

Abstract
Oxidative stress is implicated in neuronal apoptosis that occurs in physiological settings and in neurodegenerative disorders. Superoxide anion radical, produced during mitochondrial respiration, is involved in the generation of several potentially damaging reactive oxygen species including peroxynitrite. To examine directly the role of superoxide and peroxynitrite in neuronal apoptosis, we generated neural cell lines and transgenic mice that overexpress human mitochondrial manganese superoxide dismutase (MnSOD). In cultured pheochromocytoma PC6 cells, overexpression of mitochondria-localized MnSOD prevented apoptosis induced by Fe2+, amyloid β-peptide (Aβ), and nitric oxide-generating agents. Accumulations of peroxynitrite, nitrated proteins, and the membrane lipid peroxidation product 4-hydroxynonenal (HNE) after exposure to the apoptotic insults were markedly attenuated in cells expressing MnSOD. Glutathione peroxidase activity levels were increased in cells overexpressing MnSOD, suggesting a compensatory response to increased H2O2 levels. The peroxynitrite scavenger uric acid and the antioxidants propyl gallate and glutathione prevented apoptosis induced by each apoptotic insult, suggesting central roles for peroxynitrite and membrane lipid peroxidation in oxidative stress-induced apoptosis. Apoptotic insults decreased mitochondrial transmembrane potential and energy charge in control cells but not in cells overexpressing MnSOD, and cyclosporin A and caspase inhibitors protected cells against apoptosis, demonstrating roles for mitochondrial alterations and caspase activation in the apoptotic process. Membrane lipid peroxidation, protein nitration, and neuronal death after focal cerebral ischemia were significantly reduced in transgenic mice overexpressing human MnSOD. The data suggest that mitochondrial superoxide accumulation and consequent peroxynitrite production and mitochondrial dysfunction play pivotal roles in neuronal apoptosis induced by diverse insults in cell culture and in vivo.
Keywords: Alzheimer’s disease, amyloid β-peptide, cyclosporin A, hydroxynonenal, middle cerebral artery occlusion, nitric oxide, superoxide anion radical, transgenic
Reactive oxygen species (ROS) may act as mediators of cell death that occurs in mitotic cells during their normal turnover, in neurons during development of the nervous system, and in neurodegenerative disorders (Benzi and Moretti, 1995;Bredesen, 1995; Mattson et al., 1996). Examples include the killing of lymphocytes by tumor necrosis factor and chemotherapeutic drugs (Schreck et al., 1991), trophic factor withdrawal-induced apoptosis of sympathetic neurons (Greenlund et al., 1995), and neuronal apoptosis induced by amyloid β-peptide (Aβ) (Loo et al., 1993; Kruman et al., 1997). Although these kinds of studies have documented increased levels of oxidative stress in cells undergoing apoptosis, the specific ROS involved, their subcellular sources, and their mode(s) of action in inducing apoptosis are unknown.
Mitochondria, a major subcellular source of ROS (Dugan et al., 1995;Piantadosi and Zhang, 1996), may play pivotal roles in apoptosis (Kroemer et al., 1997). Alterations occur in mitochondria before nuclear manifestations of apoptosis including impairment of energy charge and redox state, disruption of mitochondrial transmembrane potential, permeability transition, and release of cytochrome C (Liu et al., 1996; Zamzami et al., 1996a). Radical scavengers, thiol reducing agents, and cyclosporin A block mitochondrial permeability transition and apoptosis in several paradigms (Sato et al., 1995; Marchetti et al., 1996; Zamzami et al., 1996b), suggesting essential roles for mitochondrial ROS generation and permeability transition in apoptosis. Superoxide anion radical (O2−⋅) is the major ROS generated in mitochondria and can interact with nitric oxide (NO) to form peroxynitrite, which may damage cells by promoting membrane lipid peroxidation and nitration of proteins on tyrosine residues (Beckman and Crow, 1993). NO donors and peroxynitrite can induce, and NO synthase inhibitors can prevent, apoptosis in many types of cultured cells including neurons (Estevez et al., 1995; Nicotera et al., 1995; Szabo, 1996; Troy et al., 1996). Superoxide accumulation is prevented by its conversion to hydrogen peroxide, a process catalyzed by the superoxide dismutases Cu/ZnSOD and MnSOD; Cu/ZnSOD is a cytoplasmic enzyme, whereas MnSOD is localized in mitochondria (Weisiger and Fridovich, 1973; Fridovich, 1975). Correlations between MnSOD expression and increased resistance to cell injury and death have been established in several paradigms, including resistance of tumor cells to killing by tumor necrosis factor-α (TNFα) (Wong and Goeddel, 1988) and resistance of cardiac myocytes treated with TNFα to ischemic injury (Nelson et al., 1995). Despite such correlations, it is not known whether and how mitochondrial MnSOD exerts an anti-apoptotic function.
NO production and lipid peroxidation are detected at early time points (minutes to hours) after ischemic and traumatic brain injuries (Bromont et al., 1989; Hall, 1995; Iadecola, 1997). Increased levels of nitrotyrosine (Smith et al., 1997) and 4-hydroxynonenal (HNE) (Montine et al., 1997; Lovell et al., 1997) are associated with degenerating neurons in Alzheimer’s disease brain, suggesting pathogenic roles for peroxynitrite and membrane lipid peroxidation in this disorder. Evidence of apoptotic neuronal death in such neurodegenerative disorders has recently emerged (for review, see Choi, 1996; Cotman and Su, 1996). To address the role of mitochondrial superoxide and peroxynitrite production in such neurodegenerative disorders, we generated neural cell lines and transgenic mice in which human MnSOD is overexpressed in mitochondria and examined their responses to various insults.
MATERIALS AND METHODS
Materials. 4-Hydroxynonenal was purchased from Cayman Chemical (Ann Arbor, MI). 3-(4,5-Dimethylthiazol-2-yl)−2,5-diphenyltetrazolium bromide (MTT), propanol, hexanal, malondialdehyde, propyl gallate, glutathione-ethyl ester (GSH), and cyclosporin A were from Sigma (St. Louis, MO). Nonaldehyde was from Aldrich (Milwaukee, WI), and trans-2-nonenal was from Waco Pure Chemical Industries. All aldehydes were prepared as 500× stocks in ethanol. Aβ25–35 was purchased from Bachem, stored lyophilized, and dissolved in PBS at a concentration of 2 mm 2 hr before experiments. Propidium iodide, 2,7-dichlorofluorescein diacetate (DCF), dihydrorhodamine 123 (DHR), and JC-1 were purchased from Molecular Probes (Eugene, OR). RPMI medium, horse serum, and fetal bovine serum were purchased from GIBCO. zVAD-fmk [benzyloxycarbonyl-Val-Ala-Asp (O-methyl) fluoromethyl ketone] was purchased from Enzyme Systems Products (Livermore, CA).
Generation, maintenance, and experimental manipulation of PC6 cell lines. Lines of PC6 pheochromocytoma cells [a variant of the PC12 clone originally described by Greene and coworkers (Black and Greene, 1982)] stably overexpressing human MnSOD were generated in the laboratory of S. Steiner. Cells were transfected with empty vector or vector containing the human MnSOD cDNA (St Clair et al., 1991). The MnSOD cDNA was ligated into the mammalian expression vector pCB6+neo (generously provided by Dr. S. Zimmer) that is under the control of the cytomegalovirus (CMV) promoter. All recombinant DNA procedures were performed according to methods described previously (Maniatis et al., 1982). The MnSOD expression vector or empty vector was transfected into subconfluent PC6 cell cultures using DOTAP, and G418 resistant clones were isolated. Analysis of human MnSOD mRNA expression was accomplished by Northern blot analysis. Total RNA was extracted from cells as described previously (Chomcynzski and Sacchi, 1987). RNA was size fractioned by electrophoresis on a 1% formaldehyde–agarose gel and transferred to nitrocellulose. The RNA was UV cross-linked to the filter and hybridized overnight with a32P-labeled human MnSOD cDNA in 50% formamide at 45°C. The filter was washed 3 times in 1× SSC containing 0.1% SDS at room temperature for 15 min, followed by one wash in 0.25% SSC containing 0.1% SDS at 55°C for 30 min. Cells were grown in RPMI containing 10% horse serum and 5% fetal bovine serum. Cultures were treated with NGF (100 ng/ml) for 48–72 hr before experimental treatment. Immediately before experimental treatment, the culture medium was replaced with RPMI medium lacking serum.
Quantification of apoptosis. Cells were fixed in 4% paraformaldehyde, membranes were permeabilized (5 min incubation in 0.2% Triton X-100 in PBS), and cells were stained with the fluorescent DNA-binding dye propidium iodide (Kruman et al., 1997). Images of propidium iodide-stained cells were acquired using a confocal laser scanning microscope (488 nm excitation and 510 nm barrier filter) using a 60× oil immersion objective. The percentage of apoptotic cells (cells with condensed and fragmented DNA) in each culture was determined (100–150 cells/culture were counted, and counts were made in at least four separate cultures per treatment condition; analyses were performed without knowledge of the treatment history of the cultures).
