

Abstract
Studies in non-neural cells have suggested that a fall in mitochondrial membrane potential (ΔΨM) is one of the earliest events in apoptosis. It is not known whether neural apoptosis caused by nerve growth factor (NGF) and serum withdrawal involves a decrease in ΔΨM. We used epifluorescence and laser confocal microscopy with the mitochondrial potentiometric dyes chloromethyl-tetramethylrosamine methyl ester and 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethybenzimidazol carbocyanine iodide to estimate ΔΨM. PC12 cells were differentiated in media containing serum and NGF for 6 d before withdrawal of trophic support. After washing, the cells were incubated with media containing serum and NGF (M/S+N), media without serum and NGF, or media with the “trophic-like” monoamine oxidase B inhibitor, (−)-deprenyl. Mitochondria in cells without trophic support underwent a progressive shift to lower ΔΨM values that was significant by 3 hr after washing. The percentages of cells with nuclear chromatin condensation or nuclear DNA fragmentation were not significantly increased above those for cells in M/S+N until 6 hr after washing. Replacement of cells into M/S+N or treatment with (−)-deprenyl markedly reduced the proportion of mitochondria with decreased ΔΨM. Measurements of cytoplasmic peroxyl radical levels with 2′,7′-dihydrodichlorofluorescein fluorescence and intramitochondrial Ca2+ with dihydro-rhodamine-2-acetylmethyl ester indicated that cytoplasmic peroxyl radical levels were not increased until after 6 hr, whereas increases in intramitochondrial Ca2+ paralleled the decreases in ΔΨM. (−)-Deprenyl appeared to alter the relationship between intramitochondrial Ca2+ levels and ΔΨM, possibly through its reported capacity to increase the synthesis of proteins such as BCL-2.
Keywords: apoptosis, mitochondrial membrane potential, DNA cleavage, chromatin condensation, JC-1, CMTMR, Rhod-2AM, DCFH
Neuronal apoptosis in the developing nervous system results from inadequate neurotrophic support (Lo et al., 1995). Apoptosis may also contribute to human neurodegeneration (Chalmers-Redman et al., 1997), possibly because of increases in reactive oxygen species (ROS) produced by defective mitochondrial energy production (Richter et al., 1995; Beal, 1996). Various findings link inadequate neurotrophic support and mitochondrial dysfunction in the genesis of neuronal death (Frim et al., 1993; Mattson et al., 1993;Kirschner et al., 1996).
A loss of mitochondrial membrane potential (ΔΨM) may be a critical mediator of apoptosis. The outward pumping of protons across the inner mitochondrial membrane produces a proton gradient that drives the conversion of ADP to ATP and is reflected by ΔΨM (Sherratt, 1991). Measurements of whole-cell potentiometric dye fluorescence have indicated that ΔΨMis reduced before the appearance of apoptotic nuclear changes in a variety of blood, hepatic, and immune cell models (Susin et al., 1996b). Similarly, decreased potentiometric dye uptake has suggested a fall in ΔΨM in sympathetoblasts after nerve growth factor (NGF) withdrawal (Deckwerth and Johnson, 1993). Accordingly, neurotrophic withdrawal may reduce ΔΨM.
The relationship between ΔΨM and apoptotic initiation is uncertain. Decreased ΔΨM induces opening of the mitochondrial permeability transition pore (PTP), which may lead to the release of mitochondrial apoptosis initiation factors (AIFs) (Marchetti et al., 1996a; Susin et al., 1996a; Zamzami et al., 1996a). Cytochrome C can facilitate apoptosis when released into the cell cytoplasm (Liu et al., 1996; Kluck et al., 1997). An overall decrease in ΔΨM, however, was reported to occur late in apoptosis, well after the release of cytochrome C from mitochondria (Yang et al., 1997). It has not been determined how changes in ΔΨM progress across populations of mitochondria in a single cell. Loss of ΔΨM in a small proportion of mitochondria might be sufficient to initiate apoptosis.
(−)-Deprenyl reduces nerve cell death caused by a mitochondrial toxin (for review, see Tatton and Chalmers-Redman, 1996), possibly by increasing neurotrophic factor synthesis by astrocytes (Biagini et al., 1994; Seniuk et al., 1994; Semkova et al., 1996). (−)-Deprenyl also increases reduced ubiquinone, suggesting that a mitochondrial action may be involved in the increased neuronal survival (Koutsilieri et al., 1996).
(−)-Deprenyl reduces the apoptosis of partially neuronally differentiated PC12 cells caused by NGF and serum withdrawal (Tatton et al., 1994b). We used epifluorescence and confocal laser microscopy in that model to examine effects of NGF and serum-borne neurotrophic factors or (−)-deprenyl on ΔΨM in apoptosis. NGF and serum withdrawal progressively shifted ΔΨM across mitochondrial populations to lower values at times that preceded any increase in apoptotic nuclear events. Reexposure to NGF and serum or treatment with 10−9m (−)-deprenyl largely prevented the shifts. Increases in intramitochondrial Ca2+ levels appeared to parallel the decreases in ΔΨM, whereas increases in cytoplasmic peroxide levels were delayed relative to decreases in ΔΨM.
MATERIALS AND METHODS
Cell culture. PC12 cells (American Type Culture Collection, Rockville, MD) were propagated in minimum essential medium (MEM) (Life Technologies, Gaithersburg, MD) with 10% horse serum (Life Technologies), 5% fetal bovine serum (Life Technologies), 2 mml-glutamine (Life Technologies), 50 U/ml penicillin, and 50 μg/ml streptomycin (PC12 media).
For counts of intact nuclei, PC12 cells were differentiated in 24 well plates in the above media supplemented with 100 ng/ml 7 S NGF (Upstate Biolotechnology, Lake Placid, NY). Cells were seeded at a density of 8 × 104 cells per well to facilitate neuronal differentiation. Seeding at higher densities prevented the full elaboration of processes under the influence of NGF. After 6 d in media containing serum and NGF (M/S+N), the cells underwent three washes in HBSS to remove NGF and serum-borne trophic agents, and the media were replaced with one of the following: MEM only (M/O), MEM with 10−9m (−)-deprenyl (M/−d), or M/S+N. (−)-Deprenyl at 10−9m was used because that concentration induces the maximum survival in the partially differentiated PC12 cells (Tatton et al., 1994b) and is insufficient to cause monoamine oxidase (MAO) inhibition in PC12 cells (Youdim et al., 1986).
For imaging studies, the PC12 cells were plated on collagen-treated (Sigma, St Louis, MO) or poly-d-lysine-treated (Mr 30,000–70,000; Sigma) coverslips and differentiated in the same media as above. Cells were seeded at 1 × 104 cells per 12 mm coverslip to facilitate extensive process outgrowth (see Fig. 1A). After 6 d in M/S+N, the cells were washed three times in HBSS, and the media was replaced with M/O, M/−d, or M/S+N.
Fig. 1.

Estimation of cell survival, nuclear chromatin condensation, nuclear DNA fragmentation, and mitochondrial membrane potential. A1, Typical interference contrast micrograph of methylene blue-stained PC12 cells that were partially neuronally differentiated in M/S+N for 6 d and were then washed to remove trophic proteins and immediately replaced in M/S+N to reestablish trophic support at 12 hr before fixation for histology.A2, Similar micrograph for cells that were washed and trophically withdrawn by placement in M/O at 12 hr before fixation.B, Fluorescence photomicrographs of in situ nuclei stained with Hoechst 33258 at 12 hr after washing and placement into M/O. Normal Hoechst 33258-stained nuclei showed a diffuse, granular substructure with division by fine septate-like structures (inset 1). Apoptotic nuclei showed dense staining characterized by the formation of shrunken, intensely fluorescent lobular structures, those being apoptotic bodies (inset 2) or ring-like structures (inset 3). C, Interference contrast micrograph of cells reacted for in situ detection of nuclear DNA fragmentation using the Apop Tag method at 12 hr after washing and placement into M/O. The method revealed nuclei that were shrunken in comparison to those without evidence of DNA fragmentation. Nuclear DNA fragmentation is evident in three of the nuclei, the external plasma membranes of which appear intact using interference contrast microscopy. D, Typical epifluorescence micrograph of CMTMR fluorescence for a partially neuronally differentiated PC12 cell grown in M/S+N and fixed 24 hr after washing and replacement into M/S+N. CMTMR fluorescence from individual mitochondria was discernible using epifluorescence microscopy with 1000×, 1.3 NA objective. However, the thickness of the optical sections resulted in the superimposition of the fluorescence signals from adjacent mitochondria.E, Confocal micrograph of a cell in M/S+N at 24 hr after washing. A pinhole value of 50 produced optical sections that were sufficiently thin to allow for fluorescence measurements from individual mitochondria and the immediately adjacent cytoplasm without superimposition of fluorescence from nearby mitochondria.
Estimates of cell survival. At 3, 6, 12, and 24 hr after washing, cells were harvested by tituration from the 24 well plates and centrifuged at 500 × g for 5 min; the supernatant was removed, and the pelleted cells were lysed with 200 μl of lysing buffer (Zap-o-globin II). Intact nuclei were counted in a hemocytometer by “blinded” observers according to the method of Soto and Sonnenschein (1985). A second estimate of cell survival was obtained for the same time points by lightly staining the cells on coverslips with 1% methylene blue and counting intact cells under interference contrast optics (see Fig. 1A1,A2). Counts of intact cells on each coverslip were taken from 25 100× fields, the coordinates of which on an x–y grid were specified by two computer-generated random numbers. The random method served to count ∼50% of the coverslip area. The accuracy was assessed by performing counts of all of the cells on 5% of the coverslips and comparing them with the randomly generated counts for the same coverslips. The values differed by less than ±3% (p > 0.05).
ApopTag and 4,4-dichloro-4-bora-3a,4a-diaza-s-indacene (BODIPY)-conjugated dUTP in situ end labeling of nuclear DNA fragmentation. Two terminal deoxynucleotidyl transferase (TdT) methods were used for the in situ labeling of 3′-OH DNA ends generated by DNA cleavage: (1) the ApopTag two-step method (Oncor, Inc., Gaithersburg, MD) and (2) a BODIPY-conjugated dUTP (Molecular Probes, Eugene, OR) single-step method. After fixation, the coverslips were rinsed with PBS, placed in equilibrating buffer, and then incubated in a reaction buffer consisting of TdT, dUTP, and necessary ions for 30 min at 37°C. After rinsing, coverslips were incubated with peroxidase-conjugated anti-digoxigenin (introduced together with Triton X-100 and a blocking agent). The coverslips were then exposed to 0.5 mg/ml diaminobenzidine and 0.05% hydrogen peroxide to generate a brown reaction product and mounted onto slides with Aquamount (BDH Chemicals, Poole, UK). The percentage of cells with ApopTag-positive nuclei were counted using interference contrast optics (see Fig.1C). Negative controls were performed by substituting distilled water for the TdT enzyme. Positive controls were performed by first nicking the DNA with DNase 1 before applying the ApopTag reagents.
