

Abstract
The vertebrate myelin sheath is greatly enriched in the galactolipids galactocerebroside (GalC) and sulfatide. Mice with a disruption in the gene that encodes the biosynthetic enzyme UDP-galactose:ceramide galactosyl transferase (CGT) are incapable of synthesizing these lipids yet form myelin sheaths that exhibit major and minor dense lines with spacing comparable to controls. These CGT mutant mice exhibit a severe tremor that is accompanied by hindlimb paralysis. Furthermore, electrophysiological studies reveal nerve conduction deficits in the spinal cord of these mutants. Here, using electron microscopic techniques, we demonstrate ultrastructural myelin abnormalities in the CNS that are consistent with the electrophysiological deficits. These abnormalities include altered nodal lengths, an abundance of heminodes, an absence of transverse bands, and the presence of reversed lateral loops. In contrast to the CNS, no ultrastructural abnormalities and only modest electrophysiological deficits were observed in the peripheral nervous system. Taken together, the data presented here indicate that GalC and sulfatide are essential in proper CNS node and paranode formation and that these lipids are important in ensuring proper axo-oligodendrocyte interactions.
Keywords: node of Ranvier, galactocerebroside, sulfatide, myelin, UDP galactose:ceramide galactosyl transferase, axo-glial interaction
Processes originating from oligodendrocytes in the CNS and Schwann cells in the peripheral nervous system (PNS) align along and spiral around axons forming myelin segments known as internodes, which are divided by regularly spaced unmyelinated regions, the nodes of Ranvier (Morell et al., 1994). The paranodal region is the portion of the myelin segment adjacent to the node where the lamellae open along the major dense line, forming cytoplasmic pockets known as lateral loops. These loops turn toward and terminate on the axon with the outermost lateral loop terminating closest to the node. Furthermore, regularly arrayed densities traverse the periaxonal space between the lateral loops and the axolemma. These structures are known as transverse bands (TBs) and are common features of the axo-oligodendrocytic junctional complex (Peters et al., 1976). Working together, these myelin structures play an integral role in dramatically enhancing conduction velocity (Salzer, 1997).
Our understanding of myelin has been dramatically facilitated by the availability of mutant models in which proteins of the myelin sheath are affected (Griffiths, 1996). More recently, mouse models have been generated that target myelin lipids, which constitute >70% of the dry weight of myelin. Bosio et al. (1996) and Coetzee et al. (1996) have generated mice with a disruption in the gene that encodes UDP-galactose:ceramide galactosyl transferase (CGT), the enzyme that catalyzes an essential step in the biosynthetic pathway of the two major myelin galactolipids galactocerebroside (GalC) (Morell and Radin, 1969) and its sulfated derivative sulfatide (Fleischer and Zambrano, 1974).
Although slightly thinner in the white matter tracts of the spinal cord, myelin in young CGT-deficient mice appears normal (Coetzee et al., 1996). The mutant animals, however, have a profound tremor as early as 2 weeks of age, and electrophysiological analysis reveal conduction deficits. By 6 weeks of age the sheaths exhibit extensive regional vacuolar degeneration in the ventral columns of the spinal cord, and the animals begin to display hindlimb weakness. Treatment of spinal cords from young animals, before demyelination, with 4-aminopyridine (4-AP), a blocker of potassium channels located in the paranodal region, partially restores the amplitude and duration of the compound action potential in the GalC-deficient mice (Coetzee et al., 1996). Ordinarily, 4-AP has no effect on action potentials of myelinated fibers, because these potassium channels, which are occluded by the myelin sheath, are not accessible to the inhibitor. Thus, the findings suggest that these channels are abnormally exposed to the extracellular milieu, perhaps because of paranodal abnormalities. Therefore, we examined nodal and paranodal structures in the CGT-deficient animals. Whereas the ultrastructure of the PNS was unaffected, we found several nodal and paranodal defects in the CNS, in the absence of apparent oligodendrocyte pathology, that correlate with the physiological abnormalities. Taken together, our findings indicate that myelin galactolipids are essential in establishing axo-oligodendrocytic interactions that ensure proper nodal and paranodal formation.
MATERIALS AND METHODS
Histological analysis. Thirty- and 45-d-old CGT-deficient mice were perfused intracardially through the left ventricle with an ice-cold solution of 4% paraformaldehyde and 2.5% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4. The perfusions were followed by a 24 hr immersion fixation at 4°C in the same solution. Tissue samples from both midbrain and hindbrain, cervical (C3) spinal cord, and sciatic nerve, 0.5 cm distal to the sciatic notch, were harvested, embedded in both cross and longitudinal orientations, and processed for electron microscopic analysis.