Measurements of peroxynitrite levels and membrane lipid peroxidation. The dye DHR was used to quantify relative levels of mitochondrial ROS using methods similar to those described previously (Mark et al., 1997c). DHR localizes to mitochondria and fluoresces when oxidized to the positively charged rhodamine 123 derivative; peroxynitrite is particularly effective in oxidizing DHR (Kooy et al., 1994). Cells were incubated for 30 min in the presence of 5 μm DHR, washed 3 times with Locke’s solution, and imaged within 30 min. The thiobarbituric acid reactive substances (TBARS) fluorescence method was used as a measure of membrane lipid peroxidation in both cultured PC6 cells and in brain tissue (Goodman et al., 1996). This test estimates the amount of malondialdehyde (MDA) precursors, including hydroperoxides and endoperoxides. Cultured cells were lysed by scraping in 300 μl of ice-cold PBS, whereas brain tissue was homogenized in 10 vol of cold 0.05 m phosphate buffer, pH 7.0, containing 0.015 m NaCl and 0.145m KCl (this buffer solution had been equilibrated previously with 100% nitrogen for 1 hr before use); aliquots were removed for protein determination. Cell suspensions and tissue homogenates were added to a solution of 0.3 ml of 10% TCA, and 0.15 ml of TBARS reagent (0.335% 2-thiobarbituric acid in 50% glacial acetic acid) was added. The solution was incubated at 90°C for 60 min, and fluorescence was measured at 553 nm with an excitation wavelength of 515 nm. The amounts of TBARS were quantified using a standard curve of MDA prepared with MDA-bis-dimethylacetal and expressed as nanomoles of MDA per milligram of protein (Bromont et al., 1989).
Subcellular fractionation and Western blot and immunocytochemical analyses. PC6-V and PC6-MnSOD cells were removed from culture flasks by trypsinization and were pelleted by centrifugation at 200 × g. Cells were resuspended in breaking buffer (0.6 m mannitol and 20 mm HEPES, pH 7.4) and homogenized in a Dounce homogenizer, and phenylmethylsulfonyl fluoride was added to a final concentration of 1 mm. The homogenate was centrifuged at 200 × g for 5 min, and the supernatant was recentrifuged at 8000 × g for 10 min. The pellet was homogenized in a Dounce homogenizer and centrifuged at 200 × g for 10 min, and the supernatant was centrifuged at 8000 × g for 10 min. The 200 ×g pellets (nuclear fraction) were pooled and resuspended in PBS. The 8000 × g pellets (mitochondrial fraction) were pooled and resuspended in PBS. The supernatant from the 8000 × g spins was centrifuged at 100,000 × gfor 45 min, and the pellet (plasma membrane fraction) was resuspended in PBS and briefly sonicated. The supernatant was saved and used as the cytosolic fraction.
Methods for Western blot and immunocytochemical analysis of HNE-protein conjugates were described previously (Waeg et al., 1996; Mark et al., 1997a). Briefly, for Western blot analysis, solubilized cell proteins were separated by electrophoresis in a 10% polyacrylamide gel, transferred to a nitrocellulose sheet, and immunoreacted with primary antibody. Nitrocellulose was further processed using HRP-conjugated anti-mouse secondary antibody and a chemiluminescent system (Amersham, Arlington Heights, IL). For immunostaining, PC6 cells were fixed in a 4% paraformaldehyde solution and further permeabilized by incubation in 0.2% Triton X-100 in PBS. Cells were then incubated in blocking serum (1% normal horse or goat serum in PBS) for 1 hr. This was followed by a 3 hr incubation in the presence of either anti-HNE mouse monoclonal antibody (clone 1 g4) (Waeg et al., 1996; Kruman et al., 1997; 1:100 in PBS) or rabbit polyclonal anti-nitrotyrosine antibody (Chemicon, Temecula, CA; 1:500 in PBS). Cells were then incubated for 1 hr in PBS containing biotinylated horse anti-mouse or goat anti-rabbit secondary antibody, followed either by a 30 min incubation in the presence of ABC reagent (Vector Laboratories, Burlingame, CA) and a 5 min exposure to diaminobenzidine or by a 30 min incubation in PBS containing FITC–avidin conjugate (Vector Laboratories). Cellular immunoreactivity was visualized either by bright-field microscopy or confocal laser scanning microscopy.
For brain immunohistochemistry, wild-type (WT) and transgenic (Tg) mice were perfused with 4% paraformaldehyde, and brains were removed and cryoprotected. Coronal brain sections (10 μm) were cut with a freezing microtome, and free-floating sections were immunostained with rabbit anti-human MnSOD antibody (1:500) or with rabbit anti-nitrotyrosine antibody (Chemicon; 1:500). After overnight incubation in PBS containing primary antibody, sections were incubated sequentially in PBS containing biotinylated secondary antibody, ABC reagent, and diaminobenzidine. Bright-field images of immunostained brain sections were captured using a video camera, and relative levels of MnSOD immunoreactivity in the digitized images were quantified in cortex and hippocampus using methods described previously (Schwab et al., 1994).
SOD and GSH peroxidase activity assays. For SOD activity assays, cerebral cortex tissue was homogenized in 50 mmpotassium phosphate buffer, pH 7.8, and SOD activities in the homogenates were measured by the nitroblue tetrazolium (NBT)-bathocuproine sulfonate (BCS) reduction inhibition method described by Spitz and Oberley (1989). Sodium cyanide at 5 mm was used to inhibit Cu/ZnSOD and thus measure only MnSOD activity. BCS and sodium cyanide were purchased from Aldrich.
For GSH peroxidase (GSH-Px) activity assays, cells were homogenized in cold 0.05 m potassium phosphate buffer, pH 7.4, containing 1 mm EGTA. The incubation mixture for the GSH-Px activity assay consisted of 1 mm glutathione, 0.2 mmNADPH, and 1.4 IU of glutathione reductase in 0.05 mpotassium phosphate buffer, pH 7.0. The reaction was initiated by the simultaneous addition of cell homogenate (0.1–0.2 mg of protein) and 0.25 mm H2O2. The change in absorbance at 340 nm was followed for 4.5 min; 1 unit of GSH-Px activity was defined as the amount required to oxidize 1 μmol of NADPH per minute, based on the molar absorptivity of 6.22 × 10−6 for NADPH.
Quantification of mitochondrial function and transmembrane potential. The conversion of the dye MTT to formazan dye crystals in cells has been shown to be related to mitochondrial respiratory chain activity (Musser and Oseroff, 1994) and mitochondrial redox state (Shearman et al., 1995). Levels of cellular MTT reduction were quantified as described previously (Mattson et al., 1995). Briefly, MTT solution (5 mg/ml) was added to cultures, and cultures were incubated for 1 hr. Cells were then washed 3 times in Locke’s solution and solubilized in dimethylsulfoxide, and absorbance was quantified using a plate reader. The dye JC-1 was used to analyze mitochondrial membrane potential as described previously (White and Reynolds, 1996).
MnSOD transgenic mice and cerebral ischemia protocol. The generation and characterization of transgenic mice expressing human MnSOD under the control of the human β-actin promoter were described previously (Yen et al., 1996). These mice exhibit no overt phenotype. The middle cerebral artery occlusion model of focal cerebral ischemia has been described previously (Yang et al., 1994; Bruce et al., 1996). Briefly, the middle cerebral artery on the right side was occluded for 1 hr with a nylon thread, and then the thread was removed to allow reperfusion. Twenty-four hours later, mice were anesthetized with chloral hydrate and killed. Brain sections were cut into 2 mm coronal sections at a distance of 3 mm from the frontal pole and stained with triphenyltetrazolium chloride for 30 min at 37°C. Infarct area was determined with a Macintosh-II computer using National Institutes of Health Image Analysis software (version 1.52).
RESULTS
Mitochondrial MnSOD protects neural cells against apoptosis induced by agents that induce peroxynitrite production and membrane lipid peroxidation
Several lines of PC6 cells stably transfected with an expression vector containing human MnSOD were selected, and the levels of MnSOD expression and the subcellular localization of MnSOD were examined. Two lines of MnSOD-overexpressing cells that exhibited MnSOD activity levels three- to fivefold above basal levels were used for experiments in the present study; levels of Cu/ZnSOD activity were unchanged in these cell lines (data not shown). Western blot analysis revealed that levels of MnSOD protein were markedly increased in the selected lines (e.g., Fig. 1A). Analyses of mitochondrial and nonmitochondrial fractions showed that the MnSOD was localized to mitochondria, with little or no MnSOD present in other cellular compartments; MnSOD present in the nuclear fraction was accounted for by mitochondrial contamination of that fraction as indicated by the presence of the mitochondria-specific enzyme F1/Fo-ATPase (Fig. 1A). To examine the impact of increased mitochondrial MnSOD levels on oxidative stress-induced apoptosis, we exposed PC6-V and PC6-MnSOD cells to FeSO4and Aβ, two agents shown previously to induced membrane lipid peroxidation and apoptosis in PC12 cells and primary rodent hippocampal and cortical neurons (Loo et al., 1993; Kruman et al., 1997). Additional cultures were exposed to the NO-generating agent sodium nitroprusside (SNP), which has been shown to induce apoptosis in cultured neurons (Maiese et al., 1993; Dawson et al., 1993; Nicotera et al., 1995). Cultures of PC6-V and PC6-MnSOD cells were exposed for 24 hr to saline (control), 100 μm FeSO4, 50 μm Aβ, or 100 μm SNP, and the percentages of cells exhibiting apoptotic nuclei were determined. In control PC6-V cultures, ∼2–3% of the cells exhibited apoptotic nuclei (Fig. 1B). In cultures exposed to FeSO4, Aβ, and SNP, ∼15–30% of the cells exhibited apoptotic nuclei (Fig. 1B). In contrast, PC6 cell lines overexpressing human MnSOD were resistant to apoptosis induced by FeSO4, Aβ, and SNP (Fig.1B). In an additional experiment, we quantified apoptosis after exposure of PC6-V and PC6-MnSOD cells to 100 μmS-nitroso-N-acetylpenicillamine (SNAP), another NO generator. Percentages of cells with apoptotic nuclei 24 hr after exposure to saline or SNAP were (mean ± SEM of determinations made in six separate cultures): saline in PC6-V cells, 2 ± 1.1%; saline in PC6-MnSOD cells, 3 ± 0.8%; SNAP in PC6-V cells, 28 ± 2.1%; and SNAP in PC6-MnSOD cells, 5 ± 0.9% (p < 0.01 compared with the value for PC6-V cells exposed to SNAP; ANOVA with Scheffe’s post hoctest).