In the single-step procedure (Li et al., 1996), fixed cells on coverslips were rinsed with PBS and incubated in 50 μl of TdT reaction buffer containing the following: 10 μl of 5× concentrated buffer solution, 5 μl of 25 mm cobalt chloride, 5 μl (25 U) of TdT (Promega, Madison, WI), and BODIPY-conjugated dUTP (B-dUTP, Promega) for 60 min at 37°C. After rinsing with PBS, the coverslips were incubated in the same medium containing 0.5 nm dideoxy-dUTP instead of deoxynucleotides for 10 min. The coverslips were then treated with 15 mm EDTA and Triton X-100 in PBS. The coverslips were mounted in glycerol–PBS on microscope slides and examined using ordinary epifluoresence microscopy (excitation, 450–490 nm; long-pass emission, 515 nm) or confocal laser microscopy (excitation, 488 nm; emission 530/30 nm).
Hoechst 33258 staining of nuclear chromatin.2′-(4-Hydroxyphenyl)−5-(4-methyl-1-piperazinyl)−2–5′-bi-1H-benzimidazole trihydrochloride pentahydrate (Hoechst 33258, Molecular Probes) binds to contiguous A–T bases in DNA. Treated cells were fixed on coverslips and exposed to 1 μg/ml Hoechst 33258 in PBS for 15 min at room temperature. The cells were then washed three times in PBS and viewed with epifluorescence microscopy using 340–380 nm excitation coupled with 430 nm long-pass emission filtration. The nuclei of the partially neuronally differentiated PC12 cells appeared oval with a septate pattern of fluorescence (see Fig. 1B). Upon chromatin condensation, the nuclei decreased in size and became intensely fluorescent (see Fig. 1B1), often with ring-like structures (see Fig. 1B3) and apoptotic bodies (see Fig. 1B2).
Chloromethyl-tetramethylrosamine methyl ester estimation of ΔΨM in paraformaldehyde-fixed cells. Cationic, lipophilic dyes, such as rhodamine 1,2,3 (Rh123) and tetramethylrosamine ester (TMRE) accumulate in the negatively charged matrix of mitochondria (Johnson et al., 1981). Experiments using hepatocytes have shown that the ratio of Rh123 or TMRE fluorescence in the cytoplasmic and nuclear compartments can yield a reliable estimate of ΔΨM in single cells with an accuracy of 10–20 mV (Ubl et al., 1996) for values of up to −150 mV. With Rh123 or TMRE, ΔΨM must be measured ad hoc in living, metabolically active cells so that immunocytochemistry for specific proteins orin situ end labeling (ISEL) for DNA fragmentation cannot be applied to cells identical to those examined for ΔΨM.
Recently, fluorescent probes for ΔΨM have been developed that are retained in cells at the time of fixation and can be used to examine ΔΨM together with levels and localization of specific proteins or nuclear DNA fragmentation (seeMacho et al., 1996). One of those dyes, chloromethyl-tetramethylrosamine methyl ester (CMTMR, Mitotracker Orange; Molecular Probes) has the same basic structure as Rh123 and TMRE with the addition of a chloromethyl moiety, which reacts with thiol groups on peptides. CMTMR enters mitochondria proportionally to the difference between the negativity of the cytoplasmic compartment and mitochondrial matrix, and the chloromethyl group reacts with thiols on proteins and peptides to form aldehyde-fixable conjugates. Once ΔΨM is lost, Rh123 and TMRE are washed out of the mitochondria, but CMTMR remains bound even after permeabilization and fixation (Calarco, 1995; Chen and Cushion, 1994). Therefore, CMTMR fluorescence represents the highest level of negativity difference in the mitochondria during the period of dye exposure before fixation.
At 3, 6, 12, and 24 hr after washing, the medium in each well was supplemented with 50 nm CMTMR and incubated at 37°C for 15 min. The solution was replaced with dye-free media for a further 10 min before washing with PBS. The cells were then fixed on ice for 10 min with 4% paraformaldehyde. After fixation, the cells were rinsed with PBS, and the coverslips were mounted onto slides using Aquamount.
The mitochondria were visualized using a Reichert Polyvar II epifluorescence microscope (Leica, Heidelberg, Germany) equipped with an oil immersion, Fluotar 100×, 1.32 numerical aperture (NA) objective (see Fig. 1D). CMTMR was excited at 540 nm, and the 590 nm emission image was captured using a charge-coupled device (Javelin Electronics Inc.) attached to an integrated silicon-intensified target device (Hammamatsou Photonics, Inc.) and digitized using a Matrox Image LC video digitization board connected to a 486DX2/66 personal computer (PC) running Metamorph (Universal Imaging Corporation, West Chester, PA) or Northern Exposure (Empix Ltd.) imaging software. Images were acquired using 512 × 480 or 512 × 512 × 8 bits per pixel resolution using 32 frame averaging with no background correction (see Fig.1D).
Average CMTMR mitochondrial fluorescence intensity was estimated for each cell by acquiring intensity values from 10 different 4 μm2 regions within the cytoplasm and calculating a ratio of the average of the 10 values divided by the average fluorescence intensity within the nucleus (see Ubl et al., 1996). An intensity ratio of 1.0 represented cytoplasmic fluorescence equal to that in the nucleus. The values for 50 cells under each treatment from three different experiments were compared statistically and presented as frequency distributions.
Confocal microscopy was then used to resolve individual mitochondria labeled with CMTMR at the time points of 6 and 24 hr after washing (see Fig. 1E). A Leica true confocal scanning microscope coupled to an argon–krypton laser (Omnichrome, USA) was used. A pinhole of 50 was used with an excitation filter wavelength of 488 nm and a long-pass emission filter of 590 nm. Images were scanned using an oil immersion, 100×, 1.3 NA objective at 512 × 512 × 8 bits per pixel resolution, background offset of 0, and averaged 16–32 times in bidirectional scan mode. The images were saved in tagged image file format (TIFF) and transferred to a 100 MHz Pentium PC. Image Pro Plus for Windows software (Media Cybernetics) was used to threshold individual mitochondrial outlines and then to measure the mean intensity within each mitochondrion. The value for each mitochondrion was normalized against mean intensity for the immediately adjacent cytoplasm, and the values were presented as frequency distributions.
Immunocytochemistry for tubulin and histones. Cells grown on coverslips were fixed for 15 min with 4% paraformaldehyde at 4°C. Nonspecific binding of protein was blocked by incubating the coverslips in 10% normal goat serum for 1 hr. The cells were permeabilized simultaneously with 0.1% Triton X-100 in PBS. Incubations with primary antibodies were done overnight at 4°C. Mouse anti-tubulin (1:100, Sigma) (see Fig. 5A1) and mouse anti-histone (1:200; Boehringer Mannheim, Indianapolis, IN) (see Fig.5B1,C1) were used in the presence 0.2% normal serum and 0.05% Triton X-100. Coverslips were washed with PBS four times and incubated for 1 hr with Texas Red-labeled anti-mouse antibody (1:500) in the dark at room temperature. The coverslips were then washed four times in PBS and mounted in PBS/glycerol and sealed before viewing.
Fig. 5.

Nuclear DNA fragmentation demonstrated with BODIPY-dUTP simultaneously with CMTMR staining to estimate ΔΨM. A1, A2, Low-power epifluorescence micrographs of the same image fields of cells after 12 hr in M/O that were immunoreacted for tubulin (A1) and also reacted using the BODIPY-dUTP method for detecting nuclear DNA fragmentation (A2). Confocal laser images of identical fields for tissue dually reacted with an anti-histone antibody are shown inB1 and C1, and BODIPY-dUTP is shown inB2 and C2. D1–D3, E1–E3, Identical confocal image fields for serial image planes (separated by ∼1.0 μm) through a group of partially neuronally differentiated PC12 cells in M/O at 6 hr after washing. Cultures are stained for BODIPY-dUTP nuclear staining and CMTMR mitochondrial fluorescence, respectively.
Some of the coverslips had been exposed to CMTMR for ΔΨM measurements before fixation or were immunoreacted for tubulin or histone identification before in situlabeling of nuclear DNA fragmentation using BODIPY-conjugated dUTP.
JC-1 estimation of ΔΨM in living cells.5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethybenzimidazol carbocyanine iodide (JC-1, Molecular Probes) is a lipophilic, cationic dye that enters mitochondria in proportion to the membrane potential and forms J-aggregates at the high intramitochondrial concentrations induced by higher ΔΨM values. Formation of the J-aggregates is associated with a Stoke’s shift in emission (Smiley et al., 1991) from 527 nm for the monomer to 590 nm for the J-aggregate. The dye allows for a dual measurement of dye concentration that does not require the measurement of a nuclear or cytoplasmic reference value.
Studies using isolated cardiac myocyte mitochondria have shown that the 527 nm emission from monomeric JC-1 increases almost linearly with ΔΨM potentials ranging from −46 to −182 mV, whereas the 590 nm J-aggregate emission is less sensitive to ΔΨM values less negative than −140 mv and is strongly sensitive to potential values in the range of −140 to −182 mV (Di Lisa et al., 1995). Calculation of the 527:590 nm fluorescence ratio eliminates many of the potential pitfalls of fluorescent dye measurements, which require comparison between different cellular compartments. Because the ratio can be calculated on a pixel-for-pixel basis, it eliminates the possibility that changes in the volume of the emitter account for changes observed for one experimental condition relative to another. JC-1 imaging using a confocal microscope offers a finer resolution and sensitivity of mitochondrial potential in living cells and allows ΔΨM to be estimated for single living mitochondria.
At 6 and 24 hr after washing, the media from each well containing PC12 cells grown on coverslips was removed, and a modified medium containing 10 μg/ml JC-1 in prewarmed MEM was added. The cells were placed back into the incubator (37°C, 5% CO2, 100% humidity) for 10 min, and the cells were then washed twice with HBSS to remove unbound dye. The coverslips were then placed into a thermostatically controlled cell chamber (Fine Science Tools Ltd.) into which warmed MEM was circulated. Thermoregulation of the chamber was achieved by passing a current through an enclosed nichrome wire, which was controlled by feedback from a thermocoupler immersed in the media to maintain media temperature at 37 ± 0.1°C.