The prevalence of abnormal nodes of Ranvier in the ventral columns of the spinal cord was quantitatively assessed. The number of nodes was determined per field, defined as a single square from a 200 mesh grid (Electron Microscopy Sciences, Ft. Washington, PA, catalog #G200Cu Gilder). Counts were restricted to fields that were completely filled with longitudinally oriented processes. Because older animals exhibit extensive myelin vacuolar degeneration, only 30-d-old animals were quantitatively analyzed, therefore ensuring that the presence of the abnormal structures was not attributable to the processes of demyelination and remyelination. For the quantitative studies, four CGT-deficient mice and three littermate controls were used, and typically 8–10 fields per animal were counted. Values are presented as mean ± SE, and all counts were statistically evaluated byt tests.
Expression of myelin genes. Total RNA was isolated from the brains of CGT mutant and wild-type animals using Trizol according to the manufacturer’s specifications (Life Technologies, Grand Island, NY). RNAs were separated on 0.8% formaldehyde–agarose gels, transferred to nylon, and probed as described previously (Coetzee et al., 1996). Total protein extracts were isolated by homogenization of brains in 1% SDS for 10 sec using a Polytron homogenizer. The protein extract was then boiled for 10 min and insoluble material removed by centrifugation. Myelin was extracted as described previously (Coetzee et al., 1996). After mild delipidation, myelin proteins were resuspended in 1% SDS. Total brain and myelin proteins were separated on 12.5% polyacrylamide gels, transferred to nitrocellulose, and probed with antibodies to myelin basic protein (MBP) (SMI-99; Sternberger Monoclonals, Inc., Baltimore, MD) or proteolipid protein (PLP) (kindly provided by Dr. Joyce Benjamins, Wayne State University, Detroit, MI) (Benjamins et al., 1994). Antibodies were detected by chemiluminescence following the instructions of the manufacturer (Boehringer Mannheim, Indianapolis, IN).
Lipid analysis. Thin-layer chromatography was used to determine myelin lipid content in the PNS. Total lipids were extracted and pooled from 10 sciatic nerves from 30-d-old animals. Lipids were extracted and analyzed as described previously (Coetzee et al., 1996).
Electrophysiological analysis. Conduction properties of myelinated axons were examined in acutely isolated sciatic nerves from 60-d-old CGT-deficient and wild-type mice. Recordings were made with two different sucrose gap recording arrangements. The first, identical to that described by Coetzee et al. (1996), was used to examine compound potential amplitude and the response to 4-AP, for direct comparison with the findings in spinal cord axons in the earlier study. The second technique used two sets of bipolar silver–silver chloride wire hook electrodes to stimulate the nerve at two points, 5.5 mm apart. The difference in conduction delay of the compound action potential from stimulation at these two points was then used to calculate conduction speed in the usual manner. These two techniques provided stable recording and stimulating conditions over the course of the 1–2 hr experiments.
The animals were anesthetized with an intraperitoneal injection of ketamine HCl (60 mg/kg), xylazine (10 mg/kg), and acepromazine maleate (0.6 mg/kg). They were then decapitated, and the sciatic nerves on both sides were removed over the length from the knee to the exit from the spine. The isolated nerves were incubated in oxygenated Krebs’ solution (in mm, NaCl 124, KCl 2, KH2PO4 1.2, MgSO4 1.3, CaCl 1.2, dextrose 10, NaHCO3 26, and sodium ascorbate 6, equilibrated with 95% oxygen and 5% carbon dioxide). After a period of at least 1 hr, the nerve was removed to the recording chamber and mounted in the sucrose gap. The main part of the nerve was superfused continuously with oxygenated Krebs’ solution at a rate of 2 ml/min. 4-AP (Regis Technologies, Morton Grove, IL) was applied to the nerve in a concentration of 0.1 mm, dissolved in Krebs’ solution, with pH adjusted to 7.3, as necessary. The temperature of the chamber was controlled with a Peltier unit, and recordings were made at 37°C. Data were recorded using a Neurodata Instruments bridge amplifier, digitized, and recorded on videotape for subsequent analysis using Labview software on a Macintosh computer.
RESULTS
CGT-deficient mice form compact myelin sheaths that exhibit normal periodicity up to 1 month of age, followed by the onset of vacuolar degeneration of the CNS sheaths by 1.5 months of age. With this in mind, 30- and 45-d-old animals were analyzed, and similar ultrastructural abnormalities were observed at both time points. The major difference between the two time points was that demyelination was not yet detected by 30 d of age. Therefore, the majority of the data presented is from 30-d-old mice, thus increasing the likelihood that all structural anomalies in the CGT-deficient animals were formed during the initial process of myelination and not by demyelination or during remyelination.
