

Summary
The tangerine pathotype of the ascomycete fungus Alternaria alternata is the causal agent of citrus brown spot, which can result in significant losses of both yield and marketability for tangerines worldwide. A conditionally dispensable chromosome (CDC), which harbours the host‐selective ACT toxin gene cluster, is required for tangerine pathogenicity of A. alternata. To understand the genetic makeup and evolution of the tangerine pathotype CDC, we isolated and sequenced the CDCs of the A. alternata Z7 strain and analysed the function and evolution of their genes. The A. alternata Z7 strain has two CDCs (~1.1 and ~0.8 Mb, respectively), and the longer Z7 CDC contains all but one contig of the shorter one. Z7 CDCs contain 254 predicted protein‐coding genes, which are enriched in functional categories associated with ‘metabolic process’ (55 genes, P = 0.037). Relatively few of the CDC genes can be classified as carbohydrate‐active enzymes (CAZymes) (4) and transporters (19) and none as kinases. Evolutionary analysis of the 254 CDC proteins showed that their evolutionary conservation tends to be restricted within the genus Alternaria and that the CDC genes evolve faster than genes in the essential chromosomes, likely due to fewer selective constraints. Interestingly, phylogenetic analysis suggested that four of the 25 genes responsible for the ACT toxin production were likely transferred from Colletotrichum (Sordariomycetes). Functional experiments showed that two of them are essential for the virulence of the tangerine pathotype of A. alternata. These results provide new insights into the function and evolution of CDC genes in Alternaria.
Keywords: accessory chromosome, Dothideomycetes, evolutionary origin, horizontal gene transfer, karyotype, pathogenicity
Introduction
Alternaria alternata fungi can be ubiquitously found in soil, various plants, and decaying plant debris (Thomma, 2003). Some A. alternata strains can cause plant diseases and result in severe crop losses worldwide. Because these strains can often be morphologically very similar but exhibit substantial pathological differences, they have been defined as pathotypes of A. alternata (Tsuge et al., 2013). At least seven pathogenic A. alternata pathotypes, each producing a unique host‐selective toxin (HST) essential to pathogenicity, have been recognized to cause diseases in Japanese pear, strawberry, tangerine, apple, tomato, rough lemon and tobacco (Tsuge et al., 2013). Generally, genes required for HST biosynthesis in A. alternata are clustered on relatively small chromosomes that are 1.0–2.0 Mb in size (Tsuge et al., 2013). These accessory or conditionally dispensable chromosomes (CDCs) are highly variable among species, are generally not required for growth and reproduction on artificial media, but are required for A. alternata pathogenicity (Hatta et al., 2002; Johnson et al., 2001).
The importance of CDCs conferring pathogenicity to fruits has been demonstrated by the construction of A. alternata hybrids in laboratory conditions. More specifically, two distinct laboratory hybrids were constructed from tomato and strawberry pathotypes and separately for apple and tomato pathotypes. The resulting hybrids harboured two CDCs from their parents, allowing for the production of HSTs that caused diseases in both parental plant species (Akagi et al., 2009b; Akamatsu et al., 2001). Furthermore, these studies support the hypothesis that CDCs can be transmitted between different strains thereby facilitating the spread and evolutionary diversification of fungal phytopathogens.
Besides A. alternata, many other fungal phytopathogens have CDCs in their genomes, including Fusarium oxysporum, Nectria haematococca, Zymoseptoria tritici (previously known as Mycosphaerella graminicola and Septoria tritici) and Colletotrichum gloeosporioides (Coleman et al., 2009; Ma et al., 2010; Stukenbrock et al., 2010). Because CDCs are typically found in some, but not all, strains, they have been proposed to have different evolutionary origins than the essential chromosomes (Covert, 1998; Tsuge et al., 2013), e.g. through horizontal transfer from distantly related species (Covert, 1998; Hatta et al., 2002). The possibility of horizontal transfer of CDCs between species has been demonstrated in several studies. For example, a 2 Mb chromosome was transferred between two different biotypes of C. gloeosporioides during vegetative co‐cultivation in the laboratory (He et al., 1998). Importantly, CDC acquisition can facilitate the transition from the non‐pathogenic to pathogenic phenotype in the recipient organism. For example, co‐incubation of non‐pathogenic and pathogenic genotypes of F. oxysporum revealed that the transfer of a CDC from the pathogenic (donor) to non‐pathogenic genotype (recipient) enabled the recipient to become pathogenic to tomatoes (Ma et al., 2010).
Previously, the ~1.0 Mb CDC of the tomato pathotype of A. alternata was identified and characterized (Hu et al., 2012). Genes in that CDC are abundant in the categories of ‘metabolic process’ and ‘biosynthetic process’, and include 36 polyketide and non‐ribosomal peptide synthetase domain‐containing genes. Furthermore, the G+C content of third codon positions, codon usage bias and repeat region load in the CDC are different from those in the essential chromosomes (Hu et al., 2012), raising the hypothesis that the A. arborescens CDC was acquired through horizontal transfer from an unrelated fungus (Hu et al., 2012).
The tangerine pathotype of A. alternata, which can cause citrus brown spot on tangerines and tangerine hybrids, was demonstrated to harbour an additional chromosome of about 1.9–2.0 Mb by pulsed‐field gel electrophoresis studies (Miyamoto et al., 2008, 2009). Seven genes, ACTTR, ACTT2, ACTT3, ACTT5, ACTT6, ACTTS2 and ACTTS3, were located on this chromosome and found to be required for the biosynthesis of the ACT toxin, a unique HST produced by the tangerine pathotype (Tsuge et al., 2013). However, relatively little is known about the content, potential biological functions and evolution of the other genes in the CDC of the tangerine pathotype of A. alternata.
To address this question, we isolated and sequenced the CDCs of the tangerine pathotype strain Z7 of A. alternata isolated from Zhejiang, China (Huang et al., 2015). We found that the Z7 strain contains two different CDCs, 1.1 and 0.8 Mb in size; in contrast, another strain of the same pathotype from Florida was described to have one CDC (Miyamoto et al., 2009). Interestingly, the smaller CDC is nearly identical in sequence to part of the larger CDC in the Z7 strain. Examination of the functional annotation of all 254 genes in the Z7 CDCs showed that 55 out of the 254 CDC genes are enriched in functions associated with ‘metabolic process’. Furthermore, we found that conservation for the majority of the 254 Z7 CDC genes was restricted to the genus Alternaria and that four CDC genes belonging to the ACT toxin gene cluster likely originated via horizontal gene transfer (HGT) events from other fungi. The recombination mutants of two HGT genes showed a dramatic reduction in virulence to citrus leaves. These results suggest that Alternaria CDCs exhibit chromosomal polymorphisms, that CDC genes are involved in processes associated with metabolism, that they are rapidly evolving and that the characteristics associated with their virulence sometimes originate via HGT.
Results
Identification and sequencing of the CDCs
The tangerine pathotype strain of A. alternata from Florida contains one CDC (Miyamoto et al., 2009), which led us to reason that the A. alternata Z7 strain would also contain one CDC. To evaluate the genomic size of the CDC in A. alternata Z7, we performed pulsed‐field gel electrophoresis using Z7 protoplast cells. Unexpectedly, we found that the strain Z7 possesses two CDCs, one that is ~1.1 Mb in size and another that is ~0.8 Mb (Fig. 1). The electrophoretic karyotype of A. alternata Z7 is different from that of the tangerine pathotype strains isolated from Florida, USA, which has only one ~1.9–2.0 Mb CDC (Miyamoto et al., 2009). These results suggest that Alternaria strains within the same pathotype exhibit variability in both the number and size of their CDCs.
Figure 1.
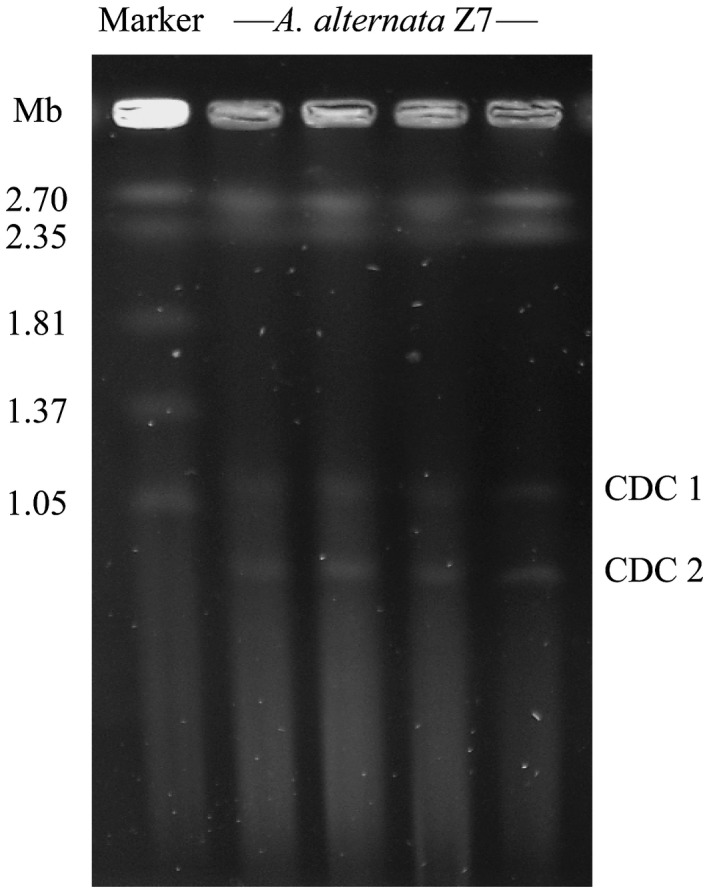
Electrophoretic karyotype of the Alternaria alternata Z7 conditionally dispensable chromosomes (CDCs). Chromosomal bands were separated by CHEF gel electrophoresis. The size marker is Hansenula wingei.
