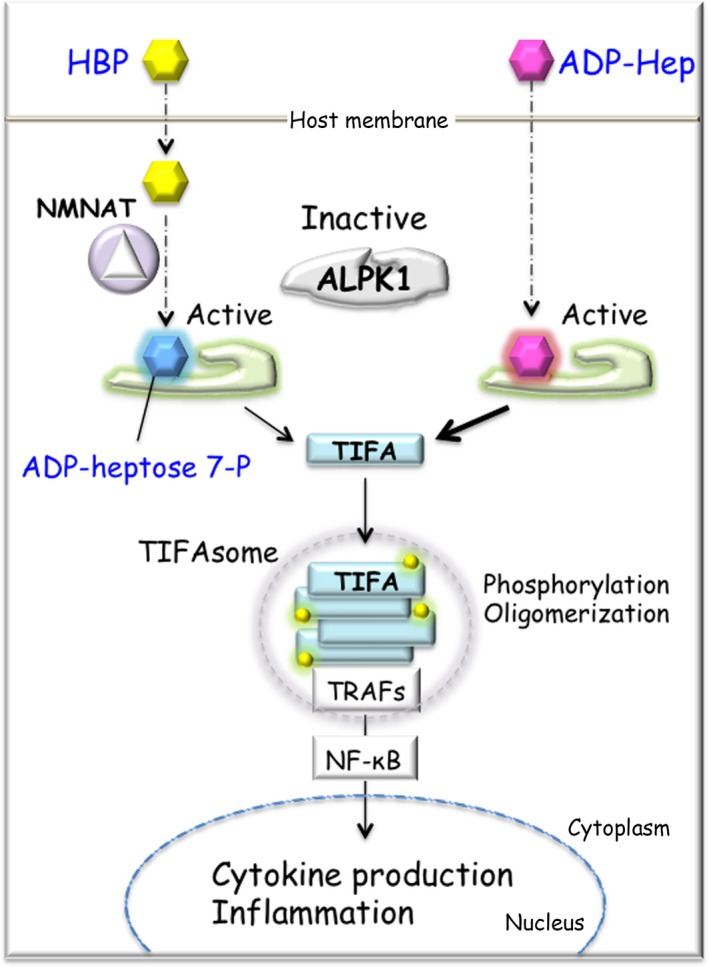
ALPK1‐TIFA‐NF‐κB axis. Once HBP and ADP‐Hep, bacterial products of the LPS biosynthetic pathway, are transported into the host cell, both types of sugars activate NF‐κB signaling pathway. ADP‐heptose 7‐P, which is converted from HBP by host enzyme NMNAT, interacts to N‐terminus of ALPK1. Activated ALPK1 phosphorylates TIFA and induces the TIFA oligomerization with TRAFs, named TIFAsome. ADP‐Hep can also interact with ALPK1 in the same fashion, leading to the activation of NF‐κB and inflammation. ADP‐Hep has much more potent NF‐κB activator than HBP. ADP‐Hep, ADP‐β‐D‐manno‐heptose; ALPK1, alpha‐kinase 1; HBP, D‐glycero‐β‐D‐manno‐heptose 1,7‐bisphosphate; LPS, lipopolysaccharide; NMNAT, nicotinamide mononucleotide adenylyltransferase; TIFA, TRAF‐interacting protein with forkhead‐associated domain; TRAF, TNF receptor‐associated factor
10. CONCLUSION
Over 20 years, we have witnessed a remarkable advance in understanding the mechanism of pathogen recognition by the innate immune system. New players involved in the innate immune system continue to be reported. However, despite enormous efforts, our knowledge of how the innate immune system is involved in the development of allergic diseases is still limited, and feasible target molecules or pathways are yet to be discovered. It is necessary to determine how pathogen recognition molecules and subsequent signaling pathways are involved in the development of allergic diseases.
CONFLICTS OF INTEREST
Kazuhiko Maeda has research collaboration with Otsuka Pharmaceutical Co., Ltd. Shizuo Akira has research support from Chugai Pharmaceutical Co., Ltd. The terms of this arrangement have been reviewed and approved by Osaka University in accordance with its policy on objectivity in research.
Maeda K, Caldez MJ, Akira S. Innate immunity in allergy. Allergy. 2019;74:1660–1674. 10.1111/all.13788
Funding information
Work from our laboratory group is supported in part by the Grant‐in‐Aid for Scientific Research (C) (18K07173 to KM) and Specially Promoted Research (15H05704 to SA) of the Japan Society for the Promotion of Science (JSPS).
