

Abstract
Selected clones of the sympathetic precursor-like cell line PC12 (rCl8) are resistant to oxidative cell death induced by the Alzheimer’s disease-associated amyloid β protein (Aβ) and hydrogen peroxide (H2O2). Here, we show that the transcriptional activity and DNA binding activity of the redox-sensitive transcription factor NF-κB and its nuclear expression are constitutively increased in rCl8 cells compared with their nonresistant parental PC12 cell (PC12p) counterpart. Suppression of the transcriptional activity of NF-κB in rCl8 cells with the synthetic glucocorticoid dexamethasone or by direct overexpression of a super-repressor mutant form of IκBα, a specific inhibitor of NF-κB, reversed the oxidative stress resistance phenotype of these cells and ultimately led to increased cell death after the challenge with H2O2. Dexamethasone treatment also caused an increase in the protein level of IκBα. Our data show that an increased baseline of NF-κB activity may mediate the resistance of these cells of neuronal origin to oxidative stress. Therefore, the presented model may help to identify possible neuronal target genes of NF-κB and to further elucidate the molecular basis of the differential sensitivity of neurons in neurodegenerative conditions associated with an increased oxidative burden, such as in Alzheimer’s disease.
Keywords: NF-κB, Alzheimer’s disease, amyloid β protein, oxidative stress, antioxidant enzymes, glucocorticoids, neuroprotection
Oxidative stress describes the imbalance between the generation of free radicals or reactive oxygen species (ROS) and various enzymatic and nonenzymatic antioxidant defense systems and may be involved in the pathogenesis of various neurodegenerative disorders, including Alzheimer’s disease (AD) (Halliwell and Gutteridge, 1989; Coyle and Puttfarcken, 1993; Olanow, 1993). Amyloid plaques are a characteristic feature of AD and are composed mainly of the 39–43 amino acid amyloid β protein (Aβ) (Glenner and Wong, 1984; Masters et al., 1985). Recently, it has been shown that Aβ can cause oxidative stress via the intracellular messenger hydrogen peroxide (H2O2), the precursor of highly reactive hydroxyl radicals (Behl et al., 1994), adding to the growing evidence that oxidative events may play a causative role in the pathogenesis of AD (Colye and Puttfarcken, 1993;Behl, 1997; Markesbery, 1997).
NF-κB was the first eukaryotic transcription factor described to respond directly to oxidative stress as induced by ROS and H2O2 (Schreck et al., 1991; Schmidt et al., 1995). It is a nuclear transcription factor, initially identified as a lymphoid-specific protein that binds to the κ-light chain gene intronic enhancer and resembles a heterodimeric protein composed of a 50 kDa subunit and a 65 kDa subunit (Sen and Baltimore, 1986). Typically, NF-κB is sequestered in the cytoplasm by the specific inhibitory protein IκB (Bäuerle and Baltimore, 1988; Israel, 1995; Verma et al., 1995; Baldwin, 1996). Activation and regulation of NF-κB transition into the nucleus, where it can induce the transcription of the target genes of NF-κB, is tightly controlled by IκB proteins (Israel, 1995; Baldwin, 1996). Until now, a wide range of inducers of NF-κB, including UV irradiation, growth factors, and viral infections, have been described (Grilli et al., 1993;Bäuerle and Henkel, 1994). Although the primary role for NF-κB in immune cells has always been thought to be the activation of defense genes during the inflammatory response, a potential function of NF-κB during cell death has also been suggested (Wu et al., 1996). It has been shown that the activation of NF-κB protects cells of a fibrosarcoma cell line and also immune cells against tumor necrosis factor-α (TNF-α)-induced apoptotic cell death (Beg and Baltimore, 1996; Liu et al., 1996; Van Antwerp et al., 1996; Wang et al., 1996).
Because most stimuli that can induce NF-κB activity are known to induce ROS, this transcription factor has recently gained attention for playing a possible role in the pathogenesis of oxidative stress-associated neurodegenerative disorders. Three major findings strongly suggest an involvement of NF-κB in AD. (1) Aβ can activate NF-κB (Behl et al., 1994), (2) antioxidants that block activation of NF-κB (Schreck et al., 1991; Meyer et al., 1993) can protect neurons against oxidative stress-induced cell death (Behl et al., 1994;Kaltschmidt et al., 1997), and (3) two NF-κB DNA binding sites are present in the regulatory region of the amyloid β protein precursor (AβPP) gene (Grilli et al., 1995), which is rapidly induced in response to stress conditions (Siman et al., 1989). Secreted AβPP can protect nerve cells from glutamate and Aβ toxicity (Mattson et al., 1993; Schubert and Behl, 1993).
Recently, we isolated clones of the sympathetic precursor-like cell line PC12 that are resistant to Aβ and other oxidative stressors, such as H2O2, and that have increased activities of catalase and glutathione peroxidase (Behl et al., 1994;Sagara et al., 1996). Here, we show that these oxidative stress-resistant cells have a constitutively increased NF-κB baseline activity and that the suppression of its activity resulted in a reversal of the resistance phenotype, suggesting that high levels of NF-κB activity may mediate resistance of neuronal cells against oxidative stress.
MATERIALS AND METHODS
Materials, cell lines, and cell culture. PC12 parental cells (PC12p) and the Aβ-resistant PC12 clone rCl8 were cultivated as described (Behl et al., 1994; Sagara et al., 1996). For glucocorticoid treatment, either medium containing steroid-free charcoal-stripped FCS or N2-medium was used. All media, media supplements, and sera were from Life Technologies (Eggenstein, Germany). The amyloid β protein used (fragment 25–35) was from Bachem/Saxon (Hannover, Germany). Polyethylenimine was from Aldrich (Deisenhofen, Germany). RU486 (mifepristone) was a kind gift from Dr. E. Baulieu. All other chemicals were from Sigma (Deisenhofen, Germany) unless stated otherwise.