To obtain the genomic sequence of the A. alternata Z7 CDCs, we isolated the corresponding CDC bands from the pulsed‐field electrophoresis gel and performed Illumina resequencing. We generated a total of 2.6 and 2.9 Gb of mate‐pair read sequences for CDCs 1 and 2, respectively. Then, 236 Mb (CDC 1) and 249 Mb (CDC 2) of sequence reads were successfully mapped to the Z7 reference genome, the covered percentage of each contig was calculated and those contigs with more than 50% of their sequences covered by CDC sequencing reads were considered to be parts of the A. alternata Z7 CDC (Table S1). Finally, we obtained the 945 kb sequence of Z7 CDC 1 composed of 30 contigs and a 447 kb sequence of Z7 CDC 2 composed of 16 contigs. Intriguingly, CDC 1 contains all but one contig of CDC 2, suggesting that chromosomal duplication may be involved in the formation of the CDCs in A. alternata Z7 (Table S1).
General features of CDCs
The overall G + C content of the CDCs (~50%) was comparable with that (51%) of the essential chromosomes (ECs). The percentages of repetitive sequences on the Z7 CDC 1 and CDC 2 were 3.95% and 3.86%, respectively, around 8‐fold of that of ECs (0.51%). The actual percentage of repetitive sequences on the Z7 CDCs is expected to be larger as repetitive sequences are the major factor contributing to the fragmented assembly of the CDCs. The average gene length and gene density of CDCs were smaller than those of ECs, and the percentage of intronless genes in CDCs (~36%) was higher than that of genes in the ECs (~24%) (Table 1).
Table 1.
General features of the essential chromosomes (ECs) and conditionally dispensable chromosomes (CDCs) in Alternaria alternata strain Z7.
| Features | ECs | CDC 1 | CDC 2 |
|---|---|---|---|
| Sequenced genome size (kb) | 33329 | 945 | 447 |
| Number of contigs | 124 | 30 | 16 |
| G+C content (%) | 51.0 | 50.1 | 50.1 |
| Protein‐coding genes | 11785 | 238 | 117 |
| Gene density (number of genes per Mb) | 354 | 252 | 262 |
| Mean gene length (bp) | 1731 | 1530 | 1497 |
| Mean number of exons per gene | 2.8 | 2.4 | 2.4 |
| Mean length of exons | 558 | 547 | 544 |
| Mean number of introns per gene | 1.8 | 1.4 | 1.4 |
| Mean length of introns (bp) | 93 | 132 | 127 |
| Percentage of genes without intron (%) | 24 | 35 | 36 |
| Repeat rate (%)* | 0.51 | 3.95 | 3.86 |
| tRNA genes | 111 | 4 | 2 |
The fragmented assembly of the CDCs might give an under‐representation of the repetitive content.
Functional annotation of the genes in the CDC
The CDC 1 was predicted to comprise 238 protein‐coding genes from 22 out of 30 contigs while the CDC 2 was predicted to comprise 117 protein‐coding genes from 9 out of 16 contigs. Both CDCs share 101 genes and they have a total of 254 unique genes. The remaining 16 genes of CDC 2 are located in the one contig that was not shared with CDC 1. To predict their functions, gene ontology (GO) analysis was performed on these 254 CDC genes and 111 genes were assigned to 77 GO terms related to ‘biological process’. There are 55 genes assigned to the GO term ‘metabolic process’ (P = 0.037) and two are assigned to ‘divalent inorganic cation transport’ (P = 0.018) (Fig. S1). Within ‘metabolic process’, the GO term ‘regulation of metabolic process’ (10 genes, P = 0.016) was also significantly enriched (Fig. S1).
To functionally annotate the 254 genes in the CDCs, we classified their protein products into protein families using several different approaches. Based on Pfam domain characterization, 127/254 genes belonged to 92 protein families (Table S2). We identified four carbohydrate‐active enzyme (CAZyme) genes in the CDCs, accounting for 1.07% (4/373) of the total CAZymes of Z7. These include two glycosyl transferases (GTs), one glycoside hydrolase (GH) and one auxiliary activity (AA) (Table S2). A total of four secreted proteins were predicted in the CDCs, including three small secreted cysteine‐rich proteins (SSCPs) (Table S2). No kinase was found in the CDCs. We identified 19 transcription factors in the CDCs, which can be divided into six subfamilies: zinc finger Zn2‐Cys6 (6), zinc finger C2H2 (5), Myb‐like DNA‐binding (2), helix‐turn‐helix Psq (3), high mobility group box (2) and centromere protein B (1) (Table S2). Twelve transporter‐encoding genes were found in the CDCs (Table S2). Based on comparisons with proteins in the PHI database, 13 CDCs proteins are related to pathogenicity (Table S2). Finally, the CDCs contained only one secondary metabolite gene cluster, the ACT toxin biosynthetic gene cluster present only in the tangerine pathotype of A. alternata, which has been described in detail previously (Wang et al., 2016).
The evolutionary origin of the Z7 CDCs
To explore the evolutionary origin of the Z7 CDCs, we compared the sequence similarity (calculated by % BLAST identity score * % query coverage) of each of the 254 CDC and EC proteins to the protein sequences from the genomes of other species in the Dothideomycetes. In the species phylogeny, Alternaria species are grouped into three clades (I , II, III), which coincides with a previously constructed phylogeny based on 200 conserved single‐copy orthologues (Wang et al., 2016). Proteins in the Z7 EC showed a very high sequence similarity values (median > 75%, average = 81.0%) to proteins in other Dothideomycetes while proteins in the Z7 CDCs showed a wide range of sequence similarity values (median > 35%, average = 40.4%) to proteins in other Dothideomycetes (Fig. 2), suggesting that the level of variation of CDC genes is much larger than that of EC genes. As expected, the highest degree of similarity was with proteins of other Alternaria species (Fig. 2). For example, we found that 216/254 (85.0%) of the Z7 CDC proteins showed >50% similarity to proteins in A. turkisafria (Fig. 2), which can also cause citrus brown spot. However, A. citriarbusti and A. tangelonis can also cause citrus brown spot but these exhibited much lower numbers (130/254, 51.2% and 124/254, 48.8%, respectively) of proteins with >50% similarity to proteins in Z7 CDCs (Fig. 2). These results suggest that the gene content and sequence similarity on CDCs can be highly variable among strains of the same pathotype, which coincides with the pulsed‐field gel electrophoresis (PFGE) results (Fig. 1).
Figure 2.
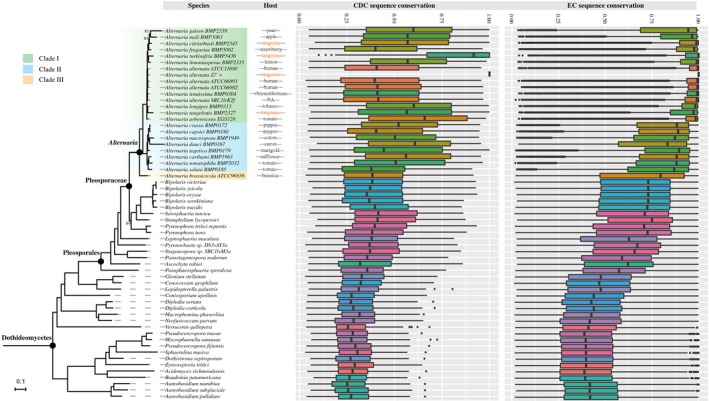
Sequence conservation of Z7 CDC genes across the phylogeny of Alternaria and representative species of Dothideomycetes. The phylogeny on the left depicts the evolutionary relationships of species of Alternaria and representative Dothideomycetes. The maximum‐likelihood phylogeny was inferred from the concatenation‐based analysis of an amino acid data matrix composed of 1754 single‐copy BUSCO genes under the LG+R10 substitution model. Branches with bootstrap support values of 100% are not shown; branches with bootstrap values <100% are shown near each branch. The heat map on the right was constructed using the sequence conservation value (% BLAST identity * % query coverage) of each Z7 CDC protein to its best counterpart in each of the species included in this analysis. The bar plot shows the degree of sequence conservation of Z7 CDC and EC proteins to the proteomes of other species in the Dothideomycetes. The sequence conservation value of each Z7 CDC protein to its best counterpart in each of the species was calculated by % BLAST identity * % query coverage. The column next to the Alternaria species’ name indicates the corresponding host to each isolate. NA, not applicable.
To examine if any of the Z7 CDC proteins were more similar to proteins outside those found in genomes from the genus Alternaria, a BLASTp search of those 254 protein sequences against the NCBI non‐redundant proteins database (without the Alternaria database included) was performed and the result was filtered using the E value < 1e−10 and sequence identity >30% criteria. We found that the best matches for 245/254 proteins were proteins from other Alternaria species, three proteins had their best matches to be proteins found in non‐Alternaria members from the family Pleosporaceae (class Dothideomycetes) and six have no hits to proteins in other species.
To further dissect the evolutionary history of Z7 CDC genes, we reconstructed the phylogenetic tree for each Z7 CDC gene with their orthologues from other species in the Dothideomycetes. There were too few orthologues to construct phylogenetic trees for two CDC genes; among the remaining 252 gene trees, 185 showed monophyly within the genus Alternaria. Taken together, our results suggest that most of the A. alternata Z7 CDC genes were likely present in the Alternaria ancestor and that some of them were likely independently lost in some of the species during the diversification of the genus.
