

Abstract
Microglia are the major inflammatory cells in the brain, in which microglial inflammatory responses are modulated by interactions with other brain cells. Here, we show that astrocytes, the most abundant cells in the brain, can secrete one or more factors capable of modulating microglial activation by regulating the microglial levels of reactive oxygen species (ROS). Treatment of microglia with astrocyte culture-conditioned media (ACM) increased the expression level and activity of hemeoxygenase-1 (HO-1). ACM also induced nuclear translocation of the nuclear factor E2-related factor 2 transcription factor, increased the binding activity of the antioxidant response element (ARE), and enhanced HO-1 promoter activity in an ARE-dependent manner. Furthermore, treatment with ACM suppressed interferon-γ (IFN-γ)-induced ROS production, leading to reduced inducible nitric oxide synthase (iNOS) expression and nitric oxide (NO) release. In agreement with these results, mimickers of HO-1 products, such as bilirubin, ferrous iron, and a carbon monoxide-releasing molecule, reduced IFN-γ-induced iNOS expression and/or NO release. Finally, we found that the active component(s) in ACM was heat labile and smaller than 3 kDa. Together, these results suggest that astrocytes could cooperate with microglia to prevent excessive inflammatory responses in the brain by regulating microglial expression of HO-1 and production of ROS.
Keywords: brain inflammation, hemeoxygenase-1, cell–cell interaction, microglia, astrocyte, reactive oxygen species
Introduction
After brain injury, brain macrophages called microglia are activated, leading to production of inflammatory mediators such as nitric oxide (NO), tumor necrosis factor-α (TNF-α), and prostaglandins. Although inflammation is an indispensable defense mechanism against pathogens, it often damages surrounding tissues. Therefore, the extent of inflammation should be tightly controlled to maximize the antipathogenic effect while minimizing tissue damage.
Studies have shown that a number of cell types act in concert to regulate inflammation via cell–cell interaction. Neutrophils were recently shown to suppress TNF-α and interleukin-6 (IL-6) release from macrophages in a mouse wound model, indicating that neutrophils can negatively regulate inflammation. This type of coordinated inflammatory regulation is also found in the brain, in which microglia act as the major inflammatory cells in concert with the highly abundant astrocytes. Previous studies have reported that astrocytes suppress the expression of IL-12 and inducible nitric oxide synthase (iNOS) in activated microglia (Aloisi et al., 1997; Vincent et al., 1997; Pyo et al., 2003). However, little is known regarding how astrocytes exert this effect.
Our group and others reported that reactive oxygen species (ROS) are one of the major signaling molecules capable of mediating microglial activation induced by several inflammatory stimulators (Min et al., 2003, 2004; Qin et al., 2004). Therefore, microglial activation could be regulated by modulating intracellular ROS level. Antioxidant enzymes such as heme oxygenase-1 (HO-1) could be considered as candidate molecules to regulate intracellular ROS. HO-1 catalyzes the degradation of heme, resulting in production of biliverdin, carbon monoxide (CO), and ferrous iron (Fe2+) (Maines, 1988). Many studies have reported the anti-inflammatory as well as antioxidant roles of HO-1. Biliverdin reductase converts biliverdin into bilirubin, and bilirubin alleviates oxidative damage at injury sites in experimental autoimmune encephalomyelitis models and inhibits lipopolysaccharide-induced NO production in macrophages (Liu et al., 2003; Wang et al., 2004). Carbon monoxide attenuates bronchial inflammation induced by aeroallergen and prevents liver inflammation (Chapman et al., 2001; Ott et al., 2005). And finally, ferrous iron released by HO activity rapidly induces expression of ferritin, protecting cells under oxidizing conditions by sequestering free cytosolic iron (Ward et al., 1994).
The induction of HO-1 is regulated at the level of transcription. Nuclear factor E2-related factor 2 (Nrf2), a basic leucine zipper transcription factor, plays an important role in the expression of HO-1 in response to diverse HO-1 inducers, including the HO-1 substrate heme, various environmental and industrial toxins, and even antioxidants (for review, see Alam and Cook, 2003). In response to these inducers, Nrf2 translocates to the nucleus and binds to the antioxidant response element (ARE) in the promoter region of HO-1 gene (Itoh et al., 1997).
Here, we examined whether astrocytes modulate microglial inflammatory responses through decreasing microglial ROS level. The results in this study demonstrated that astrocyte culture conditioned media (ACM) induced nuclear translocation of Nrf2, increased the ARE binding activity of Nrf2, and enhanced HO-1 expression and activity in microglia. These responses might lead to decreased microglial intracellular ROS levels and suppression of interferon-γ (IFN-γ)-induced inflammatory responses, indicating that excessive microglial brain inflammation and subsequent tissue damage might be prevented by interactions between microglia and astrocytes.
Materials and Methods
Materials.
H2O2 was purchase from Duksan Pure Chemical (Ansan, Korea). Interferon-γ was purchased from Peprotech (Rocky Hill, NJ). Hemin was purchased from Porphyrin Products (Logan, UT). PCR primers were obtained from Bioneer (Daejeon, Korea). The others were purchased from Sigma (St. Louis, MO).
Cell culture.
Primary microglia were cultured from the cerebral cortices of 1- to 3-d-old Sprague Dawley rats as described previously (Giulian and Baker, 1986; Pyo et al., 1998). Briefly, cortices were triturated into single cells in minimal essential media (MEM) (Sigma) containing 10% fetal bovine serum (FBS) (JBI, Taegu, Korea), plated in 75 cm2 T-flasks (0.5 hemisphere/flask), and incubated for 2 weeks. Microglia were detached from the flasks by mild shaking and filtered through a nylon mesh to remove astrocytes and cell clumps. Microglia were cultured in MEM containing 5% FBS. Primary astrocytes remaining in the flask were harvested with 0.1% trypsin. Astrocytes were plated in 100 mm dishes and cultured in MEM supplemented with 10% FBS. BV2 cells (murine microglial cells) were cultured in DMEM (Invitrogen, Carlsbad, CA) supplemented with 5% FBS.
Preparation of ACM.
Astrocytes were grown to confluence. On reaching confluence, culture media were changed with MEM without FBS. ACM were collected 3 d after the media changed and stored at −70°C until use.
Measurement of ROS.
For measurement of intracellular ROS levels, cells were incubated with 10 μm 5-(and-6)-chloromethyl-2′,7′-dichlorodihydro-fluorescein diacetate [CM-H2DCF-DA (herein referred to as DCF) (Invitrogen)] for 10 min. The cells were then washed with D-PBS (in mm: 2.68 KCl, 1.47 KH2PO4, 136.89 NaCl, and 8.1 Na2HPO4), and the fluorescence and differential interference contrast (DIC) images were taken using a Zeiss (Oberkochen, Germany) laser confocal microscope. DCF intensities of cells were also counted using LSM 5 Image Browser (Zeiss).
HO-1 activity assay.
