

Abstract
To determine the stability of β-amyloid peptide (Aβ) and the glial and neuronal changes induced by Aβ in the CNS in vivo, we made single injections of fibrillar Aβ (fAβ), soluble Aβ (sAβ), or vehicle into the rat striatum. Injected fAβ is stable in vivo for at least 30 d after injection, whereas sAβ is primarily cleared within 1 d. After injection of fAβ, microglia phagocytize fAβ aggregates, whereas nearby astrocytes form a virtual wall between fAβ-containing microglia and the surrounding neuropil. Similar glial changes are not observed after sAβ injection. Microglia and astrocytes near the injected fAβ show a significant increase in inducible nitric oxide synthase (iNOS) expression compared with that seen with sAβ or vehicle injection. Injection of fAβ but not sAβ or vehicle induces a significant loss of parvalbumin- and neuronal nitric oxide synthase-immunoreactive neurons, whereas the number of calbindin-immunoreactive neurons remains unchanged. These data demonstrate that fAβ is remarkably stable in the CNS in vivo and suggest that fAβ neurotoxicity is mediated in large part by factors released from activated microglia and astrocytes, as opposed to direct interaction between Aβ fibrils and neurons.
Keywords: Alzheimer’s disease, glia, microglia, astrocyte, neurotoxicity, nitric oxide synthase, inducible nitric oxide synthase
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by progressive memory loss and dementia. The pathological hallmarks of AD are extracellular plaques containing β-amyloid (Aβ), dystrophic neurites, activated microglia, reactive astrocytes, and neuronal loss (Selkoe, 1991). Several lines of evidence suggest that Aβ is directly involved in the neuropathology observed in AD. Aβ deposition is an invariant feature of AD, and there is a strong correlation between amyloid burden at death and the degree of dementia in life (Selkoe, 1994; Cummings and Cotman, 1995). Most importantly, several genetically heritable forms of AD are tightly linked to mutations of the amyloid precursor protein gene on chromosome 21 (Goate et al., 1991), which cause an increase in Aβ production (Citron et al., 1992). Heritable forms of AD linked to mutations on chromosomes 14 and 1 also seem to alter Aβ processing (Scheuner et al., 1996; Selkoe, 1997).
Aβ is a 39–43-amino-acid hydrophobic peptide encoded by the gene for the much larger amyloid precursor protein (Glenner and Wong, 1984; Kang et al., 1987). Aβ is constitutively produced in cells, and soluble Aβ (sAβ) is present at similar concentrations (10−9m) in normal and AD CSF, indicating that Aβ is a natural rather than a pathogenic product (Haass et al., 1992; Seubert et al., 1992; Shoji et al., 1992;Busciglio et al., 1993; van Gool et al., 1995). Several studies have shown that sAβ will spontaneously form insoluble aggregates at high concentrations (10−3 to 10−5m) and that the presence of other factors, such as metals, proteoglycans, or apoliopoprotein E4, can influence Aβ aggregation (Fraser et al., 1992; Mantyh et al., 1993; Bush et al., 1994; Ma et al., 1994; Sanan et al., 1994; Wisniewski et al., 1994; Evans et al., 1995; Esler et al., 1996).
Conversion of sAβ to insoluble, fibrillar Aβ (fAβ), an Aβ conformation similar to that found in the AD brain, has been reported to increase greatly Aβ toxicity in neuronal cultures (Pike et al., 1991a,b, 1993, 1995; Mattson et al., 1992). However, other studies have reported that sAβ at concentrations near that found in human CNS (10−9m) has marked neurotoxic and/or neuroprotective effects on neurons in vitro (Koh et al., 1990; Yankner et al., 1990; Harrigan et al., 1995; Kelly et al., 1996). Although much of the above data suggest that Aβ is neurotoxicin vitro, substantial uncertainty remains as to the form of Aβ that is neurotoxic in vivo, the long-term stability of Aβ, and the mechanism(s) via which Aβ may induce neurotoxicityin vivo.
In the present study, we use microglia, astrocyte, and neuronal markers to study the glial and neuronal changes induced by soluble and fibrillar forms of Aβ in the CNS in vivo. The use of microglia and astrocyte markers allows us to address the question of direct versus indirect toxicity of Aβ and to determine whether microglia and astrocytes are differentially involved in the CNS response to Aβ. We chose the striatum as the CNS area in which to examine the effects of Aβ, because this forebrain area is affected in AD (Selden et al., 1994), is a relatively homogeneous brain area that allows quantification of Aβ-induced cellular changes, and has well defined neuronal subpopulations that are readily quantifiable using immunohistochemical methods (Kawaguchi et al., 1995). In the present study, we have also used confocal microscopy, because this imaging technique allows us to visualize simultaneously several fluorescent markers while providing unequivocal single-cell and intracellular resolution of the changes that Aβ induces in the CNS.
MATERIALS AND METHODS
Preparation and characterization of fibrillar and soluble Aβ. Aβ1–40, synthesized by fluorenylmethoxycarbonyl chemistry and purified to near homogeneity (>98%), was obtained (Quality Control Biochemicals, Hopkinton, MA) and stored lyophilized at −20°C. Peptides used to produce fAβ and sAβ were characterized by reverse phase (RP)-HPLC, laser desorption mass spectroscopy (LD-MS), amino acid analysis (Benson et al., 1981), and size exclusion fast-protein liquid chromatography (SE-FPLC) (Soreghan et al., 1994).
Fibrillar Aβ was prepared from solutions of 10−4m Aβ in filtered PBS (10.0 mmNaH2PO4/Na2HPO4and 100.0 mm NaCl, pH 7.5). Peptide solutions were initially clear, with no evidence of flocculation or incomplete dissolution; visible precipitate appeared only after extended agitation. The fresh Aβ solution was allowed to incubate under vigorous agitation (Teflon-coated stir bar at 800 rpm) (Jarrett and Lansbury, 1992; Jarrett et al., 1993, 1994; Evans et al., 1995) at 23°C for 26–36 hr. After this incubation, the Aβ solution was distinctly turbid, and >80% of the peptide could be sedimented by centrifugation (15,000 × g; 10 min). The fAβ solution was aliquoted (100 μl per vial), frozen on dry ice, and stored at −20°C until use.
To better characterize the fAβ assembly reaction, we performed time course experiments in triplicate with mixtures of 125I-Aβ and unlabeled Aβ in microcentrifuge tubes (Evans et al., 1995) under conditions similar to those described above. At each time point, a 15 μl aliquot was taken from each tube and centrifuged (15,000 ×g; 10 min), and the amount of 125I-Aβ in each of two 3 μl aliquots of the resulting supernatants was quantified using a gamma counter. Fibrillar Aβ assembly followed a distinctly nonlinear time course with a lag time, indicative of a nucleation-dependent aggregation process (Jarrett and Lansbury, 1993). Fibrillar Aβ from the preparative procedure described above was characterized by thioflavin S, Congo red, and anti-Aβ immunohistochemical staining and then was examined for size distribution using light-field or fluorescence microscopy and image analysis. Individual fAβ aggregates had a median diameter of 12.3 μm, which compares favorably with the diameter of plaque cores (5–30 μm) purified from human AD brain (Selkoe and Abraham, 1986). Like amyloid plaques found in the AD brain, fAβ prepared in this manner displays typical Congo red birefringence under polarized light and is thioflavin S-positive, establishing that Aβ prepared in this manner has a fibrillar morphology (Kirschner et al., 1986, 1987; Evans et al., 1995). Additionally, fAβ prepared in this manner serves as a template for Aβ deposition in vitro in a manner similar to Aβ deposition onto authentic AD tissue amyloid (Esler et al., 1997). Preparations of Aβ not deliberately aggregated (sAβ) did not display Congo red birefringence or thioflavin S staining.
Fibrillar Aβ prelabeled with thioflavin S was prepared from unlabeled fAβ described above. Aliquots were thawed, centrifuged (15,000 × g; 10 min), resuspended in 100 μl of thioflavin S solution (1% in distilled water; Sigma, St. Louis, MO), and allowed to incubate overnight on a shaking platform at 23°C. The solution was then centrifuged as described above, and the fAβ was resuspended and washed twice in 70% ethanol to remove excess dye. This thioflavin S-labeled fAβ was then prepared for injection by resuspending, after the second ethanol wash, in sterile artificial CSF (aCSF; 128.6 mm NaCl, 2.6 mm KCl, 2.0 mmMgCl2, and 1.4 mmCaCl2, pH 7.4) to a total Aβ concentration of 10−4m. Unlabeled fAβ was prepared for injection by pelleting the unstained fAβ as described above and resuspending in sterile aCSF at an fAβ concentration of 10−4m. Soluble Aβ solution was prepared by dissolving Aβ (Quality Control Biochemicals) in sterile water to a concentration of 10−4m and was stored at −20°C in 100 μl aliquots until injection. No evidence of Aβ aggregation was observed in the sAβ aliquots before or during the injection process.
Injection of Aβ and preparation of brain tissue. Sprague Dawley rats (male; 200–250 gm; Harlan Sprague Dawley, Indianapolis, IN) were deeply anesthetized with sodium pentobarbital (60 mg/kg; Abbott Labs, Irving, TX) and then mounted in a small-animal stereotaxic instrument. The fAβ solution (10 μl) was injected stereotaxically into the striatum (anterior, 0.5 mm; lateral, 3.0 mm; and ventral, 6.5 mm) using a 26 gauge needle (Fig.1A). Stereotaxic coordinates were measured from bregma (Paxinos and Watson, 1986). Injections of vehicle or 10−4m sAβ (10 μl) were made into the contralateral striatum. Each 10 μl injection of fAβ contained ∼2400 individual aggregates.