Fig. 1.

Expression of human MnSOD in PC6 cells: localization to the mitochondria and protection against oxidative stress-induced apoptosis. A, Western blot analysis and subcellular fractionation of MnSOD and F1/Fo-ATPase levels in PC6-V and PC6-MnSOD cells are shown. W, Whole cells;Mi, mitochondrial fraction; N, nuclear fraction; Me, membrane fraction; S, soluble fraction (cytosol). Fifty nanograms of protein/lane (for both PC6-V and PC6-MnSOD cells) were separated by SDS-PAGE, transferred to a nitrocellulose sheet, and immunoreacted with antibodies to either MnSOD (upper) or the mitochondrial enzyme F1/Fo-ATPase (lower). The upper and lower blots are two separate blots using the same sample preparations; the upper blot was reacted with the MnSOD antibody, and the lower blot was reacted with the F1/Fo-ATPase antibody. Note that MnSOD is localized almost exclusively in the mitochondrial fraction. B, Cultures of PC6-V and two different lines of PC6-MnSOD cells were exposed for 24 hr to saline (vehicle), 100 μm FeSO4, 50 μm Aβ, or 100 μm SNP, and the percentages of cells exhibiting apoptotic nuclei were determined. Values are the mean ± SEM of determinations made in eight cultures; *p < 0.01 compared with the value for vehicle-treated cultures and with each value in PC6-MnSOD cells (ANOVA with Scheffe’s post hoc tests).
Mitochondrial MnSOD suppresses peroxynitrite production, protein tyrosine nitration and membrane lipid peroxidation
Peroxynitrite, which is generated in cells by interaction of mitochondria-derived O2−⋅ with NO, can be detected and quantified in living cells using the fluorescent probe DHR (Kooy et al., 1994; Mark et al., 1997c). Exposure of PC6-V cells to 100 μm FeSO4, 50 μm Aβ, or 100 μm SNP resulted in large three- to fivefold increases in cellular DHR fluorescence (Fig.2A). In contrast, only small (40–50%) increases in DHR fluorescence occurred in PC6-MnSOD cells exposed to FeSO4, Aβ, or SNP (Fig.2A). As expected, the peroxynitrite scavenger uric acid (Szabo, 1996; Hooper et al., 1997; Mattson et al., 1997) blocked the increase in DHR fluorescence induced by FeSO4, Aβ, and SNP (data not shown). A consequence of NO production and peroxynitrite accumulation in cells is that proteins can be nitrated on tyrosine residues; such protein tyrosine nitration can be detected using antibodies generated against nitrated proteins (Beckmann et al., 1994). Exposure of PC6-V cells to FeSO4, Aβ, and SNP resulted in significant increases in nitrotyrosine immunoreactivity during 6–12 hr exposure periods, with the increase being greatest in cells exposed to SNP (Fig. 2B). The nitrotyrosine immunoreactivity appeared to be most concentrated in organelles with a cytoplasmic distribution consistent with mitochondria (Fig.2C). Levels of nitrotyrosine immunoreactivity after exposure to Aβ or SNP were significantly lower in cells overexpressing mitochondrial MnSOD compared with levels in PC6-V cells (Fig.2B). The increase in nitrotyrosine levels after exposure of PC6-MnSOD cells to FeSO4 was also less than the increase in PC6-V cells.
Fig. 2.

Mitochondrial MnSOD suppresses peroxynitrite accumulation and protein tyrosine nitration. A, Levels of DHR fluorescence were quantified 18 hr after exposure of PC6-V and PC6-MnSOD cells to vehicle, 100 μmFeSO4, 50 μm Aβ25–35, or 100 μm SNP. Values are the mean ± SEM of determinations made in six cultures; *p < 0.01 compared with the value for vehicle-treated control cultures; **p < 0.01 compared with the corresponding value in PC6-V cells.B, Levels of nitrotyrosine immunoreactivity were quantified 6 and 12 hr after exposure to vehicle, 100 μmFeSO4, 50 μm Aβ25–35, or 100 μm SNP. Values are the mean ± SEM of determinations made in six cultures. At both the 6 and 12 hr time points in PC6-V cells, the values for cultures exposed to FeSO4, Aβ25–35, and SNP were significantly greater than were the values for vehicle-treated cultures (p < 0.01). At both the 6 and 12 hr time points, the value for PC6-MnSOD cells exposed to SNP was significantly less than was the value for PC6-V cells exposed to SNP (p < 0.01). At the 12 hr time point, the values for PC6-MnSOD cells exposed to FeSO4(p < 0.05) or Aβ25–35 (p < 0.01) were significantly less than were the corresponding values in PC6-V cells (ANOVA with Scheffe’spost hoc tests). C, Confocal laser scanning microscope images of nitrotyrosine immunoreactivity in cultured PC6-V and PC6-MnSOD cells exposed to vehicle (Control), 50 μm Aβ25–35, or 100 μm SNP are shown. Aβ and SNP induced large increases in nitrotyrosine immunoreactivity in PC6-V cells (e.g., arrowheads) but not in PC6-MnSOD cells. Scale bar, 5 μm.
Fe2+ (Zhang et al., 1993), Aβ (Behl et al., 1994;Butterfield et al., 1994; Goodman et al., 1996), and peroxynitrite (Beckman and Crow, 1993) have been shown to induce membrane lipid peroxidation. As expected, FeSO4, Aβ, and SNP each induced progressive increases in lipid peroxidation in PC6-V cells during 3–24 hr exposure periods as detected using the TBARS assay (Fig.3A,B). The magnitude of the increase in TBARS fluorescence was greatest in PC6-V cells exposed to FeSO4 (two- to threefold increases) and least in cells exposed to SNP (30–40% increase). Lipid peroxidation induced by each of the oxidative insults was primarily suppressed in PC6 cells expressing MnSOD (Fig.3A,B). Uric acid completely blocked SNP-induced lipid peroxidation (Fig. 3B), suggesting that peroxynitrite mediated the induction of lipid peroxidation by SNP. A potentially toxic aldehyde released when membrane lipids are peroxidized is HNE (Esterbauer et al., 1991). HNE covalently binds to proteins, and such HNE–protein adducts can be detected using specific antibodies (Waeg et al., 1996; Mark et al., 1997a,b). Exposure of PC6-V cells to FeSO4 and Aβ resulted in accumulation of HNE immunoreactive proteins in the cells (Fig. 3C). Levels of HNE immunoreactivity in PC6-MnSOD cells exposed to FeSO4and Aβ were considerably less than the levels in PC6-V cells exposed to the same insults (Fig. 3C). MnSOD overexpression also suppressed the accumulation of HNE–protein conjugates in cells exposed to SNP (data not shown).
Fig. 3.

Mitochondrial MnSOD suppresses lipid peroxidation induced by apoptotic insults. A, Levels of TBARS fluorescence were quantified in PC6-V and PC6-MnSOD cells at the indicated time points after exposure to vehicle (Cont), 100 μm FeSO4, or 50 μmAβ. Values are the mean ± SEM of determinations made in eight cultures. At each time point, the value for PC6-MnSOD cells exposed to FeSO4 or Aβ was significantly less than was the corresponding value for PC6-V cells exposed to FeSO4 or Aβ (p < 0.01; ANOVA with Scheffe’spost hoc tests). B, Cultures of PC6-V cells (control and uric acid; 200 μm uric acid) and PC6-MnSOD cells (MnSOD) were exposed for 6 hr to either vehicle or 100 μm SNP. Levels of TBARS fluorescence were quantified, and the values represent the mean ± SEM of determinations made in eight separate cultures; *p < 0.01 compared with the value for vehicle-treated cultures. C, Cells were exposed for 12 hr to vehicle (Cont), 100 μm FeSO4, or 50 μmAβ25–35. The cells were then immunostained with HNE antibody, and confocal laser scanning microscope images of cellular immunofluorescence were acquired. Note that both FeSO4 and Aβ induced a large increase in HNE immunoreactivity in PC6-V cells but not in PC6-MnSOD cells.