Abnormal node and paranode formation in the CNS
Because the electrophysiological deficits in the spinal cord of the CGT−/− mice were suggestive of nodal and paranodal abnormalities (Coetzee et al., 1996), we used electron microscopic techniques to analyze longitudinal sections of these regions. Our results revealed a significantly higher incidence of aberrantly formed nodal and paranodal structures. In the CGT-deficient animals, 3.10 ± 0.04 abnormal nodes and paranodes per field were observed compared with 0.29 ± 0.03 per field in the littermate controls (p < 0.0001). Typically, nodes were longer in the mutant mice than in the controls (Fig. 1, compareA,B), and heminodes, paranodes juxtaposed to extended unmyelinated axonal regions, were present (Fig. 1C). Adjacent myelin sheaths with overlapping paranodal regions that completely occluded the formation of the node were also occasionally observed (Fig.1D). Moreover, in the mutant mice, the periaxonal space between the lateral loops and the axolemma lacked TBs (Fig.2A,B). Furthermore, paranodal regions were frequently configured in a reversed orientation with the lateral loops facing away from the axon and with the outermost lamella terminating farthest from the node (Fig. 2C,D). Finally, astrocytic processes, which were identified by the presence of bundles of 10 nm filaments, presumptive glial fibrillary acidic protein, and glycogen particles, were observed in the periaxonal space of <5% of the myelinated axons (Fig.3).
Fig. 1.
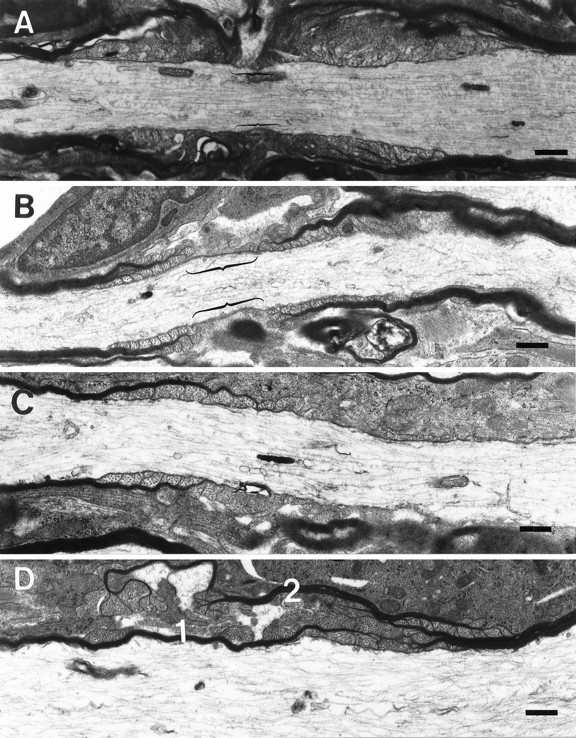
Nodal and paranodal abnormalities in the CGT-deficient mice. Myelinated processes from the cervical region of the spinal cord of 30-d-old CGT-deficient mice demonstrating ultrastructurally normal (A) and abnormal nodes of Ranvier (B–D). B, An aberrantly formed node with lateral loops of the outermost lamellae forming farther from the node than the more medial lamellae, and these loops are facing away from the axon. Furthermore, the length of the nodes (bracketed) in the CGT-deficient mice is typically longer than in the age-matched wild types (bracketed inA). C, The most commonly observed aberrant nodal structure was the heminode, a myelinated segment adjacent to a nonmyelinated region of the axon. In addition, heminodes are the extreme example of node elongation. D, Less frequently observed was the overlapping of paranodal regions. Two myelin sheaths (1, 2) failed to form a node of Ranvier, because the regions of lateral loop formation overlap, thus excluding the nodal region. Scale bar, 1.0 μm.
Fig. 2.
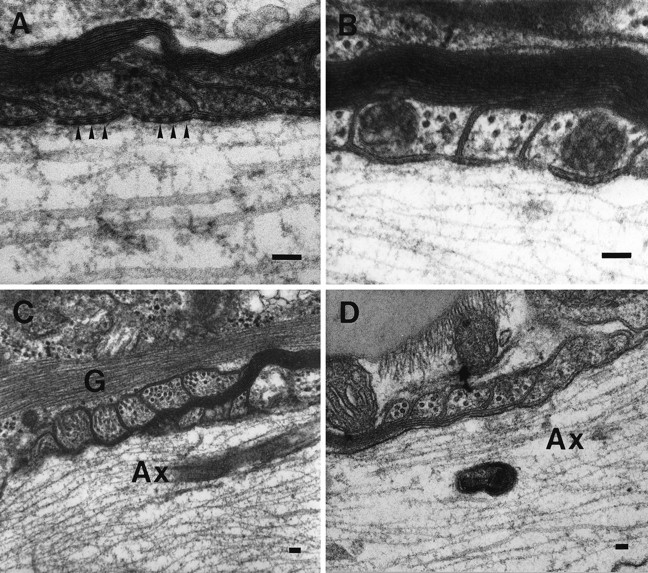
High magnification of aberrant paranodal structures in the CGT mutant animals. A, A paranodal region from a 30-d-old littermate control displays a periaxonal space with regularly arrayed densities known as transverse bands (arrowheads). These structures are prominent features of the junctional complex normally formed between the axolemma and the myelin sheath. B, Transverse bands were not found in age-matched CGT-deficient mice. Lateral loops in both the spinal cord (C) and brain (D) frequently are reversed and face away from the axon. Furthermore, the outer most lamella terminates farther from the node. Ax, Axon; G, glial fibrillary acid protein. Scale bar, 0.1 μm.
