

Aberrant PP2Cδ inhibits p300-mediated p53 acetylation and activation via the ATM/BRCA1 pathway, leading to cancer progression.
Abstract
Although nuclear type 2C protein phosphatase (PP2Cδ) has been demonstrated to be pro-oncogenic with an important role in tumorigenesis, the underlying mechanisms that link aberrant PP2Cδ levels with cancer development remain elusive. Here, we found that aberrant PP2Cδ activity decreases p53 acetylation and its transcriptional activity and suppresses doxorubicin-induced cell apoptosis. Mechanistically, we show that BRCA1 facilitates p300-mediated p53 acetylation by complexing with these two proteins and that S1423/1524 phosphorylation is indispensable for this regulatory process. PP2Cδ, via dephosphorylation of ATM, suppresses DNA damage–induced BRCA1 phosphorylation, leading to inhibition of p300-mediated p53 acetylation. Furthermore, PP2Cδ levels correlate with histological grade and are inversely associated with BRCA1 phosphorylation and p53 acetylation in breast cancer specimens. C23, our newly developed PP2Cδ inhibitor, promotes the anticancer effect of doxorubicin in MCF-7 xenograft–bearing nude mice. Together, our data indicate that PP2Cδ impairs p53 acetylation and DNA damage response by compromising BRCA1 function.
INTRODUCTION
The serine-threonine protein phosphatase PP2Cδ (also known as WIP1 or PPM1D) is a nuclear type 2C protein phosphatase (PP2C) that is overexpressed and amplified in many types of cancers such as breast cancer, ovarian clear cell adenocarcinoma, gastric carcinoma, and pancreatic adenocarcinoma (1). PP2Cδ is unique in that its transcription is induced in response to DNA damaging agents in a p53-dependent manner. It dephosphorylates and inactivates several proteins critical for cellular stress responses, including p38 mitogen-activated protein kinase (MAPK) (2), Chk1 (3), Chk2 (4), ataxia telangiectasia mutated (ATM) (5), and p53 (3). As a negative regulator of these signaling proteins, PP2Cδ acts, together with the corresponding upstream kinases, to accurately control the magnitude and duration of their phosphorylation and activity.
PP2Cδ has been demonstrated to have clear oncogenic properties and to play an important role in tumorigenesis (6). It is involved in the regulation of several key cellular processes, such as cell cycle and apoptosis, which are vital for tumor development and progression. Inappropriate activation of PP2Cδ can inactivate p53 and RB pathways and therefore results in the stimulation of cell cycle and tumorigenesis (3). The overexpression of PP2Cδ increases mammary cell transformation in a breast tumor–susceptible animal model (7). Conversely, PP2Cδ−/− mice show a lower incidence of spontaneously occurring cancers (8) and resistance to oncogene-induced transformation (7, 9). For example, PP2Cδ deficiency notably postpones the onset of Myc-induced lymphomas in an ATM- and p53-dependent manner (9). Therefore, PP2Cδ is an attractive drug target for the treatment of cancers, and inhibition of its expression or activity could be a new vital strategy for therapeutic intervention to halt the progression of several different cancers.
The tumor suppressor p53 is a crucial transcription factor in cellular stress response pathways (10). In response to genotoxic agents, such as ultraviolet (UV) light, γ-irradiation, chemical carcinogens, and chemotherapeutic agents, p53 is stabilized and activated as a transcriptional regulator. The activated p53 primarily induces expression of genes crucial for cell growth arrest and apoptosis (11), thus preventing tumor initiation and/or progression (12). Although the exact mechanisms of p53 activation are not completely understood, it is generally believed that they need posttranslational modifications such as phosphorylation, acetylation, and methylation of the p53 polypeptide (13, 14). Among these, acetylation of p53 is the most important reversible enzymatic process that occurs upon genotoxic stress and is critical for p53 stability, sequence-specific DNA binding, and transcriptional activity (15). p53 acetylation at several C-terminal Lys residues is mainly mediated by the p300 and CBP acetyltransferases in vivo (16). Without a functional p53 pathway, the cell could potentially progress through DNA replication but could not stop the cell cycle in the event of DNA damage, thereby leading to potentially favorable mutations for the cancer cell to survive as well as tissue malignancies. Thus, loss or weakening of p53 function (even in a twofold modest alteration) increases cancer risk and progression. For instance, loss of one p53 allele in mice (p53+/−) leads to early development of tumors (17). In view of the close relationship between p53 and PP2Cδ, two important tumor-associated proteins, more in-depth investigation is needed to further elucidate the signaling regulatory mechanism between p53 function and aberrant expression of PP2Cδ. Further research on the regulatory effect of PP2Cδ on p53 acetylation in response to DNA damage stress will contribute to a greater understanding of mechanisms that regulate cell fate in addition to developing therapies for refractory or chemoresistant tumors.
Here, we demonstrate a novel mechanism of p53 inactivation by PP2Cδ. We present evidence to support the notion that BRCA1 facilitates p300-mediated p53 acetylation and activation by complexing with these two proteins and that S1423/1524 phosphorylation is indispensable for this regulatory process. Aberrant PP2Cδ activity, by directly dephosphorylating ATM, suppresses DNA damage–induced BRCA1 phosphorylation, leading to inhibition of p300-mediated p53 acetylation and activation. We also observed that high PP2Cδ expression in human breast cancer tissues correlates with more advanced histological grade and is inversely associated with BRCA1 phosphorylation and p53 acetylation. Knockdown or inhibition of PP2Cδ potentiates the antineoplastic effects of DNA damage–based treatment in vitro and in vivo, proposing exciting opportunities for combinatorial therapy.
RESULTS
Overexpression of PP2Cδ attenuates ATM and BRCA1 phosphorylation, p53 acetylation, and DNA damage–induced apoptosis in MCF-7 cells
PP2Cδ has been demonstrated to have clear oncogenic properties and to play an important role in tumorigenesis (6). We first examined the potential effect of PP2Cδ overexpression on BRCA1 phosphorylation and p53 acetylation, which are important in apoptosis, cell cycle, and tumorigenesis. As shown in the immunoblot depicted in Fig. 1A, the levels of PP2Cδ protein are substantially higher in MCF-7 breast cancer cells than those of human normal mammary epithelial cell line (MCF-10A), which is accompanied by decreased phosphorylation of BRCA1 and its upstream kinase ATM, as well as by p53 acetylation. These results imply that overexpression of PP2Cδ in cancer cells may compromise BRCA1 and p53 function, thereby resulting in inhibition of apoptosis. To characterize the PP2Cδ-mediated protective effect on DNA damage/p53–induced apoptosis, we overexpressed PP2Cδ in MCF-10A cells and assessed the effects of PP2Cδ overexpression on DNA damage–induced apoptosis. Cells were exposed to a genotoxic drug, doxorubicin (Dox) (0.1 μM), after the transfection. Twenty-four hours after drug treatment, cells were then collected for apoptotic cell analysis. As shown in Fig. 1B, PP2Cδ overexpression considerably reduces the percentage of dead cells under the treatment of Dox. The transfection of human wild-type (WT) PP2Cδ expression plasmid (hWIP1 FLAG) was effective to overexpress PP2Cδ in MCF-10A cells, as confirmed by Western blot analysis (Fig. 1C). Intriguingly, PP2Cδ overexpression markedly suppresses phosphorylation of BRCA1 and ATM, as well as p53 acetylation. Furthermore, the death-reducing effect of PP2Cδ is accompanied by a significant reduction in cleavage of procaspase-3 to the active caspase-3 (Fig. 1D).
