

Abstract
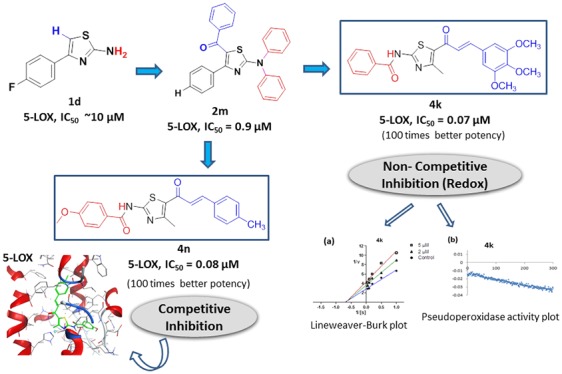
A hybrid pharmacophore approach is used to design and synthesize novel chalcone-thiazole hybrid molecules. Herein, thiazole has been hybridized with chalcone to obtain a new class of 5-LOX inhibitors. In vitro biological evaluation showed that most of the compounds were better 5-LOX inhibitors than the positive control, Zileuton (IC50 = 1.05 ± 0.03 μM). The best compounds in the series, namely, 4k, 4n, and 4v (4k: IC50 = 0.07 ± 0.02 μM, 4n: IC50 = 0.08 ± 0.05 μM, 4v: 0.12 ± 0.04 μM) are found to be 10 times more active than previously reported 2-amino thiazole (2m: IC50 = 0.9 ± 0.1 μM) by us. Further, 4k has redox (noncompetitive) while 4n and 4v act through a competitive inhibition mechanism. SAR indicated that the presence of methoxy/methyl either in the vicinity of chalcone or both thiazole and chalcone contributed to the synergistic inhibitory effect.
Keywords: Chalcone, thiazole, 5-LOX, pharmacophore, design, hybrid, synthesis, pseudoperoxidase
5-Lipoxygenase (5-LOX) is an important enzyme which catalyzes arachidonic acid (AA) for the production of several leukotrienes (LTs) which are well-known mediators of inflammation related disorders such as rheumatoid arthritis, allergic rhinitis, atherosclerosis, and cardiovascular diseases among which asthma is the major pathophysiological implication associated with it.1,2 5-LOX oxygenates essential polyunsaturated fatty acid (AA) to 5(S)-hydroperoxy-6-trans-8,11,14-cis-eicosatetraenoic acid (5-HpETE) and further dehydrates it to the unstable epoxide leukotriene A4 (LTA4). So far, Zileuton is the only marketed 5-LOX inhibitor against asthma.2 But it has side-effects of hepatotoxicity and poor pharmacokinetics. Its hepatotoxicity is associated with its mechanism involving a redox process in which N-hydroxyurea is involved in electron transfer through free radical-mediated lipid peroxidation of cell membranes and thiophene forms chemically reactive metabolites resulting in liver serum enzyme elevation and toxicity.2 So, there is a need to develop inhibitors against 5-LOX.
Thiazoles are active constituents of various naturally occurring biologically significant compounds such as thiamine, mycothiazole, as well as synthetic drugs which act as antimicrobial, anti-inflammatory, anticancer, antiviral, and antitubercular.3,4 Thiazol-4(5H)-one derivatives, namely darbufelone and CI-987, are reported as dual COX/LOX inhibitors.5 Recently, thiazole bearing scaffolds, namely 2-amino-4-aryl thiazole-5-phenylmethanones,6,7 N-aryl thiazole-2-amines,8 and 5-benzylidene-2-phenylthiazolinones,9 are identified as selective 5-LOX whereas aminothiazole pirinixic acids10 as dual 5-LOX/mPGES1 inhibitors.
Chalcone (α,β-unsaturated ketone, collectively defined as an aromatic ketone and enone) based analogues are found in many natural compounds including turmeric in the form of curcumin, liquorice as isoliquiritigenin, and synthetic drugs such as homobutein.11 Chalcones are known to exhibit biological properties, such as antibacterial, anti-inflammatory, antioxidant, anticancer, antimalarial, and as carbonic anhydrase inhibitor.11,12 Diarylsulfonylurea-chalcones,12 and di-o-prenylated chalcones13 have been reported as 5-LOX whereas chalcone-triazole hybrids14 as 15-LOX inhibitors. Phenylsulphonyl urenyl chalcones15 and 3,4-dihydroxychalcones16 act as dual 5-LOX/COX inhibitors.
