

Abstract
Studies of the developing nervous system led to the general view that growth factors promote neuronal survival in a “retrograde” manner. For example, release of NGF from postsynaptic peripheral targets followed by uptake and retrograde transport by presynaptic neurons provided a widely accepted conceptual framework for the action of neurotrophins. In contrast, although presynaptic or “anterograde” influences on the survival of developing neurons have been recognized for some time, the mechanisms by which afferent input regulates the survival of postsynaptic cells have received considerably less attention. In the forebrain network for learned vocal behavior in zebra finches, lesions of a cortical region for song control, the lateral magnocellular nucleus of the anterior neostriatum (lMAN), remove presynaptic input to a motor-cortical song region, the robust nucleus of the archistriatum (RA), and cause massive RA neuron death in young birds that are entering the sensitive period for song learning. Here we report that lesions of lMAN followed by infusions of neurotrophins directly into RA completely suppress neuronal apoptosis in RA. Moreover, we show that lMAN neurons are able to transport neurotrophins in the anterograde direction to RA, that neurotrophin-like immunoreactivity is present in cells in lMAN and RA, and that neurotrophin receptor-like immunoreactivity is present in RA. Expression of neurotrophins in lMAN and RA suggests that lMAN presynaptic input could regulate RA neuron survival by synthesizing, transporting, and releasing neurotrophins anterogradely or by regulating the auto/paracrine release of neurotrophins within RA, or perhaps by both. These data provide the first in vivodemonstration that neurotrophins can prevent the death of deafferented cortical neurons, and they raise the possibility that nonretrograde signaling by neurotrophins may be a common means of promoting neuronal survival in the vertebrate telencephalon. Anterograde and auto/paracrine neurotrophin signaling, along with the more established view that neurotrophins regulate neuron survival via retrograde mechanisms, suggests multidirectional neurotrophin signaling in the vertebrate telencephalon.
Keywords: songbird, cortex, afferent, apoptosis, neurotrophins, Trk receptors
The elaborate morphology of the vertebrate brain is sculpted in part by a developmental phase of abundant but selective neuron death (Jacobson, 1991). The survival of young neurons during this phase depends on molecular signaling interactions with other cells. Studies of the signals that regulate neuron survival in the developing brain have traditionally emphasized retrograde signaling mechanisms. In this model, trophic factors are released in limited amounts by postsynaptic (target) cells and taken up by presynaptic (afferent) neurons to promote survival of the latter (Oppenheim, 1991;Thoenen, 1995). It is clear, however, that presynaptic input also regulates the survival of target neurons during brain development (Levi-Montalcini, 1949; Rubel et al., 1990; Clarke, 1992), yet by mechanisms that remain to be fully elucidated. Although depolarization of postsynaptic cells is perhaps the most obvious means by which presynaptic input could influence neuronal survival, electrical activity alone is apparently insufficient to maintain developing neurons. For example, studies of chick brainstem and optic tectum indicate that activity of postsynaptic neurons per se is insufficient to prevent cell death and that presynaptic release of trophic molecules that act either alone or in combination with electrical activity is important for neuronal survival (Hyson and Rubel, 1989, 1995; Catsicas et al., 1992). Recently, neurotrophins have emerged as factors that are likely to influence activity-dependent synaptic plasticity as well as neuronal survival and differentiation (see Lo, 1995; Bonhoeffer, 1996). Because it is clear that neurotrophins can exert potent effects on morphological and physiological properties of postsynaptic neurons (Lohof et al., 1993; Kang and Schuman, 1995; McAllister et al., 1995), we reasoned that they may constitute one class of signaling molecules from presynaptic neurons onto their targets.
The neural circuitry that underlies the development of vocal learning and behavior in songbirds provides a model system for examining cell–cell interactions in the developing brain, in part because song-control brain regions tend to make and receive a limited number of axonal connections (for review, see Bottjer and Arnold, 1997). For example, during early stages of song development, the robust nucleus of the archistriatum (RA), a motor cortical region for song control, receives a single major source of afferent input from a premotor cortical nucleus, the lateral magnocellular nucleus of the anterior neostriatum (lMAN), (Fig. 1; cf. Konishi and Akutagawa, 1985; Mooney, 1992). We have shown previously that removal of afferent input from lMAN to RA induces massive neuronal death at the onset of song learning in juvenile male zebra finches (Johnson and Bottjer, 1994). By implication, lMAN neurons that are presynaptic to RA must synthesize, anterogradely transport, and release a factor that directly promotes the survival of developing RA neurons or one that regulates the release of survival-promoting factors within RA. To determine whether neurotrophin growth factors play a role in the afferent regulation of cortical neuron survival, we tested members of the neurotrophin family for the ability to prevent the death of RA neurons after removal of presynaptic input from lMAN (Fig. 1).
Fig. 1.

Dorsal view of the zebra finch brain showing the intracortical pathway from lMAN to RA on both sides; each pathway is strictly ipsilateral. lMAN and RA form part of the neural substrate for learned vocal behavior in oscine passerine birds and are necessary for normal vocal development (Nottebohm et al., 1976; Bottjer et al., 1984). Bilateral lMAN lesions (each marked with anX) were made to obtain a bilateral deafferentation of RA. Infusions of neurotrophin were made into left RA, and right RA served as a control for the effects of removing afferent input. lMAN, Lateral magnocellular nucleus of the anterior neostriatum; RA, robust nucleus of the archistriatum.
Our findings demonstrate that infusion of BDNF, NT-3, or NT-4/5 (but not NGF) can rescue neuron number in RA after removal of presynaptic input from lMAN. Moreover, we show that lMAN neurons have the ability to transport BDNF and NT-3 (but not NT-4/5) in the anterograde direction, and we provide evidence that neurotrophins and their receptors are differentially expressed within this circuit. That is, many lMAN neurons contain BDNF-like and/or NT-3-like immunoreactivity, and many RA neurons contain NT-3-like immunoreactivity. RA also contains intense somal, neuropil, and fiber immunoreactivity for TrkB, the receptor for BDNF and NT-4/5 (which also binds NT-3 with high affinity), as well as sparse fiber immunoreactivity for TrkC, the NT-3 receptor. Our results are compatible with two nonretrograde mechanisms for neurotrophin signaling in the vertebrate telencephalon: (1) lMAN neurons synthesize, anterogradely transport, and release BDNF and/or NT-3 into RA, and (2) lMAN afferent input regulates the auto/paracrine release of NT-3 and/or NT-4/5 by RA neurons. These multiple modes of neurotrophin signaling are not mutually exclusive and could function in a complementary manner to organize the development of RA.