Fig. 1. PP2Cδ negatively regulates DNA damage–induced p53 acetylation and apoptosis.
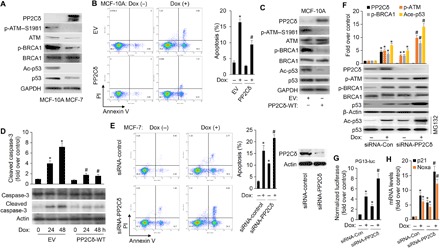
(A) Western blot analysis of PP2Cδ, phosphorylation and protein levels of ATM and BRCA1, and acetylation and protein levels of p53 in MCF-10A and MCF-7 cells. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (B) Human normal mammary epithelial cells (MCF-10A) transfected with empty vector (EV) or plasmid expressing wild-type (WT) PP2Cδ (PP2Cδ-WT) were exposed to Dox (0.1 μM) for 24 hours. Cells were then collected and processed for apoptotic cell analysis using flow cytometry after annexin V–fluorescein isothiocyanate (FITC)/propidium iodide (PI) staining. An average from three replicates for each treatment (±SD) is shown. *P < 0.05 versus EV/Dox (−); #P < 0.05 versus EV/Dox (+). (C) MCF-10A cells were transfected with EV or plasmid expressing WT PP2Cδ. Cells were lysed and subjected to Western blot analysis with the indicated antibodies. (D) MCF-10A cells transfected with EV or plasmid expressing WT PP2Cδ were exposed to Dox (0.1 μM) for 24 and 48 hours. Whole-cell lysates were collected, resolved by SDS–polyacrylamide gel electrophoresis (PAGE), and immunoblotted with antibodies specific for caspase-3 and cleaved caspase-3, which is an apoptotic indicator. Equal loading was confirmed by β-actin immunoblot. The bar graphs above are densitometry analyses of the bands. Data presented are mean ± SD from three independent experiments, with nontreated controls set to 1. *P < 0.05 versus EV/Dox (−); #P < 0.05 versus their corresponding EV/Dox (+). (E) MCF-7 cells were transfected with control siRNA or PP2Cδ siRNA for 24 hours, followed by incubation with Dox (1.0 μM) for 48 hours. Cells were then collected and processed for apoptotic cell analysis using flow cytometry after annexin V–FITC/PI staining. The down-regulation of PP2Cδ expression by siRNA was confirmed by Western blot analysis (right). An average from three replicates for each treatment (±SD) is shown. *P < 0.05 versus siRNA-control/Dox (−); #P < 0.05 versus siRNA-control/Dox (+). (F) MCF-7 cells were transfected with control siRNA or PP2Cδ siRNA for 24 hours, followed by incubation with vehicle or Dox (0.5 μM) for 24 hours. Cell lysates underwent immunoblotting for the proteins as indicated. *P < 0.05 versus siRNA-control/Dox (−); #P < 0.05 versus siRNA-control/Dox (+). (G) MCF-7 cells were transiently transfected with 0.5 μg of pG13-LUC reporter plasmid. About 6 hours after transfection, cells were treated as in (F). Luciferase activity was determined from the transfected cell extracts. Values (mean ± SD) are expressed as fold over untreated control. *P < 0.05 versus siRNA-control/Dox (−); #P < 0.05 versus siRNA-control/Dox (+). (H) The p21 and Noxa mRNA for each treatment were analyzed by reverse transcription quantitative PCR (RT-qPCR). All mRNAs are normalized to PUM1 and presented as fold (mean ± SD) over untreated cells based on three experiments. *P < 0.05 versus siRNA-control/Dox (−); #P < 0.05 versus siRNA-control/Dox (+).
To further substantiate the protective role of PP2Cδ against DNA damage–induced apoptosis, we used PP2Cδ small interfering RNAs (siRNAs) to knock down PP2Cδ expression and our newly developed PP2Cδ inhibitor C23 (18) in MCF-7 cells. As indicated in Fig. 1E and fig. S1A, PP2Cδ knockdown or C23 significantly promotes the basal and Dox-induced cancer cell apoptosis. In addition to apoptosis, PP2Cδ knockdown with siRNA potently stimulates other p53 pathways such as cell cycle arrest and senescence (fig. S1, B and C). To address the potential mechanisms of the augmented apoptosis in PP2Cδ knockdown, we examined the function of apoptosis-related gene p53. PP2Cδ knockdown markedly increases p53 acetylation and its transcriptional regulation activity, as indicated by PG13-luc p53 reporter gene assays and transcription of its target genes p21 and Noxa, compared with levels in controls. This increase in p53 activity is coincident with an increase in BRCA1 and ATM phosphorylation (Fig. 1, F to H). Together, these results demonstrate that PP2Cδ suppresses p53 transcriptional activity and basal and DNA damage–induced apoptosis, which may have a causal relationship with ATM/BRCA1.
BRCA1 facilitates p53 acetylation by p300 in vivo
Our studies suggested that the decreased BRCA1 phosphorylation coincided with reduced p53 acetylation levels (Fig. 1). To delineate the role of BRCA1 in the regulation of p53 acetylation, MCF-7 breast cancer cells were transfected with BRCA1 siRNA, followed by UV treatment. We use UV irradiation to induce DNA damage because it has been shown to elicit robust p53 acetylation in MCF-7 cells (Fig. 2A). BRCA1 knockdown markedly reduced both basal and UV-induced p53 acetylation, indicating a critical role of BRCA1 in the regulation of p53 acetylation. In addition to UV, BRCA1 is required for acetylation of p53 induced by other major genotoxin stress such as DNA cross-linker cisplatin and topoisomerase I inhibitor topotecan (fig. S2, A and B). Prime candidates for the p53 acetylases are p300 and its family member CBP (19). However, how BRCA1 enhances p53 acetylation by p300 is still unknown. To address this, we first define the protein interaction between BRCA1, p300, and p53 by undertaking immunoprecipitation-immunoblotting studies. Both p53 and p300 were coprecipitated with BRCA1 (Fig. 2B, lane 2). Reciprocally, both BRCA1 and p300 were found in p53 immunoprecipitates (Fig. 2B, lane 3). Together, these observations suggest that BRCA1 interacts with both p300 and p53. These data are also supported by results reported by Pao et al. (20) and Zhang et al. (21).
Fig. 2. BRCA1 facilitates p300-dependent p53 acetylation.