Combining two bioactive pharmacophores to design a hybrid molecule has considerably attracted the interest of medicinal chemists. This molecular hybridization is a useful tool to generate lead molecules with synergetic and superior biological activities.17 Considering the anti-inflammatory potencies of thiazole and chalcone, it is envisaged that integrating both these moieties in one molecular platform could possibly produce compounds with synergistic properties. Therefore, in the present study, as an extension of our earlier work in the field of development of LT inhibitors, an attempt has been made to design and synthesize hitherto novel chalcone-thiazole structural hybrids along with elaboration of their mechanism to understand their redox and nonredox behavior to overcome pharmacokinetic problems as potential 5-LOX inhibitors.
Rational Design and Pharmacophore Elucidation
The molecular hybridization approach on the concept of combining two bioactive pharmacophores to achieve superior biological activities forms the basis of the designed prototype (A) in this study (Figure 1). Optimization for hybrid molecules (A) containing both chalcone and thiazole scaffolds has been pursued via pharmacophore elucidation toward 5-LOX inhibition. The pharmacophore is a group of essential features, namely an H-bond acceptor, a donor, and a hydrophobic and aromatic center for a particular biological activity required for enzyme–ligand interaction.18 To elucidate the structure activity relationship (SAR) of the designed structural hybrid (A), a pharmacophore model (Ph model) was generated from the set of known and reported inhibitors of 5-LOX (Figure 2). It included a known drug: Zileuton, clinical trial inhibitors: Atreleuton and Siteleuton,19 and reported inhibitors: di-o-prenylated and diarylsulfonylurea chalcones.12,13
Figure 1.
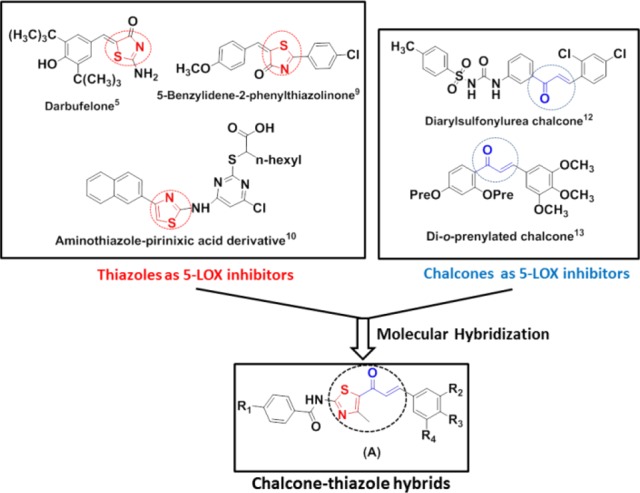
Design of chalcone–thiazole hybrids via molecular hybridization.
Figure 2.
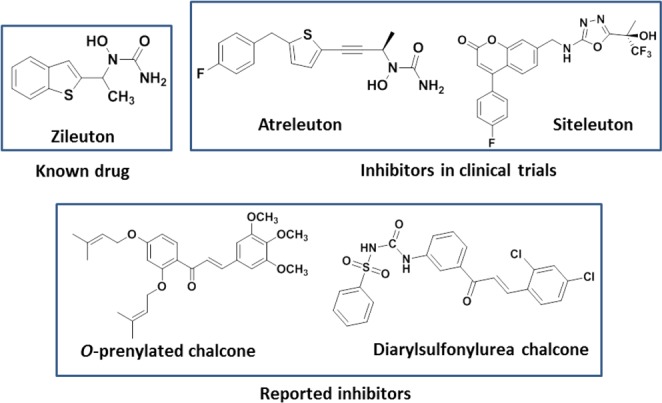
Structures of reported 5-LOX inhibitors.
The Ph model obtained gave a four-point pharmacophoric feature consisting of three hydrophobic centroids and one H-bond acceptor (Figure 3a, b). All the in-house designed compounds are used as query molecules and subjected to conformational search to create a library database. A pharmacophore search is performed for the in-house database against the Ph model to examine the mapping of the designed molecules to get the hit molecules. Surprisingly, all the 23 compounds designed in the series hit the query and were found to map exactly with the four-point common features of the Ph model such as 4k and 4n (Figure 3c, d). It can be noticed that the double bond of chalcone is one of the important reasons for the generation of hydrophobic centroids, contributing a synergistic anti-inflammatory effect. Thus, pharmacophore elucidation gave a preliminary indication that this designed hybrid (A) could be a good lead against 5-LOX, and hence, these were synthesized and biologically tested.
Figure 3.