HO enzyme activity was measured as described previously (Kutty and Maines, 1982). Briefly, microsomes from BV2 cells and primary microglia were added to a reaction mixture containing NADPH (0.8 mm), rat liver cytosol (2 mg) as a source of biliverdin reductase, the substrate hemin (10 μm), glucose-6-phosphate (2 mm), and glucose-6-phosphate dehydrogenase (0.2 U). The reaction was performed in the dark for 1 h at 37°C and terminated by the addition of 500 μl of chloroform. The amount of bilirubin left in the chloroform layer was measured by calculating the differences in optical densities at 450 and 540 nm.
Immunocytochemistry.
Cells were fixed with 100% methanol at −20°C for 20 min, washed with PBS, and incubated with 10% FBS for 30 min. The cells were then incubated overnight at 4°C with an anti-Nrf2 antibody (Santa Cruz Biotechnology, Santa Cruz, CA), washed with PBS, and reacted with a fluorescence-conjugated secondary antibody (Cappel, Durham, NC) for 1 h at room temperature. The cells were then washed, stained for 5 min with 20 μm Hoechst 33342 (Invitrogen), and finally examined under a Zeiss laser confocal microscope. Phase images were taken under a Nikon (Tokyo, Japan) Diaphot 300.
Measurement of NO.
The amount of nitrite formed from NO was measured by mixing the culture medium (50 μl) with an equal volume of Griess reagent (0.1% naphthylethylene diamine, 1% sulfanilamide, and 2.5% H3PO4). The optical density was measured at 540 nm (Ding et al., 1988).
Reverse transcription-PCR.
Total RNA was isolated using RNAzol B (iNtRON, Sungnam, Korea), and cDNA was prepared using the Avian Myeloblastosis Virus reverse transcriptase (Promega, Madison, WI), according to the instructions of the manufacturer. Reverse transcription (RT)-PCR was performed using primers specific for the HO-1, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and actin genes (Table 1). The amplified products were separated by electrophoresis on a 1.5% agarose gel and detected under UV light.
Table 1.
Primers for RT-PCR of HO-1, GAPDH, and actin
| Sense | Antisense | |
|---|---|---|
| Rat HO-1 | 5′-ACTTTCAGAAGGGTCAGGTGTCC-3′ | 5′-TTGAGCAGGAAGGCGGTCTTAG-3′ |
| Rat GAPDH | 5′-TCCCTCAAGATTGTCAGCAA-3′ | 5′-AGATCCACAACGGATACATT-3′ |
| Mouse HO-1 | 5′-CTATGTAAAGCGTCTCCA-3′ | 5′-GTCTTTGTGTTCCTCTGTC-3’ |
| Mouse actin | 5′-CATGTTTGAGACCTTCAACACCCC-3′ | 5′-GCCATCTCCTGCTCGAAGTCTAG-3′ |
Western blot analysis.
Cells were washed twice with cold PBS and lysed on ice in modified radioimmunoprecipitation assay buffer (50 mm Tris-HCl, pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mm NaCl, 1 mm Na3VO4, and 1 mm NaF) containing protease inhibitors [2 mm phenylmethylsulfonyl fluoride (PMSF), 10 μg/ml leupeptin, 10 μg/ml pepstatin, and 2 mm EDTA]. Each lysate was centrifuged at 10,000 × g for 10 min at 4°C, and the supernatants were collected. Proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was incubated with antibodies against iNOS (Upstate Biotechnology, Lake Placid, NY), HO-1 (Stressgen Biotechnologies, Victoria, British Columbia, Canada), or actin (Santa Cruz Biotechnology), followed by incubation with peroxidase-conjugated secondary antibodies (Zymed, San Francisco, CA) and visualization using an enhanced chemiluminescence system.
Plasmid.
The wild-type (E1 and E2) and ARE site-mutated (E1-M739 and E2-M45) HO-1 promoter/luciferase fusion constructs were kindly provided by Dr. Alam (Ochsner Clinic Foundation, New Orleans, LA) (Alam et al., 2003). E1 and E2 are mouse HO-1 promoters containing three ARE sequences, and E1-M739 and E2-M45 have mutations in three ARE core sequences.
Transfections and luciferase assay.
Transient transfections were performed in triplicate on 35 mm dishes. One day before transfection, BV2 cells were plated to maintain ∼60–80% confluence. The cells were transiently transfected with the plasmid E1, E2, E1-M739, or E2-M45 using Lipofectamine Plus reagents and Lipofectamine as instructed by the manufacturer (Invitrogen). After overnight transfection, cells were treated with ACM. Luciferase assay was performed according to the instructions of the manufacturer (Promega).
Measurement of ARE binding activity by electrophoresis mobility shift assay and supershift assay.
ARE binding activities were measured using electrophoresis mobility shift assay (EMSA), as described previously (Pier et al., 1981; Ryu et al., 2000). BV2 cells (1 × 106) and primary microglia (1.5 × 106) were harvested and suspended in 900 μl of hypotonic solution [in mm: 10 HEPES, 10 KCl, 0.1 EDTA, 0.1 EGTA, 1 dithiothreitol (DTT), and 0.5 PMSF, pH 7.9] for 15 min, followed by incubation in hypotonic solution containing 0.5% NP-40 for 5 min. The cells were then centrifuged at 500 × g for 10 min at 4°C, and the pellet containing the nuclear fraction was resuspended in buffer containing 20 mm HEPES, 20% glycerol, 0.4 m NaCl, 1 mm EDTA, 1 mm EGTA, 1 mm DTT, and 1 mm PMSF, pH 7.9. The samples were incubated on ice for 60 min with gentle shaking and then centrifuged at 10,000 × g for 10 min. The crude nuclear fractions (supernatants) were collected and stored at −70°C until use. Oligonucleotides representing the ARE [5′-TGGGGAACCTGTGCTGAGTCACTGGAG-3′; core ARE sequence is underlined) (Santa Cruz Biotechnology) (Suh et al., 2004)] and its complementary sequence were end labeled using T4 polynucleotide kinase (Promega) and [γ-32P]dATP. The labeled DNA probe (∼0.5 ng) was incubated for 30 min with 1 μg of nuclear proteins in a reaction mixture containing 21.4 mm EDTA, 21.4 mm EGTA, 20% glycerol, 0.29 mm ZnSO4, 10 ng/ml poly(dI-dC), 1 mm DTT, 0.4 mg/ml bovine serum albumin, and 8 mm MgCl2. The reaction mixture was resolved on a 6% polyacrylamide gel, the gel was dried, and the results were subjected to autoradiography. For supershift assays, the nuclear extracts were preincubated with 3 μg of anti-Nrf2 antibody (Santa Cruz Biotechnology) for 3 h at 4°C before the addition of the probe, and the reaction mixture was subjected to electrophoresis on a 6% polyacrylamide gel.
Fractionation of ACM.
ACM was fractionated using Amicon (Beverly, MA) Centriplus YM-3 centrifugal filter devices with a 3000 nominal molecular weight limit (Millipore, Bedford, MA), using ultrafiltration at 3000 × g for 140 min. The fractions above and below the filter were saved separately.