Fig. 1.
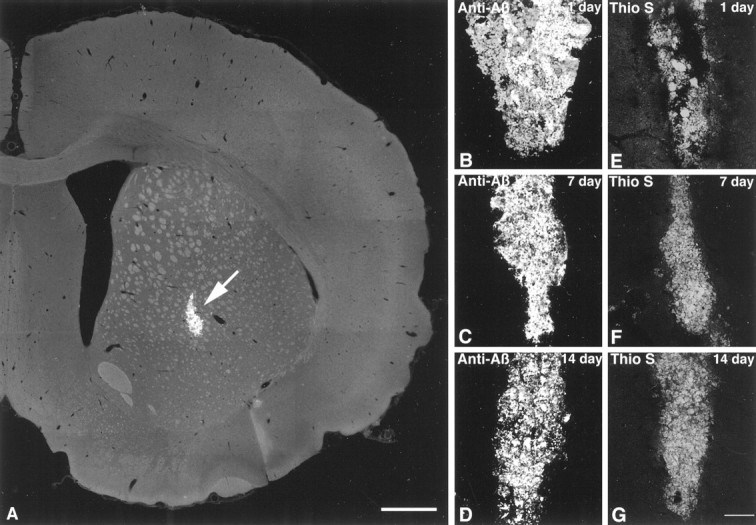
A, A confocal photomicrograph shows the location of fAβ injection into the rat striatum. The fAβ shown here (arrow) was prelabeled with thioflavin S before injection anterior 0.5 mm, lateral 3.0 mm, and ventral 6.5 mm relative to bregma (Paxinos and Watson, 1986). The image was constructed from six optical sections acquired at 1.0 μm intervals using a 4× lens.B–G, Fluorescent confocal images of injected fAβ in the striatum show that fAβ is stable in vivo.B–D, Injected fAβ is identified in tissue by labeling with anti-Aβ. E–G, Injected fAβ prelabeled with thioflavin S is also readily observable in the striatum. Note that, although variability in the shape of the injection site is evident in the images, there is little decrease in the total amount of injected fAβ present over time. Also, no significant decrease in thioflavin S fluorescence is observed over time, suggesting injected fAβ monomers are not being replaced with endogenous rat Aβ. Images were projected from six optical sections acquired at 1.0 μm intervals using a 10× lens. Scale bars: A, 1.0 mm; B–G, 75 μm.
Seventy-five animals were used in the present study. Injections of either unlabeled or thioflavin S-prelabeled fAβ were made into the striatum, with injections into the contralateral striatum of either sAβ or vehicle (aCSF or sterile water). The effects of fAβ, sAβ, and vehicle injections were analyzed 1, 7, 14, and 30 d after injection (n ≥ 3 for each treatment group). None of the animals used in the above experimental procedures showed signs of infection in the injection area before killing or after histological examination. Animals were group-housed with food and water availablead libitum in an American Association for Accreditation of Laboratory Animal Care-approved animal research facility. After the appropriate survival time, rats were killed by CO2asphyxiation and then rapidly perfused through the ascending aorta with 200 ml of PBS (3.0 mmNa2HPO4, 0.9 mmKH2PO4, and 154.0 mm NaCl, pH 7.4; 23°C) followed by 500 ml of PBS containing 4% formaldehyde (Sigma, 4°C). After perfusion, the brain was removed, blocked in the transverse plane, post-fixed in 4% formaldehyde (4°C; 1 d), and then placed in a 30% sucrose solution (4°C; 1 d). The brains were then serially sectioned at 20 μm using a cryostat microtome (Model OTF/AS; Bright Instrument Co.) and collected in PBS.
Immunohistochemical examination of Aβ, glia, and neuronal markers. Immunohistochemical identification of Aβ was performed by washing sections in Tris-buffered saline (TBS; 140 mmNaCl, 2.7 mm KCl, and 24.8 mm Tris base, pH 7.4) for 20 min and then in ethanol/distilled water (1:1) for 10 min. Sections were washed three times for 10 min each in TBS, washed in blocking solution containing TBS and 10% normal goat serum (NGS) for 30 min, and then incubated overnight at 23°C in TBS containing 1% NGS and polyclonal anti-Aβ (R1282; 1:1500; a kind gift from Dr. D. Selkoe) raised against uncoupled synthetic Aβ1–40 (Haass et al., 1992). Sections were washed three times for 10 min each in TBS and then incubated 1 hr at 23°C in TBS containing 1% NGS and biotinylated donkey anti-rabbit IgG (1:500; Jackson ImmunoResearch, West Grove, PA). Sections were washed three times for 10 min each in TBS and then incubated 45 min at 23°C in TBS containing 1% NGS and fluorescein (FITC)-conjugated avidin (1:200; Jackson ImmunoResearch). Sections were washed three times for 10 min each in TBS, then mounted by floating onto gelatin-coated slides, and coverslipped using PBS/glycerol (1:1).
Immunohistochemical identification of microglia was performed by washing sections in blocking solution containing PBS and 1% NGS for 30 min and then incubating overnight at 23°C in PBS containing 1% NGS and monoclonal anti-CD11b equivalent (OX-42; 1:10; Serotec, Indianapolis, IN). OX-42 is specific for the C3bicomplement protein receptor that is expressed in the CNS primarily, if not exclusively, by microglia. Sections were washed three times for 10 min each in PBS and then incubated 3 hr at 23°C in PBS containing 1% NGS and cyanine 3.18 (Cy3)-conjugated donkey anti-mouse IgG (1:600; Jackson ImmunoResearch). Sections were washed three times for 10 min each in PBS and then mounted and coverslipped as described above.
Immunohistochemical identification of astrocytes was performed by washing sections in blocking solution containing PBS, 1% NGS, and 0.3% Triton X-100 (Sigma) for 30 min and then incubating overnight at 23°C in PBS containing 1% NGS, 0.3% Triton X-100, and either monoclonal or polyclonal anti-glial fibrillary acidic protein (GFAP; 1:400; Sigma). Sections were washed three times for 10 min each in PBS and then incubated 3 hr at 23°C in PBS containing 1% NGS, 0.3% Triton X-100, and cyanine 5.18 (Cy5)-, Cy3-, or FITC-conjugated donkey anti-mouse or anti-rabbit IgG (Jackson ImmunoResearch) at concentrations of 1:600 (Cy3), 1:450 (Cy5), or 1:150 (FITC). Sections were washed three times for 10 min each in PBS and then mounted and coverslipped as described above.
Immunohistochemical identification of inducible nitric oxide synthase (iNOS) was performed by washing sections in blocking solution containing PBS and 10% bovine serum albumin (BSA; Sigma) for 1 hr and then incubating overnight at 4°C in PBS containing 10% BSA and polyclonal anti-iNOS (1:200; Biomol, Plymouth Meeting, PA), which is specific for the iNOS 130 kDa protein with no advertised cross-reactivity to endothelial nitric oxide synthase or neuronal nitric oxide synthase (nNOS). Cross-reactivity of the iNOS antibody was not tested in this study. Sections were washed three times for 10 min each in PBS and then incubated 3 hr at 23°C in PBS containing 10% BSA and Cy3-conjugated donkey anti-rabbit IgG (1:600; Jackson ImmunoResearch). Sections were washed three times for 10 min each in PBS and then mounted and coverslipped as described above.
Immunohistochemical examination of parvalbumin, nNOS, and calbindin neurons were performed by washing sections in blocking solution containing PBS, 1% NGS, and 0.3% Triton X-100 (Sigma) for 30 min and then incubating overnight at 23°C in PBS containing 1% NGS, 0.3% Triton X-100, and either monoclonal anti-parvalbumin (1:1000; Sigma), polyclonal anti-nNOS (1:500; Chemicon, Temecula, CA), or monoclonal anti-calbindin (1:300; Sigma). Sections were washed three times for 10 min each in PBS and then incubated 3 hr at 23°C in PBS containing 1% NGS, 0.3% Triton X-100, and Cy3-conjugated donkey anti-mouse or anti-rabbit IgG (1:600; Jackson ImmunoResearch). Sections were washed three times for 10 min each in PBS and then mounted and coverslipped as described above.
Image analysis. Size characterization of fAβ was done using a Leitz Orthoplan II microscope equipped for epifluorescence (Leitz, Wetzlar, Germany). Fibrillar Aβ was labeled with thioflavin S as described above; then 10 μl of this solution was placed on a microscope slide, coverslipped, and visualized under fluorescence. The size distribution of 120 randomly selected fAβ aggregates was analyzed using the National Institutes of Health Image version 1.51 software program (National Institutes of Health, Bethesda, MD).
Immunofluorescence in brain tissue was examined using a MRC-1024 Confocal Imaging System equipped with a krypton/argon ion laser (Bio-Rad, Hercules, CA) in conjunction with an Olympus BX-75 microscope equipped for epifluorescence (Olympus Immunochemicals, Lake Success, NY). Labeled sections were imaged using filters appropriate for the specific visualization of fluorescein, cyanine 3.18, and cyanine 5.18 (Brelje et al., 1993; Kennedy et al., 1994). Images were acquired using two different methods. The first method involved collecting multiple scans of optical sections (z-series) that were acquired at 1.0 μm intervals. The second method involved collecting multiple scans of a single optical section using the Kalman imaging program, which allowed collection of low power confocal images while conserving the intensity and resolution of the original tissue staining. The optical sections were then projected into single images using image-processing software provided with the confocal system (Bio-Rad).