MATERIALS AND METHODS
Deafferentation of RA and neurotrophin treatments.Subjects were juvenile male zebra finches (18–22 d old) from our breeding colony. Birds were deeply anesthetized with Equithesin, secured in a stereotaxic instrument, and then given bilateral electrolytic lesions of lMAN to deafferent RA (each lMAN, 100 μA for 3 min; nichrome wire electrode, 210 μm diameter) (see Fig. 1). Although RA receives a second source of afferent input from the high vocal center, the development of this pathway is delayed until ∼35 d of age (Konishi and Akutagawa, 1985). Immediately after lesioning, an intraparenchymal infusion of neurotrophin was made into left RA using a glass micropipette (outer tip diameter = 10–20 μm) attached to a pressure injection system (Picospritzer II, General Valve Corp). A second infusion into RA was given at 12 hr postlesion. Each infusion consisted of 1.0–2.5 μl of neurotrophin in PBS that was slowly infused into RA over a 10 min interval. Infusions were always given at 0 and 12 hr postlesion. Before filling the micropipettes with neurotrophin, volume calibration marks were made along the barrel of each micropipette. By observing the movement of the fluid level with a dissecting microscope, the dose of neurotrophin was measured accurately and extrusion of neurotrophin into brain verified. Doses of neurotrophin were as follows: murine NGF (Upstate Biotechnology, Lake Placid, NY), 1.0 μg/1.0 μl PBS; recombinant human BDNF, 1.0 μg/1.0 μl PBS or 2.5 μg/2.5 μl PBS; recombinant human NT-3, 1.0 μg/1.0 μl PBS or 2.5 μg/2.5 μl PBS; recombinant human NT-4/5, 1.0 μg/2.5 μl PBS (BDNF, NT-3, and NT-4/5 were gifts of Amgen/Regeneron Partnership). Some birds received infusions of equine cytochrome-c (Sigma, St. Louis, MO; 2.5 μg/2.5 μl PBS) into right RA (n = 2 in 2.5 μg BDNF and NT-3 groups) as a control; RA neuron number in these birds was not different from untreated RA in other birds, and therefore cytochrome-c data were included with untreated RA data as a single control group. Birds were killed at 24 hr postlesion, because extensive neuron death is present in deafferented RA by this time (see Johnson and Bottjer, 1994 and Fig. 2). After overdose with Equithesin, birds were perfused intracardially with bird saline and 10% buffered formalin, after which brains were removed and allowed to post-fix for 2 d in 10% buffered formalin. After post-fixation, brains were transferred to 20% buffered sucrose for 24 hr (for cryoprotection) and then frozen-sectioned in the coronal plane (section thickness, 20 or 30 μm). Brain sections were mounted directly on slides coated with pig gelatin, after which the sections were counterstained with thionin and coverslipped.
Fig. 2.

Intraparenchymal infusions of neurotrophins prevent apoptotic cell death in RA after removal of afferent input. Cell counts are shown for the various neurotrophin treatment and dose groups as a function of neurotrophin-treated versus untreated (control) RA. For comparison, normative RA neuron data from Johnson and Bottjer (1994) are shown in the top panels of Aand B. A, Top panel, Neuron counts in groups receiving a 1 μg dosage showed that BDNF, NT-3, and NT-4/5 each rescued significant numbers of RA neurons. By comparison, NGF treatment did not attenuate neuron loss from RA.A, Bottom panel, BDNF-, NT-3-, and NT-4/5-treated RA had significantly fewer apoptotic cells than untreated RA, indicating that the rescue of neurons shown in the top panel of A was attributable to a suppression of neuron death. NGF treatment did not suppress the appearance of apoptotic cells (NS, compare Fig. 3A, B).B, Top panel, When compared with normative data, birds that received 2.5 μg doses of BDNF or NT-3 showed a complete rescue of RA neuron number. B,Bottom panel, Deafferented RA treated with 2.5 μg doses of BDNF or NT-3 showed a low incidence of apoptosis, indicating that these neurotrophin treatments rescued RA neuron number by preventing neuron death.
Micropipette tracks were clearly visible in the counterstained tissue, and brains were initially analyzed to determine whether the neurotrophin infusions penetrated the lamina surrounding RA. Birds with neurotrophin infusions that were near RA but failed to penetrate the lamina surrounding RA showed no prevention of RA neuron death; these birds served as an important control showing that the infusions were effective only if they were in the direct vicinity of RA somata and that the neurotrophins were not working via a retrograde mechanism after diffusion through the brain parenchyma to an efferent target of RA (i.e., nXIIts or DM/ICo). Final treatment group sizes for analyses were all n = 4 and included only birds in which the infusions were within RA. The incidence of neurons and apoptotic cells in neurotrophin-treated and control RA was measured by sampling the density of neurons and apoptotic cells in RA at high magnification (1000×) using an eyepiece grid. Neuronal nucleoli were the unit of count for neurons, because nucleoli are very small with respect to section thickness (1–2 vs 20–30 μm, respectively). Neurons with two nuclei were only counted once, but the number of neurons with two nucleoli was low (5–10%) and did not differ among groups. Apoptotic cells were easily identified on the basis of their overall pyknotic morphology and the presence of large particles of basophilic material within the nucleus (Fig.2A,C,E,F); apoptotic cells were only counted if the entire cell could be visualized within the section by focusing up and down (numbers of apoptotic cells therefore likely represent an underestimation). The number of neurons and apoptotic cells was then divided by the total sampled volume to obtain RA cell density values. Density values were multiplied by the total volume of RA to obtain total numbers of neurons and apoptotic cells in RA (see Johnson and Bottjer, 1994). Neuron and apoptotic cell number data were analyzed using an ANOVA followed by multiple comparison tests (Student–Newman–Keuls) at 0.05 and 0.01 significance levels.
Infusions of 125I-labeled neurotrophins.125I-labeled neurotrophins were infused directly into lMAN to test whether lMAN neurons are able to transport neurotrophins in the anterograde direction to RA. Neurotrophins were iodinated to a specific activity of 2413–3659 cpm/fmol as described previously (DiStefano et al., 1992). Juvenile male zebra finches from our breeding colony (ranging between 20 and 30 d old) were deeply anesthetized and given 100 nl pressure injections of [125I]BDNF, [125I]NT-3, or [125I]NT-4/5 into lMAN bilaterally (n = 4 birds in each group,n = 8 lMAN infusions per group). One day later, birds received a second 100 nl pressure injection of 125I-labeled neurotrophin. Birds were overdosed with anesthetic and perfused intracardially with bird saline and 4% paraformaldehyde 15–24 hr after the second injection. Brains were post-fixed for 3–5 d, transferred to 20% buffered sucrose for 24 hr, sectioned on a crytostat (20-μm-thick coronal sections), and mounted directly onto slides coated with pig gelatin. Autoradiograms were prepared by dipping the slides in emulsion (Kodak NTB2, Eastman Kodak, Rochester, NY) and then storing the slides in lightproof boxes at 4°C. Autoradiograms were developed and fixed after an exposure time of 8 weeks and were counterstained with thionin and then coverslipped. Sections containing lMAN were analyzed first to determine whether lMAN had been successfully targeted by the 125I-labeled neurotrophin infusions. RA was then examined visually and photographed to document any specific accumulation of silver grains. There was no specific accumulation of silver grains over RA when the infusions missed lMAN (n = 2 missed lMAN infusions).