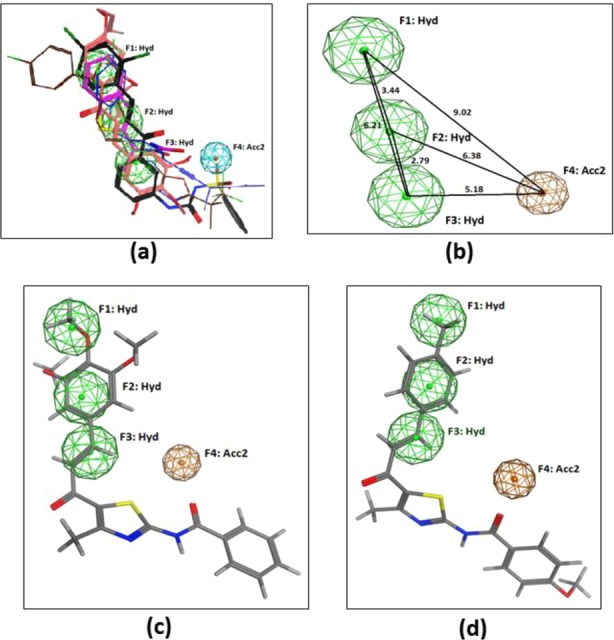
(a) Pharmacophoric model (Ph model) generated from known 5-LOX inhibitors in software MOE 2016.0801; (b) Ph model with geometric distances (A0) in a 3D spatial relationship; pharmacophore mapping of (c) 4k and (d) 4n with a Ph model. Pharmacophoric features: Hydrophobic (Hyd - green), H-bond acceptor (Acc2 - orange) and (blue in (Figure 3a)). Inhibitors superimposed in (Figure 3a) are Zileuton - magenta, Atreleuton - navy blue, Siteleuton - brown, di-o-prenylated - pink, and diarylsulfonylurea chalcone - black.
The synthetic pathways for the novel chalcone-thiazoles are illustrated in Scheme 1. The key intermediates (2a–d) and (3a–d) are prepared as per previously reported procedures with some modifications.20,21 Briefly, benzoyl isothiocyanates (1a–d) in the first step are synthesized by treating substituted benzoyl chloride with ammonium thiocyanate-acetonitrile. The product (1) in the acetonitrile (ACN) layer in the second step is treated with the required amount of ammonium hydroxide at 0 °C to yield various N-carbamothioyl substituted benzamides (2a–d). In the third step, 3-chloropentane-2,4-dione is added to the second step intermediate, N-carbamothioyl substituted benzamide (2), followed by reflux to afford the desired intermediates, N-(5-acetyl-4-methylthiazol-2-yl) substituted benzamides (3a–d) via Hantzsch thiazole synthesis. The final step for the synthesis of the target chalcone-thiazole hybrids (4a–w) is achieved using base catalyzed Claisen–Schmidt condensation reaction between benzamide thiazoyl ketones (3a–d) and various substituted aromatic/heteroaromatic aldehydes in ethanol and 2.5 N NaOH.14 All the structures were confirmed using 1H NMR, 13C NMR, and HRMS (Supporting Information).
Scheme 1. Synthesis of Chalcone-Thiazole Hybrid (4a–w) Derivatives.

All the compounds (4a–w) at a concentration of 10 μM are screened for 5-LOX inhibition activity in vitro with Zileuton as a reference drug in a cell free system using human recombinant 5-LOX enzyme (Tables 1 and 2). The inhibition is measured by determining the conversion of substrate AA to product, 5-HPETE, at λ236.22 2% DMSO was taken as negative control which did not show any inhibition while standard drug, Zileuton showed an IC50 of 1.05 ± 0.03 μM as expected according to the literature18 (Table 3). Most of the compounds exhibited good inhibition. The best active compounds were tested further at different concentrations (below 10 μM) against 5-LOX keeping substrate concentration constant to find out their IC50 (using GraphPad Prism, 5.01). It is heartening to note that many compounds exhibited better potency than the reference drug, with IC50s ranging from 0.05 to 0.5 μM (4k, 4n, and 4v, being the most potent in the series) (Table 3).
Table 1. 5-LOX Inhibition and DPPH Radical Scavenging Activities of N-(5-(3-(Substituted phenyl)acryloyl)-4-methylthiazol-2-yl) Benzamide Derivatives (4a–q) at 10 μMa.


Data is expressed as means ± SD from three independent experiments.
Table 2. 5-LOX Inhibition and DPPH Radical Scavenging Activities of N-(5-(3-Substituted Acryloyl)-4-methylthiazol-2-yl) Benzamide Derivatives (4r–w) at 10 μMa.
![]()

Data is expressed as means ± SD from three independent experiments.
Table 3. 5-LOX Inhibition IC50, Lipinski Parameters, Pseudoperoxidase Activity, and Type of Mechanism for the Best Compounds of the Series.

Determined experimentally.
Calculated using ChemBioDraw Ultra 11.0.
Calculated using Swiss ADME software.
Determined experimentally. Negative values indicate % decrease in absorbance due to consumption of 13(S)-HpODE, and positive values indicate % increase in absorbance.