The JC-1-treated coverslips were excited at 488 nm, and emission was recorded simultaneously at 527 and 590 nm into independent detectors. Images were scanned using an oil immersion, 100×, 1.3 NA objective at 256 × 256 or 512 × 512 × 8 bits per pixel resolution, background offset of 0, and averaged 16–32 times in a bidirectional scan mode. The confocal pinhole aperture was set to 50, and the voltage to the photomultiplier tubes of each channel was maintained at equal values. Illumination was limited to periods of image acquisition. In this way, the two images were exactly in phase and represented the amount of monomeric and J-aggregate JC-1 fluorescence (see Fig. 7 for images generated by combining the 527 and 590 nm images into a single image). For each time point, 10–20 mitochondria were measured from 50 to 70 cells from each of three to six coverslips taken from two duplicate experiments.
Fig. 7.
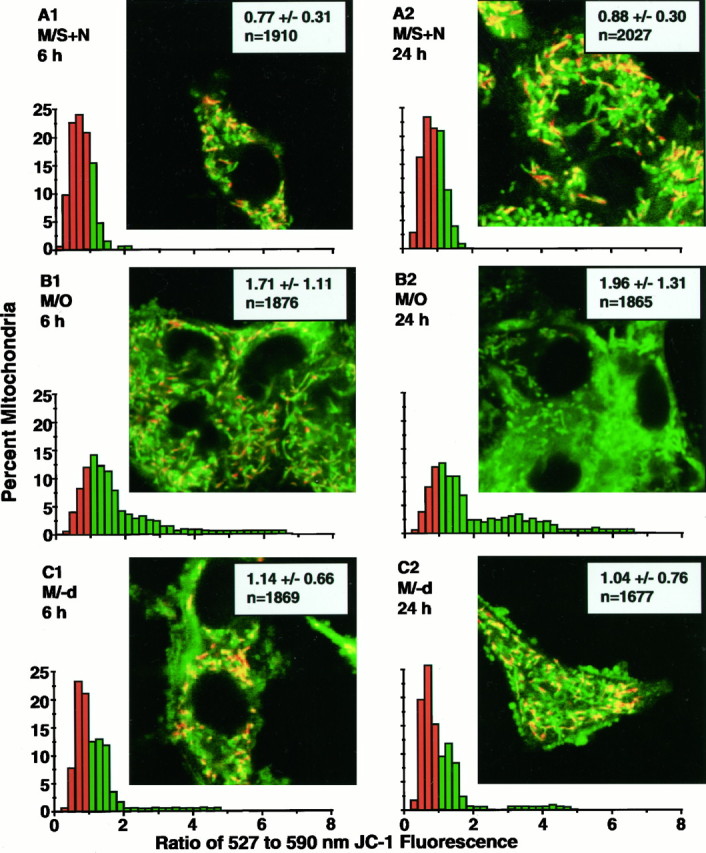
Dual-emission images for JC-1 fluorescence. Eachinset image shows the effect of superimposition of the recolored 527 and 590 nm images by the algebraic addition of corresponding pixels: regions of mitochondria with predominant 527 nm emission color green, those with overlapping 527 and 590 nm emission color yellow, and those with higher 590 nm emission color orange–red in the superimposed images. Each panel shows typical examples of superimposed JC-1 images and 527:590 nm emission ratio distributions for the pixel domains of individual mitochondria for the three treatment conditions at 6 and 24 hr after washing. A1, A2, C1, C2, For cells in M/S+N and M/−d, respectively, show a mixture of green,yellow, and orange–red mitochondria in a single cell and appear to indicate a wide range of ΔΨMvalues for mitochondria within a single cell. The corresponding distributions for these panels show that the majority of mitochondria have 527:590 nm emission ratios that are <1.0, indicating high levels of ΔΨM. B1, Cells in M/O at 6 hr and relative decrease in orange–red mitochondria and a preponderance of yellow and greenmitochondria found in those cells. As shown in the accompanying distribution, those changes reflect a shift in the mitochondrial 527:590 nm emission ratios to higher values, with a majority of the mitochondria showing ratios of >1.0. B2, Cells in M/O at 24 hr and little, if any, yellow ororange–red. The accompanying distribution shows a further shift of the emission ratios to values of >1.0.
Each image pair was analyzed in TIFF format using Image Pro Plus for Windows software (Media Cybernetics) by conversion from 8 bit format to floating point format to avoid division errors. The 527:590 nm ratio image was calculated by dividing the pixel values of the 527 nm image with its corresponding pixel value of the 590 nm image and then scaling the result by 50. Using the ratio image, individual mitochondria were digitally traced from the corresponding 527 nm image, and the average intensity was calculated by integrating all of the intensity values within the bound area and dividing by the number of pixels. The mitochondrial data from all cells within a single treatment group were pooled and plotted as an aggregate frequency distribution histogram.
Dihydro-rhodamine-2-acetylmethyl ester estimation of mitochondrial free calcium. Dihydro-rhodamine-2-acetylmethyl ester (Rhod-2AM, Molecular Probes) is structurally related to Rh123 and undergoes a 3.4-fold enhancement of fluorescence on binding to Ca2+ (Minta et al., 1989). The rhodamine portion of the molecule causes the Rhod-2AM to be concentrated in mitochondria and thereby serves as an indicator of mitochondrial Ca2+levels. Cells were incubated at 37°C for 20 min in media containing 5 mm Rhod-2AM in MEM. The cells were washed with two changes of HBSS and transferred to a temperature-controlled chamber (see above). Confocal images of the cellular fluorescence were obtained using a 100×, 1.3 NA oil immersion objective, excitation wavelength of 488 nm, emission wavelength of 575 nm, pinhole setting of 90, and averaging of 16 images (see Fig. 8). Individual mitochondrial-like organelles within each cell were digitally traced, and the mean fluorescence was determined using Image Pro Plus for Windows software similar to the procedures above for CMTMR. Data for each treatment group were plotted as a frequency distribution histogram.
Fig. 8.

Rhod-2AM as an indicator of mitochondrial Ca2+. A1–A3, B1–B3, insets, Typical confocal microscopic images of living cells stained with Rhod-2AM for the three treatments at 6 and 24 hr after washing. The accompanyinggraphs show the distributions for the Rhod-2AM fluorescence for subcellular structures likely to correspond to mitochondria at the two time points. Values above each distribution present the mean ± SD and the numbers of mitochondria examined in each group. The distributions show that Ca2+ levels in mitochondrial-like organelles were markedly elevated at both 6 and 24 hr after washing and placement in M/O and were only partially reduced by treatment with (−)-deprenyl.
2′,7′-Dihydrodichlorofluorescein estimation of cytoplasmic peroxide radicals. 2′,7′-Dihydrodichlorofluorescein (DCFH2) is a sensitive fluorometric probe for peroxide radicals. The addition of the acetate moieties gives the molecule lipophilic properties that enables the use of dihydrodichlorofluorescein diacetate (DCFH2-DA) as a probe for H2O2 production in living cells (LeBel et al., 1992). DCFH2-DA permeates the cell membrane and is hydrolyzed into DCFH2, a nonfluorescent compound that remains trapped within the cell. Oxidation of DCFH2 is induced by H2O2 in a stoichiometric reaction catalyzed by peroxidases, yielding the fluorescent product 2′,7′-dichlorofluorescein.
Stock DCFH2-DA solutions were prepared by dissolving the solid in ethanol to a concentration of 1 mg/ml. Aliquots were stored in the dark at −80°C. At 24 hr after washing, the media were replaced with 100 μm DCFH2-DA in MEM. The cells were incubated at 37°C, 5% CO2, and 100% humidity in the dark for 30 min and then washed three times in HBSS before placement into a temperature-controlled cell chamber. Confocal images of the cellular fluorescence were obtained using a 100×, 1.3 NA oil immersion objective, excitation wavelength of 488 nm, emission wavelength of 530 nm, pinhole setting of 90, and line averaging of 16 images.
Cellular fluorescence was quantitated by measuring the average intensity within the non-nuclear cytoplasm of each cell using Image Pro Plus for Windows software and calculating the average intensity of the cell body cytoplasmic area for each cell.
Statistical evaluation. To evaluate the data, the individual measurements from different treatment groups were first analyzed using Statistica software (StatSoft) to perform two-tailed independent samplet testing. Levene’s testing for homogeneity of variances showed that most pairs of samples were not homogeneous, and χ2 evaluation of the distributions showed that most did not fit a normal distribution (see Siegel, 1956). If the data were nonhomogeneous and did not fit to a normal distribution, analysis with parametric methods such as the t test might not provide valid results. The data were therefore rank-ordered and compared in a pairwise manner using Statistica software to perform nonparametric Mann–Whitney U testing (Siegel, 1956). The method depends on permutations to calculate significance values and therefore does not require homogeneity of variances, that the underlying distributions for the data be known, or that the values are linearly related.
RESULTS
Cell death, nuclear chromatin condensation, and DNA strand breakage increases at 6 hr
As illustrated in Figure1A2, cells that underwent NGF and serum withdrawal and survived beyond 6 hr showed a marked reduction in process number and length compared with cells replaced into M/S+N (Fig. 1A1). Counts of intact nuclei and intact methylene blue-stained cells (Fig.2A) revealed statistically insignificant losses of between 5 and 10% at 3 hr for cells in M/O (p > 0.05 compared with M/S+N). The losses increased to 10–15% at 6 hr (p < 0.05 compared with M/S+N), 60–70% at 12 hr (p< 0.0001), and 65–80% at 24 hr (p < 0.0001). Hence about two-thirds of the total cell death found over 24 hr occurred between 6 and 12 hr after trophic withdrawal. The addition of M/−d did not alter the nuclear or cell loss at 6 hr (p > 0.05 compared with M/O) but increased both the percentages of intact cells (Fig. 2A) and nuclei (Fig. 2B) at both 12 and 24 hr (p < 0.001 compared with M/O).
Fig. 2.

Estimates of the time course of cell death and the appearance of the nuclear stigmata (chromatin condensation and DNA fragmentation) of apoptosis. A, Values at four time points after washing for the two methods that we used to estimate cell survival: counts of intact nuclei after plasma membrane lysing using a hemocytometer, and counts of intact cells from coverslips after fixation and methylene blue staining. B, Percentages of cells with nuclear chromatin condensation and nuclear DNA fragmentation at the same four time points.