Statistical analysis.
Data in Figure 2C were analyzed by t test, and others were analyzed by one-way ANOVA followed by post hoc comparisons (Student–Newman–Keuls test) using the Statistical Package for Social Sciences 8.0 (SPSS, Chicago, IL).
Figure 2.

ACM induces HO-1 expression in microglia. Primary microglia (A–C) and BV2 cells (D–F) were treated with ACM for the indicated times. HO-1 mRNA and protein expression were determined using RT-PCR (A, D) and Western blotting (B, E). C, F, HO-1 activity was assayed as described in Materials and Methods. Values were mean ± SEM of three samples. #p < 0.05; *p < 0.01 compared with the control. Data are representative of at least three independent experiments.
Results
ACM suppresses ROS levels in microglia
Astrocytes are known to suppress the expression of inflammatory mediators in activated microglia (Vincent et al., 1996; Aloisi et al., 1997; Pyo et al., 2003). Because ROS are important signaling molecules for mediation of microglial activation induced by several stimulators (Min et al., 2003, 2004; Qin et al., 2004), we hypothesized that astrocytes might be capable of regulating microglial intracellular ROS levels. First, we examined whether soluble factors produced from astrocytes could regulate microglial ROS levels using ACM. When microglia were treated with H2O2 (100 nm), the intracellular ROS levels increased within 5 min and decreased to basal levels by 60 min (Fig. 1A,B). Next, we measured the intracellular ROS levels induced by H2O2 in microglia pretreated with ACM for 3–12 h. Interestingly, ROS levels were significantly reduced in cells pretreated with ACM for 6 and 12 h but not in cells pretreated with ACM for 3 h (Fig. 1C,D). These results suggested that soluble factors from astrocytes could regulate microglial ROS levels and that these soluble factors do not have direct ROS scavenging effect.
Figure 1.

ACM inhibits H2O2-induced ROS production in microglia. Primary microglia were treated with 100 nm H2O2 for the indicated time periods (A, B) or pretreated (P) with ACM for the indicated times and then treated with 100 nm H2O2 for 5 min (C, D). Intracellular ROS levels were assayed using 10 μm DCF as described in Materials and Methods. A, C, Fluorescence (DCF) images; DIC images were taken using a Zeiss laser confocal microscope. Scale bars, 50 μm. B, D, DCF intensities of cells in A and C were counted using LSM 5 Image Browser (Zeiss). Values are mean ± SEM of 10–15 cells. *p < 0.01 compared with H2O2. The data are representative of at least three independent experiments.
ACM increases HO-1 expression
Because antioxidant enzymes are considered good candidate molecules for modulating intracellular ROS levels, we examined whether ACM could induce expression of an antioxidant enzyme, HO-1. In primary cultured microglia, ACM significantly increased HO-1 mRNA and protein expression within 6 h, and the expression levels increased at 12 h and were sustained and/or further increased for up to 24 h (Fig. 2A,B). ACM also increased HO-1 activity at 12 h (Fig. 2C).
In BV2 microglia, ACM also induced HO-1 mRNA and protein expression. However, BV2 cells and primary cultured microglia showed slight differences in the time course of HO-1 mRNA and protein expression. In BV2 cells, both mRNA and protein levels increased within 3–6 h, a little earlier than in primary cultured microglia (data not shown), and slightly decreased at 24 h (Fig. 2D,E). ACM also increased HO-1 activity in BV2 cells. Within 6 h, HO-1 activity was increased 1.5- to 3-fold depending on experiments, and the activity was sustained for up to 24 h (Fig. 2F). These results collectively suggested that ACM could modulate microglial ROS levels through the expression of HO-1.
ACM activates Nrf2
Nrf2 is a key transcription factor that mediates expression of HO-1 (Itoh et al., 1997). Therefore, we examined whether ACM could induce Nrf2 activation, as measured by translocation of Nrf2 from the cytosol to the nucleus (Itoh et al., 1999). In primary cultured microglia, nuclear translocation of Nrf2 occurred within 15 min after ACM treatment, and Nrf2 was located in the nuclei for up to 60 min after ACM treatment (Fig. 3A). ACM also changed microglial morphology. In response to ACM, microglia became flat within 15 min and then became ramified at ∼60 min (Fig. 3B) but returned to the round shape within 12 h (Fig. 1C).
Figure 3.

ACM induces nuclear translocation of Nrf2. Primary microglia were treated with ACM for the indicated times. A, Cells were fixed and labeled with an anti-Nrf2 antibody, followed by an FITC-conjugated secondary antibody. The nuclei were visualized with Hoechst staining as described in Materials and Methods. The fluorescence images were obtained using a Zeiss laser confocal microscope. B, Phase-contrast images were obtained using a Nikon Diaphot 300. Scale bar, 50 μm. Data are representative of two independent experiments.
An EMSA revealed that ACM treatment increased the binding activities of the cellular nuclear extract to ARE-containing oligonucleotides within 5–15 min and that this activity was maintained for up to 60 min in rat primary microglia (Fig. 4A, top) and mouse BV2 cells (Fig. 4A, bottom). We used a supershift assay to confirm that Nrf2 bound to ARE-containing oligonucleotides. In the presence of Nrf2 antibodies, the intensity of the shifted band was significantly reduced (Fig. 4B).
Figure 4.
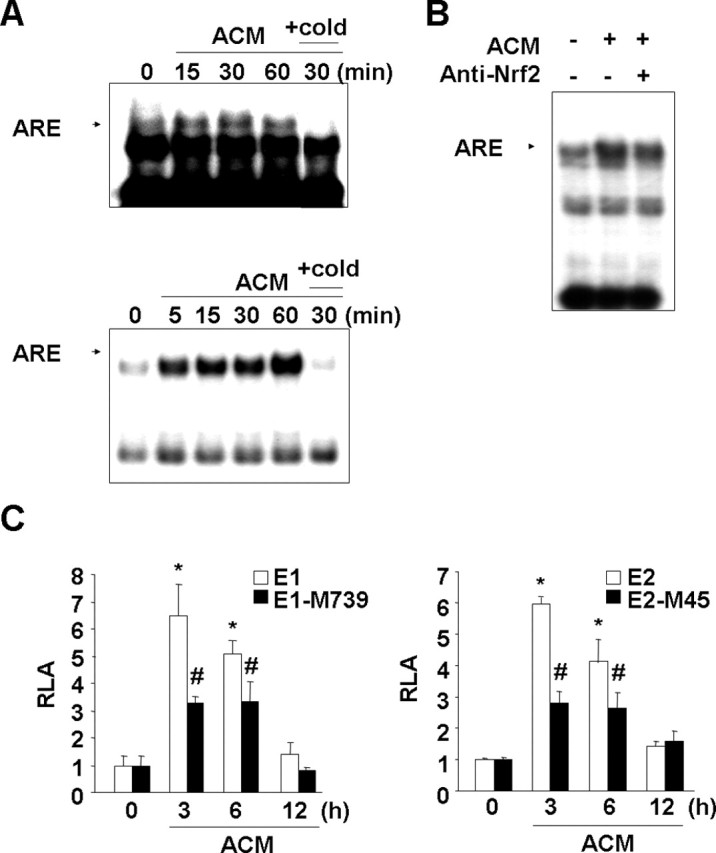
ACM induces ARE binding activity in nuclear extracts and enhances HO-1 promoter activity in an ARE-dependent manner. A, Primary microglia (top) and BV2 cells (bottom) were treated with ACM for the indicated times. Nuclear extracts were prepared, and the ARE-specific oligonucleotide–protein complexes were detected with an electrophoresis mobility shift assay. Addition of excess (X20) unlabeled oligonucleotide (cold) was used to determine specific binding of labeled oligonucleotides to the nuclear protein. B, For the supershift assay, nuclear extracts were prepared from BV2 cells treated with ACM for 30 min, mixed with 3 μg of Nrf2 antibody, and assayed for ARE-specific oligonucleotide–protein complexes as described in Materials and Methods. C, HO-1 promoter/luciferase fusion constructs, E1 and E2, and ARE site-mutated E1-M739 and E2-M45 constructs were transfected into BV2 cells. The transfected cells were treated with ACM for the indicated times and then assayed for luciferase activity as described in Materials and Methods. Values in C are mean ± SEM of three samples and represent relative luciferase activity (RLA). *p < 0.01 compared with the vector control. #p < 0.01 compared with the E1 or E2. Data are representative of three independent experiments.