In Vitro 5-LOX Inhibition and SAR
To explore the influence of hydrophobic centroids of chalcones with H-bond acceptor property of the substituted thiazoles on 5-LOX inhibition, a series of hybrids (A), (4a–w) have been synthesized. Substitutions on both thiazole (at R1) and chalcone (at R2, R3, R4) pharmacophores of prototype (A) are done to analyze their effect on the inhibition and develop a tentative SAR profile.
The basic chalcone–thiazole core, 4a, having no substitutions exhibited moderate inhibition, 73.5 ± 2.8%, at 10 μM. It is interesting to note that the introduction of highly electronegative electron withdrawing halogen, fluoro at the para position (R3) in the phenyl ring attached to chalcone, 4b enhanced the inhibition to 98.0 ± 0.7% (Table 1) with IC50 of 0.14 ± 0.06 μM (Table 3). The activities for other para halogen substituted compounds, 4c (63.7 ± 2.8%) and 4d (52.3 ± 2.1%), are found in the decreasing order of electronegativity of halogens indicative 4b > 4c > 4d (p-F > p-Cl > p-Br). Again a slight decrease in the potency is observed when p-Br is replaced with m-Br, 4e (42.5 ± 3.6%) as compared to 4d. Insertion of electron donating substituent, methoxy at R1 in the phenyl ring of benzamido group attached to thiazole, 4f (IC50 = 0.56 ± 0.15 μM, 88.2 ± 7.8%) resulted in a fold decrease in the efficiency with respect to 4b (IC50 = 0.14 ± 0.06 μM). However, even addition of electron withdrawing group, -NO2 at R1, 4g (48.8 ± 0.7%) could not improve the potency, instead reduced it by 1.3-fold, as compared to 4c. Surprisingly, replacing bromo by another electron withdrawing group, -NO2, at the meta position, 4h (IC50 = 0.24 ± 0.07 μM, 98.0 ± 2.1%) increased the inhibitory activity to more than double when compared to 4e (42.5 ± 3.6%). The inhibition activity seems to increase with increasing number of electron donating methoxy substituent (R2, R3, R4) in the phenyl ring adjacent to the vicinity of chalcone, 4i < 4j < 4k which might be due to increase in electron density on the substituted phenyl. Trimethoxy substituted compound, 4k, is a highly active inhibitor (IC50 of 0.07 ± 0.02 μM). The existence of electron donating substituents, -OCH3 (R1) and -CH3 (R3) in the vicinity of both thiazole and chalcone phenyl rings respectively is well tolerated in 4n which contributed to a synergistic effect exhibiting an IC50 of 0.08 ± 0.05 μM. A marginal decrease in potency is observed with the replacement of -OCH3 by an electron withdrawing fluoro (R1), 4m (0.54 ± 0.10 μM).
It was encouraging to substitute targeted hybrid (A) with heteroaromatic groups in the vicinity of chalcone. The methylenedioxy group is known to be widely found in natural products and drugs. Addition of the methylenedioxy bromophenyl group at R2 yielded 4v (Table 2) which resulted in significantly promising inhibitory activity (IC50 of 0.12 ± 0.04 μM). But with replacement of -H at R1 with fluoro, 4w (50.6 ± 1.4%) is not tolerated and potency reduced to half. Other heteroaromatic groups such as furan, thiophene substitutions at R2 resulted in moderate to good inhibition (60 to 80%).
5-LOX Kinetic Studies
The mode of action of the best active compounds (4k, 4n, and 4v) is estimated by measuring the initial rate of formation of product for different substrate (AA) concentrations with three concentrations of these inhibitors (0, 2, and 5 μM).23 A Lineweaver–Burke (L-B) plot indicates that the most active analogue, 4k, acts as a noncompetitive inhibitor against the enzyme (Figure 4a) with constant Km (1.9 ± 0.4, 1.8 ± 0.4, 2.1 ± 0.4 μM) and decreasing Vmax (0.38 ± 0.02, 0.29 ± 0.02, 0.25 ± 0.01 nmol/min) with increase in inhibitor concentration (0, 2, and 5 μM respectively). So, it can be concluded that 4k can bind either enzyme or enzyme–substrate complex with equal affinity. L-B plots for the other two compounds, 4n and 4v, showed competitive inhibition (Figure 4b, c) namely at these three different concentrations; the lines intersect the y-axis indicating constant Vmax (4n: 0.33 ± 0.01, 0.32 ± 0.01, 0.31 ± 0.01; 4v: 0.45 ± 0.01, 0.43 ± 0.02, 0.42 ± 0.02 nmol/min) while Km increases for both compounds (4n: 0.35 ± 0.10, 0.43 ± 0.10, 0.53 ± 0.13; 4v: 0.31 ± 0.08, 1.41 ± 0.33, 2.91 ± 0.55 μM with increasing concentrations 0, 2, and 5 μM respectively). It suggested that both these inhibitors probably bind to the enzyme’s active site and thus hinder the substrate binding site.