Transfection, luciferase assay, and plasmids. Transient transfections using polyethylenimine (PEI) were performed exactly as described (Boussif et al., 1995). Each transfection experiment was performed in quadruplicate, repeated three times, and normalized for identical amounts of protein using the Bio-Rad protein reagent to determine protein concentrations of the samples (Bio-Rad, München, Germany). Extracts of transfected cells were assayed for luciferase activities exactly as described (Behl et al., 1997). The plasmid constructs were generously provided by the following: NF-κB-Luc and Tk-Luc (containing only the thymidine kinase promoter linked to a luciferase construct as control plasmid) by Dr. P. Bäuerle (Tularik Inc., San Francisco, CA), pRShGRα by Dr. R. M. Evans (The Salk Institute, San Diego, CA), Iκβα super-repressor by Dr. D. W. Ballard (Vanderbilt University, Nashville, TN), and CMV-β galactosidase by Dr. D. Spengler (Max-Planck-Institute of Psychiatry, Munich, Germany).
Immunocytochemistry. Immunocytochemistry was performed as described previously (Brugg et al., 1996). Briefly, cells were plated in LabTek chamber slices (Nunc, Dannstadt, Germany), fixed with 4% formaldehyde, and rinsed in 1× PBS. They were incubated with an anti-p65 polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) at 1:500 dilution for 1 d. Specific antibody binding was detected using a biotinylated antiserum (1:500) (Amersham, Braunschweig, Germany). Immunolabeling was revealed with streptavidin sulforhodamine (Boehringer Mannheim, Penzberg, Germany) diluted at 1:500. After a final PBS wash, cells were visualized by phase contrast and fluorescence microscopy and photographed.
Cell survival analysis. Cell viability was assessed using a modified 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay as described (Behl et al., 1994). In addition, trypan blue exclusion assays in combination with cell counting, using morphological criteria for cell death, were performed as described previously (Behl et al., 1994). Hydrogen peroxide-induced DNA degradation and cell death were further detected, using terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick-end labeling (TUNEL) staining according to the manufacturer’s instructions (Boehringer Mannheim). All survival assays were repeated four times in triplicate. In the transient transfection experiments, using the β-galactosidase expression vector, the fractions of dead and alive blue (β-galactosidase-expression) cells were counted. For each experimental determination, at least five optical fields were observed, and cellular survival was determined. One optical field consisted of >200 cells.
Electrophoretic mobility shift assay (EMSA). Cytoplasmic and nuclear extracts for the EMSAs were prepared by a mini-extraction protocol (Schreiber et al., 1989). The NF-κB double-stranded oligonucleotide corresponding to the NF-κB consensus sequence in the κ light chain enhancer in B cells (5′-AGT TGA GGG GAC TTT CCC AGG C-3′), and oligonucleotides for AP-1 and Oct-1 were from Promega/Serva (Heidelberg, Germany). The oligonucleotides were end-labeled with γ-[32P]ATP (3000 Ci/mmol) (Amersham) and T4 polynucleotide kinase (Promega/Serva) and purified on a G-25 column. Nuclear extracts (8–12 μg) were incubated for 20 min at room temperature with 20 μl of 2 μg of poly(dI.dC) (Pharmacia, Freiburg, Germany), 10% glycerol, 100 mm NaCl, 1 mm EDTA, 1 mm dithiothreitol, 0.5 mm phenylmethylsulfonyl fluoride, and 15,000–25,000 counts per min of 32P-oligonucleotides. For the reaction with specific antibodies, the nuclear extracts and the labeled probe were coincubated for an additional 30 min at room temperature with 1.5 μl of either p50 or p65 antibody stocks (Santa Cruz). For competition studies, before the addition of NF-κB-labeled probe, nuclear extracts were preincubated for 10 min at room temperature with a 100-fold excess of unlabeled NF-κB oligonucleotides. DNA–protein complexes were resolved on a 6% nondenaturing polyacrylamide gel at 20 mA for 3 hr in 0.5× TBE (45 mm Tris-borate and 1 mm EDTA). Gels were vacuum-dried and exposed to Fuji x-ray films at −80°C for 12–24 hr.
Western blotting. Western blottings were performed exactly as described previously (Sagara et al., 1996). The anti-p65 antibody and the anti-IκBα antibody were from Santa Cruz, and the anti-actin antibody was purchased from Boehringer Mannheim. Densitometer readings of the autoradiographs of the Western blots (and also EMSAs) were performed, using a Beckmann photometer.
Total intracellular glutathione (GSH) and enzyme assays.Cells were washed twice with ice-cold PBS, collected by scraping, and lysed with 3% sulfosalicylic acid. Lysates were incubated on ice for 10 min, and supernatants were collected after centrifugation in a microcentrifuge. After neutralization of supernatants with triethanolamine, total GSH (reduced and oxidized) concentration was determined by the method described originally (Tietze, 1969). Pure GSH was used to obtain the standard curve. To determine the enzymatic activity of catalase and glutathione peroxidase, standard procedures were used (Aebi, 1974; Gunzler and Flohe, 1985).
β-galactosidase expression. Resistant Cl8 cells were cultured in 24-well plates (Life Technologies). Plasmids encoding β-galactosidase (2 μg/well) and indicated plasmids were transfected, using PEI as described above, and stained for β-galactosidase activity as described (Yao et al., 1995a).
Statistical analysis. For statistical comparisons ANOVA followed by an appropriate post hoc test was used as indicated.
RESULTS
Transcriptional activity and DNA binding activity of NF-κB and the nuclear expression of p65 are constitutively increased in rCl8 cells
After transient transfections with an NF-κB reporter plasmid, containing 6 NF-κB-binding DNA consensus sites linked to a luciferase reporter gene (NF-κB-Luc), we found that the baseline transcriptional activity of NF-κB was approximately 20 times higher in rCl8 cells than in PC12p cells (Fig.1A). In addition, the DNA binding activity of this transcription factor was increased in rCl8 cells, compared with the parental cells as detected by EMSAs, using a DNA probe that represented the κB motif (Fig.2A–C). A direct comparison of the DNA binding activity of NF-κB in rCl8 and PC12p cells under nonstimulated basal conditions revealed a 3.5-fold increase in rCl8 cells (Fig. 2C). Although the DNA binding activity of Oct-1 is not significantly different in the two cell lines, the DNA binding activity of AP-1 was increased in PC12p compared with rCl8 (Fig. 2C), indicating that there is not a general upregulation of various transcription factors in rCl8 cells.
Fig. 1.