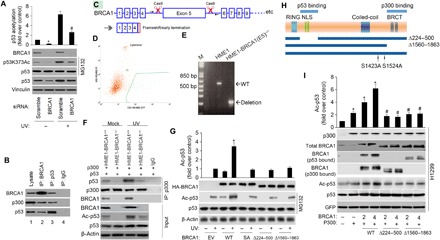
(A) p53 acetylation was decreased upon BRCA1 knockdown. MCF-7 cells were transfected with scramble or BRCA1 siRNA, followed by UV treatment (20 J/m2 for an 8-hour recovery). p53 protein level was normalized by MG132 (20 μM). p53 acetylation level on Lys373 and levels of BRCA1, p53, and vinculin were detected with specific antibodies. The data represent mean ± SD from three separate experiments. *P < 0.05 versus control; #P < 0.05 versus UV + scramble siRNA. (B) Coimmunoprecipitation of BRCA1, p300, and p53 in MCF-7 cells transfected with the WT BRCA1 expression plasmid (WT BRCA1). Aliquots of cellular lysate were subjected to immunoprecipitations using anti-BRCA1, p53 antibodies, or control immunoglobulin G (IgG), followed by immunoblotting with antibodies against BRCA1, p300, or p53. (C) Schematic outline of CRISPR-Cas9 genome editing design to knock out BRCA1 exon 5. sgRNA1/2 specifically bind the introns before and after exon 5. The arrows represent location of primers for deletion PCR. Deletion of exon 5 results in frameshift, with early translational termination, mimicking a known BRCA1 pathogenic mutation. (D) Sorting for Cas9/guide transfected (GFP+) cells. (E) PCR confirms the deletion of BRCA1 exon 5 in the hTERT-HME1-BRCA1 (E5)−/− line. bp, base pairs. (F) BRCA1 KO decreases basal and UV-induced p300’s binding to total p53 and p53 acetylation. Twenty-four hours after cotransfection of the indicated plasmids, cells were treated with or without UV. Aliquots of cellular lysate were subjected to immunoprecipitations (IP) using anti-p300 antibody or control IgG, followed by immunoblotting with antibodies against p53 or p300. BRCA1, p53, and Ac-p53 were measured by immunoblotting. (G) HME1-BRCA1−/− cells were transfected with 2 μg of hemagglutinin (HA)–tagged WT, S1423A (SA), ∆224-500, and ∆1560-1863 mutant. Twenty-four hours after transfection of the indicated plasmids, cells were treated with or without UV. Cell extracts were prepared and underwent immunoblotting for the proteins as indicated, and signal intensity was quantified. Steady-state levels of transfected protein were determined by Western blot analysis using antibody against the HA epitope tag (α-HA). The data represent mean ± SD from three separate experiments. *P < 0.05 versus UV/EV. (H) Schematic diagram of BRCA1 deletion mutants used in (D). (I) H1299 cells were transfected with p53 WT and internal control GFP (lane 1) or cotransfected with c-myc–tagged p300 (lane 2) or c-myc–tagged p300 and the indicated amounts of BRCA1 WT (lanes 3 and 4), ∆224-500 mutant (lanes 5 and 6), and ∆1560-1863 mutant (lanes 7 and 8). Cell extracts were prepared (36 hours after transfection), and the levels of acetylation and total p53 protein were determined by Western blotting as described in Materials and Methods. p300 levels were determined by anti-p300 (RW128). The data represent mean ± SD from three separate experiments. *P < 0.05 versus control; #P < 0.05 versus corresponding controls in WT.
To further substantiate the role of BRCA1 in facilitating p300-mediated p53 acetylation, we generated a homozygous BRCA1 deletion hTERT-HME1 line by targeting exon 5 of the BRCA1 gene using the CRISPR-Cas9 gene editing system. Deletion of exon 5 results in frameshift and early translational termination, simulating a known BRCA1 pathogenic mutation (Fig. 2C). hTERT-HME1 cells were transfected with double Cas9/green fluorescent protein (GFP)–guide RNA (gRNA) plasmids carrying single-guide RNAs (sgRNAs) targeting exon 5 of the BRCA1 gene. GFP-positive cells were FACS (fluorescence-activated cell sorting)–sorted (Fig. 2D), and individual cell clones were expanded and characterized. The BRCA1-targeted regions were analyzed by polymerase chain reaction (PCR) to show that two alleles were deleted (Fig. 2E). Primers surrounding exon 5 were designed, and the genetic region amplified by PCR was analyzed by Sanger sequencing. The dual allelic BRCA1 exon 5 deletion was confirmed (fig. S2, C and D). Immunoblotting results also validated the BRCA1 knockout (KO) in the hTERT-HME1-BRCA1−/− line (Fig. 2F). As shown in Fig. 2F, BRCA1 KO notably decreases basal and UV-induced p53-p300 association and p53 acetylation compared to the levels in parental hTERT-HME1-BRCA1+/+ cells. Thus, the presence of BRCA1 is important to stimulate p53 interaction with p300 and its subsequent acetylation. Reintroducing WT BRCA1, but not a S1423A mutant (SA), ∆224-500, and ∆1560-1863 mutant in HME1-BRCA1−/− cells, restores the basal and UV-induced p53-p300 association and p53 acetylation (Fig. 2G). Together, these results suggest that BRCA1 plays a critical role in facilitating p300-mediated p53 acetylation.
To further study how BRCA1 promotes p53 acetylation, we analyzed different BRCA1 mutants with specific functional domains deleted (Fig. 2H). Explicitly, we tested BRCA1 mutants that are deficient in p53 binding (∆224-500) or p300 binding (∆1560-1863). As illustrated in Fig. 2I, after transfection into H1299 cells, all these BRCA1 variants were expressed (second panel). Nevertheless, when compared to WT BRCA1, both the p53-binding mutant (∆224-500) and the p300-binding mutant (∆1560-1863) were defective as they fail to promote p53 acetylation even when expressed at a higher level. These results demonstrate that physical combination with both p53 and p300 is essential for full activity of BRCA1 to promote p53 acetylation. Together, we conclude that BRCA1 can actively facilitate p300-induced p53 acetylation and that this activity needs physical binding to both p300 and p53 in vivo.
Phosphorylation of BRCA1 at Ser1423/1524 positively regulates p300-mediated p53 acetylation and activation
To address whether BRCA1 phosphorylation enhances p53 acetylation, we determined that BRCA1 proteins with different phosphorylation statuses could directly affect p300/CBP-mediated p53 acetylation in vitro. As shown in Fig. 3A, although recombinant WT BRCA1 had no effect on p300 autoacetylation, it efficiently increased p53 acetylation in a dose-dependent manner. The phosphomimicking mutant BRCA1 (S1423/1524D) had a stronger stimulating effect on p53 acetylation than WT BRCA1. In contrast, phosphorylation-deficient mutants (S1423/1524A) had no effect on p300-induced p53 acetylation. These observations suggest that the ability of BRCA1 to facilitate p53 acetylation is attributed to its phosphorylation levels. We hypothesize that phosphorylation induces a conformational change in BRCA1, facilitating interactions of p300 with p53. To further confirm that S1423/1524 phosphorylation directly affects the influence of BRCA1 on p300-mediated p53 acetylation and activation, we cotransfected H1299 cells with p53, p300, and various BRCA1 expression plasmids, including WT BRCA1 and S1423/1524 phosphorylation mutants (S1423/1524A or S1423/1524D). WT BRCA1 and S1423/1524D promote p53 acetylation (Fig. 3B) and its transcriptional regulation activity, according to our PG13-luc p53 reporter gene assays (Fig. 3C) and transcription of its target genes p21 and Noxa (Fig. 3D), more effectively than S1423/1524A. These results suggest that S1423/1524 phosphorylation is indispensable for the regulatory effect of BRCA1 on p300-mediated p53 acetylation and activation. To further confirm this point, we next manipulated the expression and function of BRCA1 and p300 and assayed for effects on apoptosis in culture. Knockdown of BRCA1 and p300 partially block the induction of apoptosis seen with PP2Cδ inhibition in MCF-7 cells (fig. S3A). Overexpression of a BRCA1 phosphomimetic allele (S1423D/1524D) that does not rely on ATM overcomes the suppression of apoptosis by PP2Cδ overexpression in MCF-10A cells (fig. S3B). These results further substantiate the role of BRCA1 phosphorylation and p300 in p53 activation and apoptosis induced by PP2Cδ inhibition.
