

Abstract
Activation of the Drosophila photoresponse is a rapid process that results in plasma membrane Ca2+and Na+ conductances. Ca2+functions in negative feedback regulation of Drosophilavision including deactivation. Protein kinase C (PKC) binds directly to Ca2+ and is required for deactivation. However, the consequences of disrupting phosphorylation of any individual PKC substrate in the Drosophila retina have not been addressed. In the current work, we show that NINAC p174, which consists of a protein kinase domain joined to the head region of myosin heavy chain, is a phosphoprotein and is phosphorylated in vitro by PKC. Mutation of either of two PKC sites in the p174 tail resulted in an unusual defect in deactivation that had not been detected previously for other ninaC alleles or other loci. After cessation of the light stimulus, there appeared to be a transient reactivation of the visual cascade. This phenotype suggests that a mechanism exists to prevent reactivation of the visual cascade and that p174 participates in this process.
Keywords: NINAC, protein kinase C, myosin, Drosophila, phototransduction, vision, deactivation
Activation of theDrosophila visual cascade is extremely rapid and results in opening of the cation influx channels transient receptor potential (TRP) and transient receptor potential-like (TRPL) within ∼10–20 msec of photostimulation of rhodopsin (Ranganathan et al., 1991). The G-protein–signaling cascade that leads to opening of the ion channels has been extensively characterized and is known to involve the inositol phospholipid–signaling system (for review, see Montell, 1998). Termination of the photoresponse, after cessation of the light stimulus, is also rapid and is a Ca2+-regulated process (Hardie, 1995); however, our understanding of the mechanism by which Ca2+ contributes to termination of the photoresponse is quite incomplete.
Several proteins have been identified that seem to mediate Ca2+-dependent termination of phototransduction. These include the Ca2+-binding regulatory protein calmodulin that functions in both light adaptation and termination of the light response (Porter et al., 1993, 1995; Arnon et al., 1997;Scott et al., 1997). The ninaC (neither inactivation nor afterpotential C) locus, which encodes two isoforms, p132 and p174, each of which consists of a protein kinase domain fused to a myosin head domain (Montell and Rubin, 1988), also functions in negative feedback regulation of the photoresponse (Porter et al., 1995). The two NINAC proteins differ because of unique C-terminal ends. p174 is localized exclusively to the microvillar portion of the photoreceptors, the rhabdomeres, and p132 is restricted to the cell bodies (Porter et al., 1992). Null mutations in ninaC cause defects in adaptation and response termination (Porter et al., 1992, 1995). These functions are caused by p174 because elimination of p174, but not p132, causes each of these phenotypes (Porter et al., 1992).
Because negative feedback regulation seems to be mediated by Ca2+ (Hardie, 1995), it is plausible that p174 is regulated by Ca2+. However, p174 does not contain a known Ca2+-binding motif, such as an EF hand or C2 domain, and there is no evidence that it binds Ca2+ directly. Thus, p174 seems to respond to the light-dependent Ca2+ flux indirectly. One NINAC Ca2+ sensor is calmodulin because NINAC binds to calmodulin (Porter et al., 1993) and the NINAC–calmodulin interaction is required for both adaptation and termination (Porter et al., 1993, 1995; Arnon et al., 1997). NINAC might also be regulated by Ca2+-dependent phosphorylation because p174 contains multiple protein kinase C (PKC) consensus sites including several in its unique C-terminal tail. Moreover, mutation of an eye-specific PKC (ePKC) causes perturbations in adaptation and termination (Smith et al., 1991; Hardie et al., 1993). The role of PKC in negative feedback regulation may be more significant than that indicated by mutation of ePKC because a second PKC, brain PKC (brPKC), is known to be enriched in the Drosophila retina and a third PKC, PKC98F, is highly expressed in adult heads (Schaeffer et al., 1989). Two retinal substrates for PKC have been identified. These are the TRP cation influx channel (Huber et al., 1998) and the PSD95, DLG, and ZO-1 (PDZ)-containing protein inactivation, no afterpotential D (INAD) (Huber et al., 1998), which binds to most of the proteins that function in phototransduction and organizes a supramolecular signaling complex (for review, see Montell, 1998). However, the consequences of disrupting PKC phosphorylation of any retinal substrate that functions in Drosophila vision have not been determined.
In the current report, we show that p174 is a phosphoprotein and is phosphorylated in vitro by PKC. Mutation of the PKC sites in the p174 unique tail resulted in an unusual phenotype not described previously. Specifically, after cessation of the light stimulus, there appeared to be an oscillation in the termination of the photoresponse. The initial termination of the photoresponse was short-lived and was followed by one or more transient depolarizations of decreasing amplitude. Thus, phosphorylation of the p174 tail seems to be required for stable termination of the visual cascade.