Although overexpression of MnSOD would be expected to decrease superoxide anion levels and peroxynitrite production, it would also be expected to increase H2O2 levels and thereby potentially promote hydroxyl radical production. However, the observation that levels of lipid peroxidation after exposure of cells to FeSO4 were reduced in cells overexpressing MnSOD suggested that mechanisms for removal of H2O2may be enhanced. To test this possibility, we quantified levels of GSH-Px activity in PC6-V cells and in two different lines of PC6-MnSOD cells with the following results (mean ± SEM of determinations made in four separate cultures): PC6-V cells, 653 ± 10 mU/mg protein; PC6-MnSOD1, 904 ± 24 mU/mg protein (p < 0.001 compared with the value for PC6-V cells); and PC6-MnSOD2, 935 ± 55 mU/mg protein (p < 0.005 compared with the value for PC6-V cells).
Agents that suppress membrane lipid peroxidation, peroxynitrite accumulation, and caspase activation prevent apoptosis
The decreased peroxynitrite accumulation and lipid peroxidation after exposure to oxidative insults in PC6 cells overexpressing mitochondrial MnSOD suggested a causal link between these ROS and apoptosis. To determine whether suppression of peroxynitrite production and/or lipid peroxidation was sufficient to prevent apoptosis, we pretreated PC6-V cells with antioxidants that suppress membrane lipid peroxidation (propyl gallate; Behl et al., 1994), detoxify 4-hydroxynonenal (glutathione ethyl ester; Kruman et al., 1997), or scavenge peroxynitrite (uric acid; Hooper et al., 1997), and these cells were then exposed to the oxidative insults. Each antioxidant primarily prevented apoptosis induced by FeSO4, Aβ, and SNP (Fig. 4). Each antioxidant also suppressed the increase in TBARS fluorescence induced by FeSO4, Aβ, and SNP (data not shown), demonstrating that the reduced apoptosis was indeed correlated with reduced membrane lipid peroxidation. Caspases have been shown to play a key role in apoptotic cascades induced by a variety of insults including exposure of cells to oxidative stress (for review, see Miura and Yuan, 1996;Schwartz and Milligan, 1996). Pretreatment of PC6-V cells with the broad-spectrum caspase inhibitor zVAD-fmk completely prevented apoptosis induced by FeSO4, Aβ, and SNP (Fig.4).
Fig. 4.

A caspase inhibitor and antioxidants prevent apoptosis induced by FeSO4, Aβ25–35, and SNP. Cultures of PC6-V cells were pretreated for 2 hr with 50 μm zVAD-fmk (ZVAD), 2 mmglutathione ethyl ester (GSH), 50 μm propyl gallate (PG), or 200 μm uric acid (UA). Cultures were then exposed for 24 hr to 100 μm FeSO4, 50 μm Aβ25–35, or 100 μm SNP, and percentages of cells with apoptotic nuclei were quantified. Values are the mean ± SEM of determinations made in six cultures; *p < 0.001 compared with the value for vehicle-treated (Cont) cultures and with the values for cultures pretreated with ZVAD, GSH,PG, and UA (ANOVA with Scheffe’spost hoc tests).
Mitochondrial dysfunction and permeability transition linked to oxidative stress-induced apoptosis
Mitochondrial changes occur in cells undergoing apoptosis, and some of the alterations may be causally involved in the cell death process (for review, see Kroemer et al., 1997). Two alterations that occur in mitochondria in cells that proceed to undergo apoptosis are a decrease in membrane potential and a permeability transition associated with the opening of large pores in the mitochondrial membrane. Confocal laser scanning microscope analysis of cellular JC-1 fluorescence was used as a measure of mitochondrial transmembrane potential (Reers et al., 1995). Exposure of PC6-V cells to FeSO4, Aβ, and SNP for 12 hr resulted in highly significant decreases in JC-1 fluorescence (Fig. 5A). In contrast, mitochondrial transmembrane potential was maintained in PC6-MnSOD cells exposed to each insult. In addition, levels of MTT reduction (a measure of mitochondrial energy charge) were significantly reduced in PC6-V cells but not in PC6-MnSOD cells after exposure to FeSO4, Aβ, and SNP (data not shown). To determine whether mitochondrial permeability transition played a role in oxidative stress-induced apoptosis, we treated PC6-V cells with cyclosporin A, an agent that prevents the permeability transition (Petronilli et al., 1994) and can protect cells against elevated calcium levels and oxidative stress (Broekemeier et al., 1992). Cyclosporin A primarily prevented apoptosis induced by FeSO4, Aβ, and SNP (Fig. 5B).
Fig. 5.
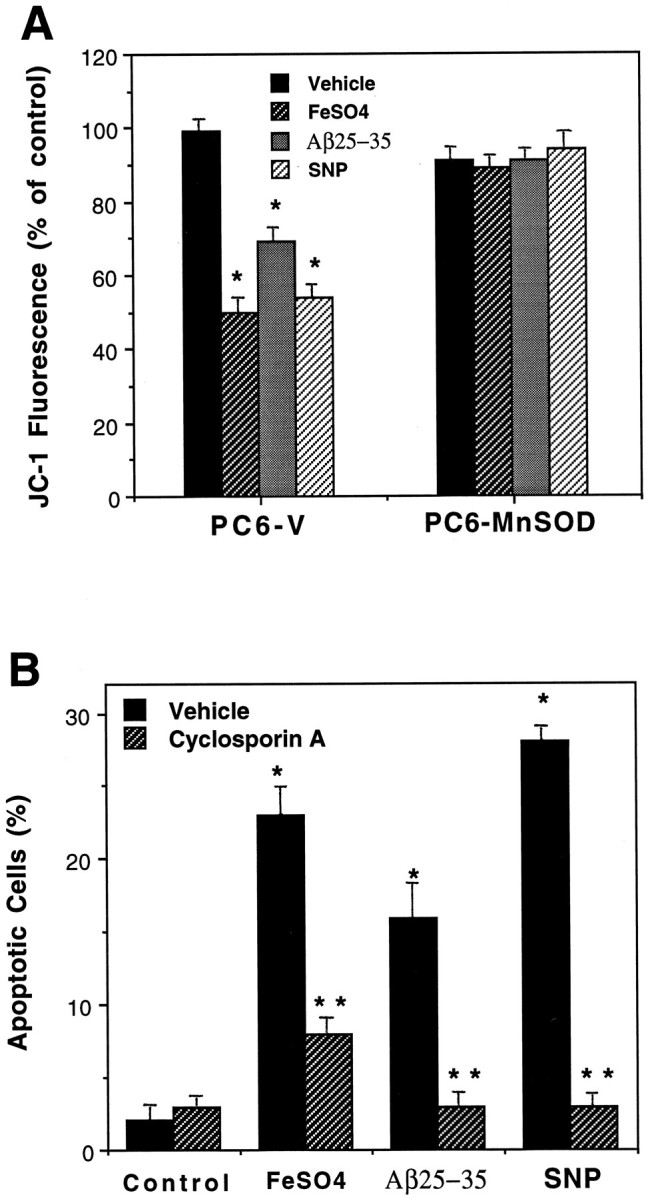
Evidence that peroxynitrite-induced mitochondrial dysfunction mediates oxidative stress-induced apoptosis.A, PC6-V and PC12-MnSOD cells were exposed to vehicle, 100 μm FeSO4, 50 μmAβ, or 100 μm SNP for 12 hr, and levels of JC-1 fluorescence were quantified. Values are the mean ± SEM of determinations made in six separate cultures; *p < 0.01 compared with the value for vehicle-treated PC6-V cells and with each value in PC6-MnSOD cells. B, Cultures were pretreated for 2 hr with vehicle or cyclosporin A and were then exposed for 20 hr to 100 μm FeSO4, 50 μm Aβ25–35, or 100 μm SNP. Values are the mean ± SEM of determinations made in six cultures; *p < 0.01 compared with the control value; **p < 0.01 compared with the corresponding vehicle value.
Overexpression of human mitochondrial MnSOD in transgenic mice protects brain cells against ischemic injury
To examine the impact of MnSOD on neuronal vulnerability in vivo, we used Tg mice expressing human MnSOD under the control of a human β-actin promoter (Yen et al., 1996). Northern blot analysis demonstrated the expression of the human MnSOD transcript in brain tissue (Fig. 6A). Immunocytochemical analysis (Fig. 6B) and enzyme activity assays (data not shown) revealed that MnSOD protein and activity levels were each increased approximately twofold in brain tissue from the transgenic mice. Consistent with previous studies of MnSOD immunoreactivity in adult rats (Liu et al., 1994), the majority of MnSOD was localized to neurons in the cortex and hippocampus of both WT and MnSOD Tg mice (data not shown).
Fig. 6.