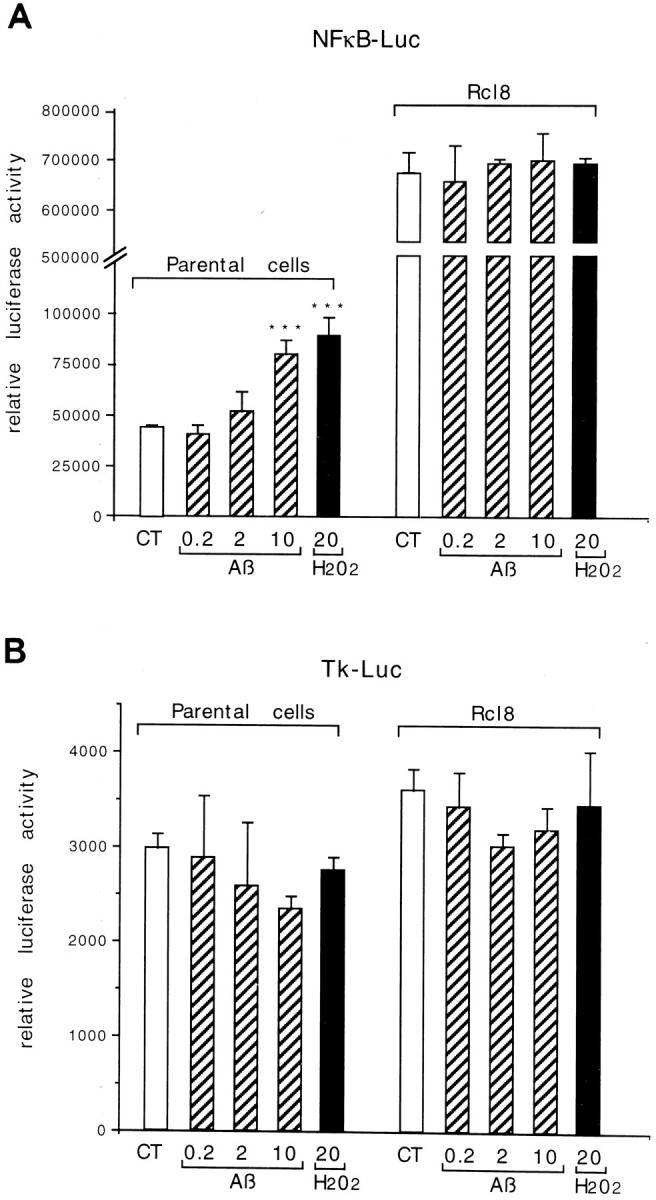
Transcriptional activity of NF-κB in PC12p and rCl8 cells. Cells were transfected with 1 μg of NF-κB-Luc plasmid (A) or Tk-Luc control vector (B) and were then exposed to increasing micromolar concentrations of Aβ or 20 μmH2O2 for 12–15 hr before harvesting. Results are shown in arbitrary units of luciferase activity (relative luciferase activity) corrected for identical protein amounts and are representative of four independent experiments.
Fig. 2.

DNA binding activity of NF-κB in PC12p and rCl8 cells. Nuclear extracts were prepared from PC12p (A) and rCl8 (B) cells, and EMSAs were performed. Autoradiograph of a native gel is shown. The effects of Aβ and H2O2 on NF-κB DNA binding activity in PC12p (A, lanes 1–6) and rCl8 cells (B, lanes 1–6) are also depicted. Cell cultures were treated with 10 μm Aβ (A, lanes 2–5; B, lanes 2–5) for the indicated times or with 150 μm H2O2 for 2 hr (A, lane 6; B, lane 6).Lane 7 represents the reaction mixture containing 100-fold excess unlabeled NF-κB oligonucleotides as competitor. InC, a direct comparison of the DNA binding activity of NF-κB in PC12p (lane 1) and rCl8 (lane 2) is shown. DNA binding activity of AP-1 and Oct-1 in PC12p (lanes 3 and 6) and rCl8 (lanes 4 and 7) is also depicted;lanes 5 and 8 represent the reaction mixture with a 100-fold excess of the corresponding oligonucleotides as competitors. In D, nuclear extracts of rCl8 cells were analyzed after the reaction with an antibody against either p50 or p65. Nuclear extracts after reaction with a 100-fold excess of an unlabeled NF-κB probe are also shown (NS). Filled arrowheads indicate the position of specific NF-κB/DNA complexes, circles depict the position of nonspecific complexes, and open arrowheads show the positions of the free nonbound DNA probe. CT, Untreated control cells;O.N., overnight incubation; NS, nonspecific. The dried gels were exposed to autoradiography for 24 hr in A, B, and C, and for 12 hr in D.
Although incubation of the cells with 10 μm Aβ or 20 μm H2O2 led to an approximately twofold activation of the κB-dependent reporter construct in PC12p, no further increase in the luciferase activity was detected in rCl8 (Fig. 1A). Higher concentrations of H2O2 did not further increase the luciferase activity under these experimental conditions (data not shown). The transcription of the Tk-Luc control plasmid was not altered after addition of Aβ or H2O2 (Fig.1B). Similar luciferase activities after transfection of the two cell lines with Tk-Luc under nonstimulated baseline conditions suggest comparable transfection efficiencies. Consistent with the induction of the transcriptional activity of NF-κB on stimulation in PC12p, the DNA binding activity of NF-κB also could be enhanced in PC12p, with a maximum increase after a 7 hr treatment with Aβ and after a 2 hr incubation with 150 μmH2O2 (Fig. 2A, lanes 3 and 6). Again, no further increase could be observed in rCl8 (Fig. 2B).
To identify the proteins involved in binding the labeled oligonucleotide in the EMSAs, nuclear extracts were incubated with antibodies against p50 or p65. In our experimental conditions, both antibodies caused a decrease in the specific band and consistently an increase in the nonspecific band (Fig. 2D), indicating that the NF-κB–DNA complex is composed of a p50–p65 heterodimer. Antisera against proteins not related to NF-κB did not produce any effect (data not shown). The lack of a clear supershift has been found to be a common effect in PC12 cells, when the binding of the antibody interferes with the oligonucleotide-binding site on NF-κB, as reported previously (Taglialatela et al., 1997). The specificity of NF-κB DNA binding was further demonstrated by the addition of a 100-fold excess of unlabeled cold NF-κB oligonucleotide probe (Fig.2B,D).