Relative evolutionary rate of Z7 CDCs
To figure out if genes in the CDCs and genes in the ECs in Z7 evolve differently, we built a phylogenetic tree for each Z7 gene that has orthologues in more than 11 (50% of total number) species within the Alternaria genus. A total of 146 CDC and 11 340 EC genes were used to calculate the average branch lengths for each tree. The average branch length for most genes in both the CDCs and ECs is very low (Fig. 3), which indicates that most genes are highly conserved within the genus Alternaria. However, as a whole, we found that EC genes had lower average branch lengths than CDC genes (P < 2.2e‐16, Fig. 3), suggesting that the Z7 CDC genes are evolving faster than the Z7 EC genes.
Figure 3.
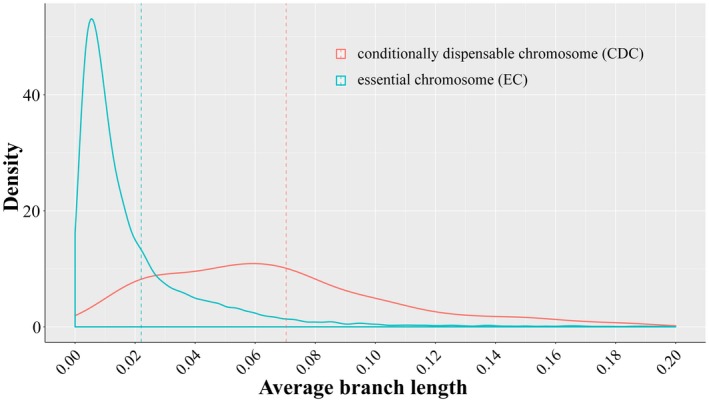
Distribution of the average branch length from all individual Z7 conditionally dispensable chromosome (CDC) (red line) or essential chromosome (EC) (blue line) gene trees constructed from groups of orthologous genes within Alternaria. The dashed lines denote the mean values of the two distributions.
To figure out if genes in the CDCs may have different selective pressure from genes in the ECs, we calculated the dN/dS value for all the above‐mentioned CDC and EC gene orthologs. Overall, we found that CDC genes had higher dN/dS value than EC genes (P < 2.2e−16, Fig. 4), indicating that CDC genes have less selective constraints than EC genes.
Figure 4.
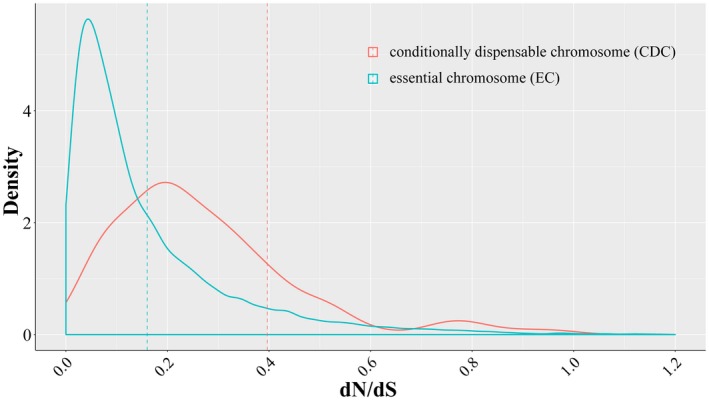
Distribution of the dN/dS value from all individual Z7 conditionally dispensable chromosome (CDC) (red line) or essential chromosome (EC) (blue line) gene trees constructed from groups of orthologous genes within Alternaria. The dashed lines denote the mean values of the two distributions.
HGT of genes in the CDC
To examine whether any of the A. alternata Z7 CDC genes originated via HGT, we calculated the Alien Index (AI) (Gladyshev et al., 2008; Wisecaver et al., 2016) of all 254 genes. A total of 11 genes show AI > 0 and at least 80% of their top 200 BLASTp hits with a taxonomic classification other than Dothideomycetes. The validity of these 11 HGT candidates was further examined phylogenetically. The phylogenetic trees for most of these 11 HGT candidates were weakly supported, but the evolutionary origin of four of them was strongly supported to be outside Dothideomycetes (Table 2 and Figs [Link], [Link], [Link], [Link]). The approximately unbiased (AU) test for each of the four genes significantly rejected the hypothesis that they formed a monophyletic group with the rest of the sequences from Dothideomycetes (Table 2).
Table 2.
Summary list of all horizontal gene transfer candidates in Alternaria alternata Z7 conditionally dispensable chromosomes (CDCs).
| Gene ID | Protein length (amino acids) | Closest sequence | Closest species | Proteome identity (%) | Protein identity (%) | Description | Distribution in Alternaria * | AU test † P value |
|---|---|---|---|---|---|---|---|---|
| AALT_g12032 | 349 | KZL71680.1 | Colletotrichum tofieldiae | 51 | 72 | Acyl‐CoA dehydrogenase | Three pathotypes | 3E−73 |
| AALT_g11755 | 296 | KZL71655.1 | Colletotrichum tofieldiae | 51 | 64 | HMG‐CoA hydrolase | Three pathotypes | 2E−50 |
| AALT_g11757 | 2349 | OLN84341.1 | Colletotrichum chlorophyti | 50 | 59 | Polyketide synthase | Three pathotypes | 3E−52 |
| AALT_g11758 | 439 | OLN84339.1 | Colletotrichum chlorophyti | 50 | 74 | Cytochrome P450 | Three pathotypes | 2E−56 |
Three pathotypes: the Japanese pear, strawberry and tangerine pathotypes.
AU, approximately unbiased.
The ACT toxin is essential for the pathogenicity of A. alternata Z7 to citrus leaves, the synthesis of which is predicted to be controlled by a cluster composed of about 25 genes (Wang et al., 2016). The ACT toxin gene cluster is only located in the CDC and most of their genes have multiple copies (Miyamoto et al., 2009, 2008; Wang et al., 2016). Surprisingly, 4/25 of the ACT cluster genes contained in the Z7 CDC strain are always grouped together with sequences from the unrelated Colletotrichum (Sordariomycetes) in their gene phylogenies (Fig. 5A and Figs [Link], [Link], [Link], [Link]). Interestingly, the orthologous genes in Colletotrichum tofieldiae are physically linked with each other and are part of a secondary metabolite biosynthetic gene cluster predicted by antiSMASH 4.0 (Blin et al., 2017) (Fig. 5B), although the gene order and orientation of the two clusters is different (Fig. 5B). Besides A .alternata Z7 (the tangerine pathotype), this cluster is also present in the Japanese pear pathotype and the strawberry pathotype (Wang et al., 2016); thus we infer that the HGT events occurred before the divergence of the Z7 strain from the other Alternaria pathotypes examined and not after. The four HGT genes encode an acyl‐CoA dehydrogenase, a 3‐hydroxy‐3‐methylglutaryl (HMG)‐CoA hydrolase, a polyketide synthase and a cytochrome P450 (Table 2). To know the expression pattern of these four HGT genes during infection related to time course, we performed real‐time PCR at 1, 3, 6, 9 and 12 h post‐infection for each of these four genes. The results showed that all four genes were expressed during the infection process. Though the magnitude of fold changes differed, expression of AALT_g12032, AALT_g11755 and AALT_g11757 was highest at 6 h post‐inoculation while expression of AALT_g11758 progressively increased during the tested time course (Fig. 6).
Figure 5.
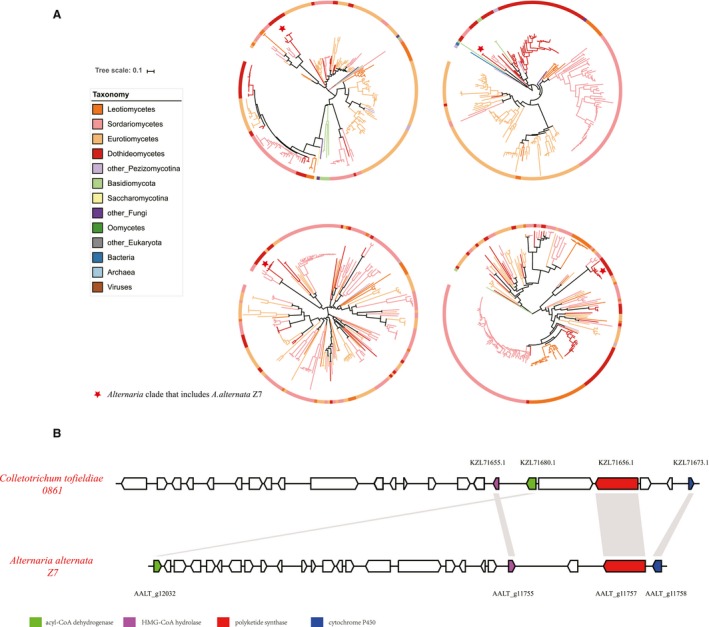
Horizontal transfer of four genes in the ACT toxin gene cluster in the tangerine pathotype of Alternaria alternata Z7 conditionally dispenable chromosome (CDC). (A) Phylogenetic evidence of the horizontal gene transfer of these genes. For each gene, the maximum‐likelihood phylogeny was inferred under the best substitution model automatically selected by ModelFinder, as implemented in IQ‐TREE 1.5.4. Branch colours indicate the taxonomic lineages to which the different taxa included in each phylogeny belong. The red asterisk indicates the Alternaria clade on each phylogeny. The full phylogenetic trees of the individual genes can be found in Figs [Link], [Link], [Link], [Link]. (B) Conservation of synteny among the ACT toxin gene cluster in A. alternata Z7 and a predicted secondary metabolic gene cluster in Colletotrichum tofieldiae 0861. Orthologues among different species are marked with same colour and homologous regions are shown by the grey boxes. Arrows indicate gene direction.
