

Abstract
Light‐inducible approaches provide a means to control biological systems with spatial and temporal resolution that is unmatched by traditional genetic perturbations. Recent developments of optogenetic and chemo‐optogenetic systems for induced proximity in cells facilitate rapid and reversible manipulation of highly dynamic cellular processes and have become valuable tools in diverse biological applications. New expansions of the toolbox facilitate control of signal transduction, genome editing, “painting” patterns of active molecules onto cellular membranes, and light‐induced cell cycle control. A combination of light‐ and chemically induced dimerization approaches have also seen interesting progress. Herein, an overview of optogenetic systems and emerging chemo‐optogenetic systems is provided, and recent applications in tackling complex biological problems are discussed.
Keywords: chemo-optogenetics, dimerization, optogenetics, photochemistry, proteins
Proteins under the spotlight: Various optogenetic systems that involve photosensitive proteins are summarized and emerging chemo‐optogenetic systems with caged or photocleavable chemical dimerizers are reviewed (see figure). The advantages and disadvantages of these systems are discussed. Recent biological applications of these strategies are presented.
1. Introduction
Proximity is an essential regulatory factor in biological processes, which can be controlled by induced dimerization approaches. The applications of induced proximity range from manipulation of protein folding, activation, localization, and degradation to controlling gene transcription or cell therapy.1 Although novel tools are continuously emerging, the underlying principle of induced dimerization remains constant: two proteins are artificially brought into close proximity upon stimulation by chemicals or light. In contrast to traditional genetic perturbation, manipulation by induced dimerization approaches features superior spatiotemporal resolution and is thus better suited for studying dynamics and spatiotemporally confined processes.2
Induced dimerization systems differ mostly according to their dimerization trigger. Chemically induced dimerization (CID) systems are based on small molecules interacting with two identical proteins (homodimerization) or two different proteins (heterodimerization).2b, 3 Optogenetic dimerization systems employ photosensitive proteins that undergo a conformational change upon illumination, and consequently, induce protein interaction.4 Chemo‐optogenetic dimerization systems use photoactivatable and/or ‐cleavable small‐molecule dimerizers, so that proximity can be induced and/or disrupted by light.
An ideal dimerization system features high specificity and binding affinity, bioorthogonality, reversibility, simple operation, and high spatiotemporal resolution. CID systems are facile to operate by adding compounds and are tunable by controlling the dosage of compounds.5 Chemical dimerizers can penetrate tissues inaccessible to light. The dimerization kinetics range from seconds to minutes. Whereas first‐generation dimerizers, such as rapamycin and rapalogs,6 suffer from poor reversibility and bioorthogonality, more recent CID systems, such as radical‐induced cell death protein 1 (rCD1) and SLF′‐TMP (SLF′=a derivative of a synthetic ligand of FKBP12(F36V); TMP=trimethoprim), have largely overcome these limitations.7
However, light‐induced dimerization systems confer a spatiotemporal resolution that is unmatched by chemical approaches. Dimerization kinetics are not limited by the diffusion rate into the cell and photoactivation can be performed in a spatially defined manner at a subcellular level. Still, the photoactivation conditions, that is, wavelength and intensity, are also decisive factors because short wavelength leads to poor tissue penetration of light and intensive illumination can cause phototoxicity to cells. We summarize different strategies of optogenetic systems and emerging chemo‐optogenetic systems and present their recent applications in biological research.
2. Optogenetic Systems
Light‐sensitive regulatory proteins occur naturally throughout all kingdoms of life, orchestrating processes such as plant development, gene expression, and circadian rhythm and facilitating the communication of visual information. Optogenetic systems based on natural photoreceptors have been employed to control intracellular processes. These photoreceptors contain a chromophore that undergoes isomerization or formation of a chemical bond upon absorption of a photon, leading to a conformational change in the photoreceptor that is eventually propagated to the effector domain.8 Although some photoreceptors, such as rhodopsins, integrate both sensory and effector functions, most photoreceptors, such as light–oxygen–voltage (LOV) proteins, cryptochromes (CRYs), and phytochromes, mediate intra‐ or intermolecular interactions in response to light.9
LOV domains are flavin mononucleotide (FMN) binding photosensors and form a transient covalent bond to FMN molecules upon blue‐light activation that may remain stable for seconds to days.10 The first LOV‐based dimerization system involving the LOV domain‐containing protein FKF1 and the interacting GIGANTEA (GI) protein from Arabidopsis thaliana suffer from slow association and dissociation kinetics.11 The LOV2 domain from Avena sativa phototropin was employed to build photoactivatable Rac1 GTPase for control of localized cell protrusion.12 The LOV2 domain features a characteristic C‐terminal helical extension, termed Jα. In the dark, the helix associates with LOV2, which enables association of LOV2 with Rac1 and steric inhibition of Rac1 interaction with its effectors. Upon photoactivation with blue light, Jα unwinds and dissociates from the LOV2 domain rapidly. Another light‐induced dimerization system, termed a tunable light‐inducible dimerization tag (TULIP), is based on the interaction between LOV2 and variants of the engineered PDZ domain (ePDZ). Upon photoactivation, the ePDZ‐binding peptide is exposed and facilitates dimerization of the LOVpep domain and the ePDZ domain.13 The LOV2–peptide interaction system has been optimized through an improved light‐induced dimer (iLID) approach by using the bacterial SsrA peptide and its binding partner SspB, which displays more than 50‐fold change in binding affinity upon light activation.14 Engineering of the LOV domain led to LOV variants with half‐lives of thermal reversion in the dark ranging from 28 s to 50 h and with improved dynamic range.15 Recently, an approach called LOV2 trap and release of protein (LOVTRAP) was used to control protein release, in which the Zdark (Zdk) protein binds to the LOV2 domain in the dark and dissociates upon photoactivation with >150‐fold change in dissociation constant.16 Vivid (VVD), the smallest LOV‐containing protein from fungi, and its engineered variants (Magnets) have been used to control homo‐ and heterodimerization, respectively.17