Immunohistochemistry for neurotrophins and Trk receptors.The brains of five male juvenile birds (15–20 d old) were stained with polyclonal antibodies to the extracellular domain of TrkB (courtesy of Dr. S. Feinstein) (cf. Fryer et al., 1996), the extracellular domain of TrkC (courtesy of Dr. F. Lefcort) (cf. Lefcort et al., 1996), the extracellular domain of p75 (the low-affinity neurotrophin receptor, courtesy of Dr. L. F. Reichardt) (cf. Weskamp and Reichardt, 1991), BDNF (courtesy of Amgen/Regeneron Partnership), or NT-3 (courtesy of Dr. R. Rush) (cf. Zhou and Rush, 1994), all raised in rabbit. Birds were overdosed with Equithesin and perfused with avian saline and buffered in 4% paraformaldehyde with 0.4% glutaraldehyde (experimentation with different fixation protocols revealed that the addition of 0.4% glutaraldehyde significantly enhanced staining of both Trk and neurotrophin antibodies in zebra finch brain). The brains were removed and post-fixed in 4% paraformaldehyde for 48 hr before being submerged in 20% sucrose. Brains were frozen-sectioned in the coronal plane at a thickness of 20 μm, and four alternate series of sections were collected into staining trays (free-floating) for immunohistochemical staining. Brain sections containing lMAN and RA were stained for TrkB, TrkC, p75, BDNF, or NT-3. Sections were rinsed three times in PBS and pretreated with 1.0% hydrogen peroxide followed by 5% normal goat serum in 0.3% Triton X-100. The tissue was then incubated in the primary antiserum with 2% normal goat serum, 0.3% Triton X-100, and 0.1% sodium azide at room temperature on a shaker for ∼12–24 hr. Dilution of antibodies was optimized for best contrast and quality of staining. Final dilutions were as follows: anti-TrkB, 1:2500; anti-TrkC, 1:7500 to 1:8000; anti-p75, 1:5000; anti-BDNF and anti-NT-3, 1:1000. After overnight incubation in primary antibody, the tissue was rinsed three times in PBS and reacted using standard avidin–biotin techniques, although some sections were processed with nickel intensification (Elite ABC kit, Vector Labs, Burlingame, CA). After treatment with biotinylated anti-rabbit IgG and the avidin–biotin complex reagent, the tissue was developed in a chromagen solution of 0.05% DAB and 0.01% H2O2. Sections were then rinsed three times in PBS, mounted immediately, and coverslipped.
Specificity of staining was assessed by verifying that omission of the primary antibody resulted in no specific accumulation of reaction product. Typically, three to five control sections per brain were removed from the reaction dish just before treatment with primary antibody; they were stored in 5% normal goat serum in 0.3% Triton X-100. The control sections were returned to the reaction dish after the remaining tissue had been incubated in the primary antibody, just before the third rinse in PBS. For the sake of convenience, we will refer to the staining observed as being against TrkB, TrkC, p75, BDNF, or NT-3 for the remainder of this report.
RESULTS
Neurotrophins preserve neuron number in deafferented RA
Twenty-four hours after lesioning of lMAN, massive cell death and neuronal loss were observed in RA on the untreated (control) side (Fig.2; cf. Johnson and Bottjer, 1994). However, infusions of BDNF, NT-3, or NT-4/5 all enhanced RA neuronal survival 24 hr after the removal of presynaptic input from lMAN (Fig. 2A, top panel). RA neuron counts in birds that received 1 μg doses showed that neurotrophin-treated RA had more neurons relative to contralateral (control) RA (main effect of treated vs control RA:F(1,12) = 68.29, p < 0.0001) and that the neurotrophins differed in their ability to rescue neuron number in deafferented RA (main effect of neurotrophin:F(3,12) = 5.92, p = 0.01). Comparisons between neurotrophin-treated and control RA within each neurotrophin treatment group showed that BDNF, NT-3, and NT-4/5 each rescued RA neurons (p < 0.01 in all cases), whereas NGF did not (NS; see Fig.3E,F). Another group of birds treated with ciliary neurotrophic factor also showed no prevention of neuron death in deafferented RA at a 1 μg dose (data not shown). In addition, infusions that missed RA failed to rescue RA neuron number (see Materials and Methods), which is significant, because this demonstrates that the neurotrophins did not rescue via a systemic or nonspecific mechanism.
Fig. 3.

High-power digitized videomicrographs from the central region of left and right RA in three birds that received bilateral lMAN lesions 24 hr earlier. Left panels show the high incidence of apoptotic cells in control RA 24 hr after deafferentation (A, C, E). RA neurons that died after deafferentation show clear morphological indications of apoptotic cell death; they were shrunken (pyknotic), their nuclei contained large particles of condensed chromatin material, and membrane “blebbing” (formation of apoptotic bodies) was frequently observed at the cell perimeter (Clarke, 1990). Right panels show the ability of 2.5 μg doses of BDNF or NT-3 infusions to suppress apoptotic neuron death and completely rescue RA neuron number (B, D). NGF infusions failed to prevent apoptosis and rescue neurons (F). Arrowheads indicate examples of apoptotic cells, and arrows show neurons. Scale bar, 10 μm.
When compared with the number of RA neurons normally present in juvenile male zebra finches (see “normative” in top panel of Fig. 2A), birds treated with the 1 μg dose of BDNF, NT-3, or NT-4/5 appeared to have fewer RA neurons. As one way of evaluating this trend, we conducted a t test using combined RA data from BDNF-, NT-3-, and NT-4/5-treated RAs (n = 12) and normative unilateral RA data from a group of age-matched birds (n = 14) (from Johnson and Bottjer, 1994; these birds received unilateral lesions of lMAN to assess the incidence of cell death in RA, and the procedures for tissue preparation and analysis were identical to those in the present study). The t test showed that deafferented RA treated with 1 μg doses of BDNF, NT-3, or NT-4/5 had significantly fewer RA neurons than are normally present at this age (t24 = −3.23,p = 0.004). Thus, the 1 μg doses did not produce 100% rescue of RA neuron number after the removal of presynaptic input from lMAN. To determine whether a more comprehensive rescue of RA neurons was possible, additional lMAN-lesioned birds received increased doses (2.5 μg) of BDNF and NT-3. Analysis of birds that received 2.5 μg doses of BDNF or NT-3 into deafferented RA revealed a complete saving of RA neurons (Fig, 2B, top panel); that is, there was no difference between the number of neurons in deafferented RA treated with 2.5 μg doses of BDNF or NT-3 and the number of neurons normally present in RA (t20 = 1.13, p = 0.27;t test conducted using combined data from the 2.5 μg BDNF and NT-3 birds, n = 8, and age-matched birds fromJohnson and Bottjer, 1994, n = 14). Both neurotrophins preserved neuron number in RA equally well at the 2.5 μg dose (main effect of neurotrophin, p = 0.28), and both BDNF and NT-3 produced a larger number of neurons in neurotrophin-treated RA relative to the contralateral (control) side (main effect of treated vs control RA: F(1,6) = 50.418, p = 0.0004, see top panel in Figs. 2B,3A–D).
Neuron death in deafferented RA occurs rapidly and is apoptotic
In our previous study (Johnson and Bottjer, 1994), RA neuron number was assessed at 2, 4, or 6 d after lesioning of lMAN; this analysis revealed a 30–50% loss of neurons by 2 d postlesion and a slight trend of additional loss at 4 and 6 d postlesion. We also observed significant numbers of apoptotic cells in RA at 2, 4, or 6 d postlesion (with a small peak at 4 d), suggesting that RA neuron death after deafferentation occurred gradually over the course of several days. As is typically the case, counts of dying cells were always small in absolute number and never accounted for more than 4–5% of the total amount of neuron loss, a pattern that has encouraged the view that neurons dying in vivo undergo rapid death and clearance by phagocytosis and thereby largely evade efforts to count them (Oppenheim, 1981).