Figure 4.

Lineweaver–Burk plots for inhibitors at 0, 2, and 5 μM concentrations: (a) 4k, (b) 4n, (c) 4v. [S] = concentration of the substrate (AA, in μM), V = reaction rate.
5-LOX Pseudoperoxidase Activity and Mechanism
Known mechanisms for 5-LOX inhibition are redox, nonredox, and iron chelating.10 The inhibitors acting via redox mechanism disrupt the redox cycle by affecting the iron’s ionic state from Fe3+ to Fe2+. Deactivated ferrous ion has a tendency to consume lipid peroxide for activating the redox cycle of 5-LOX. Here, the substrate, 13(S)-hydroperoxyoctadecadienoic acid (13(S) HpODE) in the pseudoperoxidase assay is consumed in the presence of a redox inhibitor. The decrease in absorbance (at 234 nm) with respect to time is the measure of consumption of the substrate.7 Pseudoperoxidase activities of 4k, 4n, 4v, 4b, 4h, 4m, 4f, and 4p are performed and reported here (Figure 5). The most active inhibitor, 4k, decreases 56.6% of absorbance in 300 s (Table 3, Figure 5b) depicting significant consumption of the hydroperoxide, exhibiting redox nature. Zileuton is a well-known reported redox inhibitor. When analyzed experimentally it showed 63.4% hydroperoxide consumption (Figure 5a). 4h and 4m also show significant decrease in the peroxide by 40.2 and 56.3%, respectively, suggesting they also interrupt the redox cycle of the enzyme, while compounds 4n, 4v, 4b, 4f, and 4p do not show significant fall (Table 3) in the absorbance and the lines are almost parallel to the x-axis, suggesting that they act by nonredox process (Figure 5c, d, e, h, i). Therefore, these compounds probably interact in the active site. These pseudoperoxidase activity results are supported by L-B plots also.
Figure 5.
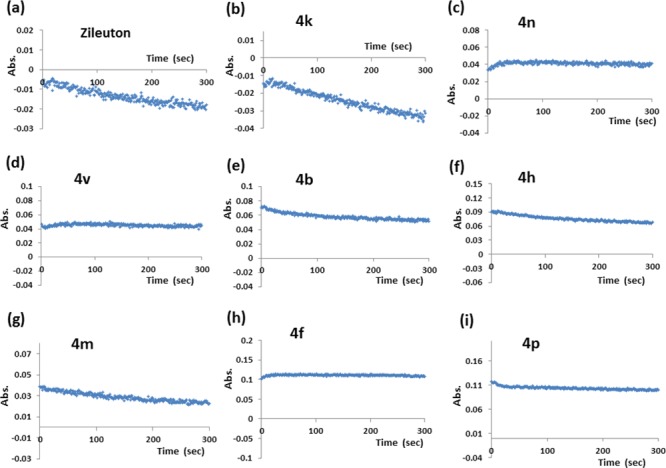
Pseudoperoxidase activity plots for the consumption of 13(S)-HpODE (at 234 nm; control subtracted) by (a) Zileuton, (b) 4k, (c) 4n, (d) 4v, (e) 4b, (f) 4h, (g) 4m, (h) 4f, and (i) 4p as a function of time (in sec).
DPPH Radical Scavenging Activity
5-LOX contains Fe3+ in the activated state, and most of the 5-LOX inhibitors are found to be good scavenging radicals. Therefore, the scavenging property of all the derivatives is determined by 2,2-diphenyl-1-picrylhydrazyl (DPPH) methodology as per a previously reported procedure.24 Positive control (ascorbic acid) showed 60.5 ± 2.3% antioxidant activity (Table 1). Results showed that almost all compounds are poor radical scavengers, showing below ∼10% activity and Zileuton exhibited 10.3 ± 0.5% at 10 μM. Whereas 4m possesses about 15% of antioxidant activity for which pseudoperoxidase assay also suggested that it acts by disrupting the redox cycle of iron. The other best inhibitors, namely 4n and 4v, showed antioxidant activities in the range of ∼5%. In support, pseudoperoxidase assay also indicated that both compounds act through a nonredox mechanism.