CRY proteins are photoreceptors that contain a conserved N‐terminal photolyase homology region (PHR) that binds a flavin adenine dinucleotide (FAD) chromophore.18 A light‐induced dimerization system was developed based on the CRY2 domain from A. thaliana, which bound CRY‐interacting basic‐helix‐loop‐helix (CIB1) or its shorter N‐terminal variant (CIBN) in its photoexcited state.19 The light‐induced dimerization of CRY2 with CIBN is complete within 10 s and slowly reverses over 12 min in the dark. New engineered variants of CRY2 have been developed to improve the dynamic range (reduced dark activity) and to alter photocycle kinetics with longer or shorter half‐lives for CIB1 binding.20 However, CRY2 tends to form oligomeric clusters in response to blue light. CRY2 or its E490G mutant alone was also employed to reversibly control clustering of proteins in live cells.21
Whereas LOV‐ or CRY‐based systems rely on blue‐light‐sensitive photoreceptors, phytochromes with a tetrapyrrole chromophore are red‐light sensitive. Red light can penetrate deeper into tissues and has a lower energy, and thus, produces less phototoxicity. In this regard, it can be more beneficial for optogenetic control in organisms. Phytochromes allosterically switch between the red‐sensitive Pr and far‐red‐sensitive Pfr conformational states. The conformational change from Pr to Pfr is triggered by photoactivation at λ=650 nm, and can be reversed by illumination at λ=750 nm.9a The A. thaliana phytochrome B (PhyB) protein binds to a downstream transcription factor, phytochrome interaction factor 3 (PIF3), only in the Pfr state, but shows no measurable binding affinity in the Pr state. Thus, dimerization can be controlled by red and far‐red light with rapid on/off kinetics in seconds.22 However, in contrast to LOV and CRY systems that use endogenous flavin chromophores, mammalian cells do not produce tetrapyrroles, such as phytochrombilin or phycocyanobilin (PCB). The chromophore has to be added exogenously, which may lead to a fluorescent background.22 Alternatively, bacterial phytochrome photoreceptors (BpHPs) that use endogenous biliverdin chromophores can be employed.23 The interaction of Rhodopseudomonas palustris BphP1 with its binding partner PpsR2 (or the Q‐PAS1 domain of PpsR2) can be switched on and off by illumination at λ=740 and 650 nm, respectively.24
Other photosensitive proteins with absorption at different wavelengths, such as UVR8; the fluorescent protein (FP) Dronpa; and cobalamin (vitamin B12) binding domains (CBDs) have been added to the optogenetic toolbox. Dronpa variants switch from fluorescent tetra‐ or dimers to non‐fluorescent monomers under violet light (λ≈400 nm) and are switched off under cyan light (λ≈500 nm).25 However, UVR8 dedimerizes or binds to COP1 irreversibly upon illumination by UV‐B light.26 The chromophore of CBDs, 5′‐deoxyadenosylcobalamin (AdoCbl), absorbs green light, which fills the gap of the optogenetic spectrum. Oligomerization of CBDs in the dark was disrupted by illumination with green light (λ=545 nm).27
Optogenetic systems that use photosensitive proteins are powerful tools (Figure 1); however, several considerations should be taken into account when choosing and operating these systems. In the case of the PhyB‐PIF system, constant illumination with a ratio of stimulatory light and inhibitory light has to be used to maintain activation at a certain level.28 However, this requires simultaneous operation of λ=650 and 750 nm light through a digital micromirror device, which substantially complicates the experimental setup.
Figure 1.
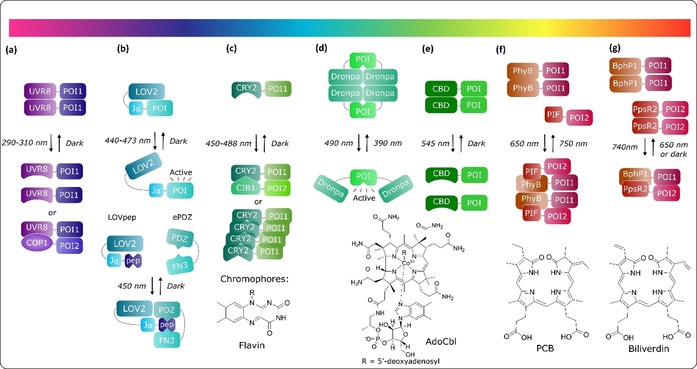
Optogenetic dimerization systems. a) UVR8 proteins homodimerize in the dark, and dissociate to monomers and bind to COP1 upon photoinduced (λ=290–315 nm) conformational change. b) Illumination of LOV2 at λ=440–473 nm causes unwinding of the Jα helix and uncaging of the protein of interest (POI). c) Photoactivation of CRY2 at λ=450–488 nm causes a conformational change, leading to oligomerization of CRY2 or interaction with CIB1. d) The fluorescent Dronpa proteins form tetra‐ or homodimers in the dark and dissociate after illumination at λ=490 nm. The process is reversible by illumination at λ=400 nm. e) CBDs oligomerize in the dark and dissociate upon green‐light illumination. f) PhyB interacts with PIF3/6 under illumination at λ=650 nm, which can be reversed by illumination at λ=750 nm. g) BphP1 interacts with PpsR2 under illumination at λ=750 nm, which can be reversed by illumination at λ=650 nm or in the dark. Chromophores: flavin for LOV2 and CRY2, AdoCbl for CBD, PCB for PhyB and Cph1, and biliverdin for BphP1.
Photosensitive proteins exist in equilibrium between active and inactive conformations. Variants that return rapidly to the off state in the dark require more frequent pulses of illumination, whereas those with a slow off rate are essentially irreversible. To ensure photosensitive proteins are constantly active, pulses of illumination are often required. However, longer exposure of cells to light may lead to phototoxicity.
Optogenetic systems also vary in their dynamic range, which is determined by the basal activity of photosensitive proteins in the dark and the affinity of binding pairs. The basal activation in the PhyB system could be minimized by illumination with inactivating light.22 The optogenetic binding modules also suffer from non‐zero affinity in the dark state, leading to preactivation of the POI. In theory, improved binding affinity results in a kinetically slower system and enhances the binding of optogenetic modules in the dark.29
The absorption spectra of photosensitive proteins (e.g., LOV: λ=400–500 nm, Cry2: λ=390–500 nm, PhyB: λ=550–800 nm, Dronpa: λ=350–530 nm) overlap with commonly used FPs. As a result, the number of FPs that can be used in optogenetic experiments is greatly limited because light wavelengths for imaging and perturbation have to be orthogonal to each other, that is, the absorption spectra have to be sufficiently separated or the excitation wavelength of the FP does not perturb the photosensitive protein. Therefore, photosensitive proteins that absorb far‐red and near‐infrared (NIR) light, such as the BphP1‐PpsR2 system would be more beneficial in this regard.24a Light at wavelengths of 514 and 405 nm is orthogonal to the perturbation light in the PhyB‐PIF system.28, 30 Accordingly, FPs or organic dyes with orthogonal absorption to the photosensitive protein would be a solution.
A summary of the properties of selected optogenetic dimerization systems is provided in Table 1.
Table 1.