REFERENCES
- 1. Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol. 2009;21:317‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Janeway CA Jr, Medzhitov R. Constructing and contesting legitimacy and accountability in polycentric regulatory regimes. Annu Rev Immunol. 2008;20(2): 197‐216. [Google Scholar]
- 3. Stefka AT, Feehley T, Tripathi P, et al. When institutions matter: union growth and decline in Western Europe, 1950‐1995. Eur Sociol Rev. 1999;15(2):135‐158. [Google Scholar]
- 4. Lambrecht BN, Hammad H. The immunology of the allergy epidemic and the hygiene hypothesis. Nat Immunol. 2017;18:1076‐1083. [DOI] [PubMed] [Google Scholar]
- 5. Conrad ML, Ferstl R, Teich R, et al. Maternal TLR signaling is required for prenatal asthma protection by the nonpathogenic microbe Acinetobacter lwoffii F78. J Exp Med . 2009;206:2869‐2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fujimura KE, Lynch SV. Microbiota in allergy and asthma and the emerging relationship with the gut microbiome. Cell Host Microbe . 2015;17:592‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. West CE, Renz H, Jenmalm MC, et al. Bacteriology in the service of sanitation: the factory environment and the regulation of industrial anthrax in late‐victorian Britain. Soc Hist Med. 2012;25:343‐361. [Google Scholar]
- 8. Walton KL, Galanko JA, Balfour Sartor R, Fisher NC. T cell‐mediated oral tolerance is intact in germ‐free mice. Clin Exp Immunol. 2006;143:503‐512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sudo N, Sawamura S, Tanaka K, Aiba Y, Kubo C, Koga Y. The requirement of intestinal bacterial flora for the development of an IgE production system fully susceptible to oral tolerance induction. J Immunol. 1997;159:1739‐1745. [PubMed] [Google Scholar]
- 10. Torii I, Shimizu S, Daimon T, et al. Exposure to high doses of lipopolysaccharide during ovalbumin sensitization prevents the development of allergic Th2 responses to a dietary antigen. J Toxicol Pathol. 2014;27:205‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y, Zhou X, Zhou B. DC‐derived TSLP promotes Th2 polarization in LPS‐primed allergic airway inflammation. Eur J Immunol. 2012;42:1735‐1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou B, Comeau MR, Smedt TD, et al. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat Immunol. 2005;6:1047‐1053. [DOI] [PubMed] [Google Scholar]
- 13. Comeau MR, Ziegler SF. The influence of TSLP on the allergic response. Mucosal Immunol. 2010;3:138‐147. [DOI] [PubMed] [Google Scholar]
- 14. Fort MM, Cheung J, Yen D, et al. IL‐25 induces IL‐4, IL‐5, and IL‐13 and Th2‐associated pathologies in vivo. Immunity. 2001;15:985‐995. [DOI] [PubMed] [Google Scholar]
- 15. Angkasekwinai P, Park H, Wang Y‐H, et al. Interleukin 25 promotes the initiation of proallergic type 2 responses. J Exp Med. 2007;204:1509‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schmitz J, Owyang A, Oldham E, et al. IL‐33, an interleukin‐1‐like cytokine that signals via the IL‐1 receptor‐related protein ST2 and induces T helper type 2‐associated cytokines. Immunity. 2005;23:479‐490. [DOI] [PubMed] [Google Scholar]
- 17. Willart MA, Deswarte K, Pouliot P, et al. Interleukin‐1alpha controls allergic sensitization to inhaled house dust mite via the epithelial release of GM‐CSF and IL‐33. J Exp Med. 2012;209:1505‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martinez‐Gonzalez I, Steer CA, Takei F. Lung ILC2s link innate and adaptive responses in allergic inflammation. Trends Immunol. 2015;36:189‐195. [DOI] [PubMed] [Google Scholar]
- 19. Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol. 2016;17:765‐774. [DOI] [PubMed] [Google Scholar]
- 20. Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. 2015;16:45‐56. [DOI] [PubMed] [Google Scholar]
- 21. Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973‐983. [DOI] [PubMed] [Google Scholar]
- 22. Medzhitov R, Preston‐Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394‐397. [DOI] [PubMed] [Google Scholar]
- 23. Kawai T, Akira S. The role of pattern‐recognition receptors in innate immunity: update on Toll‐like receptors. Nat Immunol. 2010;11:373‐384. [DOI] [PubMed] [Google Scholar]
- 24. Kawai T, Akira S. Toll‐like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637‐650. [DOI] [PubMed] [Google Scholar]
- 25. Pandey S, Kawai T, Akira S. Microbial sensing by Toll‐like receptors and intracellular nucleic acid sensors. Cold Spring Harb Perspect Biol. 2014;7:a016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783‐801. [DOI] [PubMed] [Google Scholar]
- 27. Lin SC, Lo YC, Wu H. Helical assembly in the MyD88‐IRAK4‐IRAK2 complex in TLR/IL‐1R signalling. Nature. 2010;465:885‐890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tesse R, Pandey RC, Kabesch M. Genetic variations in toll‐like receptor pathway genes influence asthma and atopy. Allergy. 2011;66:307‐316. [DOI] [PubMed] [Google Scholar]