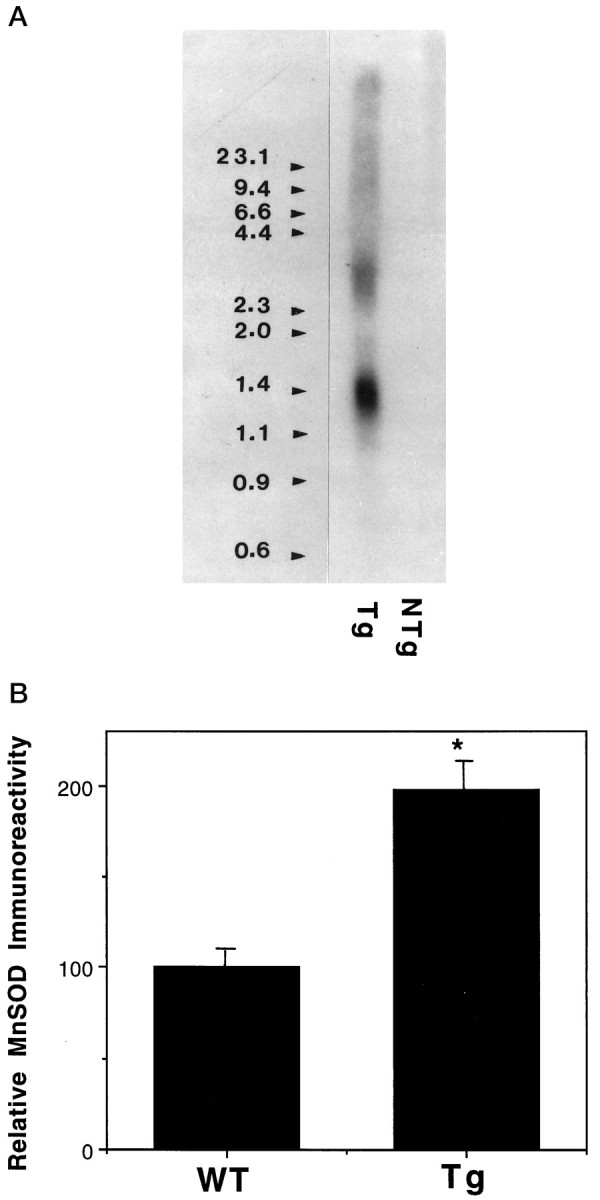
Characterization of MnSOD expression in brain tissue of MnSOD transgenic mice. A, Northern blot analysis of MnSOD mRNA in brain tissue from Tg and wild-type (NTg) mice is shown. Poly(A+)RNA from the brains of Tg and NTg mice was separated on a 1.1% formaldehyde–agarose gel and, after transfer to nylon membrane, probed with a 32P-labeled human MnSOD cDNA.B, Relative levels of MnSOD immunoreactivity in cerebral cortex were quantified in brain tissue sections (see Materials and Methods). Values are the mean ± SEM (n = 4 mice/group); *p < 0.01 compared with the corresponding WT value.
Six MnSOD Tg and six WT mice were subjected to 1 hr middle cerebral artery (MCA) occlusion, followed by 24 hr of reperfusion, and cortical infarct volumes were quantified. The infarct volume was significantly reduced in MnSOD Tg mice compared with WT mice (Fig.7A). To determine whether overexpression of MnSOD conferred resistance to membrane lipid peroxidation in vivo, we subjected MnSOD Tg and WT mice to MCA occlusion, and levels of lipid peroxidation in the ischemic cortex were quantified by TBARS analysis. After ischemia, TBARS values in the MnSOD Tg mice were significantly less than the values in the WT mice (Fig. 7B). Interestingly, basal levels of lipid peroxidation were also lower in the MnSOD Tg mice, suggesting a lower basal level of oxidative stress in the brains of MnSOD Tg mice. Examination of brain sections from WT and MnSOD Tg mice showed that ischemia induced a clear increase in nitrotyrosine immunoreactivity in the region of the injured cortex and that the extent of the nitrotyrosine immunoreactivity was markedly decreased in MnSOD Tg mice (Fig.8).
Fig. 7.
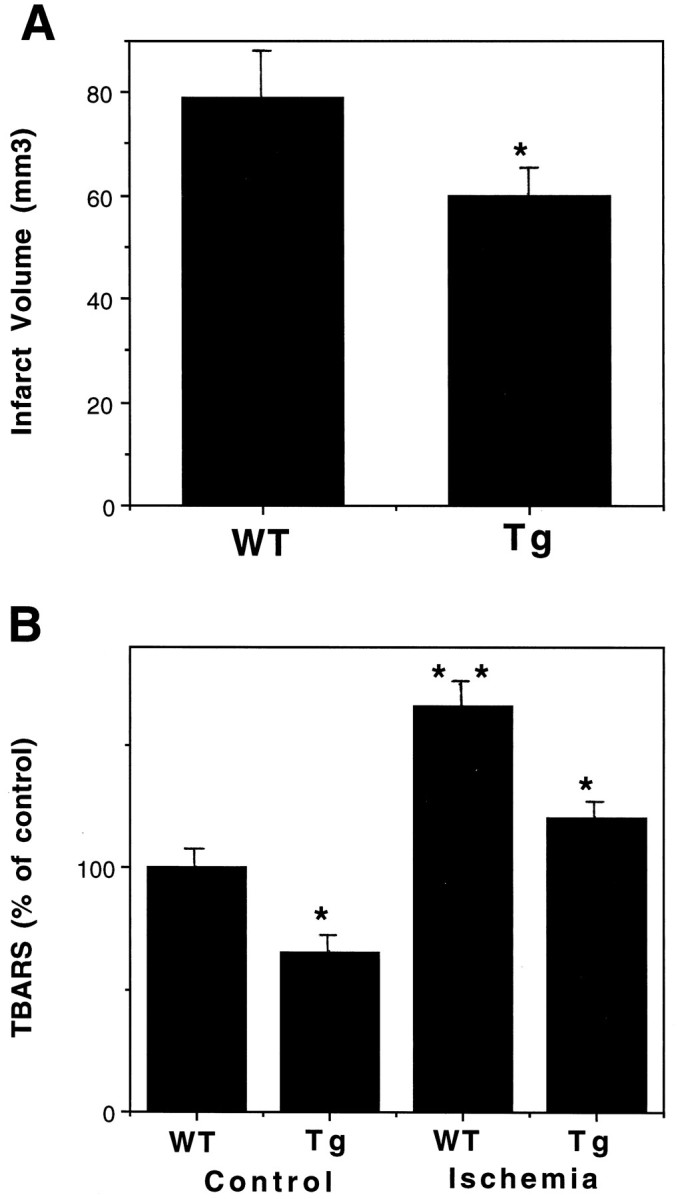
Levels of cellular injury and lipid peroxidation are reduced in MnSOD Tg mice after cerebral ischemia. A, Cortical infarct volumes were quantified 24 hr after MCA occlusion in WT and MnSOD Tg mice. Values are the mean ± SEM (n = 6 mice in each group); *p< 0.05 compared with the WT value. B, TBARS levels were quantified in infarcted cortical tissue 24 hr after MCA occlusion in WT and MnSOD Tg mice. Values are the mean ± SEM (n = 4 mice in each group); *p< 0.01 compared with the corresponding WT value; **p < 0.01 compared with the WT control value.
Fig. 8.
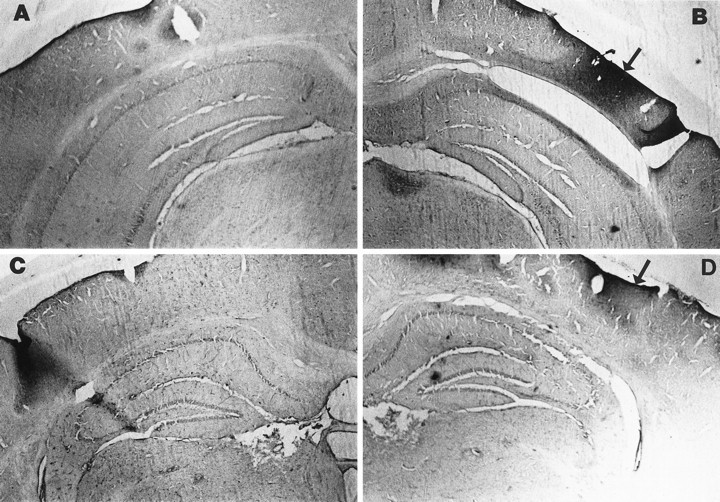
Ischemia-induced protein tyrosine nitration is reduced in MnSOD Tg mice. Bright-field micrographs of nitrotyrosine immunoreactivity in coronal brain sections from mice killed 24 hr after sham surgery (A, wild-type; C, MnSOD Tg) or after MCA occlusion ischemia (B, wild-type;D, MnSOD Tg). Note the great increase in nitrotyrosine immunoreactivity in the injured cortex of the wild-type mouse (B, arrow) relative to the MnSOD Tg mouse that showed less of an increase in nitrotyrosine immunoreactivity (D, arrow). Similar differences were observed in each of four wild-type and four MnSOD Tg mice examined.
DISCUSSION
Our findings suggest that superoxide production and resulting peroxynitrite formation plays a pivotal role in the mitochondrial dysfunction and apoptosis induced by diverse insults. Because MnSOD is specific for the removal of superoxide radicals (Fridovich, 1975) and is localized exclusively to the mitochondria, we interpret our data as direct evidence that superoxide radicals play a primary role in neuronal apoptosis induced by three different insults (Fe2+, Aβ, and the NO generators SNP and SNAP). Cell death resulting from exposure to each insult manifests as apoptosis, indicated by nuclear DNA condensation and fragmentation and prevention of cell death by the caspase inhibitor zVAD-fmk. Although each apoptotic insult initiates oxidative stress by a different mechanism (Fe2+ catalyzes hydroxyl radical production from H2O2, Aβ induces peroxide accumulation and membrane lipid peroxidation, and SNP generates NO), they each induced peroxynitrite accumulation as demonstrated by increases in cellular DHR fluorescence and nitrotyrosine immunoreactivity. However, it should be noted that DHR fluorescence is not a specific indicator of mitochondrial peroxynitrite levels because it can localize to other cellular compartments and can be oxidized by agents other than peroxynitrite and because its cellular retention is affected by mitochondrial membrane potential. Nevertheless, a necessary role for peroxynitrite production in apoptosis is indicated by the ability of uric acid, a peroxynitrite scavenger, to protect the neural cells against apoptosis induced by each insult. Peroxynitrite apparently mediated mitochondrial changes associated with apoptosis because SNP caused a decrease of mitochondrial transmembrane potential and because uric acid prevented mitochondrial alterations and apoptosis otherwise induced by SNP. Although previous studies demonstrated that NO donors and exogenous peroxynitrite can induce apoptosis (Estevez et al., 1995; Nicotera et al., 1995), the present findings suggest that mitochondrial peroxynitrite formation plays an important role in the apoptotic process induced by diverse oxidative insults.