MATERIALS AND METHODS
In vivo labeling and detection of NINAC proteins. Proteins were labeled in adult flies according to procedures similar to those described previously (Matsumoto et al., 1982). One hundred young adult flies (<5 d old) of each genotype were starved for 24 hr in an empty vial placed in a humidified chamber. One hundred microliters of 32P-labeled orthophosphate in water (1 mCi/ml) were mixed with 50 μl of sucrose (1 m) and dispensed onto a 1 inch × 2 inch square of Whatmann No. 3MM paper set in an otherwise empty vial. The starved flies were transferred to another vial containing orthophosphate and were allowed to feed on the sucrose mixture for 24 hr in a humidified chamber. The NINAC proteins were extracted in imidazole buffer (10 mmimidazole, pH 7.35, 300 mm NaCl, 50 mm NaF, 1 mm DTT, 10 μg/ml PMSF, and 1 μg/ml leupeptin) and immunoprecipitated using protein A beads and 25 μl of antiserum to p174 (αp174) or p132 (αp132). The immunocomplexes were then fractionated by SDS-PAGE and detected by autoradiography.
In vitro expression of NINAC and phosphorylation by PKC. To express full-length p174 in Sf9 cells (derived fromSpodoptera frugiperda), we subcloned aSmaI-SpeI fragment from pcninaC15R (Montell and Rubin, 1988) between the SmaI-XbaI sites of the baculovirus transfer vector pVL1393 (PharMingen, San Diego, CA). A cDNA encoding full-length Drosophilacalmodulin (Yamanaka et al., 1987) was also subcloned into pVL1393 by transferring a BamHI and EcoRI (blunt-ended) fragment from pJFM34 to the BamHI and SalI (blunt-ended) sites of the transfer vector (pBac-CaM). The p174 and calmodulin clones were introduced into baculovirus by cotransfecting the transfer vector clones and the linearized baculovirus DNA in Sf9 using the BaculoGold transfection kit (PharMingen). Recombinant baculovirus clones were isolated, and the proteins were expressed in Sf9 as described by the manufacturer. p174 was extracted from Sf9 cells by lysing the cells in 20 mm imidazole, pH 8.0, 1 mm EDTA, 1 mm PMSF, 10% sucrose, 0.5%N-lauroylsarkosyl, and 160 mm NaCl. To determine whether p174 was a PKC substrate, we purified p174 by first fractionating the extracts on a gel filtration column (Sephacryl S-400 HR) and passing the pooled fractions over a Q-Sepharose column. NINAC eluted in a sharp peak using a KCl gradient, and the pooled fractions were then applied to calmodulin–agarose and eluted with EGTA. p174 (1 μl) was added to 99 μl of reaction mix [2 mmMgCl2, 4 mm CaCl2, 1 μl of [γ-32P]ATP (100 μCi), and 1 μl of protein kinase C catalytic subunit (∼3 ng; ∼2 units; Calbiochem, La Jolla, CA)] and incubated for 20 min at 30°C
To map the PKC sites in the p174 tail, we inserted cDNA fragments encoding amino acid residues 1281–1501, 1254–1326, and 1326–1501 into pUR288 to create the β-galactosidase fusion proteins βgNC1281, βgNC1254, and βgNC1326, respectively. Point mutations were performed using single-stranded pSL2 as described previously (Porter et al., 1992). pSL2 consists of a cDNA fragment encoding the C-terminal 222 residues of p174 (residues 1280–1501) subcloned into pBluescript KS+ (Porter et al., 1992). After the mutagenesis, the cDNA fragments were subcloned into pUR288 to create βgNC1281. All fusion proteins were expressed in Escherichia coli and extracted in lysis buffer (50 mm Tris, pH 7.5, 1 mm EDTA, 1 mm PMSF, 10% sucrose, 0.5% Triton X-100, and 160 mm NaCl). To test whether the β-galactosidase fusion proteins were substrates for phosphorylation by PKC, we combined 150 μl of total bacterial extract (∼50 μg of the fusion protein) with 150 μl of reaction mix and incubated for 20 min with ∼3 ng of PKC (∼2 units; Calbiochem) at 30°C. In some experiments, the fusion proteins were subsequently purified using p-aminophenyl β-d-thiogalactopyranoside beads (Sigma, St. Louis, MO).