Properties of optogenetic dimerization systems.
|
System |
λ [nm] |
Timescale |
Ref. |
|
|---|---|---|---|---|
|
|
Association |
Dissociation |
|
|
|
UVR8‐COP1 |
290–310 |
dark |
dimerization: s–h (depending on the diffusion and association of UVR8 with COP1) dissociation: no |
|
|
UVR8‐UVR8 |
dark |
312 |
re‐dimerization: no dissociation: t 1/2=1.9 s |
|
|
FKF1‐GI |
450 |
dark |
dimerization: min dissociation: h |
|
|
TULIPs |
440/473 |
dark |
dimerization: s |
|
|
LOVpep‐ePDZ |
dissociation: s‐min |
|||
|
iLID |
488 |
dark |
dimerization: s |
|
|
LOVSsrA‐SsrB |
dissociation: s |
|||
|
LightOn |
450 |
dark |
dimerization: min |
|
|
VVD‐VVD |
dissociation: h |
|||
|
Magnets |
470 |
dark |
dimerization: s |
|
|
pMag‐nMag (VVD variants) |
dissociation: s–h |
|||
|
LOVTRAP |
dark |
450–490 |
dimerization: t 1/2=1.7–496 s |
|
|
LOV2‐Zdk |
dissociation: <1 s |
|||
|
CRY2‐CIB1/CIBN |
450–488 (two‐photon stimulation at 860 nm) |
dark |
dimerization: s dissociation: t 1/2=5.5 min |
|
|
CRY2‐CIB1 variants |
461 |
dark |
dimerization: s dissociation: t 1/2=2.5–24 min |
|
|
CRY2‐CRY2 |
470 |
dark |
dimerization: t 1/2=16–42 s dissociation: t 1/2=5.5 min |
|
|
CRY2 olig |
488 |
dark |
dimerization: t 1/2=15–75 s |
|
|
CRY2‐CRY2 (E490G mutant) |
dissociation: t 1/2=23.1 min |
|||
|
Dronpa‐Dronpa |
390 |
490 |
dimerization: min dissociation: 20 s–2 min |
|
|
CBD‐CBD |
dark |
545 |
dimerization: min dissociation: min |
|
|
PhyB‐PIF3/6 |
650 |
750 |
dimerization: t 1/2=1.3 s dissociation: t1/2=4 s |
|
|
Cph1‐Cph1 |
630/647 |
720 or dark |
dimerization: min dissociation: min in dark |
|
|
BphP1‐PpsR2 |
740 |
650 or dark |
Pfr→Pr (740 nm): t 1/2=3.5–28 s Pr→Pfr (636 nm): t 1/2=3‐21 s (t 1/2=170 s in the dark) |
|
3. Chemo‐Optogenetic Systems
Chemo‐optogenetic systems are based on CID systems that are controlled by light. These chemo‐optogenetic systems employ caged or photocleavable chemical dimerizers that can be activated or deactivated by light.
Based on the chemical dimerizer rapamycin, three light‐inducible photocaged dimerizers have been reported: pRap, cRb, and dRap. pRap was created by introducing a photolabile α‐methyl‐6‐nitropiperonyloxycarbonyl (MeNPOC) group to the C‐40 position of rapamycin (Figure 2 c).32 To increase the caging efficacy, rapamycin was linked to a more bulky group to hinder binding. In the dRap approach, a rapamycin dimer was linked through a photocleavable methyl‐6‐nitroveratryloxycarbonyl (MeNVOC) moiety (Figure 2 b),33 whereas, in the case of cRb, rapamycin was coupled to a biotin–avidin complex through a photocleavable MeNVOC linker (Figure 2 a).34 Due to the large size of the biotin–avidin complex, cRb can cross the plasma membrane upon photocleavage of the biotin group, leading to local release of rapamycin and subsequent localized activation of Rho GTPase. The uncaging of rapamycin with λ=365 nm UV light and subsequent dimerization could be achieved within a minute. However, rapamycin also binds to endogenous proteins, leading to off‐target effects. Due to the slow off rate of rapamycin‐induced dimerization, the rapamycin‐based system is essentially irreversible.
Figure 2.
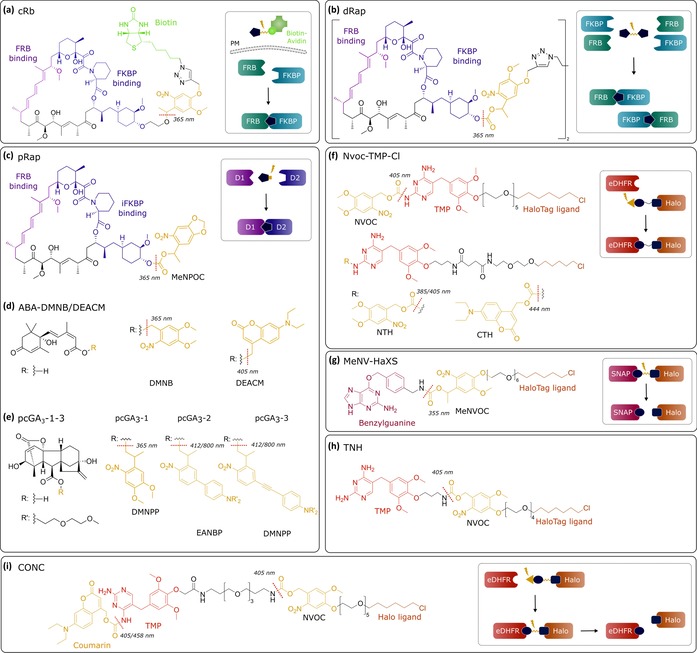
Chemo‐optogenetic dimerization systems and structures of photosensitive dimerizers. Dotted red lines indicate photocleavage sites.
Caging approaches were also applied to chemical dimerizers derived from plant hormones, which are bioorthogonal to mammalian systems. To yield caged abscisic acid (ABA), a 4,5‐dimethoxy‐2‐nitrobenzyl (DMNB) or [7‐(diethylamino)coumarin‐4‐yl]methyl (DEACM) group was attached to the carboxylic acid moiety of ABA, which could be removed upon irradiation with λ=365 or 405 nm light, respectively (Figure 2 d).35 Uncaged ABA induces the heterodimerization of ABI and PYL protein. Gibberellic acid (GA3) binds to gibberellin‐insensitive dwarf (GID1) protein and induces a conformational change that leads to interaction with gibberellin‐insensitive (GAI) protein. GA3 was modified with 2‐(4,5‐dimethoxy‐2‐nitrophenyl)propyl (DMNPP), 2‐{4′‐bis[(2‐methoxyethoxy)ethyl]amino‐4‐nitro‐[(1,1′‐biphenyl)‐3‐yl}propan‐1‐ol (EANBP), and π‐extended 2(o‐nitrophenyl)propyl caging groups to generate pcGA3‐1, ‐2, and‐3, respectively (Figure 2 e).36 Dimerization was triggered by uncaging at λ=405 nm for pcGA3‐1 and at λ=470 nm for pcGA3‐2/3. The extended π system makes pcGA3‐3 more suitable for two‐photon uncaging at λ=800 nm. Caged GA3 was rapidly photoactivated by a short illumination pulse (3 s), whereas caged ABA required a relatively long illumination time (60 s). Dimerization in cells was observed 20 s after activation of caged GA3, whereas dimerization took a few minutes after uncaging of ABA. Both systems are orthogonal to the rapamycin‐based system approaches; therefore, they could be combined in certain applications. However, similar to the rapamycin‐based system, the GA3‐based system is not reversible. The ABA‐based system can be reversed slowly (30 min) by extensive washing.