Figure 6.
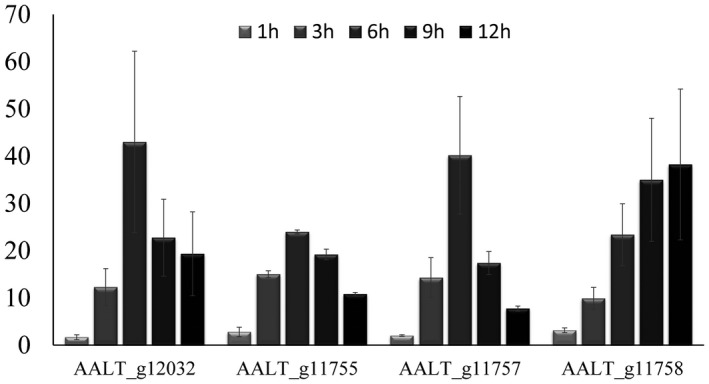
The relative transcript level of the four horizontal gene transfer candidates in the wild‐type at 1, 3, 6, 9 and 12 h post‐inoculation.
Previously, deletion of the AKT3 gene in the Japanese pear pathotype, which is the orthologue of the HMG‐CoA hydrolase gene ACTT3 (AALT_g11755), produced toxin‐deficient and non‐pathogenic mutants (Tanaka and Tsuge, 2000). To further determine the function of the remaining three HGT genes in ACT toxin biosynthesis and virulence, homologous recombination was employed to replace the target gene with the phosphotransferase B gene by protoplast transformation (Fig. S6A–C). As all of them have multiple copies in the genome, we could not generate a gene deletion mutant. In the resultant transformants, both pathogenicity‐sufficient and pathogenicity‐deficient mutants were obtained for genes AALT_g12032 and AALT_g11758 (Fig. 7A). However, pathogenicity‐deficient mutants for gene AALT_g11757 were not obtained (Fig. S6D). To further confirm the mutants, AALT_g11758‐targeted transformants were chosen to perform the Southern blot and the results showed that the pathogenicity‐deficient mutant lacks one copy of the target gene (Fig. 7B). To test whether genes AALT_g12032 and AALT_g11758 play a role in controlling ACT toxin production, we inoculated the crude ACT toxin extracts from the wild‐type and mutant strains on the citrus leaves. The results showed that the crude ACT toxin extracts of the wild‐type and pathogenicity‐sufficient transformants induced severe necrosis on the citrus leaves while that of the pathogenicity‐deficient transformants had obviously reduced toxicity (Fig. 7C). Taken together, these results show that the HGT of four genes from a lineage related to Colletotrichum have contributed to the composition of the HST gene clusters found in the CDCs of three pathotypes of Alternaria.
Figure 7.

Functional analyses of three horizontally transferred genes. (A) Necrosis symptoms in citrus leaves inoculated with the potato dextrose agar plug (48 h) of the wild‐type and mutant strains. Each leaf was inoculated with the wild‐type strain and three mutant strains. WT, wild‐type; T1, transformant 1; T2, transformant 2; T3, transformant 3. Transformants AALT_g12032‐1, AALT_g12032‐3 and AALT_g11758‐1 are pathogenicity‐sufficient and transformants AALT_g12032‐2, AALT_g11758‐2 and AALT_g11758‐3 are pathogenicity‐deficient. (B) Southern blot of the wild‐type and AALT_g11758‐targeted transformants. The genomic DNA was independently digested by three restriction enzymes: XhoI, PstI and EcoRI and then hybridized with the AALT_g11758 probe. The pathogenicity‐deficient strain (T2) lacks one copy of the AALT_g11758 gene. (C) Necrotic lesions appearing on citrus leaves inoculated with the crude ACT toxin extracts from the wild‐type and mutant strains.
Discussion
CDCs are commonly found in fungal phytopathogens and play key roles in pathogenicity (Hatta et al., 2002; Johnson et al., 2001). However, despite their importance, our knowledge about the gene content and evolutionary origin of CDCs is limited. In this study, we identified and characterized the CDCs of the tangerine pathotype strain Z7 of A. alternata and examined the function and evolutionary history of its genes. Our results suggest that tangerine pathotype of A. altenata has two CDCs that are partly identical, that several of the genes in the A. alternata CDCs are involved in processes associated with metabolism, that they are conserved within the genus Alternaria and are rapidly evolving, and that they sometimes originate via HGT. Below, we discuss our findings in the context of the genome content of Alternaria CDCs and the evolutionary origin of Alternaria CDC genes.
It has been reported that the length of CDCs in distinct pathotypes of A. alternata is very variable. For example, both the strawberry and the tomato pathotypes contain a ~1.0 Mb CDC (Hu et al., 2012; Ito et al., 2004), both the Japanese pear and the tangerine pathotypes contain a 1.9–2.0 Mb CDC (Miyamoto et al., 2009, 2008), the apple pathotype contains a 1.1–1.8 Mb CDC (Harimoto et al., 2007), and the rough lemon pathotype contains a 1.2–1.5 Mb CDC (Masunaka et al., 2005). In this study, we found that the A. alternata Z7 possesses different electrophoretic karyotypes from the Floridian tangerine pathotype strains (Fig. 1), suggesting that CDCs even in the same pathotype can be very variable (Miyamoto et al., 2009). Comparison of the Z7 CDC proteins to the proteomes of other tangerine pathotypes (A. citriarbusti and A. tangelonis), finding a relatively low protein similarity among them, also supports this conclusion (Fig. 2). In addition, the extreme chromosome dynamic was supposed to be a conserved phenomenon in the genus Zymoseptoria (Möller et al., 2018). Although the mechanism underlying the formation of these diverse CDCs in Alternaria species is largely unknown, our sequencing data demonstrated that the Z7 CDC 1 contains all but one contig of Z7 CDC 2, providing evidence that duplication can be one factor that drives chromosomal rearrangements. A recent population genomics study in Zymoseptoria tritici identified the precise breakpoint locations of insertions that give rise to the highly differentiated gene contents in Z. tritici CDCs (Croll et al., 2013). That same study also reported the occurrence of CDC losses in progeny because of non‐disjunction during meiosis as well as the emergence of a new CDC through a fusion between sister chromatids (Croll et al., 2013). Thus, the CDCs of Z. tritici were proposed to originate mainly from ancient core chromosomes through a degeneration process involving breakage–fusion–bridge cycles and large insertions (Croll et al., 2013). Although determining whether the breakage–fusion–bridge cycle model holds for Alternaria CDCs will require further sequencing of CDCs and analyses, we note that our finding of evolutionary conservation of CDC genes within Alternaria is consistent with the Z. tritici model.
In this study, we functionally annotated the CDCs of the tangerine pathotype strain Z7 of A. alternata. Although the total length of Z7 CDCs is much larger than the A. arborescens CDC, several Z7 CDC gene families are comparable to those present in the A. arborescens CDC, including CAZymes, SSCPs, transcription factors and transporters (Table S2) (Hu et al., 2012). This result suggests that there are some shared properties between CDCs from these two divergent species. Our analyses also identified differences between CDCs in A. arborescens and in A. alternata strain Z7. For example, the enrichment of ‘biosynthetic process’ genes was discovered in the A. arborescens CDC but not in the A. alternata CDCs (Hu et al., 2012). In addition, the A. arborescens CDC contains 10 SM clusters and harbours 36 polyketide and non‐ribosomal peptide synthetase genes that might be responsible for the biosynthesis of the backbone structures of several groups of secondary metabolites (Hu et al., 2012). However, in Z7 CDCs, except for the host‐selective ACT toxin gene cluster, no other gene cluster involved in the biosynthesis of secondary metabolites was identified. About 46.5% (118) of the 254 genes found in Z7 CDCs were described as hypothetical proteins according to their BLAST hits in the NCBI nr database, indicating that the functions of a great number of Z7 CDC genes are not known (Table S2).
Evolutionary analysis showed that most of the Z7 CDC proteins are conserved within the genus Alternaria and likely originated in the Alternaria last common ancestor (Fig. 2). These results contrast with those of a previous study suggesting that the A. arborescens CDC originated from an unknown species through HGT (Hu et al., 2012). The discrepancy is most likely due to the small amount of available data used in the previous study; specifically, A. arborescens protein‐coding genes were compared to only two small databases, respectively, composed of either only A. brassicicola proteins or three fungal species from other filamentous fungal genera (Leptosphaeria maculans, Pyrenophora tritici‐repentis and Aspergillus oryzae) (Hu et al., 2012). In contrast, the much larger number of Alternaria genomes examined in our study shows that the evolutionary history of most CDC genes is consistent with the species phylogeny and with vertical, not horizontal, transmission.
Horizontal transfer of CDCs between different strains of the same or closely related fungal species has been documented for Fusarium oxysporum, Colletotrichum gloeosporioides and some Alternaria species (Akagi et al., 2009a; Akamatsu et al., 2001; He et al., 1998; Ma et al., 2010). Horizontal transfer of entire CDCs may contribute not only to the introduction of CDCs in fungal populations but also to their subsequent spread and to their acquisition of virulence factors that are essential for pathogenicity. Since fungal CDCs are known to be able to transfer between closely related species, it may be very difficult to distinguish between vertical and horizontal transmission of CDCs within Alternaria. On the other hand, horizontal transfer of CDCs between distantly related species has never been demonstrated, making it a less likely explanation for the evolutionary origin of Alternaria CDCs.