A small baseline percentage (1–2%) of nuclei with chromatin condensation or nuclear DNA fragmentation were found in cells in M/S+N at all four time points (Fig. 2B). Neither percentage was increased above the baseline for cells in M/O at 3 hr (p > 0.05 compared with M/S+N), but both chromatin condensation (∼4%, p < 0.05 compared with M/S+N)) and nuclear DNA fragmentation (∼3.5%, p < 0.05 compared with M/S+N) showed small increases at 6 hr. Both increased to >20% at the 12 hr time point (p< 0.001 compared with M/S+N) and exceeded 30% at the 24 hr time point (p < 0.001 compared with M/S+N). Examples of chromatin condensation (Fig. 1B) and DNA fragmentation (Fig. 1C) in M/O cells at 9 hr after trophic withdrawal are illustrated. Accordingly, the major increase in cells showing the nuclear stigmata of apoptosis occurred after the 6 hr time point. Our previous use of DNA electrophoresis gels to determine the time course of DNA fragmentation in the partially neuronally differentiated PC12 cells after trophic withdrawal (Tatton et al., 1994b) revealed a time course similar to that found with the ISEL method (also see BODIPY-dUTP below). In those studies, DNA ladders were not detectable at 3 and 6 hr but were easily detectable at 9 and 12 hr after placement in M/O.
Figure 2B illustrates that cells in M/−d did not differ in the percentage of nuclei with chromatin condensation or DNA fragmentation from those in M/O at either 3 or 6 hr (p > 0.05 compared with M/O). The percentage of nuclei with either chromatin condensation or DNA fragmentation for cells in M/−d at 12 and 24 hr after washing exceeded those in M/S+N (p < 0.05 compared with M/S+N) but was markedly reduced compared with those in M/O (p < 0.0001 compared with M/O).
Changes In average cellular CMTMR fluorescence are present by 3 hr
Superimposition of fluorescence from adjacent mitochondria made it impossible to measure CMTMR fluorescence reliably from individual mitochondria with epifluorescence microscopy (Fig.1D). Accordingly, we compared cell body cytoplasmic fluorescence with nuclear fluorescence (cytoplasmic/nuclear ratio) as an estimate of average ΔΨM in each cell (Ubl et al., 1996). The left column of CMTMR cytoplasmic/nuclear ratio distributions in Figure 3 for cells in M/S+N at the four time points shows that the ratios ranged from 1.0 to ∼5.0 with means of 2.2–2.4. The values did not differ significantly for cells in M/S+N among any of the four time points (allp < 0.05; see Table 1for results of pairwise comparisons using both parametric and nonparametric statistical tests). In contrast to those for M/S+N, the ratio distribution for cells in M/O at 3 hr showed an overall shift to lower values when compared with M/S+N at the same time point (Fig. 3, Table 1, first row of distributions). The extent of the shift to lower ratios is illustrated by the graph labeledM/O in Figure 6A, which shows that more than 25% of the cells had average ratios of <1.6 in M/O at 3 hr, whereas only 11% of cells in M/S+N had ratios smaller than that value (see Fig. 6A, graph labeledM/S+N).
Fig. 3.

Estimation of the time course of changes in average cellular ΔΨM using epifluorescence imaging of the rhodamine derivative CMTMR. Distributions for cell body cytoplasmic CMTMR fluorescence divided by CMTMR nuclear fluorescence (cytoplasmic/nuclear ratio) as an estimate of changes in average ΔΨM after washing in each cell for the three treatments at four time points.
Table 1.
Statistical testing of data for cytoplasm/nuclear ratio of CMTMR fluorescence measurements taken using epifluorescence microscopy
| 3 h | 6 h | 12 h | 24 h | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M/O:M/SN | M/−d:M/SN | M/O:M/−d | M/O:M/SN | M/−d:M/SN | M/O:M/−d | M/O:M/SN | M/−d:M/SN | M/O:M/−d | M/O:M/SN | M/−d:M/SN | M/O:M/−d | ||||
| Independent sample t test for CMTMR cell nuclear ratio | 4.88 × 10−2 | 7.19 × 10−1 | 8.09 × 10−2 | 1.51 × 10−2* | 6.36 × 10−1 | 8.71 × 10−5* | 9.73 × 10−8* | 1.98 × 10−1 | 7.64 × 10−10 | 1.61 × 10−9* | 8.39 × 10−1 | 3.95 × 10−9* | |||
| Mann–Whitney U test for CMTMR cell nuclear ratio | 4.19 × 10−2* | 6.59 × 10−1 | 1.13 × 10−1 | 1.07 × 10−4* | 3.24 × 10−1 | 1.84 × 10−6* | 2.19 × 10−10* | 3.28 × 10−1 | 1.83 × 10−10* | 2.87 × 10−10* | 6.64 × 10−1 | 5.29 × 10−9* | |||
*Significance difference, p < 0.05.
Fig. 6.

Time course of changes in the levels of CMTMR and JC-1 fluorescence and differences in CMTMR fluorescence for cells with or without nuclear DNA fragmentation or chromatin condensation. Each plot in A–C shows percentage of cells or mitochondria with CMTMR fluorescence ratios or JC-1 527:590 nm fluorescence ratio values that are less than values shown to correspond with specific ΔΨM levels in other cellular models. D, Bar graphs of the CMTMR mitochondrial/cytoplasmic ratios and CMTMR cytoplasmic/nuclear ratios for cells in M/O at 6 hr after washing with and without evidence of nuclear DNA fragmentation or chromatin condensation shown by staining with Hoechst 33258.
By 6 hr after washing and placement in M/O, there was a marked shift in CMTMR cytoplasmic/nuclear ratios to lower values (probabilities ranging from 1.51 × 10−2 to 1.07 × 10−4 depending on the statistical test used; see Table 1). The distributions for the CMTMR cytoplasmic/nuclear ratios for cells in M/O progressively shifted to lower and more highly statistically significant values compared with those in M/S+N at 12 and 24 hr (Fig. 3, Table 1). The extent of the shift to lower ratios is illustrated in Figure 6A, showing that ∼47, 72, and 70% of cells had average ratios of <1.6 at 6, 12, and 24 hr, respectively.
Treatment with 10−9m (−)-deprenyl reduced the shift in the CMTMR cytoplasmic/nuclear ratio distributions (Fig. 3, right column of distributions; see Fig. 6A) so that ratios were significantly different for M/−d relative to M/O but not for M/−d relative to M/S+N (Table 1).
CMTMR fluorescence for individual mitochondria have shifted markedly by 6 hr
To address the possibility that changes in nuclear CMTMR binding accounted for the above findings and to determine how changes in average ΔΨM within a cell relate to shifts in the values for individual mitochondria within a single cell, we measured mitochondrial CMTMR fluorescence relative to the fluorescence in the immediately adjacent cytoplasm (mitochondrial/cytoplasmic ratio) using confocal laser microscopy (Fig. 1E).
The distributions for cells in M/S+N (Fig.4, top row) did not differ at 6 and 24 hr (p > 0.05), whereas those for cells in M/O (Fig. 4, middle row) were progressively and significantly shifted to lower values at the same time points (see Fig.6, Table 2). The mean values for the inset plots (each column of points in the insets present 15 mitochondrial/cytoplasmic ratios measured from mitochondria in a single cell) show variability for different cells in M/S+N that is similar to that found for the cytoplasmic/nuclear ratios presented above. The insets show a surprisingly high variability in CMTMR fluorescence for different mitochondria within a single cell in M/S+N. Relative CMTMR fluorescence for individual mitochondria could vary as much as sixfold within a single cell. Mitochondria in M/O showed a progressive fall and narrowing of the CMTMR ratios when 6 and 24 hr were compared (Fig. 4). (−)-Deprenyl (Fig. 4, bottom row of distributions) shifted the mitochondrial/cytoplasmic distributions toward those for cells in M/S+N. The shift was not complete because the Mann–WhitneyU test revealed a statistically significant difference between the treatments at 6 and 24 hr (Table 2).
Fig. 4.

Estimation of the changes in ΔΨMfor individual mitochondria using confocal microscopic imaging of CMTMR fluorescence. Distributions of the CMTMR mitochondrial/cytoplasmic ratios for the three treatments at the 6 and 24 hr time points together with corresponding examples (inset) of the CMTMR mitochondrial/cytoplasmic ratios for 20 mitochondria from 10 randomly chosen cells.
Table 2.
Statistical testing of data for mitochondrial/cytoplasmic ratio of CMTMR fluorescence measurements taken using laser confocal microscopy
| 6 h | 24 h | |||||
|---|---|---|---|---|---|---|
| M/O:M/SN | M/−d:M/SN | M/O:M/−d | M/O:M/SN | M/−d:M/SN | M/O:M/−d | |
| Independent sample t test for CMTMR mitochondrial cytoplasmic ratio | >1.00 × 10−15* | 1.39 × 10−1 | >1.00 × 10−15* | 0.00 × 10−10* | 1.32 × 10−3* | >1.00 × 10−15* |
| Mann–Whitney U test for CMTMR mitochondrial cytoplasmic ratio | >1.00 × 10−15* | 5.01 × 10−3* | >1.00 × 10−15* | >1.00 × 10−15* | 4.27 × 10−6* | >1.00 × 10−15* |
*Significance difference, p < 0.05.
Cells with chromatin condensation or nuclear DNA fragmentation have reduced or absent ΔΨM
The BODIPY-dUTP ISEL method for detecting nuclear DNA fragmentation provided complementary results to those for the ApopTag method. For cells in M/O, the method revealed that 1.4 ± 0.2% of nuclei showed evidence of DNA fragmentation at 3 hr and 3.1 ± 0.5, 26.2 ± 3.1, and 34.5 ± 5.2% at 6, 12 (Fig.5A1,A2), and 24 hr, respectively.
Immunoreactive nuclear histones were distributed in a diffuse, lattice-like pattern in nuclei that did not show evidence of DNA fragmentation in corresponding images for BODIPY-dUTP (Fig. 5B1,B2, respectively). In nuclei demonstrating DNA fragmentation, the histone immunoreaction was found to be marginalized to the outer portion of areas of BODIPY fluorescence, as shown in Figure 5, B1, B2, C1, and C2.
Figure 5, D1–D3 and E1–E3, shows identical confocal image fields for serial image planes (separated by ∼1.0 μm) through a group of cells in M/O at 6 hr. Figure5D1–D3 shows the fluorescence for the B-dUTP reaction and demonstrates a nucleus that has undergone DNA fragmentation and has broken into three or more pieces. The CMTMR fluorescence in the corresponding images (Fig. 5E1–E3) shows well defined mitochondrial CMTMR fluorescence in all cells that do not show evidence of nuclear DNA fragmentation, whereas the cell with nuclear DNA fragmentation shows shrunken cytoplasm with markedly reduced CMTMR fluorescence.