By immunocytochemical stainings and Western blottings, using PC12p and rCl8 cells under nonstimulated basal conditions, we also found an increased level of the p65 nuclear subunit of NF-κB in rCl8 cells (Fig. 3). These results indicate that the expression and activity of NF-κB is constitutively increased in rCl8.
Fig. 3.
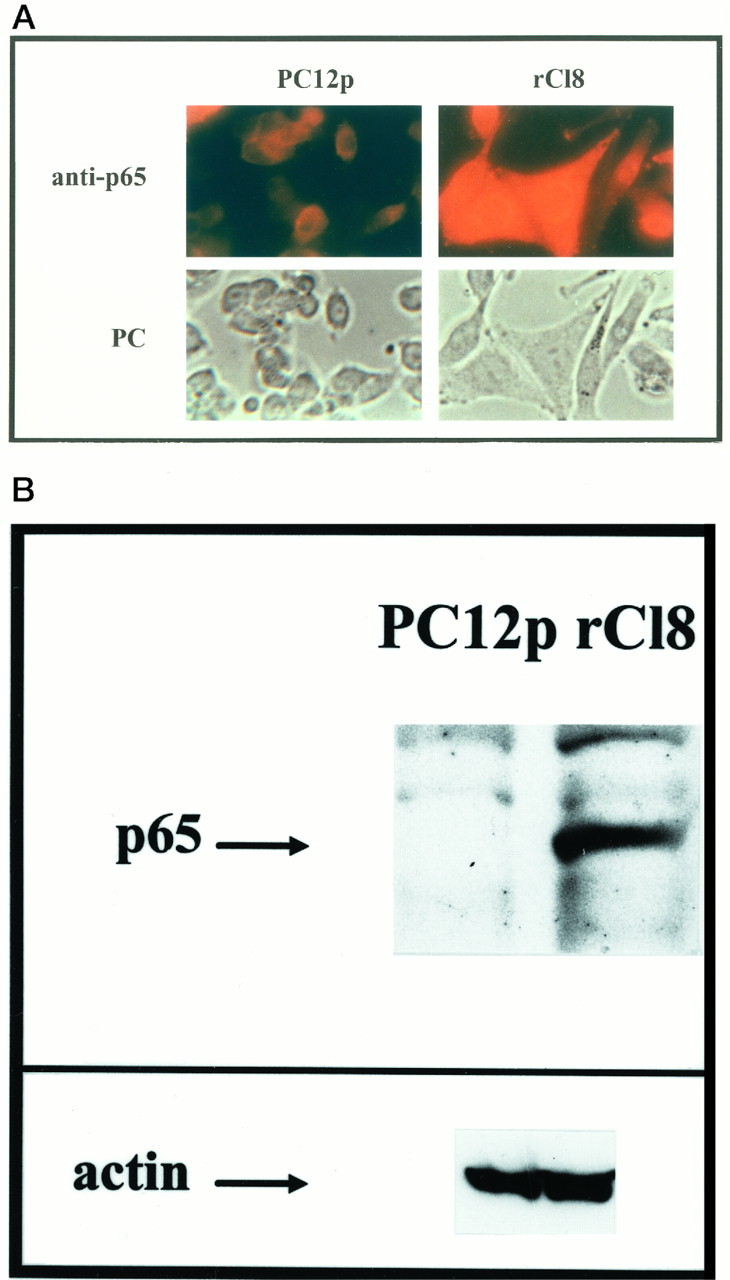
Expression of p65 in PC12p and rCl8 cells. Cells were plated, and immunocytochemistry (A) and Western blottings (B) were performed using an antibody specific for p65. Indirect immunofluorescence of cell cultures was performed as described in Materials and Methods. Corresponding phase-contrast (PC) images of the cultures are also shown. For the Western blottings, nuclear proteins of unstimulated PC12p and rCl8 cells were used, and specific binding of p65 was detected using ECL; as a control the same protein extracts were used for Western blotting with a monoclonal antibody specific for actin. A band specific for p65 could be detected in PC12p cells only after a longer time exposure (data not shown).
Dexamethasone (DEX) reverses the resistance phenotype and renders rCl8 cells more vulnerable to H2O2-induced cell death
Cells of the rCl8 clone express functional glucocorticoid receptors (GRs), which can be antagonized by the specific GR antagonist RU486 (data not shown). After a 24 hr pretreatment with DEX, the rCl8 cells were challenged with the strong oxidizing agent H2O2 at 250 μm for an additional 24 hr. Then cell survival was determined, using phase-contrast microscopy, TUNEL staining, and the MTT test (Fig.4). After DEX treatment, there was almost a 70% decrease in the survival of rCl8 cells compared with nontreated rCl8 control cells after a challenge with 250 μmH2O2 (Fig. 4A,C). Cell survival determinations by MTT assays were confirmed by trypan blue exclusion/cell countings (data not shown). When not pretreated with DEX, the morphology and integrity of the rCl8 cells were not altered after the challenge with H2O2, whereas DEX-treated cells showed cell disintegration throughout the culture and an increased DNA degradation (Fig. 4A,B). The reversal of the resistance by DEX is mediated by the activation of the GRs, because it could be blocked by the addition of RU486 (Fig.4A,B). In rCl8 cells not pretreated with DEX, a 50% killing of the cells was effected by ∼450 μmH2O2, whereas the same toxic response was achieved with only ∼150 μmH2O2 when the cells were treated with DEX before addition of H2O2 (Fig. 4C). This indicates an approximately threefold increase in the sensitivity of rCl8 cells to oxidative stress caused by the pretreatment with glucocorticoids. DEX treatment did not further enhance the sensitivity of PC12p cells for H2O2 (data not shown).
Fig. 4.