Although horizontal transfer is an unlikely explanation for the formation of CDCs in Alternaria, our study found evidence that HGT is a mechanism for the origin of some of the genes residing in its CDCs. The four genes that were likely horizontally transferred in the Z7 CDC from distantly related species formed a gene cluster (Table 2). Previously, the 23‐gene secondary metabolic gene cluster involved in the biosynthesis of the mycotoxin sterigmatocystin was shown to have been horizontally transferred from Aspergillus to Podospora (Slot and Rokas, 2011), and several other transfers of secondary metabolic gene clusters have been reported (Wisecaver and Rokas, 2015). HGT of intact gene clusters would not only contribute to fungal metabolic diversity but also potentially provide its recipient with a competitive advantage offered by the ability to synthesize a novel secondary metabolite, for example a subsequent study showed that Podospora produces sterigmatocystin (Matasyoh et al., 2011). Although the horizontally transferred gene cluster in this study contains fewer genes, the four‐gene cluster contains members of the ACT toxin gene cluster (Fig. 5B), which we showed to be expressed during the fungal infection of citrus leaves (Fig. 6). Additionally, the HMG‐CoA hydrolase coding gene AKT3 found in another transferred cluster was shown to be absolutely required for the HST production and virulence to host plant (Tanaka and Tsuge, 2000). We also functionally showed that two of the remaining three HGT genes are essential for ACT toxin biosynthesis and virulence in the tangerine pathotype of A. alternata (Fig. 7A). In this experiment, only one copy of the AALT_g11758 gene was knocked out, but it led to a dramatic effect on the phenotype. The underlying mechanism for this is yet to be solved. One possible explanation for this might be that toxin‐encoding genes are playing their roles with dosage effect and copy number is of importance for toxin biosynthesis and adaptation of the fungi to the changing environment (Steenwyk and Rokas, 2018). Taken together, these results are consistent with the view that HGT events, including ones involving the transfer of entire clusters, have played important roles over the course of the evolution of filamentous fungi (Fitzpatrick, 2012; Soanes and Richards, 2014; Wisecaver and Rokas, 2015).
Experimental Procedures
Fungal strains and culture conditions
The reference A. alternata strain, Z7, was isolated from an infected citrus leaf from Zhejiang, China (Huang et al., 2015). Z7 and its derived mutants were stored in 20% glycerol solutions at −80 °C until use. Fungi were grown on regular solid potato dextrose agar (PDA) at 25 °C and conidia were collected after incubating for 8 days. Mycelia were obtained by growing spores in liquid potato dextrose broth incubated on a rotary shaker at 160 rpm at 25 °C for 2 days.
Genomic data retrieval
The assembled A. alternata Z7 genome and proteome were downloaded from GenBank under the accession number LPVP00000000 (Wang et al., 2016). Genome data from other Alternaria species were downloaded from the Alternaria genomes database (Dang et al., 2015) and from other species of Dothideomycetes from the GenBank database (Table S3) (last access 15 April 2017).
Identification of the contigs comprising the CDC
To identify all the contigs that are part of the A. alternata Z7 CDC, pulsed‐field gel electrophoresis (PFGE) was carried out by following a previously described method with slight modifications (Akamatsu et al., 1999; Miyamoto et al., 2009). Briefly, approximately 5.0 × 108 protoplasts/mL mixed with an equal volume of molten 1.6% low melting point agarose was pipetted into plug moulds. After incubation and washing, plugs were inserted into the wells of a 0.8% (w/v) agarose gel. PFGE was carried out using a BioRad CHEF Mapper apparatus. Chromosomes were separated at 8 °C in 0.5 × TBE buffer at 5.4 V/cm with a 120–180 s pulse time gradient for 26 h. Gels were stained in GoldView (0.5 mg/mL) for 30 min immediately after the run. The CDC bands in the gel were retrieved and sequenced on an Illumina Hiseq 2500 platform, generating 150 bp paired‐end reads. Raw reads were trimmed of adapter sequences and low‐quality reads using trimmomatic v. 0.38 (Bolger et al., 2014). An index of the A. alternata Z7 reference genome was built using Bowtie 2 (Langmead and Salzberg, 2012) and cleaned reads were mapped to the reference genome using TopHat 2 (Kim et al., 2013). The covered percentage of each contig was calculated by BBMap v. 38.21 (sourceforge.net/projects/bbmap/) and those contigs with more than 50% of their sequences are covered by CDC sequencing reads were considered to be parts of the A. alternata Z7 CDCs.
The statistical reports for the EC and CDC genome were calculated by using in‐house Perl scripts. Repetitive elements were predicted by RepeatMasker v. open‐4.0.7 (http://www.repeatmasker.org) and tRNAs were identified by the tRNAscan‐SE 1.0 program (Schattner et al., 2005).
Functional annotation of the genes in the CDC
To functionally annotate the 254 genes (and their protein products) in the CDC, we performed gene ontology analysis using the topGO v. 2.28.0 (Alexa and Rahnenfuhrer, 2016) and classified the 254 proteins into protein families using the Pfam, v. 31.0 databases (last accessed 1 June 2016) (Finn et al., 2014). We predicted CDC genes that were parts of fungal secondary metabolite pathways using antiSMASH 4.0 (Blin et al., 2017). To identify CDC proteins that are CAZymes, we searched with the CAZymes Analysis Toolkit (http://www.cazy.org/, last accessed 10 July 2016) based on sequence and Pfam annotation (Lombard et al., 2014). To identify secreted proteins, we used SignalP v. 4.1 to predict transmembrane domains (Petersen et al., 2011) and we excluded non‐extracellular and glycosylphosphatidylinisotol (GPI)‐anchored proteins by using ProtComp v. 5 (Klee and Ellis, 2005) and fragAnchor (http://navet.ics.hawaii.edu/~fraganchor/NNHMM/NNHMM.html, last accessed 10 July 2016) (Poisson et al., 2007), respectively. All resulting secreted proteins that were shorter than 200 amino acids in length and contained at least four cysteine residues were considered as SSCR proteins. Kinases were searched and classified by an automated pipeline (Kosti et al., 2010). Cytochrome P450s were classified based on Pfam and the P450 database v. 1.2 (last accessed 18 June 2016) (Moktali et al., 2012). Transporters were identified by performing BLASTp against the Transporter Classification Database (last accessed 7 June 2016) (Saier et al., 2016).
Species phylogeny inference and evaluation of sequence conservation of CDC genes across Dothideomycetes
To understand the origin and evolution of genes in the A. alternata CDCs, we first constructed a species phylogeny using genomic data from all available Alternaria species as well as for representative species from other Dothideomycetes. We constructed the species phylogeny using 1754 conserved, fungal BUSCO genes as described previously (Shen et al., 2016; Simao et al., 2015). Briefly, the gene structure of each genome was predicted by AUGUSTUS 3.1 (Stanke and Waack, 2003), then the sequences of these predicted genes were aligned to the HMM alignment profile of each BUSCO gene in the OrthoDB v. 9 database (Simao et al., 2015), and the ones with alignment bit‐scores higher than 90% of the lowest bit‐score among the reference genomes in the OrthoDB v. 9 database were kept for tree construction. The isolates Alternaria porri BMP0178 and Alternaria destruens BMP0317 were excluded from downstream analyses due to their poor (<60%) coverage of BUSCO genes (Table S4). We also compared the sequence similarity of each of the 254 CDC proteins to proteins in the genomes of other Dothideomycetes by calculating the value of % BLAST identity * % query coverage.
Examination of relative evolutionary rate between genes in the CDCs and genes in the essential chromosomes
To estimate the relative evolutionary rate between Z7 CDCs and ECs, orthologue groups within the Alternaria genus containing Z7 proteins from both CDCs and ECs were extracted using OrthorMCL v. 2.0.9 and reciprocal BLASTp with identity >50% and query coverage >50% as cut‐offs (Chen et al., 2006). For each orthologue group containing more than 11 (50% of total number) species, only one sequence per isolate/species (the one that was the best hit to the Z7 protein) was kept for further analysis. The coding sequences of those orthologue proteins were then aligned with MAFFT v. 7.023b using the E‐INS‐I strategy (Yamada et al., 2016) and trimmed with trimAl v. 1.4.rev11 using its automated1 strategy (Capella‐Gutierrez et al., 2009). The maximum‐likelihood (ML) phylogenetic trees were inferred using IQ‐TREE 1.5.4, with the TEST model (Nguyen et al., 2015) and with 1000 bootstrap replicates. The non‐synonymous to synonymous rate ratio dN/dS for each gene tree was calculated using the CODEML in PAML 4.9 with the model M0 (Yang, 2007). The significance of the difference in the average total branch length and in the dN/dS value of the ML phylogenetic trees of CDC genes against that of the EC genes was determined by Wilcoxon test.
Identification of CDC genes that underwent HGT
To detect gene candidates that experienced HGT in A. alternata Z7 CDCs, we first performed a BLASTp search of the local NCBI non‐redundant protein database (nr, last accessed 21 May 2017) using Z7 CDC proteins as queries. We next selected proteins with the following characteristics as HGT candidates for further phylogenetic analyses: (a) an Alien Index (AI) score larger than 0 (Gladyshev et al., 2008; Wisecaver et al., 2016), (b) at least 80% of the top 200 BLASTp hits of the query protein are from organisms other than Dothideomycetes and (c) the sequence identity of the query protein across its entire length to its best BLASTp hit is equal to or greater than 50%.
All genes that fit these three criteria were used as query sequences in BLASTp searches against the nr database and phylogenetic trees of their most closely related sequences across the tree of life were constructed. To reduce the number of sequences used to build each phylogenetic tree, we kept only one sequence per species (the one with the best BLASTp hit to the HGT candidate), then we selected the top 200 hits from the BLAST results. The resulting sequences were used as input for multiple sequence alignment, trimming and phylogenetic inference, which were performed as described above.