In the present study, however, we observed a massive wave of apoptotic cell death in control RA at 24 hr after deafferentation (e.g., see Fig.2A,B bottom panels). These data contradict our earlier conclusion that neuron death in deafferented RA is a gradual, intermittent process lasting several days. Rather, there is a large population of RA neurons that dies rapidly and in a wholesale manner after the removal of lMAN synaptic input to RA. Particularly striking is the observation that the number of apoptotic cells in RA at 24 hr postlesion appears to account, in absolute terms, for the observed neuron loss at 24 hr postlesion. For example, within each of the neurotrophin treatment groups in Figure 2, adding the number of apoptotic cells in “control RA” (bottom panel) to the number of neurons in the corresponding “control RA” (top panel) approximates the expected unilateral value for RA neuron number in normal birds of this age (i.e., 14,000–16,000 neurons). The same result is observed if the number of apoptotic cells and neurons for “neurotrophin-treated RA” in Figure 2 are added. These data, along with the clear neuronal morphology of many of the apoptotic cells (e.g., see Fig.3E), strongly suggest that most (if not all) of the dying cells in RA at 24 hr postlesion are dying neurons.
Examination of the morphology of RA cells dying at 24 hr postdeafferentation indicated that these cells were undergoing apoptosis. Figure 3 shows examples of dying cells in RA (A,C,E,F), which demonstrate that these cells die via a process that involves overall cell condensation (pyknosis), the presence of large intranuclear particles of condensed chromatin material, and membrane “blebbing” (formation of apoptotic bodies) at the perimeter of the dying cells. The foregoing morphological characteristics are generally considered to indicate a form of apoptotic neuronal cell death (Clarke, 1990).
Neurotrophins suppress neuronal apoptosis in deafferented RA
The large population of apoptotic cells present at 24 hr postlesion in control (untreated or cytochrome-c-treated) RA enabled us to determine whether the preservation of neuron number in BDNF-, NT-3, and NT-4/5-treated RA was attributable to a suppression of apoptosis and whether the failure of NGF to rescue RA neurons was attributable to an inability to suppress apoptosis. In other in vivoexperimental models of neurodegeneration (e.g., septo-hippocampal neurons, see Hefti, 1986; locus coeruleus neurons, see Arenas and Persson, 1993), the induced death of neurons has not been correlated with a clear suppression of neuron death. As a consequence, it is often difficult to rule out change of phenotype or migration as an explanation for neurotrophic rescue of neuron number. Because of the large number of apoptotic cells present in RA at 24 hr postlesion, we were able to quantify the incidence of apoptosis in deafferented RA as a function of the various neurotrophin treatments.
Figure 2A, bottom panel, shows that compared with control RA, the 1 μg doses of BDNF, NT-3, and NT-4/5 each suppressed the appearance of apoptotic cells. The overall pattern of significance for the apoptotic cell data was similar to that obtained for neuron counts; that is, the neurotrophins differed in their ability to suppress apoptosis (main effect of neurotrophin:F(3,12) = 8.11, p = 0.003), and there were fewer apoptotic cells in neurotrophin-treated RA than in control RA (main effect of treated vs control RA:F(1,12) = 53.67, p < 0.0001). Planned comparisons of apoptotic cell numbers within neurotrophin treatment groups showed that there was a suppression of apoptosis in BDNF-, NT-3-, and NT-4/5-treated RA (p < 0.01 in all cases), whereas NGF did not suppress the appearance of apoptotic cells (NS; see also Fig.3E,F). These data demonstrate that BDNF, NT-3, and NT-4/5 preserve neuron number in deafferented RA by suppressing a program of apoptotic cell death.
An analysis of apoptotic cell data from the 2.5 μg BDNF and NT-3 dose groups showed that both of these neurotrophins suppressed apoptosis in RA to an equivalent extent (main effect of neurotrophin:p = 0.22; main effect of treated vs control RA:F(1,6) = 27.323, p = 0.002; seebottom panel in Fig. 2B and Fig.3A–D). In summary, the increased number of neurons in RA treated with various doses of BDNF, NT-3, or NT-4/5 is directly attributable to the ability of these neurotrophins to suppress neuronal apoptosis. These data represent a unique instance in which neurotrophins can be shown to prevent the apoptotic death of cortical neurons after the removal of presynaptic input.
Neurotrophins are transported in the anterograde direction by lMAN axons
Suppression of neuronal apoptosis by infusions of BDNF, NT-3, or NT-4/5 into deafferented RA suggests that these neurotrophins substitute for an endogenous anterograde signaling pathway (lMAN→RA) or for an endogenous auto/paracrine signaling pathway (RA→RA) that is under anterograde regulation by afferent input from lMAN. If lMAN axons normally release neurotrophins into RA, lMAN neurons should have the ability to transport neurotrophins from their somata, along their axons, and into RA. We examined this question by infusing125I-labeled neurotrophins (BDNF, NT-3, or NT-4/5) into lMAN of juvenile birds.
Autoradiograms in Figure 4 show examples of substantial accumulation of silver grains over RA after infusions of [125I]BDNF or [125I]NT-3 into lMAN, demonstrating that these two neurotrophins are transported in the anterograde direction by lMAN axons into RA (based on careful visual inspection BDNF, and NT-3 appeared to be transported equally well by lMAN neurons). In contrast, NT-4/5 was not anterogradely transported by lMAN axons (Fig. 4C). The failure of NT-4/5 to be transported is significant, because it indicates specificity in the uptake and/or anterograde transport mechanisms of lMAN neurons. Interestingly, none of the [125I]neurotrophins that we infused were transported in the retrograde direction by the axons of thalamic and cortical brain regions that send projections to lMAN (i.e., from the medial dorsolateral nucleus of the thalamus and from the archistriatum, pars ventralis) (see Johnson et al., 1995). These data demonstrate the selective ability of lMAN axons to anterogradely transport exogenous BDNF and NT-3.
Fig. 4.

Low-power digitized videomicrographs of autoradiograms containing coronal sections of RA from juvenile male zebra finches that received infusions of 125I-labeled neurotrophins into lMAN; dorsal is up and medial isleft. There was a clear accumulation of silver grains over RA (oval-shaped region in the center of each videomicrograph) in birds that received infusions of [125I]BDNF (A) or [125I]NT-3 (B) into lMAN, indicating anterograde transport of these neurotrophins from lMAN into RA. This pattern of silver grain labeling over RA is identical to that observed when the lMAN→RA projection is labeled with anterograde tract-tracing dyes (Bottjer et al., 1989; Johnson et al., 1995). In contrast, birds that received [125I]NT-4/5 showed no specific accumulation of silver grains over RA (C), indicating that NT-4/5 is not anterogradely transported by lMAN axons. Scale bar, 100 μm.
Immunoreactivity for neurotrophins and Trk receptors
Neurotrophins exert their cellular effects by binding to specific members of a family of receptors, the Trk family (TrkA, TrkB, TrkC; for review, see Bothwell, 1995). For example, NGF binds with high affinity to TrkA, whereas BDNF and NT-4 bind with high affinity to TrkB. NT-3 is somewhat nonselective in that it binds with high affinity to TrkA, TrkB, and TrkC, although TrkC is generally considered to be the NT-3 receptor in vivo. Based on the results of the neurotrophin infusion experiments (i.e., BDNF, NT-3, and NT-4/5 were effective, NGF was not), one prediction is that RA should contain TrkB and possibly TrkC receptors. That is, each of the three effective neurotrophins (BDNF, NT-3, NT-4/5) have the ability to bind with high affinity to TrkB receptors, whereas NT-3 might also exert an effect via high-affinity binding to TrkC receptors.