A major limitation of the aforementioned systems is rapid diffusion of the uncaged dimerizer throughout the entire cell. This makes it difficult to induce localized dimerization at a subcellular region. This issue was addressed in the recent development of synthetic ligands that were caged by a photolabile 6‐nitroveratroyloxycarbonyl (NVOC) or a DEACM group (Figure 2 f).37 The chlorohexyl moiety of NvocTMP‐Cl/NTH or CTH binds covalently to a HaloTag domain that is targeted to a subcellular compartment. Uncaging of the NVOC or DEACM group enables the interaction between the TMP group and Escherichia coli dihydrofolate reductase (eDHFR) fused to a POI. In contrast to other noncovalent dimerizers, such as rapamycin, covalent binding to a protein that anchors to a membrane or a large complex prevents the free diffusion of the dimerizer in the cell. These dimerizers can be prelocalized prior to photoactivation, and thus, provide an improved spatial resolution. NvocTMP‐Cl induced dimerization in cells within a second upon a short pulse (ms) of illumination at λ=405 nm, which was reversed within 2 min by adding the competitor TMP.37c Furthermore, these systems are bioorthogonal and may also be combined with other dimerization systems.
Whereas the photoactivatable CID (paCID) approaches mentioned above enable activation of dimerization by light, photocleavable CID (pcCID) approaches allow light‐induced control of dedimerization. A pcCID approach employs a dimerizer with bifunctional moieties that are linked by a photolabile group. MeNV‐HaXS contains a HaloTag ligand and a SNAP tag ligand that are linked by a MeNVOC group,38 whereas TNH consists of a chlorohexyl moiety and TMP that are linked by a NVOC moiety (Figure 2 g, h).37d Although dimerization induced by adding the compound takes a few minutes, dedimerization was achieved in a timeframe of seconds by a short pulse of illumination at λ=355 nm for MeNV‐HaXS and at λ=405 nm for TNH.
A photoswitchable CID (psCID) system based on CONC has been recently reported, which enables switching dimerization on and off with light (Figure 2 i).39 CONC features a chlorohexyl moiety, a TMP moiety, a coumarinyl photocaging group, and a photocleavable NVOC moiety. The coumarinyl and NVOC groups are orthogonal photocaging groups that can be independently cleaved. Accordingly, photoactivation was achieved either by illumination at λ=458 nm or by a low dose of λ=405 nm light, whereas deactivation was triggered by a high dose of λ=405 nm light. The CONC‐based system features rapid dimerization and dedimerization kinetics in the range of milliseconds and high spatial precision (μm) in the cell. Moreover, the degree of activation and deactivation can be fine‐tuned by the dose of illumination. This is very useful for emulating cellular states that are controlled by the concentration of active proteins.
A summary of the properties of selected chemo‐optogenetic dimerization systems is provided in Table 2.
Table 2.
Properties of chemo‐optogenetic dimerization systems.
|
System |
λ [nm] |
Timescale |
Ref. |
|
|---|---|---|---|---|
|
|
Association |
Dissociation |
|
|
|
pRap |
365 |
|
dimerization: 1–2 min |
|
|
FRB‐iFKBP |
dissociation: no |
|||
|
cRb |
365 |
|
dimerization: <1 min |
|
|
FRB‐FKBP12 |
dissociation: no |
|||
|
dRap |
365 |
|
dimerization: 3 min |
|
|
FRB‐FKBP12 |
dissociation: no |
|||
|
ABA‐DMNB/DEACM |
365/405 |
washout |
dimerization: min |
|
|
PYL‐ABI |
dissociation: 30 min |
|||
|
pcGA3 |
377–470 |
|
dimerization: 3–10 s |
|
|
GID‐GAI |
(two‐photon uncaging at 800 nm) |
dissociation: no |
||
|
NTH/CTH |
385–405 (NTH) |
TMP addition |
dimerization: t 1/2=15 s |
|
|
eDHFR‐HaloTag |
385/444 (CTH) |
dissociation: 2–10 min |
||
|
Nvoc‐TMP‐CI |
405 |
TMP addition |
dimerization: t 1/2=0.78 s |
|
|
eDHFR‐HaloTag |
dissociation: 2 min |
|||
|
MeNV‐HaXS |
dimerizer addition |
355 |
dimerization: min |
|
|
SNAPTag‐HaloTag |
dissociation: s |
|||
|
TNH |
dimerizer addition |
405 |
dimerization: t 1/2=2–5 min |
|
|
eDHFR‐HaloTag |
dissociation: s |
|||
|
CONC |
405/458 |
405 |
dimerization: t 1/2=0.51 s |
|
|
eDHFR‐HaloTag |
dissociation: t 1/2=0.15 s |
|||
4. Biological Applications of Light‐Induced Dimerization
Light‐induced dimerization has been largely employed to control the activity and localization of proteins in the cell, including kinases, small GTPases, guanine nucleotide exchange factors (GEFs), phosphoinositide modifying enzymes, caspases, transcriptional modulators, epigenetic modifiers, transmembrane receptors, and motor proteins. As a consequence, the associated cellular processes are manipulated by light, ranging from gene transcription, genome engineering, signaling transduction, and cytoskeleton dynamics to vesicular transport.
4.1. Gene transcription
Light‐controlled gene transcription has been achieved by recruitment of transcription activators to the respective promoter. The PhyB‐PIF3 system was first used to control gene transcription in yeast. PhyB was fused to a GAL4 DNA binding domain (GBD), whereas PIFs was fused to a GAL4 activation domain (GAD). Red‐light‐induced dimerization recruited GAD to the promoter with a GAL4 DNA binding site, and thereby activated the transcription of a desired gene. In the LightOn approach, a Gal4(65)‐VVD fusion protein forms a homodimer upon blue‐light activation. The homodimer binds to the UASG sequence and activates transcription (Figure 3 a).17a DNA transcription could also be controlled by a split Cre recombinase that was fused to CRY2 and CIB1. Light‐induced reconstitution of split Cre activated recombination of a transcriptional stop sequence flanked by loxP sites preceding a reporter gene; thus leading to activation of gene transcription.19
Figure 3.