It is important to recognize that, in addition to decreasing O2−⋅ levels, overexpression of MnSOD may elicit downstream effects that account for its neuroprotective action. Indeed, we found that levels of GSH-Px activity were significantly increased in cells overexpressing MnSOD, which is likely a compensatory response to the increased H2O2 production associated with increased superoxide dismutase activity. Increased GSH-Px activity and resulting removal of H2O2 may also account for the reduced levels of lipid peroxidation after exposure of PC6-MnSOD cells to FeSO4 and Aβ. However, although our data clearly demonstrate that overexpression of mitochondrial MnSOD results in lower levels of oxidative damage and reduced apoptosis after exposure to several different insults, they do not necessarily establish a central role for normal ambient levels of MnSOD in cytoprotection.
Our data are consistent with at least two mechanisms whereby elevated MnSOD levels may suppress membrane lipid peroxidation, one being a decrease in peroxynitrite production (Beckman and Crow, 1993) and the other being increased removal of H2O2 as the result of increased GSH-Px activity. Membrane lipid peroxidation seems to be a critical downstream event in peroxynitrite-mediated apoptosis because agents that suppress membrane lipid peroxidation (propyl gallate) and detoxify the cytotoxic lipid peroxidation product HNE (glutathione) prevented apoptosis induced by FeSO4, Aβ, and SNP. Lipid peroxidation may impair a variety of intra- and extramitochondrial membrane transport systems that may contribute to apoptotic pathways, including ion-motive ATPases (Mark et al., 1997a), glucose transporters (Mark et al., 1997b), and mitochondrial ion-transport systems (Kristal et al., 1996; Keller et al., 1997). HNE may mediate the adverse effects of lipid peroxidation on plasma and mitochondrial membrane transport functions by covalently binding to Lys, His, and Cys residues of proteins (Esterbauer et al., 1991; Mark et al., 1997b).
Mitochondrial permeability transition, which is linked to a collapse of the proton gradient across the inner membrane, can be triggered by a variety of oxidative insults (Chernyak and Bernardi, 1996; Scarlett et al., 1996). Cyclosporin A was shown previously to block permeability transition and protect non-neuronal cells against apoptosis (Waring and Beaver, 1996; Zamzami et al., 1996b). Cyclosporin A was effective in preventing apoptosis of PC6 cells induced by FeSO4, Aβ, and SNP, suggesting a role for the permeability transition in the apoptotic pathway induced by peroxynitrite and lipid peroxidation. PC6 cells expressing MnSOD were resistant to alterations in mitochondrial membrane ion permeability induced by FeSO4, Aβ, and SNP consistent with the anti-apoptotic action of MnSOD being exerted upstream of mitochondrial dysfunction.
Recently, MnSOD knock-out mice were developed, and heterozygous animals that had a reduction in MnSOD expression were shown to have increased cortical damage in response to ischemic injury (Chan et al., 1995,1996). We found that death of cerebral cortical cells after middle cerebral artery occlusion was significantly decreased in transgenic mice overexpressing MnSOD. The ability of MnSOD to protect brain cells against ischemic injury in vivo suggests a central role for superoxide radicals in ischemic cell death. Levels of membrane lipid peroxidation and protein nitration after middle cerebral artery occlusion were significantly greater in wild-type mice compared with MnSOD Tg mice, consistent with suppression of peroxynitrite formation by mitochondrial MnSOD. Apoptosis is increasingly recognized as a prominent form of neuronal death in many different experimental paradigms of human neurodegenerative conditions including ischemic stroke (Linnik et al., 1993; MacManus et al., 1993; Bredesen, 1995;Nitatori et al., 1995). NO/peroxynitrite and lipid peroxidation have been proposed to play roles in ischemic stroke (Iadecola, 1997), traumatic brain and spinal cord injuries (Hall, 1995; Springer et al., 1997), Alzheimer’s disease (Mark et al., 1996; Montine et al., 1997;Smith et al., 1997; Lovell et al., 1997), and Parkinson’s disease (Yoritaka et al., 1996). Although the initiating cause of oxidative stress may differ in each disorder, in each case a common result is peroxynitrite production and membrane lipid peroxidation. Our data suggest that MnSOD may play an important role in preventing neuronal degeneration in an array of disorders.
Because increased MnSOD expression in response to tissue injury occurs in many conditions (Wong and Goeddel, 1988; Bruce et al., 1996), our data suggest an anti-apoptotic role for MnSOD in such conditions. Previous studies have shown that a mild preconditioning ischemia can induce MnSOD expression and protect brain cells against a subsequent severe ischemic insult (Kato et al., 1995; Kirino et al., 1996). This phenomenon in vivo may have underlying mechanisms similar to those that mediate the anti-apoptotic effects of moderate stress conditions in cell culture. For example, exposure of cultured neurons to subtoxic levels of N-methyl-d-aspartate (Marini and Paul, 1993) or hyperthermia and hypoxia (Caprioli et al., 1996) confers resistance to excitotoxic cell death. Signaling cascades involving the cytokine TNFα and the transcription factor NF-κB may induce MnSOD expression under such stress conditions and thereby protect neurons against oxidative insults. As evidence, both TNFα and ceramide induce NF-κB activation and protect cultured rat hippocampal neurons against cell death induced by Aβ and Fe2+(Barger et al., 1995; Mattson et al., 1997). Moreover, excitotoxic and ischemic brain injury is enhanced, and MnSOD levels are reduced in mice lacking TNFα receptors (Bruce et al., 1996). By suppressing peroxynitrite accumulation and mitochondrial dysfunction, MnSOD may play a central role in the anti-apoptotic actions of NF-κB recently established in primary hippocampal neurons (Mattson et al., 1997). Neuroprotective strategies that target components of the apoptotic pathway elucidated in the present study (superoxide accumulation, peroxynitrite production, and lipid peroxidation) may prove beneficial in the array of degenerative disorders that involve oxidative stress.
Footnotes
This work was supported by National Institutes of Health Grants NS29001, NS35253, and AG05119 to M.P.M. and by the Kentucky Spinal Cord and Head Injury Research Trust. We thank J. Begley and W. Fu for technical assistance and G. Waeg for providing HNE antibody.
Correspondence should be addressed to Dr. Mark P. Mattson, 211 Sanders-Brown Building, University of Kentucky, Lexington, KY 40536-0230.