Fig. 3.
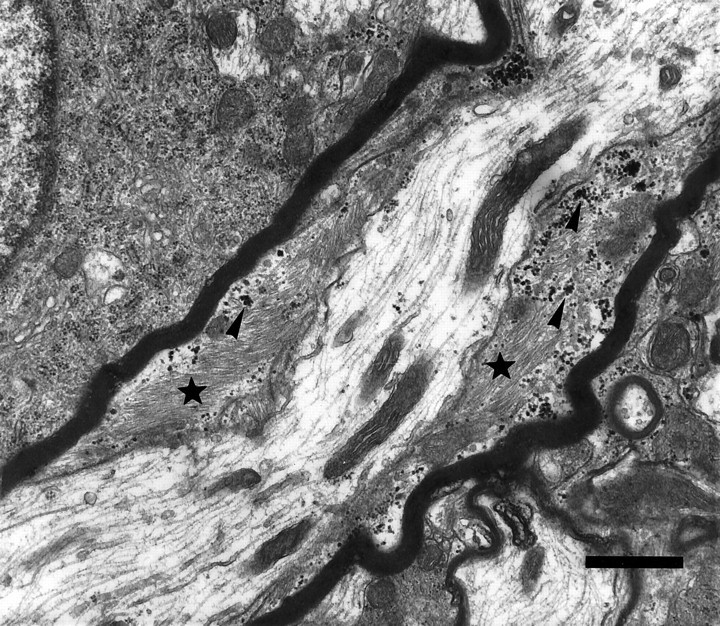
Astrocytic intrusion between the myelin sheath and the axon. Myelinated process from the spinal cord of a CGT mutant animal demonstrating the intrusion of an astrocytic process, as identified by the presence of glycogen particles (arrowheads) and bundles of filaments (stars), between the myelin sheath and the axolemma. Scale bar, 1.0 μm.
Assessment of oligodendrocyte function
Because abnormal myelin structures have been attributed to oligodendrocytic pathology (Dentinger et al., 1982; Duncan et al., 1983), we used a combination of ultrastructural and biochemical parameters to assess the function of these cells. The oligodendrocytes in the CGT-deficient mice appeared ultrastructurally normal with respect to membrane integrity and organelle structure and produced myelin sheaths that exhibit major and minor dense line spacing comparable to age-matched littermate controls (Coetzee et al., 1996; data not shown). In addition, we found that expression of MBP and PLP mRNAs were unaffected in the mutant mice relative to the littermate controls at 30 and 45 d of age (Fig.4A). Moreover, the levels of MBP and PLP protein were not significantly different between the mutants and the controls at these ages in either the total brain extracts or in myelin fractions (Fig. 4B), indicating that normal numbers of myelinating oligodendrocytes are present in the mutant animals at both time points.
Fig. 4.

Northern and Western blot analyses of myelin proteins. A, Message levels of MBP and PLP are similar between CGT-deficient mice and littermate controls at 30 and 45 d of age. B, Analysis of both total brain and isolated myelin revealed no difference in the levels of MBP and PLP at 30 and 45 d of age.
Analysis of PNS structure and function
The alterations in both the structure and function of the myelin in CNS prompted the investigation of PNS myelin. Thin-layer chromatography of the sciatic nerve demonstrated no difference in the pattern of the myelin lipid profiles between the PNS and that reported for the CNS (Coetzee et al., 1996). PNS myelin from the CGT-deficient mice lacked both normal fatty acid (NFA)- and hydroxy fatty acid (HFA)-containing GalC and sulfatide and revealed bands with mobility patterns consistent with HFA–glucocerebroside and HFA–sphingomyelin (Fig. 5).
Fig. 5.

Lipid analysis of the PNS. Both NFA- and HFA-containing GalC and sulfatide were absent in the sciatic nerves of the CGT−/− mouse. Bands with mobility patterns consistent with HFA–glucocerebroside and sphingomyelin were prominent in the mutant but not in the wild-type. Chol, Cholesterol;HFA-GalC, hydroxy fatty acid galactocerebroside;NFA-GalC, normal fatty acid galactocerebroside;HFA-GlcC, hydroxyglucocerebroside; PE, phosphatidyl ethanolamine; PC, phosphatidyl choline;SPM, sphingomyelin; HFA-SPM, hydroxy fatty acid sphingomyelin.
Given the CNS nodal and paranodal abnormalities, we examined these regions in the PNS of the CGT−/− mice. Sciatic nerves from 30-, 45-, and 90-d-old animals did not exhibit vacuolar degeneration (Fig.6A,B). In addition, heminodes were not present; lateral loops were not reversed; and node length was not altered (Fig. 6C,D).
Fig. 6.