The total number of OX-42-immunoreactive microglia in fAβ, sAβ, or vehicle injection tracks was counted using confocal microscopy by optical sectioning (2.0 μm confocal z-series) in 200 × 300 μm grids (0.06 mm2). Astrogliosis and iNOS expression were quantified using confocal microscopy by measuring the total GFAP or iNOS immunofluorescence over a similarly defined area. Data units for total immunofluorescence calculations are in gray scale pixel values, ranging from 0 (dark) to 255 (saturation level). Because individual astrocytes grouped closely together in the injection were impossible to count accurately, it was necessary to quantify astrocytes (GFAP) and iNOS expression using total fluorescence.
To determine whether microglia or astrocytes in the injection site contained phagocytized fAβ or sAβ, we imaged Aβ and OX-42 immunofluorescence from the same tissue section simultaneously. The four experimental time points were compared by averaging the percentage of total glia in the measured area (defined above) that contained phagocytized Aβ. Twenty-four animals were used for this analysis (n = 3 animals per treatment group at each time point; seven sections analyzed per animal). The average number of microglia in each confocal gridded section at 1, 7, 14, and 30 d after injection was 20, 57, 73, and 75 (fAβ) and 20, 61, 33, and 15 (sAβ). Images of iNOS immunofluorescence with OX-42 or GFAP immunofluorescence were created using pairs of confocal images (iNOS + OX-42 and iNOS + GFAP) from the same tissue section. The pairs of images were imported using the National Institutes of Health Image version 1.51 software program, and the area of colocalization was computed by finding the regions of maximum signal overlap between the two images. To determine the percentage of microglia or astrocytes that express iNOS in the fAβ injection area, we counted iNOS-immunoreactive microglia or astrocytes using the confocal imaging system (area defined above).
To quantify neurons in and surrounding the defined fAβ and sAβ injection areas, we imaged GFAP immunofluorescence (marking the astrogliosis surrounding the fAβ and sAβ needle tracks) and parvalbumin, nNOS, or calbindin immunofluorescence on the same tissue sections simultaneously. Using a gridded eyepiece on a Leitz Orthoplan II microscope, we counted neurons in bins of increasing distance on both sides of the needle track at 10× magnification. The bins measured 0–125, 126–250, and 251–375 μm outward (medial and lateral) from the margin of the GFAP-immunoreactive astrogliosis. Fifteen animals, with fAβ and sAβ injected contralaterally, were used for this analysis (total number of sections = 55; total neurons counted within defined area, 862 [nNOS], 823 [parvalbumin], and 1529 [calbindin]). The total area of quantification was the same for each section.
Statistical analysis. All data are expressed as mean ± SEM. One-way ANOVA was performed using the StatView statistical software program (Abacus Concepts, Calabasas, CA).
RESULTS
Stability of fibrillar and soluble Aβ
In the normal (noninjected) striatum or after injection of vehicle alone, no Aβ immunofluorescence, Congo red birefringence, or thioflavin S staining is observed at any time point. After injection of sAβ, Aβ immunofluorescence is only faintly visible and, when present, is almost invariably observed inside microglia immediately around the needle track at 1, 7, 14, and 30 d. At 1 d after sAβ injection, 9% of microglia within 150 μm of the needle track contain anti-Aβ-immunoreactive material. By 30 d after injection, the percentage of microglia in the same area containing Aβ-immunoreactive material increases to 19% (Fig.2), but no difference in total Aβ immunofluorescence in the area surrounding the injection site is observed at any time point (data not shown). The Aβ-immunoreactive material detectable in microglia after sAβ injection does not display Congo red birefringence or thioflavin S staining at any time point, suggesting that this Aβ is present in a nonfibrillar form.
Fig. 2.

Aβ is phagocytized by microglia over time. Fluorescent confocal images of fAβ (circles) or sAβ (triangles) labeled with anti-Aβ were merged with confocal images of OX-42-immunoreactive microglia from the same tissue sections, allowing intracellular resolution and quantification of Aβ inside microglia. Microglia actively phagocytize fAβ over time, because 93% of microglia within 150 μm of the fAβ needle track contain material that is both Aβ-immunoreactive and Congo red-birefringent at 30 d after injection. In contrast, a relatively small percentage of microglia (10–20%) within 150 μm of the sAβ needle track contain Aβ immunoreactivity at the observed time points; this Aβ-immunofluorescent material is neither Congo red-birefringent nor thioflavin S-positive (data not shown) and thus lacks the characteristics of fAβ.
After injection of fAβ or of fAβ prelabeled with thioflavin S into the striatum, intense Aβ immunoreactivity or thioflavin S fluorescence, respectively, is present at 1, 7, and 14 d after injection. Although the shape of the individual fAβ injection sites varied slightly between animals, no significant decrease in the quantity of fAβ in the striatum is observed at these time points, as measured by Aβ immunofluorescence or thioflavin S fluorescence. Also, no decrease in thioflavin S fluorescence is observed, suggesting that the injected human fAβ remains stable in vivo and is not exchanged for endogenous rat Aβ monomers at these time points (Fig.1B–G). The fAβ remains stable and detectable within the striatum up to at least 30 d after injection (Fig.3) and displays typical Congo red birefringence under polarized light at all time points (data not shown). Although the overall quantity of fAβ does not appear to decrease significantly after injection, there is a clear increase in the percentage of total fAβ that is phagocytized and concentrated within microglia over time. At 1 d after injection, the majority of fAβ remains in the extracellular space (Fig. 3A), although a significant minority of microglia (38%) within 150 μm of the needle track already contain phagocytized fAβ at this time point (Fig. 2). With increasing time after injection, nearly all of the injected fAβ is phagocytized and concentrated within microglia (Fig.3B,C). By 30 d after injection, 93% of microglia within 150 μm of the needle track contain phagocytized fAβ (Fig.2).
Fig. 3.
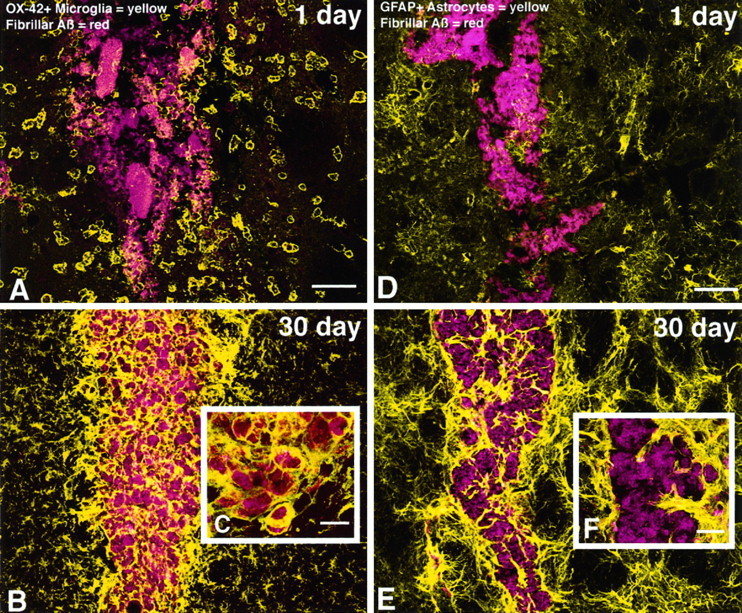
Fluorescent confocal images show that microglia actively phagocytize injected fAβ, whereas astrocytes wall-off fAβ-containing microglia from the surrounding neuropil.A–C, fAβ prelabeled with thioflavin S appearsred, and microglia labeled with anti-CD11b (OX-42) appear yellow. A, At 1 d after injection, activated microglia, characterized by short, thickened processes and marked cytoplasmic swelling, are observed surrounding injected fAβ. Note that some of these microglia are associated with injected fAβ along the outer margins of the needle track.B, C, At 30 d after injection, fAβ is not cleared from the site of injection but rather is contained almost exclusively within microglia in the needle track.D–F, fAβ prelabeled with thioflavin S appearsred, and astrocytes labeled with anti-GFAP appearyellow. D, At 1 d after injection, no significant astrogliosis is observed surrounding injected fAβ.E, F, In contrast, a marked astrogliosis, characterized by activated astrocytes with swollen processes and increased GFAP immunofluorescence, is observed at 30 d after injection. Astrocytes are shown walling-off fAβ-containing microglia from surrounding tissue. Images of fAβ and microglia or astrocytes were taken from the same double-labeled tissue sections. Images were projected from 12 optical sections acquired at 1.0 μm intervals using a 10× or 40× lens. Scale bars: A, B,D, E, 50 μm; C,F, 10 μm.
Glial responses to injections of fibrillar Aβ, soluble Aβ, or vehicle
In the normal striatum, both microglia and astrocytes display a characteristic resting morphology. Microglia detectable using OX-42 immunoreactivity are evenly distributed throughout the striatum and possess long, thin, highly ramified processes connected to a cell body that has little observable cytoplasm (Barron, 1995). Astrocytes in the normal striatum that are GFAP-immunoreactive are also relatively evenly distributed, possessing a morphology characterized by a distinctly stellate-shaped cell body with little observable cytoplasm and long, thin radially extending processes (Eddleston and Mucke, 1993).
At 1 d after injection of vehicle alone, a large percentage of microglia surrounding the needle track have an activated morphology, characterized by short, thickened processes and marked cytoplasmic swelling (Barron, 1995). A gradient of microglial activation is observed at this time point, because nearly all microglia within 100 μm of the needle track assume this activated morphology, whereas microglia that are >150 μm from the needle track display a resting morphology similar to that observed in the normal striatum. At 7 d after injection, nearly all microglia observed up to 300 μm from the needle track possess an activated morphology. At the 14 and 30 d time points, the area showing microglial activation is significantly decreased, so that by 30 d after injection, only a thin line of activated microglia marking the needle track remain in the vehicle-injected striatum (Fig.4A). This change in microglial morphology is mirrored by a change in the total number of microglia surrounding the needle track, which peaks at 7 d after injection and is followed by a gradual decline at days 14 and 30 (Fig.5A). No differences in microglial morphology or number are observed at any time point when comparing vehicle injections of aCSF and sterile water.