REFERENCES
- 1.Barger SW, Horster D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP. Tumor necrosis factors α and β protect neurons against amyloid β-peptide toxicity: evidence for involvement of a κB-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci USA. 1995;92:9328–9332. doi: 10.1073/pnas.92.20.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beckman JS, Crow JP. Pathological implications of nitric oxide, superoxide and peroxynitrite formation. Biochem Soc Trans. 1993;21:330–334. doi: 10.1042/bst0210330. [DOI] [PubMed] [Google Scholar]
- 3.Beckman J, Ye Y, Anderson P, Chen J, Accavitti M, Tarpey M, White C. Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol Chem Hoppe Seyler. 1994;375:81–88. doi: 10.1515/bchm3.1994.375.2.81. [DOI] [PubMed] [Google Scholar]
- 4.Behl C, Davis J, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid β protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 5.Benzi G, Moretti A. Are reactive oxygen species involved in Alzheimer’s disease? Neurobiol Aging. 1995;16:661–674. doi: 10.1016/0197-4580(95)00066-n. [DOI] [PubMed] [Google Scholar]
- 6.Black MM, Greene LA. Changes in the colchicine susceptibility of microtubules associated with neurite outgrowth: studies with nerve growth factor-responsive PC12 pheochromocytoma cells. J Cell Biol. 1982;95:379–386. doi: 10.1083/jcb.95.2.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bredesen DE. Neural apoptosis. Ann Neurol. 1995;38:839–851. doi: 10.1002/ana.410380604. [DOI] [PubMed] [Google Scholar]
- 8.Broekemeier KM, Carpenter-Deyo L, Reed DJ, Pfeiffer DR. Cyclosporin A protects hepatocytes subjected to high Ca2+ and oxidative stress. FEBS Lett. 1992;304:192–194. doi: 10.1016/0014-5793(92)80616-o. [DOI] [PubMed] [Google Scholar]
- 9.Bromont C, Marie C, Bralet J. Increased lipid peroxidation in vulnerable brain regions after transient forebrain ischemia in rats. Stroke. 1989;20:918–924. doi: 10.1161/01.str.20.7.918. [DOI] [PubMed] [Google Scholar]
- 10.Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- 11.Butterfield DA, Hensley K, Harris M, Mattson MP, Carney J. β-Amyloid peptide free radical fragments initiate synaptosomal lipoperoxidation in a sequence-specific fashion: implications to Alzheimer’s disease. Biochem Biophys Res Commun. 1994;200:710–715. doi: 10.1006/bbrc.1994.1508. [DOI] [PubMed] [Google Scholar]
- 12.Caprioli J, Kitano S, Morgan JE. Hyperthermia and hypoxia increase tolerance of retinal ganglion cells to anoxia and excitotoxicity. Invest Ophthalmol Vis Sci. 1996;37:2376–2381. [PubMed] [Google Scholar]
- 13.Chan PH, Epstein CJ, Li Y, Huang TT, Carlson E, Kinouchi H, Yang G, Kamii H, Mikawa S, Kondo T. Transgenic mice and knockout mutants in the study of oxidative stress in brain injury. J Neurotrauma. 1995;12:815–824. doi: 10.1089/neu.1995.12.815. [DOI] [PubMed] [Google Scholar]
- 14.Chan PH, Epstein CJ, Li Y, Huang TT, Carlson E, Mikawa S, Kondo T, Chen SF, Reola L. Studies of neuronal injury mechanism in focal stroke using mitochondrial manganese superoxide dismutase-deficient mice. In: Kriegelstein J, editor. Pharmacology of cerebral ischemia. Medpharm; Stuttgart, Germany: 1996. pp. 573–579. [Google Scholar]
- 15.Chernyak BV, Bernardi P. The mitochondrial permeability transition pore is modulated by oxidative agents through both pyridine nucleotides and glutathione at two separate sites. Eur J Biochem. 1996;238:623–630. doi: 10.1111/j.1432-1033.1996.0623w.x. [DOI] [PubMed] [Google Scholar]
- 16.Choi DW. Ischemia-induced neuronal apoptosis. Curr Opin Neurobiol. 1996;6:667–672. doi: 10.1016/s0959-4388(96)80101-2. [DOI] [PubMed] [Google Scholar]
- 17.Chomcynzski C, Sacchi B. Single step method of RNA isolation. Anal Biochem. 1987;161:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 18.Cotman CW, Su JH. Mechanisms of neuronal death in Alzheimer’s disease. Brain Pathol. 1996;6:493–506. doi: 10.1111/j.1750-3639.1996.tb00878.x. [DOI] [PubMed] [Google Scholar]
- 19.Dawson V, Dawson T, Bartley D, Uhl G, Snyder S. Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci. 1993;13:2651–2661. doi: 10.1523/JNEUROSCI.13-06-02651.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dugan LL, Sensi SL, Canzoniero LM, Handran SD, Rothman SM, Goldberg MP, Choi DW. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-d-aspartate. J Neurosci. 1995;15:6377–6388. doi: 10.1523/JNEUROSCI.15-10-06377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 22.Estevez AG, Radi R, Barbeito L, Shin JT, Thompson JA, Beckman JS. Peroxynitrite-induced cytotoxicity in PC12 cells: evidence for an apoptotic mechanism differentially modulated by neurotrophic factors. J Neurochem. 1995;65:1543–1550. doi: 10.1046/j.1471-4159.1995.65041543.x. [DOI] [PubMed] [Google Scholar]
- 23.Fridovich I. Superoxide dismutases. Biochim Biophys Acta. 1975;877:147–159. doi: 10.1146/annurev.bi.44.070175.001051. [DOI] [PubMed] [Google Scholar]
- 24.Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury and amyloid β-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66:1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- 25.Greenlund LJ, Deckwerth TL, Johnson EM., Jr Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed neuronal death. Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 26.Hall ED. Inhibition of lipid peroxidation in central nervous system trauma and ischemia. J Neurol Sci. 1995;134:79–83. doi: 10.1016/0022-510x(95)00211-j. [DOI] [PubMed] [Google Scholar]
- 27.Hooper DC, Bagasra O, Marini JC, Zborek A, Ohnishi ST, Kean R, Champion JM, Sarker AB, Bobroski L, Farber JL, Akaike T, Maeda H, Koprowski H. Prevention of experimental allergic encephalomyelitis by targeting nitric oxide and peroxynitrite: implications for the treatment of multiple sclerosis. Proc Natl Acad Sci USA. 1997;94:2528–2533. doi: 10.1073/pnas.94.6.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iadecola C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci. 1997;20:132–139. doi: 10.1016/s0166-2236(96)10074-6. [DOI] [PubMed] [Google Scholar]
- 29.Kato H, Kogure K, Araki T, Liu XH, Kato K, Itoyama Y. Immunohistochemical localization of superoxide dismutase in the hippocampus following ischemia in a gerbil model of ischemic tolerance. J Cereb Blood Flow Metab. 1995;15:60–70. doi: 10.1038/jcbfm.1995.7. [DOI] [PubMed] [Google Scholar]
- 30.Keller JN, Pang Z, Geddes JW, Begley JG, Germeyer A, Waeg G, Mattson MP. Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid β-peptide: role of the lipid peroxidation product 4-hydroxynonenal. J Neurochem. 1997;69:273–284. doi: 10.1046/j.1471-4159.1997.69010273.x. [DOI] [PubMed] [Google Scholar]
- 31.Kirino T, Nakagomi T, Kanemitsu H, Tamura A. Ischemic tolerance. Adv Neurol. 1996;71:505–511. [PubMed] [Google Scholar]
- 32.Kooy NW, Royall JA, Ischoropoulos H, Beckman JS. Peroxynitrite-mediated oxidation of dihydrorhodamine 123. Free Radic Biol Med. 1994;16:149–156. doi: 10.1016/0891-5849(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 33.Kristal BS, Park BK, Yu BP. 4-Hydroxyhexenal is a potent inducer of the mitochondrial permeability transition. J Biol Chem. 1996;271:6033–6038. doi: 10.1074/jbc.271.11.6033. [DOI] [PubMed] [Google Scholar]
- 34.Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- 35.Kruman I, Bruce AJ, Bredesen DE, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci. 1997;17:5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linnik MD, Zobrist RH, Hatfield MD. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke. 1993;24:2002–2008. doi: 10.1161/01.str.24.12.2002. [DOI] [PubMed] [Google Scholar]
- 37.Liu XH, Kato H, Araki T, Itoyama Y, Kato K, Kogure K. An immunohistochemical study of copper/zinc superoxide dismutase and manganous superoxide dismutase following focal cerebral ischemia in the rat. Brain Res. 1994;644:257–266. doi: 10.1016/0006-8993(94)91688-8. [DOI] [PubMed] [Google Scholar]
- 38.Liu X, Kim NC, Yang J, Jemmerson R, Wanf X. Induction of apoptotic program in cell free extract: requirement for dATP and cytochrome C. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 39.Loo DT, Copani A, Pike CJ, Whitmore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by β amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lovell MA, Ehmann WD, Mattson MP, Markesbery WR. Elevated 4-hydroxynonenal levels in ventricular fluid in Alzheimer’s disease. Neurobiol Aging. 1997;18:457–461. doi: 10.1016/s0197-4580(97)00108-5. [DOI] [PubMed] [Google Scholar]
- 41.MacManus JP, Buchan AM, Hill IE, Rasquinha I, Preston E. Global ischemia can cause DNA fragmentation indicative of apoptosis in rat brain. Neurosci Lett. 1993;164:89–92. doi: 10.1016/0304-3940(93)90864-h. [DOI] [PubMed] [Google Scholar]
- 42.Maiese K, Boniece I, DeMeo D, Wagner J. Peptide growth factors protect against ischemia in culture by preventing nitric oxide toxicity. J Neurosci. 1993;13:3034–3040. doi: 10.1523/JNEUROSCI.13-07-03034.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maniatis T, Fritsch EF, Sambrook J. Molecular cloning: a laboratory manual. Cold Spring Harbor; New York: 1982. [Google Scholar]
- 44.Marchetti P, Hirsch T, Zamzami N, Castedo M, Decaudin D, Susin SA, Masse B, Kroemer G. Mitochondrial permeability transition triggers lymphocyte apoptosis. J Immunol. 1996;157:4830–4836. [PubMed] [Google Scholar]