We further confirmed that ARE mediated ACM-induced HO-1 expression using mouse HO-1 promoter/luciferase fusion constructs, E1, E2, E1-M739, and E2-M45 (Alam et al., 2003). The mouse HO-1 gene has two ARE-containing enhancers, E1 and E2, and each have three intact ARE sites and several putative CCAAT/enhancer-binding protein (C/EBP) and stimulating P-1 (SP-1) sites (Alam, 1994; Alam et al., 1995, 2000). In E1-M739 and E2-M45, three ARE sites were mutated (Alam et al., 2003). In BV2 cells transfected with E1 and E2, ACM induced luciferase activities within 3 h, and the activity decreased to the basal level by 12 h (Fig. 4C). However, luciferase activities in cells transfected with E1-M739 and E2-M45 significantly decreased compared with those in cells transfected with E1 and E2, respectively (Fig. 4C). These results collectively indicated that ACM could activate Nrf2, likely allowing induction of ARE-containing genes.
ACM reduces IFN-γ-induced ROS and iNOS expression in microglia
Because previous reports showed that IFN-γ, a proinflammatory cytokine, activated microglia (Meda et al., 1995; Minghetti et al., 1998; Kang et al., 2001), we examined whether IFN-γ treatment increased microglial ROS and then tested whether ACM reduced these IFN-γ-induced ROS levels. We found that IFN-γ treatment of microglia led to an increase in intracellular ROS levels from 5 to 30 min after treatment and a decrease to basal levels at 60 min (Fig. 5A,B). ACM pretreatment for 6 and 12 h but not for 3 h significantly blocked the IFN-γ-triggered induction of ROS levels (Fig. 5C,D). Similar results were obtained when we examined IFN-γ-induced iNOS protein expression/NO release. In microglia treated with IFN-γ, iNOS protein expression was significantly suppressed in cells treated with ACM compared with non-pretreated cells (Fig. 5E, left). In addition, the levels of NO production in the media were also suppressed in ACM-treated cells, as reflected by the amount of nitrite converted from NO in the media (Fig. 5E, right). As expected, ROS scavengers also suppressed IFN-γ-induced iNOS expression/NO release. ROS scavengers such as trolox and butylated hydroxyanisole significantly inhibited IFN-γ-induced iNOS protein expression and nitrite production (Fig. 5F). These results indicated that IFN-γ induced iNOS expression through ROS-dependent signaling pathways, and ACM may increase ROS scavenging in microglia, leading to suppression of IFN-γ-induced ROS production and subsequent iNOS expression and NO release.
Figure 5.
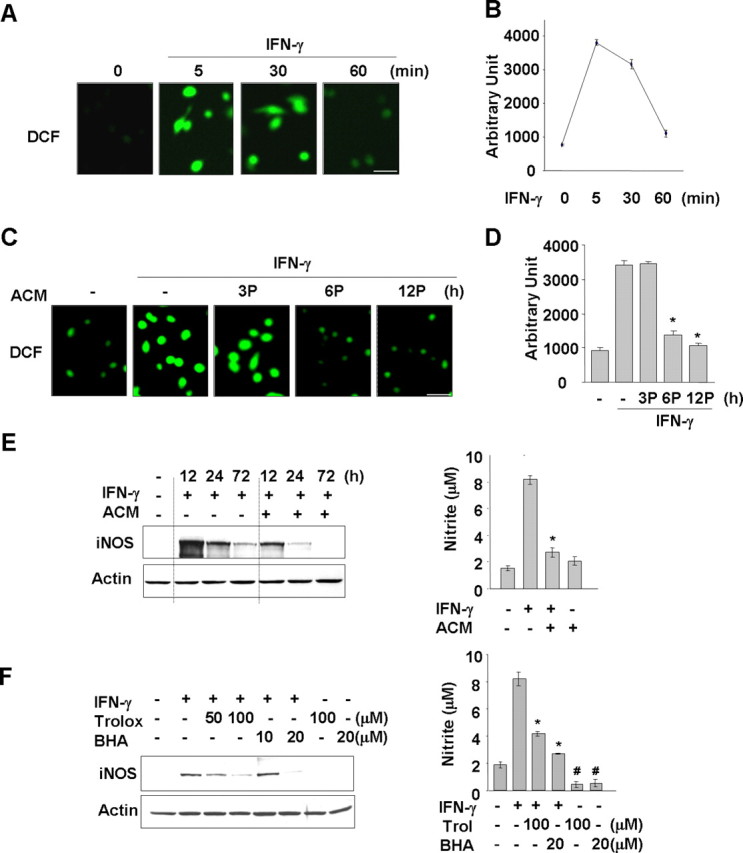
ACM suppresses IFN-γ-induced ROS production and iNOS expression/NO release. Primary microglia were treated with 20 ng/ml IFN-γ for the indicated time periods (A, B) or pretreated (P) with ACM for the indicated times and then treated with 20 ng/ml IFN-γ for 5 min (C, D). Intracellular ROS levels were assayed using 10 μm DCF as in Figure 1. A, C, Fluorescence (DCF) images. Scale bar, 50 μm. B, D, DCF intensities of cells in A and C were counted using LSM 5 Image Browser (Zeiss). Values are mean ± SEM of 10–15 cells. *p < 0.01 compared with IFN-γ. E, Primary microglia were treated with 20 ng/ml IFN-γ for the indicated times (left) or 72 h (right) in the absence or presence of ACM. F, Primary microglia were treated with 20 ng/ml IFN-γ for 12 h (left) or 72 h (right) in the absence or presence of the ROS scavenging agents trolox (Trol) or butylated hydroxyanisole (BHA). iNOS protein expression was detected by Western blotting (E, F, left). The amounts of nitrite converted from NO in the media were measured using Griess reagents, as described in Materials and Methods (E, F, right). Values in E and F are mean ± SEM of three samples. *p < 0.01 compared with IFN-γ; #p < 0.05 compared with control. The data are representative of three independent experiments.