- 29. Lee E, Kwon JW, Kim HB, et al. Association between antibiotic exposure, bronchiolitis, and TLR4 (rs1927911) polymorphisms in childhood asthma. Allergy Asthma Immunol Res. 2015;7:167‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xian M, Wawrzyniak P, Ruckert B, et al. Anionic surfactants and commercial detergents decrease tight junction barrier integrity in human keratinocytes. J Allergy Clin Immunol. 2016;138:890‐893. [DOI] [PubMed] [Google Scholar]
- 31. Guo S, Al‐Sadi R, Said HM, Ma TY. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR‐4 and CD14. Am J Pathol. 2013;182:375‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Trompette A, Divanovic S, Visintin A, et al. Allergenicity resulting from functional mimicry of a Toll‐like receptor complex protein. Nature. 2009;457:585‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Millien VO, Lu W, Shaw J, et al. Cleavage of fibrinogen by proteinases elicits allergic responses through Toll‐like receptor 4. Science. 2013;341:792‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll‐like receptor 4 triggering of airway structural cells. Nat Med. 2009;15:410‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Noval Rivas M, Chatila TA. Regulatory T cells in allergic diseases. J Allergy Clin Immunol. 2016;138:639‐652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Walker JA, McKenzie A. TH2 cell development and function. Nat Rev Immunol. 2018;18:121‐133. [DOI] [PubMed] [Google Scholar]
- 37. Hennessy EJ, Parker AE, O'Neill LA. Targeting Toll‐like receptors: emerging therapeutics? Nat Rev Drug Discov. 2010;9:293‐307. [DOI] [PubMed] [Google Scholar]
- 38. Zhu FG, Kandimalla ER, Yu D, Agrawal S. Oral administration of a synthetic agonist of Toll‐like receptor 9 potently modulates peanut‐induced allergy in mice. J Allergy Clin Immunol. 2007;120:631‐637. [DOI] [PubMed] [Google Scholar]
- 39. Aryan Z, Rezaei N. Toll‐like receptors as targets for allergen immunotherapy. Curr Opin Allergy Clin Immunol. 2015;15:568‐574. [DOI] [PubMed] [Google Scholar]
- 40. Tulic MK, Fiset P‐O, Christodoulopoulos P, et al. Amb a 1‐immunostimulatory oligodeoxynucleotide conjugate immunotherapy decreases the nasal inflammatory response. J Allergy Clin Immunol. 2004;113:235‐241. [DOI] [PubMed] [Google Scholar]
- 41. Creticos PS, Schroeder JT, Hamilton RG, et al. Immunotherapy with a ragweed‐toll‐like receptor 9 agonist vaccine for allergic rhinitis. N Engl J Med. 2006;355:1445‐1455. [DOI] [PubMed] [Google Scholar]
- 42. Senti G, Johansen P, Haug S, et al. Use of A‐type CpG oligodeoxynucleotides as an adjuvant in allergen‐specific immunotherapy in humans: a phase I/IIa clinical trial. Clin Exp Allergy. 2009;39:562‐570. [DOI] [PubMed] [Google Scholar]
- 43. Aryan Z, Holgate ST, Radzioch D, Rezaei N. A new era of targeting the ancient gatekeepers of the immune system: toll‐like agonists in the treatment of allergic rhinitis and asthma. Int Arch Allergy Immunol. 2014;164:46‐63. [DOI] [PubMed] [Google Scholar]
- 44. Dambuza IM, Brown GD. C‐type lectins in immunity: recent developments. Curr Opin Immunol. 2015;32:21‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Geijtenbeek TB, Gringhuis SI. C‐type lectin receptors in the control of T helper cell differentiation. Nat Rev Immunol. 2016;16:433‐448. [DOI] [PubMed] [Google Scholar]
- 46. Hoving JC, Wilson GJ, Brown GD. Signalling C‐type lectin receptors, microbial recognition and immunity. Cell Microbiol. 2014;16:185‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zelensky AN, Gready JE. The C‐type lectin‐like domain superfamily. FEBS J. 2005;272:6179‐6217. [DOI] [PubMed] [Google Scholar]
- 48. Saijo S, Iwakura Y. Dectin‐1 and Dectin‐2 in innate immunity against fungi. Int Immunol. 2011;23:467‐472. [DOI] [PubMed] [Google Scholar]
- 49. Plato A, Willment JA, Brown GD. C‐type lectin‐like receptors of the dectin‐1 cluster: ligands and signaling pathways. Int Rev Immunol. 2013;32:134‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kerscher B, Willment JA, Brown GD. The Dectin‐2 family of C‐type lectin‐like receptors: an update. Int Immunol. 2013;25:271‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ishikawa E, Ishikawa T, Morita YS, et al. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C‐type lectin Mincle. J Exp Med. 2009;206:2879‐2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Behler‐Janbeck F, Takano T, Maus R, et al. C‐type lectin mincle recognizes glucosyl‐diacylglycerol of Streptococcus pneumoniae and plays a protective role in pneumococcal pneumonia. Plos Pathog. 2016;12:e1006038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Imai T, Matsumura T, Mayer‐Lambertz S, et al. Lipoteichoic acid anchor triggers Mincle to drive protective immunity against invasive group A Streptococcus infection. Proc Natl Acad Sci U S A. 2018;115:E10662‐E10671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lu X, Nagata M, Yamasaki S. Mincle: 20 years of a versatile sensor of insults. Int Immunol. 2018;30:233‐239. [DOI] [PubMed] [Google Scholar]
- 55. Higashino‐Kameda M, Yabe‐Wada T, Matsuba S, et al. A critical role of Dectin‐1 in hypersensitivity pneumonitis. Inflamm Res. 2016;65:235‐244. [DOI] [PubMed] [Google Scholar]
- 56. Ito T, Hirose K, Norimoto A, et al. Dectin‐1 plays an important role in house dust mite‐induced allergic airway inflammation through the activation of CD11b+ dendritic cells. J Immunol. 2017;198:61‐70. [DOI] [PubMed] [Google Scholar]