Generation of transgenic flies expressing NINAC derivatives.To generate transgenic flies expressing altered forms of NINAC with serine-to-alanine substitutions in residues 1316 and/or 1320, we performed oligonucleotide mutagenesis as described on aXhoI-SpeI genomic fragment encoding the entire p174-specific tail (residues 1082–1501) (Porter et al., 1992). The altered restriction fragments containing the point mutations were swapped with the corresponding wild-typeXhoI-SpeI fragment in a clone, pGninaC+ (ninaC genomic sequences, coordinates 3.4–14.6, subcloned between theBamHI and EcoRI sites of pHSS7), which contains the ninaC promoter and the entire protein coding region (Porter et al., 1992). A NotI fragment containing theninaC genomic sequences was then subcloned into theNotI site of the ry+ P-element transformation vector pDM30 (Mismer and Rubin, 1987). The lysine-to-arginine mutation in residue 45 was created by oligonucleotide mutagenesis using the genomic clone pBSXX1 and was subcloned into the pDM30 clone containing the rest of theninaC gene as described (Porter et al., 1992). After injecting the DNAs (400 μg/ml) along with pπ25.7 (100 μg/ml) intoninaCP235; rosy(ry) embryos, the transformants were selected on the basis of ry+ eye color. The NINAC protein levels and the retinal morphology of the transgenic flies were checked by performing Western blots using αZB551 antiserum.
Coimmunoprecipitation of p174 and calmodulin.Coimmunoprecipitation of p174 and calmodulin with αp174 antiserum was performed as described (Porter et al., 1995). The immune complexes were fractionated by SDS-PAGE, and the Western blot was cut in half to separate the higher and lower molecular weight proteins. The upper and lower portions of the blot were probed with αp174 and calmodulin antibodies, respectively, and subsequently with125I-labeled protein A.
Electroretinogram recordings. The ERGs and prolonged depolarization afterpotentials were performed as described previously except for the substitution of a Warner Instrument Corporation Electrometer IE-210 and a MacLab/4 sec analog-to-digital converter using Chart v3.4/s (Porter et al., 1992). All flies were dark adapted for at least 2 min before recording.
RESULTS
Phosphorylation of NINAC in vivo
To determine whether the NINAC proteins were phosphoproteins, we attempted to label the proteins in vivo by feeding wild-type flies 32P-labeled orthophosphate as described previously (Matsumoto et al., 1982). The NINAC proteins were then immunoprecipitated, fractionated by SDS-PAGE, and detected by autoradiography. We found that both NINAC proteins (Fig.1A) were phosphorylatedin vivo (Fig. 1B).
Fig. 1.

NINAC proteins were phosphorylated in vivo. A, Wild-type flies were fed32P-labeled orthophosphate, and the NINAC proteins were immunoprecipitated from head extracts with antibodies to p174 (αp174) or p132 (αp132) and were fractionated by SDS-PAGE. The proteins were detected by staining with Coomassie blue. B, The same gel shown inA is shown exposed to x-ray film. C, Phosphoproteins in wild-type (WT),ninaCP235 (ninaC; null allele), and P[ninaCK45R] (K45R) flies were labeled with [32P]orthophosphate, fractionated by SDS-PAGE, and detected by staining with Coomassie blue. D, The same gel shown in C is shown exposed to x-ray film.
Because NINAC has a protein kinase domain and displays serine and threonine kinase activity in vitro (Ng et al., 1996), it was possible that the phosphorylation was caused primarily or exclusively by autophosphorylation. To ascertain whether phosphorylation of NINAC was attributable primarily to trans- or autophosphorylation, we compared the phosphorylation levels of wild-type NINAC and an altered form predicted to be devoid of protein kinase activity. We had previously generated transgenic flies, P[ninaCKD], expressing truncated derivatives of NINAC missing the entire protein kinase domain (Porter and Montell, 1993). However, the NINACKD isoform was of limited value in this analysis because the autophosphorylation sites could have resided in the kinase domain. All protein kinases contain a conserved lysine required for anchoring and orienting ATP by binding to the α and β phosphates (for review, see Johnson et al., 1996). Conservative lysine-to-arginine mutations in other protein kinases drastically reduce if not eliminate the enzymatic activity (e.g., Kamps and Sefton, 1986). Consequently, we generated transgenic flies, P[ninaCK45R], expressing a modified derivative of NINAC containing an arginine substitution in the corresponding lysine in NINAC (residue 45). The altered form of NINAC was expressed in a null background,ninaCP235, that fails to express any wild-type NINAC (Matsumoto et al., 1987; Montell and Rubin, 1988). The K45R mutation did not disrupt stability of p174 because the NINACK45R isoform was expressed at levels similar to that of the wild-type protein (Fig. 1C). Consistent with the idea that the K45R mutation affected the protein kinase activity of p174, the electrophysiological phenotype of the P[ninaCK45R] flies was indistinguishable from that observed in P[ninaCKD] flies (data not shown) (Porter et al., 1993). Of significance here, wild-type p174 and p174K45R were phosphorylated to similar extentsin vivo (Fig. 1D). Thus, it seemed that NINAC was primarily a substrate for trans- rather than autophosphorylaton.