HO-1 products mimic the anti-inflammatory roles of astrocytes
Next, we examined whether HO-1 products mimic the effect of ACM on IFN-γ-induced ROS production and iNOS expression/NO release. HO-1 has been shown to catalyze the degradation of heme into CO, ferrous ion, and bilirubin (converted from biliverdin) (Maines, 1988). Therefore, we examined the effect of these HO-1 products on IFN-γ-induced iNOS expression. The addition of bilirubin and an iron donor (FeSO4) reduced iNOS expression and the level of nitrite in IFN-γ-treated microglia (Fig. 6A,B). In contrast, tricarbonyldichlororuthenium (II) dimer [a CO-releasing molecule (CORM)] reduced the level of nitrite but had little effect on iNOS expression (Fig. 6A,B). Together, these results suggested that HO-1 could be a candidate ACM-induced ROS scavenging molecule in this context.
Figure 6.
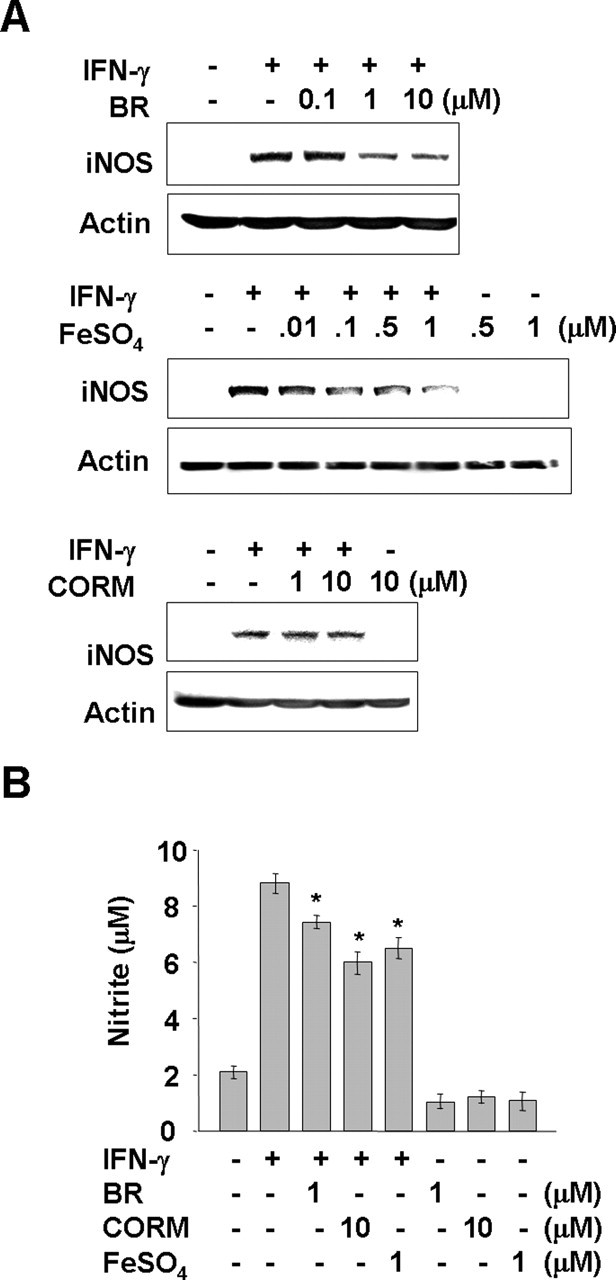
Effects of HO-1 products, bilirubin, CO, and Fe2+ on IFN-γ-induced iNOS expression/NO release. Primary microglia were treated with 20 ng/ml IFN-γ for 12 h (A) or 72 h (B) in the absence or presence of the indicated amount of BR, CORM, and FeSO4. iNOS protein expression was measured by Western blotting (A) and the amount of nitrite converted from NO in the media was measured using the Griess reagents (B). Values in B are mean ± SEM of three samples. *p < 0.05 compared with IFN-γ. The data are representative of three independent experiments.
The active component(s) in ACM is heat labile and smaller than 3 kDa
As an attempt to narrow down the candidate molecule(s) in the ACM responsible for HO-1 expression and suppression of IFN-γ-induced iNOS expression and NO release in microglia, we examined the heat sensitivity of ACM effect. ACM heated at 70°C for 15 min lost the ability to induce HO-1 expression and to inhibit IFN-γ-induced iNOS expression (Fig. 7A,B), indicating that the relevant molecule(s) is heat sensitive. These findings indicated that heat labile component(s) in the ACM was capable of actively regulating microglial sensitivity to inflammatory stimulators. Next, ACM was fractionated into molecules below or above molecular weight 3 kDa. Interestingly, the fraction including molecules <3 kDa induced HO-1 expression and inhibited IFN-γ-induced iNOS expression, whereas the fraction including molecules larger than 3 kDa neither induced HO-1 expression nor inhibited IFN-γ-induced iNOS expression (Fig. 7C,D). These results indicated that the active component(s) in ACM is smaller than 3 kDa and heat labile. Together, our novel findings in this study suggest that soluble factors released from astrocytes and acting on microglia could be important regulators capable of controlling brain inflammation.
Figure 7.

The active component(s) in ACM is heat labile and smaller than 3 kDa. A, Primary microglia were treated with ACM or heated ACM (70°C for 15 min) for 12 h. B, Primary microglia were treated with 20 ng/ml IFN-γ for 12 h in the absence or presence of ACM or heated ACM. C, BV2 cells were treated with ACM components smaller than 3 kDa (3↓) or ACM components larger than 3 kDa (3↑) for 12 h. D, BV2 cells were treated with 20 ng/ml IFN-γ for 12 h in the absence or presence of ACM components smaller than 3 kDa (3↓) or ACM components larger than 3 kDa (3↑). ACM components were fractionated as described in Materials and Methods. HO-1 (A, C) and iNOS (B, D) expression were measured by Western blotting. The data are representative of three independent experiments.
Discussion
Accumulating evidences indicate that brain inflammation could be a risk factor for the onset and progression of brain diseases such as neurodegenerative diseases, depression, and even autism (Maes, 1995; Gonzalez-Scarano and Baltuch, 1999; Jyonouchi et al., 2001; Vila et al., 2001; Vargas et al., 2005). Many studies have examined the activation of brain inflammation in separately cultured microglia and astrocytes. However, brain inflammation is the end result of integrated signaling across all brain cells. Therefore, we herein examined the possibility of interactions between astrocytes and microglia by examining whether astrocytes could modulate microglial inflammatory responses. Indeed, we found that soluble factor(s) from astrocytes suppressed IFN-γ-induced microglial inflammatory responses, and HO-1 could play a role in the action of the soluble factor(s). These results suggested that astrocytes could act as physiological regulators to prevent excessive inflammatory microglial responses.