Fig. 3. Phosphorylation of BRCA1 Ser1423/1524 promotes p53 acetylation and activation mediated by p300 in vivo.
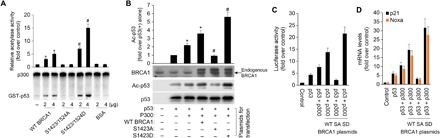
(A) Glutathione S-transferase (GST)–p53 was acetylated by recombinant p300 in the presence of the indicated amounts of purified BRCA1 or bovine serum albumin (BSA) and analyzed by SDS ± PAGE followed by autoradiography. The intensity of the acetylated GST-p53 was quantified by phosphoimager analysis and plotted. The intensity of acetylated GST-p53 in the absence of BRCA1 or BSA was set to 1. The data represent mean ± SD from three separate experiments. *P < 0.05 versus control; #P < 0.05 versus WT BRCA1. (B) H1299 cells were transfected with plasmids encoding p53 (0.3 μg), p300 (0.5 μg), and/or WT BRCA1, BRCA1 S1423A, and BRCA1 S1423D (1 μg) as indicated at the bottom. Cell lysates were prepared 36 hours after transfection for Western blot analysis; 200 μg of proteins was loaded onto a 10% SDS gel. (C) As indicated, plasmids encoding no protein as a control (1 μg; control), p53 (50 ng) alone, or with p300 (0.15 μg) or with BRCA1 (WT, S1423A, or S1423D), and with a pG13-LUC plasmid (0.2 μg) were introduced into H1299 cells by using Lipofectamine. Forty-eight hours after transfection, cells were harvested for luciferase assays. Each column represents the mean data of three experiments. (D) The p21 and Noxa mRNAs for each treatment were analyzed by RT-qPCR. All mRNAs are normalized to PUM1 and presented as fold (mean ± SD) over untreated cells based on three experiments. *P < 0.05 versus p53(+); #P < 0.05 versus p53(+)/p300(+)/WT BRCA1(+).
ATM partially mediates DNA damage–induced BRCA1 phosphorylation in human mammary epithelial cells
To explore the potential role of ATM in DNA damage–induced BRCA1 phosphorylation, we examined whether ATM inhibition affects BRCA1 phosphorylation in response to exposure of human mammary epithelial cells to UV. We used siRNA-mediated ATM depletion and the pharmaceutical inhibitor KU-55933, which specifically suppresses the ATM kinase activity at a very low micromolar concentration (22). As expected, ATM inhibition significantly reduced UV-triggered phosphorylation of BRCA1 and p53 acetylation in MCF-10A cells (Fig. 4, A and B). This result is also supported by previous studies showing that ATM is required for DNA damage–induced BRCA1 phosphorylation (23, 24). In addition, cellular inhibition of ATM by KU-55933 prevents efficient UV-induced phosphorylation of BRCA1 and acetylation of p53 (Fig. 4C) in MCF-7 cells. Most notably, inhibition of PP2Cδ by C23 further promotes UV-induced BRCA1 phosphorylation and p53 acetylation. Collectively, these observations demonstrate that ATM mediates DNA damage–induced BRCA1 phosphorylation and p53 function in human mammary epithelial cells and that PP2Cδ may negatively regulate this process.
Fig. 4. ATM is partially responsible for DNA damage–induced BRCA1 phosphorylation.
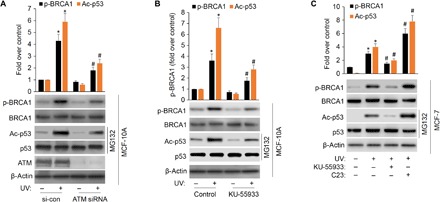
(A) MCF-10A cells were transfected with scramble or ATM siRNA, followed by UV treatment (20 J/m2 for an 8-hour recovery). BRCA1 phosphorylation level on Ser1423 and levels of BRCA1, p53 acetylation, ATM, and actin were detected with specific antibodies. The data represent mean ± SD from three separate experiments. *P < 0.05 versus control; #P < 0.05 versus UV+ scramble siRNA. (B) MCF-10A cells were pretreated with 10 μM KU-55933 for 1 hour, followed by UV treatment. BRCA1 phosphorylation and p53 acetylation were detected with specific antibodies. The data represent mean ± SD from three separate experiments. *P < 0.05 versus control; #P < 0.05 versus UV alone. (C) MCF-7 cells were pretreated with 10 μM KU-55933 or 2.5 μM C23 for 1 hour, followed by UV treatment. p53 protein level was normalized by MG132 (20 μM). Treated cells were lysed and Western blotted against indicated antibodies. The data represent mean ± SD from three separate experiments. *P < 0.05 versus control; #P < 0.05 versus UV alone.
PP2Cδ dephosphorylates ATM in human mammary epithelial cells
We next questioned whether PP2Cδ directly dephosphorylates BRCA1 or indirectly dephosphorylates it by directly dephosphorylating ATM. To show that PP2Cδ dephosphorylates the intact ATM protein in vitro, we carried out in vitro phosphatase reactions on immune-purified ATM and demonstrated that increasing PP2Cδ results in decreased Ser1981 phosphorylation. In the absence of Mg2+, the Mg2+-dependent PP2Cδ cannot dephosphorylate ATM (Fig. 5A). We next performed a second set of in vitro phosphatase assay to substantiate/determine whether PP2Cδ dephosphorylates ATM or BRCA1. Purified PP2Cδ was incubated with BRCA1-derived phosphopeptide containing Ser1423 or ATM-derived phosphopeptide (Ser1981). PP2Cδ shows high levels of dephosphorylation of p-ATM and a positive control peptide containing phospho-Thr180 from p38 MAPK, which is a putative PP2Cδ target (2, 3). In contrast, PP2Cδ has minimal effects on p-BRCA1, excluding the direct dephosphorylation of BRCA1 by PP2Cδ. The negative control, a phosphopeptide derived from the repair protein UNG2(pT31) (25), fails to show PP2Cδ dephosphorylation (Fig. 5B). Magnesium-free phosphatase reactions also result in almost no phosphatase activity, as expected for the Mg2+-dependent PP2Cδ. Hence, we demonstrate that PP2Cδ can directly dephosphorylate ATM instead of BRCA1.
Fig. 5. PP2Cδ negatively regulates basal and UV damage–induced ATM phosphorylation.