Fig. 4.
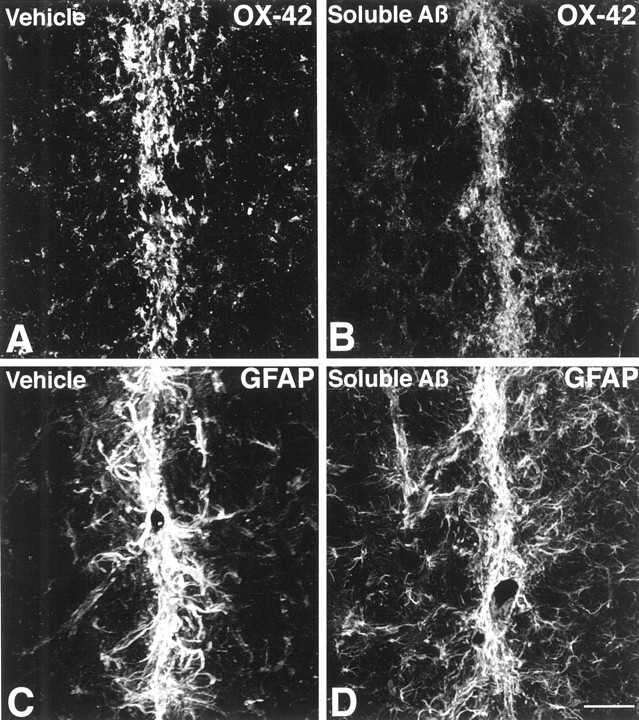
Microglia and astrocyte responses 30 d after vehicle or sAβ injection are shown. A,B, No difference in OX-42 immunofluorescence is observed in the needle track after vehicle or sAβ injection. C,D, Similarly, no difference in GFAP immunofluorescence is observed in the needle track after vehicle or sAβ injection. Images are Kalman averages of a single optical section acquired using a 20× lens. Scale bar, 50 μm.
Fig. 5.

Injected fAβ induces a larger and more-sustained gliosis compared with that seen with injected sAβ or vehicle alone.A, The number of OX-42-immunoreactive microglia, quantified in 0.06 mm2 grids centered on fAβ (open circles), sAβ (closed triangles), or vehicle (open squares) needle tracks, is similar at 1 and 7 d after injection but is significantly increased in the fAβ injection area relative to the sAβ and vehicle injection areas at 14 and 30 d after injection. Note that at 1 d after injection, the number of microglia in the fAβ, sAβ, or vehicle injection area does not differ significantly from the number of microglia present in a comparable area of normal striatum.B, Astrogliosis, measured by computing the total intensity of GFAP immunofluorescence in 0.06 mm2grids centered on fAβ (open circles), sAβ (closed triangles), or vehicle (open squares) needle tracks, is similar at 1, 7, and 14 d after injection. At 30 d after injection, GFAP immunofluorescence is significantly increased in the fAβ injection area relative to the sAβ and vehicle injection areas. Images used for microglia counts and GFAP immunofluorescence measurements were Kalman averages of a single optical section acquired using a 10× lens (*fAβ vs sAβ or vehicle, both p < 0.05).
Injection of vehicle alone also induces changes in the morphology and GFAP immunofluorescence of astrocytes surrounding the needle track. Astrocytes undergo a swelling of processes and show an increase in GFAP immunofluorescence, indicative of activation (Eddleston and Mucke, 1993; O’Callaghan et al., 1995). Evidence of astrocyte activation is faintly observable at 1 d after injection and is clearly evident at days 7 and 14. At 30 d after injection, this astrocyte activation decreases so that only a thin line of activated astrocytes marking the needle track remain in the vehicle-injected striatum (Fig.4C). This change in astrocyte morphology is mirrored by a change in GFAP immunofluorescence, which peaks at 14 d after injection and is followed by a marked decline at day 30 (Fig.5B). No differences in astrocyte morphology or GFAP fluorescence are observed at any time point when comparing vehicle injections of aCSF and sterile water.
Striatal injection of sAβ induces microglial and astrocyte changes that are nearly indistinguishable from those observed after injection of vehicle alone. However, one significant difference between the injection of sAβ and vehicle is that although vehicle injection alone never induces any detectable accumulation of Aβ, injection of sAβ results in accumulation of anti-Aβ immunoreactivity in ∼10–20% of microglia in the needle track at the time points examined (Fig. 2). Those microglia that contain Aβ immunoreactivity invariably have an activated morphology and maintain this morphology up to at least 30 d after injection. Even with the presence of microglia containing Aβ immunoreactivity after sAβ injection, only a thin line of activated microglia and astrocytes, similar in appearance to the vehicle-injected striatum, remain at 30 d after sAβ injection (Fig. 4B,D). Neither the number of microglia nor the GFAP immunofluorescence surrounding the needle track differs significantly at any time point when comparing sAβ with vehicle injection (Fig. 5A,B).
Injection of fAβ induces several changes in microglia and astrocytes that are significantly different from those observed after injection of either vehicle or sAβ. The earliest difference observed when comparing fAβ and sAβ injection sites is in the percentage of microglia that contain Aβ immunoreactivity. At 1 d after injection of fAβ, 38% of microglia within 150 μm of the needle track contain phagocytized fAβ. The percentage of microglia containing fAβ continues to rise until day 30, at which point 93% of microglia in the same area contain detectable concentrations of Aβ immunoreactivity (Figs. 2, 3). At all time points examined, microglia that contain fAβ invariably have an activated morphology. No significant increase in astrocyte activation is observed at 1 d after fAβ injection (Fig. 3D). However, at 30 d after injection, a marked astrogliosis, characterized by activated astrocytes with swollen processes and increased GFAP immunofluorescence, is observed in the fAβ injection area. A virtual wall of activated, GFAP-immunoreactive astrocytes is observed surrounding the fAβ-containing microglia at 30 d after injection (Fig.3E,F). Although changes in the number of microglia and in the GFAP immunofluorescence induced by injection of fAβ may initially be masked by changes induced by the damage associated with needle placement alone, there are significantly greater numbers of microglia within 150 μm of the needle track after fAβ compared with sAβ or vehicle injection at days 14 and 30 (Fig. 5A). Similarly, there is a significantly greater concentration of GFAP immunofluorescence within 150 μm of the needle track after fAβ compared with sAβ or vehicle injection at day 30 (Fig.5B).
Fibrillar Aβ induces iNOS expression in microglia and astrocytes
In the normal striatum, <20% of the microglia and <5% of astrocytes display iNOS immunoreactivity, and those microglia and astrocytes that do express iNOS do so at low levels. Within 150 μm of the needle track, there is a significant increase in iNOS immunofluorescence above those levels observed in the normal striatum at 30 d after vehicle (64% increase) or sAβ (75% increase) injection, but this difference between vehicle and sAβ injection did not reach statistical significance (data not shown). Those microglia and astrocytes that express iNOS at 30 d after vehicle or sAβ injection seem to be from the same small population of microglia and astrocytes that remain activated and mark the thin line of the needle track. No increase in iNOS immunofluorescence above the level in the normal striatum is observed in tissue areas outside of the vehicle or sAβ needle tracks.
Compared with that in the normal striatum, iNOS immunofluorescence is 121% higher within 150 μm of the needle track at 30 d after fAβ injection. Injection of fAβ also promotes an increase in iNOS expression that significantly exceeds the iNOS expression observed after vehicle or sAβ injection (data not shown). Simultaneous imaging of iNOS and OX-42 or GFAP immunofluorescence on the same tissue sections confirms that fAβ induces iNOS expression in both microglia and astrocytes (Fig. 6). At 30 d after injection, nearly 100% of microglia that contain phagocytized fAβ are also iNOS-immunoreactive. Similarly, nearly 100% of astrocytes within 100 μm of microglia containing Aβ are iNOS-immunoreactive. Outside of this area, iNOS expression closely resembles that of the normal striatum described above.
Fig. 6.
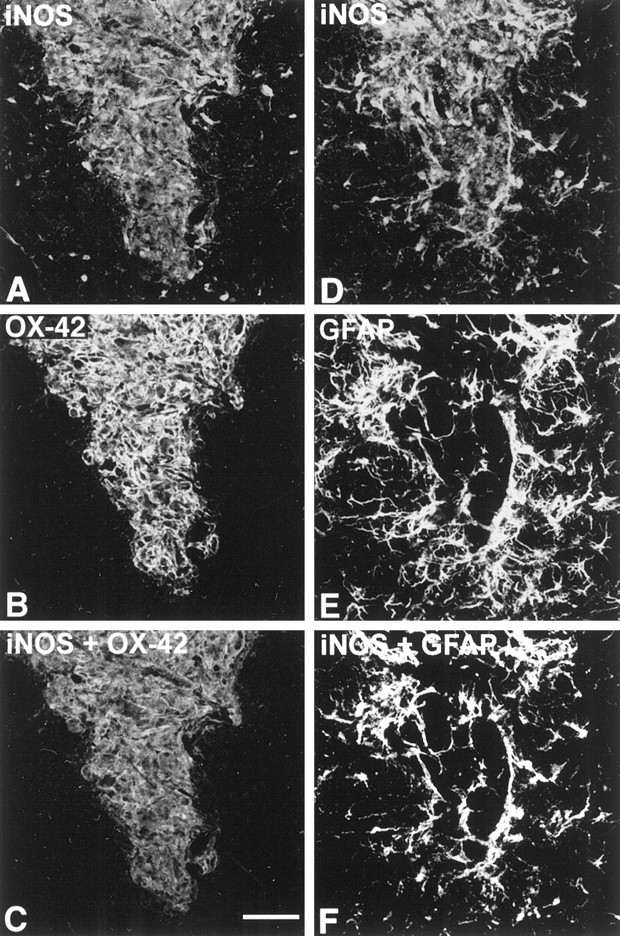
iNOS expression by microglia and astrocytes is increased 30 d after fAβ injection. A,B, D, E, Tissue sections were labeled with anti-iNOS and with either OX-42 or anti-GFAP.C, F, Using the National Institutes of Health Image 1.51 software program, we merged iNOS and OX-42 or GFAP images, and the area of maximum signal overlap between the images was computed to show immunofluorescence colocalization. Note that microglia in the fAβ injection area, which were shown in Figure 3 to contain fAβ, also express iNOS at 30 d after injection, whereas most microglia outside the injection area do not. Also note that only those astrocytes forming a virtual wall around the injected area, within 100 μm of the Aβ-containing microglia, express high levels of iNOS. The iNOS, OX-42, and GFAP images were taken from the same double-labeled tissue sections and are Kalman averages of a single optical section acquired using a 10× lens. Scale bar, 50 μm.