Cells in M/O at 6 hr after washing that showed evidence of nuclear DNA fragmentation showed average mitochondrial/cytoplasmic ratios of 1.15 (Fig. 6D, left). Ratios of 1.0–1.4 would fall among the lowest values in the 6 hr M/O distribution in Figure 4 and would appear to represent ∼20% of the mitochondria in comparison to <2% of the mitochondria for cells in M/S+N (Fig. 6B). Similarly, cells without evidence of chromatin condensation had average CMTMR cytoplasmic/nuclear ratios (Fig. 6D, right) that were not significantly different (p > 0.05) from those previously found for cells in M/S+N at the same time point (Fig. 3). Those with well defined chromatin condensation had significantly reduced CMTMR cytoplasmic/nuclear ratios (p < 0.001) that were in the lower range of those previously found for cells in M/O (Figs. 3, 6A).
Decreases in mitochondrial JC-1 fluorescence in living cells parallel those found with CMTMR
To address the possibilities that changes in mitochondrial size accounted for some or all of the changes in fluorescence or that fixation altered the cytoplasmic/nuclear or mitochondrial/cytoplasmic ratios to differing extents for the three treatments, we maintained living cells in a temperature-controlled bath and used dual simultaneous detection confocal laser microscopy to measure the 527 and 590 nm fluorescence of the JC-1 monomer and J-aggregate, respectively. Pixel-for-pixel algebraic addition of the 527 nm emission images colored green and 590 nm emission images colored red allowed us to visualize mitochondria with low (green motochondria), medium (yellow mitochondria), and high (red mitochondria) ΔΨM levels in single images (Fig. 7,insets). A mixture of green, yellow, and orange–red mitochondria within single cells in M/S+N or M/−d (Fig.7A1,A2,C1,C2, insets) seemed to show the same wide range of ΔΨM for different mitochondria in single cells indicated by CMTMR mitochondrial/cytoplasmic ratios (Fig. 4,insets for M/S+N and M/−d). A progressive reduction in orange–red and then yellow mitochondria in cells in M/O at 6 and 24 hr (Fig. 7B1,B2) revealed the same fall and narrowing of ΔΨM for the mitochondrial population in a single cell as found with the CMTMR mitochondrial/cytoplasmic ratios.
It should be noted that shifts of distributions of CMTMR cytoplasmic/nuclear (Fig. 3) or mitochondrial/cytoplasmic (Fig. 4) ratios to lower values indicate a decrease in ΔΨM. In contrast, shifts of the distributions (Fig. 7) of the 527:590 nm ratios to lower values indicate an increase in ΔΨM, whereas increased ratios indicate decreased ΔΨM values (Di Lisa et al., 1995). Mitochondria in cells in M/O showed a significant shift in the 527:590 ratio to higher values compared with those in M/S+N at both 6 and 24 hr (Figs. 6C, 7, Table3). The distributions for cells in M/−d revealed a maintenance of the ratio for a majority of the mitochondria but similar to that found for the CMTMR mitochondrial/cytoplasmic ratios (Figs. 4, 6B); a distinct subpopulation of mitochondria with ratios of >2.0 did not appear to respond to (−)-deprenyl treatment at either 6 or 24 hr (Fig.7C1,C2).
Table 3.
Statistical testing of data for 527:590 nm ratio of JC-1 fluorescence measurements taken using laser confocal microscopy
| 6 h | 24 h | |||||
|---|---|---|---|---|---|---|
| M/O:M/SN | M/−d:M/SN | M/O:M/−d | M/O:M/SN | M/−d:M/SN | M/O:M/−d | |
| Independent sample t test for JC-1 527:590 nm ratio | >1.00 × 10−15* | >1.00 × 10−15* | >1.00 × 10−15* | >1.00 × 10−15* | >1.00 × 10−15* | >1.00 × 10−15* |
| Mann–Whitney U test for JC-1 527:590 nm ratio | >1.00 × 10−15* | 1.74 × 10−6* | >1.00 × 10−15* | >1.00 × 10−15* | 3.64 × 10−2* | >1.00 × 10−15* |
*Significance difference, p < 0.05.
CMTMR cytoplasmic/nuclear ratio measured with epifluorescence microscopy or whole cell cytofluorescence measurements with a single-wavelength fluorescence emitter offer values that depend on both ΔΨM and the cytoplasmic density of mitochondria. Cell shrinkage would increase the number of mitochondria per unit of cross-sectional area and therefore the measurements. An increase in the number of mitochondria per unit of volume or an increase in mitochondrial size would increase whole cell cytofluorometric measurements independently of ΔΨM when using a single-wavelength emitter dye (Castedo et al., 1996). In contrast, the use of CMTMR mitochondrial/cytoplasmic ratios or JC-1 with confocal images provides measures of ΔΨM that should be uninfluenced by changes in mitochondrial mass.
Increased calcium levels in mitochondrial-like organelles parallel preapoptotic decreases in ΔΨM
Increases in intramitochondrial Ca2+ secondary to high cytoplasmic Ca2+ levels and increases in ROS (Richter et al., 1995) have been considered as primary factors in causing reductions of ΔΨM. To estimate mitochondrial Ca2+ levels and cytoplasmic ROS levels in PC12 cells that survived for 6 and 24 hr after NGF and serum withdrawal, we used the fluorescent dye Rhod-2AM. The Rhod-2AM images showed localized fluorescence in a subcellular component that appeared to correspond to mitochondria. Both the intensity of fluorescence in individual components and the number of visibly fluorescent components increased greatly for cells in M/O (Fig.8A2,B2) compared with those in M/S+N (Fig. 8A1,B1). Cells in M/O often showed a localized region of nuclear fluorescence that appeared to correspond to the nucleolus, and high-resolution confocal images revealed increased fluorescence in the background areas surrounding the subcellular components.
Distributions for the fluorescence in the subcellular component revealed a significant shift to higher values for cells in M/O (Fig.8A2,B2) compared with those in M/S+N (Fig.8A1,B1) with an 80% average increase in fluorescence at 6 hr and 92% average increase at 24 hr (p < 0.001 for the t test and Mann–Whitney statistical comparisons). Addition of (−)-deprenyl to the MEM (Fig.8A3,B3) shifted the distribution back toward the lower values found for cells in M/S+N, but the mean values for the distributions were increased by 62 and 53% above M/S+N values at 6 and 24 hr, respectively (p > 0.01).
Cytoplasmic peroxyl radical levels increase after decreases in ΔΨM
Cells in M/S+N showed low levels of DCFH2 fluorescence (Fig.9A1,B1). DCFH2cytoplasmic fluorescence was not significantly increased at 6 hr in cells in M/O (Fig. 9A2; p > 0.05) but was increased in cells surviving to 24 hr (Fig. 9B2, p > 0.05). As illustrated by the image and the distribution in Figure 9B2, nearby cells could show markedly different levels of cytoplasmic DCFH2 fluorescence after trophic withdrawal. (−)-Deprenyl treatment at 24 hr returned the average cytoplasmic DCFH2 fluorescence levels (Fig.9B3) to within the range of those found in cells in M/S+N at the same time points (p > 0.05).
Fig. 9.

DCFH fluorescence as an indicator of cytoplasmic peroxyl radical levels. A1–A3, B1–B3, insets, Typical examples of DCFH fluorescence of living cells in M/S+N, M/O, and M/−d, respectively, at 6 and 24 hr after washing. The accompanyinggraphs show the distributions for the DCFH fluorescence in the cellular cytoplasm. Values above each distribution present the mean ± SD and the number of cells examined in each group. The distributions show that cytoplasmic peroxyl radical levels were not elevated at 6 hr when ΔΨM was already markedly decreased but were elevated in cells surviving to 24 hr.
DISCUSSION
Decreased mitochondrial membrane potential precedes nuclear DNA cleavage, nuclear chromatin condensation, and cell death
One third of the average decrease in our estimators of ΔΨM found at 24 hr had occurred by 3 hr, and more than half had occurred by 6 hr (Fig.10A). The percentages of cells with detectable nuclear chromatin condensation or DNA fragmentation were not increased above baseline at 3 hr and were increased by only 2–3% above baseline at 6 hr. Therefore a decrease in ΔΨM led the nuclear changes in an average cell by at least 3 hr. The relative timing of cell death, nuclear changes, and a decrease in the fluorescence of a mitochondrially accumulated dye in our study using microscopic measurements seems similar to that found using whole-cell cytofluorescence measurements in rodent embryo cells (Vayssiere et al., 1994), thymocytes (Castedo et al., 1995; Petit et al., 1995), and lymphocytes (Zamzami et al., 1995a) entering apoptosis.
Fig. 10.

Timing of cell death, the appearance of the nuclear stigmata of apoptosis, Rhod-2AM levels in mitochondrial-like organelles, cytoplasmic peroxyl radical levels, and decreases in ΔΨM in trophically withdrawn cells. A, Time course for cells in MEM only; B, time course for those in MEM with 10−9m (−)-deprenyl. The number of intact nuclei (open triangles) determined by cell lysing and direct nuclear counting and number of intact cells (inverted open triangles) determined by methylene blue staining yielded a decrease to 30–38% of MS+N values by 12 hr and ∼20% by 24 hr. Average values for the CMTMR cytoplasmic/nuclear ratio (open circles) were used to estimate ΔΨM at 3 hr. CMTMR cytoplasmic/nuclear ratio, CMTMR mitochondrial/cytoplasmic ratio (gray circles), and the reciprocal of the JC-1 527:590 nm ratio (black circles) are presented for cells in M/O at 6 hr and show decreases of 20, 39, and 53%, respectively. By 24 hr, surviving cells showed average decreases in ΔΨM of 60–70% depending on the method used. Chromatin condensation (open diamonds) and DNA cleavage (open squares) were shown to increase above baseline only after 6 hr after trophic withdrawal. Calcium accumulation into mitochondrial-like organelles (gray squares) increased to 80% of that measured in cells in MS+N, and cytoplasmic peroxyl radical levels (black diamonds) increased linearly to 65% at 24 hr. Similar measurements were performed on cells treated with M/−d. All measurements remained similar to MS+N values with the exception of calcium accumulation into mitochondrial-like organelles, in which levels increased to 60% beyond MS+N values at 6 hr and stabilized to 50% above control at 24 hr.