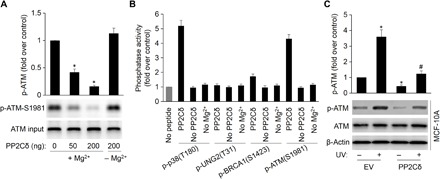
(A) PP2Cδ dephosphorylates intact ATM at phospho-Ser1981. An in vitro phosphatase assay was performed by incubating immunoprecipitated ATM and purified PP2Cδ, followed by Western blot probing with antibodies to ATM phospho-Ser1981. The data represent mean ± SD from three separate experiments. *P < 0.05 versus control. (B) Phosphopeptides from p38 MAPK (positive control), UNG2 (negative control), BRCA1 (phospho-Ser1423), and ATM (phospho-Ser1981) were incubated with PP2Cδ in an in vitro phosphatase assay. Release of free phosphate was measured by absorbance at 630 nm in the presence of molybdate dye. Reactions were also carried out without magnesium or peptide. (C) MCF-10A cells were transfected with EV or plasmid expressing WT PP2Cδ, followed by UV treatment (20 J/m2 for an 8-hour recovery). Cells were lysed, and the ATM phosphorylation and protein levels were detected by Western blotting with specific antibodies. The data represent mean ± SD from three separate experiments. *P < 0.05 versus EV/UV (−); #P < 0.05 versus EV/UV (+).
To further substantiate this notion, we overexpressed PP2Cδ in MCF-10A cells and assessed the effects of PP2Cδ overexpression on DNA damage–induced ATM phosphorylation. As shown in Fig. 5C, PP2Cδ overexpression significantly represses basal and UV-induced ATM phosphorylation. On the contrary, inhibition of PP2Cδ with C23 markedly increases basal and UV-induced ATM phosphorylation in MCF-7 cells (fig. S4). Furthermore, knockdown of PP2Cδ expression in MCF-7 cells by using PP2Cδ siRNAs markedly promotes the basal and UV-induced ATM phosphorylation (Fig. 1F). These results further argue in favor of the dephosphorylation of ATM by PP2Cδ in breast cancer cells, consistent with previous reports that inhibition of PP2Cδ up-regulates ATM in B cells and, in turn, overexpression of PP2Cδ decreases activation of ATM-dependent signaling cascades in response to DNA damage (5, 9). In summary, PP2Cδ dephosphorylates ATM at critical sites and is thus implicated as an important regulator in the ATM-dependent signaling pathways and cancer surveillance networks.
Inhibition of PP2Cδ enhances Dox-induced chemotherapy effects in breast cancer xenografts
As demonstrated by previous studies, PP2Cδ regulates BRCA1 phosphorylation and the activity of p53. To further substantiate the association of PP2Cδ and BRCA1 or p53, we examined the PP2Cδ protein expression, BRCA1 phosphorylation, and p53 acetylation in breast cancer tissues from immunocompromised mice bearing human MCF-7 xenografts by immunohistochemistry (IHC) staining. Breast cancer and paired surrounding breast adipose tissues from 12 mice were stained. Representative photomicrographs of PP2Cδ staining are shown in Fig. 6A. PP2Cδ expression is significantly higher in breast cancer than in peritumoral breast tissues. As expected, we observed a negative correlation between the expression levels of PP2Cδ and BRCA1 phosphorylation or p53 acetylation levels in the breast cancer specimens (Pearson’s R = −0.7114, P = 0.0095; R = −0.723, P = 0.0079; Fig. 6, B and C).
Fig. 6. Inhibition of PP2Cδ potentiates in vivo antitumor efficacy of Dox.
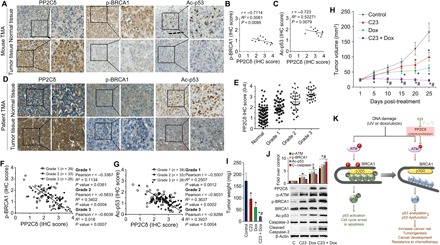
(A to C) Paraffin sections of mouse normal breast and MCF-7 xenograft tumor tissues were subjected to IHC staining using the antibodies indicated. Correlation between tumor tissue PP2Cδ level, p-BRCA1, and Ac-p53 staining score was tested by Pearson’s rank correlation analysis, with r and P values indicated. n = 12 mice per cohort. (D) Breast cancer tissue microarrays (TMAs) with adjacent normal breast tissue and breast cancer tissues from 103 breast cancer patients were subjected to IHC staining using the antibodies indicated. (E) IHC analysis of PP2Cδ staining score in normal adjacent breast tissue and breast cancer tissues. P value was calculated by one-way analysis of variance (ANOVA). *P < 0.01 versus normal tissue. (F and G) Correlation between tissue PP2Cδ level and p-BRCA1 or Ac-p53 staining score was tested by Spearman’s rank correlation analysis, with r and P values indicated. (H) Female nude mice inoculated with MCF-7 cells were treated with C23 (3 mg/kg, intraperitoneally), Dox (1.70 mg/kg, intraperitoneally; five times per week every 2 weeks), or C23 + Dox. Control animals received intraperitoneal saline injections. Values represent the average tumor size (mean ± SD) at intervals after initiation of Dox administration (n = 12 animals per treatment group). (I) Tumor weights from (H). Values represent mean ± SD; *P < 0.05 versus control; #P < 0.05 versus Dox. (J) Xenograft tumor tissues were excised from each animal and prepared for Western blot analysis. The protein expressions of PP2Cδ, p-ATM, p-BRCA1, Ac-p53, and cleaved caspase-3 were identified, and relative intensities were measured. β-Actin was used as a loading control. Data presented are mean ± SD from four independent experiments, with nontreated controls set to 1. *P < 0.05 versus control; #P < 0.05 versus Dox. (K) The identified signaling pathway in this study indicated that BRCA1 facilitates p300-mediated p53 acetylation and activation by complexing with these two proteins and that S1423/1524 phosphorylation is indispensable for this regulatory process. Aberrant PP2Cδ activity, via directly dephosphorylating ATM, suppresses DNA damage–induced BRCA1 phosphorylation and subsequently inhibits p300-mediated p53 acetylation and activation, leading to cancer development or resistance to chemotherapy. C23 represses this process by inhibiting PP2Cδ activity.
To evaluate the clinical relevance of PP2Cδ in breast cancer progression, we detected PP2Cδ expression by IHC staining of PP2Cδ in paraffin sections from 103 breast cancer cases (Fig. 6D). Compared with the paired normal tissues, 58 breast cancer specimens (78.6%) have substantially increased PP2Cδ expression. High PP2Cδ expression correlates significantly with more advanced histological grade (Fig. 6E). Consistent with previous studies, our IHC analysis demonstrates that PP2Cδ expression is negatively associated with BRCA1 phosphorylation and p53 acetylation in breast cancer tissues (Pearson’s R = −0.6102, P < 0.0001; R = −0.5735, P < 0.0001; Fig. 6, F and G). This significantly negative correlation was also observed in each grade subgroups.