- 45.Marini A, Paul SM. Induction of a neuroprotective state in cerebellar granule cells following activation of N-methyl-d-aspartate receptors. Ann NY Acad Sci. 1993;679:253–259. doi: 10.1111/j.1749-6632.1993.tb18305.x. [DOI] [PubMed] [Google Scholar]
- 46.Mark RJ, Blanc EM, Mattson MP. Amyloid β-peptide and oxidative cell injury in Alzheimer’s disease. Mol Neurobiol. 1996;12:211–224. doi: 10.1007/BF02755589. [DOI] [PubMed] [Google Scholar]
- 47.Mark RJ, Lovell MA, Markesberry WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal in disruption of ion homeostasis and neuronal death induced by amyloid β-peptide. J Neurochem. 1997a;68:255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- 48.Mark RJ, Pang Z, Geddes JW, Uchida K, Mattson MP. Amyloid β-peptide impairs glucose uptake in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. J Neurosci. 1997b;17:1046–1054. doi: 10.1523/JNEUROSCI.17-03-01046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mark RJ, Keller JN, Kruman I, Mattson MP. Basic FGF attenuates amyloid β-peptide-induced oxidative stress, mitochondrial dysfunction, and impairment of Na+/K+-ATPase activity in hippocampal neurons. Brain Res. 1997c;756:205–214. doi: 10.1016/s0006-8993(97)00196-0. [DOI] [PubMed] [Google Scholar]
- 50.Mattson MP, Barger SW, Begley JG, Mark RJ. Calcium, free radicals and excitotoxic neuronal death in primary cell culture. Methods Cell Biol. 1995;46:187–216. doi: 10.1016/s0091-679x(08)61930-5. [DOI] [PubMed] [Google Scholar]
- 51.Mattson MP, Furukawa K, Bruce AJ, Mark RJ, Blanc EM. Molecular mechanisms of dementia, pp 103–143. Humana; Totowa, NJ: 1996. Calcium homeostasis and free radical metabolism as convergence points in the pathophysiology of dementia. [Google Scholar]
- 52.Mattson MP, Goodman Y, Luo H, Fu W, Furukawa K. Activation of NF-κB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of Mn-SOD and suppression of peroxynitrite production and protein tyrosine nitration. J Neurosci Res. 1997;49:681–697. doi: 10.1002/(SICI)1097-4547(19970915)49:6<681::AID-JNR3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 53.Miura M, Yuan J. Regulation of programmed cell death by interleukin-1 β-converting enzyme family of proteases. Adv Exp Med Biol. 1996;389:165–172. doi: 10.1007/978-1-4613-0335-0_20. [DOI] [PubMed] [Google Scholar]
- 54.Montine KS, Olson SJ, Amarnath V, Whetsell WO, Graham DG, Montine TJ. Immunohistochemical detection of 4-hydroxy-2-nonenal adducts in Alzheimer’s disease is associated with inheritance of ApoE4. Am J Pathol. 1997;150:437–443. [PMC free article] [PubMed] [Google Scholar]
- 55.Musser DA, Oseroff AR. The use of tetrazolium salts to determine sites of damage to the mitochondrial electron transport chain in intact cells following in vitro photometric therapy with Photofrin 2. J Immunol. 1994;59:621–626. doi: 10.1111/j.1751-1097.1994.tb09666.x. [DOI] [PubMed] [Google Scholar]
- 56.Nelson SK, Wong GH, McCord JM. Leukemia inhibitory factor and tumor necrosis factor induce manganese superoxide dismutase and protect rabbit hearts from reperfusion injury. J Mol Cell Cardiol. 1995;27:223–229. doi: 10.1016/s0022-2828(08)80021-1. [DOI] [PubMed] [Google Scholar]
- 57.Nicotera P, Bonfoco E, Brune B. Mechanisms for nitric oxide-induced cell death: involvement of apoptosis. Adv Neuroimmunol. 1995;5:411–420. doi: 10.1016/0960-5428(95)00025-9. [DOI] [PubMed] [Google Scholar]
- 58.Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci. 1995;15:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petronilli V, Nicolli A, Costantini P, Colonna R, Bernardi P. Regulation of the permeability transition pore, a voltage-dependent mitochondrial channel inhibited by cyclosporin A. Biochim Biophys Acta. 1994;1187:255–259. doi: 10.1016/0005-2728(94)90122-8. [DOI] [PubMed] [Google Scholar]
- 60.Piantadosi CA, Zhang J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke. 1996;27:327–331. doi: 10.1161/01.str.27.2.327. [DOI] [PubMed] [Google Scholar]
- 61.Reers M, Smiley ST, Mottola-Hartshorn C, Chen A, Lin M, Chen LB. Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 1995;260:406–417. doi: 10.1016/0076-6879(95)60154-6. [DOI] [PubMed] [Google Scholar]
- 62.Sato N, Iwata S, Nakamura K, Hori T, Mori K, Yodoi J. Thiol-mediated redox regulation of apoptosis. Possible roles of cellular thiols other than glutathione in T cell apoptosis. J Immunol. 1995;154:3194–3203. [PubMed] [Google Scholar]
- 63.Scarlett JL, Packer MA, Porteous CM, Murphy MP. Alterations to glutathione and nicotinamide nucleotides during the mitochondrial permeability transition induced by peroxynitrite. Biochem Pharmacol. 1996;52:1047–1055. doi: 10.1016/0006-2952(96)99426-5. [DOI] [PubMed] [Google Scholar]
- 64.Schreck R, Albermann K, Baeuerle PA. Nuclear factor κB: an oxidative stress-responsive transcription factor of eukaryotic cells (a review). Free Radic Res Commun. 1991;17:221–237. doi: 10.3109/10715769209079515. [DOI] [PubMed] [Google Scholar]
- 65.Schwab C, Bondada V, Sparks DL, Cahan LD, Geddes JW. Postmortem changes in the levels and localization of microtubule-associated proteins (tau, MAP2 and MAP1B) in the rat and human hippocampus. Hippocampus. 1994;4:210–225. doi: 10.1002/hipo.450040212. [DOI] [PubMed] [Google Scholar]
- 66.Schwartz LM, Milligan CE. Cold thoughts of death: the role of ICE proteases in neuronal cell death. Trends Neurosci. 1996;10:555–563. doi: 10.1016/s0166-2236(96)10067-9. [DOI] [PubMed] [Google Scholar]
- 67.Shearman M, Hawthin S, Taylor V. The intracellular component of cellular 3-(4;5-dimethylthiazol-2-yl)-2:5-diphenyltetrazoliumj bromide (MTT) reduction is specifically inhibited by β-amyloid peptides. J Neurochem. 1995;65:218–227. doi: 10.1046/j.1471-4159.1995.65010218.x. [DOI] [PubMed] [Google Scholar]
- 68.Smith MA, Harris PLR, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Spitz DR, Oberley LW. An assay for superoxide dismutase activity in mammalian tissue homogenate. Anal Biochem. 1989;179:8–18. doi: 10.1016/0003-2697(89)90192-9. [DOI] [PubMed] [Google Scholar]
- 70.Springer JE, Azbill RD, Mark RJ, Begley JG, Waeg G, Mattson MP. 4-Hydroxynonenal, a lipid peroxidation product, rapidly accumulates following spinal cord injury and inhibits glutamate uptake. J Neurochem. 1997;68:2469–2476. doi: 10.1046/j.1471-4159.1997.68062469.x. [DOI] [PubMed] [Google Scholar]
- 71.St Clair DK, Oberley TD, Ho YS. Overproduction of human Mn- superoxide dismutase modulates paraquat-mediated toxicity in mammalian cells. FEBS Lett. 1991;293:199–203. doi: 10.1016/0014-5793(91)81186-c. [DOI] [PubMed] [Google Scholar]
- 72.Szabo C. DNA strand breakage and activation of poly-ADP ribosyltransferase: a cytotoxic pathway triggered by peroxynitrite. Free Radic Biol Med. 1996;21:855–869. doi: 10.1016/0891-5849(96)00170-0. [DOI] [PubMed] [Google Scholar]
- 73.Troy CM, Derossi D, Prochiantz A, Greene LA, Shelanski ML. Downregulation of Cu/Zn superoxide dismutase leads to cell death via the nitric oxide-peroxynitrite pathway. J Neurosci. 1996;16:253–261. doi: 10.1523/JNEUROSCI.16-01-00253.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Waeg G, Dimsity G, Esterbauer H. Monoclonal antibodies for detection of 4-hydroxynonenal modified proteins. Free Radic Res. 1996;25:149–159. doi: 10.3109/10715769609149920. [DOI] [PubMed] [Google Scholar]
- 75.Waring P, Beaver J. Cyclosporin A rescues thymocytes from apoptosis induced by very low concentrations of thapsigargin: effects on mitochondrial function. Exp Cell Res. 1996;227:264–276. doi: 10.1006/excr.1996.0276. [DOI] [PubMed] [Google Scholar]
- 76.Weisiger RA, Fridovich I. Superoxide dismutase organelle specificity. J Biol Chem. 1973;248:4793–4796. [PubMed] [Google Scholar]
- 77.White JR, Reynolds IJ. Mitochondrial depolarization in glutamate stimulated neurons: an early signal specific to excitotoxin exposure. J Neurosci. 1996;16:5688–5697. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wong GH, Goeddel DV. Induction of manganous superoxide dismutase by tumor necrosis factor: possible protective mechanism. Science. 1988;242:941–944. doi: 10.1126/science.3263703. [DOI] [PubMed] [Google Scholar]
- 79.Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H. Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke. 1994;25:165–170. doi: 10.1161/01.str.25.1.165. [DOI] [PubMed] [Google Scholar]
- 80.Yen HC, Oberley TD, Vichitbandha S, Ho YS, St Clair DK. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J Clin Invest. 1996;98:1253–1260. doi: 10.1172/JCI118909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci USA. 1996;93:2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zamzami N, Susin SA, Marchetti P, Hirsch T, Castedo M, Kroemer G. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996a;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zamzami N, Marchetti P, Castedo M, Hirsch T, Susin SA, Masse B, Kroemer G. Inhibitors of permeability transition interfere with the disruption of the mitochondrial transmembrane potential during apoptosis. FEBS Lett. 1996b;384:53–57. doi: 10.1016/0014-5793(96)00280-3. [DOI] [PubMed] [Google Scholar]
- 84.Zhang Y, Tatsuno T, Carney J, Mattson MP. Basic FGF, NGF, and IGFs protect hippocampal neurons against iron-induced degeneration. J Cereb Blood Flow Metab. 1993;13:378–388. doi: 10.1038/jcbfm.1993.51. [DOI] [PubMed] [Google Scholar]