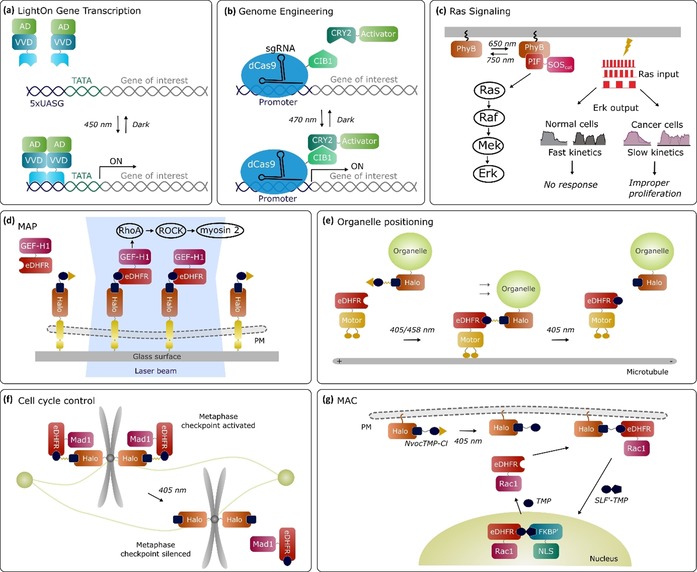
Biological applications of light‐induced dimerization systems. a) The LightOn strategy with the VVD homodimerization system to control gene transcription. b) A combination of the CYR2‐CIB1 system with dCas9 and transcriptional modulators. Blue‐light‐induced dimerization recruited the transcriptional activator to a specific site in the genome targeted by dCas9, leading to upregulation of an endogenous gene. c) The optoSOS approach with the PhyB‐PIF system generated dynamic input stimulus patterns by red light. The altered pathway output downstream of signaling was monitored. d) Nvoc‐TMP‐CI molecules are anchored to the plasma membrane through artificial receptors immobilized on the glass surface. Uncaging with a focused laser beam allows patterns of activated signaling molecules to be generated at the plasma membrane. e) Organelle transport is controlled through the CONC system. Uncaging of the coumarinyl group stimulated recruitment of motor proteins to the cargo and subsequent transport along microtubules. Photocleavage of the NVOC group led to dissociation of motor proteins and cessation of transport. f) Maintaining the spindle checkpoint protein Mad1 on a subpopulation of kinetochores through the photocleavable TNH system was sufficient to halt the transition from metaphase to anaphase. Mad1 was released from a subpopulation of kinetochores within the cell by light. g) Uncaging of NvocTMP‐Cl stimulated recruitment of Rac1 from the cytosol to the plasma membrane through dimerization between HaloTag and eDHFR. Addition of SLF′‐TMP triggered the dissociation of Rac1 and translocation to the nucleus through dimerization between eDHFR and FKBP′. Addition of TMP, which competed for binding to eDHFR, led to release of Rac1 to the cytosol. Multiple cycles could be achieved through washing out.
4.2. Genome engineering
Recently, a combination of optogenetic or chemo‐optogenetic tools with CRISPR‐Cas9 (CRISPR=clustered regularly interspaced palindromic repeat) enabled control of genome editing by light. The CRISPR‐Cas9 technology originates from the type II CRISPR‐Cas adaptive immune system in bacteria. The CRISPR associated protein Cas9 is an endonuclease that requires guide RNA molecules, crRNA‐tracrRNA, to introduce site‐specific binding and cleavage of double strands in DNA. The dual tracrRNA:crRNA was engineered as a single guide RNA (sgRNA) that targeted Cas9 to any DNA sequence of interest. The CRISPR‐Cas9 system has emerged as a revolutionarily technology for genome editing.40 In a photoactivatable Cas9 (pCas9) approach, the split Cas9 fragments were fused to the pMag‐nMag pair. Reconstitution of Cas9 activity was achieved by blue‐light‐mediated dimerization of the Magnets system.41 In another approach, Cas9 was caged by a dimeric Rhodobacter sphaeroides LOV (RsLOV) domain, dimeric Dronpa, or a MeNPOC caging group at K866; these could be activated by blue, cyan, or UV light, respectively.42
In addition to approaches that allow control over Cas9 activity, new approaches for regulating catalytically dead Cas9 (dCas9) have emerged recently. The dCas9 variant has served as a sequence‐specific DNA‐binding protein to manipulate endogenous gene expression.43 The CYR2‐CIB1 system was combined with dCas9 and transcriptional activators, such as VP64 or p65. Blue‐light‐induced dimerization recruited the transcriptional activator to a specific site in the genome targeted by dCas9, leading to upregulation of an endogenous gene (Figure 3 b).44 A combination of orthogonal GA3‐ and ABA‐based CID systems with the transcriptional activator (VPR) and repressor (KRab) in the dCas9‐based platform afforded independent regulation of different genes within the same cell. This strategy also enabled logic operations and diametric up‐ and downregulation of genes with distinct dimerizers.45 In a similar application, rapamycin and GA3 systems were combined with dCas9 and transcriptional activators to enable orthogonal regulation of multiple genes in human cells.46
In another dCas9 approach, RNA aptamer sequences were linked to sgRNA, which specifically recruited an MS2 phage coat protein fused to effectors (e.g., transcriptional activators) to the dCas9‐sgRNA complex at specific loci. This approach showed improved upregulation of endogenous genes. A recent study demonstrated photoactivatable CPT2.0 and Split‐CPT2.0 systems based on the dCas9‐aptamer platform.47 In the CPT2.0 approach, the MS2 protein and transcriptional activators (p65 and HSF1) were fused to CIB1 and CRY2, respectively. Blue‐light‐induced heterodimerization then facilitated recruitment of transcriptional activators. In the Split‐CPT2.0 strategy, split dCas9 fragments were fused to pMag and nMagHigh1. Upon illumination by blue light, reconstitution of dCas9 enables recruitment of the sgRNA with MS2‐binding sequences that associate with the MS2‐transcriptional activator fusion proteins.
The light‐inducible transcriptional effector (LITE) approach combines CRY2‐CIB1 with a customizable TALE DNA‐binding domain and a transcriptional activator or histone modifiers to control endogenous gene expression, as well as targeted epigenetic chromatin modifications.48 As shown in the LITE approach, inducible dCas9 systems could extend to other effectors, such as histone deacetylases.49
4.3. Signal transduction
The Ras‐Erk pathway regulates important cellular processes, such as cell proliferation and oncogenic mutations in the pathway that are thought to drive cancer by constitutive activation. To understand whether the altered dynamic transmission properties in signaling pathways play a role in disease, Lim and co‐workers used Ras‐activating optoSOS to generate artificial stimulations.50 To this end, the PhyB‐PIF system was used to rapidly, reversibly recruit the Ras‐activating SOS2 catalytic domain that activated Ras at the plasma membrane. OptoSOS produced different dynamic stimulus patterns that were controlled by light with high temporal resolution. This optogenetic approach facilitated systematic profiling of signal transmission in cells. They found that the kinetics of Ras‐Erk pathway inactivation were significantly (10‐fold) slower in cancer cells with B‐Raf mutations (≈20 min after switching off optoSOS) than that in normal cells (1–2 min). This resulted in signal misinterpretation of dynamic inputs, which propagated to proliferative decisions. As a result, the nonproliferative inputs that are normally filtered by the pathway can now drive proliferation (Figure 3 c).
A large number of light‐induced dimerization systems have been used to manipulate protein localization in distinct cellular compartments. The plasma membrane of eukaryotic cells is an especially important compartment that integrates external stimuli and internal states to control cellular processes. Light‐inducible approaches have been used to control a wide range of signaling molecules at the plasma membrane, including Ras and Ras signaling molecules, Rho GTPase family proteins, ion channels, transmembrane receptors, and phosphoinositide modifying enzymes.2a, 4a However, proteins localized at the plasma membrane undergo fast lateral diffusion, rendering stable targeting at the plasma membrane challenging.