When reacted with antibody against TrkB, brain sections containing RA showed heavy staining of fibers, neuropil, and somata throughout RA. Only cell nuclei remained unstained by the antibody, giving the tissue sections a punctate appearance (Fig. 5A). Thus, the presence of TrkB immunoreactivity in RA stands in good agreement with the infusion results, which showed that BDNF, NT-3, and NT-4/5 (i.e., neurotrophins able to bind with high affinity to TrkB) were able to maintain neuron number in deafferented RA. Antibody staining against TrkC, the high-affinity NT-3 receptor, revealed no labeling of somata in RA, but sparse fiber labeling was located throughout the nucleus (Fig. 5C,D). A low-affinity receptor that binds all neurotrophins (p75) (for review, see Bothwell, 1995; Chao and Hempstead, 1995) was also detected immunohistochemically in RA and found to display a pattern of intense somal labeling throughout the nucleus (Fig. 5B; similar labeling for p75 was observed throughout the telencephalon).
Fig. 5.
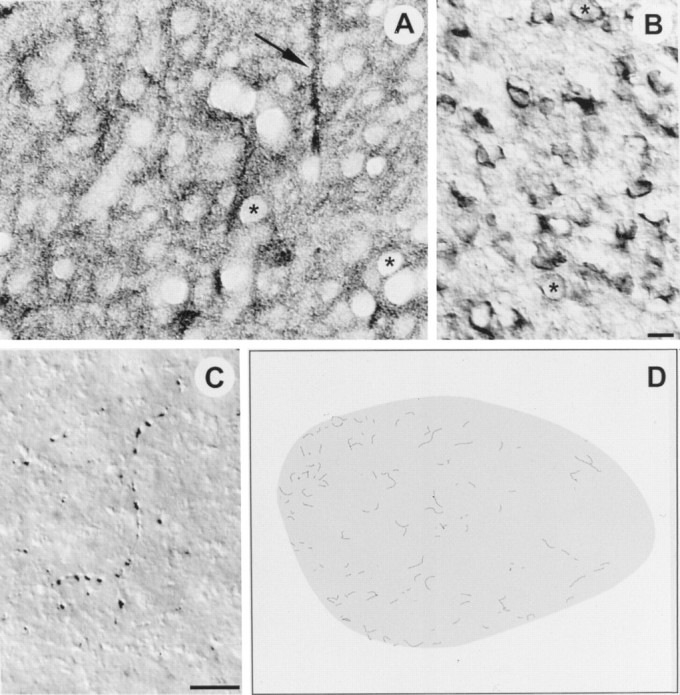
High-power digitized videomicrographs show immunoreactivity against the extracellular domains of TrkB (A), p75 (B), and TrkC (C) in RA of a juvenile male zebra finch; dorsal is up and medial is left in all images.A, TrkB immunoreactivity was found throughout RA. Fiber bundles (arrow), neuropil, and somata were all labeled by the antibody; only nuclei were left unstained, giving the tissue a punctuate appearance (asterisks show two examples of nuclei). The labeled fiber bundles (arrow) were oriented dorsoventrally, and they appeared to pass completely through RA, perhaps projecting to a target ventral to RA. These fiber bundles do not appear to be the axons of lMAN neurons, because lMAN axons enter RA dorsolaterally and laterally (Johnson et al., 1995). B, Immunoreactivity for p75 was found throughout RA and was localized primarily to neuronal somata, although fine labeled processes were sometimes observed. The asterisks show two examples of neuronal nuclei for comparison with nuclei in A.C, Antibody against TrkC only labeled fine processes in RA that seemed to be axons; these fibers often appeared to terminate by encircling somata within RA. D, The distribution of TrkC fiber labeling in RA based in a low-power camera lucida drawing. Note the TrkC labeling in RA was generally sparse but with a somewhat higher incidence of labeled fibers in medial RA. Scale bars (B,C), 10 μm.
The ability of BDNF or NT-3 to maintain neuron survival in deafferented RA and to be transported in the anterograde direction by lMAN neurons is consistent with the hypothesis that lMAN neurons normally synthesize and transport one or both of these neurotrophins to promote neuron survival in RA. However, in addition to the anterograde release of neurotrophins, presynaptic input from lMAN could regulate the auto/paracrine release of neurotrophins by cells in RA. Antibodies against BDNF and NT-3 therefore were used to begin to determine the expression of neurotrophins within the lMAN→RA pathway. When reacted with antibody against NT-3, tissue sections containing lMAN or RA revealed staining of neuronal somata throughout both nuclei, with no labeling of fibers or neuropil (Fig.6E,F); overall examination revealed NT-3-immunoreactive somata throughout the telencephalon. These data suggest that throughout the avian telencephalon (including lMAN and RA), NT-3 could be available for uptake and retrograde transport by a presynaptic population, auto/paracrine release, or anterograde transport and release.
Fig. 6.

Digitized videomicrographs of brain sections from juvenile male zebra finches reacted with antibody against BDNF (A–D) or NT-3 (E,F); dorsal is up and medial isright. A, A low-power image of lMAN shows that lMAN contains numerous BDNF-immunoreactive somata; lMAN is composed of a magnocellular core surrounded by a parvicellular shell (of which only the core projects to RA) (Johnson et al., 1995) and BDNF-immunoreactive somata were present throughout both regions (thethin line indicates outer borders of lMANcore/shell, and the thicker linesshow lamina that define the dorsal and ventral boundaries of the anterior neostriatum). B, Although this low-power image shows that RA did not contain BDNF immunoreactive somata (RA is the distinct oval-shaped region just to the right of center), RA did contain numerous BDNF immunoreactive fibers (D).C, D, High-power images from the central regions of lMANcore and RA show BDNF-immunoreactive somata in lMAN (C) and a fiber in RA (D,arrows), respectively. Because lMAN is the only input to RA that contains BDNF immunoreactive somata, the BDNF-immunoreactive fibers in RA may be the axons of lMANcore neurons (note that the BDNF-immunoreactive fibers appeared to project into RA along the same lateral and dorsolateral orientation followed by most lMANcore axons) (Johnson et al., 1995).E, F, High-power images from the central regions of lMANcore and RA, respectively, show that antibody against NT-3 labeled somata throughout both nuclei. No labeling of fibers was seen with the NT-3 antibody (sections shown inE, F were nickel-intensified). Scale bars: A, B, 100 μm; C,E, F, 16 μm; D, 10 μm.LH, Hyperstriatal lamina; LMD dorsal medullary lamina; LAD, dorsal archistriatal lamina.
Compared with the widespread distribution of NT-3 immunoreactivity, antibody against BDNF produced a more restricted pattern of labeling. BDNF-immunoreactive somata were found in lMAN (and throughout much of the anterior telencephalon) but not in RA (Fig.6A–D). In fact, the posterior telencephalon had considerably fewer BDNF-immunoreactive cells overall, although an arc of labeled cells was present in the dorsal archistriatum overlying RA (Fig. 6B). Thus, although BDNF expressed by lMAN cells could be available for uptake and retrograde transport by a presynaptic population, auto/paracrine release, or anterograde transport and release, release of BDNF by RA neurons appears to be unlikely. Although RA lacked BDNF-immunoreactive somata, RA did contain BDNF-immunoreactive fibers (Fig.6D), which in many cases appeared to terminate within the nucleus (the fibers appeared to be axons based on their length).
DISCUSSION
The present results show that neurotrophins can provide 100% neuronal rescue in a deafferented RA in vivo, and that this preservation of neuron number is directly attributable to a suppression of neuronal apoptosis. In addition, the 125I-labeled neurotrophin data demonstrate selective anterograde transport of two neurotrophins (BDNF and NT-3, but not NT-4/5) by the lMAN neurons that provide presynaptic input to RA. These data show that specific neurotrophins (BDNF and NT-3) can be anterogradely transported by telencephalic neurons to their postsynaptic target, and that these same neurotrophins can provide neuronal rescue when injected directly into the deafferented postsynaptic region. Moreover, immunohistochemical labeling within RA indicated the appropriate receptor types (both high and low affinity) for the neurotrophins that suppressed apoptosis in deafferented RA. Finally, immunohistochemistry for neurotrophins revealed a pattern that is consistent with anterograde transport and release of BDNF and/or NT-3 by lMAN neurons or lMAN-regulated auto/paracrine release of NT-3 by RA neurons, or both types of mechanisms.