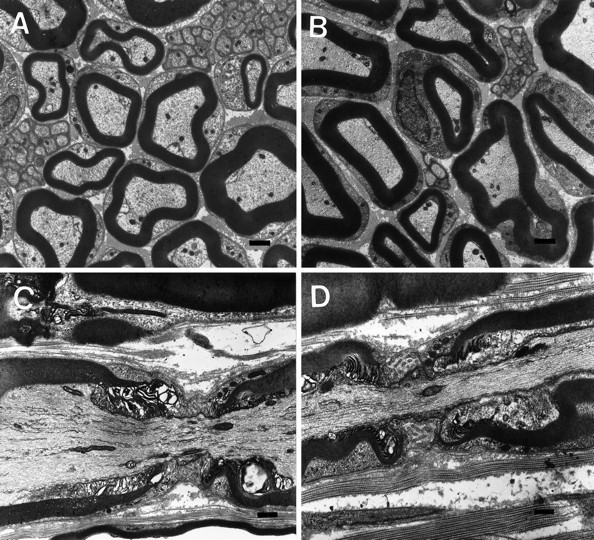
Structural analysis of the PNS. Sciatic nerve myelin sheaths revealed no differences between CGT+/+ (A, C) and CGT−/− (B, D) mice with respect to thickness (A, B) or node formation (C, D). Scale bar, 1.0 μm.
Electrophysiological parameters indicated some alteration in the function of the PNS in the CGT-deficient mice (Table1). As depicted in Figure7 and Table 1, CGT−/− animals demonstrated ∼50% reduction in compound action potential amplitude and 30–40% reduction in conduction velocity. Furthermore, a slight increase in the responsiveness to blockade of the fast potassium channels with 4-AP (Fig. 7D) was observed; however, this effect was not statistically significant as assessed by ANOVA. None of these effects was as dramatic as those seen in the CNS (Coetzee et al., 1996).
Table 1.
Characteristics of compound action potentials from the sciatic nerve of CGT+/+ and CGT−/− mice
| +/+ | −/− | |
|---|---|---|
| Amplitude at 37°C | 33.6 ± 5.5 mV | 15.9 ± 2.9 mV* |
| Conduction speed at 37°C | 32.3 ± 2.7 m/sec | 22.9 ± 2.2 m/sec* |
| Response to 4-AP (% amplitude increase) | 2.0 ± 1.7 | 3.1 ± 2.7 |
Compound action potentials were recorded from acutely isolated sciatic nerves from CGT+/+ (n = 3) and CGT−/− (n = 4) animals. Mean values ± SE are reported.
*Significant difference between values (p < 0.05, ANOVA).
Fig. 7.

Electrophysiological analysis of the PNS.A, Diagram of the recording apparatus used for measurement of conduction velocity. The isolated nerve was placed in a Plexigal chamber and superfused with oxygenated Krebs’ solution, and one end was isolated by a flowing sucrose gap in a separate chamber slowly perfused with isotonic KCl solution (120 mm). Silver–silver electrodes were used to record the potential across the gap and to stimulate the nerve at two points, 5.5 mm apart in the main chamber. An example of compound potentials recorded in response to stimulation of the nerve at proximal and distal electrode pairs is illustrated in B. The conduction time for the length of nerve between the stimulating electrodes was derived from the time between the compound potential peaks in each recording.C, Three recordings are superimposed showing the effect of superfusing a nerve from a CGT+/+ mouse with Krebs’ solution containing 100 μm 4-AP. There was a slight increase in amplitude of the response and the depolarizing afterpotential. The compound action potential increase reversed more readily with 10 min of washing in normal Krebs’ solution. D, Recordings, similar to those in C, show a slightly more pronounced effect of 4-AP on the amplitude of the compound potential and depolarizing afterpotential in a nerve from a CGT−/− mouse.
DISCUSSION
We have examined the structure of the paranode and the node of Ranvier in mice incapable of synthesizing the myelin galactolipids GalC and sulfatide. Whereas nodal structure was unaffected in the PNS, several abnormalities were observed in the CNS. Nodal length was frequently increased; TBs were absent; and paranodal orientation was reversed. In addition, heminodes were abundant. One explanation for the abundance of heminodes is the occurrence of segmental demyelination. Whereas this possibility cannot be disregarded, cross sectional examination of the spinal cords from 30-d-old mice revealed minimal signs of demyelination. Although the mechanism for proper formation of nodal and paranodal structures is unknown, the data indicate that GalC and sulfatide are essential in this process and abnormalities in these regions are the result of disruptions in axo-oligodendrocytic interactions. These CNS myelin anomalies, however, occurred in the absence of detectable alterations in oligodendrocyte form or function. In contrast, despite the absence of the myelin galactolipids, the PNS appeared structurally normal, and the electrophysiological studies revealed only modest deficits.