Fibrillar Aβ-induced loss of specific neuronal populations
To determine the effects of fAβ and sAβ injections on striatal neurons, we labeled a population of projection neurons using anti-calbindin and two distinct populations of interneurons using anti-parvalbumin and anti-nNOS. Spiny projection neurons represent ∼90% of total neurons in the striatum, and aspiny interneurons represent ∼10%. Calbindin labels nearly all projection neurons, whereas among interneurons, parvalbumin labels 40–50%, and nNOS labels 10–20% (Kawaguchi et al., 1995). These three antibodies label distinct subpopulations of neurons, with only a small percentage of interneurons showing colocalization of calbindin and nNOS immunoreactivity (Bennett and Bolam, 1993). The numbers of parvalbumin-, nNOS-, and calbindin-immunoreactive neurons were compared in tissue areas surrounding injected fAβ and sAβ at 30 d after injection and in the normal (noninjected) striatum (Figs.7, 8). Compared with levels in both the sAβ injection area and the normal striatum, a significant decrease in the number of parvalbumin-immunoreactive neurons is observed in the areas 0–125 μm (51% reduction) and 126–250 μm (24% reduction) from the margin of the astrogliosis surrounding the injected fAβ (Figs. 7, 8). Similarly, fAβ induces a significant decrease in nNOS-immunoreactive neurons in the area 0–125 μm from the margin of the injection track compared with both the sAβ injection area and the normal striatum (46% reduction). In contrast, no significant decrease in the number of calbindin-immunoreactive neurons is observed in tissue areas surrounding fAβ or sAβ injection compared with the normal striatum (Fig. 8).
Fig. 7.
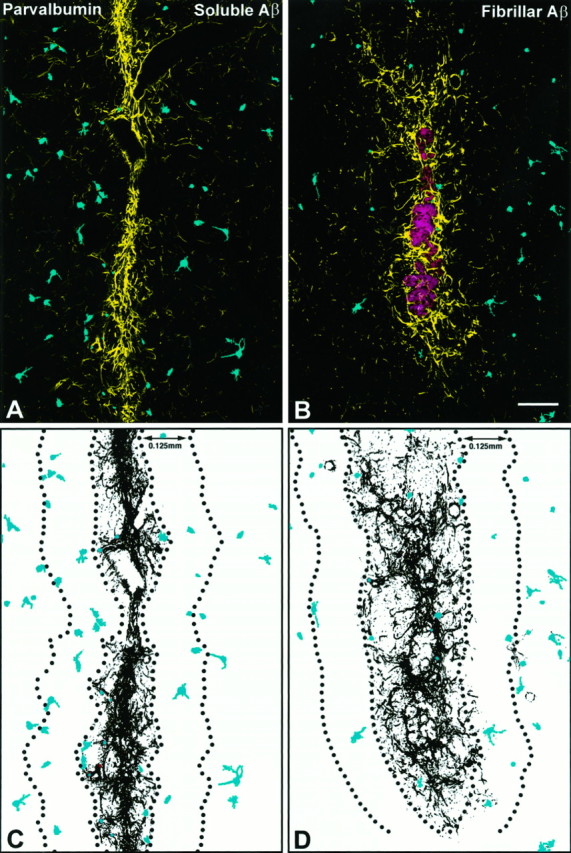
Injection of fAβ induces a reduction in the number of parvalbumin-immunoreactive neurons. A,B, Anti-parvalbumin and anti-GFAP were used to label parvalbumin neurons (blue) and the astrogliosis (yellow) surrounding sAβ and fAβ needle tracks. A, C, Note that in tissue areas surrounding injected sAβ, parvalbumin neurons are evenly distributed outward from the margin of the astrogliosis. B,D, In contrast, a significant reduction in the number of parvalbumin neurons is observed near the astrogliosis surrounding injected fAβ (red; image of fAβ from a serial section labeled with anti-Aβ). Images of parvalbumin neurons and astrocytes were taken from the same double-labeled tissue sections and are Kalman averages of a single optical section acquired using a 10× lens. Scale bar, 125 μm.
Fig. 8.
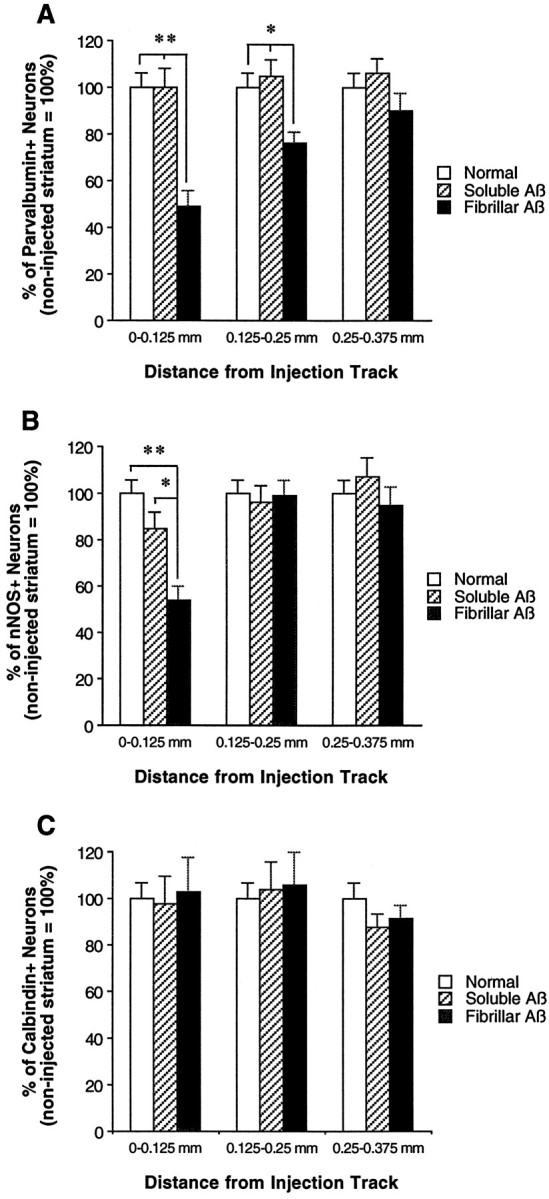
Injected fAβ induces a reduction in the number of parvalbumin- and nNOS- but not calbindin-immunoreactive neurons.A–C, Subpopulations of striatal neurons were labeled on separate tissue sections using anti-parvalbumin, anti-nNOS, or anti-calbindin, and on all sections the needle track was double-labeled using anti-GFAP. Numbers of parvalbumin-, nNOS-, and calbindin-immunoreactive neurons are compared over identical areas around sAβ and fAβ needle tracks at 30 d after injection and in the normal (noninjected) striatum. A, A significant reduction in the number of parvalbumin-immunoreactive neurons is observed up to 250 μm from the margin of the fAβ injection track relative to the sAβ injection track and normal striatum.B, In the fAβ injection area, a significant reduction is also observed in the number of nNOS-immunoreactive neurons up to 125 μm from the injection track relative to the sAβ injection track and normal striatum. C, In contrast, no significant difference in the number of calbindin-immunoreactive neurons is observed when comparing fAβ and sAβ injection areas and the normal striatum. (**p < 0.01; *p < 0.05)
DISCUSSION
Stability of fibrillar Aβ in the rat CNS in vivo
In the present study, injected fAβ is remarkably stable in the rat CNS in vivo, contrasting with previous reports that noted an apparent lack of Aβ stability after injection or infusion. An early review of AD animal models (Price et al., 1992) reported that sAβ or its fragments injected into various brain regions were either neurotoxic, nontoxic, or neurotrophic depending on the aggregation state of the peptide. One consistent feature of these studies, however, was the difficulty encountered in visualizing Aβ after injection of sAβ. In the present study, sAβ was not stable in vivo, appearing to be cleared rapidly from the site of injection. Rapid clearance of sAβ may also explain the inability to visualize injected Aβ in the previous studies.
Aggregates of injected fAβ, which are of comparable size to AD plaque cores found in the human brain, are highly stable in the present study. The stability of fAβ contrasts with a previous study that showed that human AD plaque cores injected into the rat brain were cleared by microglia from their site of injection within 30 d (Frautschy et al., 1992). This difference in stability between fAβ and AD plaque cores may be explained in part by the presence of a variety of extracellular, non-Aβ components in the plaque cores obtained from humans. For example, complement proteins C1q, C4d, and C3d, which help facilitate phagocytosis of foreign material by macrophages and microglia, are bound to Aβ in AD plaques (McGeer and McGeer, 1996). The presence of these proteins and other non-Aβ components in the injected plaque cores may explain their rapid clearance from the rat brain in the previous study. In contrast, injected fAβ aggregates that remain stable in the rat striatum up to 30 d after injection are composed of synthetic Aβ without any non-Aβ components.