Although neurotrophins were initially viewed as molecules that are released by postsynaptic tissues for uptake and retrograde transport by presynaptic cells (Oppenheim, 1991; Thoenen, 1995), the present data join a growing body of evidence that supports two nonretrograde modes of neurotrophin release: from presynaptic to postsynaptic cell (cf.Ferguson et al., 1990; von Bartheld et al., 1996), and local secretion within a given cell population (cf. Acheson et al., 1995). Previous authors have reported transcripts for neurotrophins and Trk receptors in patterns that seem to indicate auto/paracrine or anterograde neurotrophin signaling (Schecterson and Bothwell, 1992; Miranda et al., 1993). However, limited information is available on the mechanisms of action of neurotrophins in the CNS in vivo. Our results explicitly demonstrate the ability of neurotrophins to prevent the death of deafferented telencephalic neurons in vivo, the ability of telencephalic neurons to transport neurotrophins in the anterograde direction, and a pattern of neurotrophin and Trk immunoreactivity consistent with anterograde and auto/paracrine mechanisms of release.
Although it is beyond the scope of this report to determine whether the BDNF-immunoreactive fibers in RA represent a site of anterograde BDNF release, the overall context is certainly suggestive of such a mechanism: BDNF can prevent the death of deafferented RA neurons, BDNF-immunoreactivity is expressed by lMAN somata, BDNF is transported anterogradely by lMAN axons, and TrkB (a high-affinity receptor for BDNF) is expressed in RA. The presence of BDNF-immunoreactive fibers in the absence of BDNF-immunoreactive somata within RA suggests that the labeled fibers are not axons that are in the process of transporting BDNF supplied by an RA cell (otherwise RA somata would have been labeled by the antibody as well). Because lMAN is the only source of presynaptic input to RA that contains BDNF-immunoreactive somata, it is tempting to speculate that the fibers are the axons of BDNF-immunoreactive neurons in lMAN.
One neurotrophin, NGF, did not rescue RA neuron number, an outcome presumably attributable to the inability of NGF to suppress neuronal apoptosis in deafferented RA. We have also been unable to detect TrkA receptors immunohistochemically in RA (F. Johnson and S. Bottjer, unpublished observations), a finding that agrees with the inability of NGF to rescue RA neurons. However, infusion of NT-4/5 directly into RA did preserve RA neuron number, although NT-4/5 was not transported from lMAN to RA. Lack of NT-4/5 anterograde transport is important, because it indicates that lMAN neurons have uptake and transport machinery that is at least partially specific for BDNF and NT-3. In addition, absence of transport appears to rule out NT-4/5 as a neurotrophin that lMAN neurons might transport into RA and release. Nevertheless, the transport data do not address the question of whether the ability of NT-4/5 infusions to rescue RA neurons is a physiological versus a pharmacological effect. RA was intensely immunoreactive for TrkB, the receptor for NT-4/5, and it is possible that endogenous auto/paracrine release of NT-4/5 within RA could be regulated by presynaptic input from lMAN (antibodies to recognize NT-4/5 in avian brain are not presently available). With respect to the presence of multiple growth factors in RA, it is important to recognize that RA shows a complex assortment of developmental changes during song learning, including rapid overall growth that is attributable to a substantial increase in the size and spacing of neuronal somata and the delayed arrival of new axons from another cortical song region, HVC (Bottjer et al., 1985,1986; Konishi and Akutagawa, 1985; Nordeen and Nordeen, 1988; Kirn and DeVoogd, 1989; Akutagawa and Konishi, 1994). Because neurotrophins have been shown recently to regulate the growth of somata, dendritic arbors, and axon terminals (Cabelli et al., 1995; Cohen-Cory and Fraser, 1995,McAllister et al., 1995; Riddle et al., 1995), it seems likely that multiple anterograde, retrograde, and auto/paracrine signaling pathways could be involved in orchestrating the overall growth and development of RA.
It is also worth noting that although our findings indicate a role for neurotrophins in the regulation of neuron survival in avian telencephalon, recent studies of transgenic mice lacking the high-affinity neurotrophin receptors TrkA (Smeyne et al., 1994), TrkB (Klein et al., 1993), and TrkC (Klein et al., 1994) reveal no deficit in telencephalic neuron number (Barbacid, 1994), even though transcripts for Trk receptors and neurotrophins are located throughout the forebrain of normal mice (Klein et al., 1990; Barbacid, 1994;Lamballe et al., 1994). One hypothesis to explain these results is that the survival of forebrain neurons in mammalian brain is determined by multiple signaling pathways [such that the removal of one signaling pathway is compensated for by others (Snider, 1994; Meyer-Franke et al., 1995)]. The anatomical basis of redundant trophic signaling may lie in the fact that neuronal populations in the mammalian forebrain generally make and receive a large number of (often reciprocal) synaptic connections with several neuronal populations (Snider, 1994); glial and auto/paracrine mechanisms of neurotrophin release may also contribute to redundant neurotrophic signaling (Acheson et al., 1995;Yan et al., 1995). By comparison, because the song-control system in the avian forebrain contains large populations of neurons that typically contact a limited number of afferent inputs and efferent targets in a serial, nonreciprocal manner (Bottjer et al., 1989;Johnson et al., 1995; Vates and Nottebohm, 1995), the loss of even one input or target removes a major source of trophic support and can result in extensive anterograde or retrograde neurodegeneration (Johnson and Bottjer, 1993, 1994; Akutagawa and Konishi, 1994). However, even among song-control regions, the requirement for anterograde or retrograde trophic support is transient and appears to wane over the course of juvenile development (e.g., deafferentation of RA ceases to induce neuron death by 40 d of age; Johnson and Bottjer, 1994; cf. Johnson and Bottjer, 1993). Thus, redundant trophic signaling pathways may play an increasing role as the avian telencephalon matures. Because of the unique architecture and ontogeny of the songbird vocal control system, we suggest that it may provide a valuable in vivo model that can be used to begin to examine basic questions of neurotrophic signaling in the developing telencephalon.
The idea that lMAN afferent input regulates RA neuron survival via anterograde or auto/paracrine release of neurotrophins has interesting implications for the development of neural and behavioral sex dimorphisms in zebra finches. Females do not learn to sing, and many female song regions undergo naturally occurring neuron death during development (i.e., during the same phase when juvenile males are learning to sing) (Bottjer et al., 1985; Konishi and Akutagawa, 1985;Kirn and DeVoogd, 1989; Konishi and Akutagawa, 1990). One of the most dramatic degenerative events in the developing female song system is a period of neuronal death in RA that corresponds to a normally occurring loss of lMAN afferent input to RA (Kirn and DeVoogd, 1989; Nordeen et al., 1992). The present findings suggest that the naturally occurring deafferentation of RA in juvenile females may involve a loss of neurotrophin signaling in RA, thereby leading to neuronal apoptosis and the development of a female-typical RA. Although it has been known for some time that gonadal hormone treatments during juvenile life can masculinize the song system of females (Gurney, 1981), preliminary data show that direct infusions of neurotrophins into RA of juvenile females will partially “masculinize” (i.e., increase) RA neuron number (Johnson, Shim, and Bottjer, unpublished observations). These initial results suggest that sex hormones could differentially sculpt brain development by regulating the expression of neurotrophins and hence the survival of neurons in RA (cf. Toran-Allerand, 1996).