Because the proper formation of a node of Ranvier is likely dependent on communications between the axon and myelinating cells, one explanation for the abnormal formation of nodal and paranodal structures is the disruption of these cell–cell interactions. Such a disruption could lead to misalignment of the oligodendrocytic processes along the axon resulting in increased nodal lengths and the formation of heminodes. Furthermore, the presence of reversed lateral loops and the improper order of lateral loop termination also suggest a loss of cell–cell communication. Finally, the presence of myelin sheaths that appear to overlap and occlude node formation is further evidence of oligodendrocytic process misalignment during myelinogenesis. Taken together, these data indicate that the abnormal nodal formations are the result of a disruption in axo-oligodendrocytic interactions that are dependent on the presence of functional myelin galactolipids.
Although similar ultrastructural myelin abnormalities have been reported in the CNS of myelin-deficient rat (Rosenbluth, 1987) and shaking pup (Griffiths et al., 1981), these models are the products of a point mutation in the PLP gene that results in oligodendrocyte cell death (Dentinger et al., 1982; Duncan et al., 1983), apparently because of the accumulation of the mutant protein in the endoplasmic reticulum (Gow et al., 1994). In contrast, our data provide no evidence for the loss of oligodendrocytic viability in the CGT-deficient mice. The oligodendrocytes appear ultrastructurally normal, form abundant amounts of myelin with correct periodicity, and express normal levels of MBP and PLP mRNAs and protein. Therefore, the loss of CGT function and not oligodendrocyte death is the likely cause of the abnormal formation of nodal and paranodal structures in these animals.
The absence of TBs, prominent features of the axo-oligodendrocytic junctional complex in the periaxonal space of the paranode, in the CNS of the CGT mutant animals strongly supports a role for the myelin galactolipids in establishing cell–cell interactions. Rosenbluth (1987) suggested that in the absence of TBs, the lateral loops are not tightly opposed to the axon, exposing the paranodal axolemma to the extracellular milieu. Therefore, the lack of formation of TB in the CGT−/− mice likely explains our previous findings with 4-AP, an inhibitor of potassium channels positioned in the paranodal region of the axolemma. Because these channels are normally occluded by the myelin sheath, treatment of control animals with the channel blocker had little or no effect on the action potential. In the CNS of the CGT-deficient mice, however, 4-AP treatment partially restored the compound action potential amplitudes (Coetzee et al., 1996), indicating that the inhibitor is capable of gaining access to the channels via a compromised axo-oligodendrocytic junction. Moreover, further evidence for a disruption in this axo-glial junction is provided by the intrusion of astrocytic processes into the periaxonal space between the myelin sheath and the axolemma. In the presence of normal axo-oligodendrocytic junctions, astrocytes are unable to penetrate the paranodal region. In the case of the CGT-deficient mice, astrocytic penetration into the periaxonal space of the paranodal region indicates that the myelin galactolipids are essential in the formation of TBs, and thus in establishing proper axo-oligodendrocytic interactions.
The CNS myelin abnormalities also provide a structural correlate for the electrophysiological deficits previously observed (Coetzee et al., 1996). The frequency of abnormal node formation, particularly the prevalence of heminodes, likely accounts for the conduction deficits in the spinal cord of the CGT mutant animals. With regard to the heminodes, long stretches of unmyelinated axons would inhibit the efficient spreading of the depolarization down the axon, and conduction would be compromised. Moreover, the lateral loops are frequently reversed and not in contact with the axolemma. Such aberrant structures are consistent with the 4-AP studies (Coetzee et al., 1996), which indicate current leakage into the paranodal region resulting in the reduction of depolarization efficiency. In addition, the absence of TBs may alter saltatory conduction because these structures have been implicated in retarding abnormal ion fluxes and maintaining proper distribution of sodium and potassium channels (Salzer, 1997).
Although our data implicate GalC and/or sulfatide in mediating cell–cell interactions, the mechanism by which this is accomplished is unknown. Several studies, however, have indicated that galactolipids have functional properties compatible with cell–cell interactions. First, a number of proteins with adhesive properties bind sulfatidein vitro (Vos et al., 1994), and one of these, tenascin-R, facilitates adhesion of oligodendrocytes through a sulfatide-mediated mechanism (Pesheva et al., 1997). Furthermore, because some CNS neurons express tenascin-R (Fuss et al., 1993; Wintergerst et al., 1993), the interaction of this protein with sulfatide may mediate communications between the axon and the myelinating cell. Moreover, a sulfatide antibody specifically labeled nodes of Ranvier in the rabbit (Nardelli et al., 1995), indicating that sulfatide may be important in the structure and function of this region. Furthermore, implantation of anti-GalC-producing hybridomal cells inhibited tight junction formation between lateral loops (Rosenbluth et al., 1995), and sulfatide has been shown to be a prominent constituent of myelin tight junctions known as the radial component (Karthigasan et al., 1994). In addition, galactolipids may be critical in trafficking cell adhesion proteins to the myelin sheath (Kramer et al., 1997). Taken together, it is tantalizing to suggest a role for GalC/sulfatide in mediating the linkage between the axon and the oligodendrocyte at the nodal and paranodal regions and that a disruption in this linkage could yield the structural abnormalities observed in the CGT-deficient mice.