Previous studies (26) have shown that a decreased level of functional p53 would prevent cell death induced by chemotherapy or radiotherapy. In light of the inhibitory effects of PP2Cδ on BRCA1 and p53, we would like to evaluate whether inhibition of PP2Cδ would potentiate the antineoplastic effects of Dox. Specifically, MCF-7 breast cancer cells (with a high level of PP2Cδ expression) were implanted in fat pads of female athymic nude mice. When the xenograft tumors became palpable, the mice were arbitrarily assigned into four groups, which received either vehicle alone (the control group), C23, a small-molecule inhibitor of PP2Cδ (18), Dox, or C23 + Dox. After treatment, compared with controls, either C23 or Dox alone can suppress tumor growth, as assessed by tumor volume and weight. C23, in combination with Dox, exhibits more potent anticancer efficacy versus Dox treatment alone (Fig. 6, H and I). Next, to substantiate the proposed mechanism of action for C23 against breast cancer in vivo, we analyzed phosphorylation of ATM and BRCA1, p53 acetylation, and the apoptotic marker cleaved caspase-3 in xenograft tumor tissues. As shown in Fig. 6J, C23 alone or in combination with Dox significantly increased the levels of ATM and BRCA1 phosphorylation, p53 acetylation, and tumor cell apoptosis compared to untreated controls or Dox treatment alone. Together, these results support the inhibition of PP2Cδ as a promising strategy for breast cancer and a suitable approach to enhance effects of DNA damage–based chemotherapy.
DISCUSSION
The DNA damage response (DDR) is a crucial network of cellular pathways that coordinate a series of essential biochemical and cellular events upon DNA damage. Activation of these different pathways has similar consequences, including DNA repair, inhibition of overall translation, cell cycle arrest, and eventually cell survival or death. Thus, DDR forms a barrier to cancer progression. Chemotherapeutic drugs target cancer cells by triggering DNA damage directly or indirectly, thus inducing p53-mediated DDR pathways and activation of cell cycle arrest and apoptotic machinery (27). Specifically, cells containing WT p53 protein with normal function can recognize DNA damage, arrest the cell cycle, and activate the appropriate cellular machinery needed to repair the damage before the cell undergoes replication (28). On the other hand, if the DNA damage cannot be repaired, p53 is capable of initiating the apoptotic pathway, which averts the unrestrained proliferation of transformed cells. Recent studies demonstrate that apart from antagonizing oncogenic transformation, p53 can also coordinate non–cell-autonomous responses to DNA damage to clear damaged cells via the innate immune system (29, 30). Future delineation of the precise causal link between tumorigenic factors and the inhibition of the p53 function and DDR that promote tumor progression holds a key to better understand chemoresistance and develop more effective cancer therapies.
In this study, we have presented evidence that inappropriate activity of PP2Cδ attenuates p53 acetylation and DNA damage–induced apoptosis in human breast cancer cells. Clinical IHC data also show that breast cancer specimens have substantially increased PP2Cδ expression compared with the paired normal tissues, and high PP2Cδ expression correlates significantly with more advanced histological grade. PP2Cδ expression is inversely associated with BRCA1 phosphorylation and p53 acetylation in breast cancer tissues. We have also demonstrated that PP2Cδ, through direct dephosphorylation of ATM, suppresses DNA damage–induced BRCA1 phosphorylation, leading to inhibition of p300-mediated p53 acetylation and activation (Fig. 6E). Last, knockdown or inhibition of PP2Cδ markedly increases p53 acetylation and its transcriptional regulation activity and potentiates the antineoplastic effects of DNA damage–based treatment in vitro and in vivo, paving an avenue to combinatorial therapy. Given that as a phosphatase, PP2Cδ has been demonstrated to dephosphorylate p53 (3) or its upstream kinases such as p38 MAPK (2), Chk1 (3), Chk2 (4), and ATM (5), this study moves a step forward in terms of previously unknown knowledge and concepts of p53 posttranslational modification by PP2Cδ. Findings in this study provide new insights into the mechanism of p53 inhibition in response to aberrant PP2Cδ activity and may shed some light on understanding the pathogenesis and drug resistance mechanisms of breast cancer.
BRCA1 is a cancer suppressor implicated in basic cellular functions required for cell replication, DNA synthesis, transcription, DNA damage signal networking, and DNA repair and is essential for maintaining genome integrity. Decreased BRCA1 expression or epigenetic inactivation results in compromised mammary gland differentiation and augmented risk of breast tumor development. Similar to p53, BRCA1 plays an important role in DDR, which is likely to contribute to cancer suppression (31). Some intricate cross-talk between BRCA1 and p53 biochemical pathways exists, including regulation of BRCA1 transcription by p53, BRCA1 induction of p53 accumulation after γ-irradiation by regulating its phosphorylation and Mdm2 expression, and changed selectivity of p53-dependent gene activation by BRCA1 (32, 33). In the present study, we revealed a new mechanism by which BRCA1 regulates p53. Mechanistically, we demonstrated that BRCA1 facilitates p300-mediated p53 acetylation and activation by complexing with these two proteins and that S1423/1524 phosphorylation is indispensable for this regulatory process. Multiple lines of evidence support this notion. First, BRCA1 knockdown by siRNA or KO by the CRISPR-Cas9 system markedly reduced both basal and UV-induced p300-p53 interaction and p53 acetylation, indicating a critical role of BRCA1 in the regulation of p53 acetylation. Second, BRCA1 physically interacts with both p300 and p53, and association of BRCA1 with p53 or p300 is required for its promoting effect on p53 acetylation. Last, studies with the phosphorylation-deficient mutant BRCA1 (S1423/1524A) and phosphomimicking mutant (S1423/1524D) substantiate that the ability of BRCA1 to facilitate p300-mediated p53 acetylation and transcriptional activity attributed to its phosphorylation levels. We speculate that phosphorylation induces a conformational change in BRCA1, facilitating interactions of p300 with p53 (Fig. 6E). To our knowledge, this is the first direct evidence that functionally depicts an involvement of BRCA1 in p300-p53 interaction and p53 acetylation.
In addition to p53 and BRCA1, a crucial protein kinase, ATM, also plays an important role in DDR pathways. ATM kinase transduces signals that regulate cell cycle checkpoints, apoptosis, and DNA repair (34). The ATM-initiated DDR pathway is activated quickly after DNA damage and acts as a barrier that helps to postpone or thwart cancer (35). It has been shown that inhibition of PP2Cδ up-regulates ATM and suppresses tumorigenesis in B cells (5, 9). Conversely, overexpression of PP2Cδ diminishes the activation of ATM-dependent signaling cascades. Consistent with this, we show that ATM partially mediates DNA damage–induced BRCA1 phosphorylation and that PP2Cδ can directly dephosphorylate ATM instead of BRCA1 in human breast cancer cells. In addition, we directly assess the therapeutic potential of our newly developed PP2Cδ inhibitor C23 as conjunctive treatments with UV or DNA damaging therapeutic agent Dox. In cell culture, the combinatorial treatment results in increased DDR signaling compared to UV treatment alone; excitingly, in xenograft tumor models, the combination therapy more efficiently reduces the tumor size and weight than Dox alone. In conclusion, we demonstrate a novel mechanism of p53 inactivation by PP2Cδ, namely, aberrant PP2Cδ activity inhibits p300-mediated p53 acetylation and activation via the ATM/BRCA1 pathway, leading to impairment of DDR (Fig. 6K). Our findings also substantiate PP2Cδ inhibition as an attractive strategy to confer Dox sensitization in breast cancers. This concept will be further explored in preclinical and clinical aspects.