- 57. Overton NL, Simpson A, Bowyer P, Denning DW. Genetic susceptibility to severe asthma with fungal sensitization. Int J Immunogenet. 2017;44:93‐106. [DOI] [PubMed] [Google Scholar]
- 58. Barrett NA, Maekawa A, Rahman OM, Austen KF, Kanaoka Y. Dectin‐2 recognition of house dust mite triggers cysteinyl leukotriene generation by dendritic cells. J Immunol. 2009;182:1119‐1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Barrett NA, Rahman OM, Fernandez JM, et al. Dectin‐2 mediates Th2 immunity through the generation of cysteinyl leukotrienes. J Exp Med. 2011;208:593‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Parsons Mw, Li L, Wallace Am, et al. Dectin‐2 regulates the effector phase of house dust mite‐elicited pulmonary inflammation independently from its role in sensitization. J Immunol. 2014;192:1361‐1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kostarnoy AV, Gancheva PG, Lepenies B, et al. Receptor Mincle promotes skin allergies and is capable of recognizing cholesterol sulfate. Proc Natl Acad Sci U S A. 2017;114:E2758‐E2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sirvent S, Soria I, Cirauqui C, et al. Novel vaccines targeting dendritic cells by coupling allergoids to nonoxidized mannan enhance allergen uptake and induce functional regulatory T cells through programmed death ligand 1. J Allergy Clin Immunol. 2016;138:558‐567. [DOI] [PubMed] [Google Scholar]
- 63. Soria I, López‐Relaño J, Viñuela M, et al. Oral myeloid cells uptake allergoids coupled to mannan driving Th1/Treg responses upon sublingual delivery in mice. Allergy. 2018;73:875‐884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Benito‐Villalvilla C, Soria I, Subiza JL, Palomares O. Novel vaccines targeting dendritic cells by coupling allergoids to mannan. Allergo J Int. 2018;27:256‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Edelmann KH, Richardson‐Burns S, Alexopoulou L, Tyler KL, Flavell RA, Oldstone MB. Does Toll‐like receptor 3 play a biological role in virus infections? Virology. 2004;322:231‐238. [DOI] [PubMed] [Google Scholar]
- 66. Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG‐I has an essential function in double‐stranded RNA‐induced innate antiviral responses. Nat Immunol. 2004;5:730‐737. [DOI] [PubMed] [Google Scholar]
- 67. Andrejeva J, Childs KS, Young DF, et al. The V proteins of paramyxoviruses bind the IFN‐inducible RNA helicase, mda‐5, and inhibit its activation of the IFN‐beta promoter. Proc Natl Acad Sci U S A. 2004;101:17264‐17269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yoneyama M, Fujita T. RNA recognition and signal transduction by RIG‐I‐like receptors. Immunol Rev. 2009;227:54‐65. [DOI] [PubMed] [Google Scholar]
- 69. Kato H, Takahasi K, Fujita T. RIG‐I‐like receptors: cytoplasmic sensors for non‐self RNA. Immunol Rev. 2011;243:91‐98. [DOI] [PubMed] [Google Scholar]
- 70. Kato H, Takeuchi O, Sato S, et al. Differential roles of MDA5 and RIG‐I helicases in the recognition of RNA viruses. Nature. 2006;441:101‐105. [DOI] [PubMed] [Google Scholar]
- 71. Pichlmair A, Schulz O, Tan Cp, et al. RIG‐I‐mediated antiviral responses to single‐stranded RNA bearing 5'‐phosphates. Science. 2006;314:997‐1001. [DOI] [PubMed] [Google Scholar]
- 72. Hornung V, Ellegast J, Kim S, et al. 5'‐Triphosphate RNA is the ligand for RIG‐I. Science. 2006;314:994‐997. [DOI] [PubMed] [Google Scholar]
- 73. Goubau D, Schlee M, Deddouche S, et al. Antiviral immunity via RIG‐I‐mediated recognition of RNA bearing 5'‐diphosphates. Nature. 2014;514:372‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kato H, Takeuchi O, Mikamo‐Satoh E, et al. Length‐dependent recognition of double‐stranded ribonucleic acids by retinoic acid‐inducible gene‐I and melanoma differentiation‐associated gene 5. J Exp Med. 2008;205:1601‐1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF‐kappaB and IRF 3. Cell. 2005;122:669‐682. [DOI] [PubMed] [Google Scholar]
- 76. Meylan E, Curran J, Hofmann K, et al. Cardif is an adaptor protein in the RIG‐I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167‐1172. [DOI] [PubMed] [Google Scholar]
- 77. Kawai T, Takahashi K, Sato S, et al. IPS‐1, an adaptor triggering RIG‐I‐ and Mda5‐mediated type I interferon induction. Nat Immunol. 2005;6:981‐988. [DOI] [PubMed] [Google Scholar]
- 78. Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus‐triggered IFN‐beta signaling. Mol Cell. 2005;19:727‐740. [DOI] [PubMed] [Google Scholar]