Different timetables for apoptosis in partially neuronally differentiated PC12 cells and fully neuronally differentiated PC12 cells
In a previous study of PC12 cell death after trophic withdrawal (Mesner et al., 1995), cells were maintained in RPMI 1640 media with 1% horse serum and 50 ng/ml NGF. The horse serum was withdrawn after 12 d, and the NGF was withdrawn at 15 d. The major period of PC12 cell death was delayed by ∼3 hr compared with the present study, and ∼10% of nuclei in control cells were positive for DNA strand breaks, compared with 2% in our study. Changes in the expression of a variety of genes in their trophically withdrawn, fully differentiated PC12 cells appeared to occur at later times than those found in the partially neuronally differentiated PC12 cells (Tatton et al., 1996). The accelerated apoptosis in our cells may relate to the rapidity of trophic withdrawal or to the constitutive presence of proteins necessary for the progression of apoptosis. Undifferentiated PC12 cells (Rukenstein et al., 1991) and partially neuronally differentiated PC12 cells (Tatton et al., 1994b) do not require new protein synthesis for apoptosis caused by trophic withdrawal. Fully differentiated PC12 cells (Mesner et al., 1992) do require newly synthesized protein for apoptosis, which may also account for the slower progression of apoptosis.
Increased mitochondrial Ca2+ may be responsible for the decrease in ΔΨM after serum and NGF withdrawal
Several factors can account for a decrease in ΔΨM, including increased cytosolic Ca2+ levels with Ca2+ uptake into mitochondria (Richter, 1993), elevated levels of ROS (Skulachev, 1996), decreased electron transport (Krippner et al., 1996), or decreased proton pumping by the mitochondrial respiratory chain (Wolvetang et al., 1994). Our cells in M/O at 6 hr had decreased and relatively uniform ΔΨM levels but showed unchanged cytoplasmic levels of peroxyl radicals according to DCFH2 fluorescence (Fig. 10A). Hence increased cytoplasmic ROS levels did not seem essential for the early decreases in ΔΨM.
Alternately, Rhod-2AM fluorescence suggested an inverse relationship between intramitochondrial Ca2+ levels and ΔΨM in the cells at 6 and 24 hr (Fig.10A). ΔΨM should vary inversely with intramitochondrial Ca2+ levels according to a Nernstian relationship (Richter, 1993). Increases in cytosolic Ca2+ are induced by trophic withdrawal and can lead to apoptosis in PC12 cells (Mariggio et al., 1994; Fulle et al., 1997). NGF, basic fibroblast growth factor (bFGF), and insulin-like growth factor II (IGF-II) prevented increases in cytosolic Ca2+ levels and concomitant decreases in ΔΨM in cultured hippocampal neurons deprived of glucose (Mattson et al., 1993). bFGF and IGF-II reduce the death of PC12 cells caused by serum and NGF withdrawal (Rydel and Greene, 1987; Rukenstein et al., 1991) and are likely constituents of the serum used in the present study. Hence the capacity of NGF- and serum-borne trophic agents to maintain normal cytosolic Ca2+ levels could explain the maintenance of ΔΨM in our cells in M/S+N and the decrease caused by their withdrawal.
Decreases in ΔΨM induce the opening of a PTP in the mitochondrial membranes (Zoratti and Szabo, 1995) and the consequent rupture of the outer mitochondrial membrane, which may allow the release of heat labile AIFs from the intermembrane mitochondrial space (Marchetti et al., 1996a,b; Susin et al., 1996a; Zamzami et al., 1996b). Studies in cell-free systems using Xenopus egg extracts established that mitochondrial factors are essential to nuclear chromatin condensation and DNA fragmentation (Newmeyer et al., 1994). AnN-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone-inhibitable 50 kDa protein is released by decreased ΔΨM in immune cells (Susin et al., 1996a) and appears to be a mitochondrial AIF. Studies reporting that cytochrome C, another possible AIF, was found in the extramitochondrial cytoplasm before any decrease in ΔΨM in preapoptotic cells (Yang et al., 1997) did not provide ΔΨM distributions across the mitochondrial populations of single cells. Because opening of the PTP has been shown to release cytochrome C (Kantrow and Piantadosi, 1997), it remains to be determined whether ΔΨM decreases in a relatively small proportion of the mitochondria of a cell can increase cytoplasmic cytochrome C levels sufficiently.
(−)-Deprenyl maintains ΔΨM and reduces apoptosis by inducing new protein synthesis
(−)-Deprenyl decreases hydroxyl radical levels in the rat striatum exposed to 1-methyl-4-phenyl pyridinium ion (MPP+) (Wu et al., 1993) without inhibiting MAO-B. It also increases superoxide dismutase (SOD1 and SOD2) activity in rat striata (Kitani et al., 1994) and in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-damaged mouse striatum (Thiffault et al., 1995). It alters glial fibrillary acidic protein (GFAP) levels in rat striatum and facial nucleus (Biagini et al., 1993; Ju et al., 1994), bFGF protein in activated astrocytesin vivo (Biagini et al., 1994), nitric oxide synthase in rat hippocampus (Zhang et al., 1996), and BCL-2, SOD1, and SOD2 in partially neuronally differentiated PC12 cells (Tatton et al., 1996). (−)-Deprenyl increases levels of mRNA encoding for GFAP in C6 glioma cells (Li et al., 1993), ciliary neurotophic factor in process-bearing astrocytes (Seniuk et al., 1994), NGF in flat and process-bearing astrocytes (Semkova et al., 1996), neurotrophin receptor (trk C) in the rat forebrain (Ekblom et al., 1994), aromatic amino acid decarboxylase in NGF-supported PC12 cells (Li et al., 1992), and SOD1, SOD2, and BCL-2 in partially neuronally differentiated PC12 cells (Tatton et al., 1996). Translational and transcriptional inhibitors block the capacity of (−)-deprenyl to reduce PC12 cell apoptosis (Tatton et al., 1994a).
We found that (−)-deprenyl maintains ΔΨM but decreases the percentage of cells with nuclear chromatin condensation and DNA fragmentation at 6, 12, and 24 hr and decreases average cytoplasmic DCFH fluorescence at 24 hr (Fig. 9B). Increased BCL-2 levels may explain our findings. Increased bcl-2 expression in fibrosarcoid cells reduced apoptosis and increased ΔΨM (Hennet et al., 1993). Localization of BCL-2 to the outer mitochondrial membrane (Lithgow et al., 1994) seems critical to antiapoptosis (Tanaka et al., 1993; Nguyen et al., 1994). Furthermore, isolated nuclei from cells overexpressing bcl-2 undergo similar chromatin condensation and DNA fragmentation to controls (Susin et al., 1996a), whereas BCL-2 maintains ΔΨM in anucleate cells (Castedo et al., 1996;Zamzami et al., 1995a).
The PTP is composed of a voltage-dependent anion channel, the adenine nucleotide translocator, and a peripheral benzodiazepine receptor (Zoratti and Szabo, 1995). Mitochondrial BCL-2 is localized near the peripheral benzodiazepine receptor and modulates in concert with the receptor (Carayon et al., 1996; McEnery, 1992). BCL-2 seems to facilitate PTP closure (Zamzami et al., 1996a). Hence (−)-deprenyl may induce PTP closure and may maintain ΔΨM by increasing BCL-2 levels. (−)-Deprenyl seems to alter the relationship between intramitochondrial Ca2+ levels and ΔΨM, because (−)-deprenyl treatment maintained ΔΨM levels at near M/S+N values, whereas Rhod-2AM levels remained elevated (Fig. 10, compareA,B). This seems in accord with the finding that bcl-2 overexpression increases the capacity for mitochondrial Ca2+ sequestration (Murphy et al., 1996).
Increased sod1 expression induced by (−)-deprenyl might bolster an action of BCL-2 on the PTP by scavenging cytoplasmic ROS (Kowaltowski et al., 1996). Increased ROS in the presence of increased Ca2+ causes cross linking of mitochondrial protein thiols (Valle et al., 1993), which facilitates opening of the PTP (Chernyak and Bernardi, 1996).
Mitochondria did not appear to participate in apoptosis, because ρo cells lacking mitochondrial DNA died by apoptosis, which could be blocked by bcl-2 overexpression (Jacobson et al., 1993). ρo cells are now known to use glycolytic ATP to maintain normal ΔΨM levels (Skowronek et al., 1992; Marchetti et al., 1996c; Zamzami et al., 1996b). PTP opening by reduced ΔΨM in isolated ρo mitochondria releases a factor that induces the nuclear stigmata of apoptosis (Marchetti et al., 1996c).
This study shows that withdrawal of neurotrophic support causes a decrease in ΔΨM in neural-like cells, and that the decrease precedes the nuclear changes associated with apoptosis. At present, it is not known whether similar decreases in ΔΨM are operative in neuronal apoptosis found in nervous system development or in neurodegeneration.
Footnotes
This work was supported by Medical Research Council of Canada Grant MT5218, the Parkinson’s Foundation of Canada, and The Lowenstein Foundation.
Correspondence should be addressed to Dr. William G. Tatton, Mount Sinai School of Medicine, Department of Neurology, One Gustave L. Levy Place, Annenburg 1494, Box 1137, New York, NY 10029-6574.