Injection of fibrillar Aβ induces a differential response in microglia and astrocytes
In the present study, fAβ of a known quantity, size range, and fibrillar character was injected into one striatum, with injections of sAβ or vehicle made into the contralateral striatum. Using this model system, we demonstrate that fAβ alone is sufficient to induce a marked gliosis and iNOS induction in microglia and astrocytes in the rat brain in vivo. Although it cannot be excluded that, once in the brain, Aβ may be interacting with other endogenous, plaque-associated components to exert its effects, our results demonstrate that injection of synthetic fAβ alone is sufficient to cause an increase in iNOS expression and to induce a marked gliosis similar to that observed in the human AD brain (Itagaki et al., 1989;Uchihara et al., 1995). Previous reports have suggested that iNOS expression may serve as an index of reactive gliosis (Murphy et al., 1993; Brosnan et al., 1994); thus, the present results strongly suggest that injection of fAβ activates both microglia and astrocytes. One surprising observation in the present study is a low level of iNOS immunoreactivity present in a small minority of microglia and astrocytes in the normal (noninjected) striatum. A recent study (Wong et al., 1996) reported no detectable iNOS gene expression in the normal (noninduced) rat brain. The low level of iNOS immunoreactivity in the normal striatum may be caused by slight cross-reactivity between the iNOS antibody and other agents in the striatum.
In contrast to injections of fAβ, contralateral injection of sAβ produces no significant increase in gliosis and iNOS expression above that observed for injection of vehicle alone. One reason for the lack of sAβ-induced gliosis may be that sAβ is rapidly cleared from brain parenchyma, suggested here by the fact that 30 d after sAβ injection, Aβ immunofluorescence is only detected in a minority of microglia surrounding the needle track. The total amount of Aβ present within these microglia is a small fraction of the total sAβ originally injected. Furthermore, the Aβ present in microglia is neither Congo red-birefringent nor thioflavin S-positive, suggesting that it is not in a fibrillar form. Together, these results suggest that although sAβ may be neurotoxic in vitro, in the rat CNS in vivo, injected sAβ is cleared rapidly and does not induce a sustained glial response that characterizes acute and subchronic neurotoxicity (Gramsbergen and van den Berg, 1994;O’Callaghan et al., 1995).
Although both microglia and astrocytes show a dramatic upregulation of iNOS expression in response to injection of fAβ, there are marked spatial and morphological differences in the microglia and astrocyte responses to fAβ. Whereas microglia surround and phagocytize fAβ, astrocytes show no evidence of Aβ phagocytosis but rather form a virtual wall between microglia containing fAβ and the surrounding neurons. Previous studies of human AD tissue (Itagaki et al., 1989;Uchihara et al., 1995) and the brains of mice overexpressing a mutant APP gene (Games et al., 1995) have also noted that reactive microglia are concentrated toward the center of AD plaques, whereas reactive astrocytes are present at the margins of plaques. Microglia containing intracellular Aβ have been noted in AD brain (Akiyama et al., 1996), but these microglia lack detectable mRNA encoding Aβ, suggesting an extracellular Aβ origin (Scott et al., 1993). Similar findings have also been noted in vitro, where microglia have been shown to scavenge and accumulate Aβ intracellularly (Frackowiak et al., 1992;Shaffer et al., 1995; Ard et al., 1996; Paresce et al., 1996), whereas astrocytes envelop and wall-off fAβ (Canning et al., 1993; Pike et al., 1994; DeWitt and Silver, 1996).
Aβ-induced neuronal loss
In the current study, no significant difference in the number of calbindin-immunoreactive projection neurons was observed when comparing fAβ and sAβ injection areas and the normal striatum. In the AD striatum, the density of calbindin neurons has been shown previously not to differ significantly from that of normal controls (Selden et al., 1994). The current findings differ, however, from previous studies that showed a decrease in calbindin immunoreactivity in AD versus normal cortex (Ferrer et al., 1993; Nishiyama et al., 1993). In contrast, the number of parvalbumin- and nNOS-immunoreactive interneurons in the current study is significantly reduced in the fAβ injection area compared with the sAβ injection area and the normal striatum. Previous studies have shown that parvalbumin neurons in the rat striatum are more sensitive to excitotoxic injury than are calbindin neurons (Waldvogel et al., 1991). Although the results of the current study strongly suggest that fAβ induces a selective loss of neurons in the rat striatum, this effect could also be explained by a selective downregulation of parvalbumin and nNOS in neurons surrounding the injected fAβ. Aβ1–40, the peptide length examined in the present study, has also been shown to cause glial-induced inhibition of neurite outgrowth in vitro (Canning et al., 1993) and to reduce numbers of choline acetyltransferase-immunoreactive neurons after injection in vivo (Giovannelli et al., 1995).
Mechanisms of Aβ toxicity
After injection of fAβ, microglia and astrocytes rapidly separate fAβ from the surrounding neuropil, suggesting that Aβ-induced neurotoxicity may not result from direct contact between Aβ fibrils and surrounding neurons. If fAβ is not directly toxic to neurons, how might the intervening glia mediate fAβ toxicity? Recent studies have demonstrated that the receptor for advanced glycation end products (RAGE) and class A and B scavenger receptors on microglia bind Aβ, which leads to microglial activation (El Khoury et al., 1996;Paresce et al., 1996; Yan et al., 1996). Glial activation, in turn, results in enhanced expression of a variety of bioactive molecules, including neurotoxic phenolic amines, cytokines, growth factors, protease inhibitors, adhesion molecules, and nitric oxide (Boje and Arora, 1992; Dickson et al., 1992; Ishii and Haga, 1992; Haga et al., 1993; Giulian et al., 1995; Mrak et al., 1995). Of particular interest is a recent study (Giulian et al., 1996) showing that human Aβ1–40 and Aβ1–42, but not rodent Aβ, induce microglial adherence of Aβ and killing of neurons in vitro. This finding, together with those of the present study in vivo, underscores the importance of using the human Aβ peptide to induce neuronal toxicity in rodent models of AD.
Aβ-induced NOS expression and NO production in cultured microglia and macrophages have also been reported previously (Klegeris et al., 1994;Goodwin et al., 1995; Meda et al., 1995), and NOS induction has also been linked to neuronal impairment and death in several neurological disorders involving multiple sclerosis and acquired immunodeficiency syndrome dementia (Bo et al., 1994; Adamson et al., 1996). The present study demonstrates that fAβ induces neuronal loss and a significant increase in iNOS expression by microglia and astrocytes in vivo, suggesting that it is the release of bioactive molecules like nitric oxide by microglia and astrocytes, rather than direct contact between Aβ fibrils and neurons, that mediates Aβ neurotoxicity in AD. Note, however, that the expression of NOS by human microglia remains controversial, because evidence supporting induction of reactive nitrogen species in human microglia seems strongly dependent on the experimental methodologies used (Brosnan et al., 1994).
A critical question that must be addressed in examining any animal model of a human disease is how well the animal model mimics the mechanisms and ultimate pathology observed in the human disease. Clearly, there are differences between the present rat model and human AD. First, whereas in the present study gliosis and pathology are observed over a time course of 30 d, in human AD the time course is on the order of decades. Second, the amyloid load per unit of brain area is generally higher and the amyloid distribution more extensive in the human AD brain than in our rat model. However, a key difficulty in addressing the mechanisms of pathology in human AD is that one rarely looks at human brain tissue at the initiation of the disease but rather examines the AD brain at the end stage of the disease, long after the initial mechanisms of pathology occur. A major advantage of the present rat model of fAβ-induced gliosis and neurotoxicity is that it provides a time- and space-compressed view of the initial changes that fAβ induces in the CNS in vivo. Thus, determining how glia serve as intermediaries in the apparent fAβ-induced neurotoxicity observed in the present model should shed significant light on the mechanisms of fAβ-induced neurotoxicity and allow one to test whether therapeutic interventions can block this effect.
Footnotes
This work was supported by National Institutes of Health Grants AG11852, NS23970, and AG12853, by the Veterans Administration Merit Review, and by Alzheimer’s Association Grant PRG94-194. We thank Dr. Dennis Selkoe for the contribution of anti-Aβ R1282 and Evelyn Stimson for preparation of the 125I-Aβ tracer.
Correspondence should be addressed to Dr. Patrick W. Mantyh, Department of Preventive Sciences, University of Minnesota, 515 Delaware Street, Minneapolis, MN 55455.