- 79. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion‐like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Holtzman MJ, Byers DE, Alexander‐Brett J, Wang X. The role of airway epithelial cells and innate immune cells in chronic respiratory disease. Nat Rev Immunol. 2014;14:686‐698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lamborn IT, Jing H, Zhang Yu, et al. Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J Exp Med. 2017;214:1949‐1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Asgari S, Schlapbach LJ, Anchisi S, et al. Severe viral respiratory infections in children with IFIH1 loss‐of‐function mutations. Proc Natl Acad Sci U S A. 2017;114:8342‐8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Diogo D, Tian C, Franklin CS, et al. Phenome‐wide association studies across large population cohorts support drug target validation. Nat Commun. 2018;9:4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mahmutovic Persson I, Akbarshahi H, Menzel M, Brandelius A, Uller L. Increased expression of upstream TH2‐cytokines in a mouse model of viral‐induced asthma exacerbation. J Transl Med. 2016;14:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Beale J, Jayaraman A, Jackson DJ, et al. Rhinovirus‐induced IL‐25 in asthma exacerbation drives type 2 immunity and allergic pulmonary inflammation. Sci Transl Med. 2014;6:256ra134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mahmutovic‐Persson I, Akbarshahi H, Bartlett Nw, et al. Inhaled dsRNA and rhinovirus evoke neutrophilic exacerbation and lung expression of thymic stromal lymphopoietin in allergic mice with established experimental asthma. Allergy. 2014;69:348‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Luecke S, Holleufer A, Christensen MH, et al. cGAS is activated by DNA in a length‐dependent manner. Embo Rep. 2017;18:1707‐1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ablasser A, Goldeck M, Cavlar T, et al. cGAS produces a 2'‐5'‐linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Diner E, Burdette D, Wilson S, et al. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 2013;3:1355‐1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gao Pu, Ascano M, Wu Y, et al. Cyclic [G(2',5')pA(3',5')p] is the metazoan second messenger produced by DNA‐activated cyclic GMP‐AMP synthase. Cell. 2013;153:1094‐1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP‐AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wu J, Sun L, Chen X, et al. Cyclic GMP‐AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826‐830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zhong B, Yang Y, Li S, et al. The adaptor protein MITA links virus‐sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538‐550. [DOI] [PubMed] [Google Scholar]
- 95. Sun W, Li Y, Chen L, et al. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci U S A. 2009;106:8653‐8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Liu S, Cai X, Wu J, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347:aaa2630. [DOI] [PubMed] [Google Scholar]
- 97. Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. 2012;5:ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gao D, Wu J, Wu Y‐t, et al. Cyclic GMP‐AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341:903‐906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal roles of cGAS‐cGAMP signaling in antiviral defense and immune adjuvant effects. Science. 2013;341:1390‐1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Mackenzie KJ, Carroll P, Martin C‐A, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548:461‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548:466‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Chen G, Shaw MH, Kim YG, Nunez G. NOD‐like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol. 2009;4:365‐398. [DOI] [PubMed] [Google Scholar]
- 103. Caruso R, Warner N, Inohara N, Nunez G. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity. 2014;41:898‐908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Humphries F, Yang S, Wang B, Moynagh PN. RIP kinases: key decision makers in cell death and innate immunity. Cell Death Differ. 2015;22:225‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Penack O, Smith OM, Cunningham‐Bussel A, et al. NOD2 regulates hematopoietic cell function during graft‐versus‐host disease. J Exp Med. 2009;206:2101‐2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ogura Y, Lala S, Xin W, et al. Expression of NOD2 in Paneth cells: a possible link to Crohn's ileitis. Gut. 2003;52:1591‐1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Pashenkov MV, Balyasova LS, Dagil YA, Pinegin BV. The role of the p38‐MNK‐eIF4E signaling axis in TNF production downstream of the NOD1 receptor. J Immunol. 2017;198:1638‐1648. [DOI] [PubMed] [Google Scholar]
- 108. Rastogi R, Du W, Ju D, et al. Dysregulation of p38 and MKP‐1 in response to NOD1/TLR4 stimulation in sarcoid bronchoalveolar cells. Am J Respir Crit Care Med. 2011;183:500‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Fritz JH, Le Bourhis L, Sellge G, et al. Nod1‐mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity. 2007;26:445‐459. [DOI] [PubMed] [Google Scholar]
- 110. Duan W, Mehta AK, Magalhaes JG, et al. Innate signals from Nod2 block respiratory tolerance and program T(H)2‐driven allergic inflammation. J Allergy Clin Immunol. 2010;126:1284‐1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ramasamy A, Curjuric I, Coin LJ, et al. A genome‐wide meta‐analysis of genetic variants associated with allergic rhinitis and grass sensitization and their interaction with birth order. J Allergy Clin Immunol. 2011;128:996‐1005. [DOI] [PubMed] [Google Scholar]