In this study, we found that two HO-1 products (bilirubin and ferrous iron) reduced iNOS expression and NO production in IFN-γ-treated microglia. In contrast, CORM attenuated NO production but had little effect on iNOS expression. Similar results have been reported in macrophages, in which CORM treatment reduced NO production but not iNOS expression (Sawle et al., 2005). Mechanistically, it is thought that decreased NO production in the absence of iNOS changes in the presence of CORM could be associated with changes in arginine (substrate of iNOS) supply, enzyme dimerization, phosphorylation, protein stability, or cofactor binding (for review, see Aktan, 2004). In the brain, HO-1 expression is dramatically increased under several pathophysiological conditions. After transient occlusion of the middle cerebral artery, HO-1-immunoreactivity was seen in microglia in the infarct area, and in astrocytes and microglia in the penumbra region (Koistinaho et al., 1996). After traumatic brain injury, accumulation of HO-1-positive microglia was detected for up to 6 months (Beschorner et al., 2000). It is possible that HO-1 acts to protect tissues from damage under these pathophysiological conditions, because HO-1 has protective antioxidant, antiapoptotic, and anti-inflammatory functions (Lee et al., 1996; Willis et al., 1996; Dennery et al., 1997). For example, human pulmonary epithelial cells overexpressing HO-1 are resistant to hyperoxia-induced cell death (Lee et al., 1996), whereas HO-1-deficient fibroblast cells are vulnerable to oxidative stress (Dennery et al., 1997). The anti-inflammatory roles of HO-1 were clearly demonstrated in HO-1-deficient mice, which showed chronic renal inflammation (Nath et al., 2001) and enhanced secretion of proinflammatory cytokines from splenocytes (Nath et al., 2001; Kapturczak et al., 2004). Consistent with these findings, cerebral ischemia models using transgenic mice overexpressing HO-1 showed significant reductions in lipid peroxidation end products and infarct sizes (Panahian et al., 1999). These and other findings strongly suggest that HO-1 expression protects against oxidative stress (Koistinaho et al., 1996; Beschorner et al., 2000) and inflammation (Yamauchi et al., 2004).
Astrocytes play vital roles in the maintenance of normal brain function; they regulate blood–brain barrier formation, brain homeostasis, neuronal activities, synaptic transmission, neuronal survival, synaptogenesis, and neurogenesis (Kesslak et al., 1986; Dani et al., 1992; Vesce et al., 1999; Lim and Alvarez-Buylla, 1999). In injured brain, astrocytes regulate neuronal injury in both direct and indirect manners. Astrocytes directly modulate neuronal survival by producing growth factors (Dhandapani et al., 2003) and regulate expression of the NMDA receptor subunit (Daniels and Brown, 2001) and the glutamate transporter EAAC1 (excitatory amino acid carrier) (Canolle et al., 2004), which influences neuronal sensitivity to glutamate toxicity. In the presence of astrocytes, neurons become resistant to oxidative stress (Langeveld et al., 1995; Tanaka et al., 1999). Indirectly, astrocytes affect neuronal injury by modulating inflammation via decreases in the expression of microglial inflammatory mediators (Vincent et al., 1996, 1997; Aloisi et al., 1997; Pyo et al., 2003).
The results in this study showed that soluble factor(s) from astrocytes suppressed IFN-γ-induced microglial inflammatory responses through the expression of HO-1. ACM induced nuclear translocation of Nrf2 and increased the binding activity of ARE, which plays a major role in the expression of HO-1 (Itoh et al., 1997). The experimental results using HO-1 promoter/luciferase fusion constructs also showed that ARE sites are important in HO-1 expression in ACM-treated microglia because ARE mutation (E1-M739 and E2-M45) significantly decreased luciferase activity. However, we could not exclude the possible involvement of other transcription factor(s) such as C/EBP and SP-1 in the regulation of HO-1 expression because ARE mutation did not completely abolish luciferase activity and the luciferase activity was detected a little earlier than HO-1 protein expression.
ACM is known to contain a number of factors such as transforming growth factor-β (TGF-β) (Tran et al., 1999), S100β (Eriksen and Druse, 2001), fibroblast growth factor-2 (Reuss and Unsicker, 2000), plasminogen activator inhibitor-1 (Kimura et al., 2000), and others. Among them, we examined possible involvement of TGF-β in ACM-induced HO-1 expression, because TGF-β has been shown to induce HO-1 expression in epithelial cells (Hill-Kapturczak et al., 2000). Although TGF-β treatment (10 ng/ml) induced HO-1 expression in microglia, the TGF-β content in ACM was in the range of 300–500 pg/ml, which is not enough to induce HO-1 (data not shown). Furthermore, TGF-β treatment did not trigger translocation of Nrf2 into nuclei, and a TGF-β blocking antibody did not abolish ACM-induced HO-1 expression (data not shown). We further showed that the factor(s) involved in the latter pathway is heat labile and smaller than 3 kDa. Daley et al. (2005) reported recently that neutrophil-producing soluble factor(s) that is smaller than 3 kDa is capable of modifying the inflammatory responses of macrophages. Thus, astrocytes and neutrophils may produce similar soluble factor(s) that modify brain inflammation and systemic inflammation, respectively. These findings suggest that, in vivo, cell–cell interactions could be a critical mechanism for inducing efficient inflammatory responses while concurrently minimizing tissue damage. Thus, we herein show that soluble astrocyte-based factor(s) modulate microglial inflammatory responses and propose that such factors could function as important natural anti-inflammatory agents in the brain.
Footnotes
This work was supported by the Korea Science and Engineering Foundation (KOSEF) through the Brain Disease Research Center at Ajou University and Grant M103KV010006 03K2201 00650 from the Brain Research Center of the 21st Century Frontier Research Program funded by the Ministry of Science and Technology, Republic of Korea (E.-h.J.), and KOSEF through Chronic Inflammatory Disease Research Center at Ajou University Grant R13-2003-019 (I.J.).