REFERENCES
- 1.Beal MF. Mitochondria, free radicals, and neurodegeneration. Curr Opin Neurobiol. 1996;6:661–666. doi: 10.1016/s0959-4388(96)80100-0. [DOI] [PubMed] [Google Scholar]
- 2.Biagini G, Zoli M, Fuxe K, Agnati LF. l-Deprenyl increases GFAP immunoreactivity selectively in activated astrocytes in rat brain. NeuroReport. 1993;4:955–958. doi: 10.1097/00001756-199307000-00030. [DOI] [PubMed] [Google Scholar]
- 3.Biagini G, Frasoldati A, Fuxe K, Agnati LF. The concept of astrocyte-kinetic drug in the treatment of neurodegenerative diseases: evidence for l-deprenyl-induced activation of reactive astrocytes. Neurochem Int. 1994;25:17–22. doi: 10.1016/0197-0186(94)90047-7. [DOI] [PubMed] [Google Scholar]
- 4.Calarco PG. Polarization of mitochondria in the fertilized mouse oocyte. Dev Genet. 1995;16:36–43. doi: 10.1002/dvg.1020160108. [DOI] [PubMed] [Google Scholar]
- 5.Carayon P, Portier M, Dussossoy D, Bord A, Petipretre G, Canat X, Le Fur G, Casellas P. Involvement of peripheral benzodiazepine receptors in the protection of hematopoietic cells against oxygen radical damage. Blood. 1996;87:3170–3178. [PubMed] [Google Scholar]
- 6.Castedo M, Macho A, Zamzami N, Hirsch T, Marchetti P, Uriel J, Kroemer G. Mitochondrial perturbations define lymphocytes undergoing apoptotic depletion in vivo. Eur J Immunol. 1995;25:3277–3284. doi: 10.1002/eji.1830251212. [DOI] [PubMed] [Google Scholar]
- 7.Castedo M, Hirsch T, Susin SA, Zamzami N, Marchetti P, Macho A, Kroemer G. Sequential acquisition of mitochondrial and plasma membrane alterations during early lymphocyte apoptosis. J Immunol. 1996;157:512–521. [PubMed] [Google Scholar]
- 8.Chalmers-Redman RME, Fraser AD, Ju WYH, Wadia J, Tatton NA, Tatton WG. Mechanisms of nerve cell death: apoptosis or necrosis after cerebral ischaemia. In: Green AR, Cross AJ, editors. Neuroprotective agents and cerebral ischaemia. Academic; San Diego: 1997. pp. 1–25. [DOI] [PubMed] [Google Scholar]
- 9.Chen F, Cushion MT. Use of fluoresent probes to investigate the metabolic state of Pneumocystis carinii mitochondria. J Eukaryot Microbiol. 1994;41:79S. [PubMed] [Google Scholar]
- 10.Chernyak BV, Bernardi P. The mitochondrial permeability pore is modulated by oxidative agents through both pyridine nucleotides and glutathione at two separate sites. Eur J Biochem. 1996;238:623–630. doi: 10.1111/j.1432-1033.1996.0623w.x. [DOI] [PubMed] [Google Scholar]
- 11.Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Lisa F, Blank PS, Colonna R, Gambassi G, Siverman HS, Stern MD, Hansford RG. Mitochondrial membrane potential in single living adult rat cardiac myocytes exposed to anoxia or metabolic inhibition. J Physiol (Lond) 1995;486:1–13. doi: 10.1113/jphysiol.1995.sp020786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ekblom J, Jossan SS, Ebendal T, Soderstrom S, Oreland L, Aquilonius SM. mRNA expression of neurotrophins and members of the trk family in the rat brain after treatment with l-deprenyl. Acta Neurol Scand. 1994;89:147–148. doi: 10.1111/j.1600-0404.1994.tb01651.x. [DOI] [PubMed] [Google Scholar]
- 14.Frim DM, Simpson J, Uhler TA, Short MP, Bossi SR, Breakfield XO, Isacson O. Striatal degeneration induced by mitochondrial blockade is prevented by biologically delivered NGF. J Neurosci Res. 1993;35:452–458. doi: 10.1002/jnr.490350413. [DOI] [PubMed] [Google Scholar]
- 15.Fulle S, Mariggio MA, Belia S, Nicoletti I, Fano G. Nerve growth factor inhibits apoptosis induced by S-100 binding in neuronal PC12 cells. Neuroscience. 1997;76:159–166. doi: 10.1016/s0306-4522(96)00352-1. [DOI] [PubMed] [Google Scholar]
- 16.Hennet TG, Bertoni G, Richter C, Peterhans E. Expression of Bcl-2 protein enhances the survival of mouse fibrosarcoid cells in tumor necrosis factor-mediated cytotoxicity. Cancer Res. 1993;53:1456–1460. [PubMed] [Google Scholar]
- 17.Jacobson MD, Burne JF, King MP, Miyashita T, Reed JC, Raff MC. Bcl-2 blocks apoptosis in cells lacking mitochondrial DNA. Nature. 1993;361:365–369. doi: 10.1038/361365a0. [DOI] [PubMed] [Google Scholar]
- 18.Johnson LV, Walsh ML, Bockus BJ, Chen IB. Monitoring of relative mitochondrial membrane potential in living cells by fluoresence microscopy. J Cell Biol. 1981;88:526–535. doi: 10.1083/jcb.88.3.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ju WY, Holland DP, Tatton WG. (−)-Deprenyl alters the time course of death of axotomized facial motoneurons and the hypertrophy of neighboring astrocytes in immature rats. Exp Neurol. 1994;126:233–246. doi: 10.1006/exnr.1994.1061. [DOI] [PubMed] [Google Scholar]
- 20.Kantrow SP, Piantadosi CA. Release of cytochrome c from liver mitochondria during permeability transition. Biochem Biophys Res Commun. 1997;232:669–671. doi: 10.1006/bbrc.1997.6353. [DOI] [PubMed] [Google Scholar]
- 21.Kirschner PB, Jenkins BG, Schulz JB, Finkelstein SP, Matthews RT, Rosen BR, Beal MF. NGF, BDNF and NT-5, but not NT-3 protect against MPP(+) toxicity and oxidative stress in neonatal animals. Brain Res. 1996;713:178–185. doi: 10.1016/0006-8993(95)01513-2. [DOI] [PubMed] [Google Scholar]
- 22.Kitani K, Kanai S, Carrillo MC, Ivy GO. (−)-Deprenyl increases the life span as well as activities of superoxide dismutase and catalase but not of glutathione peroxidase in selective brain regions in Fischer rats. Ann NY Acad Sci. 1994;717:60–71. doi: 10.1111/j.1749-6632.1994.tb12073.x. [DOI] [PubMed] [Google Scholar]
- 23.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome C from mitochondria: a primary site for BCL-2 regulation. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 24.Koutsilieri E, Chen TS, Rausch WD, Riederer P. Selegiline is neuroprotective in primary brain cultures treated with 1-methyl-4-phenylpyridinium. Eur J Pharmacol. 1996;306:181–186. doi: 10.1016/0014-2999(96)00184-7. [DOI] [PubMed] [Google Scholar]
- 25.Kowaltowski AJ, Castilho RF, Vercesi AE. Opening of the mitochondrial permeability pore by uncoupling or inorganic phosphate in the presence of Ca2+ is dependent on mitochondrial-generated reactive oxygen species. FEBS Lett. 1996;378:150–152. doi: 10.1016/0014-5793(95)01449-7. [DOI] [PubMed] [Google Scholar]
- 26.Krippner A, Matsuno-Yagi A, Gottlieb RA, Babior BM. Loss of function of cytochrome c in Jurkat cells undergoing Fas-mediated apoptosis. J Biol Chem. 1996;271:21629–21636. doi: 10.1074/jbc.271.35.21629. [DOI] [PubMed] [Google Scholar]
- 27.LeBel CP, Ischiropoulos H, Bondy SC. Evaluation of the probe 2′,7′-dichlorofluorescein as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol. 1992;5:227–231. doi: 10.1021/tx00026a012. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Melamed MR, Darzynkiewicz Z. Detection of apoptosis and DNA replication by differential labeling of DNA strand breaks with fluorochromes of different color. Exp Cell Res. 1996;222:28–37. doi: 10.1006/excr.1996.0004. [DOI] [PubMed] [Google Scholar]
- 29.Li XM, Juorio AV, Paterson IA, Zhu MY, Boulton AA. Specific irreversible monoamine oxidase-B inhibitors stimulate gene expression of aromatic l-amino acid decarboxylase in PC12 cells. J Neurochem. 1992;59:2324–2327. doi: 10.1111/j.1471-4159.1992.tb10127.x. [DOI] [PubMed] [Google Scholar]
- 30.Li XM, Qi J, Juorio AV, Boulton AA. Reduction in glial fibrillary acidic protein messenger RNA abundance induced by (−)-deprenyl and other monoamine oxidase B inhibitors in C6 glioma cells. J Neurochem. 1993;61:1573–1576. doi: 10.1111/j.1471-4159.1993.tb13660.x. [DOI] [PubMed] [Google Scholar]
- 31.Lithgow T, van Driel R, Bertram JF, Strasser A. The protein product of the oncogene bcl-2 is a component of the nuclear envelope, the endoplasmic reticulum, and the outer mitochondrial membrane. Cell Growth Differ. 1994;5:411–417. [PubMed] [Google Scholar]
- 32.Liu XS, Kim CN, Yang J, Jemmerson R, Wang XD. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 33.Lo AC, Houenou LJ, Oppenheim RW. Apoptosis in the nervous system: morphological features, methods, pathology, and prevention. Arch Histol Cytol. 1995;58:139–149. doi: 10.1679/aohc.58.139. [DOI] [PubMed] [Google Scholar]
- 34.Macho A, Decaudin D, Castedo M, Hirsch T, Susin SA, Zamzami N, Kroemer G. Chloromethyl-X-rosamine is an aldehyde-fixable potential-sensitive fluorochrome for the detection of early apoptosis. Cytometry. 1996;25:333–340. doi: 10.1002/(SICI)1097-0320(19961201)25:4<333::AID-CYTO4>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 35.Marchetti P, Castedo M, Susin SA, Zamzami N, Hirsch T, Macho A, Haeffner A, Hirsch F, Geuskens M, Kroemer G. Mitochondrial permeability transition is a central coordinating event of apoptosis. J Exp Med. 1996a;184:1155–1160. doi: 10.1084/jem.184.3.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marchetti P, Hirsch T, Zamzami N, Castedo M, Decaudin D, Susin SA, Masse B, Kroemer G. Mitochondrial permeability transition triggers lymphocyte apoptosis. J Immunol. 1996b;157:4830–4836. [PubMed] [Google Scholar]
- 37.Marchetti P, Susin SA, Decaudin D, Gamen S, Castedo M, Hirsch T, Zamzami N, Naval J, Senik A, Kroemer G. Apoptosis-associated derangement of mitochondrial function in cells lacking mitochondrial DNA. Cancer Res. 1996c;56:2033–2038. [PubMed] [Google Scholar]
- 38.Mariggio MA, Fulle S, Calissano P, Nicoletti I, Fano G. The brain protein S-100ab induces apoptosis in PC12 cells. Neuroscience. 1994;60:29–35. doi: 10.1016/0306-4522(94)90201-1. [DOI] [PubMed] [Google Scholar]
- 39.Mattson MP, Zhang Y, Bose S. Growth factors prevent mitochondrial dysfunction, loss of Ca2+ homeostasis, and cell injury, but not ATP depletion in hippocampal neurons deprived of glucose. Exp Neurol. 1993;121:1–13. doi: 10.1006/exnr.1993.1066. [DOI] [PubMed] [Google Scholar]