Identification of PKC sites specific to p174
The observation that NINAC is a phosphoprotein and that the phosphorylation was not detectably reduced in P[ninaCK45R] flies indicated that NINAC was principally phosphorylated by other protein kinases. PKC might phosphorylate NINAC because both proteins function in adaptation and deactivation of the photoresponse (Smith et al., 1991; Hardie et al., 1993; Porter et al., 1995). Moreover, p174 contains 21 potential PKC sites including five that are specific to p174 because they are in its unique C-terminal tail (Fig. 2). To determine whether p174 was an in vitro substrate for PKC, we expressed the full-length protein in Sf9 cells using a baculovirus expression vector. The vast majority of p174 expressed in the cells was insoluble; however, we found that coexpression of p174 with calmodulin greatly increased the solubility (data not shown). p174 was then partially purified (see Materials and Methods) and tested as anin vitro substrate for PKC. The results of the analysis indicated that p174 (Fig. 3A) was phosphorylated in vitro by PKC (Fig. 3B). However, another partially purified protein, E. coliβ-galactosidase (Fig. 3A), did not serve as a PKC substrate (Fig. 3B). Because p174 contained multiple potential PKC sites, most of which were common to both NINAC isoforms, we focused on the sites specific to p174 because this is the isoform that is required for phototransduction (Porter et al., 1992). Thus, phosphorylation of p174 in the C-terminal tail may regulate a function specific to p174. This would potentially include calmodulin binding to the site unique to p174 or localization to the rhabdomeres.
Fig. 2.

Consensus PKC phosphorylation sites are indicated by the small vertical lines above the schematics of p132 and p174. The protein kinase, the myosin heavy chain head, and the tail domains specific to each protein are represented by different shading. The positions of the calmodulin (CaM) binding sites are indicated by thearrowheads. Shown at the bottom are schematics of three fragments of the p174 tail (residues 1281–1501, 1254–1326, and 1326–1501) that were fused to β-galactosidase to create the fusion proteins βgNC1281, βgNC1254, and βgNC1326, respectively. aa, Amino acids.
Fig. 3.

p174 is phosphorylated by PKC on serine residues 1316 and 1320. A, p174 expressed in Sf9 cells. Total protein extracts from untransfected Sf9 cells (Sf9), total protein extracts from Sf9 cells transfected with p174 baculovirus-transfected Sf9 cells (Sf9/p174), and p174 purified from Sf9 cells (Purif. p174) were fractionated by SDS-PAGE, and protein was detected by staining with Coomassie blue. In addition, β-galactosidase was expressed inE. coli and purified using p-aminophenyl β-d-thiogalactopyranoside (APTG) beads (Purif. β-gal). B, The purified p174 protein, shown in A, in vitrophosphorylated with PKC and detected by autoradiography. The β-galactosidase, shown in A, did not serve as anin vitro substrate for PKC. C, In vitro phosphorylation of β-galactosidase fusion proteins (see Fig. 2). The fusion proteins (indicated by the dots to the left of the bands) were expressed in BL21 bacteria and labeled with [γ-32P]ATP, and PKC and the fusion proteins were purified with APTG beads.Top, An SDS-PAGE gel stained with Coomassie blue.Bottom, The corresponding autoradiograph. Thelower band in the βgNC1281 lane is a degradation product. D, Expression of derivatives of the βgNC1281 fusion protein in BL21 bacteria. The Coomassie-stained gel shows total bacterial extracts from cells containing unmodified βgNC1281 (WT) minus and plus isopropyl thiogalactoside (IPTG) induction and the total proteins from cells expressing the βgNC1281 derivatives with alanine substitutions at the following sites: both 1316 and 1320 (Both), 1316, and 1320. Total proteins from cells expressing β-galactosidase (β-gal) after IPTG induction are also shown. The fusion protein (Fusion) andβ-gal are indicated. E, PKC phosphorylation assay of βgNC1281 and indicated mutant derivatives. Total bacterial extracts were fractionated by SDS-PAGE, and the phosphoproteins were detected by autoradiography. The fusion proteins (Fusion) and PKC are indicated. The fusion proteins were detected as doublets because of protein degradation that occurred during the course of the phosphorylation assay.
To determine whether PKC phosphorylated any sites in the p174 tail, we expressed in E. coli a β-galactosidase fusion protein that included all five p174-specific sites (βgNC1281; Fig. 2). Total bacterial extracts were then incubated with PKC and [γ-32P]ATP; the fusion protein was purified usingp-aminophenyl β-d-thiogalactopyranoside beads and was fractionated by SDS-PAGE. βgNC1281, but not β-galactosidase, was phosphorylated by PKC (Fig. 3C). Moreover, in total bacterial extracts containing β-galactosidase–NINAC fusion proteins, only the fusion proteins and none of the endogenous proteins were labeled (Fig.3D,E). In addition, the PKC added to the extracts was autophosphorylated.
