

Abstract
The mechanism by which microtubule-based axonal transport regulates activity-dependent presynaptic plasticity in developing neurons remains mostly unknown. Our previous studies established that syntabulin is an adaptor capable of conjoining the kinesin family member 5B (KIF5B) motor and syntaxin-1. We now report that the complex of syntaxin-1–syntabulin–KIF5B mediates axonal transport of the active zone (AZ) components essential for presynaptic assembly. Syntabulin associates with AZ precursor carriers and colocalizes and comigrates with green fluorescent protein (GFP)-Bassoon-labeled AZ transport cargos within developing axons. Knock-down of syntabulin or disruption of the syntaxin-1–syntabulin–KIF5B complex impairs the anterograde transport of GFP-Bassoon out of the soma and reduces the axonal densities of synaptic vesicle (SV) clusters and FM4-64 [N-(3-triethylammoniumpropyl)-4-(p-dibutylaminostyryl)pyridinium, dibromide] loading. Furthermore, syntabulin loss of function results in a reduction in both the amplitude of postsynaptic currents and the frequency of asynchronous quantal events, and abolishes the activity-induced recruitment of new GFP-Bassoon into the axons and subsequent coclustering with SVs. Consequently, syntabulin loss of function blocks the formation of new presynaptic boutons during activity-dependent synaptic plasticity in developing neurons. These studies establish that a kinesin motor–adaptor complex is critical for the anterograde axonal transport of AZ components, thus contributing to activity-dependent presynaptic assembly during neuronal development.
Keywords: axonal transport, microtubules, presynaptic assembly, synaptogenesis, presynaptic plasticity, active zone precursor, kinesin-1 motor
Introduction
The formation of new synapses or remodeling of existing synapses requires the targeted delivery of synaptic components to the sites of axodendritic contact, a process that begins with the transport of synaptic carrier vesicles along the secretory pathway (Horton and Ehlers, 2004). Presynaptic components, including precursors of synaptic vesicle (SV) and active zone (AZ) compartments, mitochondria, and proteins essential for SV release, are transported to the nerve terminals by kinesin motors moving along microtubules (MT) (Bradke and Dotti, 1998; Hirokawa, 1998; Goldstein and Philp, 1999; Roos and Kelly, 2000). On arrival at the terminal, cargo-loaded transport vesicles undergo fusion with the plasma membrane to assemble AZs and reconstitute SVs (Ahmari et al., 2000; Sampo et al., 2003; Shapira et al., 2003).
Activity-dependent neuronal plasticity includes subcellular target selection, synapse formation, and modulation of neuronal circuits (Katz and Crowley, 2002; Hua and Smith, 2004). Long-term synaptic plasticity represents both functional and morphological changes of synapses with contributions from both presynaptic and postsynaptic mechanisms, particularly during neuronal development (Bailey and Kandel, 1993; Collingridge and Bliss, 1995; Nicoll and Malenka, 1995; Sanes and Lichtman, 1999; Hansel et al., 2001). Remodeling of preexisting synapses and the formation of new synapses might play an important role in the various forms of synaptic plasticity of complex neuronal networks (Antonova et al., 2001; Kim et al., 2003; Udo et al., 2005; Wang et al., 2005). Reorganization of the local actin network at synapses contributes to integrating molecular and morphological changes associated with activity-dependent synaptic plasticity (Colicos et al., 2001; Zhang and Benson, 2001; Huntley et al., 2002; Okamoto et al., 2004; Shen et al., 2006; Yao et al., 2006). However, whether and how the MT-based axonal transport of AZ precursors contributes to the activity-induced formation of new synapses in developing neurons remains mostly unknown.
Syntabulin is a syntaxin-binding and kinesin-1 family member 5B (KIF5B) motor adaptor protein that mediates anterograde transport of syntaxin-1 to neuronal processes (Su et al., 2004; Zhai and Bellen, 2004). Here, we report that the syntabulin–KIF5B transport complex plays a critical role in axonal delivery of the AZ components essential for presynaptic assembly during long-term synaptic plasticity induced by repetitive-spaced stimulation. Using time-lapse imaging in live hippocampal neurons, we demonstrate that syntabulin colocalizes and comigrates with green fluorescent protein (GFP)-Bassoon-labeled AZ precursor vesicles along axonal processes. The knockdown of syntabulin or interference of its interactions with either KIF5B or syntaxin-1 in developing neurons results in the deficient trafficking of the AZ components to nerve terminals, reduces the density of release sites, and impairs synaptic transmission. In contrast, syntabulin loss of function does not affect the axonal delivery of the SV proteins synaptophysin and synapsin-I. Furthermore, expression of syntabulin dominant-negative mutants or suppression of syntabulin expression in neurons inhibits the activity-induced recruitment of GFP-Bassoon at release sites and subsequent formation of new presynaptic boutons. Our findings suggest a mechanism through which long-term presynaptic plasticity is regulated by the syntabulin–KIF5B-mediated axonal transport of the AZ components in developing neurons.
Materials and Methods
Materials and additional methods can be found in the supplemental material (available at www.jneurosci.org).
Functional labeling of presynaptic boutons with N-(3-triethylammoniumpropyl)-4-(p-dibutylaminostyryl)pyridinium, dibromide.
FM dye imaging was performed on hippocampal cultures 2–6 d after transfection. Presynaptic terminals were loaded with 15 μm fluorescent styryl dye N-(3-triethylammoniumpropyl)-4-(p-dibutylaminostyryl)pyridinium, dibromide (FM4-64) (Invitrogen, San Diego, CA) in a high K+ solution, pH 7.4, containing the following (in mm): 95 NaCl, 50 KCl, 10 glucose, 10 HEPES, 1.2 CaCl2, 1.2 MgCl2, and 0.5 kynurenic acid for 2 min at room temperature and washed for 15 min with a calcium-free Tyrode's solution (in mm: 145 NaCl, 3 KCl, 1.2 MgCl2, 15 glucose, and 10 HEPES, pH 7.4) to remove nonspecific membrane bound FM4-64. Depolarization-dependent destaining was obtained by application of the high K+ solution after a 15 min perfusion with calcium-free Tyrode's solution. Nerve terminals of the transfected neurons were identified by tracing the optically distinguishable axons expressing GFP encoded in the small interfering RNA (siRNA) construct or GFP-tagged transgenes. The imaging was performed using a confocal microscope (LSM 510; Zeiss, Oberkochen, Germany) with a 40× C apochromatic water immersion lens (1.2 numerical aperture). Fluorescence of GFP and FM dye was excited at 488 and 543 nm, respectively. All images were collected at 1024 × 1024 pixel resolution and 2.0–4.0× software zoom, using a z-series projection of 8–11 images taken at 0.8 μm depth intervals. Those fluorescence-labeled axonal terminals capable of dye uptake were considered functional release sites. Only the fluorescent puncta of FM4-64 with an area of at least 1.5 × 1.5 μm2 were included in the analysis.
Image acquisition and analysis.
Transfected hippocampal neurons were imaged using a Zeiss LSM 510 oil immersion 40× objective with sequential-acquisition setting. Images were captured using the same settings below saturation at a resolution of 1024 × 1024 pixels (12 bit) for the same exposure time. Eight to 10 sections were taken from top to bottom of the specimen, and brightest point projections were made. The selected images were then exported to Adobe Photoshop and/or Image-Pro Plus analysis software (Media Cybernetics, Silver Spring, MD) or NIH ImageJ.
For time-lapse analysis, transfected neurons were imaged with 1% of the intensity of the argon laser to minimize bleaching and damage to neurons; maximum pinhole opening was set; at least three sections were taken from top to bottom of the specimen for brightest point projections to cover most of the thickness of the processes; and 1.4–4.0× digital zoom was used for better viewing. Images of 512 × 512 pixel resolution (12 bit) were taken at ∼10 s intervals (4–5 s of scanning and 5 s cycle delay) for a total of 100 frames. The short interval was used to minimize laser-induced damage to the neurons. All recordings started 6 min after the coverslip was placed in the chamber.
For repetitive stimulation, the normal Tyrode's solution was replaced with Tyrode's containing 50 mm KCl for 2 min, followed by 6 min recovery in Tyrode's solution, for a total of six repeats. The same fields were imaged and quantified before and after the repetitive stimulation using the same confocal settings.
For morphometric measurements of immunofluorescence staining, the mean intensity of the soma in both transfected and untransfected neurons in the same images was measured using Image-Pro Plus analysis software. Intensity measurements were expressed in arbitrary units of fluorescence per square area.
To quantify the number and size of puncta along a transfected axon, all neuronal images were first imported into Image-Pro Plus analysis software. The transfected axon in low-density culture was randomly selected, usually at least 100 μm away from the cell body, and manually traced on the basis of GFP fluorescence, and then the traced regions were generated with the software. Puncta along the traced axonal processes were identified and analyzed with a threshold function of the Image-Pro by setting the threshold intensity to include as many puncta as possible and exclude structures other than puncta as background. Finally, the data were automatically scored and obtained by the software based on the fluorescence intensity profile. The axon of transfected neurons was identified based on the morphology and by immunostaining for the dendritic marker microtubule-associated protein 2 (MAP2), and at least >1500 μm total axon length per condition was pooled together for quantification. Transfected neurons were randomly chosen from more than six chambers of Lab-Tek in at least three independent experiments for each DNA construct. The number of neurons and the length of axons used for quantification are indicated in the text and figure legends. Measured data were then exported to Excel software for analysis. All statistical analyses were performed using the Student's t test, and all measurements are presented as mean ± SEM.
Electrophysiology.
Dual patch-clamp recordings were performed with whole-cell configuration at room temperature. Neurons were clamped at −70 mV and membrane currents were digitized by Digidata 1322A and acquired using a MultiClamp 700B amplifier driven by pCLAMP 9.2 software (Molecular Devices, Palo Alto, CA). Data were analyzed with Clampfit 9.2 (Molecular Devices). Extracellular recording solution contained the following (in mm): 145 NaCl, 3 KCl, 10 HEPES, 3 CaCl2, 8 glucose, and 2 MgCl2, pH 7.3, and 300 mOsm. External CaCl2 concentration was adjusted to 2 mm for paired-pulse test to increase sensitivity of the fusion machinery. Patch pipettes (Sutter Instruments, Novato, CA) with resistance of 4–8 MΩ were first tip-filled and then backfilled with internal solution containing the following (in mm): 146.5 K-gluconate, 7.5 KCl, 9 NaCl, 1 MgCl2, 10 HEPES, and 0.2 EGTA and 2 Na2-ATP, pH 7.2 and 300 mOsm. Transfected presynaptic neurons [marked by an enhanced GFP (EGFP) fluorescence] forming synapses with the untransfected neurons were selected for recordings. Signals were filtered at 10 kHz, and only recordings with access resistance <20 MΩ and leak current <100 pA were included in analysis. During recording of postsynaptic currents (PSCs), a hyperpolarization (from −70 to −75 mV; 40 ms) was injected at the end of each trial of stimulation to monitor the steady state of input resistance. To record asynchronous release, CaCl2 was substituted with the same concentration of SrCl2 in the extracellular solution, and data were analyzed with Mini Analysis program, version 6.0.3 (Synaptosoft, Decatur, GA). A brief depolarization (from −70 to +30 mV for 2 ms) was given to the transfected presynaptic neuron at the frequency of 0.05 Hz. Small synaptic currents after the major initial postsynaptic current were collected within 200 ms to calculate the frequency and amplitude of asynchronous events. The data collecting criteria were the same as PSC recordings described above.