A recent strategy, termed molecular activity painting (MAP), facilitates the study of signal transduction at the plasma membrane.37c The MAP approach combines the photoactivatable Nvoc‐TMP‐CI system with immobilized artificial receptors. To mitigate lateral diffusion of the dimerization domain anchored to the plasma membrane, HaloTag was fused to artificial receptors that were immobilized through interactions with surface‐immobilized antibodies. Consequently, a single, focused pulse of λ=405 nm light rapidly and persistently targeted the Rho‐specific activator GEF‐H1 protein to the region of photoactivation (ROP) at the plasma membrane. Immediately after the recruitment of GEF‐H1, the association kinetics of downstream signaling molecules, RhoA and myosin 2, were monitored at the ROP. The fast onset and long‐lasting persistence of the switch‐like perturbation affords a straightforward interpretation of the cellular response kinetics, which is invaluable for studying many plasma membrane associated processes. Moreover, the technique also allows patterns of activated proteins to be “painted” onto the plasma membrane (Figure 3 d).
4.4. Organelle positioning
Organelle distribution plays an important role in the spatial organization of diverse cellular processes, including cell signaling, cell polarization, and neurite outgrowth. For example, bidirectional transport along axonal microtubules plays a central role in the proper subcellular distribution of cellular cargos and its misregulation is thought to play an important role in neurodegenerative diseases. The combination of CID or light‐induced dimerization systems with motor proteins such as kinesin, dynein, and myosin has been used to control cargo transport in the cell. A constitutively active motor domain or a motor adaptor was fused to a dimerization module. The other dimerization module was targeted to a specific cargo. Upon induced dimerization, motor proteins were loaded onto the cargo and subsequently promoted the transport of cargo into the cell. Depending on the type of motor, cargo moves along microtubules or actin filaments towards the cell center or periphery. The optogenetic LOVpep‐ePDZ system was employed to control positioning of peroxisomes, recycling endosomes (REs), and mitochondria in neurons.51 This study showed that kinesin‐driven local enrichment of REs at axonal growth cones promoted axon growth, whereas dynein‐driven removal of REs inhibited axon growth. Other optogenetic and chemo‐optogenetic systems, such as CRY2‐CIB1, NTH, and CONC, have been applied in mammalian cells to control positioning of various organelles, including mitochondria, lysosomes, endosomes, and peroxisomes (Figure 3 e).37b, 39, 52
4.5. Cell cycle
Kinetochores are large regulatory complexes that coordinate multiple processes during cell division, mostly through recruitment of specific regulatory proteins. By using chemo‐optogenetic tools to recruit or remove specific regulatory proteins to kinetochores, several interesting questions related to regulation of kinetochore function have been elucidated.37d Release of the spindle checkpoint protein Mad1 from a subpopulation of kinetochores through the photocleavable TNH system was able to halt the transition from metaphase to anaphase, which suggested that Mad1 localization to a few kinetochores was sufficient for checkpoint activity. By using the photoactivatable CTH system, the kinetochore motor CENP‐E or non‐kinetochore motor kinesin‐1 was recruited to kinetochores under different conditions. The results demonstrated that CENP‐E transported chromosomes from poles to the equator during congression and then maintained attachment of bioriented sister kinetochores to microtubule ends during metaphase oscillations (Figure 3 f).
In another application, the spatial precision of the photoactivatable CID approach made it possible to control spindle asymmetry during meiosis.53 Cdc42 was recruited to one spindle pole by local uncaging of the CTH dimerizer, which induced spindle asymmetry by increasing tyrosinated α‐tubulin signals on the recruited site. The results showed that tyrosinated α‐tubulin asymmetry enabled stronger centromeres to interact differentially with the two sides of the spindle to preferentially orient toward the egg pole. This study provides new insights into the mechanisms underlying spindle asymmetry that drive asymmetric cell division during female meiosis.
4.6. Multidirectional activity control
The optogenetic and chemo‐optogenetic approaches typically afford a single layer of activity control, such as turning on/off a protein by targeting it to a specific cellular compartment to study relatively simple cellular processes. A recent development of the multidirectional activity control (MAC) strategy is beneficial for the analysis of more complex processes that involve cycling, trafficking, or shuttling of signal molecules between different cell compartments.5 Here, the photoactivatable Nvoc‐TMP‐CI system was combined with the SLF′‐TMP system to afford a photoactivatable, dual‐chemically induced dimerization (pdCID) system. The parallel MAC approach was used to tune background activity before light‐induced control of Rac1 protein function in the cell. The competitive MAC approach was employed to manipulate multiple cycles of Rac1 shuttling among the cytosol, plasma membrane, and nucleus. This is useful to investigate the nucleocytoplasmic shuttling of Rac1, which plays an important role in tumor invasion (Figure 3 g). Moreover, the competitive MAC approach was used to control bidirectional transport of organelles by targeting kinesin and dynein motors with opposite directions of transport along microtubules. Peroxisomes were transported to the cell periphery and then to the cell body, or vice versa, within a single cell.
5. Summary and Outlook
New variations and applications of light‐induced dimerization approaches have been continuously growing in recent years. These offer many opportunities for scientists to choose the most suited strategy for their individual applications. Chemo‐optogenetic dimerization approaches can be a valuable complement to optogenetic systems. First, chemo‐optogenetic dimerization systems can be fine‐tuned by the dose of illumination. Therefore, the number of molecules in the on/off state can be quantitatively controlled. In contrast, the optogenetic PhyB‐PIF system, for instance, is experimentally more challenging in terms of tunability. Second, in contrast to many photosensitive proteins that suffer from basal activity in the dark state and relatively low affinity (K d=0.1–10 μm) in the on state, there is no binding between the caged dimerizer and the dimerization module, whereas the uncaged dimerizer usually binds in high affinity (K d in the nanomolar range). Hence, the dynamic range of chemo‐optogenetic systems is significantly improved. However, current chemo‐optogenetic dimerization systems are subjected to only one‐time or one‐round control by light, whereas optogenetic systems undergo multiple cycles. Current chemo‐optogenetic dimerization systems are largely stimulated by UV or blue light, which is not favorable for application on organisms due to phototoxicity and limited tissue penetration depths. However, chemo‐optogenetic systems are facile for chemical modifications. Future developments could expand to caging groups that can be cleaved by longer wavelength light.54
In conclusion, there remains a high demand for new light‐induced dimerization tools, which will pave the way to solve increasingly complex biological problems. Importantly, a combination of novel light‐inducible strategies with biological engineering technologies is needed to provide innovative solutions to cell‐based therapy and the manufacture of bioproducts.55 The spatial and temporal precision of optogenetic and chemo‐optogenetic systems will be beneficial not only to subcellular regulation, but also to subtissue control, which opens up new avenues to understand intercellular communication.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Yao‐Wen Wu received his BS in chemistry from Sun Yat‐sen University in 2001 and his MS in organic chemistry from Tsinghua University, P.R. China, in 2004. After graduating as Dr rer. nat. (2008) from the TU Dortmund, working at the Max Planck Institute of Molecular Physiology in Germany, and a postdoctoral study in cell biology at King's College London, UK, he has been leader of an Otto‐Hahn group at the Max Planck Institute in Dortmund since 2010. Since 2012, he has been group leader of the Chemical Genomics Centre of the Max Planck Society. Since 2018, he has been full Professor of Biochemistry at the Department of Chemistry, Umeå University, Sweden. His research group develops small molecules and new chemical methods to modify proteins or manipulate protein function in the context of biological systems with a particular focus on molecular mechanisms in membrane trafficking and autophagy.