- 40.McEnery MW. Isolation of the mitochondrial benzodiazepine receptor: association with the voltage dependent anion channel and the adenine nucleotide carrier. Proc Natl Acad Sci USA. 1992;89:3170–3174. doi: 10.1073/pnas.89.8.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mesner PW, Winters TR, Green SH. Nerve growth factor withdrawal-induced cell death in neuronal PC12 cells resembles that in sympathetic neurons. J Cell Biol. 1992;119:1669–1680. doi: 10.1083/jcb.119.6.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mesner PW, Epting CL, Hegarty JL, Green SH. A timetable of events during programmed cell death induced by trophic factor withdrawal from neuronal PC12 cells. J Neurosci. 1995;15:7357–7366. doi: 10.1523/JNEUROSCI.15-11-07357.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Minta A, Kao J, Tsien R. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J Biol Chem. 1989;264:8171–8185. [PubMed] [Google Scholar]
- 44.Murphy AN, Bredesen DE, Cortopassi G, Wang E, Fiskum G. Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc Natl Acad Sci USA. 1996;93:9893–9898. doi: 10.1073/pnas.93.18.9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Newmeyer DD, Farschon DM, Reed JC. Cell-free apoptosis in Xenopus egg extracts: inhibition by Bcl-2 and requirement for an organelle fraction enriched in mitochondria. Cell. 1994;79:353–364. doi: 10.1016/0092-8674(94)90203-8. [DOI] [PubMed] [Google Scholar]
- 46.Nguyen M, Branton PE, Walton PA, Oltvai ZN, Korsmeyer SJ, Shore GC. Role of membrane anchor domain of Bcl-2 in suppression of apoptosis caused by E1B-defective adenovirus. J Biol Chem. 1994;269:16521–16524. [PubMed] [Google Scholar]
- 47.Petit PX, Lecoeur H, Zorn E, Dauguet C, Mignotte B, Gougeon ML. Alterations in mitochondrial structure and function are early events of dexamethasone-induced thymocyte apoptosis. J Cell Biol. 1995;130:157–167. doi: 10.1083/jcb.130.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richter C. Pro-oxidants and mitochondrial Ca2+: their relationship to apoptosis and oncogenesis. FEBS Lett. 1993;325:104–107. doi: 10.1016/0014-5793(93)81423-w. [DOI] [PubMed] [Google Scholar]
- 49.Richter C, Gogvadze V, Laffranchi R, Schlapbach R, Schweizer M, Suter M, Walter P, Yaffee M. Oxidants in mitochondria: from physiology to diseases. Biochim Biophys Acta. 1995;1271:67–74. doi: 10.1016/0925-4439(95)00012-s. [DOI] [PubMed] [Google Scholar]
- 50.Rukenstein A, Rydel RE, Greene LA. Multiple agents rescue PC12 cells from serum-free cell death by translation- and transcription-independent mechanisms. J Neurosci. 1991;11:2552–2563. doi: 10.1523/JNEUROSCI.11-08-02552.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rydel RE, Greene LA. Acidic and basic fibroblast growth factors promote stable neurite outgrowth and neuronal differentiation in cultures of PC12 cells. J Neurosci. 1987;7:3639–3653. doi: 10.1523/JNEUROSCI.07-11-03639.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Semkova I, Wolz P, Schilling M, Krieglstein J. Selegiline enhances NGF synthesis and protects central nervous system neurons from excitotoxic and ischemic damage. Eur J Pharmacol. 1996;315:19–30. doi: 10.1016/s0014-2999(96)00593-6. [DOI] [PubMed] [Google Scholar]
- 53.Seniuk NA, Henderson JT, Tatton WG, Roder JC. Increased CNTF gene expression in process-bearing astrocytes following injury is augmented by R(−)-deprenyl. J Neurosci Res. 1994;37:278–286. doi: 10.1002/jnr.490370213. [DOI] [PubMed] [Google Scholar]
- 54.Sherratt HSA. Mitochondria: structure and function. Rev Neurol (Paris) 1991;147:417–430. [PubMed] [Google Scholar]
- 55.Siegel S. Non-parametric statistics for the behavioral sciences, pp 127–136. McGraw-Hill; New York: 1956. [Google Scholar]
- 56.Skowronek P, Haferkamp O, Rodel G. A fluorescence-microscopic and flow-cytometric study of HeLa cells with an experimentally induced respiratory deficiency. Biochem Biophys Res Commun. 1992;187:991–998. doi: 10.1016/0006-291x(92)91295-2. [DOI] [PubMed] [Google Scholar]
- 57.Skulachev VP. Why are mitochondria involved in apoptosis? Permeability transition pores and apoptosis as selective mechanisms to eliminate superoxide-producing mitochondria and cell. FEBS Lett. 1996;397:7–10. doi: 10.1016/0014-5793(96)00989-1. [DOI] [PubMed] [Google Scholar]
- 58.Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith T, Steele GD, Chen L. Intracellular heterogeneity in mitochondrial membrane potentials revealed by J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci USA. 1991;88:3671–3675. doi: 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Soto AM, Sonnenschein C. The role of estrogens on the proliferation of human breast tumor cells (MCF-7). J Steroid Biochem. 1985;23:87–94. doi: 10.1016/0022-4731(85)90265-1. [DOI] [PubMed] [Google Scholar]
- 60.Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med. 1996a;184:1331–1341. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Susin SA, Zamzami N, Kroemer G. The cell biology of apoptosis: evidence for the implication of mitochondria. Apoptosis. 1996b;1:231–242. [Google Scholar]
- 62.Tanaka S, Saito K, Reed JC. Structure-function analysis of the Bcl-2 oncoprotein. Addition of a heterologous transmembrane domain to portions of the Bcl-2 beta protein restores function as a regulator of cell survival. J Biol Chem. 1993;268:10920–10926. [PubMed] [Google Scholar]
- 63.Tatton WG, Chalmers-Redman RME. Modulation of gene expression rather than monoamine oxidase inhibition: (−)-deprenyl-related compounds in controlling neurodegeneration. Neurology. 1996;47:S171–S183. doi: 10.1212/wnl.47.6_suppl_3.171s. [DOI] [PubMed] [Google Scholar]
- 64.Tatton WG, Ju W, Wadia J, Ansari K, Zhang F, Buys Y, Seniuk N. (−)-Deprenyl reduces neuronal apoptosis by maintaining Bcl-2 synthesis and mitochondrial membrane potential. Mov Disord. 1994a;9:4. [Google Scholar]
- 65.Tatton WG, Ju WY, Holland DP, Tai C, Kwan M. (−)-Deprenyl reduces PC12 cell apoptosis by inducing new protein synthesis. J Neurochem. 1994b;63:1572–1575. doi: 10.1046/j.1471-4159.1994.63041572.x. [DOI] [PubMed] [Google Scholar]
- 66.Tatton WG, Ju WJH, Wadia J, Tatton NA. Reduction of neuronal apoptosis by small molecules: promise for new approaches to neurological therapy. In: Olanow CW, Jenner P, Youdim M, editors. Neurodegeneration and neuroprotection in parkinson’s disease. Academic; London: 1996. pp. 209–229. [Google Scholar]
- 67.Thiffault C, Aumont N, Quirion R, Poirier J. Effect of MPTP and l-deprenyl on antioxidant enzymes and lipid peroxidation levels in mouse brain. J Neurochem. 1995;65:2725–2733. doi: 10.1046/j.1471-4159.1995.65062725.x. [DOI] [PubMed] [Google Scholar]
- 68.Ubl JJ, Chatton J-Y, S C, Stucki JW. A critical evaluation of in situ measurement of mitochondrial potentials in single hepatocytes. Biochim Biophys Acta. 1996;1276:124–132. doi: 10.1016/0005-2728(96)00067-9. [DOI] [PubMed] [Google Scholar]
- 69.Valle VGR, Fagian MM, Parentoni LS, Meinicke AR, Vercesi AE. The participation of reactive oxygen species and protein thiols in the mechanism of mitochondrial inner membrane permeabilization by calcium plus prooxidants. Arch Biochem Biophys. 1993;307:1–7. doi: 10.1006/abbi.1993.1551. [DOI] [PubMed] [Google Scholar]
- 70.Vayssiere JL, Petit PX, Risler Y, Mignotte B. Commitment to apoptosis is associated with changes in mitochondrial biogenesis and activity in cell lines conditionally immortalized with simian virus 40. Proc Natl Acad Sci USA. 1994;91:11752–11756. doi: 10.1073/pnas.91.24.11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wolvetang EJ, Johnson KL, Krauer K, Ralph SJ, Linnane AW. Mitochondrial respiratory chain inhibitors induce apoptosis. FEBS Lett. 1994;339:40–44. doi: 10.1016/0014-5793(94)80380-3. [DOI] [PubMed] [Google Scholar]
- 72.Wu RM, Chiueh CC, Pert A, Murphy DL. Apparent antioxidant effect of l-deprenyl on hydroxyl radical formation and nigral injury elicited by MPP+ in vivo. Eur J Pharmacol. 1993;243:241–247. doi: 10.1016/0014-2999(93)90181-g. [DOI] [PubMed] [Google Scholar]
- 73.Yang J, Liu XS, Bhalla K, Kim CN, Ibrado AM, Cai JY, Peng TI, Jones DP, Wang XD. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 74.Youdim MB, Heldman E, Pollard HB, Fleming P, McHugh ESO. Contrasting monoamine oxidase activity and tyramine induced catecholamine release in PC12 and chromaffin cells. Neuroscience. 1986;19:1311–1318. doi: 10.1016/0306-4522(86)90145-4. [DOI] [PubMed] [Google Scholar]
- 75.Zamzami N, Marchetti P, Castedo M, Zanin C, Vayssiere JL, Petit PX, Kroemer G. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J Exp Med. 1995a;181:1661–1672. doi: 10.1084/jem.181.5.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zamzami N, Marchetti P, Castedo M, Hirsh T, Susin SA, Masse B, Kroemer G. Inhibitors of permeability transition interfere with the disruption of mitochondrial transmembrane potential during apoptosis. FEBS Lett. 1996a;384:53–57. doi: 10.1016/0014-5793(96)00280-3. [DOI] [PubMed] [Google Scholar]
- 77.Zamzami N, Susin SA, Marchetti P, Hirsch T, Gomez-Monterrey I, Castedo M, Kroemer G. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996b;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang X, Zuo DM, Davis BA, Boulton AA, Yu PH. Immunohistochemical evidence of neuroprotection by R(−)-deprenyl and N-(2-hexyl)-N-methylpropargylamine on DSP-4-induced degeneration of rat brain noradrenergic axons and terminals. J Neurosci Res. 1996;43:482–489. doi: 10.1002/(SICI)1097-4547(19960215)43:4<482::AID-JNR9>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 79.Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]