- 112. Hysi P, Kabesch M, Moffatt MF, et al. NOD1 variation, immunoglobulin E and asthma. Hum Mol Genet. 2005;14:935‐941. [DOI] [PubMed] [Google Scholar]
- 113. Macaluso F, Nothnagel M, Parwez Q, et al. Polymorphisms in NACHT‐LRR (NLR) genes in atopic dermatitis. Exp Dermatol. 2007;16:692‐698. [DOI] [PubMed] [Google Scholar]
- 114. Bursztejn Ac, Romano A, Guéant‐Rodriguez Rm, et al. Allergy to betalactams and nucleotide‐binding oligomerization domain (NOD) gene polymorphisms. Allergy. 2013;68:1076‐1080. [DOI] [PubMed] [Google Scholar]
- 115. Kabesch M, Peters W, Carr D, Leupold W, Weiland SK, von Mutius E. Association between polymorphisms in caspase recruitment domain containing protein 15 and allergy in two German populations. J Allergy Clin Immunol. 2003;111:813‐817. [DOI] [PubMed] [Google Scholar]
- 116. Keestra‐Gounder AM, Byndloss MX, Seyffert N, et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature. 2016;532:394‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407‐420. [DOI] [PubMed] [Google Scholar]
- 118. Dorfleutner A, Chu L, Stehlik C. Inhibiting the inflammasome: one domain at a time. Immunol Rev. 2015;265:205‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Indramohan M, Stehlik C, Dorfleutner A. COPs and POPs patrol inflammasome activation. J Mol Biol. 2018;430:153‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev. 2015;265:6‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Franchi L, Munoz‐Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13:325‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Cookson BT, Brennan MA. Pro‐inflammatory programmed cell death. Trends Microbiol. 2001;9:113‐114. [DOI] [PubMed] [Google Scholar]
- 123. Nadiri A, Wolinski MK, Saleh M. The inflammatory caspases: key players in the host response to pathogenic invasion and sepsis. J Immunol. 2006;177:4239‐4245. [DOI] [PubMed] [Google Scholar]
- 124. He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41:1012‐1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826‐837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Abais JM, Xia M, Zhang Y, Boini KM, Li PL. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal. 2015;22:1111‐1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Netea MG, van de Veerdonk FL, van der Meer JW, Dinarello CA, Joosten LA. Inflammasome‐independent regulation of IL‐1‐family cytokines. Annu Rev Immunol. 2015;33:49‐77. [DOI] [PubMed] [Google Scholar]
- 128. Carta S, Penco F, Lavieri R, et al. Cell stress increases ATP release in NLRP3 inflammasome‐mediated autoinflammatory diseases, resulting in cytokine imbalance. Proc Natl Acad Sci U S A. 2015;112:2835‐2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Chung Y, Chang SH, Martinez GJ, et al. Critical regulation of early Th17 cell differentiation by interleukin‐1 signaling. Immunity. 2009;30:576‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Banuelos J, Cao Y, Shin SC, Lu NZ. Immunopathology alters Th17 cell glucocorticoid sensitivity. Allergy. 2017;72:331‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Xiao Y, Xu W, Su W. NLRP3 inflammasome: a likely target for the treatment of allergic diseases. Clin Exp Allergy. 2018;48:1080‐1091. [DOI] [PubMed] [Google Scholar]
- 132. Choy DF, Hart KM, Borthwick LA, et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med. 2015;7:301ra129. [DOI] [PubMed] [Google Scholar]
- 133. Unterholzner L, Keating SE, Baran M, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11:997‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Jin T, Perry A, Jiang J, et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity. 2012;36:561‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Jin T, Perry A, Smith P, Jiang J, Xiao TS. Structure of the absent in melanoma 2 (AIM2) pyrin domain provides insights into the mechanisms of AIM2 autoinhibition and inflammasome assembly. J Biol Chem. 2013;288:13225‐13235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Hornung V, Ablasser A, Charrel‐Dennis M, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase‐1‐activating inflammasome with ASC. Nature. 2009;458:514‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Fernandes‐Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Bürckstümmer T, Baumann C, Blüml S, et al. An orthogonal proteomic‐genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009;10:266‐272. [DOI] [PubMed] [Google Scholar]
- 139. Roberts Tl, Idris A, Dunn Ja, et al. HIN‐200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057‐1060. [DOI] [PubMed] [Google Scholar]
- 140. Man SM, Karki R, Kanneganti TD. AIM2 inflammasome in infection, cancer, and autoimmunity: role in DNA sensing, inflammation, and innate immunity. Eur J Immunol. 2016;46:269‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Su S, Zhao J, Xing Y, et al. Immune checkpoint inhibition overcomes ADCP‐induced immunosuppression by macrophages. Cell. 2018;175:442‐457. [DOI] [PubMed] [Google Scholar]
- 142. Wang Y, Ning X, Gao P, et al. Inflammasome activation triggers caspase‐1‐mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity. 2017;46:393‐404. [DOI] [PubMed] [Google Scholar]