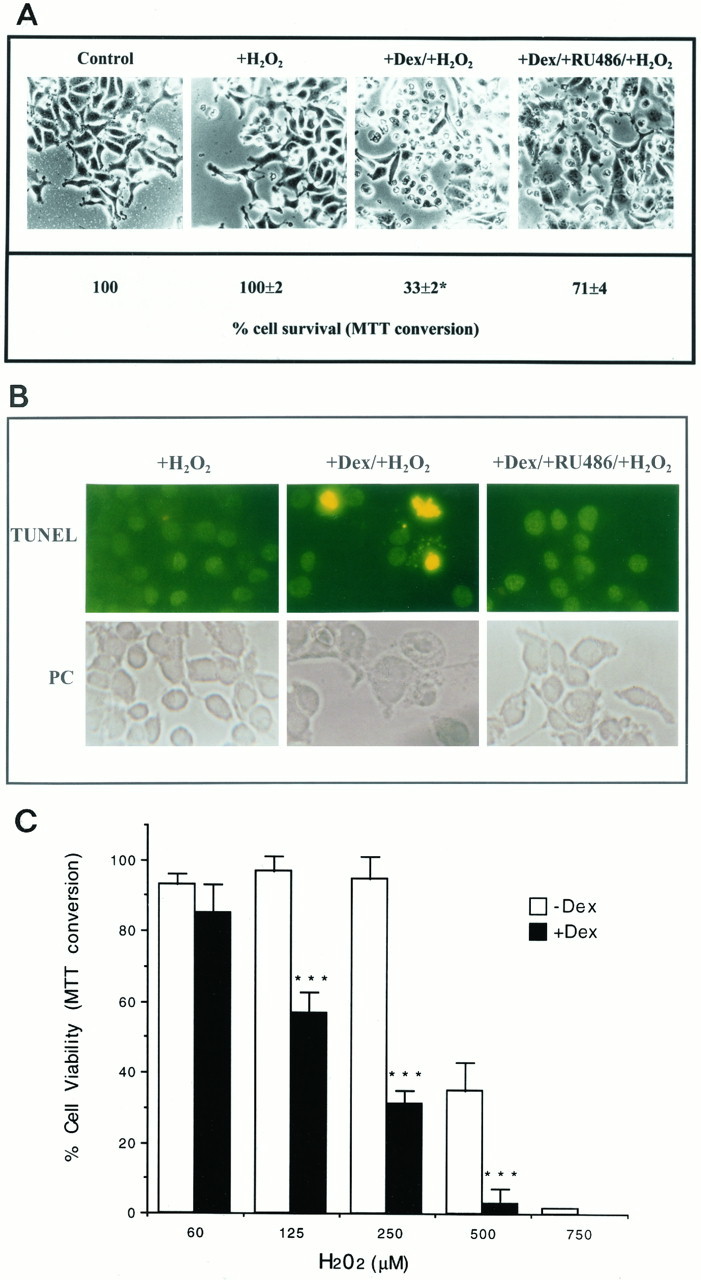
In A, rCl8 cells were plated and incubated with 10−7m DEX with or without 10−6m RU486 for 24 hr before the addition of 250 μm H2O2. After 20 hr, cultures were first photographed and then MTT assays were performed. MTT data presented are the mean ± SEM for triplicate determinations. The viability of the untreated resistant control cells was defined as 100%. *p < 0.001 (cell viability after incubation with DEX followed by the toxin was compared with cell viability after coadministration of DEX and RU486 followed by the toxin) was considered significant. Incubation of the cells with DEX or RU486 alone did not affect cell survival. In B, H2O2-induced DNA degradation and cell death was further detected in rCl8 cells pretreated with 10−7m DEX using TUNEL staining. Without pretreatment and after the GRs were blocked with RU486, rCl8 cells were resistant against H2O2. Magnifications were 100-fold inA and 200-fold in B. In C, rCl8 cells were pretreated with 10−7mDEX and then challenged with increasing concentrations of H2O2 for 20 hr. Cell survival was determined by MTT assays, and data presented are the mean ± SEM for triplicate determinations. The viability of the untreated resistant control cells was defined as 100%. ***p < 0.001 (cell viability after incubation with DEX followed by H2O2 was compared with cell viability after administration of H2O2 alone) was considered significant.
DEX suppresses the transcriptional activity of NF-κB and increases the cytoplasmic level of IκBα protein in rCl8 cells
In cells treated with increasing concentrations of DEX, the transcriptional activity of NF-κB decreased dose dependently (Fig.5A). The DNA binding activity of NF-κB did not decrease after DEX treatment of the rCl8 cells at various time points (Fig. 5B). Nevertheless, we found that the cytoplasmic level of the NF-κB inhibitory protein IκBα was increased up to 3.5-fold after a 4 hr treatment with DEX (Fig.5C).
Fig. 5.

Dexamethasone (DEX) suppresses the transcriptional activity of NF-κB and increases IκBα expression in rCl8.A, Downregulation of the transcriptional activity of NF-κB by DEX. Resistant clone 8 cells were transfected with 1 μg of NF-κB-Luc and incubated for 16 hr with increasing concentrations of DEX. The luciferase activity of untreated rCl8 cells is considered as 100%. Results are shown in relative luciferase activity corrected for identical protein amounts and represent an average of three independent experiments. Data presented are the mean ± SEM (**p < 0.01 and ***p < 0.001 compared with control values; Student’s t test).B, DNA binding activity of NF-κB is not altered by DEX. Resistant clone 8 cultures were treated with 10−7m DEX for 2 hr (lane 2), 4 hr (lane 3), 6 hr (lane 4), or overnight (lane 5) and analyzed by EMSAs. Untreated rCl8 cells represent the control (lane 1). The filled arrowhead indicates the position of specific NF-κB/DNA complexes, the circle represents the position of nonspecific complexes, and the open arrowhead represents the position of the free probe.C, DEX increases IκBα protein levels in rCl8. Cultures were treated with 10−7m DEX for 2 hr (lane 2), 4 hr (lane 3), 6 hr (lane 4), or overnight (lane 5). The overnight control is depicted in lane 6. Cytoplasmic extracts were analyzed by immunoblotting with IκBα antibody; as a control, the same protein extracts were used for Western blotting with a monoclonal antibody specific for actin.
Overexpression of IκBα also suppresses NF-κB-dependent transcriptional activity and reverses the resistant phenotype of rCl8 cells
After transfection of rCl8 cells with a super-repressor form of IκBα, which is resistant to both phosphorylation and proteolytic degradation and therefore permanently prevents the nuclear translocation of NF-κB (Brockman et al., 1995), the transcriptional activity of NF-κB was reduced by 85 ± 1% (p < 0.001; comparison of cultures transfected with the IκBα super-repressor with control-transfected cultures). Forty-eight hours after the transfection, cells were challenged with increasing concentrations of H2O2, and cell survival was determined by cell counting (Fig.6). Resistant Cl8 cells cotransfected with a β-galactosidase vector (to identify transfected cells) and a vector encoding the IκBα super-repressor became sensitive to H2O2, as shown morphologically for the challenge with 450 μm H2O2 in Figure 6A. β-Galactosidase-expressing blue cells cotransfected with a control vector missing the IκBα super-repressor coding sequence were still less sensitive to H2O2 (Fig. 6B,C). The quantification by cell counting revealed a significant dose-dependent difference in the sensitivity for H2O2 of the rCl8 cells overexpressing the super-repressor, when compared with rCl8 cells transfected with the control vector (Fig. 6C).