In contrast to the CNS, the PNS showed no structural deficit. Nevertheless, some functional impairment was observed. It is important to note, however, that in wild-type animals, galactolipid content in the PNS is lower than in the CNS (Norton and Cammer, 1984). Whether this difference can account for the heterogeneity in the tolerance of GalC and sulfatide elimination between the Schwann cells and the oligodendrocytes is unclear. The reduction in amplitude and conduction velocity in the PNS may be attributed to a combination of factors, including an overall reduction in tissue size in the mutant (data not shown), slight (undetectable) ultrastructural abnormalities, or changes in the lipid composition and electrical characteristics of myelin. The changes in physiology would be consistent with a lowered resistivity of the myelin sheath. Furthermore, 4-AP had only a slight effect, indicating a fairly tight paranodal junction.
Collectively, the nodal abnormalities also provide an explanation for the vacuolar degeneration that occurs in the CGT−/− mice after 6 weeks of age (Coetzee et al., 1996). A prominent feature of this degenerative process is separation of lamellae along the intraperiod line. Most likely this is the result of penetration of fluid between the lateral loops. The absence of TBs and the inversion of lateral loops provide a means for fluid seepage between the lamellae. Indeed, the ability of 4-AP to penetrate the periaxonal space provides evidence for leakage into the paranodal region. Abnormal entrance of fluid, as indicated by the large vacuoles associated with the demyelinated axons, into the periaxonal space may initiate the vacuolar degeneration in older animals (Coetzee et al., 1996). In support of this possibility, the PNS of the CGT mutant animals does not show nodal abnormalities or vacuolar degeneration of myelin in older animals.
In summary, we demonstrate the presence of abnormal nodal and paranodal structures in the CNS, but not in the PNS, of mice that lack the myelin galactolipids GalC and sulfatide. These defects include altered node length, high incidence of heminodes, absence of TBs, and reversed lateral loops. These findings indicate that GalC and sulfatide mediate interactions between the axon and oligodendrocyte that are essential for proper CNS node and paranode formation.
Footnotes
This work was supported by Grants NS27336 (B.P.), NS24453 (K.S.), and NS33687 (A.B.) from the National Institutes of Health and Mental Retardation Research Center Core Grant HD03110 from the National Institutes of Health. J.D. was supported by Training Grant HD07201 from the National Institutes of Health and an Advanced Postdoctoral Fellowship from the National Multiple Sclerosis Society. T.C. is a recipient of an Advanced Postdoctoral Fellowship from the National Multiple Sclerosis Society. B.P. is the recipient of Research Career Development Award NS01637 from the National Institutes of Health. We thank Clarita Langaman and Tom Kelly for their superb technical assistance with the electron microscopic and electrophysiological analysis, respectively. We also thank Dr. Jack Rosenbluth for his insightful critique of the electron micrographs and Dr. Kunihiko Suzuki for his assistance in the interpretation of the lipid analysis.
Correspondence should be addressed to Dr. Brian Popko, Neuroscience Center, CB 7250, University of North Carolina, Chapel Hill, NC 27599-7250.
REFERENCES
- 1.Benjamins JA, Studzinski DM, Skoff RP. Analysis of myelin proteolipid protein and F0 ATPase subunit 9 in normal and jimpy CNS. Neurochem Res. 1994;19:1013–1022. doi: 10.1007/BF00968711. [DOI] [PubMed] [Google Scholar]
- 2.Bosio A, Binczek E, Stoffel W. Functional breakdown of the lipid bilayer of the myelin membrane in central and peripheral nervous system by disrupted galactocerebroside synthesis. Proc Natl Acad Sci USA. 1996;93:13280–13285. doi: 10.1073/pnas.93.23.13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, Popko B. Myelination in the absence of galactocerebroside and sulfatide: normal structure and abnormal function and regional instability. Cell. 1996;86:209–219. doi: 10.1016/s0092-8674(00)80093-8. [DOI] [PubMed] [Google Scholar]
- 4.Dentinger MP, Barron KD, Csiza CK. Ultrastructure of the central nervous system in a myelin deficient rat. J Neurocytol. 1982;11:671–691. doi: 10.1007/BF01262431. [DOI] [PubMed] [Google Scholar]