The phylogenetic tree of each HGT candidate was manually inspected and only those trees that were evidently incongruent with the species phylogeny and strongly supported (bootstrap value > 95%) were retained as HGT candidates. For those HGT candidates, we used the Consel software, v. V0.1i (Shimodaira, 2002; Shimodaira and Hasegawa, 2001) to perform the AU comparative topology test between the unconstrained ML tree and the constrained ML tree in which the Alternaria gene sequence was forced to be monophyletic with the rest of the sequences from Dothideomycetes. All phylogenetic trees were visualized using ITOL v. 3.0 (Letunic and Bork, 2016).
qRT‐PCR analysis
To investigate the regulatory role of the four HGT genes in pathogenesis, the mycelia from the A. alternata Z7 were inoculated on the citrus leaves and harvested at 1, 3, 6, 9 and 12 h post‐inoculation. Collected mycelia from each time course were then ground in liquid nitrogen and total RNA was extracted using an AxyPrepTM multisource total RNA miniprep kit. Then 5 μg of each RNA sample was used for reverse transcription with the Prime Script RT reagent kit (TakaRa Biotechnology, Co., Dalian, China). The relative transcript level of the four genes was quantified on a Bio‐Rad CFX96 Real‐Time PCR system and repeated three times. The primers used in this study are listed in Table S5. The tubulin‐encoding gene (AALT_g4466) was used as an internal control and the resulting data were normalized using the comparative 2−ΔΔCT as described previously (Wang et al., 2014).
Functional analyses of horizontally transferred genes
One or more copies of the AALTg12032, AALTg11757 and AALTg11758 genes were knocked out using a fungal protoplast transformation protocol, as described previously (Chen et al., 2017). Briefly, the two flanking 600–900 bp fragments and a bacterial phosphotransferase B gene were fused together and the resulting fragment was then introduced into fungal protoplasts using polyethylene glycol and CaCl2. The transformants growing on a medium supplemented with 150 μg/mL hygromycin were selected and examined by PCR with specific primer pairs. All the primers used in this study are listed in Table S5.
Wild‐type and mutant strains were grown on PDA at 25 °C and fungal virulence was assessed on Citrus clementina leaves inoculated by placing a 5 mm plug taken from the media for 2 days. The fungal ACT toxin was extracted using the Amberlite XAD‐2 resin and ethyl acetate from culture filtrates, and its toxicity was assessed by inoculating 10 µL ethyl acetate extracts onto citrus leaves (Kohmoto et al., 1993, Wang et al., 2018). Each strain was tested on at least four leaves and experiments were repeated twice.
Accession Numbers
The raw sequence reads of the A. alternata Z7 CDCs can be accessed at the NCBI SRA database with the accession number SRP154962.
Supporting information
Fig. S1 GO enrichment analysis of the Z7 CDC genes in the category 'Biological Process'. Significantly enriched GO terms (P < 0.05) are illustrated by rectangles. Rectangle colour corresponds to degree of statistical significance and ranges from bright yellow (least significant) to dark red (most significant). For each node, the GO identifier, the GO term name, and P‐value of each functional category is shown. The final line inside each node shows the number of genes that belong to the functional category in the CDC and in the whole genome of A. alternata Z7, respectively.
Fig. S2 Full ML phylogenetic trees of HGT gene homologues across the tree of life. Each gene’s maximum‐likelihood phylogeny was inferred under the best substitution model automatically selected by ModelFinder, as implemented in IQ‐TREE 1.5.4. Branch colours indicate the taxonomic lineages to which the different taxa included in each phylogeny belong. The A. alternata Z7 CDC sequence that was used as a query in the BLAST search is shown in red font. Bootstrap values are shown near each branch. AALT_g12032.
Fig. S3 Full ML phylogenetic trees of HGT gene homologues across the tree of life. Each gene’s maximum‐likelihood phylogeny was inferred under the best substitution model automatically selected by ModelFinder, as implemented in IQ‐TREE 1.5.4. Branch colours indicate the taxonomic lineages to which the different taxa included in each phylogeny belong. The A. alternata Z7 CDC sequence that was used as a query in the BLAST search is shown in red font. Bootstrap values are shown near each branch. AALT_g11755.
Fig. S4 Full ML phylogenetic trees of HGT gene homologues across the tree of life. Each gene’s maximum‐likelihood phylogeny was inferred under the best substitution model automatically selected by ModelFinder, as implemented in IQ‐TREE 1.5.4. Branch colours indicate the taxonomic lineages to which the different taxa included in each phylogeny belong. The A. alternata Z7 CDC sequence that was used as a query in the BLAST search is shown in red font. Bootstrap values are shown near each branch. AALT_g11757.
Fig. S5 Full ML phylogenetic trees of HGT gene homologues across the tree of life. Each gene’s maximum‐likelihood phylogeny was inferred under the best substitution model automatically selected by ModelFinder, as implemented in IQ‐TREE 1.5.4. Branch colours indicate the taxonomic lineages to which the different taxa included in each phylogeny belong. The A. alternata Z7 CDC sequence that was used as a query in the BLAST search is shown in red font. Bootstrap values are shown near each branch. AALT_g11758.
Fig. S6 Disruption of one or more copies of the 3 HGT genes. (A) Schematic depiction of gene disruption of AALT_g12032, AALT_g11757, and AALT_g11758 via homologous recombination. Numbers denote primers listed in supplementary Table S5. (B) PCR verification of the AALT_g12032, AALT_g11757, and AALT_g11758‐targeted transformant. Long bands can be only amplified in the mutants using the outside PCR primer pairs. The short band can be amplified from both the wild‐type (WT) and mutant strains using the inner PCR primer pairs, indicating that A. alternata Z7 has multiple copies of these genes. Primers used were listed in Table S5. T1: transformant 1; T2: transformant 2. (C) Vegetative growth of the wild‐type and mutant strains on potato dextrose agar (PDA). (D) Necrosis symptoms in citrus leaves inoculated with the PDA plug (48 h) of the wild‐type and AALT_g11757‐targeted mutant strains. For each leaf, the wild‐type strain was inoculated on the left while the AALT_g11757‐targeted transformant strain was on the right.
Table S1 Statistical information for each contig in the Alternaria alternata Z7 genome uniquely matched by the CDC resequencing reads.
Table S2 Annotation information for all Z7 CDC genes.
Table S3 GenBank assembly accessions for genome data used in this study.
Table S4 Coverage of BUSCO genes for each genome used in this study.
Table S5 Primers used in this study.
Acknowledgements
We thank members of the Rokas laboratory for helpful discussions. We thank Abigail Leavitt Labella and Jacob Steenwyk for their critical comments on this paper. We thank Nicholas Pun and Professor Jianping Xu for their kind help in analysing the data. We thank Professor Chuanxi Zhang at Zhejiang University for providing us with the apparatus to perform the PFGE experiments. This work was conducted in part using the resources of the Advanced Computing Center for Research and Education at Vanderbilt University. This work was supported by the National Foundation of Natural Science of China (31571948 to H.L.), the earmarked fund for China Agriculture Research System (CARS‐27 to H.L.), the China Postdoctoral Science Foundation (2016M601945 to M.W.) and the US National Science Foundation (DEB‐1442113 to A.R.). The authors declare that there are no conflicts of interest.
Data Availability Statement
The raw sequence reads of the A. alternata Z7 CDCs can be accessed at the NCBI SRA database with the accession number SRP154962. All the other data generated in this study, including CDC contigs, CDC gene annotation, multiple sequence alignments and phylogenetic trees, have been deposited on the figshare repository at https://doi.org/10.6084/m9.figshare.8313794.