Fig. 6.

Overexpression of IκBα super-repressor increases the sensitivity of rCl8 to H2O2. rCl8 cells were cotransfected with CMV-β-galactosidase (2 μg/well) and either an expression vector coding for the IκBα super-repressor (2 μg/well) (A) or a control vector missing the IκBα cDNA (B). After 48 hr, the cells were challenged with 450 μm H2O2 for 6 hr and stained for β-galactosidase. Although most of the β-galactosidase-expressing cells transfected with the IκBα super-repressor are dead (A, filled arrowheads), all of the β-galactosidase-expressing cells transfected with the control vector are resistant and viable (B, filled arrowheads). The surrounding cells that are not transfected are resistant and viable (A, open arrowhead). Different optical fields of the transfected cultures were randomly observed, and the figure shows one representative image. Scale bar, 50 μm. In C, the cell survival data are shown with increasing concentrations of H2O2; cell countings were performed as described in Material and Methods. ***p < 0.001 (rCl8 cell viability after transfection of the IκBα super-repressor and exposure to H2O2 was compared with the viability of rCl8 transfected with the control vector and challenged with H2O2) was considered significant.
DISCUSSION
The molecular events and genetic programs that can be activated by oxidative challenges or in response to oxidative stress, and that might eventually lead to a resistance against oxidative insults, are poorly understood. Considering the fact that some neuronal populations can survive the accumulating oxidative challenges and degenerative processes during the development of AD (Braak and Braak, 1991), an understanding of the molecular mechanisms that can decrease the vulnerability of neurons and consequently can increase their resistance to oxidative stress conditions are of great interest.
The AD-associated neurotoxic Aβ can induce NF-κB activation via the intracellular accumulation of H2O2 in neuronal cell lines in primary neuronal culture and, most interestingly, also in neurons and astroglia present in postmortem sections of brains from AD patients (Behl et al., 1994; Kaltschmidt et al., 1997). In the neuronal cell clone rCl8 that is resistant against Aβ and also H2O2, we found (1) that the nuclear expression of the transcription factor NF-κB and also its transcriptional activity are constitutively increased compared with their parental PC12 cells and (2) that the suppression of the transcriptional activity of NF-κB reverses the oxidative stress-resistance phenotype of rCl8 cells. We speculate that the transcription factor NF-κB is part of a defense program that enables these neuronal cells to protect themselves against oxidative stressors.
Cells of the rCl8 clone have been shown to express high levels of antioxidant enzymes, such as catalase and GSH peroxidase (Sagara et al., 1996). Antioxidants and antioxidant enzymes that can inhibit NF-κB activation (Meyer et al., 1993) may prevent the accumulation of ROS and peroxides in response to Aβ treatment. Therefore, these antioxidant enzymes may prevent the accumulation of ROS and peroxides in response to Aβ treatment. Consequently, a further increase in activation of NF-κB is inhibited. Our findings that treatment of rCl8 with Aβ and one of its intracellular messengers H2O2 cannot further enhance NF-κB activity are consistent with earlier observations of other catalase- and GSH peroxidase-overexpressing cellular systems (Schmidt et al., 1995;Kretz-Remy et al., 1996). However, NF-κB baseline activity remains high in rCl8 despite the basic increase in antioxidant enzymes. We speculate that in rCl8 there could be an enhanced degradation of IκBα, leading to constitutive NF-κB activity as proposed for mature murine B-cells (Miyamoto et al., 1994). Another possibility could be that in such cells specific nuclear coactivators could boost NF-κB expression and activity.
The suppression of the transcriptional activity of NF-κB in rCl8 cells was effected by (1) glucocorticoid treatment and (2) the overexpression of IκBα. A direct inhibition of NF-κB activity by glucocorticoids, such as DEX, has been shown primarily for immune cells (Auphan et al., 1995; Scheinman et al., 1995). Although most recently it has been reported that DEX can attenuate NF-κB DNA binding activity in rat brain in vivo (Unlap and Jope, 1997), we found no reduction of its DNA binding activity in rCl8 cells after DEX treatment. In our approach, we rather observed an effective block of the transcriptional activity of NF-κB that was followed by the reversal of the oxidative stress-resistance phenotype of the rCl8. Such an increased vulnerability of neuronal cells after DEX treatment is consistent with the observation that glucocorticoid treatment can enhance neurotoxic insults (Sapolsky, 1987; Behl et al., 1997). A glucocorticoid-mediated repression of NF-κB-dependent transcription, without affecting the NF-κB–DNA complex, has also been reported recently for murine fibroblasts and endothelial cells (De Bosscher et al., 1997). Whether the DEX-induced increase in the cytoplasmic level of IκBα protein in the rCl8 cells contributes to the reversal of the resistance phenotype caused by DEX remains to be elucidated. A glucocorticoid-induced increase in IκBα protein has previously been found in immune cells (Auphan et al., 1995; Scheinman et al., 1995). Interestingly, it has been shown for fibroblasts that an increased synthesis of IκBα is neither required nor sufficient for a glucocorticoid-mediated downregulation of NF-κB activity (Heck et al., 1997). Independent of the level of IκBα, a direct physical interaction between the activated GRs and the RelA protein of the NF-κB complex has to be considered as one mechanism for the inhibition of NF-κB activity by glucocorticoids, as suggested byCaldenhoven et al. (1995) and Van der Burg et al., (1997).
With respect to possible neuronal target genes, which could be controlled by NF-κB, we found (1) that neither the expression nor the activity of catalase and glutathione peroxidase was affected by DEX treatment in rCl8 cells (data not shown), but (2) that there was a minor decrease in the level of the intracellular antioxidant GSH and in the expression of its synthesizing enzyme γ-glutamylcysteine synthetase (γ-GCS) (data not shown). Whether such a drop in the intracellular GSH content is sufficient to reverse the resistance phenotype of the rCl8 cells remains to be determined. Exogenous addition of GSH has been shown to protect cells against oxidative stress (Halliwell and Gutteridge, 1989), and a drop in intracellular GSH levels mediates oxidative glutamate toxicity in neurons (Murphy et al., 1989). Interestingly, NF-κB binding sites are present in the promoter of the γ-GCS synthetase gene (Yao et al., 1995b).