References
- Aktan F (2004). iNOS-mediated nitric oxide production and its regulation. Life Sci 75:639–653. [DOI] [PubMed] [Google Scholar]
- Alam J (1994). Multiple elements within the 5′ distal enhancer of the mouse heme oxygenase-1 gene mediate induction by heavy metals. J Biol Chem 269:25049–25056. [PubMed] [Google Scholar]
- Alam J, Cook JL (2003). Transcriptional regulation of the heme oxygenase-1 gene via the stress response element pathway. Curr Pharm Des 9:2499–2511. [DOI] [PubMed] [Google Scholar]
- Alam J, Camhi S, Choi AM (1995). Identification of a second region upstream of the mouse heme oxygenase-1 gene that functions as a basal level and inducer-dependent transcription enhancer. J Biol Chem 270:11977–11984. [DOI] [PubMed] [Google Scholar]
- Alam J, Wicks C, Stewart D, Gong P, Touchard C, Otterbein S, Choi AM, Burow ME, Tou J (2000). Mechanism of heme oxygenase-1 gene activation by cadmium in MCF-7 mammary epithelial cells. Role of p38 kinase and Nrf2 transcription factor. J Biol Chem 275:27694–27702. [DOI] [PubMed] [Google Scholar]
- Alam J, Killeen E, Gong P, Naquin R, Hu B, Stewart D, Ingelfinger JR, Nath KA (2003). Heme activates the heme oxygenase-1 gene in renal epithelial cells by stabilizing Nrf2. Am J Physiol Renal Physiol 284:F743–F752. [DOI] [PubMed] [Google Scholar]
- Aloisi F, Penna G, Cerase J, Menendez IB, Adorini L (1997). IL-12 production by central nervous system microglia is inhibited by astrocytes. J Immunol 159:1604–1612. [PubMed] [Google Scholar]
- Beschorner R, Adjodah D, Schwab JM, Mittelbronn M, Pedal I, Mattern R, Schluesener HJ, Meyermann R (2000). Long-term expression of heme oxygenase-1 (HO-1, HSP-32) following focal cerebral infarctions and traumatic brain injury in humans. Acta Neuropathol (Berl) 100:377–384. [DOI] [PubMed] [Google Scholar]
- Canolle B, Masmejean F, Melon C, Nieoullon A, Pisano P, Lortet S (2004). Glial soluble factors regulate the activity and expression of the neuronal glutamate transporter EAAC1: implication of cholesterol. J Neurochem 88:1521–1532. [DOI] [PubMed] [Google Scholar]
- Chapman JT, Otterbein LE, Elias JA, Choi AM (2001). Carbon monoxide attenuates aeroallergen-induced inflammation in mice. Am J Physiol Lung Cell Mol Physiol 281:L209–L216. [DOI] [PubMed] [Google Scholar]
- Daley JM, Reichner JS, Mahoney EJ, Manfield L, Henry WL Jr, Mastrofrancesco B, Albina JE (2005). Modulation of macrophage phenotype by soluble product(s) released from neutrophils. J Immunol 174:2265–2272. [DOI] [PubMed] [Google Scholar]
- Dani JW, Chernjavsky A, Smith SJ (1992). Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron 8:429–440. [DOI] [PubMed] [Google Scholar]
- Daniels M, Brown DR (2001). Astrocytes regulate N-methyl-d-aspartate receptor subunit composition increasing neuronal sensitivity to excitotoxicity. J Biol Chem 276:22446–22452. [DOI] [PubMed] [Google Scholar]
- Dennery PA, Sridhar KJ, Lee CS, Wong HE, Shokoohi V, Rodgers PA, Spitz DR (1997). Heme oxygenase-mediated resistance to oxygen toxicity in hamster fibroblasts. J Biol Chem 272:14937–14942. [DOI] [PubMed] [Google Scholar]
- Dhandapani KM, Hadman M, De SL, Wade MF, Mahesh VB, Brann DW (2003). Astrocyte protection of neurons: role of transforming growth factor-beta signaling via a c-Jun-AP-1 protective pathway. J Biol Chem 278:43329–43339. [DOI] [PubMed] [Google Scholar]
- Ding AH, Nathan CF, Stuehr DJ (1988). Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J Immunol 141:2407–2412. [PubMed] [Google Scholar]
- Eriksen JL, Druse MJ (2001). Astrocyte-mediated trophic support of developing serotonin neurons: effects of ethanol, buspirone, and S100B. Brain Res Dev Brain Res 131:9–15. [DOI] [PubMed] [Google Scholar]
- Giulian D, Baker TJ (1986). Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci 6:2163–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Baltuch G (1999). Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci 22:219–240. [DOI] [PubMed] [Google Scholar]
- Hill-Kapturczak N, Truong L, Thamilselvan V, Visner GA, Nick HS, Agarwal A (2000). Smad7-dependent regulation of heme oxygenase-1 by transforming growth factor-beta in human renal epithelial cells. J Biol Chem 275:40904–40909. [DOI] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y (1997). An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236:313–322. [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M (1999). Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 13:76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jyonouchi H, Sun S, Le H (2001). Proinflammatory and regulatory cytokine production associated with innate and adaptive immune responses in children with autism spectrum disorders and developmental regression. J Neuroimmunol 120:170–179. [DOI] [PubMed] [Google Scholar]
- Kang J, Yang M, Jou I, Joe E (2001). Identification of protein kinase C isoforms involved in interferon-gamma-induced expression of inducible nitric oxide synthase in murine BV2 microglia. Neurosci Lett 299:205–208. [DOI] [PubMed] [Google Scholar]
- Kapturczak MH, Wasserfall C, Brusko T, Campbell-Thompson M, Ellis TM, Atkinson MA, Agarwal A (2004). Heme oxygenase-1 modulates early inflammatory responses: evidence from the heme oxygenase-1-deficient mouse. Am J Pathol 165:1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesslak JP, Nieto-Sampedro M, Globus J, Cotman CW (1986). Transplants of purified astrocytes promote behavioral recovery after frontal cortex ablation. Exp Neurol 92:377–390. [DOI] [PubMed] [Google Scholar]
- Kimura M, Soeda S, Oda M, Ochiai T, Kihara T, Ono N, Shimeno H (2000). Release of plasminogen activator inhibitor-1 from human astrocytes is regulated by intracellular ceramide. J Neurosci Res 62:781–788. [DOI] [PubMed] [Google Scholar]
- Koistinaho J, Miettinen S, Keinanen R, Vartiainen N, Roivainen R, Laitinen JT (1996). Long-term induction of haem oxygenase-1 (HSP-32) in astrocytes and microglia following transient focal brain ischaemia in the rat. Eur J Neurosci 8:2265–2272. [DOI] [PubMed] [Google Scholar]
- Kutty RK, Maines MD (1982). Oxidation of heme c derivatives by purified heme oxygenase. Evidence for the presence of one molecular species of heme oxygenase in the rat liver. J Biol Chem 257:9944–9952. [PubMed] [Google Scholar]
- Langeveld CH, Jongenelen CA, Schepens E, Stoof JC, Bast A, Drukarch B (1995). Cultured rat striatal and cortical astrocytes protect mesencephalic dopaminergic neurons against hydrogen peroxide toxicity independent of their effect on neuronal development. Neurosci Lett 192:13–16. [DOI] [PubMed] [Google Scholar]