REFERENCES
- 1.Adamson DC, Wildemann B, Sasaki M, Glass JD, McArthur JC, Christov VI, Dawson TM, Dawson VL. Immunologic NO synthase–elevation in severe AIDS dementia and induction by HIV-1 Gp41. Science. 1996;274:1917–1921. doi: 10.1126/science.274.5294.1917. [DOI] [PubMed] [Google Scholar]
- 2.Akiyama H, Schwab C, Kondo H, Mori H, Kametani F, Ikeda K, McGeer PL. Granules in glial cells of patients with Alzheimer’s disease are immunopositive for C-terminal sequences of beta-amyloid protein. Neurosci Lett. 1996;206:169–172. doi: 10.1016/s0304-3940(96)12474-5. [DOI] [PubMed] [Google Scholar]
- 3.Ard MD, Cole GM, Wei J, Mehrle AP, Fratkin JD. Scavenging of Alzheimer’s amyloid beta-protein by microglia in culture. J Neurosci Res. 1996;43:190–202. doi: 10.1002/(SICI)1097-4547(19960115)43:2<190::AID-JNR7>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 4.Barron KD. The microglial cell. A historical review. J Neurol Sci. 1995;134:57–68. doi: 10.1016/0022-510x(95)00209-k. [DOI] [PubMed] [Google Scholar]
- 5.Bennett BD, Bolam JP. Two populations of calbindin D28k-immunoreactive neurones in the striatum of the rat. Brain Res. 1993;610:305–310. doi: 10.1016/0006-8993(93)91414-n. [DOI] [PubMed] [Google Scholar]
- 6.Benson JR, Louie PC, Bradshaw RA. Amino acid analysis of peptides. In: Gross E, Mienhofer J, editors. The peptides, Vol 4. Academic; New York: 1981. pp. 217–260. [Google Scholar]
- 7.Bo L, Dawson TM, Wesselingh S, Mork S, Choi S, Kong PA, Hanley D, Trapp BD. Induction of nitric oxide synthase in demyelinating regions of multiple sclerosis brains. Ann Neurol. 1994;36:778–786. doi: 10.1002/ana.410360515. [DOI] [PubMed] [Google Scholar]
- 8.Boje KM, Arora PK. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992;587:250–256. doi: 10.1016/0006-8993(92)91004-x. [DOI] [PubMed] [Google Scholar]
- 9.Brelje TC, Wessendorf M, Sorenson RL. Multicolor laser scanning confocal immunofluorescence microscopy: practical application and limitations. In: Matsumoto B, editor. Methods in cell biology, Vol 38. Academic; San Diego: 1993. pp. 97–181. [DOI] [PubMed] [Google Scholar]
- 10.Brosnan CF, Battistini L, Raine CS, Dickson DW, Casadevall A, Lee SC. Reactive nitrogen intermediates in human neuropathology: an overview. Dev Neurosci. 1994;16:152–161. doi: 10.1159/000112102. [DOI] [PubMed] [Google Scholar]
- 11.Busciglio J, Gabuzda DH, Matsudaira P, Yankner BA. Generation of beta-amyloid in the secretory pathway in neuronal and nonneuronal cells. Proc Natl Acad Sci USA. 1993;90:2092–2096. doi: 10.1073/pnas.90.5.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bush AI, Pettingell WH, Multhaup G, Paradis M, Vonsattel JP, Gusella JF, Beyreuther K, Masters CL, Tanzi RE. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science. 1994;265:1464–1467. doi: 10.1126/science.8073293. [DOI] [PubMed] [Google Scholar]
- 13.Canning DR, McKeon RJ, DeWitt DA, Perry G, Wujek JR, Frederickson RC, Silver J. Beta-amyloid of Alzheimer’s disease induces reactive gliosis that inhibits axonal outgrowth. Exp Neurol. 1993;124:289–298. doi: 10.1006/exnr.1993.1199. [DOI] [PubMed] [Google Scholar]
- 14.Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 15.Cummings BJ, Cotman CW. Image analysis of beta-amyloid load in Alzheimer’s disease and relation to dementia severity. Lancet. 1995;346:1524–1528. doi: 10.1016/s0140-6736(95)92053-6. [DOI] [PubMed] [Google Scholar]
- 16.DeWitt DA, Silver J. Regenerative failure: a potential mechanism for neuritic dystrophy in Alzheimer’s disease. Exp Neurol. 1996;142:103–110. doi: 10.1006/exnr.1996.0182. [DOI] [PubMed] [Google Scholar]
- 17.Dickson DW, Ksiezak-Reding H, Liu WK, Davies P, Crowe A, Yen SH. Immunocytochemistry of neurofibrillary tangles with antibodies to subregions of tau protein: identification of hidden and cleaved tau epitopes and a new phosphorylation site. Acta Neuropathol (Berl) 1992;84:596–605. doi: 10.1007/BF00227736. [DOI] [PubMed] [Google Scholar]
- 18.Eddleston M, Mucke L. Molecular profile of reactive astrocytes–implications for their role in neurologic disease. Neuroscience. 1993;54:15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature. 1996;382:716–719. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- 20.Esler WP, Stimson ER, Jennings JM, Ghilardi JR, Mantyh PW, Maggio JE. Zinc-induced aggregation of human and rat beta-amyloid peptides in vitro. J Neurochem. 1996;66:723–732. doi: 10.1046/j.1471-4159.1996.66020723.x. [DOI] [PubMed] [Google Scholar]
- 21.Esler WP, Stimson ER, Ghilardi JR, Felix AM, Lu Y, Vinters HV, Mantyh PW, Maggio JE. A-beta deposition inhibitor screen using synthetic amyloid. Nature Biotech. 1997;15:258–263. doi: 10.1038/nbt0397-258. [DOI] [PubMed] [Google Scholar]
- 22.Evans KC, Berger EP, Cho CG, Weisgraber KH, Lansbury PT., Jr Apolipoprotein E is a kinetic but not a thermodynamic inhibitor of amyloid formation: implications for the pathogenesis and treatment of Alzheimer disease. Proc Natl Acad Sci USA. 1995;92:763–767. doi: 10.1073/pnas.92.3.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferrer I, Tunon T, Soriano E, del Rio A, Iraizoz I, Fonseca M, Guionnet N. Calbindin D-28k immunoreactivity in the temporal neocortex in patients with Alzheimer’s disease. Clin Neuropathol. 1993;12:53–58. [PubMed] [Google Scholar]
- 24.Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol (Berl) 1992;84:225–233. doi: 10.1007/BF00227813. [DOI] [PubMed] [Google Scholar]
- 25.Fraser PE, Nguyen JT, Chin DT, Kirschner DA. Effects of sulfate ions on Alzheimer beta/A4 peptide assemblies: implications for amyloid fibril-proteoglycan interactions. J Neurochem. 1992;59:1531–1540. doi: 10.1111/j.1471-4159.1992.tb08470.x. [DOI] [PubMed] [Google Scholar]
- 26.Frautschy SA, Cole GM, Baird A. Phagocytosis and deposition of vascular beta-amyloid in rat brains injected with Alzheimer beta-amyloid. Am J Pathol. 1992;140:1389–1399. [PMC free article] [PubMed] [Google Scholar]
- 27.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 28.Giovannelli L, Casamenti F, Scali C, Bartolini L, Pepeu G. Differential effects of amyloid peptides β-(1–40) and β-(25–35) injections into the rat nucleus basalis. Neuroscience. 1995;66:781–792. doi: 10.1016/0306-4522(94)00610-h. [DOI] [PubMed] [Google Scholar]
- 29.Giulian D, Haverkamp LJ, Li J, Karshin WL, Yu J, Tom D, Li X, Kirkpatrick JB. Senile plaques stimulate microglia to release a neurotoxin found in Alzheimer brain. Neurochem Int. 1995;27:119–137. doi: 10.1016/0197-0186(95)00067-i. [DOI] [PubMed] [Google Scholar]
- 30.Giulian D, Haverkamp LJ, Yu JH, Karshin W, Tom D, Li J, Kirkpatrick J, Kuo YM, Roher AE. Specific domains of beta-amyloid from Alzheimer plaques elicit neuron killing in human microglia. J Neurosci. 1996;16:6021–6037. doi: 10.1523/JNEUROSCI.16-19-06021.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glenner GG, Wong CW. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 32.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Williamson R, Rossor M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 33.Goodwin JL, Uemura E, Cunnick JE. Microglial release of nitric oxide by the synergistic action of beta-amyloid and IFN-gamma. Brain Res. 1995;692:207–214. doi: 10.1016/0006-8993(95)00646-8. [DOI] [PubMed] [Google Scholar]
- 34.Gramsbergen JB, van den Berg KJ. Regional and temporal profiles of calcium accumulation and glial fibrillary acidic protein levels in rat brain after systemic injection of kainic acid. Brain Res. 1994;667:216–228. doi: 10.1016/0006-8993(94)91499-0. [DOI] [PubMed] [Google Scholar]
- 35.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, Selkoe DJ. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 36.Haga S, Ikeda K, Sato M, Ishii T. Synthetic Alzheimer amyloid beta/A4 peptides enhance production of complement C3 component by cultured microglial cells. Brain Res. 1993;601:88–94. doi: 10.1016/0006-8993(93)91698-r. [DOI] [PubMed] [Google Scholar]
- 37.Harrigan MR, Kunkel DD, Nguyen LB, Malouf AT. Beta amyloid is neurotoxic in hippocampal slice cultures. Neurobiol Aging. 1995;16:779–789. doi: 10.1016/0197-4580(95)00095-v. [DOI] [PubMed] [Google Scholar]
- 38.Ishii T, Haga S. Complements, microglial cells and amyloid fibril formation. Res Immunol. 1992;143:614–616. doi: 10.1016/0923-2494(92)80043-k. [DOI] [PubMed] [Google Scholar]
- 39.Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989;24:173–182. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- 40.Jarrett JT, Lansbury PT., Jr Amyloid fibril formation requires a chemically discriminating nucleation event: studies of an amyloidogenic sequence from the bacterial protein OsmB. Biochemistry. 1992;31:12345–12352. doi: 10.1021/bi00164a008. [DOI] [PubMed] [Google Scholar]
- 41.Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 42.Jarrett JT, Berger EP, Lansbury PT., Jr The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 43.Jarrett JT, Costa PR, Griffin RG, Lansbury PT., Jr Models of the beta protein C-terminus: differences in amyloid structure may lead to segregation of “long” and “short” fibrils. J Am Chem Soc. 1994;116:9741–9742. [Google Scholar]