Docking Studies
The binding interactions of competitive inhibitors, 4n and 4b, were evaluated by docking them into the active site of 5-LOX. The Molecular Operating Environment (MOE 2017.0801) software was used, and 5-LOX protein (pdb ID 3O8Y) was obtained from Protein Data Bank (PDB). 4n showed a binding score of −9.09 kcal/mol along with H-bond and pi-H interactions (Figure 6a,b). The carbon in the double bond of chalcone interacted with the polar amino acids, Gln363, forming a H-bond donor with a distance of 2.62 Å. Thiazole ring forms a pi-H interaction with the Leu414 (4.7 Å) in the hydrophobic pocket of the enzyme. 4v with a binding score of −8.03 kcal/mol showed side chain H-bond interaction with the polar Gln557 (3.25 Å) and bromo group of benzo ethylenedioxide substituent. Other two pi-H interactions are seen with the polar residues of thiazole (Leu414) and benzene rings of the benzoamide group attached to thiazole (Tyr181) (Figure 6c,d).
Figure 6.
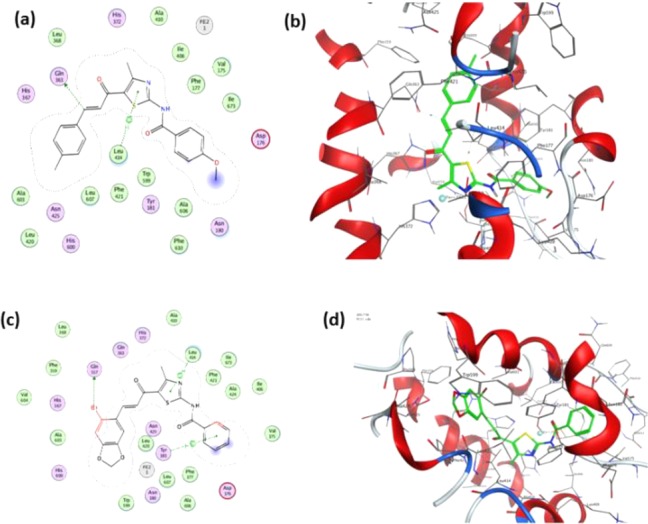
Binding modes of compounds with amino acid residues of 5-LOX (pdb ID 3O8Y). (a) Ligplot interaction of 4n. (b) Docked pose of 4n. (c) Ligplot interaction of 4v. (d) Docked pose of 4v. Dotted lines indicate interactions. Colors depicted are hydrogen: cyan, carbon: gray, nitrogen: blue, oxygen: red, sulfur: yellow.
The active inhibitors 4k, 4n, and 4v have higher total polar surface area (TPSA) (Table 3) than Zileuton indicating chances of lesser adverse effects (www.asteris-app.com/technical-info/core-properties/logp.htm). The Swiss ADME prediction indicated that these inhibitors satisfy the Lipinski rule of five as well as Ghose, Veber, Egan, and Muegge rules signifying that they have good druglikeness properties along with C log P values more than that of Zileuton indicating their higher lipophilic nature.25
In conclusion, a series of novel chalcone-thiazole molecules were rationally designed and synthesized using a pharmacophore hybridization approach and evaluated against enzyme 5-LOX. All the 23 compounds synthesized are completely new molecules, verified by Scifinder. In analogy to our previous study,7 it was possible to incorporate the chalcone group along with thiazole for 5-LOX inhibition with a synergistic effect. As a result, molecules (4k: IC50 = 0.07 ± 0.02 μM, 4n: IC50 = 0.08 ± 0.05 μM) synthesized here are found to be 10–100 times better potent than previously reported thiazole molecules (2m: IC50 = 0.9 ± 0.1 μM, 1d: IC50 = 10 μM) by us and also better than Zileuton (IC50 = 1.05 ± 0.03 μM).6,7 SAR indicated that the presence of a greater number of electron donating groups, namely, methoxy and methyl, either in the vicinity of chalcone or both thiazole and chalcone contributed a synergistic effect and yielded highly active molecules. Pharmacophore elucidation revealed that the double bond of chalcone is one of the important reasons for hydrophobic centroid generation for inhibition. Further, pseudoperoxidase assay and kinetic studies revealed the mechanism of 4k has a redox type (noncompetitive) while 4n and 4v act through competitive inhibition. Docking studies of 4n and 4v gave molecular binding interactions with the active site amino acids. Therefore, the results have laid a foundation for the discovery of novel hybrids which will help further to develop a new class of next generation 5-LOX inhibitors targeting inflammation.
Acknowledgments
Shweta Sinha acknowledges DST, New Delhi, WOS-A (SR/WOS-A/CS-126/2013), GoI for funding and fellowship for the research. The authors thank SAIF, IIT-Madras for NMR and IR spectral analysis and Department of Biotechnology, IIT-Madras for HRMS analysis. The authors thank VIT, Vellore for providing a “VIT Seed Grant” for the research.