- 143. Gaidt MM, Ebert TS, Chauhan D, et al. The DNA inflammasome in human myeloid cells is initiated by a STING‐cell death program upstream of NLRP3. Cell. 2017;171:1110‐1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Swanson KV, Junkins RD, Kurkjian CJ, et al. A noncanonical function of cGAMP in inflammasome priming and activation. J Exp Med. 2017;214:3611‐3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Duncan JA, Canna SW. The NLRC4 inflammasome. Immunol Rev. 2018;281:115‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Reyes Ruiz VM, Ramirez J, Naseer N, et al. Broad detection of bacterial type III secretion system and flagellin proteins by the human NAIP/NLRC4 inflammasome. Proc Natl Acad Sci U S A. 2017;114:13242‐13247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. 2011;477:592‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Zhao Y, Yang J, Shi J, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477:596‐600. [DOI] [PubMed] [Google Scholar]
- 149. Zhao Y, Shi J, Shi X, Wang Y, Wang F, Shao F. Genetic functions of the NAIP family of inflammasome receptors for bacterial ligands in mice. J Exp Med. 2016;213:647‐656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Rauch I, Tenthorey JL, Nichols RD, et al. NAIP proteins are required for cytosolic detection of specific bacterial ligands in vivo. J Exp Med. 2016;213:657‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Karki R, Lee E, Place D, et al. IRF8 regulates transcription of naips for NLRC4 inflammasome activation. Cell. 2018;173:920‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Hiruma J, Harada K, Motoyama A, et al. Key component of inflammasome, NLRC4, was identified in the lesional epidermis of psoriatic patients. J Dermatol. 2018;45:971‐977. [DOI] [PubMed] [Google Scholar]
- 153. Uh ST, Park JS, Koo SM, et al. Association of genetic variants of NLRP4 with exacerbation of asthma: the effect of smoking. DNA Cell Biol. 2019;38:76‐84. [DOI] [PubMed] [Google Scholar]
- 154. Rathinam V, Vanaja S, Waggoner L, et al. TRIF licenses caspase‐11‐dependent NLRP3 inflammasome activation by gram‐negative bacteria. Cell. 2012;150:606‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Kayagaki N, Warming S, Lamkanfi M, et al. Non‐canonical inflammasome activation targets caspase‐11. Nature. 2011;479:117‐121. [DOI] [PubMed] [Google Scholar]
- 156. Akhter A, Caution K, Abu Khweek A, et al. Caspase‐11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity. 2012;37:35‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Case Cl, Kohler Lj, Lima Jb, et al. Caspase‐11 stimulates rapid flagellin‐independent pyroptosis in response to Legionella pneumophila . Proc Natl Acad Sci U S A. 2013;110:1851‐1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Aachoui Y, Leaf Ia, Hagar Ja, et al. Caspase‐11 protects against bacteria that escape the vacuole. Science. 2013;339:975‐978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase‐11: implications in TLR4‐independent endotoxic shock. Science. 2013;341:1250‐1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Kayagaki N, Wong Mt, Stowe Ib, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246‐1249. [DOI] [PubMed] [Google Scholar]
- 161. Shi J, Zhao Y, Wang Y, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187‐192. [DOI] [PubMed] [Google Scholar]
- 162. Stowe I, Lee B, Kayagaki N. Caspase‐11: arming the guards against bacterial infection. Immunol Rev. 2015;265:75‐84. [DOI] [PubMed] [Google Scholar]
- 163. Hara H, Seregin SS, Yang D, et al. The NLRP6 inflammasome recognizes lipoteichoic acid and regulates gram‐positive pathogen infection. Cell. 2018;175:1651‐1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Rieber N, Singh A, Öz H, et al. Pathogenic fungi regulate immunity by inducing neutrophilic myeloid‐derived suppressor cells. Cell Host Microbe. 2015;17:507‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Gringhuis SI, Kaptein TM, Wevers BA, et al. Dectin‐1 is an extracellular pathogen sensor for the induction and processing of IL‐1beta via a noncanonical caspase‐8 inflammasome. Nat Immunol. 2012;13:246‐254. [DOI] [PubMed] [Google Scholar]
- 166. Ganesan S, Rathinam V, Bossaller L, et al. Caspase‐8 modulates dectin‐1 and complement receptor 3‐driven IL‐1beta production in response to beta‐glucans and the fungal pathogen, Candida albicans . J Immunol. 2014;193:2519‐2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Gurung P, Anand Pk, Malireddi R, et al. FADD and caspase‐8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol. 2014;192:1835‐1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Qi X, Gurung P, Malireddi R, et al. Critical role of caspase‐8‐mediated IL‐1 signaling in promoting Th2 responses during asthma pathogenesis. Mucosal Immunol. 2017;10:128‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Zhong FL, Mamai O, Sborgi L, et al. Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell. 2016;167:187‐202. [DOI] [PubMed] [Google Scholar]