Several lines of evidence in addition to a possible direct oxidative neurotoxicity of Aβ and the involvement of inflammatory reactions support the oxidative stress hypothesis of the etiopathogenesis of AD (Rogers et al., 1992; Coyle and Puttfarcken, 1993; Smith et al., 1995;Behl, 1997; Markesbery, 1997). Because oxidative stress caused by Aβ can directly induce NF-κB activity in neurons, we tried to elucidate a possible role for the activation of this redox-sensitive transcription factor in neuronal cells resistant against Aβ and other oxidative stressors. The reversal of the resistance phenotype at the specific suppression of NF-κB activity strongly supports a protective role for this factor in neuronal rCl8 cells. Our hypothesis is supported by observations that demonstrate that after overexpression of IκBα, immune cells become vulnerable to TNF-α-induced apoptosis (Beg and Baltimore, 1996; Liu et al., 1996; Van Antwerp et al., 1996;Wang et al., 1996), but it is in contrast to a recent report by Grilli et al. (1996) showing that the suppression of NF-κB activity by anti-inflammatory drugs can protect neurons against glutamate toxicity. However, in this latter paper no direct modulation in NF-κB activity was demonstrated to support the conclusion of the authors.
In summary, the results of our study suggest that the transcription factor NF-κB may directly mediate the resistance of clones of the sympathetic precursor-like PC12 cell line against oxidative stress-induced cell death. The activity of NF-κB may drive defense programs in response to oxidative attacks, which afford protection against oxidative insults. As a next step, it will be important to identify neuroprotective target genes that are driven by the transcriptional activity of NF-κB and to investigate their exogenous inducibility to increase the survival of neurons under oxidative stress.
Footnotes
This work was supported by a Max-Planck fellowship to F.L. and by a National Institutes of Health fellowship (Grant 1 F32 NS10032) to Y. S. We thank M. Gräf, B. Berning, S. Engert, and R. Dargusch for expert technical assistance. We are grateful to Dr. E. C. Hirsch and S. Hunot for advice in immunocytochemistry. We thank Dr. B. Lutz for critically reading this manuscript.
Correspondence should be addressed to Dr. Christian Behl, Max-Planck-Institute of Psychiatry, Kraepelinstrasse 10, 80804 Munich, Germany.
REFERENCES
- 1.Aebi H. Methods of enzymatic analysis. In: Bergmeyer H, editor. Catalase, Vol I. Catalase Academic; New York: 1974. pp. 673–684. [Google Scholar]
- 2.Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-κB activity through induction of IκB synthesis. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- 3.Baldwin AS. The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 4.Bäuerle P, Baltimore D. IκB: a specific inhibitor of the NF-κB transcription factor. Science. 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- 5.Bäuerle P, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 6.Beg AA, Baltimore D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 7.Behl C. Amyloid β-protein toxicity and oxidative stress in Alzheimer’s disease (review). Cell Tissue Res. 1997;296:471–480. doi: 10.1007/s004410050955. [DOI] [PubMed] [Google Scholar]
- 8.Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid β protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 9.Behl C, Lezoualc’h F, Trapp T, Widmann M, Skutella T, Holsboer F. Glucocorticoids enhance oxidative stress-induced cell death in hippocampal neurons in vitro. Endocrinology. 1997;138:101–106. doi: 10.1210/endo.138.1.4835. [DOI] [PubMed] [Google Scholar]
- 10.Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 12.Brockman JA, Scherer DC, Mckinsey TA, Hall SM, Qi XX, Lee WY, Ballard DW. Coupling of a signal response domain in IkBa to multiple pathways for NF-κB activation. Mol Cell Biol. 1995;15:2809–2818. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brugg B, Michel PP, Agid Y, Ruberg M, Ceramide Ceramide induces apoptosis in cultured mesencephalic neurons. J Neurochem. 1996;66:733–739. doi: 10.1046/j.1471-4159.1996.66020733.x. [DOI] [PubMed] [Google Scholar]
- 14.Caldenhoven E, Liden J, Wissink S, Vandestolpe A, Raaijmakers J, Koenderman L, Okret S, Gustafsson JA, Vandersaag PT. Negative cross-talk between RelA and the glucocorticoid receptor: a possible mechanism for the antiinflammatory action of glucocorticoids. Mol Endocrinol. 1995;9:401–412. doi: 10.1210/mend.9.4.7659084. [DOI] [PubMed] [Google Scholar]
- 15.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 16.De Bosscher K, Schmitz ML, Berghe WV, Plaisance S, Fiers W, Haegeman G. Glucocorticoid-mediated repression of nuclear factor-κB-dependent transcription involves direct interference with transactivation. Proc Natl Acad Sci USA. 1997;94:13504–13509. doi: 10.1073/pnas.94.25.13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 18.Grilli M, Chiu JS, Lenardo M. NF-κB and Rel: participation in a multiform transcriptional regulatory system. Int Rev Cytol. 1993;143:1–62. doi: 10.1016/s0074-7696(08)61873-2. [DOI] [PubMed] [Google Scholar]
- 19.Grilli M, Ribola M, Alberici A, Valerio A, Memo M, Spano P. Identification and characterization of a κB/Rel binding site in the regulatory region of the APP gene. J Biol Chem. 1995;270:26774–26777. doi: 10.1074/jbc.270.45.26774. [DOI] [PubMed] [Google Scholar]
- 20.Grilli M, Pizzi M, Memo M, Spano P. Neuroprotection by aspirin and sodium salicylate through blockade of NF-κB activation. Science. 1996;274:1383–1385. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- 21.Gunzler W, Flohe L. Glutathione peroxidase. In: Greenwald R, editor. Handbook of methods for oxygen radical research. CRC; Boca Raton, FL: 1985. pp. 285–290. [Google Scholar]
- 22.Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. Oxford UP; Oxford: 1989. [Google Scholar]