The sites in the p174 tail phosphorylated by PKC were mapped by performing additional in vitro phosphorylation analyses using NINAC fusion proteins containing subsets of the five consensus motifs specific to the p174 tail. PKC phosphorylated a β-galactosidase fusion protein, βgNC1254, that contained two of the consensus PKC sites, residues 1316 and 1320, but not a different fusion, βgNC1326, that included the remaining three C-terminal sites at residues 1426, 1446, and 1457 (Fig. 3C). Substitution of an alanine residue at position 1316 or 1320 reduced the level of phosphorylation approximately twofold, and mutation of both sites eliminated all detectable phosphorylation of the β-galactosidase fusion protein (Fig. 3E). Thus, p174 was an in vitro substrate for PKC at sites 1316 or 1320.
In vivo consequences of mutation of p174-specific PKC sites
The effects of mutating the PKC sites in the p174 tail, 1316 and 1320, were addressed by generating transgenic flies in which one or both phosphorylation sites (BP) were mutated: P[ninaC1316], P[ninaC1320], and P[ninaCBP]. Each of the transgenes was introduced into a ninaCP235 background to prevent expression of any wild-type NINAC. All three transgenic lines are collectively referred to as P[ninaC174PS] (p174 PKC sites) because they exhibited similar phenotypes (see below). Mutation of either 1316, 1320, or both sites had no deleterious effects on the stability of p174 (Fig.4A), on calmodulin binding (Fig. 4B), or on rhabdomere localization of p174 (data not shown). Furthermore, the P[ninaC174PS] flies did not exhibit retinal degeneration (data not shown).
Fig. 4.

p174 derivatives expressed in the transgenic flies expressed normal levels of proteins and coimmunoprecipitated with calmodulin. A, Total head extracts fromninaCP235 (ninaC null allele), Canton S (WT), and transgenic flies expressing the indicated p174 derivatives were fractionated by SDS-PAGE, and a Western blot was probed with αZB551 antiserum and125I-labeled protein A. B, Coimmunoprecipitation of calmodulin with p174 from fly head extracts is shown. The p174 was immunoprecipitated with αp174 antiserum in the presence or absence of Ca2+, and a Western blot of the immune complexes was divided in half. Bottom, Probed with anti-calmodulin antibodies. Top, Probed with αZB551 antiserum.
To explore whether there were any effects on negative feedback regulation resulting from mutation of the PKC sites, we performed electroretinogram recordings (ERGs). ERGs measure the light responses of all the cells in the retina. In wild type, the corneal negative response to light is followed by a rapid return to baseline after cessation of the light stimulus (Fig. 5). Elimination of p174 or both NINAC proteins results in a defect in adaptation as indicated by an initial corneal negative deflection that is larger than normal (Fig. 5) (Porter et al., 1992). In addition, the termination of the photoresponse appears to be disrupted inninaC because the return to the baseline after cessation of the light stimulus is slower than that in wild type (Fig. 5) (Porter et al., 1992).
Fig. 5.
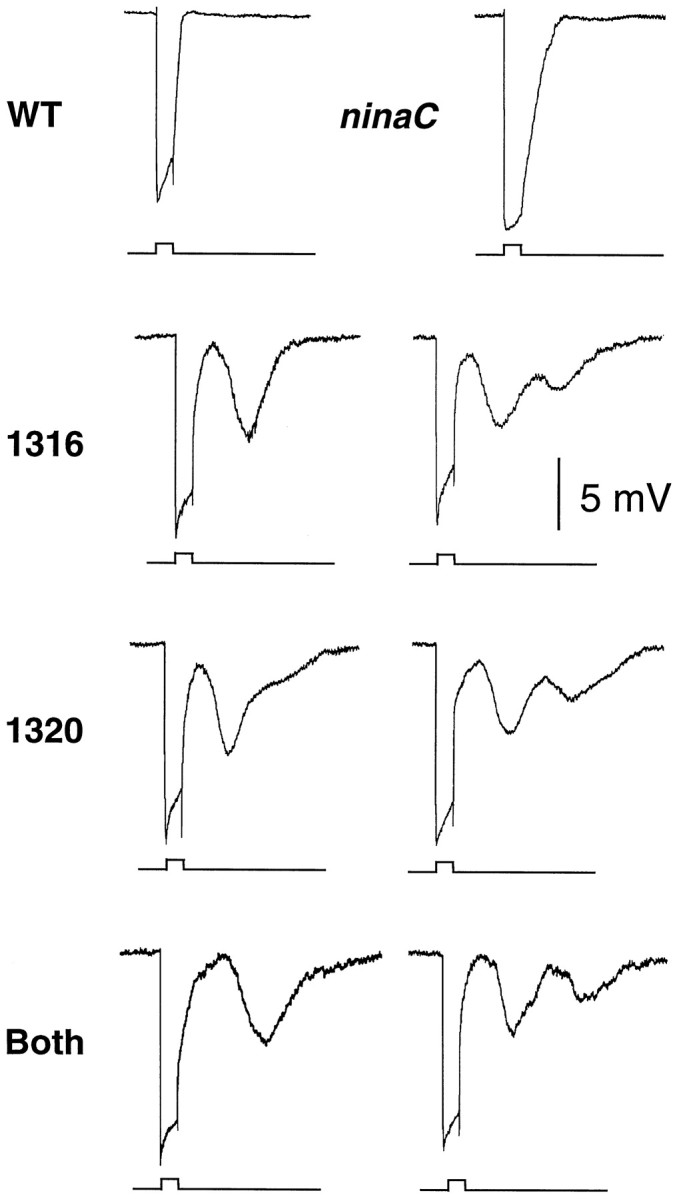
ERGs to a single pulse of white light (5 sec duration). The ERGs were performed onw+ flies that were 7–14 d old. An event marker indicating the initiation and cessation of the light stimulus is shown (bottom of eachrecording). Shown are one or two representative ERGs obtained using each of the following flies: wild-type (WT), ninaCP235null mutant (ninaC), P[ninaC1316] (1316), P[ninaC1320] (1320), and P[ninaCBP] (Both) transgenic flies.