Results
Syntabulin associates with motile AZ cargos along developing axonal processes
Our previous studies suggested that syntabulin acts as an adaptor capable of conjoining syntaxin-1 and the KIF5B motor, thereby mediating the trafficking of syntaxin-1 cargos to neuronal processes (Su et al., 2004). To determine whether syntabulin and KIF5B also associate with AZ components comprising a subset of syntaxin-1 carrier packets, we performed immunoisolation from light membrane fractions of fetal rat brain using magnetic beads coated with the antibody against Bassoon or syntabulin. The purified membranous vesicles were evaluated by Western blot. Syntabulin and KIF5B were copurified with the AZ precursors containing Bassoon, Piccolo, N-cadherin, soluble N-ethylmaleimide-sensitive factor attachment protein 25 (SNAP-25), and syntaxin-1 (Fig. 1 A). Furthermore, using beads coated with syntabulin antibody, we coisolated Bassoon, N-cadherin, β-catenin, SNAP-25, syntaxin-1, and KIF5B from the syntabulin-containing membranous vesicles (Fig. 1 B). None of these proteins was isolated with beads coated with IgG control. The relative purity of the AZ cargos was confirmed by absence of the markers for other membranous organelles including early endosomal antigen 1 (EEA1) (endosomes), p115 (Golgi), and synaptophysin (SVs). Thus, immunoisolation with antibody against Bassoon or syntabulin consistently showed that both syntabulin and KIF5B physically attach to AZ precursor vesicles.
Figure 1.

Syntabulin and KIF5B associate with immunoisolated AZ precursors containing Bassoon and syntaxin-1. A, B, Bassoon- or syntabulin-associated membranous organelles were immunoisolated from light membrane fractions of fetal rat brain with Dyna magnetic beads coated with either anti-Bassoon (A) or anti-syntabulin (B) antibody, or normal IgG as controls. The bead-bound membranous organelles were solubilized and resolved by 3–8% Tris-acetate PAGE (top three panels) or 4–12% Bis-Tris PAGE (other panels) and sequentially detected by Western blot with antibodies as indicated in the same membranes after stripping between each application of antibody. Relative purity of the isolated Bassoon- or syntabulin-associated membranous vesicles was assessed by immunoblotting the AZ components (Bassoon, Piccolo, N-cadherin, syntaxin-1, SNAP-25, KIF5B, or β-catenin) and the markers for other membranous organelles including EEA1 (endosomes), p115 (Golgi), and synaptophysin (SVs).
Bassoon is synthesized and packaged into the AZ precursor cargos in the soma and temporally transported within the axons of developing neurons as early as day 4 in vitro (DIV4), and by DIV10, large synaptic clusters are observed along dendritic profiles (Zhai et al., 2001; Shapira et al., 2003; Dresbach et al., 2006). We examined intracellular distribution and colocalization of syntabulin with Bassoon in hippocampal neurons at DIV7. Endogenous syntabulin appeared as vesicular structures, was widely distributed in both the soma and along the developing neuronal processes, and was relatively enriched in MAP2-negative axons (Fig. 2 A,B). Because a large proportion of Bassoon within the axons at DIV7 is not colocalized with SV markers (Zhai et al., 2001; Shapira et al., 2003), the colocalized puncta of syntabulin with Bassoon along the axons at DIV6 (Fig. 2 C) likely represent axonal trafficking packets of AZ precursors. To address whether the colocalization of syntabulin with AZ precursor vesicles is temporally regulated in developing neurons, we further coimmunostained cultured neurons between DIV4 and DIV11 with antibodies against syntabulin, synaptophysin, and Piccolo (a marker of AZ precursor carrier). We found that the percentage of Piccolo puncta that colocalized with syntabulin dramatically decreased during the course of neuronal maturation: 71.3 ± 1.8% (DIV4), 55.4 ± 4.2% (DIV6), 41.9 ± 1.5% (DIV8), and 12.7 ± 1.4% (DIV11). In contrast, the percentage of Piccolo puncta that colocalized with synaptophysin steadily increased as follows: 14.6 ± 1.8% (DIV4), 35.7 ± 4% (DIV6), 54.6 ± 2.7% (DIV8), and 60.8 ± 7.8% (DIV11) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These temporal and spatial patterns of colocalization are consistent with previous reports that AZ precursors finally target to and cluster at synapses of mature neurons (Zhai et al., 2001; Shapira et al., 2003).
Figure 2.

Syntabulin and Bassoon colocalize and comigrate as vesicular structures along developing axonal processes. A, B, Intracellular distribution of syntabulin in cultured developing hippocampal neurons at DIV7. Low-density neurons were coimmunostained with antibodies against syntabulin (green) and MAP2 (red), and the two-color image is shown in merged differential interference contrast (A). B, Close-up view of the boxed region in A. C, The partial colocalization of syntabulin (green) and Bassoon (red) along developing axon at DIV6. The arrows indicate colocalized puncta of both proteins, whereas the arrowheads point to Bassoon-unlabeled syntabulin structures. D, Syntabulin colocalizes with motile Bassoon along the axonal processes. Hippocampal neurons at DIV7 were cotransfected with GFP-Bassoon and mRFP-syntabulin followed by time-lapse imaging 18 h after transfection for the selected axonal process (D, white box). The arrows indicate GFP-Bassoon-labeled puncta, which comigrate anterogradely with mRFP-syntabulin to the distal end of the axon at an average velocity between 0.1 and 0.3 μm/s. Relative stationary vesicles are marked as asterisks (*). Scale bars, 10 μm.
To determine whether syntabulin attaches to motile AZ precursor vesicles, we conducted time-lapse imaging in live developing neurons coexpressing monomeric red fluorescent protein (mRFP)-syntabulin and EGFP-Bassoon. EGFP-Bassoon is a widely used marker for monitoring AZ precursors, because the labeled vesicles displayed high motility typical of transport vesicles, colocalized with endogenous Bassoon, and were effectively recruited to synapses (Dresbach et al., 2003, 2006). Our study showed that EGFP-Bassoon-labeled AZ cargos colocalize with mRFP-syntabulin (Fig. 2 D), and that some of the colabeled puncta move anterogradely along axons. Consistent with our biochemical findings that syntabulin and KIF5B physically attach to AZ cargos (Fig. 1), live-cell imaging further suggests that syntabulin colocalizes and comigrates with motile AZ cargos along developing axonal processes.
Syntabulin is essential for the axonal transport of the AZ components
To address whether syntabulin is involved in the axonal transport of AZ components, we suppressed syntabulin expression with a small interfering siRNA (STB-siRNA), which was rigorously and extensively tested for its specificity and efficiency in our previous study (Su et al., 2004). A scrambled siRNA (con-siRNA) that was not homologous to any sequence in the GenBank was used as the control. To rescue the RNA interference (RNAi) phenotype, we constructed a syntabulin mutant (STB*) by altering the nucleotide sequence targeted by STB-siRNA without changing the amino acid sequence of syntabulin. Expression of STB* but not wild-type syntabulin was resistant to RNAi in both COS cells (Fig. 3 A) and hippocampal neurons (data not shown). Neurons at DIV3 were transfected with STB-siRNA, or rescued by cotransfection with STB*, followed by immunostaining for Bassoon and MAP2. After the depletion of syntabulin, Bassoon puncta accumulated in the perinuclear region of the cell body, representing a 40% increase in the relative immunofluorescence intensity (146.8 ± 11.3%; n = 15) compared with that of neurons transfected with con-siRNA (105.1 ± 4.9%; n = 15; p < 0.01) (data not shown). Consistently, the RNAi reduced the number of Bassoon clusters per 10 μm of axon length by 36% (3.55 ± 0.35 for STB-siRNA relative to 5.50 ± 0.40 for con-siRNA; n = 15; p < 0.001) (Fig. 3 B,C). In addition, coexpression of the siRNA-resistant mutant STB* rescued the phenotype, further indicating that syntabulin is essential for transport of the AZ components from the soma into axons.
Figure 3.
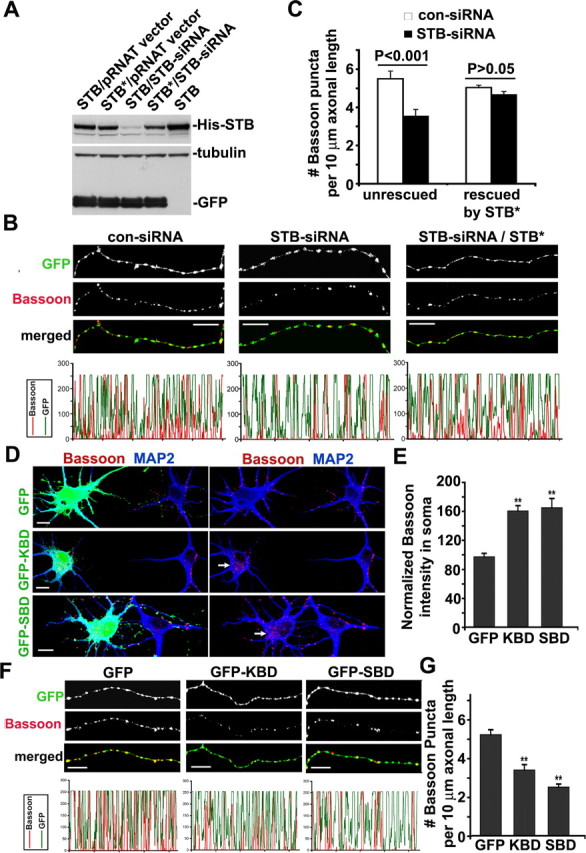
Knockdown of syntabulin or expression of its dominant-negative transgenes reduces the density of Bassoon clusters within axons. A, Syntabulin knockdown is rescued by the RNAi-resistant mutant of syntabulin (STB*). COS7 cells were cotransfected with siRNA constructs with cDNAs expressing His-STB or His-STB*. The expression of syntabulin was determined by immunoblotting of cell lysates prepared 4 d after transfection. To monitor transfection efficiency and equal protein loading (15 μg), GFP and β-tubulin were immunoblotted after stripping the same blot. B, Representative axonal images of hippocampal neurons transfected at DIV3 with con-siRNA or STB-siRNA, or cotransfected with siRNA constructs and HA-STB*, followed by immunostaining for Bassoon and MAP2. A cGFP was encoded in the siRNA vector to visualize the morphology of transfected neurons. MAP2-negative axonal segments are shown in grayscale for individual channels and in color for merged channels. The corresponding profiles reflect the relative distribution (density) of Bassoon clusters (red) along GFP-filled axonal processes. C, Relative density of Bassoon puncta per 10 μm axonal length. Total axon length measured was 3541.82 μm (con-siRNA), 4007.47 μm (STB-siRNA), 5994.11 μm (con-siRNA and HA-STB*), and 4384.79 μm (STB-siRNA and HA-STB*). D–G, Hippocampal neurons were transfected at DIV5 with GFP-KBD, GFP-SBD, or GFP alone, followed by immunostaining at DIV7 for Bassoon (red) and MAP2 (blue). D, Representative images of the cell body for each transfection condition. The arrows point to Bassoon accumulated within the somata of transfected neurons. E, The relative mean intensity of Bassoon in the somata, normalized by that of untransfected cells in the same image. F, Representative MAP2-negative axonal images shown in grayscale for individual channels and in color for merged channels. G, Reduced density of Bassoon puncta within the axons of developing neurons expressing either GFP-KBD or GFP-SBD. Total axon length measured was 4037.98 μm (GFP-KBD), 4323.81 μm (GFP-SBD), or 4169.57 μm (GFP). The data were collected from 15 neurons cultured on three different coverslips for each construct. Error bars indicate SEM (**p < 0.001). Scale bars, 10 μm.