References
- Akagi, Y. , Akamatsu, H. , Otani, H. and Kodama, M. (2009a) Horizontal chromosome transfer, a mechanism for the evolution and differentiation of a plant‐pathogenic fungus. Eukaryot. Cell, 8, 1732–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akagi, Y. , Taga, M. , Yamamoto, M. , Tsuge, T. , Fukumasa‐Nakai, Y. , Otani, H. and Kodama, M. (2009b) Chromosome constitution of hybrid strains constructed by protoplast fusion between the tomato and strawberry pathotypes of Alternaria alternata . J. Gen. Plant Pathol. 75, 101–109. [Google Scholar]
- Akamatsu, H. , Taga, M. , Kodama, M. , Johnson, R. , Otani, H. and Kohmoto, K. (1999) Molecular karyotypes for Alternaria plant pathogens known to produce host‐specific toxins. Curr. Genet. 35, 647–656. [DOI] [PubMed] [Google Scholar]
- Akamatsu, H. , Fukumasa‐Nakai, Y. , Otani, H. and Kodama, M. (2001) Construction and genetic analysis of hybrid strains between apple and tomato pathotypes of Alternaria alternata by protoplast fusion. J. Gen. Plant Pathol. 67, 97–105. [Google Scholar]
- Alexa, A. and Rahnenfuhrer, J. (2016) topGO: Enrichment Analysis for Gene Ontology. [Google Scholar]
- Blin, K. , Wolf, T. , Chevrette, M.G. , Lu, X. , Schwalen, C.J. , Kautsar, S.A. , Suarez Duran, H.G. , de Los Santos, E.L.C. , Kim, H.U. , Nave, M. , Dickschat, J.S. , Mitchell, D.A. , Shelest, E. , Breitling, R. , Takano, E. , Lee, S.Y. , Weber, T. and Medema, M.H. (2017) antiSMASH 4.0‐improvements in chemistry prediction and gene cluster boundary identification. Nucl. Acids Res. 45, W36–W41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella‐Gutierrez, S. , Silla‐Martinez, J.M. and Gabaldon, T. (2009) trimAl: a tool for automated alignment trimming in large‐scale phylogenetic analyses. Bioinformatics, 25, 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, F. , Mackey, A.J. , Stoeckert, C.J. Jr and Roos, D.S. (2006) OrthoMCL‐DB: querying a comprehensive multi‐species collection of ortholog groups. Nucl. Acids Res. 34, D363–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L.H. , Tsai, H.C. , Yu, P.L. and Chung, K.R. (2017) A major facilitator superfamily transporter‐mediated resistance to oxidative stress and fungicides requires Yap1, Skn7, and MAP kinases in the citrus fungal pathogen Alternaria alternata . PLoS One, 12, e0169103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman, J.J. , Rounsley, S.D. , Rodriguez‐Carres, M. , Kuo, A. , Wasmann, C.C. , Grimwood, J. , Schmutz, J. , Taga, M. , White, G.J. , Zhou, S. , Schwartz, D.C. , Freitag, M. , Ma, L.‐j. , Danchin, E.G.J. , Henrissat, B. , Coutinho, P.M. , Nelson, D.R. , Straney, D. , Napoli, C.A. , Barker, B.M. , Gribskov, M. , Rep, M. , Kroken, S. , Molnár, I. , Rensing, C. , Kennell, J.C. , Zamora, J. , Farman, M.L. , Selker, E.U. , Salamov, A. , Shapiro, H. , Pangilinan, J. , Lindquist, E. , Lamers, C. , Grigoriev, I.V. , Geiser, D.M. , Covert, S.F. , Temporini, E. and VanEtten, H.D. (2009) The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. PLoS Genet. 5, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covert, S.F. (1998) Supernumerary chromosomes in filamentous fungi. Curr. Genet. 33, 311–319. [DOI] [PubMed] [Google Scholar]
- Croll, D. , Zala, M. and McDonald, B.A. (2013) Breakage‐fusion‐bridge cycles and large insertions contribute to the rapid evolution of accessory chromosomes in a fungal pathogen. PLoS Genet. 9, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang, H.X. , Pryor, B. , Peever, T. and Lawrence, C.B. (2015) The Alternaria genomes database: a comprehensive resource for a fungal genus comprised of saprophytes, plant pathogens, and allergenic species. BMC Genom. 16, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, R.D. , Bateman, A. , Clements, J. , Coggill, P. , Eberhardt, R.Y. , Eddy, S.R. , Heger, A. , Hetherington, K. , Holm, L. , Mistry, J. , Sonnhammer, E.L.L. , Tate, J. and Punta, M . ( 2014) Pfam: the protein families database. Nucleic Acids Res. 42, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick, D.A. (2012) Horizontal gene transfer in fungi. FEMS Microbiol. Lett. 329, 1–8. [DOI] [PubMed] [Google Scholar]
- Gladyshev, E.A. , Meselson, M. and Arkhipova, I.R. (2008) Massive horizontal gene transfer in bdelloid rotifers. Science, 320, 1210–1213. [DOI] [PubMed] [Google Scholar]
- Harimoto, Y. , Hatta, R. , Kodama, M. , Yamamoto, M. , Otani, H. and Tsuge, T. (2007) Expression profiles of genes encoded by the supernumerary chromosome controlling AM‐toxin biosynthesis and pathogenicity in the apple pathotype of Alternaria alternata . Mol. Plant‐Microbe Interact. 20, 1463–1476. [DOI] [PubMed] [Google Scholar]
- Hatta, R. , Ito, K. , Hosaki, Y. , Tanaka, T. , Tanaka, A. , Yamamoto, M. , Akimitsu, K. and Tsuge, T. (2002) A conditionally dispensable chromosome controls host‐specific pathogenicity in the fungal plant pathogen Alternaria alternata . Genetics, 161, 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, C. , Rusu, A.G. , Poplawski, A.M. , Irwin, J.A. and Manners, J.M. (1998) Transfer of a supernumerary chromosome between vegetatively incompatible biotypes of the fungus Colletotrichum gloeosporioides . Genetics, 150, 1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J. , Chen, C. , Peever, T. , Dang, H. , Lawrence, C. and Mitchell, T. (2012) Genomic characterization of the conditionally dispensable chromosome in Alternaria arborescens provides evidence for horizontal gene transfer. BMC Genom. 13, 1471–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, F. , Fu, Y. , Nie, D. , Stewart, J.E. , Peever, T.L. and Li, H. (2015) Identification of a novel phylogenetic lineage of Alternaria alternata causing citrus brown spot in China. Fungal Biol. 119, 320–330. [DOI] [PubMed] [Google Scholar]
- Ito, K. , Tanaka, T. , Hatta, R. , Yamamoto, M. , Akimitsu, K. and Tsuge, T. (2004) Dissection of the host range of the fungal plant pathogen Alternaria alternata by modification of secondary metabolism. Mol. Microbiol. 52, 399–411. [DOI] [PubMed] [Google Scholar]
- Johnson, L.J. , Johnson, R.D. , Akamatsu, H. , Salamiah, A. , Otani, H. , Kohmoto, K. and Kodama, M . ( 2001) Spontaneous loss of a conditionally dispensable chromosome from the Alternaria alternata apple pathotype leads to loss of toxin production and pathogenicity. Curr. Genet. 40, 65–72. [DOI] [PubMed] [Google Scholar]
- Kim, D. , Pertea, G. , Trapnell, C. , Pimentel, H. , Kelley, R. and Salzberg, S.L. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, 2013–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klee, E.W. and Ellis, L.B. (2005) Evaluating eukaryotic secreted protein prediction. BMC Bioinform. 6, 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohmoto, K. , Itoh, Y. , Shimomura, N. , Kondoh, Y. , Otani, H. , Kodama, M. , Nishimura, S. and Nakatsuka, S. (1993) Isolation and biological activities of two host‐specific toxins from the tangerine pathotype of Alternaria alternata . Phytopathology, 83, 495–502. [Google Scholar]
- Kosti, I. , Mandel‐Gutfreund, Y. , Glaser, F. and Horwitz, B.A. (2010) Comparative analysis of fungal protein kinases and associated domains. BMC Genom. 11, 1471–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. and Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I. and Bork, P. (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucl. Acids Res. 44, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard, V. , Golaconda Ramulu, H. , Drula, E. , Coutinho, P.M. and Henrissat, B. (2014) The carbohydrate‐active enzymes database (CAZy) in 2013. Nucl. Acids Res. 42, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, L.J. , van der Does, H.C. , Borkovich, K.A. , Coleman, J.J. , Daboussi, M.J. , Di Pietro, A. , Dufresne, M. , Freitag, M. , Grabherr, M. , Henrissat, B. , Houterman, P.M. , Kang, S. , Shim, W.‐B. , Woloshuk, C. , Xie, X. , Xu, J.‐R. , Antoniw, J. , Baker, S.E. , Bluhm, B.H. , Breakspear, A. , Brown, D.W. , Butchko, R.A.E. , Chapman, S. , Coulson, R. , Coutinho, P.M. , Danchin, E.G.J. , Diener, A. , Gale, L.R. , Gardiner, D.M. , Goff, S. , Hammond‐Kosack, K.E. , Hilburn, K. , Hua‐Van, A. , Jonkers, W. , Kazan, K. , Kodira, C.D. , Koehrsen, M. , Kumar, L. , Lee, Y.‐H. , Li, L. , Manners, J.M. , Miranda‐Saavedra, D. , Mukherjee, M. , Park, G. , Park, J. , Park, S.‐Y. , Proctor, R.H. , Regev, A. , Ruiz‐Roldan, M.C. , Sain, D. , Sakthikumar, S. , Sykes, S. , Schwartz, D.C. , Turgeon, B.G. , Wapinski, I. , Yoder, O. , Young, S. , Zeng, Q. , Zhou, S. , Galagan, J. , Cuomo, C.A. , Kistler, H.C. and Rep, M. (2010) Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium . Nature, 464, 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masunaka, A. , Ohtani, K. , Peever, T.L. , Timmer, L.W. , Tsuge, T. , Yamamoto, M. , Yamamoto, H. and Akimitsu, K. (2005) An isolate of Alternaria alternata that is pathogenic to both tangerines and rough lemon and produces two host‐selective toxins, ACT‐ and ACR‐toxins. Phytopathology, 95, 241–247. [DOI] [PubMed] [Google Scholar]
- Matasyoh, J.C. , Dittrich, B. , Schueffler, A. and Laatsch, H. (2011) Larvicidal activity of metabolites from the endophytic Podospora sp. against the malaria vector Anopheles gambiae . Parasitol. Res. 108, 561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto, Y. , Masunaka, A. , Tsuge, T. , Yamamoto, M. , Ohtani, K. , Fukumoto, T. , Gomi, K. , Peever, T.L. and Akimitsu, K. (2008) Functional analysis of a multicopy host‐selective ACT‐toxin biosynthesis gene in the tangerine pathotype of Alternaria alternata using RNA silencing. Mol. Plant‐Microbe Interact. 21, 1591–1599. [DOI] [PubMed] [Google Scholar]
- Miyamoto, Y. , Ishii, Y. , Honda, A. , Masunaka, A. , Tsuge, T. , Yamamoto, M. , Ohtani, K. , Fukumoto, T. , Gomi, K. , Peever, T.L. and Akimitsu, K. (2009) Function of genes encoding acyl‐CoA synthetase and enoyl‐CoA hydratase for host‐selective ACT‐toxin biosynthesis in the tangerine pathotype of Alternaria alternata . Phytopathology, 99, 369–377. [DOI] [PubMed] [Google Scholar]