- 44.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 45.Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18:527–535. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- 46.Kelly JF, Furukawa K, Barger SW, Rengen MR, Mark RJ, Blanc EM, Roth GS, Mattson MP. Amyloid beta-peptide disrupts carbachol-induced muscarinic cholinergic signal transduction in cortical neurons. Proc Natl Acad Sci USA. 1996;93:6753–6758. doi: 10.1073/pnas.93.13.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kennedy WR, Wendelschafer-Crabb G, Brelje TC. Innervation and vasculature of human sweat glands: an immunohistochemistry–laser scanning confocal fluorescence microscopy study. J Neurosci. 1994;14:6825–6833. doi: 10.1523/JNEUROSCI.14-11-06825.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kirschner DA, Abraham C, Selkoe DJ. X-ray diffraction from intraneuronal paired helical filaments and extraneuronal amyloid fibers in Alzheimer disease indicates cross-beta conformation. Proc Natl Acad Sci USA. 1986;83:503–507. doi: 10.1073/pnas.83.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kirschner DA, Inouye H, Duffy LK, Sinclair A, Lind M, Selkoe DJ. Synthetic peptide homologous to beta protein from Alzheimer disease forms amyloid-like fibrils in vitro. Proc Natl Acad Sci USA. 1987;84:6953–6957. doi: 10.1073/pnas.84.19.6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klegeris A, Walker DG, McGeer PL. Activation of macrophages by Alzheimer beta amyloid peptide. Biochem Biophys Res Commun. 1994;199:984–991. doi: 10.1006/bbrc.1994.1326. [DOI] [PubMed] [Google Scholar]
- 51.Koh JY, Yang LL, Cotman CW. Beta-amyloid protein increases the vulnerability of cultured cortical neurons to excitotoxic damage. Brain Res. 1990;533:315–320. doi: 10.1016/0006-8993(90)91355-k. [DOI] [PubMed] [Google Scholar]
- 52.Ma J, Yee A, Brewer HB, Jr, Das S, Potter H. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature. 1994;372:92–94. doi: 10.1038/372092a0. [DOI] [PubMed] [Google Scholar]
- 53.Mantyh PW, Ghilardi JR, Rogers S, DeMaster E, Allen CJ, Stimson ER, Maggio JE. Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of beta-amyloid peptide. J Neurochem. 1993;61:1171–1174. doi: 10.1111/j.1471-4159.1993.tb03639.x. [DOI] [PubMed] [Google Scholar]
- 54.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. Beta-amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McGeer PL, McGeer EG. Anti-inflammatory drugs in the fight against Alzheimer’s disease. Ann NY Acad Sci. 1996;777:213–220. doi: 10.1111/j.1749-6632.1996.tb34421.x. [DOI] [PubMed] [Google Scholar]
- 56.Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 57.Mrak RE, Sheng JG, Griffin WS. Glial cytokines in Alzheimer’s disease: review and pathogenic implications. Hum Pathol. 1995;26:816–823. doi: 10.1016/0046-8177(95)90001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Murphy S, Simmons ML, Agullo L, Garcia A, Feinstein DL, Galea E, Reis DJ, Minc-Golomb D, Schwartz JP. Synthesis of nitric oxide in CNS glial cells. Trends Neurosci. 1993;16:323–328. doi: 10.1016/0166-2236(93)90109-y. [DOI] [PubMed] [Google Scholar]
- 59.Nishiyama E, Ohwada J, Iwamoto N, Arai H. Selective loss of calbindin D28K-immunoreactive neurons in the cortical layer II in brains of Alzheimer’s disease: a morphometric study. Neurosci Lett. 1993;163:223–226. doi: 10.1016/0304-3940(93)90388-2. [DOI] [PubMed] [Google Scholar]
- 60.O’Callaghan JP, Jensen KF, Miller DB. Quantitative aspects of drug and toxicant-induced astrogliosis. Neurochem Int. 1995;26:115–124. doi: 10.1016/0197-0186(94)00106-5. [DOI] [PubMed] [Google Scholar]
- 61.Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–565. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 62.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; North Ryde, New South Wales, Australia: 1986. [Google Scholar]
- 63.Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. Aggregation-related toxicity of synthetic beta-amyloid protein in hippocampal cultures. Eur J Pharmacol. 1991a;207:367–368. doi: 10.1016/0922-4106(91)90014-9. [DOI] [PubMed] [Google Scholar]
- 64.Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. In vitro aging of beta-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 1991b;563:311–314. doi: 10.1016/0006-8993(91)91553-d. [DOI] [PubMed] [Google Scholar]
- 65.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by beta-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pike CJ, Cummings BJ, Monzavi R, Cotman CW. Beta-amyloid-induced changes in cultured astrocytes parallel reactive astrocytosis associated with senile plaques in Alzheimer’s disease. Neuroscience. 1994;63:517–531. doi: 10.1016/0306-4522(94)90547-9. [DOI] [PubMed] [Google Scholar]
- 67.Pike CJ, Walencewicz-Wasserman AJ, Kosmoski J, Cribbs DH, Glabe CG, Cotman CW. Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25–35 region to aggregation and neurotoxicity. J Neurochem. 1995;64:253–265. doi: 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- 68.Price DL, Borchelt DR, Walker LC, Sisodia SS. Toxicity of synthetic Aβ peptides and modeling of Alzheimer’s disease. Neurobiology of Aging. 1992;13:623–625. doi: 10.1016/0197-4580(92)90069-a. [DOI] [PubMed] [Google Scholar]
- 69.Sanan DA, Weisgraber KH, Russell SJ, Mahley RW, Huang D, Saunders A, Schmechel D, Wisniewski T, Frangione B, Roses AD, Strittmatter WJ. Apolipoprotein E associates with beta amyloid peptide of Alzheimer’s disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J Clin Invest. 1994;94:860–869. doi: 10.1172/JCI117407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 71.Scott SA, Johnson SA, Zarow C, Perlmutter LS. Inability to detect beta-amyloid protein precursor mRNA in Alzheimer plaque-associated microglia. Exp Neurol. 1993;121:113–118. doi: 10.1006/exnr.1993.1076. [DOI] [PubMed] [Google Scholar]
- 72.Selden N, Geula C, Hersh L, Mesulam MM. Human striatum: chemoarchitecture of the caudate nucleus, putamen and ventral striatum in health and Alzheimer’s disease. Neuroscience. 1994;60:621–636. doi: 10.1016/0306-4522(94)90491-x. [DOI] [PubMed] [Google Scholar]
- 73.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 74.Selkoe DJ. Alzheimer’s disease: a central role for amyloid. J Neuropathol Exp Neurol. 1994;53:438–447. doi: 10.1097/00005072-199409000-00003. [DOI] [PubMed] [Google Scholar]
- 75.Selkoe DJ. Alzheimer’s disease–genotypes, phenotype, and treatments. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 76.Selkoe DJ, Abraham CR. Isolation of paired helical filaments and amyloid fibers from human brain. Methods Enzymol. 1986;134:388–404. doi: 10.1016/0076-6879(86)34105-3. [DOI] [PubMed] [Google Scholar]
- 77.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, McCormack R, Wolfert R, Selkoe D, Lieberburg I, Schenk D. Isolation and quantification of soluble Alzheimer’s beta-peptide from biological fluids. Nature. 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 78.Shaffer LM, Dority MD, Gupta-Bansal R, Frederickson RC, Younkin SG, Brunden KR. Amyloid beta protein (A beta) removal by neuroglial cells in culture. Neurobiol Aging. 1995;16:737–745. doi: 10.1016/0197-4580(95)00055-j. [DOI] [PubMed] [Google Scholar]
- 79.Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B, Younkin SG. Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science. 1992;258:126–129. doi: 10.1126/science.1439760. [DOI] [PubMed] [Google Scholar]
- 80.Soreghan B, Kosmoski J, Glabe C. Surfactant properties of Alzheimer’s A beta peptides and the mechanism of amyloid aggregation. J Biol Chem. 1994;269:28551–28554. [PubMed] [Google Scholar]
- 81.Uchihara T, Kondo H, Akiyama H, Ikeda K. Single-laser three-color immunolabeling of a histological section by laser scanning microscopy: application to senile plaque-related structures in post-mortem human brain tissue. J Histochem Cytochem. 1995;43:103–106. doi: 10.1177/43.1.7822758. [DOI] [PubMed] [Google Scholar]
- 82.van Gool WA, Kuiper MA, Walstra GJ, Wolters EC, Bolhuis PA. Concentrations of amyloid beta protein in cerebrospinal fluid of patients with Alzheimer’s disease. Ann Neurol. 1995;37:277–279. doi: 10.1002/ana.410370221. [DOI] [PubMed] [Google Scholar]
- 83.Waldvogel HJ, Faull RL, Williams MN, Dragunow M. Differential sensitivity of calbindin and parvalbumin immunoreactive cells in the striatum to excitotoxins. Brain Res. 1991;546:329–335. doi: 10.1016/0006-8993(91)91497-o. [DOI] [PubMed] [Google Scholar]
- 84.Wisniewski T, Castano EM, Golabek A, Vogel T, Frangione B. Acceleration of Alzheimer’s fibril formation by apolipoprotein E in vitro. Am J Pathol. 1994;145:1030–1035. [PMC free article] [PubMed] [Google Scholar]
- 85.Wong M-L, Rettori V, Al-Shekhlee A, Bongiorno PB, Canteros G, McCann SM, Gold PW, Licinio J. Inducible nitric oxide synthase gene expression in the brain during systemic inflammation. Nat Med. 1996;2:581–584. doi: 10.1038/nm0596-581. [DOI] [PubMed] [Google Scholar]
- 86.Yan SD, Chen X, Fu J, Chen M, Zhu HJ, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 87.Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]