Biographical Information
Laura Klewer did her PhD research at the Max‐Planck‐Institute of Molecular Physiology and the Chemical Genomics Centre in Dortmund, Germany, under the supervision of Prof. Dr. Yao‐Wen Wu. She was awarded her PhD degree from the Technical University Dortmund in 2017. During her PhD project in chemical biology, she focused on manipulating autophagy with chemically induced dimerization and light‐induced dimerization approaches, and implemented novel approaches to control highly dynamic cellular processes.

Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft, DFG (grant no. SPP 1623); the European Research Council, ERC (ChemBioAP); the Knut and Alice Wallenberg Foundation; and Vetenskapsrådet (no. 2018‐04585) to Y.W.W.
L. Klewer, Y.-W. Wu, Chem. Eur. J. 2019, 25, 12452.
References
- 1. Stanton B. Z., Chory E. J., Crabtree G. R., Science 2018, 359, eaao5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Rost B. R., Schneider-Warme F., Schmitz D., Hegemann P., Neuron 2017, 96, 572–603; [DOI] [PubMed] [Google Scholar]
- 2b. Voss S., Klewer L., Wu Y.-W., Curr. Opin. Chem. Biol. 2015, 28, 194–201. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. DeRose R., Miyamoto T., Inoue T., Pflugers Arch. 2013, 465, 409–417; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Fegan A., White B., Carlson J. C., Wagner C. R., Chem. Rev. 2010, 110, 3315–3336; [DOI] [PubMed] [Google Scholar]
- 3c. Putyrski M., Schultz C., FEBS Lett. 2012, 586, 2097–2105. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Weitzman M., Hahn K. M., Curr. Opin Cell Biol. 2014, 30, 112–120; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Tischer D., Weiner O. D., Nat. Rev. Mol. Cell Biol. 2014, 15, 551–558; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Zhang K., Cui B., Trends Biotechnol. 2015, 33, 92–100; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Losi A., Gardner K. H., Moglich A., Chem. Rev. 2018, 118, 10659–10709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen X., Venkatachalapathy M., Dehmelt L., Wu Y.-W., Angew. Chem. Int. Ed. 2018, 57, 11993–11997; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12169–12173. [Google Scholar]
- 6. Liberles S. D., Diver S. T., Austin D. J., Schreiber S. L., Proc. Natl. Acad. Sci. USA 1997, 94, 7825–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Feng S., Laketa V., Stein F., Rutkowska A., MacNamara A., Depner S., Klingmuller U., Saez-Rodriguez J., Schultz C., Angew. Chem. Int. Ed. 2014, 53, 6720–6723; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6838–6841; [Google Scholar]
- 7b. Liu P., Calderon A., Konstantinidis G., Hou J., Voss S., Chen X., Li F., Banerjee S., Hoffmann J. E., Theiss C., Dehmelt L., Wu Y. W., Angew. Chem. Int. Ed. 2014, 53, 10049–10055; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10213–10219. [Google Scholar]
- 8. Miesenböck G., Annu. Rev. Cell Dev. Biol. 2011, 27, 731–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Shimizu-Sato S., Huq E., Tepperman J. M., Quail P. H., Nat. Biotechnol. 2002, 20, 1041–1044; [DOI] [PubMed] [Google Scholar]
- 9b. Zemelman B. V., Lee G. A., Ng M., Miesenböck G., Neuron 2002, 33, 15–22. [DOI] [PubMed] [Google Scholar]
- 10. Lee J., Natarajan M., Nashine V. C., Socolich M., Vo T., Russ W. P., Benkovic S. J., Ranganathan R., Science 2008, 322, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yazawa M., Sadaghiani A. M., Hsueh B., Dolmetsch R. E., Nat. Biotechnol. 2009, 27, 941–945. [DOI] [PubMed] [Google Scholar]
- 12. Wu Y. I., Frey D., Lungu O. I., Jaehrig A., Schlichting I., Kuhlman B., Hahn K. M., Nature 2009, 461, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Strickland D., Lin Y., Wagner E., Hope C. M., Zayner J., Antoniou C., Sosnick T. R., Weiss E. L., Glotzer M., Nat. Methods 2012, 9, 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guntas G., Hallett R. A., Zimmerman S. P., Williams T., Yumerefendi H., Bear J. E., Kuhlman B., Proc. Natl. Acad. Sci. USA 2015, 112, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Christie J. M., Corchnoy S. B., Swartz T. E., Hokenson M., Han I. S., Briggs W. R., Bogomolni R. A., Biochemistry 2007, 46, 9310–9319; [DOI] [PubMed] [Google Scholar]
- 15b. Zoltowski B. D., Vaccaro B., Crane B. R., Nat. Chem. Biol. 2009, 5, 827–834; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Strickland D., Yao X., Gawlak G., Rosen M. K., Gardner K. H., Sosnick T. R., Nat. Methods 2010, 7, 623–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang H., Vilela M., Winkler A., Tarnawski M., Schlichting I., Yumerefendi H., Kuhlman B., Liu R., Danuser G., Hahn K. M., Nat. Methods 2016, 13, 755–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Wang X., Chen X., Yang Y., Nat. Methods 2012, 9, 266–269; [DOI] [PubMed] [Google Scholar]
- 17b. Kawano F., Suzuki H., Furuya A., Sato M., Nat. Commun. 2015, 6, 6256. [DOI] [PubMed] [Google Scholar]
- 18. Liu H., Yu X., Li K., Klejnot J., Yang H., Lisiero D., Lin C., Science 2008, 322, 1535–1539. [DOI] [PubMed] [Google Scholar]
- 19. Kennedy M. J., Hughes R. M., Peteya L. A., Schwartz J. W., Ehlers M. D., Tucker C. L., Nat. Methods 2010, 7, 973–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Taslimi A., Zoltowski B., Miranda J. G., Pathak G. P., Hughes R. M., Tucker C. L., Nat. Chem. Biol. 2016, 12, 425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Bugaj L. J., Choksi A. T., Mesuda C. K., Kane R. S., Schaffer D. V., Nat. Methods 2013, 10, 249–252; [DOI] [PubMed] [Google Scholar]
- 21b. Taslimi A., Vrana J. D., Chen D., Borinskaya S., Mayer B. J., Kennedy M. J., Tucker C. L., Nat. Commun. 2014, 5, 4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Levskaya A., Weiner O. D., Lim W. A., Voigt C. A., Nature 2009, 461, 997–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gasser C., Taiber S., Yeh C. M., Wittig C. H., Hegemann P., Ryu S., Wunder F., Moglich A., Proc. Natl. Acad. Sci. USA 2014, 111, 8803–8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. Kaberniuk A. A., Shemetov A. A., Verkhusha V. V., Nat. Methods 2016, 13, 591–597; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24b. Redchuk T. A., Omelina E. S., Chernov K. G., Verkhusha V. V., Nat. Chem. Biol. 2017, 13, 633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou X. X., Chung H. K., Lam A. J., Lin M. Z., Science 2012, 338, 810–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.
- 26a. Chen D., Gibson E. S., Kennedy M. J., J. Cell Biol. 2013, 201, 631–640; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b. Crefcoeur R. P., Yin R., Ulm R., Halazonetis T. D., Nat. Commun. 2013, 4, 1779. [DOI] [PubMed] [Google Scholar]
- 27. Kainrath S., Stadler M., Reichhart E., Distel M., Janovjak H., Angew. Chem. Int. Ed. Angew. Chem. Int. Ed. Engl. 2017, 56, 4608–4611; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4679–4682. [Google Scholar]
- 28. Toettcher J. E., Gong D., Lim W. A., Weiner O. D., Nat. Methods 2011, 8, 837–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Niu J., Ben Johny M., Dick I. E., Inoue T., Biophys. J. 2016, 111, 1132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Toettcher J. E., Weiner O. D., Lim W. A., Cell 2013, 155, 1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reichhart E., Ingles-Prieto A., Tichy A. M., McKenzie C., Janovjak H., Angew. Chem. Int. Ed. 2016, 55, 6339–6342; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6447–6450. [Google Scholar]
- 32. Karginov A. V., Zou Y., Shirvanyants D., Kota P., Dokholyan N. V., Young D. D., Hahn K. M., Deiters A., J. Am. Chem. Soc. 2011, 133, 420–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brown K. A., Zou Y., Shirvanyants D., Zhang J., Samanta S., Mantravadi P. K., Dokholyan N. V., Deiters A., Chem. Commun. 2015, 51, 5702–5705. [DOI] [PubMed] [Google Scholar]
- 34. Umeda N., Ueno T., Pohlmeyer C., Nagano T., Inoue T., J. Am. Chem. Soc. 2011, 133, 12–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wright C. W., Guo Z. F., Liang F. S., Chembiochem 2015, 16, 254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schelkle K. M., Griesbaum T., Ollech D., Becht S., Buckup T., Hamburger M., Wombacher R., Angew. Chem. Int. Ed. 2015, 54, 2825–2829. [DOI] [PubMed] [Google Scholar]
- 37.
- 37a. Ballister E. R., Aonbangkhen C., Mayo A. M., Lampson M. A., Chenoweth D. M., Nat. Commun. 2014, 5, 5475; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37b. Ballister E. R., Ayloo S., Chenoweth D. M., Lampson M. A., Holzbaur E. L., Curr. Biol. 2015, 25, R407–408; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37c. Chen X., Venkatachalapathy M., Kamps D., Weigel S., Kumar R., Orlich M., Garrecht R., Hirtz M., Niemeyer C. M., Wu Y.-W., Dehmelt L., Angew. Chem. Int. Ed. 2017, 56, 5916–5920; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 6010–6014; [Google Scholar]
- 37d. Zhang H., Aonbangkhen C., Tarasovetc E. V., Ballister E. R., Chenoweth D. M., Lampson M. A., Nat. Chem. Biol. 2017, 13, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zimmermann M., Cal R., Janett E., Hoffmann V., Bochet C. G., Constable E., Beaufils F., Wymann M. P., Angew. Chem. Int. Ed. 2014, 53, 4717–4720; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4808–4812. [Google Scholar]
- 39. Chen X., Wu Y.-W., Angew. Chem. Int. Ed. 2018, 57, 6796–6799; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6912–6915. [Google Scholar]
- 40. Doudna J. A., Charpentier E., Science 2014, 346, 1258096. [DOI] [PubMed] [Google Scholar]
- 41. Nihongaki Y., Kawano F., Nakajima T., Sato M., Nat. Biotechnol. 2015, 33, 755–760. [DOI] [PubMed] [Google Scholar]
- 42.
- 42a. Hemphill J., Borchardt E. K., Brown K., Asokan A., Deiters A., J. Am. Chem. Soc. 2015, 137, 5642–5645; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42b. Richter F., Fonfara I., Gelfert R., Nack J., Charpentier E., Moglich A., Curr. Opin. Biotechnol. 2017, 48, 119–126; [DOI] [PubMed] [Google Scholar]
- 42c. Zhou X. X., Zou X., Chung H. K., Gao Y., Liu Y., Qi L. S., Lin M. Z., ACS Chem. Biol. 2018, 13, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dominguez A. A., Lim W. A., Qi L. S., Nat. Rev. Mol. Cell Biol. 2016, 17, 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.
- 44a. Nihongaki Y., Yamamoto S., Kawano F., Suzuki H., Sato M., Chem. Biol. 2015, 22, 169–174; [DOI] [PubMed] [Google Scholar]
- 44b. Polstein L. R., Gersbach C. A., Nat. Chem. Biol. 2015, 11, 198–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gao Y., Xiong X., Wong S., Charles E. J., Lim W. A., Qi L. S., Nat. Methods 2016, 13, 1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bao Z., Jain S., Jaroenpuntaruk V., Zhao H., ACS Synth. Biol. 2017, 6, 686–693. [DOI] [PubMed] [Google Scholar]
- 47. Nihongaki Y., Furuhata Y., Otabe T., Hasegawa S., Yoshimoto K., Sato M., Nat. Methods 2017, 14, 963–966. [DOI] [PubMed] [Google Scholar]
- 48. Konermann S., Brigham M. D., Trevino A. E., Hsu P. D., Heidenreich M., Cong L., Platt R. J., Scott D. A., Church G. M., Zhang F., Nature 2013, 500, 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kwon D. Y., Zhao Y. T., Lamonica J. M., Zhou Z., Nat. Commun. 2017, 8, 15315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bugaj L. J., Sabnis A. J., Mitchell A., Garbarino J. E., Toettcher J. E., Bivona T. G., Lim W. A., Science 2018, 361, eaao3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van Bergeijk P., Adrian M., Hoogenraad C. C., Kapitein L. C., Nature 2015, 518, 111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Duan L., Che D., Zhang K., Ong Q., Guo S., Cui B., Chem. Biol. 2015, 22, 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Akera T., Chmatal L., Trimm E., Yang K., Aonbangkhen C., Chenoweth D. M., Janke C., Schultz R. M., Lampson M. A., Science 2017, 358, 668–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ankenbruck N., Courtney T., Naro Y., Deiters A., Angew. Chem. Int. Ed. 2018, 57, 2768–2798; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2816–2848. [Google Scholar]
- 55. Mansouri M., Strittmatter T., Fussenegger M., Adv. Sci. 2019, 6, 1800952. [DOI] [PMC free article] [PubMed] [Google Scholar]