Of primary significance in the current report, mutation of either one or both PKC sites was found to result in an unusual defect in deactivation of the photoresponse that had not been detected previously. In the transgenic flies, the initial corneal negative deflection in the ERGs was indistinguishable between wild type and P[ninaC174PS]. Thus, there was no apparent defect in adaptation or activation. Furthermore, after cessation of the light stimulus, the return of the maintained component to baseline was not significantly slower in P[ninaC174PS] than in wild type. However, subsequent corneal negative deflections were observed in the dark (dark responses). A dark response, with a peak amplitude ∼30–50% as large as the initial light response, was typically detected. In addition, a second dark response was detected in ∼20% of the ERGs. Although the maximum peak amplitude of the second dark response was ∼50% of the second component, the amplitude was usually <25% as large as the first dark response. The peak amplitude of the first dark response occurred ∼14 sec after cessation of the light stimulus, and the maximum amplitude of the second dark response was ∼17 sec after the first dark response. Thus, the temporal appearance of the first and second dark responses as well as the gradual slopes of the waveforms was similar. The relative amplitudes of each successive peak, the shapes of the waveforms, and the temporal relationship of each component of the ERG did not vary appreciatively with the intensity of the light pulse (data not shown).
DISCUSSION
The P[ninaC174PS] phenotype is unique among phototransduction mutants in the production of corneal negative deflections in the ERG after a seemingly normal light-dependent activation and return to the baseline. This phenotype indicates that absence of light is insufficient to prevent transient reactivation of the visual cascade after cessation of the light stimulus. Stable termination seems to require an active mechanism that involves NINAC p174. Absence of this mechanism seems to result in a dampening oscillatory deactivation of the cascade. The specific components in the phototransduction machinery that require p174 for stable termination remain to be identified. However, the recent observation that p174 binds to the PDZ domain-containing protein INAD (P. D. Wes, X.-Z. S. Xu, H.-S. Li, F. Chien, S. K. Doberstein, and C. Montell, unpublished observations), which tethers most of the proteins critical in Drosophilaphototransduction together (for review, see Montell, 1998), indicates that p174 is located in close proximity to many signaling components. These include rhodopsin, phospholipase C, PKC, and the TRP and TRPL cation channels as well as other components that function in phototransduction (Huber et al., 1996; Shieh and Zhu, 1996; Chevesich et al., 1997; Tsunoda et al., 1997; Xu et al., 1998).
There were two significant differences in the ERG responses of P[ninaC174PS] and flies that do not express p174, such as P[ninaCΔ174]. First, the initial termination of the photoresponse is delayed in P[ninaCΔ174] (Porter et al., 1992) but appears normal in P[ninaC174PS]. One possibility is that the delay is a consequence of the reduction in the rhabdomeral concentration of calmodulin, which occurs in P[ninaCΔ174] (Porter et al., 1993) but not in P[ninaC174PS]. Ca2+/calmodulin functions in deactivation (Scott et al., 1997); thus, P[ninaC174PS] may not display any initial defect in termination because the rhabdomeral concentration of calmodulin appears to be normal in these flies.