MATERIALS AND METHODS
Experimental design
Research objectives
The objectives of this research were to elucidate the underlying mechanisms that link aberrant PP2Cδ levels with breast cancer development. A prespecified hypothesis was that PP2Cδ impairs p53 acetylation and DDR by compromising BRCA1 function. Once we confirmed the cross-talk between PP2Cδ and BRCA1/p300-p53 pathway, we investigated the clinical IHC data to explore the relationships between PP2Cδ levels and BRCA1 phosphorylation or p53 acetylation in human breast cancer specimens, as well as the correlation with patients’ histological grade. Furthermore, we sought to evaluate whether inhibition of PP2Cδ with C23, our newly developed PP2Cδ inhibitor, would potentiate the antineoplastic effects of Dox in MCF-7 xenograft–bearing nude mice.
Ethics statement
All animal studies were performed in accordance with the guidelines approved by the Institutional Animal Care and Use Committee of Charles R. Drew University of Medicine and Science.
Study design
To explore the molecular mechanisms underlying PP2Cδ inhibition of p53 acetylation, genetic ablation, or chemical inhibition, single base mutation, deletion mutation, and gene overexpression techniques were used. To evaluate whether inhibition of PP2Cδ would potentiate the antineoplastic effects of Dox, MCF-7 xenograft–bearing nude mice were used in these studies. We designed the studies to test the different agents with 10 mice per treatment group (one xenograft per mouse). Mice were randomized on the basis of tumor volume. With 10 mice per group, an effect size of 1.3 with 80% power could be detected, which was reasonable for a controlled animal experiment. The drugs used were coded so that researchers did not know which drug or combination treatments were used until experiments and analysis are completed.
Cell lines, cell culture, and DNA damaging agents
Human cell lines, including MCF-10A, MCF-7, H1299, and hTERT-HME1, were obtained from the American Type Culture Collection (Rockville, MD). The hTERT-HME1 cell line was cultured in MEBM supplemented with MEGM Mammary Epithelial Cell Growth Medium SingleQuots Supplements and Growth Factors. Cultures were maintained in a humidified incubator at 37°C with 5% CO2. These cells were authenticated by Laragen Inc. (Culver City, CA) by short tandem repeat profiling and monitoring cell morphology and biological behavior and tested to exclude mycoplasma contamination before experiments. For UV-induced damage, Stratalinker 1800 (Stratagene, La Jolla, CA) was used at 20 J/m2. Another agent used to induce DNA damage was Dox (Sigma-Aldrich) at 0.1 μM for MCF-10A and 0.5 or 1.0 μM for MCF-7. Standard cell culture and reverse transcription PCR were carried out as described previously (18, 36, 37).
Immunoprecipitations and Western blot analyses
Immunoprecipitations, Western blot analysis, and immunoprecipitation Westerns were carried out by standard methods described before (18, 25). Densitometry was performed using Scion Image software (Scion Corp., Frederick, MD). Antibodies were obtained from commercial sources listed below. Anti-PP2Cδ, anti-ATM, anti-ATM (pS1981), anti–caspase-3, horseradish peroxidase (HRP)–anti-mouse immunoglobulin G (IgG), and HRP–anti-rabbit IgG were obtained from Santa Cruz Biotechnology; anti-p53, anti-p53 (acetyl K382), anti-p53 (acetyl K373), and anti-BRCA1 (pS1423) were provided by Abcam; and anti-BRCA1, anti-p300, anti–cleaved caspase-3, anti-actin, and anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were purchased from Cell Signaling Technology.
Apoptosis assay
Human normal mammary epithelial cells (MCF-10A) transfected with empty vector (EV) or plasmid expressing WT PP2Cδ (PP2Cδ-WT) were exposed to Dox (0.1 μM) for 24 hours. MCF-7 cells were transfected with control siRNA or PP2Cδ siRNA for 24 hours, followed by incubation with Dox (1.0 μM) for 48 hours. Apoptosis was determined by measuring the membrane changes (phosphatidylserine based) with the APC Annexin V Apoptosis Detection Kit and PI (BioLegend Inc., San Diego, CA), as previously described (38). Fluorescence was detected using a BD FACSCanto flow cytometer (Becton Dickinson, Mountain View, CA) and FlowJo software version 10.4 (TreeStar, Ashland, OR). The experiments were repeated three times.
sgRNAs, plasmids, and cloning
To effectively target the BRCA1 gene for KO, CRISPR guide sequences were selected using the website http://crispor.tefor.net based on the following criteria: The sgRNA sequence should have a high efficiency score and a high specificity score (low off-target effects). Sequences for sgRNA oligos can be found in table S1.
sgRNA oligos were cloned into vectors according to the provided protocols from Bauer et al.’s paper (39). The pSpCas9(BB)-2A-GFP plasmid, which contained both Cas9 endonuclease and enhanced GFP (EGFP) expression cassettes, was purchased from Addgene.
Transfection and CRISPR genome editing
Cells were transfected with double Cas9/GFP-gRNA plasmids using Lipofectamine 2000, as recommended by the manufacturer. Twenty-four hours after transfection, GFP-positive cells were FACS-sorted on a GFP+/PI− cell sorter. Single-cell clones were grown and screened by PCR and immunoblotting for BRCA1 KO.
Sequencing
Regions surrounding sgRNA target/off-target sites were amplified by PCR using forward primer and reverse primer. PCRs were cleaned using the QIAquick PCR Purification Kit (Qiagen Inc.). Amplicons were then analyzed by Sanger sequencing (Quintara Biosciences Inc.). The following primers were used: caatgcattatatctgctgtgga (forward) and tgaaaaatgtgcaccttacga (reverse).
In vitro p53 acetylation assay
Recombinant p300 protein (1 μg) purified from baculovirus was preincubated with the indicated amounts of purified bacterially expressed BRCA1 protein or bovine serum albumin (BSA) for 10 min at room temperature. After preincubation, 1 μg of GST-p53 was added and p53 acetylation was measured as described previously (16).
Cell transfection with plasmids or siRNAs
The human WT PP2Cδ expression plasmid (hWIP1 FLAG) was a gift from L. Donehower (Addgene plasmid no. 28105). The plasmid pBABEpuro HA Brca1 encoding HA-tagged WT BRCA1 and plasmid pBABEpuro Brca1 S1423A S1524A HA encoding HA-tagged BRCA1 S1423A/S1524A mutant were gifts from S. Elledge (Addgene plasmid nos. 14999 and 41968). pBABEpuro HA Brca1-S1423D,S1524D encoding HA-tagged BRCA1 S1423D/S1524D mutant, pBABEpuro HA Brca1-∆224-500 encoding BRCA1 ∆224-500 mutants deleted for amino acids 224 to 500, which could not bind to p53, and pBABEpuro HA Brca1-∆1560-1863 encoding BRCA1 ∆1560-1863 mutants, which could not bind to p300, were constructed by Harmonious One Biotech Co. Ltd. (Shanghai, China). Cell transfection with plasmid DNA was carried out using Lipofectamine and PLUS Reagent (Invitrogen). For siRNA transfection, BioT transfection reagent (Bioland Scientific LLC) was used. ATM siRNAs from Santa Cruz Biotechnology, PP2Cδ siRNAs from Sigma-Aldrich Co., and negative control siRNA from Ambion (Austin, TX) were purchased.
p53 reporter luciferase assays
A p53-responsive reporter plasmid containing 13 copies of the p53-binding consensus sequence, pG13-LUC, was a gift from B. Vogelstein (Addgene plasmid no. 16442). Cells were transfected with pG13-LUC plasmid, and luciferase assays were carried out as previously described (40).