- 170. Cunha LD, Ribeiro JM, Fernandes TD, et al. Inhibition of inflammasome activation by Coxiella burnetii type IV secretion system effector IcaA. Nat Commun. 2015;6:10205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Malireddi R, Gurung P, Mavuluri J, et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J Exp Med. 2018;215:1023‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Orning P, Weng D, Starheim K, et al. Pathogen blockade of TAK1 triggers caspase‐8‐dependent cleavage of gasdermin D and cell death. Science. 2018;362:1064‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Xu D, Jin T, Zhu H, et al. TBK1 suppresses RIPK1‐driven apoptosis and inflammation during development and in aging. Cell. 2018;174:1477‐1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Lamkanfi M, Dixit VM. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe. 2010;8:44‐54. [DOI] [PubMed] [Google Scholar]
- 175. Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532‐1535. [DOI] [PubMed] [Google Scholar]
- 176. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. 2017;23:279‐287. [DOI] [PubMed] [Google Scholar]
- 177. Toussaint M, Jackson DJ, Swieboda D, et al. Host DNA released by NETosis promotes rhinovirus‐induced type‐2 allergic asthma exacerbation. Nat Med. 2017;23:681‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Toussaint M, Jackson DJ, Swieboda D, et al. Corrigendum: host DNA released by NETosis promotes rhinovirus‐induced type‐2 allergic asthma exacerbation. Nat Med. 2017;23:1384. [DOI] [PubMed] [Google Scholar]
- 179. Feng S, Fox D, Man SM. Mechanisms of gasdermin family members in inflammasome signaling and cell death. J Mol Biol. 2018;430:3068‐3080. [DOI] [PubMed] [Google Scholar]
- 180. Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660‐665. [DOI] [PubMed] [Google Scholar]
- 181. Liu X, Zhang Z, Ruan J, et al. Inflammasome‐activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. He WT, Wan H, Hu L, et al. Gasdermin D is an executor of pyroptosis and required for interleukin‐1beta secretion. Cell Res. 2015;25:1285‐1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Ding J, Wang K, Liu W, et al. Pore‐forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111‐116. [DOI] [PubMed] [Google Scholar]
- 184. Aglietti RA, Estevez A, Gupta A, et al. GsdmD p30 elicited by caspase‐11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A. 2016;113:7858‐7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Sborgi L, Rühl S, Mulvihill E, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35:1766‐1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Chen X, He W‐T, Hu L, et al. Pyroptosis is driven by non‐selective gasdermin‐D pore and its morphology is different from MLKL channel‐mediated necroptosis. Cell Res. 2016;26:1007‐1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Sollberger G, Choidas A, Burn GL, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3:eaar6689. [DOI] [PubMed] [Google Scholar]
- 188. Rathkey JK, Zhao J, Liu Z, et al. Chemical disruption of the pyroptotic pore‐forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol. 2018;3:eaat2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Chen KW, Monteleone M, Boucher D, et al. Noncanonical inflammasome signaling elicits gasdermin D‐dependent neutrophil extracellular traps. Sci Immunol. 2018;3:eaar6676. [DOI] [PubMed] [Google Scholar]
- 190. Moffatt MF, Kabesch M, Liang L, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470‐473. [DOI] [PubMed] [Google Scholar]
- 191. Moffatt MF, Gut IG, Demenais F, et al. A large‐scale, consortium‐based genomewide association study of asthma. N Engl J Med. 2010;363:1211‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Das S, Miller M, Beppu AK, et al. GSDMB induces an asthma phenotype characterized by increased airway responsiveness and remodeling without lung inflammation. Proc Natl Acad Sci U S A. 2016;113:13132‐13137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Gaudet Rg, Sintsova A, Buckwalter Cm, et al. Cytosolic detection of the bacterial metabolite HBP activates TIFA‐dependent innate immunity. Science. 2015;348:1251‐1255. [DOI] [PubMed] [Google Scholar]
- 194. Milivojevic M, Dangeard AS, Kasper CA, et al. ALPK1 controls TIFA/TRAF6‐dependent innate immunity against heptose‐1,7‐bisphosphate of gram‐negative bacteria. Plos Pathog. 2017;13:e1006224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Zimmermann S, Pfannkuch L, Al‐Zeer MA, et al. ALPK1‐ and TIFA‐dependent innate immune response triggered by the Helicobacter pylori type IV secretion system. Cell Rep. 2017;20:2384‐2395. [DOI] [PubMed] [Google Scholar]
- 196. Kanamori M, Suzuki H, Saito R, Muramatsu M, Hayashizaki Y. T2BP, a novel TRAF2 binding protein, can activate NF‐kappaB and AP‐1 without TNF stimulation. Biochem Biophys Res Commun. 2002;290:1108‐1113. [DOI] [PubMed] [Google Scholar]
- 197. Zhou P, She Y, Dong Na, et al. Alpha‐kinase 1 is a cytosolic innate immune receptor for bacterial ADP‐heptose. Nature. 2018;561:122‐126. [DOI] [PubMed] [Google Scholar]
- 198. García‐Weber D, Dangeard AS, Cornil J, et al. ADP‐heptose is a newly identified pathogen‐associated molecular pattern of Shigella flexneri . EMBO Rep. 2018;19:e46943. [DOI] [PMC free article] [PubMed] [Google Scholar]