- 5.Duncan ID, Griffiths IR, Munz M. “Shaking pup”: a disorder of central myelination in the spaniel dog. III. Quantitative aspects of the glia and myelin in the spinal cord and optic nerve. Neuropathol Appl Neurobiol. 1983;9:355–368. doi: 10.1111/j.1365-2990.1983.tb00121.x. [DOI] [PubMed] [Google Scholar]
- 6.Fleischer B, Zambrano F. Golgi apparatus of rat kidney: preparation and role in sulfatide formation. J Biol Chem. 1974;249:5995–6003. [PubMed] [Google Scholar]
- 7.Fuss B, Wintergerst ES, Bartsch U, Schachner M. Molecular characterization and in situ messenger RNA localization of the neural recognition molecule J1–160/180-a modular structure similar to tenascin. J Cell Biol. 1993;120:1237–1249. doi: 10.1083/jcb.120.5.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gow A, Friedrich VL, Jr, Lazzarini RA. Many naturally occurring mutations of myelin proteolipid protein impair its intracellular transport. J Neurosci Res. 1994;37:574–583. doi: 10.1002/jnr.490370504. [DOI] [PubMed] [Google Scholar]
- 9.Griffiths IR. Myelin mutants: model systems for the study of normal and abnormal myelination. Bioassays. 1996;18:189–197. doi: 10.1002/bies.950181005. [DOI] [PubMed] [Google Scholar]
- 10.Griffiths IR, Duncan ID, McCulloch M, Harvey MJA. Shaking pups: a disorder of central myelination in the spaniel dog. J Neurol Sci. 1981;50:423–433. doi: 10.1016/0022-510x(81)90154-4. [DOI] [PubMed] [Google Scholar]
- 11.Karthigasan J, Kosaras B, Nguyen J, Kirschner DA. Protein and lipid composition of radial component-enriched CNS myelin. J Neurochem. 1994;62:1203–1213. doi: 10.1046/j.1471-4159.1994.62031203.x. [DOI] [PubMed] [Google Scholar]
- 12.Kramer E, Koch T, Niehaus A, Trotter J. Oligodendrocytes direct glycosyl phosphatidylinositol-anchored proteins to the myelin sheath in glycosphingolipid-rich complexes. J Biol Chem. 1997;272:8937–8945. doi: 10.1074/jbc.272.14.8937. [DOI] [PubMed] [Google Scholar]
- 13.Morell P, Radin NS. Synthesis of cerebroside by brain from uridine diphosphate galactose and ceramide containing hydroxy fatty acid. Biochemistry. 1969;8:506–512. doi: 10.1021/bi00830a008. [DOI] [PubMed] [Google Scholar]
- 14.Morell P, Quarles RH, Norton WT. Myelin formation, structure and biochemistry. In: Siegel GJ, Agranoff BW, Albers RW, Molinoff, editors. Basic neurochemistry: molecular, cellular, and medical aspects. Raven; New York: 1994. pp. 117–143. [Google Scholar]
- 15.Nardelli E, Bassi A, Mazzi G, Anzini P, Rizzuto N. Systemic passive transfer studies using IgM monoclonal antibodies to sulfatide. J Neuroimmunol. 1995;63:29–37. doi: 10.1016/0165-5728(95)00125-5. [DOI] [PubMed] [Google Scholar]
- 16.Norton WT, Cammer W. Isolation and characterization of myelin. In: Morell P, editor. Myelin. Plenum; New York: 1984. pp. 147–196. [Google Scholar]
- 17.Pesheva P, Gloor S, Schachner M, Probtmeier R. Tenascin-R is an intrinsic autocrine factor for oligodendrocyte differentiation and promotes cell adhesion by a sulfatide-mediated mechanism. J Neurosci. 1997;17:4642–4651. doi: 10.1523/JNEUROSCI.17-12-04642.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peters A, Palay SL, Webster HdeF. The fine structure of the nervous system: the neurons and supporting cells. Saunders; Philadelphia: 1976. [Google Scholar]
- 19.Rosenbluth J. Abnormal axoglial junctions in the myelin-deficient rat. J Neurocytol. 1987;16:497–509. doi: 10.1007/BF01668504. [DOI] [PubMed] [Google Scholar]
- 20.Rosenbluth J, Liang W-L, Liu Z, Guo D, Schiff R. Paranodal structural abnormalities in rat CNS myelin developing in vitro in the presence of implanted 01 hybridoma cell. J Neurocytol. 1995;24:818–824. doi: 10.1007/BF01179981. [DOI] [PubMed] [Google Scholar]
- 21.Salzer JL. Clustering sodium channels at the node of Ranvier: close encounters of the axon-glia kind. Neuron. 1997;18:843–846. doi: 10.1016/s0896-6273(00)80323-2. [DOI] [PubMed] [Google Scholar]
- 22.Vos JP, Lopes-Cardozo M, Gadella BM. Metabolic and functional aspects of sulfogalactolipids. Biochim Biophy Acta. 1994;1211:125–149. doi: 10.1016/0005-2760(94)90262-3. [DOI] [PubMed] [Google Scholar]
- 23.Wintergerst ES, Fuss B, Bartsch U. Localization of janusin mRNA in the central nervous system of the developing and adult mouse. Eur J Neurosci. 1993;5:299–309. doi: 10.1111/j.1460-9568.1993.tb00497.x. [DOI] [PubMed] [Google Scholar]