In summary, the present data suggest that nonretrograde signaling mechanisms normally contribute to the neurotrophic regulation of neuron survival in the developing cortex of the male zebra finch brain. That is, the pattern of Trk receptor and neurotrophin immunohistochemistry indicates that neurotrophins could promote neuron survival in RA by binding to TrkB (and possibly TrkC) receptors after anterograde release from lMAN axons (for BDNF or NT-3) or auto/paracrine release from within RA (for NT-3), or both. In light of evidence that depolarization augments the ability of neurotrophins to promote neuron survival (Ghosh et al., 1994; Meyer-Franke et al., 1995), that exposure to neurotrophins induces dendritic growth and remodeling (McAllister et al., 1995), and that neurotrophins potentiate synaptic transmission (Lohof et al., 1993; Kang and Schuman, 1995), anterograde and auto/paracrine release of neurotrophins would appear to represent important mechanisms for intercellular communication during telencephalic development. Interestingly, a recent study demonstrating anterograde transneuronal transport of neurotrophins in the developing retino-tectal pathway (von Bartheld et al., 1996) suggests that nonretrograde neurotrophic signaling may be a general phenomenon of the CNS that is not simply a unique feature of the cortical system we have described here.
Footnotes
This research was supported by grants to F.J. and S.W.B. from National Institutes of Health. F.J. is also supported by a Faculty Scholar Award from the Alzheimer’s Association. We thank M. Sablan and P. Shim for technical assistance.
Correspondence should be addressed to Dr. Frank Johnson, Department of Psychology, Florida State University, Tallahassee, FL 32306-1051.
REFERENCES
- 1.Acheson A, Conover JC, Fandl JP, DeChiara TM, Russell M, Thadani A, Squinto SP, Yancopoulos GD, Lindsay RM. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature. 1995;374:450–453. doi: 10.1038/374450a0. [DOI] [PubMed] [Google Scholar]
- 2.Akutagawa E, Konishi M. Two separate areas of the brain differentially guide the development of a song control nucleus in the zebra finch. Proc Natl Acad Sci USA. 1994;91:12413–12417. doi: 10.1073/pnas.91.26.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arenas E, Persson H. Neurotrophin-3 prevents the death of adult central noradrenergic neurons in vivo. Nature. 1993;367:368–371. doi: 10.1038/367368a0. [DOI] [PubMed] [Google Scholar]
- 4.Barbacid M. The trk family neurotrophin receptors. J Neurobiol. 1994;25:1368–1403. doi: 10.1002/neu.480251107. [DOI] [PubMed] [Google Scholar]
- 5.Bonhoeffer T. Neurotrophins and activity-dependent development of the neocortex. Curr Opin Neurobiol. 1996;6:119–126. doi: 10.1016/s0959-4388(96)80017-1. [DOI] [PubMed] [Google Scholar]
- 6.Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. Annu Rev Neurosci. 1995;18:223–53. doi: 10.1146/annurev.ne.18.030195.001255. [DOI] [PubMed] [Google Scholar]
- 7.Bottjer SW, Arnold AP. Developmental plasticity in neural circuits for a learned behavior. Annu Rev Neurosci. 1997;20:455–477. doi: 10.1146/annurev.neuro.20.1.459. [DOI] [PubMed] [Google Scholar]
- 8.Bottjer SW, Glaessner SL, Arnold AP. Ontogeny of brain nuclei controlling song learning and behavior in zebra finches. J Neurosci. 1985;5:1556–1562. doi: 10.1523/JNEUROSCI.05-06-01556.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bottjer SW, Miesner EA, Arnold AP. Changes in neuronal number, density and size account for increases in volume of song-control nuclei during song development in zebra finches. Neurosci Lett. 1986;67:263–268. doi: 10.1016/0304-3940(86)90319-8. [DOI] [PubMed] [Google Scholar]
- 10.Bottjer SW, Halsema KA, Brown SA, Miesner EA. Axonal connections of a forebrain nucleus involved with vocal learning in zebra finches. J Comp Neurol. 1989;379:312–326. doi: 10.1002/cne.902790211. [DOI] [PubMed] [Google Scholar]
- 11.Cabelli RJ, Hohn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- 12.Catsicas M, Pequignot Y, Clarke PGH. Rapid onset of neuronal death induced by blockade of either axoplasmic transport or action potentials in afferent fibers during brain development. J Neurosci. 1992;12:4642–4650. doi: 10.1523/JNEUROSCI.12-12-04642.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chao MV, Hempstead BL. p75 and Trk: a two-receptor system. Trends Neurosci. 1995;18:321–326. [PubMed] [Google Scholar]
- 14.Clarke PGH. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol. 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 15.Clarke PGH. Neuron death in the developing avian isthmo-optic nucleus, and its relation to the establishment of functional circuitry. J Neurobiol. 1992;23:1140–1158. doi: 10.1002/neu.480230907. [DOI] [PubMed] [Google Scholar]
- 16.Cohen-Cory S, Fraser SE. Effects of brain-derived neurotrophic factor on optic axon branching and remodeling in vivo. Nature. 1995;378:192–196. doi: 10.1038/378192a0. [DOI] [PubMed] [Google Scholar]
- 17.DiStefano PS, Friedman B, Radziejewski C, Alexander C, Boland P, Schick CM, Lindsay RM, Wiegand SJ. The neurotrophins BDNF, NT-3, and NGF display distinct patterns of retrograde axonal transport in peripheral and central neurons. Neuron. 1992;8:983–993. doi: 10.1016/0896-6273(92)90213-w. [DOI] [PubMed] [Google Scholar]
- 18.Ferguson IA, Schweitzer JB, Johnson EM., Jr Basic fibroblast growth factor: receptor-mediated internalization, metabolism, and anterograde transport in retinal ganglion cells. J Neurosci. 1990;10:2146–2189. doi: 10.1523/JNEUROSCI.10-07-02176.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fryer RH, Kaplan DR, Feinstein SC, Radeke MJ, Grayson DR, Kromer LF. Developmental and mature expression of full-length and truncated TrkB receptors in the rat forebrain. J Comp Neurol. 1996;374:21–40. doi: 10.1002/(SICI)1096-9861(19961007)374:1<21::AID-CNE2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- 21.Gurney ME. Hormonal control of cell form and number in the zebra finch song system. J Neurosci. 1981;1:658–567. doi: 10.1523/JNEUROSCI.01-06-00658.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hefti F. Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transections. J Neurosci. 1986;6:2155–2162. doi: 10.1523/JNEUROSCI.06-08-02155.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hyson RL, Rubel EW. Transneuronal regulation of protein synthesis in the brainstem auditory system of the chick requires synaptic activation. J Neurosci. 1989;9:2835–2845. doi: 10.1523/JNEUROSCI.09-08-02835.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyson RL, Rubel EW. Activity-dependent regulation of a ribosomal RNA epitope in the chick cochlear nucleus. Brain Res. 1995;672:196–204. doi: 10.1016/0006-8993(94)01390-4. [DOI] [PubMed] [Google Scholar]
- 25.Jacobson M. Developmental neurobiology. Plenum; New York: 1991. [Google Scholar]