In vitro phosphatase assays
The in vitro phosphatase assays had been previously described (25). All the phosphopeptides for the in vitro phosphatase assays were custom-synthesized by InnoPep Inc. The sequences are p-UNG2 (T31), H-AVQG-pThr-GVAGV-NH2; p-BRCA1 (S1423), H-LEQHG-pSer-QPSNS-NH2; p-p38 (T180/Y182), H-TDDEM-pThr-GpYVAT-NH2; and p-ATM (S1981), H-AFEEG-pSer-QSTTI-NH2.
Xenograft model of human breast cancer
Female athymic nude mice (Foxn1nu) (The Jackson Laboratory, Bar Harbor, ME) at 5 to 7 weeks of age were used for the xenograft studies. E2 pellets (Innovative Research of America, Sarasota, FL) were implanted 7 days before cancer cell injection to augment the endogenous E2 production. The cultured MCF-7 cells in the logarithmic phase were collected and diluted to 3 × 107 cells/ml. Then, the cancer cell suspension (in Matrigel) was inoculated into the second mammary fat pad on the right side of mice (100 μl per injection). Approximately 14 days after inoculation, the animals were randomly assigned to treatment groups including vehicle only controls, poly(lactic-co-glycolic acid) nanoparticles of C23 (3 mg/kg, intraperitoneally) (18), Dox (1.70 mg/kg, intraperitoneally, five times per week every 2 weeks), or C23 + Dox (n = 10). Electronic calipers were used to determine the length and width of each tumor at intervals after initiation of Dox administration. The following equation was used to calculate tumor volumes: volume = 0.5(length × width2). At the end of the experiment, the animals were sacrificed and the tumors were harvested. The fresh tumor tissues were instantaneously placed in 4% paraformaldehyde for further IHC analysis.
Human tissue microarray and IHC staining
Tissue microarray (TMA) slides containing primary breast cancers (ER+) and adjacent normal tissue specimens from 103 breast cancer cases were purchased from U.S. Biomax (Rockville, MD) (BR243u) and Alena Biotechnology Ltd. Co. (Xian, China) (BR1921c). Representative areas of each invasive carcinoma were identified on the corresponding hematoxylin and eosin (H&E)–stained slides. The TMA sections were immunohistochemically stained for PP2Cδ, p-BRCA1, and acetyl-p53 with a standard immunostaining protocol as previously described (41). The assessment of staining scores was described previously (42). In brief, the percentages of staining-positive cells were scored into four categories: 0 to 1 (0 to 25%), 1 to 2 (26 to 50%), 2 to 3 (51 to 75%), and 3 to 4 (76 to 100%). The staining intensities were scored into four grades: 0 (none), 1 (weak), 2 (moderate), 3 (strong), and 4 (very strong). The final staining score was defined as the product of the percentage and intensity scores. Correlations between tissue PP2Cδ level and p-BRCA1 or acetyl-p53 staining score were tested by Pearson’s rank correlation analysis, with R and P values indicated.
Statistical analysis
Statistical analysis in the present study was performed using SPSS version 18.0 software (SPSS Inc.). Results were expressed as mean value ± SD. The significance of mean values between two groups was determined by Student’s t test. All differences were two-sided. The significance of the results from patient specimens was analyzed by the χ2 test or the Pearson’s correlation coefficient test. The effects of treatments on tumor size were evaluated using two-way analysis of variance (ANOVA) and Fisher’s post hoc test. A P value of <0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank M. Y. Keung (Charles R. Drew University) for assistance in editing this manuscript. Funding: This work was supported, in part, by NIH-NIMHD U54MD007598, NIH/NCI1U54CA14393, and U56 CA101599-01; Department of Defense Breast Cancer Research Program grant BC043180, NIH/NCATS CTSI UL1TR000124 (to J.V.); and Accelerating Excellence in Translational Science Pilot Grants G0812D05, NIH/NCI SC1CA200517 (to Y.W.). We are also grateful to CSUPERB New Investigator Award and Research Development Award (to Q.-H.C.). This study was partially supported by Key Disciplines Group Construction Project of Pudong Health Bureau of Shanghai (grant no. PWZxq2017-13), the International Postdoctoral Fellowship Program (grant 20150069 to Q.L.), and Stanford Cancer Institute Cancer Innovation Award (to J.R.P.). Author contributions: Y.W. and J.V. conceived and designed the experiments. Q.H., W.C., J.L., and K.W. performed most of the experiments and data analysis. K.W., W.C., and J.L. performed xenograft implantation experiments. Q.L. and Q.H. performed studies on molecular mechanism and function. K.W., Q.H., and Y.E. performed studies on tissue IHC of animal samples. Q.L., Q.H., and J.R.P. performed CRISPR genome editing. Q.-H.C. and G.C. synthesized C23. D.Z. synthesized drug-loaded poly(lactic-co-glycolic acid) nanoparticles and performed nanoparticle characterization. Y.W. wrote the manuscript with the help of all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaaw8417/DC1
Fig. S1. Effect of PP2Cδ inhibition on apoptosis, cell cycle progression, and senescence in MCF-7 cells.
Fig. S2. p53 acetylation was decreased upon BRCA1 knockdown.
Fig. S3. Phosphorylation of BRCA1 on Ser1423/1524 mediates apoptosis triggered by PP2Cδ inhibition.
Fig. S4. C23 markedly increases basal and UV-induced ATM phosphorylation in MCF-7 cells.
Table S1. Sequences for sgRNA oligos.
REFERENCES AND NOTES
- 1.Lu X., Nguyen T.-A., Moon S.-H., Darlington Y., Sommer M., Donehower L. A., The type 2C phosphatase Wip1: An oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev. 27, 123–135 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takekawa M., Adachi M., Nakahata A., Nakayama I., Itoh F., Tsukuda H., Taya Y., Imai K., p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 19, 6517–6526 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu X., Nannenga B., Donehower L. A., PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 19, 1162–1174 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oliva-Trastoy M., Berthonaud V., Chevalier A., Ducrot C., Marsolier-Kergoat M.-C., Mann C., Leteurtre F., The Wip1 phosphatase (PPM1D) antagonizes activation of the Chk2 tumour suppressor kinase. Oncogene 26, 1449–1458 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Shreeram S., Demidov N., Hee W. K., Yamaguchi H., Onishi N., Kek C., Timofeev O. N., Dudgeon C., Fornace A. J., Anderson C. W., Minami Y., Appella E., