

Abstract
Many fungi produce multiple lytic polysaccharide monooxygenases (LPMOs) with seemingly similar functions, but the biological reason for this multiplicity remains unknown. To address this question, here we carried out comparative structural and functional characterizations of three cellulose-active C4-oxidizing family AA9 LPMOs from the fungus Neurospora crassa, NcLPMO9A (NCU02240), NcLPMO9C (NCU02916), and NcLPMO9D (NCU01050). We solved the three-dimensional structure of copper-bound NcLPMO9A at 1.6-Å resolution and found that NcLPMO9A and NcLPMO9C, containing a CBM1 carbohydrate-binding module, bind cellulose more strongly and were less susceptible to inactivation than NcLPMO9D, which lacks a CBM. All three LPMOs were active on tamarind xyloglucan and konjac glucomannan, generating similar products but clearly differing in activity levels. Importantly, in some cases, the addition of phosphoric acid–swollen cellulose (PASC) had a major effect on activity: NcLPMO9A was active on xyloglucan only in the presence of PASC, and PASC enhanced NcLPMO9D activity on glucomannan. Interestingly, the three enzymes also exhibited large differences in their interactions with enzymatic electron donors, which could reflect that they are optimized to act with different reducing partners. All three enzymes efficiently used H2O2 as a cosubstrate, yielding product profiles identical to those obtained in O2-driven reactions with PASC, xyloglucan, or glucomannan. Our results indicate that seemingly similar LPMOs act preferentially on different types of copolymeric substructures in the plant cell wall, possibly because these LPMOs are functionally adapted to distinct niches differing in the types of available reductants.
Keywords: plant cell wall, substrate specificity, protein stability, ascorbic acid, dehydrogenase, hydrogen peroxide, AA9, gene multiplicity, lytic polysaccharide monooxygenase (LPMO), substrate binding
Introduction
The filamentous fungus Neurospora crassa is commonly found in nature where it grows on dead plant material, particularly grasses. Its cellulolytic potential has been known for decades (1–3). The N. crassa genome encodes a similar number of glycoside hydrolase (GH)2 enzymes as Hypocrea jecorina, the main industrial source of enzymes for biomass depolymerization (4). The N. crassa genome also encodes 14 genes encoding lytic polysaccharide monooxygenases (LPMOs) belonging to CAZy family AA9 (5). Transcriptome studies done at a time when these LPMOs were erroneously thought to be glycoside hydrolases belonging to CAZy family GH61 showed transcripts for 13 of the AA9-encoding genes (6). LPMO-encoding genes tend to be abundant in the genomes of biomass-degrading fungi, indicating an important role in biomass degradation and raising questions as to the evolutionary drivers of this high multiplicity (7–11).
LPMOs are monocopper enzymes (12, 13) that were identified in 2010 (14) and that oxidatively cleave glycosidic bonds in polysaccharides. The discovery of LPMOs has been followed by extensive research related to their structure, function, mechanism, diversity, and industrial application (11, 15–20). LPMOs have become an important ingredient of commercial enzyme mixtures for industrial biomass conversion (21–24). Although LPMO action usually is considered in light of the enzymatic deconstruction of cellulose and chitin, some LPMOs act on other substrates, including xyloglucan and other (1,4)-linked β-glucans (25, 26), starch (27, 28), and xylan (29, 30). LPMO action requires reduction of the copper by an enzymatic or nonenzymatic electron donor (9, 31, 32). Subsequently, the enzymes use either molecular oxygen (14, 33) or hydrogen peroxide (34, 35) to generate an oxygen species at the copper site that is capable of abstracting a hydrogen atom from the C1 or the C4 of the scissile glycosidic bond.
The biological reason for the multiplicity of LPMOs remains unresolved. For example, all of the nine biochemically characterized NcLPMOs (25, 36–38) are known to act on cellulose, seemingly varying only in terms of their oxidative regioselectivity: NcLPMO9F (NCU03328), NcLPMO9E (NCU08760), NCU02344, and NcLPMO9G (NCU00836) are strictly C1-oxidizing; NcLPMO9A (NCU02240), NcLPMO9C (NCU02916), and NcLPMO9D (NCU01050) are strictly C4-oxidizing; and NcLPMO9M (NCU07898) and NcLPMO9B (NCU07760) show mixed C1/C4 oxidation. For NcLPMO9C, activity on other β-glucans and soluble cellodextrins has been demonstrated (25). Next to variation in oxidative regioselectivity, the LPMOs also vary in terms of the absence or presence of appended carbohydrate-binding modules (CBMs).
For more insight into potential differences between LPMOs, we have studied and compared the three strictly C4-oxidizing LPMOs of N. crassa, NcLPMO9A (NCU02240), NcLPMO9C (NCU02916) (39), and NcLPMO9D (NCU01050) (37). Of these, NcLPMO9A and NcLPMO9C contain a family 1 carbohydrate-binding module (CBM1; known for binding both crystalline and amorphous cellulose (40, 41)). To allow structural comparisons, we solved the crystal structure of the catalytic domain of NcLPMO9A. Functional analyses were inspired by the rich functional data set that was already available for NcLPMO9C (25, 39, 42, 43). We have assessed properties such as substrate binding and specificity, ability to recruit electrons from enzymatic and nonenzymatic electron donors, operational stability, the ability to generate H2O2 in the absence of substrate (36, 42), and potential differences between O2-driven and H2O2-driven catalysis.
Results
Crystal structure and initial characterization of NcLPMO9A
The three-dimensional structure of NcLPMO9A with copper bound was solved to 1.6-Å resolution (Table S1) and revealed a typical LPMO structure, both with regard to the overall fold and the copper-containing active site (Fig. 1). Product analysis after incubation of NcLPMO9A with phosphoric acid–swollen cellulose (PASC) using chromatographic methods (44), MS/MS analysis (42), and identification of characteristic products obtained upon reduction (33) showed that NcLPMO9A exclusively oxidizes C4, confirming results of Vu et al. (38) (results not shown).
Figure 1.

Sequence alignment and structural representation of C4-oxidizing NcLPMOs. A, sequence alignment of NcLPMO9A, NcLPMO9C, and NcLPMO9D with fully conserved residues shown in white (on a red background). The sequence numbering refers to NcLPMO9A. Blue frames indicate residues with similar physicochemical properties (printed in red on a white background). The loop regions that potentially contribute to functional variation among LPMOs, named L2, L3, LS, and LC, are marked with yellow, purple, red, and blue horizontal bars, respectively. Family 1 carbohydrate-binding modules and linkers are marked by black and gray horizontal bars, respectively. Asterisks indicate amino acid residues whose amide NMR signals shift upon binding to oligocellulose or xyloglucan (i.e. every tested substrate so far) to NcLPMO9C (46); these include His-1 and His-81, which make up the histidine brace. The hashtag indicates a histidine residue in a surface loop that includes the L3 region, which NMR studies showed is involved in binding of xyloglucan. B–D, cartoon representation of the catalytic domain structures of NcLPMO9A (B), NcLPMO9C (C), and NcLPMO9D (D) with copper depicted as a cyan sphere and L2, L3, LS, and LC loops colored in yellow, purple, red, and blue, respectively.
Overall structural comparison
Fig. 1 shows a sequence alignment and structural representations of the three C4-oxidizing LPMOs from N. crassa, NcLPMO9A, NcLPMO9C, and NcLPMO9D. The pairwise sequence identities of the catalytic domains range from 50 to 60%. Regions that are believed to be involved in substrate binding in family AA9 LPMOs are known as the L2, L3, LS, and LC loops (18, 45). Apart from the L3 loop, sequence conservation in these loops is similar to sequence conservation in the rest of the protein. The L3 loop shows higher sequence variability, and both the L3 and the LC loops are somewhat shorter in NcLPMO9A and NcLPMO9D compared with NcLPMO9C (Fig. 1A). Previous NMR studies on NcLPMO9C have shown that His-1, Ala-80, His-83, and His-155 (Fig. 1A, marked with * at positions 1, 78, 81, and 153, respectively) are involved in binding of both oligocellulose and polymeric xyloglucan (46), and three of these four residues are conserved among the three NcLPMOs. The residue at position 78 is predicted to take part in forming the −1 subsite (subsite prediction based on Courtade et al. (46) and Frandsen et al. (47)) varies; it is an Ala (Ala-80) in NcLPMO9C and an Asp in NcLPMO9A (Asp-78) and NcLPMO9D (Asp-81). Superposition of the three NcLPMO structures with that of an AA9 LPMO from Lentinus similis (LsAA9A) in complex with cellohexaose (PDB code 5ACI (47)) (Fig. 2) shows that, apart from position 78, residues in subsites −1 to −3 are highly conserved among the three N. crassa enzymes, whereas more variation occurs in the +1 and +2 subsites as well as in the −4 subsite. These variations reside in the L2/L3 and LC regions (Fig. 1A) of the protein, respectively.
Figure 2.

Amino acids potentially involved in substrate binding in NcLPMO9A (green), NcLPMO9C (magenta), and NcLPMO9D (blue). Substrate binding was predicted based on superposition of the crystal structures of NcLPMO9A (PDB code 5FOH; this study), NcLPMO9C (PDB code 4D7U (39)), and NcLPMO9D (PDB code 4EIR (37)) with LsAA9A in complex with cellohexaose (PDB code 5ACI (47)); the picture shows the side chains of the three NcLPMO9s. Amino acids are numbered based on NcLPMO9A (see Fig. 1A). Fully conserved residues have black labels, whereas not fully conserved residues have multiple labels that are colored according to the corresponding LPMO. The numbers within the sugar units (shown in light gray) indicate subsites. For clarity, the catalytic copper of only NcLPMO9A and only one of the conformations for the side chains of the residues Tyr-25 and Ala-77 in NcLPMO9D are shown.
Courtade et al. (46) showed that the L3 region is involved in the interaction with xyloglucan, and both the sequence alignment and the structures (Figs. 1 and 2) show considerable variation in this region. Although Asp-76 in the L3 loop of NcLPMO9C (position 74 in the alignment in Fig. 1A) is likely to interact with the C6-hydroxyl of the glucose unit in subsite +1, the corresponding residues in NcLPMO9A (Asp-74) and NcLPMO9D (Ala-77) seem not to be capable of such an interaction (Fig. 2). There is also considerable variation, i.e. at positions 67 and 68 in the L3 region and 25 and 26 in the L2 region. Position 68 in the L3 loop is of particular interest. The glutamate (Glu-65) in NcLPMO9C is likely to contribute to substrate binding at the +2 subsite (1.9–2.2 Å from the C1- and C2-hydroxyls), similarly to Asn-67 in LsAA9A (47). The side chains of the corresponding residues at this position in NcLPMO9A (Thr-68) and NcLPMO9D (Met-68) are less suitable to hydrogen bond with the substrate and seem to be located further away (3.7–4.4 Å) from the glucose unit in the +2 subsite. Another clear difference concerns position 25 in the L2 region where both NcLPMO9A and NcLPMO9D have a tyrosine, whereas NcLPMO9C has an aspartate. A tyrosine could be engaged in stacking interactions with the substrate and may potentially extend the substrate-binding surface, perhaps forming a +3 subsite. The tyrosine at position 204 in the +3/+4 subsite is highly conserved in all AA9 LPMOs and well-known to be important for binding cello-oligomers (47) and xyloglucan (46). Interestingly, adjacent to this Tyr residue NcLPMO9D carries a Trp (Trp-207), which could contribute to substrate binding through aromatic stacking.
Binding to PASC
Fig. 3A shows that in the absence of ascorbic acid (AscA) binding of all three NcLPMOs reached equilibrium in 30 min after mixing with PASC. The strongest binding was observed for the two NcLPMOs containing a CBM1, NcLPMO9C and NcLPMO9A, with ∼80 and ∼50% binding, respectively. Binding of NcLPMO9D was much weaker, with only ∼15% binding. The presence of AscA had an initial positive effect on binding observed for all three NcLPMOs, but during the next hour the bound fractions of all three LPMOs gradually decreased (Fig. 3B). When equilibrium was reached, the fractions of bound protein were lower than those in the reactions without AscA. Analysis of the experiments shown in Fig. 3 by SDS-PAGE of nonbound protein showed similar trends (Fig. S1).
Figure 3.

Time-dependent binding of C4-oxidizing NcLPMOs to PASC in the absence (A) and presence (B) of 1 mm AscA. The percentage of unbound LPMO was determined by measuring the reduction in the concentration of LPMO in solution over time. The experiments were carried out using 2 mg·ml−1 PASC and 5 μm LPMO in 50 mm Bis-Tris (pH 6.5) at 45 °C and 1000 rpm. Error bars represent S.D. of triplicate samples. Analysis of the same experiments by SDS-PAGE showed similar trends (Fig. S1).
Substrate specificity
It has previously been shown that NcLPMO9A and -9D are active on PASC (38), whereas a broader substrate specificity has been demonstrated for NcLPMO9C, which acts on cellooligosaccharides with degree of polymerization ≥5, xyloglucan, glucomannan, and β-glucan (25, 42). To gain further information about substrate specificity of each of the three NcLPMOs, their activities were tested in the presence of AscA and single hemicelluloses but also on more complex substrates whereby PASC was premixed with different hemicelluloses. Product mixtures were analyzed by high-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD) and MALDI-TOF MS.
When using PASC as a substrate, HPAEC-PAD analysis showed, as expected, only products oxidized at the C4 position for all three LPMOs (Fig. S2). Considerable amounts of native products were also detected, which is due in part to on-column conversion of C4-oxidized cellodextrins to native compound with one less sugar (the oxidized sugar is chemically removed (48)). Of note, native species emerge when LPMOs act close to chain ends of cellulose, which is more pronounced on amorphous substrates (44). In this case, the ratio of native-to-oxidized species is relatively high at low conversion levels as visible in Fig. S2.
Activity on tamarind xyloglucan (TXG) was detected for NcLPMO9C and NcLPMO9D but not for NcLPMO9A. The product profile generated by NcLPMO9C was as reported previously (25). NcLPMO9D yielded less intense peaks (Fig. S3A), indicating weaker activity of this LPMO on TXG or possibly reflecting that this LPMO is rapidly inactivated in reactions with TXG. MALDI-TOF MS spectra confirmed the TXG activity of NcLPMO9C and NcLPMO9D, showing characteristic clusters of products where each cluster contains products where the number of pentoses equals a multitude of 3 (Fig. 4A). According to the MS data, the products released by NcLPMO9C and NcLPMO9D were identical (Fig. 4B), and the clustered product profiles indicate that for both LPMOs the lytic reaction occurs at the nonsubstituted glucosyl unit.
Figure 4.

Reaction products generated from TXG and from TXG coated on PASC. The figures show MALDI-TOF MS spectra of products generated from TXG (A) with a close-up of the HexnPen3ox region (B) or TXG coated on PASC (C) with a close-up of the HexnPen3ox region (D) by NcLPMO9A (black), NcLPMO9C (red), and NcLPMO9D (blue). Due to the substitution pattern of TXG, where blocks of three substituted glucose units are interspersed with one nonsubstituted glucose, the products appear in clusters where in each cluster the number of pentoses equals a multitude of 3. Variation in the number of hexoses is due to further substitution of the xyloses. Abbreviations (for details see Refs. 25 and 79) used are: Hex, hexose (+162 Da); Pen, pentose (+132 Da); Glc, glucose; X, α-d-Xyl-(1→6)-β-d-Glc; L, β-d-Gal-(1→2)-α-d-Xyl-(1→6)-β-d-Glc; ox, oxidized. Note that in product spectra generated from TXG (B), only products characteristic for xyloglucan appear: GlcoxXXX, GlcoxXXL, and GlcoxXLL. Product spectra generated from TXG coated on PASC (D) also show oxidized products derived from cellulose: Glcox(Glc)5, Glcox(Glc)6, and Glcox(Glc)7.
Remarkably, when incubating NcLPMO9A with a mixture of TXG and PASC, this LPMO showed activity on TXG, yielding products similar to the other two LPMOs (Figs. 4, C and D, and S3B). Although quantitative interpretation of MS data requires caution, it is worth noting the differences between the LPMOs that are visible in Fig. 4D: whereas the product spectrum for NcLPMO9C almost exclusively shows xyloglucan-derived products, the product spectra of the other two show considerable amounts of PASC-derived products.
HPAEC-PAD chromatograms showed that all three LPMOs generated were active on konjac glucomannan (KGM), with NcLPMO9D being the least efficient (Fig. 5A). Analysis of the reaction products by MALDI-TOF MS confirmed oxidative activity on KGM, as oxidized hexose oligomers as well as their acetylated and double acetylated forms (characteristic for KGM) appeared in the spectra (Fig. S4). HPAEC-PAD chromatograms further showed that the activity on KGM was promoted by the presence of PASC, in particular for NcLPMO9D (compare Fig. 5, A and B).
Figure 5.

HPAEC-PAD profiles of soluble reaction products generated by C4-oxidizing NcLPMOs from KGM (A) and KGM coated on PASC (B). Reaction mixtures contained 1 μm NcLPMO9A (black), 1 μm NcLPMO9C (red), or 1 μm NcLPMO9D (blue) and 2 mg·ml−1 KGM (A) or 2 mg·ml−1 KGM and 2 mg·ml−1 PASC (B) with (solid lines) or without (dashed lines) AscA. Note that the LPMO activity not only leads to generation of KGM-derived products eluting between 24 and 40 min but also to a shift of the broad substrate peak that is visible in the control reactions without AscA. Peaks eluting prior to 24 min include oxidized AscA and, in B, oxidized cello-oligomers.
Activity on cellopentaose was confirmed only for NcLPMO9C as reported before (42), whereas only trace activities were detected for both NcLPMO9A and NcLPMO9D (Fig. S5). No activity for any LPMO was observed toward birchwood xylan and ivory nut mannan, either in the absence or in the presence of PASC (data not shown). In the absence of an electron donor, no activity was detected for any substrate.
Variation of electron donors
The dependence of the activity of the NcLPMOs on the nature of the reductant was tested in the reactions with PASC using AscA as a nonenzymatic electron donor and cellobiose dehydrogenase from Myriococcum thermophilum (MtCDH) or pyrroloquinoline quinone (PQQ)-dependent pyranose dehydrogenase from Coprinopsis cinerea (CcPDH) as enzymatic electron donors.
Fig. 6 shows clear dose-response effects upon varying the concentration of AscA. Of note, in these reactions one molecule of AscA is expected to be consumed per substrate oxidation, and the amount of substrate oxidations may be underestimated, likely by a factor of approximately 2 (49), because only solubilized products are detected. In the reactions with 0.3 mm AscA, the maximum amount of released oxidized product was similar for all three LPMOs and in the order of 100 μm (Fig. 6A). In this case, the observed termination of product formation is likely due to depletion of the reductant. Higher concentrations of reductant (Fig. 6, B–D) gave faster initial rates and higher total yields of oxidized products but also increased enzyme inactivation. The latter is suggested by the fact that product formation stopped at product concentrations far below the concentration of AscA (e.g. at 0.6 mm in the reaction with NcLPMO9A and 10 mm AscA). Control experiments in which the reactions with 3.3 mm AscA were supplied with all possible combinations of fresh AscA, PASC, and/or LPMO after 240 min (i.e. at the end points shown in Fig. 6C) showed that only additions containing fresh enzyme led to restoration of product formation (Fig. S6). This confirms that the cessation of product formation indeed is due to enzyme inactivation. In terms of inactivation, the three LPMOs showed clear differences (Fig. 6): although NcLPMO9D became rapidly inactivated and yielded low product levels, NcLPMO9A and, even more so, NcLPMO9C were less sensitive to increased AscA levels, stayed active for a longer time, and reached higher product levels.
Figure 6.

Time course for release of oxidized products during incubation of 1 μmNcLPMO with 2 mg·ml−1 PASC and 0.3 (A), 1 (B), 3.3 (C), or 10 mm (D) AscA. NcLPMO9A (black), NcLPMO9C (red), and NcLPMO9D were incubated with PASC and different concentrations of AscA in 50 mm Bis-Tris (pH 6.5) at 45 °C and 1000 rpm. Solubilized oxidized products were enzymatically converted to Glc4gemGlc using 1 μm TrCel7A. The concentrations of Glc4gemGlc were determined by HPAEC-PAD using a Glc4gemGlc standard prepared as described previously (22). Error bars represent S.D. of triplicate samples.
MtCDH is known to be able to oxidize oligocellulose and cellulose, whereas the PQQ-dependent CcPDH preferably oxidizes monosaccharides with a 1C4 chair conformation such as l-fucose and d-arabinose (50, 51). Therefore, the reactions with CcPDH were fueled by addition of 1 mm l-fucose. Due to the ability of MtCDH to oxidize products released upon LPMO action on PASC, in the reactions with this electron donor it was not possible to calculate the concentration of products oxidized by LPMOs themselves. We could only assess the amount of C1-oxidized sites, which again reflects the degree of substrate solubilization by the LPMO.
Product formation in reactions with these enzymatic donors (Fig. 7) was remarkably different from the results obtained with AscA (Fig. 6). In all cases, more or less linear progress curves were obtained, and the enzyme performing best with AscA, NcLPMO9C, was clearly the least active in the reactions with MtCDH or CcPDH (Fig. 7). NcLPMO9D performed very well, and the reaction with CcPDH yielded product levels higher than in any of the AscA reactions without showing signs of enzyme inactivation. A control experiment using the conditions of Fig. 6C showed that the activity of NcLPMO9D was not improved by addition of 0.5 μm BSA (data not shown), indicating that the good performance of this enzyme in the presence of enzymatic electron donors is not due to the mere presence of additional protein. NcLPMO9A performed well with both AscA and the enzymatic electron donors.
Figure 7.

Time course for release of oxidized products during incubation of 1 μmNcLPMO with 2 mg·ml−1 PASC and 0.5 μmMtCDH (A) or 0.5 μmCcPDH (B). NcLPMO9A (black), NcLPMO9C (red), and NcLPMO9D (blue) were incubated with PASC and MtCDH or CcPDH in 50 mm Bis-Tris (pH 6.5) at 45 °C and 1000 rpm. In the reactions with MtCDH, the amount of released soluble products is expressed as the sum of the integrated peak areas (nC × min) for C1-oxidized (C1-ox) products only (in this case, C4-oxidized products cannot be detected as they are oxidized at C1 position by CDH). In the reactions with CcPDH, the released oxidized products for each sample were enzymatically converted to Glc4gemGlc using 1 μm TrCel7A. The concentrations of Glc4gemGlc in every sample were then determined by HPAEC-PAD using Glc4gemGlc standard prepared as described previously (22). Consequently, values on the y axis in A and B should not be compared; quantitative assessment is only valid for comparing the NcLPMOs within the same panel. Results from the reactions with CcPDH may be compared with the results obtained in the reactions with different AscA concentrations (Fig. 6). Error bars represent S.D. of triplicate samples.
H2O2 production
Reduced LPMOs can carry out an oxidase reaction leading to the production of H2O2 (36, 42), which is a cosubstrate for LPMOs (35). It has been suggested that generation of H2O2 may be rate-limiting in most “standard” LPMO reactions (O2 and e.g. AscA) (11, 35), but this remains somewhat controversial (52). We tested the ability of the three NcLPMO9s to produce H2O2 in the presence of 50 μm AscA as reductant and absence of substrate. The measured initial H2O2 production rates were 1.13 ± 0.05, 0.60 ± 0.08, and 0.91 ± 0.02 min−1 for NcLPMO9A, NcLPMO9C, and NcLPMO9D, respectively (Fig. S7). It is worth noting that these rates are in the same order as initial catalytic rates that could be estimated from the reactions with 0.3 mm AscA (Fig. 6A) and that the ranking of the LPMOs in terms of speed is the same.
H2O2-driven substrate degradation
Next, we assessed whether reactions with the three LPMOs could be fueled by H2O2. To do so, reaction mixtures were fed with low amounts of AscA, to secure reduction of the LPMO, and low amounts of H2O2 that were low enough to prevent enzyme inactivation. In the control reactions, only AscA was added, in the same concentrations as in the reactions with H2O2.
HPAEC and MALDI-TOF MS analyses of generated products (Figs. 8, S8, and S9) did not reveal any differences compared with the reactions performed with 1 mm AscA and no added H2O2 (see Fig. S10 for a direct comparison). In the reactions with PASC, only C4-oxidized products were detected, and, as observed above, NcLPMO9D released slightly longer products compared with NcLPMO9A and NcLPMO9C (Fig. 8A). Reactions without added H2O2 gave almost no products, as one would expect at these very low AscA concentrations (see dose-response curve for AscA in Fig. 6). Likewise, reactions with TXG (Fig. 8B) gave the same results as the reactions with high AscA discussed above (Fig. S3A): NcLPMO9C showed high activity toward TXG, whereas NcLPMO9D showed less activity, and NcLPMO9A seemed inactive. As above, when TXG was premixed with PASC (Fig. 4, C–D), NcLPMO9A showed activity on TXG in the H2O2-fueled reaction (Fig. S8). MS data showed that for all three LPMOs the cleavage pattern of TXG was the same in O2- and H2O2-fueled reactions (Figs. 8D and S8 versus Fig. 4). The results of H2O2-fueled reactions with KGM were also very similar to the results obtained under standard conditions (1 mm AscA and no added H2O2; Fig. 5): NcLPMO9A and -9C showed high activity, whereas the activity of NcLPMO9D on this substrate was barely detectable unless PASC was also present (Fig. S9, A and B). MS analysis showed similar product profiles (Fig. S9, C and D) as in the reactions with high AscA and no added H2O2 (Fig. S4).
Figure 8.

Reaction products generated by C4-oxidizing NcLPMOs from PASC or TXG in reactions with H2O2 as a cosubstrate. A and B, HPAEC-PAD analyses of products generated by 1 μm NcLPMO9A (black line), 1 μm NcLPMO9C (red line), or 1 μm NcLPMO9D (blue line) in reactions with 2 mg·ml−1 PASC (A) or 2 mg·ml−1 TXG (B) in 50 mm Bis-Tris (pH 6.5) at 45 °C and 1000 rpm with addition of ∼45 μm H2O2 to the reactions every 15 min for 4 h. Prior to every addition of H2O2, ∼12 μm AscA was added to ensure reduction of the LPMO. Control reactions (dashed lines) were done in the absence of H2O2, meaning that only ∼12 μm AscA was added every 15 min for 4 h. The labeling of oxidized cello-oligosaccharides in A is based on previous work (42); the identity of the peaks in B is unknown due to lack of standards (but see Ref. 25 for some indications). C, MALDI-TOF MS spectra of oligosaccharides with DP6 and DP7 released from PASC by NcLPMO9A (black line), NcLPMO9C (red line), or NcLPMO9D (blue line). Peaks representing sodium adducts in the DP6 cluster are labeled, and this cluster shows the native product, (Glc)6 and the oxidized hexamer in its keto form, Glcox(Glc)5, and in its gem-diol form, Glc#ox(Glc)5. D, MALDI-TOF MS spectra of products generated from TXG by NcLPMO9A (black), NcLPMO9C (red), and NcLPMO9D (blue). Clusters of products where the number of pentoses equals a multitude of 3 are indicated.
Product release during the H2O2-driven degradation of PASC
Fig. 9 shows that under the conditions used above, i.e. gradual addition of H2O2, product formation from PASC was similar for all three LPMOs, suggesting that the amount of added H2O2 was limiting product formation under these reaction conditions. NcLPMO9A and -9C showed a linear progress curve throughout the reaction, whereas NcLPMO9D showed signs of inactivation after 3 h, in accordance with the apparent lower operational stability of this enzyme that was also observed in e.g. the dose-response study with AscA (Fig. 6). In the linear phase of the reactions, the ratio between the amount of oxidized products and the cumulative amount of added H2O2 was between 40 and 60%, which is compatible with close to stoichiometric incorporation of H2O2 in oxidized products because some 50% of oxidized sites likely remain in the insoluble material (49).
Figure 9.
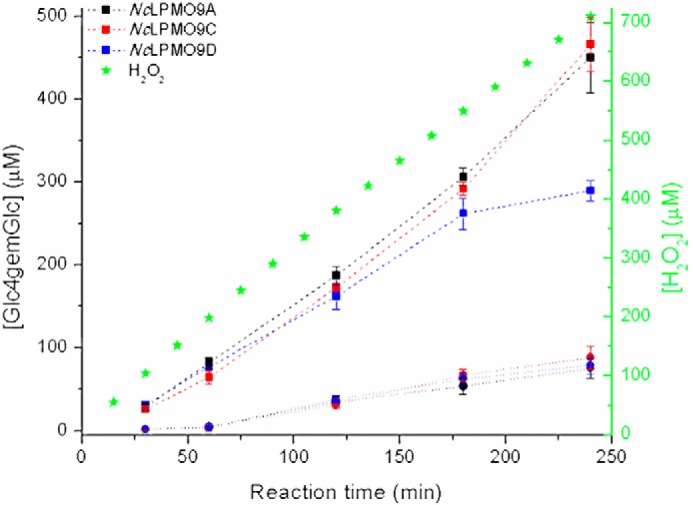
Time course for release of oxidized products during incubation of 2 mg·ml−1 PASC with 1 μm C4-oxidizing NcLPMO in reactions with H2O2 (dashed lines). NcLPMO9A (black squares), NcLPMO9C (red squares), and NcLPMO9D (blue squares) were incubated with 2 mg·ml−1 PASC in 50 mm Bis-Tris (pH 6.5) at 45 °C and 1000 rpm, and ∼45 μm H2O2 was added to the reactions every 15 min. Prior to every addition of H2O2, ∼12 μm AscA was added to ensure reduction of the LPMO. Control reactions (●, connected with dotted lines) were done in the absence of H2O2, meaning that only ∼12 μm AscA was added every 15 min for 4 h. Solubilized oxidized products were enzymatically converted to Glc4gemGlc using 1 μm TrCel7A. The concentrations of Glc4gemGlc, shown on the left y axis, were determined by HPAEC-PAD using a Glc4gemGlc standard prepared as described previously (22). Error bars represent S.D. of triplicate samples. Note that only soluble oxidized products were measured and that the nonsoluble material contains a significant fraction of the oxidized sites. The cumulative amount of H2O2 that was added to the reactions is indicated by green asterisks, and the concentration values are shown on the right y axis.
Discussion
N. crassa is a well-known model organism that also happens to be a proficient plant cell-wall degrader (3). It contains a large arsenal of different CAZymes involved in plant biomass degradation. Annotation of the genome shows that family AA9 LPMOs have the highest multiplicity of all CAZymes in N. crassa, with 14 genes predicted to encode such enzymes (6). For nine of these 14 LPMO9s, activity on cellulose has been demonstrated, and three of these (NcLPMO9A, -9C, and -9D) are strict C4 oxidizers. As structural comparisons and sequence alignments alone cannot provide a biological rationale for the multiplicity of lpmo9 genes, such as different roles during plant cell wall degradation, we carried out functional comparison of the three C4-oxidizing NcLPMOs.
Binding of LPMOs to their substrates may be enhanced by CBMs as in other fungal plant cell wall–degrading enzymes. LPMOs with reduced active-site copper and not bound to a substrate are prone to autocatalytic inactivation through nonproductive reactions with O2 or H2O2 (35), and it has indeed been shown that removal of the CBM not only reduces substrate affinity but also LPMO stability (49, 53). Accordingly, the present results show that stability of the three NcLPMO9s correlates with substrate affinity. NcLPMO9D, without a CBM1 and showing the weakest substrate binding, became rapidly inactivated, yielded low product levels, and was more sensitive to even low AscA levels than NcLPMO9A and NcLPMO9C (both containing a CBM1). In contrast, NcLPMO9C, showing the strongest substrate binding, was the most stable of the three LPMOs. The high operational stability observed for NcLPMO9C under standard conditions (high AscA with O2) may also be due to a lower rate of H2O2 production (Fig. S7), which could reduce the chance of potentially destructive encounters of nonsubstrate-bound LPMO and H2O2. It is noteworthy that the considerable differences in substrate binding between the three LPMOs are not reflected in major differences in the initial catalytic rates, indicating the importance of other factors, such as enzyme mobility on the substrate surface, in enzyme catalysis.
Our results confirm previous observations that reduction of the active-site copper promotes substrate binding (43, 46). The gradual decrease in substrate binding over time, which is visible in Figs. 3 and S1, likely reflects oxidative damage of the LPMO catalytic site and not just reoxidation of the copper, as over time the bound fraction of LPMOs in the presence of AscA was less than in the reactions without AscA.
Importantly, in-depth functional characterization of the three C4-oxidizing NcLPMO9s revealed clear differences in substrate preferences. Only NcLPMO9C is active on shorter cello-oligosaccharides, whereas only NcLPMO9C and NcLPMO9D are active on TXG. Interestingly, NcLPMO9A did show activity on TXG in reactions that also contained cellulose. It is likely that hemicelluloses such as TXG associate with cellulose (54–56), creating junction zones between cellulose and hemicelluloses that enable the LPMO to act also on the now “stabilized” hemicellulose (see also Ref. 30). Further illustrating differences between the LPMOs, in reactions with PASC–TXG mixtures, PASC was still the preferred substrate for NcLPMO9A and NcLPMO9D, whereas NcLPMO9C primarily acted on TXG. Addition of cellulose also promoted activity of the LPMOs on KGM, in particular for NcLPMO9D, which was hardly active on KGM alone. It is worth noting that in reactions with TXG the effect of adding cellulose was largest for NcLPMO9A, whereas it was largest for NcLPMO9D in reactions with KGM. Although further studies on true copolymeric substrates, such as intact plant cell walls, are needed, the present findings clearly show that the three seemingly similar NcLPMO9s have different substrate specificities that may relate to the complex structure of the plant cell wall.
Structural comparisons do not provide obvious explanations for the differences in substrate specificity. In accordance with previous studies on the binding of polymeric substrates (13, 46, 57), it may seem that a larger part of the putative substrate-binding surface is involved in determining substrate preferences. It is known that the L3 region is involved in binding xyloglucan, and Figs. 1 and 2 show that the three LPMOs exhibit relatively much variation in this region, affecting e.g. subsite +2. It is worth noting that the LPMO without a CBM, NcLPMO9D, contains relatively many aromatic residues near the substrate-binding surface (Tyr at positions 25 and 67, subsite +2; Trp at position 205, subsite −4; see Fig. 1A) that may add to substrate affinity and could thus, perhaps, compensate for the lack of a CBM. However, mutagenesis studies on the proposed residues will need to be performed before conclusions on the role of these residues in substrate binding can be drawn.
It has previously been shown that LPMOs differ when it comes to their interactions with small-molecule reductants (58, 59). Here, we analyzed both a small-molecule reductant, AscA, and two enzymatic electron donors, MtCDH and CcPDH. The three LPMOs showed remarkable differences in operational stability and ability to recruit electrons from these varying electron donors. Slower, controlled activation of the LPMOs with an enzymatic donor increased the operational stability of NcLPMO9D, whereas AscA was the better electron donor for NcLPMO9C. Interestingly, in the reactions with enzymatic electron donors, the efficiency of the LPMOs was very different from that in reactions with AscA and inversely correlated to the LPMOs' ability to bind substrate. For NcLPMO9C, it has been shown that the presence of substrate reduces the ability of CDH to reduce the LPMO (46), which could explain why tight substrate binders are less easily reduced by an enzymatic electron donor. In line with this, Várnai et al. (60) have shown that difference in the activation of different LPMOs by CcPDH can be abolished by addition of a small redox mediator, which can more easily access the active site of an LPMO bound to a substrate. The operational stability of the LPMO in the presence of various reductants is determined in part by the extent to which the LPMO is close to the substrate as it becomes reduced. The results with NcLPMO9D seem to support the notion that LPMO reduction close to the substrate is favorable: this LPMO, showing weak substrate binding and low operational stability when reduced by AscA, fully maintained operational stability when fueled by the CBM-containing and thus likely substrate-bound CcPDH.
The results presented above lend further support to the recent claims that H2O2 is a preferred, if not the only, cosubstrate of LPMOs (34, 35). All three NcLPMO9s could efficiently use added H2O2 as a cosubstrate, whereas their activity under standard conditions (1 mm AscA with no added H2O2) was correlated with their ability to produce H2O2. It has been proposed that H2O2-driven LPMO reactions are less specific than O2-driven reactions because oxidative damage in the LPMO, claimed to only occur in the H2O2-driven reaction, would compromise enzyme specificity (52). Although reduction of specificity due to oxidative damage in the active site certainly is conceivable, we would argue that this possible process is not related to the nature of the cosubstrate used. Rather, the degree of oxidative damage, for example caused by a surplus of H2O2 reacting with nonsubstrate-bound reduced LPMOs, is due to an imbalance between the various reactants that leads to nonproductive and potentially damaging LPMO reactions. Here, we show that in well-controlled reactions O2- and H2O2-fueled reactions yield essentially identical product profiles for all three LPMOs with PASC, xyloglucan, and glucomannan. Fig. 9 shows that under controlled conditions one may achieve stoichiometric incorporation of H2O2, whereas the enzymes show good stability. These results show that the specificity of the LPMO is independent of the nature of the cosubstrate used to drive the reaction.
All in all, the present study shows that the three C4-oxidizing cellulose-active LPMOs of N. crassa exhibit a variety of functional differences that may relate to their biological roles. Most importantly, although the true natural substrates of these LPMOs, such as, possibly, certain substructures of the plant cell wall, still need to be discovered, our results clearly show that the three enzymes have different substrate specificities. Another major difference concerns the interplay with varying reductants, which is interesting because the availability of both enzymatic and nonenzymatic electron donors will vary during plant cell wall degradation both in space and time (9, 61). Thus, it is conceivable that these three LPMOs have distinctive roles during degradation of the complex cell wall, which could explain the multiplicity of LPMOs in plant cell wall–degrading fungi.
Experimental procedures
Chemicals
Bis-Tris buffer salt was purchased from VWR (Radnor, PA); hydrogen peroxide (30% aqueous solution) was purchased from Merck; ascorbic acid, l-fucose, Avicel® PH-101, xylan from birch wood, horseradish peroxidase, and 10-acetyl-3,7-dihydroxyphenoxazine (AmplexRed) were purchased from Sigma-Aldrich. Cellopentaose (purity >95%), xyloglucan from tamarind seeds (purity ∼95%), konjac glucomannan (purity >98%), and ivory nut mannan (purity >98%) were purchased from Megazyme (Bray, Ireland). PASC was prepared from Avicel PH-101 as described before (62).
Enzymes
MtCDH and PQQ-dependent CcPDH were expressed in Pichia pastoris and purified as reported previously (63, 64). The enzyme concentrations of MtCDH and CcPDH preparations were determined by measuring absorbance at 280 nm and using molar extinction coefficients of 159,000 and 146,500 M−1·cm−1, respectively, determined using the ExPASy ProtParam tool (65).
Cloning, expression, and purification of LPMOs from N. crassa
The gene encoding NcLPMO9A with its native signal peptide was codon-optimized for expression in P. pastoris and ordered from GenScript (Piscataway, NJ) in a pUC57 vector. A Kozak sequence (GAAACG) was inserted upstream of the start codon, and EcoRI and Acc65I restriction sites were introduced for cloning in the 5′ and 3′ ends, respectively. The pUC57 vector containing the codon-optimized sequence was digested by EcoRI and Acc65I, and the gene fragment was ligated into EcoRI/Acc65I-digested pPINK-GAP_TaCel5A (66), yielding pPINK-GAP_2240. The expression vector was transformed into Escherichia coli TOP10 (Invitrogen), and transformants were selected on brain–heart infusion agar with 200 μg·ml−1 ampicillin. The pPINK-GAP_2240 plasmid was purified, linearized with AflII, and transformed into electrocompetent cells of P. pastoris PichiaPinkTM Strain 4. Electrocompetent P. pastoris cells were prepared by following the manufacturer's procedure. The electrocompetent cells were electroporated using a Bio-Rad Gene Pulser II electroporation unit (Bio-Rad Laboratories) at 1.8 kV, 25 microfarads, 200 ohms. The transformed cells were incubated in yeast extract–peptone–dextrose medium containing 1 m sorbitol overnight and spread on Pichia adenine-dropout selection plates, which were incubated at 30 °C.
For purification, the production strain was grown in 20 ml of buffered complex glycerol medium containing 1% (v/v) glycerol in a 100-ml shake flask at 29 °C and 200 rpm for 16 h. Subsequently, this preculture was used to inoculate 0.5 liter of buffered complex glycerol medium containing 1% (v/v) glycerol in a 2-liter shake flask followed by incubation at 29 °C and 200 rpm for 48 h. After 24 h, the culture was supplemented with 1% (v/v) glycerol. The cells were removed by centrifugation at 7,000 × g for 15 min at 4 °C. The supernatant was collected and dialyzed against 50 mm Bis-Tris buffer (pH 6.5) and concentrated to 100 ml using a VivaFlow 200 tangential cross-flow concentrator (molecular mass cutoff, 10 kDa; Sartorius Stedim Biotech GmbH, Germany). Ammonium sulfate was added to the concentrated supernatant to a final concentration of 1.42 m after which the solution was loaded onto a 5-ml HiTrap Phenyl FF column (GE Healthcare) equilibrated with 50 mm Bis-Tris buffer (pH 6.5) containing 1.42 m ammonium sulfate. Proteins bound to the column were eluted using a 25-ml linear gradient from 1.42 to 0 m ammonium sulfate in 50 mm Bis-Tris buffer (pH 6.5) using a flow rate of 1 ml·min−1. Collected fractions were analyzed by SDS-PAGE, and the fractions containing NcLPMO9A were pooled and subsequently concentrated down to 2 ml using Amicon Ultra centrifugal filters (molecular mass cutoff, 10 kDa; Merck Millipore). The protein solution was loaded onto a HiLoad 16/60 Superdex 75 size-exclusion column (GE Healthcare) in 50 mm Bis-Tris buffer (pH 6.5) containing 150 mm NaCl using a flow rate of 0.75 ml·min−1. Fractions containing pure NcLPMO9A were identified using SDS-PAGE and subsequently pooled and concentrated using Amicon Ultra centrifugal filters (molecular mass cutoff, 10 kDa).
NcLPMO9C (UniProt accession number Q7SHI8) was cloned in P. pastoris (67) and expressed and purified following a previously published protocol (36). NcLPMO9D (UniProt accession number Q1K8B6) was cloned as described earlier (23) using a protocol similar to that described above for NcLPMO9A and a synthetic gene that encoded the protein with its native signal sequence. The enzyme was purified as described above for NcLPMO9A.
The protein concentrations of NcLPMO9A, NcLPMO9C, and NcLPMO9D preparations were determined by measuring absorbance at 280 nm and using molar extinction coefficients of 45,000, 46,900, and 35,660 m−1·cm−1, respectively, determined using the ExPASy ProtParam tool (65). The enzymes were saturated with Cu(II) by incubating with an excess of Cu(II)SO4 (at an ∼3:1 molar ratio of copper:enzyme) for 90 min in 50 mm Bis-Tris (pH 6.5) at room temperature as described previously (68). The Cu(II)-loaded sample of NcLPMO9A was buffer-exchanged to 50 mm Bis-Tris buffer (pH 6.5) using Amicon Ultra centrifugal filters (molecular mass cutoff, 3 kDa). The resulting solution with purified protein was filtered through a 0.22-μm Millex®-GV filter (Merck Millipore) and stored at 4 °C.
Crystallization and X-ray data collection
Purified NcLPMO9A was cleaved with papain (papain from papaya latex, Sigma-Aldrich) at an LPMO:papain ratio of 33:1 (w/w) to remove the CBM1 that is appended to the catalytic LPMO9 domain. The reaction was carried out in 100 mm sodium acetate buffer (pH 5.0) for 48 h at 37 °C, and its outcome was assessed by SDS-PAGE. For the isolation of the cleaved catalytic domain, size-exclusion chromatography was applied using a Superdex 75 16/600 column (GE Healthcare) equilibrated in 50 mm MES (pH 6.5) and 150 mm NaCl. The purified catalytic domain (NCU02240-N) was buffer-exchanged into 20 mm MES (pH 6.5) and concentrated to 15 mg·ml−1 for crystallization trials. A wide range of crystallization conditions were tested using a Mosquito crystallization robot (TTP Labtech, UK) and commercially available 96-well kits. Crystals of NCU02240-N grew at room temperature in the presence of 20% PEG3350 and 0.2 m Li2SO4 using the sitting-drop vapor-diffusion technique. Before being flash frozen in liquid N2, a single crystal was cryoprotected by transferring to mother liquor containing 35% PEG3350. X-ray diffraction data were collected to 1.6-Å resolution on beamline ID23-2 (European Synchrotron Radiation Facility, Grenoble, France) under cryogenic conditions. The wavelength of the X-ray beam was 0.8726 Å, and the oscillation range was 0.05°. The resulting data set was processed using XDS (69), and the structure was solved by molecular replacement using Phaser (70). The molecular replacement search model was produced by CHAINSAW (71) using NcLPMO9D (PDB code 4EIR) (37) as the template structure. Iterative rounds of model building and refinement of the structure were performed using Coot (72) and REFMAC (73) from the CCP4i program suite. Solvent molecules were added using Coot and checked manually. The quality of the final structure model was evaluated using MolProbity (74). The final structure model and the structure factors are deposited at the PDB under accession code 5FOH. Data collection and refinement statistics are summarized in Table S1. All structure figures were prepared using the UCSF Chimera package (75).
Binding of LPMOs to PASC
Binding studies with PASC were performed as described before (76) in the presence or absence of 1 mm AscA. The reaction mixtures contained 2 mg·ml−1 PASC or 2 mg·ml−1 PASC premixed with 1 mm AscA and 5 μm enzyme in 50 mm Bis-Tris buffer (pH 6.5) and were incubated at 45 °C with shaking at 1000 rpm in an Eppendorf ThermoMixer. At various time points (5, 15, 30, 60, and 120 min), samples were taken, and insoluble substrate and substrate-bound enzyme were removed by filtering using a 96-well filter plate (Merck Millipore) operated by a Millipore vacuum manifold. The concentration of the enzyme in the filtrate was determined by measuring A280 (Eppendorf BioPhotometer). Furthermore, the filtrates from each time point were mixed with an equal volume of SDS sample buffer and analyzed by SDS-PAGE by loading exactly 5 μl of such prepared samples per well.
LPMO reactions with AscA
Unless otherwise stated, reaction mixtures contained 2 mg·ml−1 substrate, 1 μm NcLPMO, and 1 mm AscA in 50 mm Bis-Tris buffer (pH 6.5). Reactions were performed in 2-ml Eppendorf tubes containing 100-μl total reaction volume and incubated at 45 °C with shaking at 1000 rpm in an Eppendorf ThermoMixer. Reactions were stopped by boiling for 10 min. Subsequently, separation of soluble and insoluble fractions was done by centrifugation at 11,000 × g for 10 min. As controls, reactions without added reductant were also analyzed.
Detection of oxidized products
Oxidized products were analyzed using HPAEC-PAD and by MALDI-TOF MS. HPAEC was performed on a Dionex ICS5000 system equipped with a CarboPac PA1 analytical column (2 × 250 mm) and a CarboPac PA1 guard column (2 × 50 mm) using a 50-min gradient (44) for cellulosic and a 75-min gradient (25) for hemicellulosic substrates. Chromatograms were recorded with Chromeleon and analyzed using Origin 9.1 software (OriginLab, Northampton, MA).
MALDI-TOF MS was performed on an Ultraflex MALDI-TOF/TOF instrument (Bruker Daltonik GmbH, Bremen, Germany) equipped with a nitrogen 337 nm laser. The instrument was operated in positive-linear acquisition mode. The samples were sodium-saturated by mixing 5 μl of sample with 5 μl of 50 mm sodium acetate. 1 μl of the saturated sample was spotted on an MTP 384 ground steel MALDI target and mixed immediately with an equal volume of 20 mg·ml−1 2,5-dihydroxybenzoic acid in 30% acetonitrile and 0.1% (v/v) TFA. The data were analyzed using mMass software (77). Baseline correction and Gaussian smoothing (window size, 0.3 m/z) were applied to all spectra.
Quantitative assessment of products released from PASC with varying electron donors
Reaction mixtures (600 μl) with nonenzymatic electron donors contained 2 mg·ml−1 PASC, 1 μm NcLPMO, and 0.33, 1, 3.3, or 10 mm AscA in 50 mm Bis-Tris buffer (pH 6.5). Control reactions to check for substrate, enzyme, and reductant depletion were set up using the same conditions with 3.3 mm AscA in 100 μl; after 240 min, 100 μl of the reaction mixtures were supplemented with 50 μl of buffer (50 mm Bis-Tris (pH 6.5)) or with 50 μl of the same buffer containing various combinations of 2 mg·ml−1 substrate, 1 μm LPMO, and 3.3 mm AscA followed by further incubation for 120 min.
Reaction mixtures (600 μl) with an enzymatic electron donor contained 2 mg·ml−1 PASC, 1 μm NcLPMO, and 1 μm MtCDH (63) or 1 μm CcPDH (64). The reactions with CcPDH also contained 1 mm l-fucose, acting as a substrate for CcPDH.
All reactions were incubated at 45 °C with shaking at 1000 rpm in an Eppendorf ThermoMixer. At various time points (20, 40, 60, 120, and 240 min), 120-μl samples were taken and boiled for 10 min. Separation of soluble and insoluble fractions was achieved by centrifugation at 11,000 × g for 10 min. Prior to product quantification, soluble fractions (32 μl) were mixed with 31 μl of 150 mm sodium acetate buffer (pH 4.5) and 1 μl of TrCel7A (∼1 μm) and incubated for 16 h at 37 °C. The treatment with TrCel7A converts oligomeric products to a mixture of glucose, cellobiose, and Glc4gemGlc (C4-oxidized cellobiose) as the only oxidized product. Soluble fractions treated in this way were subsequently analyzed using HPAEC-PAD. The amount of released oxidized products was quantified using 4-hydroxy-β-d-xylo-Hexp-(1→4)-β-d-Glcp (Glc4gemGlc), prepared as described previously (22), as a standard. In the reactions with MtCDH, the sum of integrated peak areas of C1-oxidized products (nC × min) was used as a proxy for LPMO activity.
LPMO reactions with H2O2
Unless otherwise stated, reaction mixtures (200 μl) contained 2 mg·ml−1 substrate and 1 μm NcLPMO in 50 mm Bis-Tris buffer (pH 6.5). Reactions were initiated by adding 2 μl of 1.5 mm AscA immediately followed by addition of 2 μl of 5 mm H2O2. Every 15 min, an additional 2 μl of 1.5 mm AscA and 2 μl of 5 mm H2O2 (in this order) were added to the reaction mixtures (15 additions in total over a period of 4 h). Reactions were stopped by boiling for 10 min followed by separation of soluble and insoluble material by centrifugation at 11,000 × g for 10 min. Control reactions without added H2O2 were also performed.
Quantitative assessment of products released from PASC in reactions with H2O2
Reaction mixtures with H2O2 (600-μl final volume after initiation of the reaction) contained 2 mg·ml−1 PASC and 1 μm NcLPMO in 50 mm Bis-Tris buffer (pH 6.5). Reactions were initiated by adding 6 μl of 1.5 mm AscA (final concentration, 15 μm) followed by addition of 6 μl of 5 mm H2O2 (final concentration, 50 μm). After exactly 15 min, an additional 6 μl of 1.5 mm AscA and 5 mm H2O2 were added. 30 min after the reactions were initiated, a 122-μl sample was taken from the reaction mixture and boiled for 10 min. To the rest of the reaction mixture (490 μl), 5 μl of 1.5 mm AscA (15 μm) and 5 μl of 5 mm H2O2 (50 μm) were added. The same volumes of 1.5 mm AscA and 5 mm H2O2 were also added at 45 min after reaction initiation. At 60 min, a 118-μl sample was taken and boiled for 10 min. To the rest of reaction mixture (392 μl), 4 μl of 1.5 mm AscA (15 μm) and 4 μl of 5 mm H2O2 (50 μm) were added. The same volumes of 1.5 mm AscA and 5 mm H2O2 were also added at 75, 90, and 115 min after the reactions were initiated. After 120 min, 130-μl samples were taken and boiled for 10 min. To the rest of reaction mixture (294 μl), 3 μl of 1.5 mm AscA (15 μm) and 3 μl of 5 mm H2O2 (50 μm) were added. The same volumes of 1.5 mm AscA and 5 mm H2O2 were added at 135, 150, and 165 min after reaction initiation. After 180 min, 122-μl samples were taken and boiled for 10 min. To the rest of the reaction mixture (196 μl), 2 μl of 1.5 mm AscA (15 μm) and 2 μl of 5 mm H2O2 (50 μm) were added, and this was repeated at 195, 210, and 225 min after reaction initiation. After 240 min, the reactions were stopped by boiling for 10 min.
Separation of soluble and insoluble materials was achieved by centrifugation at 11,000 × g for 10 min. Samples of the soluble fractions (32 μl) were mixed with 31 μl of 150 mm sodium acetate buffer (pH 4.5) and 1 μl of TrCel7A (∼1 μm) and incubated for 16 h at 37 °C. The oxidized products were subsequently detected and quantified using HPAEC-PAD and Glc4gemGlc as a standard as described above.
H2O2 production
Measurement of H2O2 production was done as described previously (78). A reaction mixture (180 μl) containing 1 μm LPMO, 5 units·ml−1 horseradish peroxidase, and 100 μm AmplexRed in 50 mm Bis-Tris buffer (pH 6.5) was incubated for 5 min at 40 °C in a 96-well microtiter plate in a plate reader (MultiskanTM FC microplate photometer, Thermo Fisher Scientific, Bremen Germany). The reaction was initiated by the addition of 20 μl of 500 μm AscA (50 μm final concentration) in each well, and the production of resorufin was monitored at 540 nm. Control reactions in the absence of LPMO were carried out to obtain the LPMO-independent resorufin production rate. This control reaction provided a background signal equal to 0.3% of the LPMO-catalyzed reaction and was subtracted from the latter. An H2O2 standard curve was prepared using the same conditions (without AscA and LPMO). The reactions were monitored for 45 min, and H2O2 production rates were derived from data points in the linear region between 0 and 8.5 min.
Author contributions
D. M. P., A. V., M. D., G. M., M. S., B. W., and V. G. H. E. conceptualization; D. M. P., M. D., and M. S. data curation; D. M. P., A. V., and M. D. formal analysis; D. M. P. and M. D. investigation; D. M. P. and A. V. visualization; D. M. P., A. V., and V. G. H. E. writing-original draft; A. V., G. M., M. S., B. W., and V. G. H. E. supervision; M. D., G. M., M. S., B. W., and V. G. H. E. writing-review and editing; M. S. and V. G. H. E. funding acquisition.
Supplementary Material
This work was supported by Norwegian Research Council Grants 214613, 244259, and 243663 (to D. M. P., A. V., B. W., V. G. H. E.) and Swedish Energy Agency Project 40144-1 (to M. S.). This work was also supported by VINNOVA (Swedish Governmental Agency for Innovation Systems Grant 2014-01453 (to M. D.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 5FOH) have been deposited in the Protein Data Bank (http://wwpdb.org/).
This article contains Table S1 and Figs. S1–S10.
- GH
- glycoside hydrolase
- AscA
- ascorbic acid
- CBM
- carbohydrate-binding module
- CDH
- cellobiose dehydrogenase
- HPAEC-PAD
- high-performance anion-exchange chromatography with pulsed amperometric detection
- KGM
- konjac glucomannan
- LPMO
- lytic polysaccharide monooxygenase
- PASC
- phosphoric acid–swollen cellulose
- PQQ
- pyrroloquinoline quinone
- PDH
- pyranose dehydrogenase
- TXG
- tamarind xyloglucan
- CAZyme
- carbohydrate-active enzyme
- Nc
- N. crassa
- Ls
- L. similis
- PDB
- Protein Data Bank
- Mt
- M. thermophilum
- Cc
- C. cinerea
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- Tr
- Trichoderma reesei
- gem
- geminal
- nC
- nanocoulombs
- DP
- degree of polymerization.
References
- 1. Eberhart B. M., Beck R. S., and Goolsby K. M. (1977) Cellulase of Neurospora crassa. J. Bacteriol. 130, 181–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yazdi M. T., Radford A., Keen J. N., and Woodward J. R. (1990) Cellulase production by Neurospora crassa: purification and characterization of cellulolytic enzymes. Enzyme Microb. Technol. 12, 120–123 10.1016/0141-0229(90)90084-4 [DOI] [PubMed] [Google Scholar]
- 3. Davis R. H., and Perkins D. D. (2002) Timeline: Neurospora: a model of model microbes. Nat. Rev. Genet. 3, 397–403 10.1038/nrg797 [DOI] [PubMed] [Google Scholar]
- 4. Martinez D., Berka R. M., Henrissat B., Saloheimo M., Arvas M., Baker S. E., Chapman J., Chertkov O., Coutinho P. M., Cullen D., Danchin E. G., Grigoriev I. V., Harris P., Jackson M., Kubicek C. P., et al. (2008) Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat. Biotechnol. 26, 553–560 10.1038/nbt1403 [DOI] [PubMed] [Google Scholar]
- 5. Levasseur A., Drula E., Lombard V., Coutinho P. M., and Henrissat B. (2013) Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 6, 41 10.1186/1754-6834-6-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tian C., Beeson W. T., Iavarone A. T., Sun J., Marletta M. A., Cate J. H., and Glass N. L. (2009) Systems analysis of plant cell wall degradation by the model filamentous fungus Neurospora crassa. Proc. Natl. Acad. Sci. U.S.A. 106, 22157–22162 10.1073/pnas.0906810106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Horn S. J., Vaaje-Kolstad G., Westereng B., and Eijsink V. (2012) Novel enzymes for the degradation of cellulose. Biotechnol. Biofuels 5, 45 10.1186/1754-6834-5-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mba Medie F., Davies G. J., Drancourt M., and Henrissat B. (2012) Genome analyses highlight the different biological roles of cellulases. Nat. Rev. Microbiol. 10, 227–234 10.1038/nrmicro2729 [DOI] [PubMed] [Google Scholar]
- 9. Kracher D., Scheiblbrandner S., Felice A. K., Breslmayr E., Preims M., Ludwicka K., Haltrich D., Eijsink V. G., and Ludwig R. (2016) Extracellular electron transfer systems fuel cellulose oxidative degradation. Science 352, 1098–1101 10.1126/science.aaf3165 [DOI] [PubMed] [Google Scholar]
- 10. Lenfant N., Hainaut M., Terrapon N., Drula E., Lombard V., and Henrissat B. (2017) A bioinformatics analysis of 3400 lytic polysaccharide oxidases from family AA9. Carbohydr. Res. 448, 166–174 10.1016/j.carres.2017.04.012 [DOI] [PubMed] [Google Scholar]
- 11. Bissaro B., Várnai A., Røhr Å. K., and Eijsink V. G. H. (2018) Oxidoreductases and reactive oxygen species in conversion of lignocellulosic biomass. Microbiol. Mol. Biol. Rev. 82, e00029–18 10.1128/MMBR.00029-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Quinlan R. J., Sweeney M. D., Lo Leggio L., Otten H., Poulsen J. C., Johansen K. S., Krogh K. B., Jørgensen C. I., Tovborg M., Anthonsen A., Tryfona T., Walter C. P., Dupree P., Xu F., Davies G. J., et al. (2011) Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. U.S.A. 108, 15079–15084 10.1073/pnas.1105776108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aachmann F. L., Sørlie M., Skjåk-Bræk G., Eijsink V. G., and Vaaje-Kolstad G. (2012) NMR structure of a lytic polysaccharide monooxygenase provides insight into copper binding, protein dynamics, and substrate interactions. Proc. Natl. Acad. Sci. U.S.A. 109, 18779–18784 10.1073/pnas.1208822109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vaaje-Kolstad G., Westereng B., Horn S. J., Liu Z., Zhai H., Sørlie M., and Eijsink V. G. (2010) An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330, 219–222 10.1126/science.1192231 [DOI] [PubMed] [Google Scholar]
- 15. Beeson W. T., Vu V. V., Span E. A., Phillips C. M., and Marletta M. A. (2015) Cellulose degradation by polysaccharide monooxygenases. Annu. Rev. Biochem. 84, 923–946 10.1146/annurev-biochem-060614-034439 [DOI] [PubMed] [Google Scholar]
- 16. Hemsworth G. R., Johnston E. M., Davies G. J., and Walton P. H. (2015) Lytic polysaccharide monooxygenases in biomass conversion. Trends Biotechnol. 33, 747–761 10.1016/j.tibtech.2015.09.006 [DOI] [PubMed] [Google Scholar]
- 17. Johansen K. S. (2016) Lytic polysaccharide monooxygenases: the microbial power tool for lignocellulose degradation. Trends Plant Sci. 21, 926–936 10.1016/j.tplants.2016.07.012 [DOI] [PubMed] [Google Scholar]
- 18. Vaaje-Kolstad G., Forsberg Z., Loose J. S., Bissaro B., and Eijsink V. G. (2017) Structural diversity of lytic polysaccharide monooxygenases. Curr. Opin. Struct. Biol. 44, 67–76 10.1016/j.sbi.2016.12.012 [DOI] [PubMed] [Google Scholar]
- 19. Meier K. K., Jones S. M., Kaper T., Hansson H., Koetsier M. J., Karkehabadi S., Solomon E. I., Sandgren M., and Kelemen B. (2018) Oxygen activation by Cu LPMOs in recalcitrant carbohydrate polysaccharide conversion to monomer sugars. Chem. Rev. 118, 2593–2635 10.1021/acs.chemrev.7b00421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tandrup T., Frandsen K. E. H., Johansen K. S., Berrin J. G., and Lo Leggio L. (2018) Recent insights into lytic polysaccharide monooxygenases (LPMOs). Biochem. Soc. Trans. 46, 1431–1447 10.1042/BST20170549 [DOI] [PubMed] [Google Scholar]
- 21. Hu J., Chandra R., Arantes V., Gourlay K., Susan van Dyk J., and Saddler J. N. (2015) The addition of accessory enzymes enhances the hydrolytic performance of cellulase enzymes at high solid loadings. Bioresour. Technol. 186, 149–153 10.1016/j.biortech.2015.03.055 [DOI] [PubMed] [Google Scholar]
- 22. Müller G., Várnai A., Johansen K. S., Eijsink V. G., and Horn S. J. (2015) Harnessing the potential of LPMO-containing cellulase cocktails poses new demands on processing conditions. Biotechnol. Biofuels 8, 187 10.1186/s13068-015-0376-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chylenski P., Petrović D. M., Müller G., Dahlström M., Bengtsson O., Lersch M., Siika-Aho M., Horn S. J., and Eijsink V. G. H. (2017) Enzymatic degradation of sulfite-pulped softwoods and the role of LPMOs. Biotechnol. Biofuels 10, 177 10.1186/s13068-017-0862-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Müller G., Chylenski P., Bissaro B., Eijsink V. G. H., and Horn S. J. (2018) The impact of hydrogen peroxide supply on LPMO activity and overall saccharification efficiency of a commercial cellulase cocktail. Biotechnol. Biofuels 11, 209 10.1186/s13068-018-1199-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Agger J. W., Isaksen T., Várnai A., Vidal-Melgosa S., Willats W. G., Ludwig R., Horn S. J., Eijsink V. G., and Westereng B. (2014) Discovery of LPMO activity on hemicelluloses shows the importance of oxidative processes in plant cell wall degradation. Proc. Natl. Acad. Sci. U.S.A. 111, 6287–6292 10.1073/pnas.1323629111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bennati-Granier C., Garajova S., Champion C., Grisel S., Haon M., Zhou S., Fanuel M., Ropartz D., Rogniaux H., Gimbert I., Record E., and Berrin J. G. (2015) Substrate specificity and regioselectivity of fungal AA9 lytic polysaccharide monooxygenases secreted by Podospora anserina. Biotechnol. Biofuels 8, 90 10.1186/s13068-015-0274-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vu V. V., Beeson W. T., Span E. A., Farquhar E. R., and Marletta M. A. (2014) A family of starch-active polysaccharide monooxygenases. Proc. Natl. Acad. Sci. U.S.A. 111, 13822–13827 10.1073/pnas.1408090111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lo Leggio L., Simmons T. J., Poulsen J. C., Frandsen K. E., Hemsworth G. R., Stringer M. A., von Freiesleben P., Tovborg M., Johansen K. S., De Maria L., Harris P. V., Soong C. L., Dupree P., Tryfona T., Lenfant N., et al. (2015) Structure and boosting activity of a starch-degrading lytic polysaccharide monooxygenase. Nat. Commun. 6, 5961 10.1038/ncomms6961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frommhagen M., Sforza S., Westphal A. H., Visser J., Hinz S. W., Koetsier M. J., van Berkel W. J., Gruppen H., and Kabel M. A. (2015) Discovery of the combined oxidative cleavage of plant xylan and cellulose by a new fungal polysaccharide monooxygenase. Biotechnol. Biofuels 8, 101 10.1186/s13068-015-0284-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Couturier M., Ladevèze S., Sulzenbacher G., Ciano L., Fanuel M., Moreau C., Villares A., Cathala B., Chaspoul F., Frandsen K. E., Labourel A., Herpoël-Gimbert I., Grisel S., Haon M., Lenfant N., et al. (2018) Lytic xylan oxidases from wood-decay fungi unlock biomass degradation. Nat. Chem. Biol. 14, 306–310 10.1038/nchembio.2558 [DOI] [PubMed] [Google Scholar]
- 31. Phillips C. M., Beeson W. T., Cate J. H., and Marletta M. A. (2011) Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem. Biol. 6, 1399–1406 10.1021/cb200351y [DOI] [PubMed] [Google Scholar]
- 32. Westereng B., Cannella D., Wittrup Agger J., Jørgensen H., Larsen Andersen M., Eijsink V. G., and Felby C. (2015) Enzymatic cellulose oxidation is linked to lignin by long-range electron transfer. Sci. Rep. 5, 18561 10.1038/srep18561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beeson W. T., Phillips C. M., Cate J. H., and Marletta M. A. (2012) Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J. Am. Chem. Soc. 134, 890–892 10.1021/ja210657t [DOI] [PubMed] [Google Scholar]
- 34. Bissaro B., Røhr Å. K., Skaugen M., Forsberg Z., Horn S. J., Vaaje-Kolstad G., and Eijsink V. (2016) Fenton-type chemistry by a copper enzyme: molecular mechanism of polysaccharide oxidative cleavage. bioRxiv 10.1101/097022 [DOI] [Google Scholar]
- 35. Bissaro B., Røhr Å. K., Müller G., Chylenski P., Skaugen M., Forsberg Z., Horn S. J., Vaaje-Kolstad G., and Eijsink V. G. H. (2017) Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2. Nat. Chem. Biol. 13, 1123–1128 10.1038/nchembio.2470 [DOI] [PubMed] [Google Scholar]
- 36. Kittl R., Kracher D., Burgstaller D., Haltrich D., and Ludwig R. (2012) Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay. Biotechnol. Biofuels 5, 79 10.1186/1754-6834-5-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li X., Beeson W. T. 4th, Phillips C. M., Marletta M. A., and Cate J. H. (2012) Structural basis for substrate targeting and catalysis by fungal polysaccharide monooxygenases. Structure 20, 1051–1061 10.1016/j.str.2012.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vu V. V., Beeson W. T., Phillips C. M., Cate J. H., and Marletta M. A. (2014) Determinants of regioselective hydroxylation in the fungal polysaccharide monooxygenases. J. Am. Chem. Soc. 136, 562–565 10.1021/ja409384b [DOI] [PubMed] [Google Scholar]
- 39. Borisova A. S., Isaksen T., Dimarogona M., Kognole A. A., Mathiesen G., Várnai A., Røhr Å. K., Payne C. M., Sørlie M., Sandgren M., and Eijsink V. G. (2015) Structural and functional characterization of a lytic polysaccharide monooxygenase with broad substrate specificity. J. Biol. Chem. 290, 22955–22969 10.1074/jbc.M115.660183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mattinen M. L., Linder M., Drakenberg T., and Annila A. (1998) Solution structure of the cellulose-binding domain of endoglucanase I from Trichoderma reesei and its interaction with cello-oligosaccharides. Eur. J. Biochem. 256, 279–286 10.1046/j.1432-1327.1998.2560279.x [DOI] [PubMed] [Google Scholar]
- 41. Boraston A. B., Bolam D. N., Gilbert H. J., and Davies G. J. (2004) Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem. J. 382, 769–781 10.1042/BJ20040892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Isaksen T., Westereng B., Aachmann F. L., Agger J. W., Kracher D., Kittl R., Ludwig R., Haltrich D., Eijsink V. G., and Horn S. J. (2014) A C4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J. Biol. Chem. 289, 2632–2642 10.1074/jbc.M113.530196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kracher D., Andlar M., Furtmüller P. G., and Ludwig R. (2018) Active-site copper reduction promotes substrate binding of fungal lytic polysaccharide monooxygenase and reduces stability. J. Biol. Chem. 293, 1676–1687 10.1074/jbc.RA117.000109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Westereng B., Agger J. W., Horn S. J., Vaaje-Kolstad G., Aachmann F. L., Stenstrøm Y. H., and Eijsink V. G. (2013) Efficient separation of oxidized cello-oligosaccharides generated by cellulose degrading lytic polysaccharide monooxygenases. J. Chromatogr. A 1271, 144–152 10.1016/j.chroma.2012.11.048 [DOI] [PubMed] [Google Scholar]
- 45. Wu M., Beckham G. T., Larsson A. M., Ishida T., Kim S., Payne C. M., Himmel M. E., Crowley M. F., Horn S. J., Westereng B., Igarashi K., Samejima M., Ståhlberg J., Eijsink V. G., and Sandgren M. (2013) Crystal structure and computational characterization of the lytic polysaccharide monooxygenase GH61D from the Basidiomycota fungus Phanerochaete chrysosporium. J. Biol. Chem. 288, 12828–12839 10.1074/jbc.M113.459396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Courtade G., Wimmer R., Røhr Å. K., Preims M., Felice A. K., Dimarogona M., Vaaje-Kolstad G., Sørlie M., Sandgren M., Ludwig R., Eijsink V. G., and Aachmann F. L. (2016) Interactions of a fungal lytic polysaccharide monooxygenase with beta-glucan substrates and cellobiose dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 113, 5922–5927 10.1073/pnas.1602566113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Frandsen K. E., Simmons T. J., Dupree P., Poulsen J. C., Hemsworth G. R., Ciano L., Johnston E. M., Tovborg M., Johansen K. S., von Freiesleben P., Marmuse L., Fort S., Cottaz S., Driguez H., Henrissat B., et al. (2016) The molecular basis of polysaccharide cleavage by lytic polysaccharide monooxygenases. Nat. Chem. Biol. 12, 298–303 10.1038/nchembio.2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Westereng B., Arntzen M. Ø., Aachmann F. L., Várnai A., Eijsink V. G., and Agger J. W. (2016) Simultaneous analysis of C1 and C4 oxidized oligosaccharides, the products of lytic polysaccharide monooxygenases acting on cellulose. J. Chromatogr. A 1445, 46–54 10.1016/j.chroma.2016.03.064 [DOI] [PubMed] [Google Scholar]
- 49. Courtade G., Forsberg Z., Heggset E. B., Eijsink V. G. H., and Aachmann F. L. (2018) The carbohydrate-binding module and linker of a modular lytic polysaccharide monooxygenase promote localized cellulose oxidation. J. Biol. Chem. 293, 13006–13015 10.1074/jbc.RA118.004269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pricelius S., Ludwig R., Lant N., Haltrich D., and Guebitz G. M. (2009) Substrate specificity of Myriococcum thermophilum cellobiose dehydrogenase on mono-, oligo-, and polysaccharides related to in situ production of H2O2. Appl. Microbiol. Biotechnol. 85, 75–83 10.1007/s00253-009-2062-0 [DOI] [PubMed] [Google Scholar]
- 51. Takeda K., Matsumura H., Ishida T., Samejima M., Ohno H., Yoshida M., Igarashi K., and Nakamura N. (2015) Characterization of a novel PQQ-dependent quinohemoprotein pyranose dehydrogenase from Coprinopsis cinerea classified into auxiliary activities family 12 in carbohydrate-active enzymes. PLoS One 10, e0115722 10.1371/journal.pone.0115722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hangasky J. A., Iavarone A. T., and Marletta M. A. (2018) Reactivity of O2 versus H2O2 with polysaccharide monooxygenases. Proc. Natl. Acad. Sci. U.S.A. 115, 4915–4920 10.1073/pnas.1801153115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Forsberg Z., Bissaro B., Gullesen J., Dalhus B., Vaaje-Kolstad G., and Eijsink V. G. H. (2018) Structural determinants of bacterial lytic polysaccharide monooxygenase functionality. J. Biol. Chem. 293, 1397–1412 10.1074/jbc.M117.817130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Carpita N. C., and Gibeaut D. M. (1993) Structural models of primary cell walls in flowering plants: consistency of molecular structure with the physical properties of the walls during growth. Plant J. 3, 1–30 10.1111/j.1365-313X.1993.tb00007.x [DOI] [PubMed] [Google Scholar]
- 55. Burton R. A., Gidley M. J., and Fincher G. B. (2010) Heterogeneity in the chemistry, structure and function of plant cell walls. Nat. Chem. Biol. 6, 724–732 10.1038/nchembio.439 [DOI] [PubMed] [Google Scholar]
- 56. Park Y. B., and Cosgrove D. J. (2015) Xyloglucan and its interactions with other components of the growing cell wall. Plant Cell Physiol. 56, 180–194 10.1093/pcp/pcu204 [DOI] [PubMed] [Google Scholar]
- 57. Bissaro B., Isaksen I., Vaaje-Kolstad G., Eijsink V. G. H., and Røhr Å. K. (2018) How a lytic polysaccharide monooxygenase binds crystalline chitin. Biochemistry 57, 1893–1906 10.1021/acs.biochem.8b00138 [DOI] [PubMed] [Google Scholar]
- 58. Frommhagen M., Koetsier M. J., Westphal A. H., Visser J., Hinz S. W., Vincken J. P., van Berkel W. J., Kabel M. A., and Gruppen H. (2016) Lytic polysaccharide monooxygenases from Myceliophthora thermophila C1 differ in substrate preference and reducing agent specificity. Biotechnol. Biofuels 9, 186 10.1186/s13068-016-0594-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Frommhagen M., Westphal A. H., van Berkel W. J. H., and Kabel M. A. (2018) Distinct substrate specificities and electron-donating systems of fungal lytic polysaccharide monooxygenases. Front. Microbiol. 9, 1080 10.3389/fmicb.2018.01080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Várnai A., Umezawa K., Yoshida M., and Eijsink V. G. H. (2018) The pyrroloquinoline-quinone dependent pyranose dehydrogenase from Coprinopsis cinerea (CcPDH) drives lytic polysaccharide monooxygenase (LPMO) action. Appl. Environ. Microbiol. 84, e00156–18 10.1128/AEM.00156-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang J., Presley G. N., Hammel K. E., Ryu J. S., Menke J. R., Figueroa M., Hu D., Orr G., and Schilling J. S. (2016) Localizing gene regulation reveals a staggered wood decay mechanism for the brown rot fungus Postia placenta. Proc. Natl. Acad. Sci. U.S.A. 113, 10968–10973 10.1073/pnas.1608454113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wood T. M. (1988) Preparation of crystalline, amorphous, and dyed cellulase substrates, in Methods in Enzymology (Wood W. A., and Kellogg S. T., eds) pp. 19–25, Academic Press, San Diego [Google Scholar]
- 63. Zámocký M., Schümann C., Sygmund C., O'Callaghan J., Dobson A. D., Ludwig R., Haltrich D., and Peterbauer C. K. (2008) Cloning, sequence analysis and heterologous expression in Pichia pastoris of a gene encoding a thermostable cellobiose dehydrogenase from Myriococcum thermophilum. Protein Expr. Purif. 59, 258–265 10.1016/j.pep.2008.02.007 [DOI] [PubMed] [Google Scholar]
- 64. Matsumura H., Umezawa K., Takeda K., Sugimoto N., Ishida T., Samejima M., Ohno H., Yoshida M., Igarashi K., and Nakamura N. (2014) Discovery of a eukaryotic pyrroloquinoline quinone-dependent oxidoreductase belonging to a new auxiliary activity family in the database of carbohydrate-active enzymes. PLoS One 9, e104851 10.1371/journal.pone.0104851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gasteiger E., Hoogland C., Gattiker A., Duvaud S. e., Wilkins M. R., Appel R. D., and Bairoch A. (2005) Protein identification and analysis tools on the ExPASy server, in The Proteomics Protocols Handbook (Walker J. M., ed) pp. 571–607, Humana Press, Totowa, NJ [Google Scholar]
- 66. Várnai A., Tang C., Bengtsson O., Atterton A., Mathiesen G., and Eijsink V. G. (2014) Expression of endoglucanases in Pichia pastoris under control of the GAP promoter. Microb. Cell Fact. 13, 57 10.1186/1475-2859-13-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sygmund C., Kracher D., Scheiblbrandner S., Zahma K., Felice A. K., Harreither W., Kittl R., and Ludwig R. (2012) Characterization of the two Neurospora crassa cellobiose dehydrogenases and their connection to oxidative cellulose degradation. Appl. Environ. Microbiol. 78, 6161–6171 10.1128/AEM.01503-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Loose J. S., Forsberg Z., Fraaije M. W., Eijsink V. G., and Vaaje-Kolstad G. (2014) A rapid quantitative activity assay shows that the Vibrio cholerae colonization factor GbpA is an active lytic polysaccharide monooxygenase. FEBS Lett. 588, 3435–3440 10.1016/j.febslet.2014.07.036 [DOI] [PubMed] [Google Scholar]
- 69. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Stein N. (2008) CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J. Appl. Crystallogr. 41, 641–643 10.1107/S0021889808006985 [DOI] [Google Scholar]
- 72. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 74. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 76. Forsberg Z., Nelson C. E., Dalhus B., Mekasha S., Loose J. S., Crouch L. I., Røhr Å. K., Gardner J. G., Eijsink V. G., and Vaaje-Kolstad G. (2016) Structural and functional analysis of a lytic polysaccharide monooxygenase important for efficient utilization of chitin in Cellvibrio japonicus. J. Biol. Chem. 291, 7300–7312 10.1074/jbc.M115.700161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Strohalm M., Kavan D., Novák P., Volný M., and Havlícek V. (2010) mMass 3: a cross-platform software environment for precise analysis of mass spectrometric data. Anal. Chem. 82, 4648–4651 10.1021/ac100818g [DOI] [PubMed] [Google Scholar]
- 78. Petrović D. M., Bissaro B., Chylenski P., Skaugen M., Sørlie M., Jensen M. S., Aachmann F. L., Courtade G., Várnai A., and Eijsink V. G. H. (2018) Methylation of the N-terminal histidine protects a lytic polysaccharide monooxygenase from auto-oxidative inactivation. Protein Sci. 27, 1636–1650 10.1002/pro.3451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nekiunaite L., Petrović D. M., Westereng B., Vaaje-Kolstad G., Hachem M. A., Várnai A., and Eijsink V. G. (2016) FgLPMO9A from Fusarium graminearum cleaves xyloglucan independently of the backbone substitution pattern. FEBS Lett. 590, 3346–3356 10.1002/1873-3468.12385 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.