Glossary
Abbreviations
- AA
arachidonic acid
- LT
leukotriene
- 5-LOX
5-lipoxygenase
- LTA4
leukotriene A4
- DPPH
2,2-diphenyl-1-picrylhydrazyl
- 5-HPETE
5(S) hydroperoxyeicosatetraenoic acid
- 13(S) HpODE
13(S)-hydroperoxyoctadecadienoic acid
- LB
Lineweaver–Burk
- PMSF
phenylmethyl sulphonyl fluoride
- Ph
pharmacophore
- TPSA
total polar surface area
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00193.
Synthetic procedure, characterization, experimental procedures for biological evaluation (in vitro assays), docking methods; spectral details including 1H, 13C NMR, HRMS spectra, pharmacokinetic parameters from Swiss ADME (in silico studies) (PDF)
Author Contributions
SS performed the experiments and wrore the manuscript. MD and SLM designed the experiments and corrected the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Haeggström J. Z.; Rinaldo-Matthis A.; Wheelock C. E.; Wetterholm A. Advances in eicosanoid research, novel therapeutic implications. Biochem. Biophys. Res. Commun. 2010, 396, 135–139. 10.1016/j.bbrc.2010.03.140. [DOI] [PubMed] [Google Scholar]
- Werz O.; Steinhilber D. Therapeutic options for 5-lipoxygenase inhibitors. Pharmacol. Ther. 2006, 112, 701–718. 10.1016/j.pharmthera.2006.05.009. [DOI] [PubMed] [Google Scholar]
- De Souza M. V. N. Synthesis and biological activity of natural thiazoles: An important class of heterocyclic compounds. J. Sulfur Chem. 2005, 26, 429–449. 10.1080/17415990500322792. [DOI] [Google Scholar]
- Chhabria M. T.; Patel S.; Modi P.; Brahmkshatriya P. S. Thiazole: A Review on Chemistry, Synthesis and Therapeutic Importance of its Derivatives. Curr. Top. Med. Chem. 2016, 16 (26), 2841–2862. 10.2174/1568026616666160506130731. [DOI] [PubMed] [Google Scholar]
- Liaras K.; Fesatidou M.; Geronikaki A. Thiazoles and Thiazolidinones as COX/LOX Inhibitors. Molecules 2018, 23, 685–706. 10.3390/molecules23030685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S.; Sravanthi T.; Yuvaraj S.; Manju S.; Doble M. 2-Amino-4-aryl thiazole: a promising scaffold identified as a potent 5-LOX inhibitor. RSC Adv. 2016, 6, 19271–19279. 10.1039/C5RA28187C. [DOI] [Google Scholar]
- Sinha S.; Doble M.; Manju S. Design, synthesis and identification of novel substituted 2-amino thiazole analogues as potential anti-inflammatory agents targeting 5-lipoxygenase. Eur. J. Med. Chem. 2018, 158, 34–50. 10.1016/j.ejmech.2018.08.098. [DOI] [PubMed] [Google Scholar]
- Suh J.; Yum E. K.; Cheon H. G.; Cho Y. S. Synthesis and Biological Evaluation of N-aryl-4-aryl-1, 3-Thiazole-2-Amine Derivatives as Direct 5-Lipoxygenase Inhibitors. Chem. Biol. Drug Des. 2012, 80, 89–98. 10.1111/j.1747-0285.2012.01371.x. [DOI] [PubMed] [Google Scholar]
- Hofmann B.; Barzen S.; Rödl C. B.; Kiehl A.; Borig J.; Zivkovic A.; Stark H.; Schneider G.; Steinhilber D. A class of 5-benzylidene-2-phenylthiazolinones with high potency as direct 5-lipoxygenase inhibitors. J. Med. Chem. 2011, 54, 1943–1947. 10.1021/jm101165z. [DOI] [PubMed] [Google Scholar]
- Hanke T.; Dehm F.; Liening S.; Popella S.-D.; Maczewsky J.; Pillong M.; Kunze J.; Weinigel C.; Barz D.; Kaiser A. Aminothiazole-featured pirinixic acid derivatives as dual 5-lipoxygenase and microsomal prostaglandin E2 synthase-1 inhibitors with improved potency and efficiency in vivo. J. Med. Chem. 2013, 56, 9031–9044. 10.1021/jm401557w. [DOI] [PubMed] [Google Scholar]
- Nowakowska Z. A review of anti-infective and anti-inflammatory chalcones. Eur. J. Med. Chem. 2007, 42, 125–137. 10.1016/j.ejmech.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Bugata B. K.; Dowluru S. V. G. K. K.; Avupati V. R.; Gavalapu V. R.; Nori D. L. S.; Barla S. Synthesis, characterization and in vitro biological evaluation of some new diarylsulfonylurea-chalcone hybrids as potential 5-lipoxygenase inhibitors. Eur. J. Chem. 2013, 4, 396–401. 10.5155/eurjchem.4.4.396-401.878. [DOI] [Google Scholar]