A second difference in the ERGs of P[ninaC174PS] and P[ninaCΔ174] flies is that these latter flies do not exhibit the transient corneal negative responses in the dark (Porter et al., 1992). Based on the P[ninaC174PS] mutant phenotype, one of the roles of p174 may be to prevent transient reactivation of the visual cascade after cessation of the light. However, an apparent conundrum is that the defect in stable termination was not detected in the null allele ninaCP235 or in P[ninaCΔ174] flies despite the absence of the p174 protein. One possible explanation is that the protracted deactivation in P[ninaCΔ174] causes the Ca2+ levels to remain higher than normal after cessation of the light stimulus. The higher Ca2+levels may result in stable termination of the photoresponse, independent of p174.
The phenotype in P[ninaC174PS] flies resulted from mutation of one or both in vitro PKC sites in the p174 tail. NINAC represents a candidate substrate for PKC because it is a phosphoprotein, has multiple putative PKC sites, and, like ePKC (Smith et al., 1991; Hardie et al., 1993), functions in the termination of phototransduction (Porter et al., 1995). Moreover, it was of interest to test the physiological consequences of mutating the PKC sites in p174 because the effects of disrupting PKC phosphorylation of any individual retinal protein had not been addressed. However, the observation that mutation of the ePKC does not result in a defect in stable termination of the photoresponse suggests that some other protein kinase may phosphorylate p174 at residues 1316 and 1320. Candidates include other protein kinases expressed in the retina, such as brPKC, whose potential role in phototransduction has not been investigated.
In conclusion, the termination mechanisms controllingDrosophila phototransduction seem to be more complicated than envisioned previously. In addition to a requirement for NINAC in facilitating rapid deactivation after cessation of the light stimulus, there is an additional requirement for this unconventional myosin in preventing transient reactivation of the plasma membrane conductances. Because p174 also functions in adaptation, it seems that NINAC has a central role in many aspects of negative feedback regulation of the visual cascade. Recently, a homolog of NINAC has been identified in the mammalian retina (D. Hillman, A. Dose, and B. Burnside, personal communication). Thus, it is intriguing to speculate that vertebrate NINAC also functions in negative feedback regulation and that an active mechanism may also exist in mammalian photoreceptor cells to ensure stable termination of phototransduction.
Footnotes
This work was supported by the National Eye Institute Grant EY08117 to C.M.
Correspondence should be addressed to Dr. Craig Montell, Departments of Biological Chemistry and Neuroscience, The Johns Hopkins University School of Medicine, Baltimore, MD 21205.
Dr. Porter’s present address: Ontogeny, Inc., 45 Moulton Street, Cambridge, MA 02138.
REFERENCES
- 1.Arnon A, Cook B, Gillo B, Montell C, Selinger Z, Minke B. Calmodulin regulation of light adaptation and store-operated dark current in Drosophila photoreceptors. Proc Natl Acad Sci USA. 1997;94:5894–5899. doi: 10.1073/pnas.94.11.5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chevesich J, Kreuz AJ, Montell C. Requirement for the PDZ domain protein, INAD, for localization of the TRP store-operated channel to a signaling complex. Neuron. 1997;18:95–105. doi: 10.1016/s0896-6273(01)80049-0. [DOI] [PubMed] [Google Scholar]
- 3.Hardie RC. Photolysis of caged Ca2+ facilitates and inactivates but does not directly excite light-sensitive channels in Drosophila photoreceptors. J Neurosci. 1995;15:889–902. doi: 10.1523/JNEUROSCI.15-01-00889.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hardie RC, Peretz A, Suss-Toby E, Rom-Glass A, Bishop SA, Selinger Z, Minke B. Protein kinase C is required for light adaptation in Drosophila photoreceptors. Nature. 1993;363:634–637. doi: 10.1038/363634a0. [DOI] [PubMed] [Google Scholar]
- 5.Huber A, Sander P, Paulsen R. Phosphorylation of the InaD gene product, a photoreceptor membrane protein required for recovery of visual excitation. J Biol Chem. 1996;271:11710–11717. doi: 10.1074/jbc.271.20.11710. [DOI] [PubMed] [Google Scholar]
- 6.Huber A, Sander P, Bähner M, Paulsen R. The TRP Ca2+ channel assembled in a signaling complex by the PDZ domain protein INAD is phosphorylated through the interaction with protein kinase C (ePKC). FEBS Lett. 1998;425:317–322. doi: 10.1016/s0014-5793(98)00248-8. [DOI] [PubMed] [Google Scholar]