To address whether syntabulin acts as an adaptor connecting the motor protein KIF5B with the presynaptic cargos, we took an alternative approach by interrupting the interactions of syntabulin with either KIF5B or syntaxin-1 via expression of syntabulin dominant-negative binding domain transgenes. These transgenes were derived from the syntaxin-binding domain (SBD) (310–417) and the KIF5B-binding domain (KBD) (81–230) of syntabulin and significantly inhibit the syntabulin-mediated trafficking in neurons (Su et al., 2004; Cai et al., 2005). The dominant-negative effect likely results from the competition between endogenous syntabulin and the exogenously expressed binding domains for accessing the syntaxin-containing cargos or the KIF5B motor. To evaluate the potential role of these interactions in axonal transport of the AZ components, neurons were transfected at DIV5 with the dominant-negative transgenes, followed by coimmunostaining for Bassoon and MAP2 48 h after transfection. Expression of EGFP-KBD or EGFP-SBD increased the mean intensity of Bassoon staining in the perinuclear region of the soma by 64 and 69% (160.25 ± 7.30% for EGFP-KBD; 164.90 ± 12.50% for EGFP-SBD) and significantly reduced the density of Bassoon puncta along axons by 35 and 52% (3.39 ± 0.28 for EGFP-KBD; 2.50 ± 0.16 for EGFP-SBD), relative to neurons expressing EGFP control (96.99 ± 4.85% for soma and 5.21 ± 0.26 for axons) (Fig. 3 D–G).
To examine the trafficking of AZ precursor vesicles after the interruption of the syntabulin–KIF5B complex, we monitored the movement of GFP-Bassoon-labeled AZ cargos by time-lapse imaging in living hippocampal neurons. Despite the tendency to reverse their direction, most AZ precursor vesicles in control neurons transfected with mRFP vector exhibited highly dynamic anterograde movement out of the soma and along the axonal process (supplemental Movie 1, available at www.jneurosci.org as supplemental material). In contrast, expression of mRFP-KBD resulted in a marked reduction of anterograde, but not retrograde, trafficking of the AZ precursor vesicles by ∼50% and consequently decreased their distribution in axons and accumulation in the somata (supplemental Movie 2 and Fig. 2, available at www.jneurosci.org as supplemental material). The results from live-cell imaging are consistent with the redistribution of endogenous Bassoon either under depletion of syntabulin (Fig. 3 A–C) or after disruption of the syntaxin–syntabulin–KIF5B complex (Fig. 3 D–G), and further support that syntabulin is required for anterograde axonal transport of the AZ precursors via proper interactions with both the cargo receptor syntaxin-1 and the motor KIF5B.
In addition to the well characterized AZ precursor vesicles, two types of pleiomorphic organelles have been described as SV precursors, one carrying synapsin-I, VAMP2 (vesicle-associated membrane protein 2), voltage-dependent Ca2+ channels, SV2, and amphiphysin (Ahmari et al., 2000), and the other transporting synaptophysin (Nakata et al., 1998). To address whether syntabulin loss of function has a selective effect on the trafficking of AZ precursor vesicles rather than causing a traffic jam or general transport defect in axons, we examined the intracellular distribution of synapsin-I and synaptophysin. In contrast to the redistribution observed for Bassoon (Fig. 3), neither synapsin-I nor synaptophysin accumulated within the cell body after syntabulin depletion with RNAi or after expression of the syntabulin dominant-negative mutants (supplemental Figs. 3, 4, available at www.jneurosci.org as supplemental material). In addition, our time-lapse imaging study further demonstrates that the axonal trafficking of synaptophysin-labeled synaptic vesicle precursors is not affected after expressing the syntabulin dominant-negative transgene in hippocampal neurons at DIV6 (supplemental Movies 3 and 4, available at www.jneurosci.org as supplemental material). Together with the immunoisolation findings that syntabulin was not copurified with SV proteins (Fig. 1), our studies further suggest that the syntabulin–KIF5B complex is highly selective in mediating the axonal transport of AZ precursors, probably via the specific interaction with the cargo receptor syntaxin-1.
The syntaxin–syntabulin–KIF5B transport complex is essential for the assembly of functional synapses
The AZ components, including N-cadherin, β-catenin, Munc13, RIM (Rab3a-interacting molecule), CAST (CAZ-associated structural protein), Bassoon, and Piccolo, are thought to define molecularly and spatially organized sites for neurotransmitter release (Zhen and Jin, 2004; Ziv and Garner, 2004). Bassoon is among the earliest proteins to arrive at nascent synapses in cultured neurons and may subsequently trigger the assembly of scaffolding and regulatory proteins targeted to AZs. Synaptic transmission is reduced in mice deficient for Bassoon, which can be attributed to fewer active glutamatergic synapses (Altrock et al., 2003). Furthermore, loss of Bassoon in retinal photoreceptor synapses results in the inability of the ribbons to anchor at the presynaptic membrane (Dick et al., 2003; tom Dieck et al., 2005). Having established the role of syntabulin in the axonal transport of AZ components, we next performed three lines of experiments to assess the physiological relevance of syntabulin in the assembly and maintenance of functional synapses.
First, we evaluated the impact of syntabulin loss of function on the presynaptic density in cultured hippocampal neurons at DIV9–DIV10. Synaptophysin is one of the first proteins found clustered at developing CNS synapses and has been used extensively at the light microscope level to detect synapses (Bozdagi et al., 2000; Antonova et al., 2001; Bamji et al., 2003; Kim et al., 2003; Wang et al., 2005). The axonal density of synaptophysin puncta was reduced by 43% in neurons expressing STB-siRNA (2.33 ± 0.33 per 10 μm axonal length) and by 50% in neurons expressing GFP-KBD (2.58 ± 0.29) or GFP-SBD (2.55 ± 0.33), relative to neurons expressing con-siRNA (4.06 ± 0.4) or GFP (5.15 ± 0.61), respectively (Fig. 4). In addition, the normalized size of the punctual synaptophysin clusters in axons expressing STB-siRNA (n = 97; p < 0.05) was 40% smaller than those in axons transfected with con-siRNA (n = 114). Consistently, expression of the dominant-negative binding-domain transgenes reduced the size of punctual synaptophysin clusters by 50% (GFP-KBD; n = 54; p < 0.05) and 59% (GFP-SBD; n = 51; p < 0.01) relative to clusters in neurons expressing GFP (n = 110). Thus, our results indicate that the proper expression of syntabulin and its interactions with KIF5B and syntaxin-1 are not only required for the axonal transport of AZ components, but also crucial for clustering SVs at nascent presynaptic terminals. This effect is possibly attributable to impaired transport of N-cadherin and β-catenin carried by the AZ precursor vesicles (Fig. 1), because both proteins play a critical role in the accumulation of SVs at release sites in developing neurons (Togashi et al., 2002; Bozdagi et al., 2004). This assumption is supported by a recent report that synaptophysin-GFP-labeled SVs were diffusely localized along the axon of cultured neurons lacking cadherin–catenin complex, and that the number of SV in synapses was reduced in the β-catenin conditional knock-out mice (Bamji et al., 2003).
Figure 4.
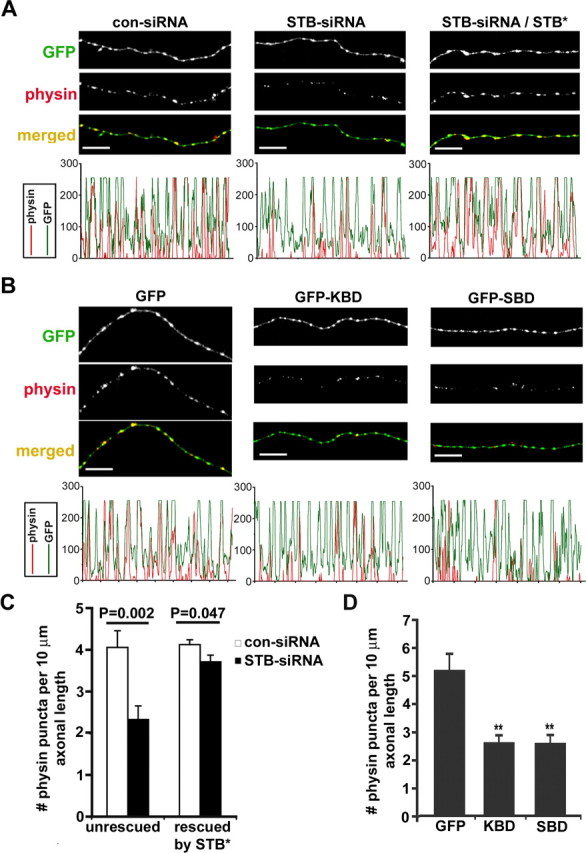
Syntabulin loss of function reduces the density of SV clusters within axons. A, Hippocampal neurons (DIV5) were transfected with either con-siRNA or STB-siRNA alone, or cotransfected with HA-STB*, followed by immunostaining for synaptophysin (physin; red) and MAP2 (blue) or HA-STB (data not shown) 4 d after transfection. The corresponding profiles within the MAP2-negative axonal segments show that STB-siRNA reduced the relative size and density of SV clusters and coexpression with HA-STB* rescued the phenotype. B, Representative axonal images of hippocampal neurons transfected at DIV7 with GFP, GFP-KBD, or GFP-SBD, followed by immunostaining at DIV9 for synaptophysin (red) and MAP2 (blue). Note that expression of the binding domain transgenes decreased both the size and density of SV clusters (red). C, D, The relative density of SV clusters within the axons expressing siRNA (C) or dominant-negative binding domain transgenes (D). Histograms represent the number of synaptophysin-labeled puncta per 10 μm axon length, and p values are calculated relative to that of neurons transfected with con-siRNA or con-siRNA/STB* (C) or GFP (D), respectively. The data were pooled from 13 neurons cultured on three separate coverslips. Total axon length measured was 1770.51 μm (con-siRNA), 1509.55 μm (STB-siRNA), 3392.95 μm (con-siRNA/STB*), 3317.92 μm (STB-siRNA/STB*), 1498.03 μm (GFP), 1809.38 μm (GFP-KBD), or 2803.15 μm (GFP-SBD). Error bars indicate SEM (**p < 0.001, Student's t test). Scale bars, 10 μm.