- Moktali, V. , Park, J. , Fedorova‐Abrams, N.D. , Park, B. , Choi, J. , Lee, Y.‐H. and Kang, S . ( 2012) Systematic and searchable classification of cytochrome P450 proteins encoded by fungal and oomycete genomes. BMC Genom. 13, 1471–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller, M. , Habig, M. , Freitag, M. and Stukenbrock, E.H. (2018) Extraordinary genome instability and widespread chromosome rearrangements during vegetative growth. Genetics 210, 519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, L.‐T. , Schmidt, H.A. , von Haeseler, A. and Minh, B.Q. (2015) IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, T.N. , Brunak, S. , von Heijne, G. and Nielsen, H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods, 8, 785–786. [DOI] [PubMed] [Google Scholar]
- Poisson, G. , Chauve, C. , Chen, X. and Bergeron, A. (2007) FragAnchor: a large‐scale predictor of glycosylphosphatidylinositol anchors in eukaryote protein sequences by qualitative scoring. Genomics Proteomics Bioinformatics, 5, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier, M.H. Jr , Reddy, V.S. , Tsu, B.V. , Ahmed, M.S. , Li, C. and Moreno‐Hagelsieb, G. (2016) The Transporter Classification Database (TCDB): recent advances. Nucl. Acids Res. 44, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattner, P. , Brooks, A.N. and Lowe, T.M. (2005) The tRNAscan‐SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucl. Acids Res. 33, W686–W689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, X.‐X. , Zhou, X. , Kominek, J. , Kurtzman, C.P. , Hittinger, C.T. and Rokas, A. (2016) Reconstructing the backbone of the Saccharomycotina yeast phylogeny using genome‐scale data. G3, 6, 3927–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimodaira, H. (2002) An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 51, 492–508. [DOI] [PubMed] [Google Scholar]
- Shimodaira, H. and Hasegawa, M. (2001) CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics, 17, 1246–1247. [DOI] [PubMed] [Google Scholar]
- Simao, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. and Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Slot, J.C. and Rokas, A. (2011) Horizontal transfer of a large and highly toxic secondary metabolic gene cluster between fungi. Curr. Biol. 21, 134–139. [DOI] [PubMed] [Google Scholar]
- Soanes, D. and Richards, T.A. (2014) Horizontal gene transfer in eukaryotic plant pathogens. Annu. Rev. Phytopathol. 52, 583–614. [DOI] [PubMed] [Google Scholar]
- Stanke, M. and Waack, S. (2003) Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics, 19, ii215–ii225. [DOI] [PubMed] [Google Scholar]
- Steenwyk, J.L. and Rokas, A. (2018) Copy number variation in fungi and its implications for wine yeast genetic diversity and adaptation. Front. Microbiol. 9, 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stukenbrock, E.H. , Jorgensen, F.G. , Zala, M. , Hansen, T.T. , McDonald, B.A. and Schierup, M.H. (2010) Whole‐genome and chromosome evolution associated with host adaptation and speciation of the wheat pathogen Mycosphaerella graminicola . PLoS Genet. 6, e1001189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, A. and Tsuge, T. (2000) Structural and functional complexity of the genomic region controlling AK‐toxin biosynthesis and pathogenicity in the Japanese pear pathotype of Alternaria alternata . Mol. Plant‐Microbe Interact. 13, 975–986. [DOI] [PubMed] [Google Scholar]
- Thomma, B.P. (2003) Alternaria spp.: from general saprophyte to specific parasite. Mol. Plant Pathol. 4, 225–236. [DOI] [PubMed] [Google Scholar]
- Tsuge, T. , Harimoto, Y. , Akimitsu, K. , Ohtani, K. , Kodama, M. , Akagi, Y. , Egusa, M. , Yamamoto, M. , Otani, H. (2013) Host‐selective toxins produced by the plant pathogenic fungus Alternaria alternata . FEMS Microbiol. Rev. 37, 44–66. [DOI] [PubMed] [Google Scholar]
- Wang, M. , Chen, C. , Zhu, C. , Sun, X. , Ruan, R. and Li, H. (2014) Os2 MAP kinase‐mediated osmostress tolerance in Penicillium digitatum is associated with its positive regulation on glycerol synthesis and negative regulation on ergosterol synthesis. Microbiol. Res. 169, 511–521. [DOI] [PubMed] [Google Scholar]
- Wang, M. , Sun, X. , Yu, D. , Xu, J. , Chung, K. and Li, H. (2016) Genomic and transcriptomic analyses of the tangerine pathotype of Alternaria alternata in response to oxidative stress. Sci. Rep. 6, 32437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Yang, X. , Ruan, R. , Fu, H. and Li, H. (2018) Csn5 is required for the conidiogenesis and pathogenesis of the Alternaria alternata tangerine pathotype. Front. Microbiol. 9, 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisecaver, J.H. and Rokas, A. (2015) Fungal metabolic gene clusters‐caravans traveling across genomes and environments. Front. Microbiol. 6, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisecaver, J.H. , Alexander, W.G. , King, S.B. , Hittinger, C.T. and Rokas, A. (2016) Dynamic evolution of nitric oxide detoxifying flavohemoglobins, a family of single‐protein metabolic modules in bacteria and eukaryotes. Mol. Biol. Evol. 33, 1979–1987. [DOI] [PubMed] [Google Scholar]
- Yamada, K.D. , Tomii, K. and Katoh, K. (2016) Application of the MAFFT sequence alignment program to large data‐reexamination of the usefulness of chained guide trees. Bioinformatics, 32, 3246–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 GO enrichment analysis of the Z7 CDC genes in the category 'Biological Process'. Significantly enriched GO terms (P < 0.05) are illustrated by rectangles. Rectangle colour corresponds to degree of statistical significance and ranges from bright yellow (least significant) to dark red (most significant). For each node, the GO identifier, the GO term name, and P‐value of each functional category is shown. The final line inside each node shows the number of genes that belong to the functional category in the CDC and in the whole genome of A. alternata Z7, respectively.
Fig. S2 Full ML phylogenetic trees of HGT gene homologues across the tree of life. Each gene’s maximum‐likelihood phylogeny was inferred under the best substitution model automatically selected by ModelFinder, as implemented in IQ‐TREE 1.5.4. Branch colours indicate the taxonomic lineages to which the different taxa included in each phylogeny belong. The A. alternata Z7 CDC sequence that was used as a query in the BLAST search is shown in red font. Bootstrap values are shown near each branch. AALT_g12032.
Fig. S3 Full ML phylogenetic trees of HGT gene homologues across the tree of life. Each gene’s maximum‐likelihood phylogeny was inferred under the best substitution model automatically selected by ModelFinder, as implemented in IQ‐TREE 1.5.4. Branch colours indicate the taxonomic lineages to which the different taxa included in each phylogeny belong. The A. alternata Z7 CDC sequence that was used as a query in the BLAST search is shown in red font. Bootstrap values are shown near each branch. AALT_g11755.
Fig. S4 Full ML phylogenetic trees of HGT gene homologues across the tree of life. Each gene’s maximum‐likelihood phylogeny was inferred under the best substitution model automatically selected by ModelFinder, as implemented in IQ‐TREE 1.5.4. Branch colours indicate the taxonomic lineages to which the different taxa included in each phylogeny belong. The A. alternata Z7 CDC sequence that was used as a query in the BLAST search is shown in red font. Bootstrap values are shown near each branch. AALT_g11757.
Fig. S5 Full ML phylogenetic trees of HGT gene homologues across the tree of life. Each gene’s maximum‐likelihood phylogeny was inferred under the best substitution model automatically selected by ModelFinder, as implemented in IQ‐TREE 1.5.4. Branch colours indicate the taxonomic lineages to which the different taxa included in each phylogeny belong. The A. alternata Z7 CDC sequence that was used as a query in the BLAST search is shown in red font. Bootstrap values are shown near each branch. AALT_g11758.
Fig. S6 Disruption of one or more copies of the 3 HGT genes. (A) Schematic depiction of gene disruption of AALT_g12032, AALT_g11757, and AALT_g11758 via homologous recombination. Numbers denote primers listed in supplementary Table S5. (B) PCR verification of the AALT_g12032, AALT_g11757, and AALT_g11758‐targeted transformant. Long bands can be only amplified in the mutants using the outside PCR primer pairs. The short band can be amplified from both the wild‐type (WT) and mutant strains using the inner PCR primer pairs, indicating that A. alternata Z7 has multiple copies of these genes. Primers used were listed in Table S5. T1: transformant 1; T2: transformant 2. (C) Vegetative growth of the wild‐type and mutant strains on potato dextrose agar (PDA). (D) Necrosis symptoms in citrus leaves inoculated with the PDA plug (48 h) of the wild‐type and AALT_g11757‐targeted mutant strains. For each leaf, the wild‐type strain was inoculated on the left while the AALT_g11757‐targeted transformant strain was on the right.
Table S1 Statistical information for each contig in the Alternaria alternata Z7 genome uniquely matched by the CDC resequencing reads.
Table S2 Annotation information for all Z7 CDC genes.
Table S3 GenBank assembly accessions for genome data used in this study.
Table S4 Coverage of BUSCO genes for each genome used in this study.
Table S5 Primers used in this study.
Data Availability Statement
The raw sequence reads of the A. alternata Z7 CDCs can be accessed at the NCBI SRA database with the accession number SRP154962. All the other data generated in this study, including CDC contigs, CDC gene annotation, multiple sequence alignments and phylogenetic trees, have been deposited on the figshare repository at https://doi.org/10.6084/m9.figshare.8313794.