- 26.Johnson F, Bottjer SW. Induced cell death in a thalamic nucleus during a restricted period of zebra finch vocal development. J Neurosci. 1993;13:2452–2462. doi: 10.1523/JNEUROSCI.13-06-02452.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson F, Bottjer SW. Afferent influences on cell death and birth during development of a cortical nucleus necessary for learned vocal behavior in zebra finches. Development. 1994;120:13–24. doi: 10.1242/dev.120.1.13. [DOI] [PubMed] [Google Scholar]
- 28.Johnson F, Sablan MM, Bottjer SW. Topographic organization of a forebrain pathway involved with vocal learning in zebra finches. J Comp Neurol. 1995;358:260–278. doi: 10.1002/cne.903580208. [DOI] [PubMed] [Google Scholar]
- 29.Kang HJ, Schuman EM. Long-lasting enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 30.Kirn JR, DeVoogd TJ. Genesis and death of vocal control neurons during sexual differentiation in the zebra finch. J Neurosci. 1989;9:3176–3187. doi: 10.1523/JNEUROSCI.09-09-03176.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klein R, Martin-Zanca D, Barbacid M, Parada LF. Expression of the tyrosine kinase receptor gene trkB is confined to the murine embryonic and adult nervous system. Development. 1990;109:845–850. doi: 10.1242/dev.109.4.845. [DOI] [PubMed] [Google Scholar]
- 32.Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BS, Joyner AL, Barbacid M. Targeted disruption of the trkB neurotrophin receptor gene results in nervous N. system lesions and neonatal death. Cell. 1993;75:113–122. [PubMed] [Google Scholar]
- 33.Klein R, Silos-Santiago I, Smeyne RJ, Lira S, Brambrilla R, Bryant S, Zhang L, Snider D, Barbacid M. Disruption of the neurotrophin-3 receptor gene trkC eliminates Ia muscle afferents and results in abnormal movements. Nature. 1994;368:249–251. doi: 10.1038/368249a0. [DOI] [PubMed] [Google Scholar]
- 34.Konishi M, Akutagawa E. Neuronal growth, atrophy and death in a sexually dimorphic song nucleus in the zebra finch brain. Nature. 1985;315:145–147. doi: 10.1038/315145a0. [DOI] [PubMed] [Google Scholar]
- 35.Konishi M, Akutagawa E. Growth and atrophy of neurons labeled at their birth in a song nucleus of the zebra finch. Proc Natl Acad Sci USA. 1990;87:3538–3541. doi: 10.1073/pnas.87.9.3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lamballe F, Smeyne RJ, Barbacid M. Developmental expression of trkC, the neurotrophin-3 receptor, in the mammalian nervous system. J Neurosci. 1994;14:14–28. doi: 10.1523/JNEUROSCI.14-01-00014.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lefcort F, Clary DO, Rusoff AC, Reichardt LF. Inhibition of the NT-3 receptor TrkC, early in chick embryogenesis, results in severe reductions in multiple neuronal subpopulations in the dorsal root ganglia. J Neurosci. 1996;16:3704–3713. doi: 10.1523/JNEUROSCI.16-11-03704.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levi-Montalcini R. The development of the acoustico-vestibular centers in the chick embryo in the absence of the afferent root fibers of the descending tracts. J Comp Neurol. 1949;91:209–242. doi: 10.1002/cne.900910204. [DOI] [PubMed] [Google Scholar]
- 39.Lo DC. Neurotrophic factors and synaptic plasticity. Neuron. 1995;15:979–981. doi: 10.1016/0896-6273(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 40.Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- 41.McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- 42.Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–819. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- 43.Miranda RC, Sohrabji F, Toran-Allerand CD. Neuronal colocalization of mRNAs for neurotrophins and their receptors in the developing central nervous system suggests a potential for autocrine interactions. Proc Natl Acad Sci USA. 1993;90:6439–6443. doi: 10.1073/pnas.90.14.6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mooney R. Synaptic basis for developmental plasticity in a birdsong nucleus. J Neurosci. 1992;12:2464–2477. doi: 10.1523/JNEUROSCI.12-07-02464.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nordeen EJ, Grace A, Burek MJ, Nordeen KW. Sex-dependent loss of projection neurons involved in avian song leaning. J Neurobiol. 1992;23:671–679. doi: 10.1002/neu.480230606. [DOI] [PubMed] [Google Scholar]
- 46.Nordeen KW, Nordeen EJ. Projection neurons within a vocal motor pathway are born during song learning in zebra finches. Nature. 1988;334:149–151. doi: 10.1038/334149a0. [DOI] [PubMed] [Google Scholar]
- 47.Oppenheim RW. Neuronal cell death and some related regressive phenomena during neurogenesis. A selective historical review and progress report. In: Cowan WM, editor. Studies in developmental neurobiology, essays in honor of viktor hamburger. Oxford UP; New York: 1981. pp. 74–133. [Google Scholar]
- 48.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 49.Riddle DR, Lo DC, Katz LC. NT-4-mediated rescue of lateral geniculate neurons from effects of monocular deprivation. Nature. 1995;378:189–191. doi: 10.1038/378189a0. [DOI] [PubMed] [Google Scholar]
- 50.Rubel EW, Hyson RL, Durham D. Afferent regulation of neurons in the brain stem auditory system. J Neurobiol. 1990;21:169–196. doi: 10.1002/neu.480210112. [DOI] [PubMed] [Google Scholar]
- 51.Schecterson LC, Bothwell M. Novel roles for neurotrophins are suggested by BDNF and NT-3 mRNA expression in developing neurons. Neuron. 1992;9:449–463. doi: 10.1016/0896-6273(92)90183-e. [DOI] [PubMed] [Google Scholar]
- 52.Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, Barbacid M. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/MGF receptor gene. Nature. 1994;368:246–249. doi: 10.1038/368246a0. [DOI] [PubMed] [Google Scholar]
- 53.Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are telling us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 54.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–597. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 55.Toran-Allerand CD. Mechanisms of estrogen action during neural development: mediation by interactions with the neurotrophins and their receptors? J Steroid Biochem Mol Biol. 1996;56:169–178. doi: 10.1016/0960-0760(95)00234-0. [DOI] [PubMed] [Google Scholar]
- 56.Vates GE, Nottebohm F. Feedback circuitry within a song-learning pathway. Proc Natl Acad Sci USA. 1995;92:5139–5143. doi: 10.1073/pnas.92.11.5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.von Bartheld CS, Byers ML, Williams R, Bothwell M. Anterograde transport of neurotrophins and axo-dendritic transfer in the developing visual system. Nature. 1996;379:830–833. doi: 10.1038/379830a0. [DOI] [PubMed] [Google Scholar]
- 58.Weskamp G, Reichardt LF. Evidence that biological activity of NGF is mediated through a novel subclass of high affinity receptors. Neuron. 1991;6:649–663. doi: 10.1016/0896-6273(91)90067-a. [DOI] [PubMed] [Google Scholar]
- 59.Yan Q, Matheson C, Lopez OT. In vivo neurotrophic effects of GDNF on neonatal and adult facial motor neurons. Nature. 1995;373:6512–6514. doi: 10.1038/373341a0. [DOI] [PubMed] [Google Scholar]
- 60.Zhou XF, Rush RA. Localization of neurotrophin-3-like immunoreactivity in the rat central nervous system. Brain Res. 1994;643:162–172. doi: 10.1016/0006-8993(94)90022-1. [DOI] [PubMed] [Google Scholar]