Second, we evaluated the effect of syntabulin on the assembly of functional release sites by examining styryl dye FM4-64-labeled SVs. FM dye at presynaptic boutons was taken up in an activity-dependent manner, because the majority of FM4-64-labeled presynaptic terminals at DIV9–DIV11 could be unloaded with 50 mm KCl solution (Fig. 5 A). However, the FM dye uptake was significantly suppressed by interference with syntabulin function or depletion of its expression. The number of FM dye puncta along axonal processes was reduced by 57% in neurons expressing STB-siRNA (1.12 ± 0.10 per 10 μm axonal length), and by 49 and 56% in neurons expressing GFP-KBD (1.24 ± 0.10) and GFP-SBD (1.06 ± 0.06), relative to neurons expressing con-siRNA (2.61 ± 0.17) or GFP (2.42 ± 0.19), respectively (Fig. 5 B,C). These observations are consistent with the reduced density and size of synaptophysin puncta along axons (Fig. 4), and further suggest that the recruitment of release machinery and the accumulation of the SV pool to AZs are significantly impaired after syntabulin loss of function.
Figure 5.
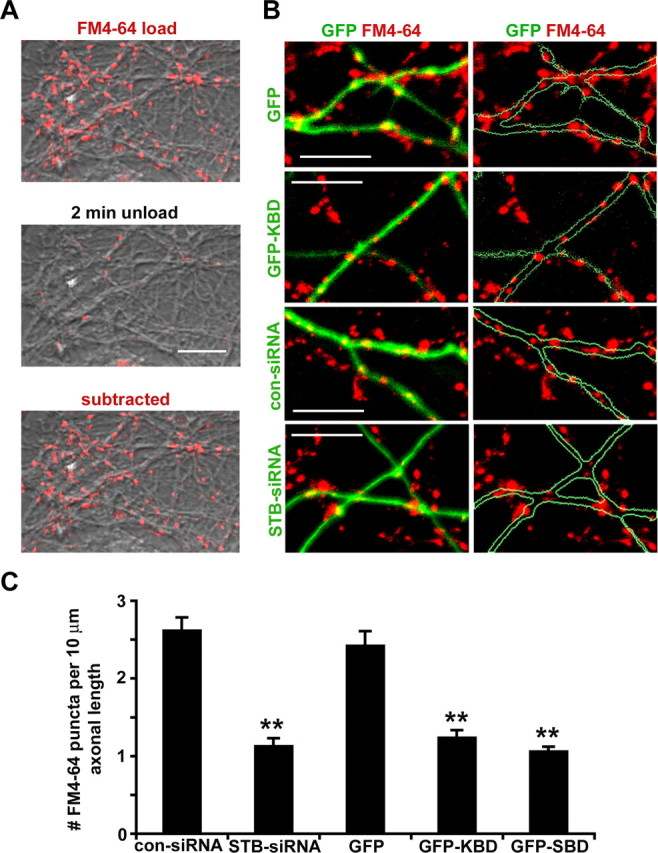
Syntabulin loss of function impairs the assembly of functional presynaptic boutons. A, Representative presynaptic terminals loaded with FM4-64 were unloaded in the presence of 50 mm KCl for 2 min. The subtracted image, the FM4-64-labeled puncta subtracted by the signal left after unloading, represents an activity-dependent uptake of FM dye at presynaptic boutons. B, Reduced FM4-64 uptake in neurons after syntabulin loss of function. Axon terminals at DIV9–DIV11 were loaded with FM4-64 (red) 4–6 d after transfection with con-siRNA or STB-siRNA (green), or 2 d after transfection with GFP, or GFP-KBD (green). C, Quantitative measurement of FM4-64 puncta. The fluorescent images were collected from 17–18 neurons from three separate coverslips for each DNA construct. Total axon length measured was 3379.85 μm (con-siRNA), 3350.12 μm (STB-siRNA), 3444.71 μm (GFP), 3679.06 μm (GFP-KBD), or 3946.01 μm (GFP-SBD). Error bars indicate SEM (**p < 0.001, relative to control siRNA or GFP by Student's t test, respectively). Scale bars, 10 μm.
To assess the physiological relevance of these morphological observations, we measured the amplitude of PSCs in the hippocampal synapses formed between transfected presynaptic neurons and untransfected postsynaptic neurons in culture. Given that synaptic contacts between cultured neurons are highly variable, a few contacts between paired neurons could be a concern when recording detectable postsynaptic currents, especially when neurons are grown in high density. To reduce the variability of synaptic response, we conducted recordings in low-density hippocampal cultures, where more synaptic contacts are likely formed between paired neurons next to each other. Paired whole-cell recordings were performed by stimulating presynaptic neurons (from −70 to 30 mV; 2 ms) at DIV8, 2 d after transfection. The expression of EGFP-KBD resulted in a marked reduction in the amplitude of PSCs (107.9 ± 19.7 pA; p < 0.05, grouped Student's t test) compared with the neurons expressing EGFP (210.7 ± 43.1 pA) or untransfected controls (224.4 ± 54.3 pA) (Fig. 6 A). In addition, for the neurons expressing EGFP-KBD, the frequency distribution of PSC amplitude exhibited a leftward shift relative to that expressing EGFP or untransfected controls (Fig. 6 B). The decrease in basal transmission could be attributable to both changes in the number of release sites (N) and the release probability (Pr). We then applied paired-pulse stimulation at different frequencies to examine whether expression of EGFP-KBD affects release probability. Remarkable paired-pulse facilitation was found in the neurons expressing EGFP-KBD at 50 Hz stimulation (1.54 ± 0.12), and the differences relative to EGFP transfected and untransfected neurons were even more significant (p < 0.01) at 100 Hz stimulation (Fig. 6 C), suggesting a decreased initial release probability after KBD expression. Given that only presynaptically transfected neurons were selected for dual whole-cell recordings, the observed defects in synaptic transmission reflect a specific effect of syntabulin loss of function on the assembly of release sites and fusion machinery, which is consistent with our morphological observations of the reduced densities and sizes of synaptophysin puncta and activity-dependent FM4-64 loading in the neurons with syntabulin loss of function.
Figure 6.

Syntabulin loss of function impairs synaptic transmission. A, Bar graph of averaged amplitudes of PSCs recorded from coupled neurons (DIV8) untransfected (open bar; n = 17), or transfected in presynaptic neurons with either EGFP control (gray bar; n = 16) or EGFP-KBD (black bar; n = 14; p = 0.041, grouped Student's t test). Inset, Representative PSCs. B, Frequency distribution of the PSC amplitudes in neurons untransfected, or transfected with either EGFP-KBD or EGFP. Note that presynaptic expression of GFP-KBD caused a left shift of PSC amplitude distribution relative to the patterns observed in the controls. C, Paired-pulse ratio plotted against different frequencies. The neurons expressing EGFP-KBD exhibit an increased paired-pulse ratio under 50 Hz (1.54 ± 0.12; n = 12; p < 0.05) and 100 Hz (1.58 ± 0.21; p < 0.01), relative to the neurons expressing GFP (1.12 ± 0.13 for 50 Hz; 0.83 ± 0.16 for 100 Hz; n = 14) or untransfected cells (1.11 ± 012 for 50 Hz; 0.80 ± 0.13 for 100 Hz; n = 14; grouped Student's t test). D, Representative traces of asynchronous events recorded in the presence of the Sr2+-containing extracellular solution from neurons (DIV12) transfected with either STB-siRNA or con-siRNA. Inset, Mean amplitude of the Sr2+-induced initial synaptic response. E, Curves of cumulative interevent interval and bar graph of interevent interval (inset) compared between both STB-siRNA (n = 7) and con-siRNA (n = 6) transfected neurons recorded in the presence of the Sr2+-containing extracellular solution. Note that suppression of syntabulin in presynaptic neurons resulted in a significant reduction in both the mean amplitude of the Sr2+-induced initial synapse response (p = 0.03) and the frequency of subsequent asynchronous events (p = 0.039). Calibration: A, D, 50 pA, 50 ms. Bar graphs are expressed as mean ± SEM.
Furthermore, we sought to examine the frequency of quantal events from the neurons transfected with STB-siRNA. Given that not all of the presynaptic terminals on recorded postsynaptic neurons can be successfully transfected, the frequency of miniature postsynaptic currents may not accurately reflect the number of release sites from transfected presynaptic neurons. Alternatively, we measured evoked asynchronous release by substitution of extracellular Ca2+ with strontium (Sr2+), a manipulation that increases delayed asynchronous release after a major peak current while decreasing synchronous release. Analysis of Sr2+-induced asynchronous events, which mimics the miniature postsynaptic responses, has been widely used as an approach to examine presynaptic quantal events (Choi and Lovinger, 1997; Oliet et al., 1997; Caillard et al., 1999; Xu-Friedman and Regehr, 2000; Hagler and Goda, 2001). Dual whole-cell recordings were performed on neurons (DIV12) 6 d after transfection with siRNAs, allowing us to selectively measure the frequency of asynchronous release from a single transfected presynaptic neuron. In parallel with the reduction of PSC amplitude after expression of EGFP-KBD (Fig. 6 A), we observed a significant decrease in the Sr2+-induced and evoked initial synaptic response in the STB-siRNA transfected neurons (52.64 ± 16.65 pA) relative to the con-siRNA transfected neurons (119.68 ± 18.01 pA) (Fig. 6 D, inset). However, no difference was found in the mean amplitude of the Sr2+-induced subsequent asynchronous quantal events after the initial response (data not shown). Consistent with the reduced density of FM4-64 puncta when syntabulin function was disrupted (Fig. 5), the frequency of the Sr2+-induced asynchronous events was significantly reduced in the presynaptic neurons expressing STB-siRNA (43.9 ± 10.1 ms) compared with those expressing con-siRNA (19.3 ± 3.2 ms; p < 0.05) (Fig. 6 E, inset). Reduced frequency of quantal events is further reflected by a rightward shift of the cumulative interevent interval distribution (Fig. 6 E) and could be attributed to a reduced density of the release sites or impaired presynaptic function after the depletion of syntabulin. Together, the electrophysiological data reinforce our conclusion that the syntabulin–KIF5B complex is important in maintaining the proper density and activity of presynaptic terminals.
The syntabulin–KIF5B complex contributes to presynaptic plasticity
To address whether the defective axonal transport of Bassoon after depletion of syntabulin affects the subsequent recruitment and incorporation of AZ components and SVs at nascent synapses, we applied repetitive-spaced stimulation in developing hippocampal neurons, a protocol widely used to induce long-term synaptic plasticity by activation/enhancement of preexisting synapses or formation of new presynaptic boutons (Wu et al., 2001; Li et al., 2004; Yao et al., 2006). We examined the coclustering of GFP-Bassoon and synaptophysin-mRFP in the neurons expressing wild-type syntabulin or its dominant-negative mutant KBD. Images from the same fields before and after the repetitive stimulation (Fig. 7 A) were compared with each other to identify activity-induced recruitment of new GFP-Bassoon into the axons and subsequent coclustering with SVs. Although the stimulation efficiently induced such new incorporation in control neurons (Fig. 7 B), there was a robust increase in the neurons overexpressing syntabulin (Fig. 7 D). In contrast, disruption of the syntabulin–KIF5B interaction with KBD almost abolished the new coclustering (Fig. 7 C). Thus, our study provides evidence that efficient axonal transport of the AZ precursor cargos is essential for the recruitment of the AZ components into the axons and subsequent incorporation with SVs.
Figure 7.
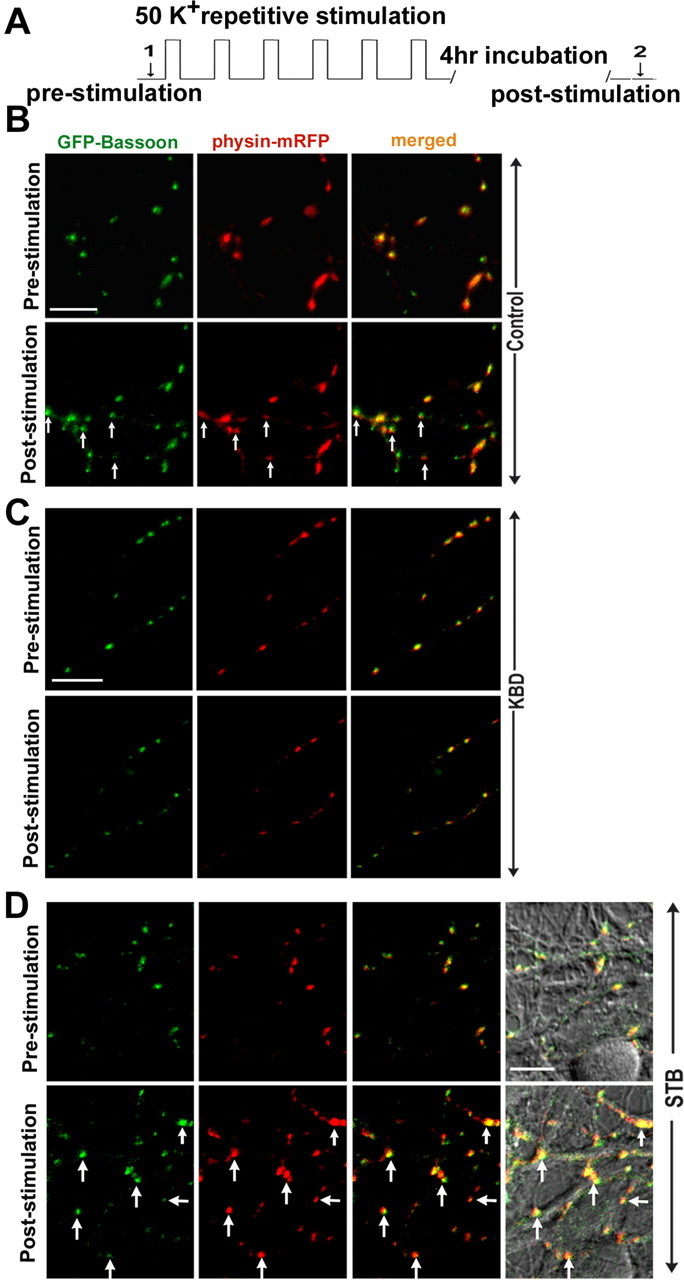
Syntabulin-mediated trafficking contributes to the incorporation of GFP-Bassoon with synaptophysin-mRFP. A, Experimental protocol for spaced repetitive high K+ stimulation. Each stimulation step includes 2 min incubation with 50 mm KCl and 6 min recovery with normal Tyrode's solution for a total of six repeats followed by 4 h incubation at 37°C. Image was acquired (arrows 1 and 2). B–D, Cultured neurons (DIV8) were cotransfected with GFP-Bassoon and synaptophysin-mRFP (B), or triply transfected together with KBD (C) or wild-type syntabulin (STB) (D). Two or 3 d after transfection, the same fields were imaged in single and merged color before and after the repetitive stimulation and incubation. Note the repetitive stimulation induces a robust increase in the coclustering of GFP-Bassoon and synaptophysin-mRFP within the axons of neurons expressing STB relative to control neurons. In contrast, expression of the dominant-negative mutant KBD almost abolishes the activity-induced incorporation of both proteins. Differential interference contrast image in D reflects the same field taken before and after the stimulation. The arrows represent new coclusters after the stimulation. Scale bars, 10 μm.
We next addressed whether the MT-based and syntabulin–KIF5B-mediated transport of the AZ precursor packets plays a critical role in the formation of new presynaptic boutons in response to neuronal activity. The neurons were transfected with synaptophysin-GFP or cotransfected with syntabulin or its dominant-negative mutant KBD at DIV8, followed by the repetitive stimulation and retrospective immunostaining for endogenous Bassoon at DIV10–DIV12. Overexpression of syntabulin causes a robust increase (80.5 ± 9.2%) in the assembly of the new presynaptic boutons in response to the repetitive stimulation, compared with a relatively mild increase in the control neurons (39.5 ± 7.9%) (Fig. 8). Almost all of these newly formed SV clusters are targeted to Bassoon-labeled AZs. In contrast, such activity-induced presynaptic assembly was almost abolished by the expression of syntabulin mutant KBD (6.07 ± 5.9%). To exclude a potential pleiotropical effect by expressing KBD transgene in neurons, we alternatively suppressed syntabulin expression with siRNA. As shown in Figure 9, although the repetitive stimulation induces new presynaptic boutons by 45.89 ± 7.11% in the neurons transfected with con-siRNA, knocking down syntabulin with STB-siRNA blocks the activity-induced formation of new presynaptic boutons (4.71 ± 3.48%). These activity-induced new presynaptic boutons represent active release sites, because the majority of them colocalize with the FM puncta (Fig. 8 A). Given that syntabulin appears as vesicular structures not primarily localized at presynaptic boutons, it is unlikely that the depletion of syntabulin directly affects the induction of activity-dependent synapse formation. Together, our findings provide new evidence for the mechanism in long-term presynaptic plasticity: a coordinated communication between neuronal activity and the MT-based anterograde axonal transport.
Figure 8.
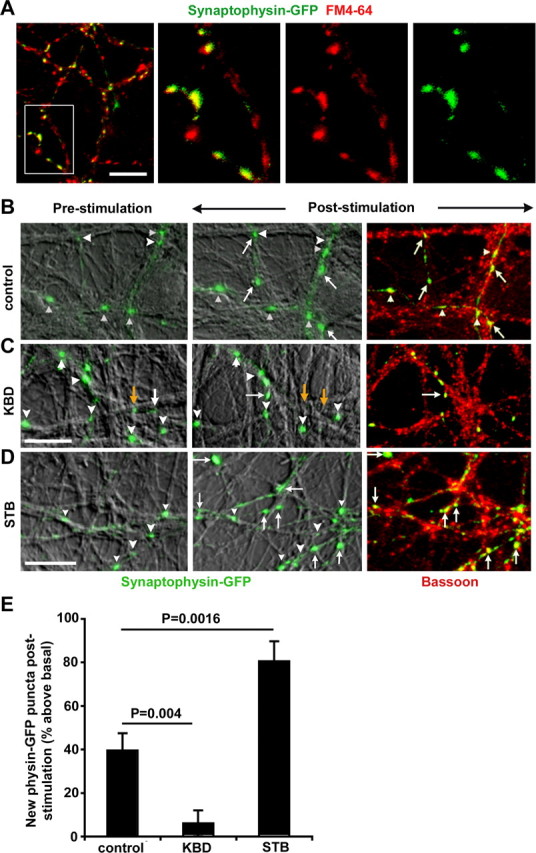
Syntabulin enhances activity-dependent assembly of presynaptic boutons. A, Synaptophysin-GFP clusters represent active presynaptic terminals. Cultured neurons (DIV8) were transfected with synaptophysin-GFP followed by activity-dependent uptake of FM4-64 dye. Note that most of the synaptophysin-GFP clusters colocalize with FM4-64 puncta, whereas synaptophysin-GFP-negative FM puncta may represent presynaptic boutons from neighboring untransfected cells. B–D, Activity-dependent presynaptic assembly in neurons transfected with synaptophysin-GFP alone (B) or cotransfected with KBD (C) or STB (D). Two to 3 d after transfection, the same fields were imaged for synaptophysin-GFP clusters before and after the repetitive stimulation/incubation, followed by retrospective immunostaining for Bassoon. Newly assembled synaptic boutons after the stimulation are labeled by white arrows, whereas previously remaining boutons are marked by white arrowheads. The yellow arrows indicate the synaptophysin clusters eliminated after the stimulation. E, Syntabulin is required for the activity-dependent formation of presynaptic boutons. The number of activity-induced synaptophysin-GFP clusters are normalized to the number of clusters before the stimulation in the same field (230 μm × 230 μm) [1531 (pre) vs 2075 (post) puncta from 12 imaging fields (control); 2502 (pre) and 2574 (post) puncta from 29 imaging fields (KBD); and 2433 (pre) and 4169 (post) puncta from 27 imaging fields (STB), in at least 3 preparations]. Error bars indicate SEM. Scale bars, 10 μm.
Figure 9.
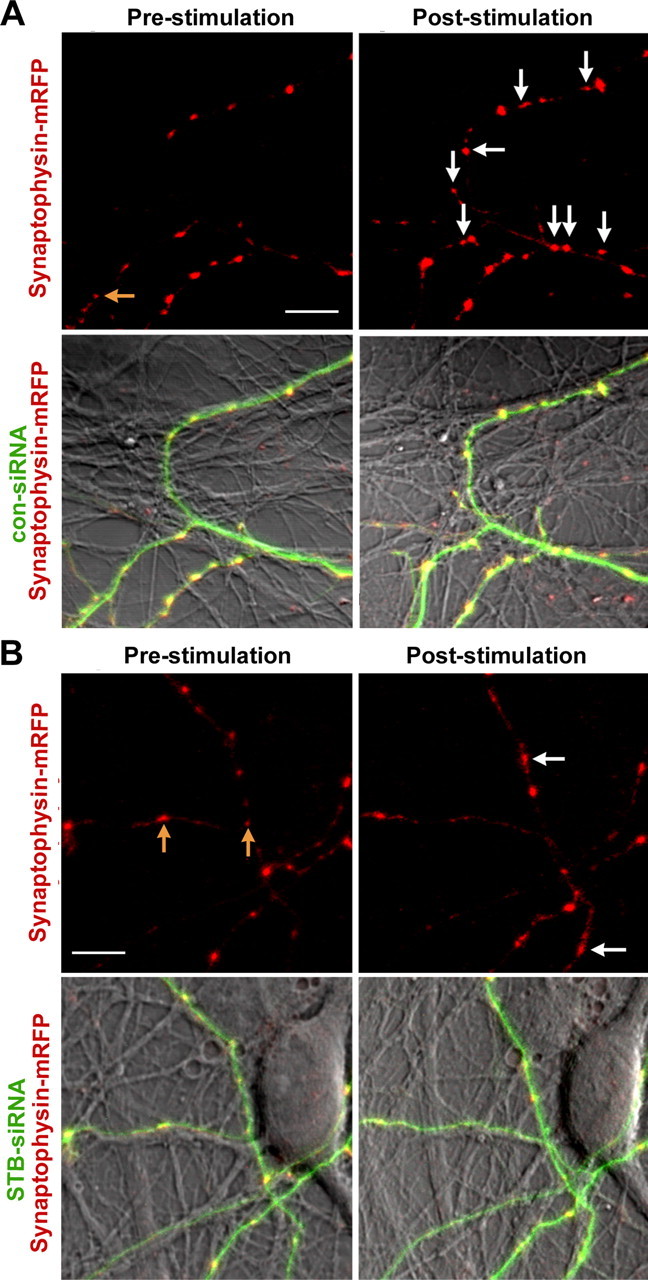
Suppression of syntabulin abolishes the activity-induced formation of new presynaptic boutons. Cultured neurons (DIV6) were cotransfected with synaptophysin-mRFP and con-siRNA (A) or STB-siRNA (B). The same fields were imaged for synaptophysin-mRFP clusters before and after the repetitive stimulation at DIV11–DIV12. Newly assembled presynaptic boutons after the stimulation are labeled by white arrows. The yellow arrows indicate the synaptophysin clusters eliminated after the stimulation. For quantitative analysis, the number of new clusters of synaptophysin-mRFP at poststimulation images is normalized to the number of clusters before the stimulation in the same field (230 μm × 230 μm) [835 (pre) vs 1201 (post) puncta from 17 imaging fields (con-siRNA); 1358 (pre) vs 1411 (post) puncta from 29 imaging fields (STB-siRNA) in at least 3 preparations]. Error bars indicate SEM. Scale bars, 10 μm.
Discussion
In this study, we discover that syntabulin and KIF5B comprise the transport machinery critical for axonal transport of the AZ precursors and contribute to activity-dependent presynaptic plasticity during neuronal development. Our study indicates that syntabulin functions as an adaptor by connecting AZ transport carriers via syntaxin-1 to KIF5B motors critical for the formation of new synapses during long-term presynaptic plasticity in developing neurons. To our knowledge, this study reveals for the first time an important anterograde transport complex capable of mediating the axonal transport of AZ precursor vesicles, thus contributing to the activity-induced presynaptic assembly.
Syntabulin acts as an adaptor linking AZ precursor vesicles to KIF5B
Newly synthesized synaptic membrane proteins are sorted into post-Golgi carriers and then transported along MTs toward the axonal or dendritic surface (Bradke and Dotti, 1998; Burack et al., 2000; Sampo et al., 2003). Substantial evidence suggests that AZ precursor carriers are generated from the trans-Golgi network (TGN) and traverse along the developing axon to nascent synapses (Friedman et al., 2000; Shapira et al., 2003; Dresbach et al., 2006). Cargo vesicles must attach to their transport motors with a high degree of specificity to preserve cargo identity and targeted trafficking (Goldstein and Yang, 2000; Manning and Snyder, 2000). The kinesin-3 family KIF1A and KIF1Bβ are candidate motor proteins mediating axonal transport of SV precursors (Yonekawa et al., 1998; Zhao et al., 2001). The kinesin-1 family KIF5B motor protein transports dendritic carriers containing GluR2/3 (glutamate receptor 2/3) of AMPA receptors through a different set of adaptor complexes (Setou et al., 2002), supporting the notion that the indirect motor–cargo linkage via a specific adaptor complex may be a general mechanism for the transport of both presynaptic and postsynaptic components in neurons. However, molecular identities of the motor–adaptor complex specific for the presynaptic AZ carriers remain unknown.
Our current study makes a significant advance by providing evidence that the syntaxin–syntabulin–KIF5B complex acts as trafficking machinery specific for the axonal transport of AZ precursor packets from the cell bodies to synapses during synaptogenesis. Disruption of this complex impairs anterograde transport of the AZ component Bassoon out of the somata, consequently resulting in its accumulation into perinuclear clusters, structures likely to be associated with the TGN (Donaldson and Lippincott-Schwartz, 2000; Allan et al., 2002). Based on our findings, we propose a candidate axonal transport machinery specific for the delivery of the AZ precursors, in which syntabulin serves as an adaptor that connects AZ precursor carriers via syntaxin-1 to the MT-based KIF5B motor protein (Fig. 10). Our previous studies showed that a significant portion of syntabulin also targets to mitochondria via its C-terminal tail, suggesting a general role for syntabulin in mediating KIF5B-driven transport of protein cargos and organelles via its unique targeting sequences (Cai et al., 2005).
Figure 10.

The MT-based axonal transport contributes to presynaptic plasticity. A, The syntaxin–syntabulin–KIF5B complex is MT-based anterograde transport machinery. Syntaxin-1 serves as a receptor on the AZ transport vesicles and syntabulin acts as an adaptor capable of conjoining syntaxin-1 and the KIF5B motor, thereby mediating trafficking of the AZ transport carriers. B, Through this motor–adaptor complex, AZ precursor carriers traverse along the axon to nascent synapses, participate in the formation of AZs, and consequently contribute to the activity-induced presynaptic plasticity in developing neurons.
It was reported that, although syntaxin-1A-null mutation impairs long-term potentiation (LTP) in hippocampal slices, it does not affect the formation of synapses and basic synaptic transmission in cultured hippocampal neurons (Fujiwara et al., 2006). Because both syntaxin-1A and -1B have similar distributions within neurons and perform similar functions as t-SNARE (target membrane-associated soluble N-ethylmaleimide-sensitive factor attachment protein receptor), syntaxin-1B can compensate for the deletion of syntaxin-1A and is able to maintain basic synaptic transmission in the syntaxin-1A mutant neurons. Similarly, the lack of change of synapse number in the syntaxin-1A mutant mice could also be attributed to the compensation by syntaxin-1B, which may constitute the syntaxin-1B–syntabulin–KIF5B complex and mediate the axonal transport of AZ precursor carriers. This assumption is supported by our previous observations that depletion of syntabulin impairs the proper distribution and trafficking of both syntaxin-1A and -1B to neuronal processes and that syntabulin binds to a homologous membrane-proximal coiled-coil domain of syntaxin-1 (Su et al., 2004).
Our study highlights the possibility that KIF5B can mediate axonal transport independent of kinesin-1 light chain (KLC), which is supported by several lines of emerging evidence. First, a recent study (Glater et al., 2006) showed that Milton and Miro can form an essential protein transport complex with kinesin-1 heavy chain (KHC) for anterograde axonal transport of mitochondria in neurons. They further found that mitochondria are properly distributed in the klc mutant photoreceptor axons. Second, KIF5B was found to preferentially associate with microtubules in the initial segment of an axon, suggesting the directional information for the KIF5 driven-polarized axonal transport (Nakata and Hirokawa, 2003). In addition, it is the motor adaptor GRIP1 (glutamate receptor-interacting protein 1), but not KIF5B itself, that determines the traffic direction of KIF5B to dendrites (Setou et al., 2002).
Syntabulin–KIF5B-mediated axonal trafficking contributes to presynaptic assembly and activity-dependent plasticity
Kinesin motor protein-driven transport of protein packets from the TGN to nascent synapses plays a critical role in the rapid assembly of synapses (Ahmari et al., 2000; Sytnyk et al., 2002; Zhen and Jin, 2004; Ziv and Garner, 2004). Mutation in Drosophila khc impairs neurotransmitter release at nerve terminals (Gho et al., 1992) and results in dramatic axonal focal swellings that are packed with axonal transport cargos including vesicles, synaptic proteins, and mitochondria, consequently causing marked reductions in the density of presynaptic boutons (Hurd and Saxton, 1996). In Caenorhabditis elegans, the mutations in motor UNC-116/KHC and motor adaptors UNC-16 or UNC-14 altered the localization of cargos containing synaptic vesicle markers (Byrd et al., 2001; Sakamoto et al., 2005). In addition, a recent genetic study in C. elegans further provides in vivo evidence for functional dependence of AZ components in synaptic assembly and subsequent accumulation of SVs (Dai et al., 2006). In vertebrates, however, kif5B knock-out mice were embryonic lethal with a severe growth retardation at 9.5–11.5 days postcoitum, which prevents additional analysis of the role for KIF5B in neuronal development and functioning (Tanaka et al., 1998). Our current studies demonstrate that syntabulin loss of function in rat hippocampal neurons results in a remarkable reduction in both the density and size of SV clusters, a decreased density of the active synapses labeled with FM4-64 uptake along developing axons, and substantial impairment in neurotransmitter release. Our results provide cellular and physiological evidence that the syntaxin–syntabulin–KIF5B complex is candidate anterograde transport machinery essential for the assembly of functional synapses in developing neurons.
Long-term synaptic plasticity is represented at the cellular level by activity-dependent modulation of both the function and structure of synaptic connections for establishing neuronal circuitry during brain development (Katz and Shatz, 1996). The mechanism underlying the changes in the formation of new synaptic boutons is still far from understood. The long-lasting late stage of synaptic plasticity coupled with new synapse formation develops more slowly and requires both gene transcription and new protein synthesis (Bailey et al., 1996; Impey et al., 1996; Qi et al., 1996; Abel et al., 1997; Luscher et al., 2000; Kim et al., 2003; Udo et al., 2005). The synthesis and recruitment of N-cadherin are significantly elevated during the induction of the late phase of LTP in hippocampal slices. Impairment of cadherin adhesion blocks the early or late LTP (Tang et al., 1998; Bozdagi et al., 2000). In the case of developmentally regulated synaptic plasticity, it takes more than an accumulation of SVs to form a release site, which also requires the release machinery, voltage-dependent calcium channels, presynaptic scaffolding and adhesion molecules (Ahmari et al., 2000; Togashi et al., 2002; Altrock et al., 2003; Bamji et al., 2003; Dick et al., 2003; Ziv and Garner, 2004; Kittel et al., 2006). Thus, efficient axonal transport of these newly synthesized key components to nascent presynaptic boutons would be critical in response to neuronal activity. Our immunoisolation analysis shows that SV fusion machinery, presynaptic scaffolding protein Bassoon, and adhesion molecules N-cadherin and β-catenin are components of the syntabulin–KIF5B-linked membranous organelles. These results support the notion that this motor–adaptor complex delivers cargos carrying the molecular elements required for the induction of activity-dependent presynaptic plasticity. Furthermore, activity-induced recruitment of the AZ components and subsequent formation of presynaptic boutons robustly increased in the neurons overexpressing wild-type syntabulin and was almost abolished in the neurons expressing syntabulin dominant-negative mutant. Our findings elucidate a mechanism by which the syntabulin–KIF5B complex mediates axonal transport of the AZ components essential for constructing release sites. Thus, in addition to previously identified mechanisms during activity-dependent long-term synaptic plasticity in developing neurons: (1) activation of transcriptional factors, (2) new protein synthesis, and (3) reorganization of the actin filaments at synapses, our studies suggest a new pathway: regulation of microtubule-based axonal transport.
Despite our progress in identifying the new motor–adaptor machinery specific for AZ components, there are still a number of questions to be addressed. In particular, does syntabulin contribute to the dendritic trafficking of postsynaptic components? Are expression and function of the syntabulin–KIF5B complex regulated in response to synaptic activity? Although syntabulin loss of function reduces activity-dependent presynaptic plasticity primarily via impairing anterograde axonal transport of presynaptic cargos, the possibility that syntabulin is also directly involved in the synapse formation cannot be completely excluded under our experimental conditions. Given that syntaxin-1 is a neuronal protein widely distributed throughout neurons, identification of the sorting signals for the axon-targeted delivery of the AZ cargos would be critical in elucidating the polarized trafficking mechanisms in neurons.
Footnotes
This work was supported by the intramural research program of National Institute of Neurological Disorders and Stroke–National Institutes of Health (NIH) (Z.-H.S.). P.-Y.P. is a graduate student of NIH–Shanghai Jiaoton University Joint PhD Program in Neuroscience. We thank the following people for their help: Gong Chen, Ling-Gang Wu, Bai Lu, Jeffrey Diamond, Jens Rettig, and Dax Hoffman for their suggestions on functional studies; Jian-Sheng Kang and Miriam Leenders for their suggestions on live-cell imaging; Claudia Gerwin and Benjamin McNeil for their critical reading of this manuscript; Eckart Gundelfinger for EGFP-Bassoon (95-3938) and guinea pig polyclonal anti-Piccolo antibody; Alaa El-Husseini for synaptophysin-GFP; and Leon Lagnado for synaptophysin-mRFP.
All animal experimentation has been conducted in accordance with the Policies on the Use of Animals and Humans in Neuroscience Research and National Institutes of Health Animal Use Guidelines.
References
- Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- Ahmari SE, Buchanan J, Smith SJ. Assembly of presynaptic active zones from cytoplasmic transport packets. Nat Neurosci. 2000;3:445–451. doi: 10.1038/74814. [DOI] [PubMed] [Google Scholar]
- Allan VJ, Thompson HM, McNiven MA. Motoring around the Golgi. Nat Cell Biol. 2002;4:236–242. doi: 10.1038/ncb1002-e236. [DOI] [PubMed] [Google Scholar]
- Altrock WD, tom Dieck S, Sokolov M, Meyer AC, Sigler A, Brakebusch C, Fassler R, Richter K, Boeckers TM, Potschka H, Brandt C, Loscher W, Grimberg D, Dresbach T, Hempelmann A, Hassan H, Balschun D, Frey JU, Brandstatter JH, Garner CC, et al. Functional inactivation of a fraction of excitatory synapses in mice deficient for the active zone protein bassoon. Neuron. 2003;37:787–800. doi: 10.1016/s0896-6273(03)00088-6. [DOI] [PubMed] [Google Scholar]
- Antonova I, Arancio O, Trillat AC, Wang HG, Zablow L, Udo H, Kandel ER, Hawkins RD. Rapid increase in clusters of presynaptic proteins at onset of long-lasting potentiation. Science. 2001;294:1547–1550. doi: 10.1126/science.1066273. [DOI] [PubMed] [Google Scholar]
- Bailey CH, Kandel ER. Structural changes accompanying memory storage. Annu Rev Physiol. 1993;55:397–426. doi: 10.1146/annurev.ph.55.030193.002145. [DOI] [PubMed] [Google Scholar]
- Bailey CH, Bartsch D, Kandel ER. Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci USA. 1996;93:13445–13452. doi: 10.1073/pnas.93.24.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamji SX, Shimazu K, Kimes N, Huelsken J, Birchmeier W, Lu B, Reichardt LF. Role of beta-catenin in synaptic vesicle localization and presynaptic assembly. Neuron. 2003;40:719–731. doi: 10.1016/s0896-6273(03)00718-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Shan W, Tanaka H, Benson DL, Huntley GW. Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron. 2000;28:245–259. doi: 10.1016/s0896-6273(00)00100-8. [DOI] [PubMed] [Google Scholar]
- Bozdagi O, Valcin M, Poskanzer K, Tanaka H, Benson DL. Temporally distinct demands for classic cadherins in synapse formation and maturation. Mol Cell Neurosci. 2004;27:509–521. doi: 10.1016/j.mcn.2004.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradke F, Dotti CG. Membrane traffic in polarized neurons. Biochim Biophys Acta. 1998;1404:245–258. doi: 10.1016/s0167-4889(98)00060-3. [DOI] [PubMed] [Google Scholar]
- Burack MA, Silverman MA, Banker G. The role of selective transport in neuronal protein sorting. Neuron. 2000;26:465–472. doi: 10.1016/s0896-6273(00)81178-2. [DOI] [PubMed] [Google Scholar]
- Byrd DT, Kawasaki M, Walcoff M, Hisamoto N, Matsumoto K, Jin Y. UNC-16, a JNK-signaling scaffold protein, regulates vesicle transport in C. elegans . Neuron. 2001;32:787–800. doi: 10.1016/s0896-6273(01)00532-3. [DOI] [PubMed] [Google Scholar]
- Cai Q, Gerwin C, Sheng ZH. Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J Cell Biol. 2005;170:959–969. doi: 10.1083/jcb.200506042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillard O, Ben-AriY , Gaiarsa JL. Mechanisms of induction and expression of long-term depression at GABAergic synapses in the neonatal rat hippocampus. J Neurosci. 1999;19:7568–7577. doi: 10.1523/JNEUROSCI.19-17-07568.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Lovinger DM. Decreased frequency but not amplitude of quantal synaptic responses associated with expression of corticostriatal long-term depression. J Neurosci. 1997;17:8613–8620. doi: 10.1523/JNEUROSCI.17-21-08613.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colicos MA, Collins BE, Sailor MJ, Goda Y. Remodeling of synaptic actin induced by photoconductive stimulation. Cell. 2001;107:605–616. doi: 10.1016/s0092-8674(01)00579-7. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Bliss TV. Memories of NMDA receptors and LTP. Trends Neurosci. 1995;18:54–56. [PubMed] [Google Scholar]
- Dai Y, Taru H, Deken SL, Grill B, Ackley B, Nonet ML, Jin Y. SYD-2 Liprin-alpha organizes presynaptic active zone formation through ELKS. Nat Neurosci. 2006;9:1479–1487. doi: 10.1038/nn1808. [DOI] [PubMed] [Google Scholar]
- Dick O, tom Dieck S, Altrock W, Ammermuller J, Weiler R, Garner CC, Gundelfinger ED, Brandstatter JH. The presynaptic active zone protein bassoon is essential for photoreceptor ribbon synapse formation in the retina. Neuron. 2003;37:775–786. doi: 10.1016/s0896-6273(03)00086-2. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Lippincott-Schwartz J. Sorting and signaling at the Golgi complex. Cell. 2000;101:693–696. doi: 10.1016/s0092-8674(00)80881-8. [DOI] [PubMed] [Google Scholar]
- Dresbach T, Hempelmann A, Spilker C, tom Dieck S, Altrock WD, Zuschratter W, Garner CC, Gundelfinger ED. Functional regions of the presynaptic cytomatrix protein bassoon: significance for synaptic targeting and cytomatrix anchoring. Mol Cell Neurosci. 2003;23:279–291. doi: 10.1016/s1044-7431(03)00015-0. [DOI] [PubMed] [Google Scholar]