- Reddy N. P.; Aparoy P.; Reddy T. C. M.; Achari C.; Sridhar P. R.; Reddanna P. Design, synthesis, and biological evaluation of prenylated chalcones as 5-LOX inhibitors. Bioorg. Med. Chem. 2010, 18, 5807–5815. 10.1016/j.bmc.2010.06.107. [DOI] [PubMed] [Google Scholar]
- Asadipour A.; Noushini S.; Moghimi S.; Mahdavi M.; Nadri H.; Moradi A.; Shabani S.; Firoozpour L.; Foroumadi A. Synthesis and biological evaluation of chalcone-triazole hybrid derivatives as 15-LOX inhibitors. Z. Naturforsch., B: J. Chem. Sci. 2018, 73, 77–83. 10.1515/znb-2017-0115. [DOI] [Google Scholar]
- Araico A.; Terencio M.; Alcaraz M.; Dominguez J.; Leon C.; Ferrandiz M. Phenylsulphonyl urenyl chalcone derivatives as dual inhibitors of cyclo-oxygenase-2 and 5-lipoxygenase. Life Sci. 2006, 78, 2911–2918. 10.1016/j.lfs.2005.11.017. [DOI] [PubMed] [Google Scholar]
- Sharma M. C. Molecular modeling studies of substituted 3, 4-dihydroxychalcone derivatives as 5-lipoxygenase and cyclooxygenase inhibitors. Med. Chem. Res. 2014, 23, 1797–1818. 10.1007/s00044-013-0745-7. [DOI] [Google Scholar]
- Viegas-Junior C.; Danuello A.; da Silva Bolzani V.; Barreiro E. J.; Fraga C. A. M. Molecular hybridization: a useful tool in the design of new drug prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. 10.2174/092986707781058805. [DOI] [PubMed] [Google Scholar]
- Wu Y.; He C.; Gao Y.; He S.; Liu Y.; Lai L. Dynamic modeling of human 5-lipoxygenase–inhibitor interactions helps to discover novel inhibitors. J. Med. Chem. 2012, 55, 2597–2605. 10.1021/jm201497k. [DOI] [PubMed] [Google Scholar]
- Sinha S.; Doble M.; Manju S.L. 5-Lipoxygenase as a drug target: A review on trends in inhibitors structural design, SAR and mechanism based approach. Bioorg. Med. Chem. 2019, 27, 3745–3759. 10.1016/j.bmc.2019.06.040. [DOI] [PubMed] [Google Scholar]
- Liaras K.; Geronikaki A.; Glamočlija J.; Ćirić A.; Soković M. Thiazole-based chalcones as potent antimicrobial agents. Synthesis and biological evaluation. Bioorg. Med. Chem. 2011, 19, 3135–3140. 10.1016/j.bmc.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Darji D. N.; Pasha T.; Bhandari A.; Molvi K.; Desai S. A.; Makwana M. V. Synthesis of some novel 2, 4, 5–trisubstituted thiazoles as possible antibacterial agents. J. Chem. Pharm. Res. 2012, 4, 2148–2152. [Google Scholar]
- Werz O.; Steinhilber D. Development of 5-lipoxygenase inhibitors—lessons from cellular enzyme regulation. Biochem. Pharmacol. 2005, 70, 327–333. 10.1016/j.bcp.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Wisastra R.; Kok P. A. M.; Eleftheriadis N.; Baumgartner M. P.; Camacho C. J.; Haisma H. J.; Dekker F. J. Discovery of a novel activator of 5-lipoxygenase from an anacardic acid derived compound collection. Bioorg. Med. Chem. 2013, 21, 7763–7778. 10.1016/j.bmc.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeberle A.; Muñoz E.; Appendino G. B.; Minassi A.; Pace S.; Rossi A.; Weinigel C.; Barz D.; Sautebin L.; Caprioglio D.; Collado J. A.; Werz O. SAR Studies on Curcumin’s Pro-inflammatory Targets: Discovery o Prenylated Pyrazolocurcuminoids as Potent and Selective Novel Inhibitors of 5-Lipoxygenase. J. Med. Chem. 2014, 57, 5638–5648. 10.1021/jm500308c. [DOI] [PubMed] [Google Scholar]
- Singh P.; Kaur J.; Singh G.; Bhatti R. Anti-inflammatory conjugates: Identification of a highly potent anti-inflammatory agent. J. Med. Chem. 2015, 58, 5989–6001. 10.1021/acs.jmedchem.5b00952. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.