- 23.Heck S, Bender K, Kullmann M, Göttlicher M, Herrlich P, Cato ACB. IκBα-independent downregulation of NF-κB activity by glucocorticoid receptor. EMBO J. 1997;16:4698–4707. doi: 10.1093/emboj/16.15.4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Israel A. A role for phosphorylation and degradation in the control of NF-κB activity. Trends Genet. 1995;11:203–205. doi: 10.1016/s0168-9525(00)89045-9. [DOI] [PubMed] [Google Scholar]
- 25.Kaltschmidt B, Uherek M, Volk B, Baeuerle PA, Kaltschmidt C. Transcription factor NF-κB-B is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:2642–2647. doi: 10.1073/pnas.94.6.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kretz-Remy C, Mehlen P, Mirault ME, Arrigo AP. Inhibition of IκB-α phosphorylation and degradation and subsequent NF-κB activation by glutathione peroxidase overexpression. J Cell Biol. 1996;133:1083–1093. doi: 10.1083/jcb.133.5.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Z, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 28.Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 29.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel R. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron. 1993;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- 31.Meyer M, Schreck R, Bäuerle PA. H2O2 and antioxidants have opposite effects on activation of NF-κB and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. EMBO J. 1993;12:2005–2015. doi: 10.1002/j.1460-2075.1993.tb05850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyamoto S, Chiao PJ, Verma IM. Enhanced IκBα degradation is responsible for constitutive NF-κB activity in mature murine B-cell lines. Mol Cell Biol. 1994;14:3276–3282. doi: 10.1128/mcb.14.5.3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- 34.Olanow CW. A radical hypothesis for neurodegeneration. Trends Neurosci. 1993;16:439–444. doi: 10.1016/0166-2236(93)90070-3. [DOI] [PubMed] [Google Scholar]
- 35.Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, Civin WH, Brachova L, Bradt B, Ward P. Complement activation by β-amyloid in Alzheimer disease. Proc Natl Acad Sci USA. 1992;89:10016–10020. doi: 10.1073/pnas.89.21.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sagara Y, Dargusch R, Klier FG, Schubert D, Behl C. Increased antioxidant enzyme activity in amyloid β protein-resistant cells. J Neurosci. 1996;16:497–505. doi: 10.1523/JNEUROSCI.16-02-00497.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sapolsky RM. Glucocorticoids and hippocampal damage. Trends Neurosci. 1987;10:346–349. [Google Scholar]
- 38.Scheinman RI, Cogswell PC, Lofquist AK, Baldwin AS., Jr Role of transcriptional activation of IκB in mediation of immunosuppression by glucocorticoids. Science. 1995;270:283–286. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt KN, Amstad P, Cerutti P, Bäuerle PA. The roles of hydrogen peroxide and superoxide as messengers in the activation of transcription factor NF-κB. Chem Biol. 1995;2:13–22. doi: 10.1016/1074-5521(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 40.Schreck R, Zorbas H, Winnacker EL, Bäuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-κB transcription factor and HIV-1. EMBO J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schreiber E, Matthias P, Müller M, Schaffner W. Rapid detection of octamer binding proteins with mini-extracts, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schubert D, Behl C. The expression of amyloid beta protein precursor protects nerve cells from β-amyloid and glutamate toxicity and alters their interaction with the extracellular matrix. Brain Res. 1993;629:275–282. doi: 10.1016/0006-8993(93)91331-l. [DOI] [PubMed] [Google Scholar]
- 43.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705–706. doi: 10.1016/0092-8674(86)90346-6. [DOI] [PubMed] [Google Scholar]
- 44.Siman R, Card JP, Nelson RB, Davis LG. Expression of β-amyloid precursor protein in reactive astrocytes following neuronal damage. Neuron. 1989;3:275–285. doi: 10.1016/0896-6273(89)90252-3. [DOI] [PubMed] [Google Scholar]
- 45.Smith MA, Syre LM, Monnier VM, Perry G. Radical ageing in Alzheimer’s disease. Trends Neurosci. 1995;18:172–176. doi: 10.1016/0166-2236(95)93897-7. [DOI] [PubMed] [Google Scholar]
- 46.Taglialatela G, Robinson R, Perezpolo JR. Inhibition of NF-κB activity induces nerve growth factor-resistant apoptosis in PC12 cells. J Neurosci Res. 1997;47:155–162. [PubMed] [Google Scholar]
- 47.Tietze F. Enzymatic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 48.Unlap MT, Jope RS. Dexamethasone attenuates NF-κB DNA binding activity without inducing IκB levels in rat brain in vivo. Mol Brain Res. 1997;45:83–89. doi: 10.1016/s0169-328x(96)00240-9. [DOI] [PubMed] [Google Scholar]
- 49.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-α-induced apoptosis. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 50.Van der Burg B, Liden J, Okret S, Delaunay F, Wissink S, van der Saag P, Gustafsson J-A. Nuclear factor-κ B repression in antiinflammation and immunosuppression by glucocorticoids. Trends Endocrinol Metab. 1997;8:152–157. doi: 10.1016/s1043-2760(97)00006-4. [DOI] [PubMed] [Google Scholar]
- 51.Verma I, Stevenson J, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-κB/IκB family: intimate tales of association and dissociation. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 52.Wang CY, Marty MW, Baldwin AS., Jr TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of apoptosis. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 53.Wu M, Lee HY, Bellas RE, Schauer SL, Arsura M, Katz D, Fitzgerald MJ, Rothstein TL, Sherr DH, Sonenshein GE. Inhibition of NF-κB/rel induces apoptosis of murine B cells. EMBO J. 1996;15:4682–4690. [PMC free article] [PubMed] [Google Scholar]
- 54.Yao H, Labudda K, Rim C, Capodieci P, Loda M, Stork PJ. Cyclic adenosine monophosphate can convert epidermal growth factor into a differentiating factor in neuronal cells. J Biol Chem. 1995a;270:20748–20753. doi: 10.1074/jbc.270.35.20748. [DOI] [PubMed] [Google Scholar]
- 55.Yao KS, Godwin AK, Johnson SW, Ozols RF, Odwyer PJ, Hamilton TC. Evidence for altered regulation of gamma-glutamylcysteine synthetase gene expression among cisplatin-sensitive and cisplatin-resistant human ovarian cancer cell lines. Cancer Res. 1995b;55:4367–4374. [PubMed] [Google Scholar]