- Dresbach T, Torres V, Wittenmayer N, Altrock WD, Zamorano P, Zuschratter W, Nawrotzki R, Ziv NE, Garner CC, Gundelfinger ED. Assembly of active zone precursor vesicles: obligatory trafficking of presynaptic cytomatrix proteins bassoon and piccolo via a trans-Golgi compartment. J Biol Chem. 2006;281:6038–6047. doi: 10.1074/jbc.M508784200. [DOI] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron. 2000;27:57–69. doi: 10.1016/s0896-6273(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Fujiwara T, Mishima T, Kofuji T, Chiba T, Tanaka K, Yamamoto A, Akagawa K. Analysis of knock-out mice to determine the role of HPC-1/syntaxin 1A in expressing synaptic plasticity. J Neurosci. 2006;26:5767–5776. doi: 10.1523/JNEUROSCI.0289-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gho M, McDonald K, Ganetzky B, Saxton WM. Effects of kinesin mutations on neuronal functions. Science. 1992;258:313–316. doi: 10.1126/science.1384131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006;173:545–557. doi: 10.1083/jcb.200601067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LS, Philp AV. The road less traveled: emerging principles of kinesin motor utilization. Annu Rev Cell Dev Biol. 1999;15:141–183. doi: 10.1146/annurev.cellbio.15.1.141. [DOI] [PubMed] [Google Scholar]
- Goldstein LS, Yang Z. Microtubule-based transport systems in neurons: the roles of kinesins and dyneins. Annu Rev Neurosci. 2000;23:39–71. doi: 10.1146/annurev.neuro.23.1.39. [DOI] [PubMed] [Google Scholar]
- Hagler DJ, Jr, Goda Y. Properties of synchronous and asynchronous release during pulse train depression in cultured hippocampal neurons. J Neurophysiol. 2001;85:2324–2334. doi: 10.1152/jn.2001.85.6.2324. [DOI] [PubMed] [Google Scholar]
- Hansel C, Linden DJ, D'Angelo E. Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat Neurosci. 2001;4:467–475. doi: 10.1038/87419. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 1998;279:519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- Horton AC, Ehlers MD. Secretory trafficking in neuronal dendrites. Nat Cell Biol. 2004;6:585–591. doi: 10.1038/ncb0704-585. [DOI] [PubMed] [Google Scholar]
- Hua JY, Smith SJ. Neural activity and the dynamics of central nervous system development. Nat Neurosci. 2004;7:327–332. doi: 10.1038/nn1218. [DOI] [PubMed] [Google Scholar]
- Huntley GW, Benson DL, Colman DR. Structural remodeling of the synapse in response to physiological activity. Cell. 2002;108:1–4. doi: 10.1016/s0092-8674(01)00631-6. [DOI] [PubMed] [Google Scholar]
- Hurd DD, Saxton WM. Kinesin mutations cause motor neuron disease phenotypes by disrupting fast axonal transport in Drosophila . Genetics. 1996;144:1075–1085. doi: 10.1093/genetics/144.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, Mark M, Villacres EC, Poser S, Chavkin C, Storm DR. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron. 1996;16:973–982. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- Katz LC, Crowley JC. Development of cortical circuits: lessons from ocular dominance columns. Nat Rev Neurosci. 2002;3:34–42. doi: 10.1038/nrn703. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kim JH, Udo H, Li HL, Youn TY, Chen M, Kandel ER, Bailey CH. Presynaptic activation of silent synapses and growth of new synapses contribute to intermediate and long-term facilitation in Aplysia . Neuron. 2003;40:151–165. doi: 10.1016/s0896-6273(03)00595-6. [DOI] [PubMed] [Google Scholar]
- Kittel RJ, Wichmann C, Rasse TM, Fouquet W, Schmidt M, Schmid A, Wagh DA, Pawlu C, Kellner RR, Willig KI, Hell SW, Buchner E, Heckmann M, Sigrist SJ. Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science. 2006;312:1051–1054. doi: 10.1126/science.1126308. [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Luscher C, Nicoll RA, Malenka RC, Muller D. Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat Neurosci. 2000;3:545–550. doi: 10.1038/75714. [DOI] [PubMed] [Google Scholar]
- Manning BD, Snyder M. Drivers and passengers wanted! The role of kinesin-associated proteins. Trends Cell Biol. 2000;10:281–289. doi: 10.1016/s0962-8924(00)01774-8. [DOI] [PubMed] [Google Scholar]
- Nakata T, Hirokawa N. Microtubules provide directional cues for polarized axonal transport through interaction with kinesin motor head. J Cell Biol. 2003;162:1045–1055. doi: 10.1083/jcb.200302175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata T, Terada S, Hirokawa N. Visualization of the dynamics of synaptic vesicle and plasma membrane proteins in living axons. J Cell Biol. 1998;140:659–674. doi: 10.1083/jcb.140.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature. 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Qi M, Zhuo M, Skalhegg BS, Brandon EP, Kandel ER, McKnight GS, Idzerda RL. Impaired hippocampal plasticity in mice lacking the Cbeta1 catalytic subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1996;93:1571–1576. doi: 10.1073/pnas.93.4.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos J, Kelly RB. Preassembly and transport of nerve terminals: a new concept of axonal transport. Nat Neurosci. 2000;3:415–417. doi: 10.1038/74773. [DOI] [PubMed] [Google Scholar]
- Sakamoto R, Byrd DT, Brown HM, Hisamoto N, Matsumoto K, Jin Y. The Caenorhabditis elegans UNC-14 RUN domain protein binds to the kinesin-1 and UNC-16 complex and regulates synaptic vesicle localization. Mol Biol Cell. 2005;16:483–496. doi: 10.1091/mbc.E04-07-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampo B, Kaech S, Kunz S, Banker G. Two distinct mechanisms target membrane proteins to the axonal surface. Neuron. 2003;37:611–624. doi: 10.1016/s0896-6273(03)00058-8. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Can molecules explain long-term potentiation? Nat Neurosci. 1999;2:597–604. doi: 10.1038/10154. [DOI] [PubMed] [Google Scholar]
- Setou M, Seog DH, Tanaka Y, Kanai Y, Takei Y, Kawagishi M, Hirokawa N. Glutamate-receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature. 2002;417:83–87. doi: 10.1038/nature743. [DOI] [PubMed] [Google Scholar]
- Shapira M, Zhai RG, Dresbach T, Bresler T, Torres VI, Gundelfinger ED, Ziv NE, Garner CC. Unitary assembly of presynaptic active zones from Piccolo-Bassoon transport vesicles. Neuron. 2003;38:237–252. doi: 10.1016/s0896-6273(03)00207-1. [DOI] [PubMed] [Google Scholar]
- Shen W, Wu B, Zhang Z, Dou Y, Rao ZR, Chen YR, Duan S. Activity-induced rapid synaptic maturation mediated by presynaptic cdc42 signaling. Neuron. 2006;50:401–414. doi: 10.1016/j.neuron.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Su Q, Cai Q, Gerwin C, Smith CL, Sheng ZH. Syntabulin is a microtubule-associated protein implicated in syntaxin transport in neurons. Nat Cell Biol. 2004;6:941–953. doi: 10.1038/ncb1169. [DOI] [PubMed] [Google Scholar]
- Sytnyk V, Leshchyns'ka I, Delling M, Dityateva G, Dityatev A, Schachner M. Neural cell adhesion molecule promotes accumulation of TGN organelles at sites of neuron-to-neuron contacts. J Cell Biol. 2002;159:649–661. doi: 10.1083/jcb.200205098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93:1147–1158. doi: 10.1016/s0092-8674(00)81459-2. [DOI] [PubMed] [Google Scholar]
- Tang L, Hung CP, Schuman EM. A role for the cadherin family of cell adhesion molecules in hippocampal long-term potentiation. Neuron. 1998;20:1165–1175. doi: 10.1016/s0896-6273(00)80497-3. [DOI] [PubMed] [Google Scholar]
- Togashi H, Abe K, Mizoguchi A, Takaoka K, Chisaka O, Takeichi M. Cadherin regulates dendritic spine morphogenesis. Neuron. 2002;35:77–89. doi: 10.1016/s0896-6273(02)00748-1. [DOI] [PubMed] [Google Scholar]
- tom Dieck S, Altrock WD, Kessels MM, Qualmann B, Regus H, Brauner D, Fejtova A, Bracko O, Gundelfinger ED, Brandstatter JH. Molecular dissection of the photoreceptor ribbon synapse: physical interaction of Bassoon and RIBEYE is essential for the assembly of the ribbon complex. J Cell Biol. 2005;168:825–836. doi: 10.1083/jcb.200408157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udo H, Jin I, Kim JH, Li HL, Youn T, Hawkins RD, Kandel ER, Bailey CH. Serotonin-induced regulation of the actin network for learning-related synaptic growth requires Cdc42, N-WASP, and PAK in Aplysia sensory neurons. Neuron. 2005;45:887–901. doi: 10.1016/j.neuron.2005.01.044. [DOI] [PubMed] [Google Scholar]
- Wang HG, Lu FM, Jin I, Udo H, Kandel ER, de Vente J, Walter U, Lohmann SM, Hawkins RD, Antonova I. Presynaptic and postsynaptic roles of NO, cGK, and RhoA in long-lasting potentiation and aggregation of synaptic proteins. Neuron. 2005;45:389–403. doi: 10.1016/j.neuron.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW. Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nat Neurosci. 2001;4:151–158. doi: 10.1038/83976. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG. Probing fundamental aspects of synaptic transmission with strontium. J Neurosci. 2000;20:4414–4422. doi: 10.1523/JNEUROSCI.20-12-04414.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Qi J, Chen G. Actin-dependent activation of presynaptic silent synapses contributes to long-term synaptic plasticity in developing hippocampal neurons. J Neurosci. 2006;26:8137–8147. doi: 10.1523/JNEUROSCI.1183-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekawa Y, Harada A, Okada Y, Funakoshi T, Kanai Y, Takei Y, Terada S, Noda T, Hirokawa N. Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J Cell Biol. 1998;141:431–441. doi: 10.1083/jcb.141.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai RG, Bellen HJ. Hauling t-SNAREs on the microtubule highway. Nat Cell Biol. 2004;6:918–919. doi: 10.1038/ncb1004-918. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Vardinon-Friedman H, Cases-Langhoff C, Becker B, Gundelfinger ED, Ziv NE, Garner CC. Assembling the presynaptic active zone: a characterization of an active one precursor vesicle. Neuron. 2001;29:131–143. doi: 10.1016/s0896-6273(01)00185-4. [DOI] [PubMed] [Google Scholar]
- Zhang W, Benson DL. Stages of synapse development defined by dependence on F-actin. J Neurosci. 2001;21:5169–5181. doi: 10.1523/JNEUROSCI.21-14-05169.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, Yang HW, Terada S, Nakata T, Takei Y, Saito M, Tsuji S, Hayashi Y, Hirokawa N. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001;105:587–597. doi: 10.1016/s0092-8674(01)00363-4. [DOI] [PubMed] [Google Scholar]
- Zhen M, Jin Y. Presynaptic terminal differentiation: transport and assembly. Curr Opin Neurobiol. 2004;14:280–287. doi: 10.1016/j.conb.2004.05.013. [DOI] [PubMed] [Google Scholar]
- Ziv NE, Garner CC. Cellular and molecular mechanisms of presynaptic assembly. Nat Rev Neurosci. 2004;5:385–399. doi: 10.1038/nrn1370. [DOI] [PubMed] [Google Scholar]