- Lee PJ, Alam J, Wiegand GW, Choi AM (1996). Overexpression of heme oxygenase-1 in human pulmonary epithelial cells results in cell growth arrest and increased resistance to hyperoxia. Proc Natl Acad Sci USA 93:10393–10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim DA, Alvarez-Buylla A (1999). Interaction between astrocytes and adult subventricular zone precursors stimulates neurogenesis. Proc Natl Acad Sci USA 96:7526–7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhu B, Wang X, Luo L, Li P, Paty DW, Cynader MS (2003). Bilirubin as a potent antioxidant suppresses experimental autoimmune encephalomyelitis: implications for the role of oxidative stress in the development of multiple sclerosis. J Neuroimmunol 139:27–35. [DOI] [PubMed] [Google Scholar]
- Maes M (1995). Evidence for an immune response in major depression: a review and hypothesis. Prog Neuropsychopharmacol Biol Psychiatry 19:11–38. [DOI] [PubMed] [Google Scholar]
- Maines MD (1988). Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J 2:2557–2568. [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L Jr, Baron P, Villalba M, Ferrari D, Rossi F (1995). Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature 374:647–650. [DOI] [PubMed] [Google Scholar]
- Min KJ, Jou I, Joe E (2003). Plasminogen-induced IL-1beta and TNF-alpha production in microglia is regulated by reactive oxygen species. Biochem Biophys Res Commun 312:969–974. [DOI] [PubMed] [Google Scholar]
- Min KJ, Pyo HK, Yang MS, Ji KA, Jou I, Joe EH (2004). Gangliosides activate microglia via protein kinase C and NADPH oxidase. Glia 48:197–206. [DOI] [PubMed] [Google Scholar]
- Minghetti L, Polazzi E, Nicolini A, Levi G (1998). Opposite regulation of prostaglandin E2 synthesis by transforming growth factor-beta1 and interleukin 10 in activated microglial cultures. J Neuroimmunol 82:31–39. [DOI] [PubMed] [Google Scholar]
- Nath KA, Vercellotti GM, Grande JP, Miyoshi H, Paya CV, Manivel JC, Haggard JJ, Croatt AJ, Payne WD, Alam J (2001). Heme protein-induced chronic renal inflammation: suppressive effect of induced heme oxygenase-1. Kidney Int 59:106–117. [DOI] [PubMed] [Google Scholar]
- Ott MC, Scott JR, Bihari A, Badhwar A, Otterbein LE, Gray DK, Harris KA, Potter RF (2005). Inhalation of carbon monoxide prevents liver injury and inflammation following hind limb ischemia/reperfusion. FASEB J 19:106–108. [DOI] [PubMed] [Google Scholar]
- Panahian N, Yoshiura M, Maines MD (1999). Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J Neurochem 72:1187–1203. [DOI] [PubMed] [Google Scholar]
- Pier GB, Markham RB, Eardley D (1981). Correlation of the biologic responses of C3H/HEJ mice to endotoxin with the chemical and structural properties of the lipopolysaccharides from Pseudomonas aeruginosa and Escherichia coli. J Immunol 127:184–191. [PubMed] [Google Scholar]
- Pyo H, Jou I, Jung S, Hong S, Joe EH (1998). Mitogen-activated protein kinases activated by lipopolysaccharide and beta-amyloid in cultured rat microglia. NeuroReport 9:871–874. [DOI] [PubMed] [Google Scholar]
- Pyo H, Yang MS, Jou I, Joe EH (2003). Wortmannin enhances lipopolysaccharide-induced inducible nitric oxide synthase expression in microglia in the presence of astrocytes in rats. Neurosci Lett 346:141–144. [DOI] [PubMed] [Google Scholar]
- Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS (2004). NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem 279:1415–1421. [DOI] [PubMed] [Google Scholar]
- Reuss B, Unsicker K (2000). Survival and differentiation of dopaminergic mesencephalic neurons are promoted by dopamine-mediated induction of FGF-2 in striatal astroglial cells. Mol Cell Neurosci 16:781–792. [DOI] [PubMed] [Google Scholar]
- Ryu J, Pyo H, Jou I, Joe E (2000). Thrombin induces NO release from cultured rat microglia via protein kinase C, mitogen-activated protein kinase, and NF-kappa B. J Biol Chem 275:29955–29959. [DOI] [PubMed] [Google Scholar]
- Sawle P, Foresti R, Mann BE, Johnson TR, Green CJ, Motterlini R (2005). Carbon monoxide-releasing molecules (CO-RMs) attenuate the inflammatory response elicited by lipopolysaccharide in RAW264.7 murine macrophages. Br J Pharmacol 145:800–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh JH, Shenvi SV, Dixon BM, Liu H, Jaiswal AK, Liu RM, Hagen TM (2004). Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci USA 101:3381–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka J, Toku K, Zhang B, Ishihara K, Sakanaka M, Maeda N (1999). Astrocytes prevent neuronal death induced by reactive oxygen and nitrogen species. Glia 28:85–96. [DOI] [PubMed] [Google Scholar]
- Tran ND, Correale J, Schreiber SS, Fisher M (1999). Transforming growth factor-beta mediates astrocyte-specific regulation of brain endothelial anticoagulant factors. Stroke 30:1671–1678. [DOI] [PubMed] [Google Scholar]
- Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA (2005). Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol 57:67–81. [DOI] [PubMed] [Google Scholar]
- Vesce S, Bezzi P, Volterra A (1999). The active role of astrocytes in synaptic transmission. Cell Mol Life Sci 56:991–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila M, Jackson-Lewis V, Guegan C, Wu DC, Teismann P, Choi DK, Tieu K, Przedborski S (2001). The role of glial cells in Parkinson’s disease. Curr Opin Neurol 14:483–489. [DOI] [PubMed] [Google Scholar]
- Vincent VA, Van Dam AM, Persoons JH, Schotanus K, Steinbusch HW, Schoffelmeer AN, Berkenbosch F (1996). Gradual inhibition of inducible nitric oxide synthase but not of interleukin-1 beta production in rat microglial cells of endotoxin-treated mixed glial cell cultures. Glia 17:94–102. [DOI] [PubMed] [Google Scholar]
- Vincent VA, Tilders FJ, Van Dam AM (1997). Inhibition of endotoxin-induced nitric oxide synthase production in microglial cells by the presence of astroglial cells: a role for transforming growth factor beta. Glia 19:190–198. [DOI] [PubMed] [Google Scholar]
- Wang WW, Smith DL, Zucker SD (2004). Bilirubin inhibits iNOS expression and NO production in response to endotoxin in rats. Hepatology 40:424–433. [DOI] [PubMed] [Google Scholar]
- Ward RJ, Kuhn LC, Kaldy P, Florence A, Peters TJ, Crichton RR (1994). Control of cellular iron homeostasis by iron-responsive elements in vivo. Eur J Biochem 220:927–931. [DOI] [PubMed] [Google Scholar]
- Willis D, Moore AR, Frederick R, Willoughby DA (1996). Heme oxygenase: a novel target for the modulation of the inflammatory response. Nat Med 2:87–90. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Lin Y, Sharp FR, Noble-Haeusslein LJ (2004). Hemin induces heme oxygenase-1 in spinal cord vasculature and attenuates barrier disruption and neutrophil infiltration in the injured murine spinal cord. J Neurotrauma 21:1017–1030. [DOI] [PubMed] [Google Scholar]