- 7.Johnson LN, Noble MEM, Owen DJ. Active and inactive protein kinases: structural basis for regulation. Cell. 1996;85:149–158. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 8.Kamps MP, Sefton BM. Neither arginine nor histidine can carry out the function of lysine-295 in the ATP-binding site of p60src. Mol Cell Biol. 1986;6:751–757. doi: 10.1128/mcb.6.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsumoto H, O’Tousa JE, Pak WL. Light-induced modification of Drosophila retinal polypeptides in vivo. Science. 1982;217:839–841. doi: 10.1126/science.7100927. [DOI] [PubMed] [Google Scholar]
- 10.Matsumoto H, Isono K, Pye Q, Pak WL. Gene encoding cytoskeletal proteins in Drosophila rhabdomeres. Proc Natl Acad Sci USA. 1987;84:985–989. doi: 10.1073/pnas.84.4.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mismer D, Rubin GM. Analysis of the promoter of the ninaE opsin gene in Drosophila melanogaster. Genetics. 1987;116:565–578. doi: 10.1093/genetics/116.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montell C. TRP trapped in fly signaling web. Curr Opin Neurobiol. 1998;8:389–397. doi: 10.1016/s0959-4388(98)80066-4. [DOI] [PubMed] [Google Scholar]
- 13.Montell C, Rubin GM. The Drosophila ninaC locus encodes two photoreceptor cell specific proteins with domains homologous to protein kinases and the myosin heavy chain head. Cell. 1988;52:757–772. doi: 10.1016/0092-8674(88)90413-8. [DOI] [PubMed] [Google Scholar]
- 14.Ng KP, Kambara T, Matusuura M, Burke M, Ikebe M. Identification of myosin III as protein kinase. Biochemistry. 1996;33:9392–9399. doi: 10.1021/bi960181a. [DOI] [PubMed] [Google Scholar]
- 15.Porter JA, Montell C. Distinct roles of the Drosophila ninaC kinase and myosin domains revealed by systematic mutagenesis. J Cell Biol. 1993;122:601–612. doi: 10.1083/jcb.122.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Porter JA, Hicks JL, Williams DS, Montell C. Differential localizations of and requirements for the two Drosophila ninaC kinase/myosins in photoreceptor cells. J Cell Biol. 1992;116:683–693. doi: 10.1083/jcb.116.3.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Porter JA, Yu M, Doberstein SK, Pollard TS, Montell C. Dependence of calmodulin localization in the retina on the ninaC unconventional myosin. Science. 1993;262:1038–1042. doi: 10.1126/science.8235618. [DOI] [PubMed] [Google Scholar]
- 18.Porter JA, Minke B, Montell C. Calmodulin binding to Drosophila NinaC required for termination of phototransduction. EMBO J. 1995;18:4450–4459. doi: 10.1002/j.1460-2075.1995.tb00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ranganathan R, Harris GL, Stevens CF, Zuker CS. A Drosophila mutant defective in extracellular calcium-dependent photoreceptor deactivation and desensitization. Nature. 1991;354:230–232. doi: 10.1038/354230a0. [DOI] [PubMed] [Google Scholar]
- 20.Schaeffer E, Smith D, Mardon G, Quinn W, Zuker C. Isolation and characterization of two new Drosophila protein kinase C genes, including one specifically expressed in photoreceptor cells. Cell. 1989;57:403–412. doi: 10.1016/0092-8674(89)90915-x. [DOI] [PubMed] [Google Scholar]
- 21.Scott K, Sun Y, Beckingham K, Zuker CS. Calmodulin regulation of Drosophila light-activated channels and receptor function mediates termination of the light response in vivo. Cell. 1997;91:375–383. doi: 10.1016/s0092-8674(00)80421-3. [DOI] [PubMed] [Google Scholar]
- 22.Shieh B-H, Zhu M-Y. Regulation of the TRP Ca2+ channel by INAD in Drosophila photoreceptors. Neuron. 1996;16:991–998. doi: 10.1016/s0896-6273(00)80122-1. [DOI] [PubMed] [Google Scholar]
- 23.Smith DP, Ranganathan R, Hardy RW, Marx J, Tsuchida T, Zuker CS. Photoreceptor deactivation and retinal degeneration mediated by a photoreceptor-specific protein kinase C. Science. 1991;254:1478–1484. doi: 10.1126/science.1962207. [DOI] [PubMed] [Google Scholar]
- 24.Tsunoda S, Sierralta J, Sun Y, Bodner R, Suzuki E, Becker A, Socolich M, Zuker CS. A multivalent PDZ-domain protein assembles signalling complexes in a G-protein-coupled cascade. Nature. 1997;388:243–249. doi: 10.1038/40805. [DOI] [PubMed] [Google Scholar]
- 25.Xu X-Z, Choudhury A, Li X, Montell C. Coordination of an array of signaling proteins through homo- and heteromeric interactions between PDZ domains and target proteins. J Cell Biol. 1998;142:545–555. doi: 10.1083/jcb.142.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamanaka MK, Saugstad JA, Hanson-Painton O, McCarthy BJ, Tobin SL. Structure and expression of the Drosophila calmodulin gene. Nucleic Acids Res. 1987;15:3335–3348. doi: 10.1093/nar/15.8.3335. [DOI] [PMC free article] [PubMed] [Google Scholar]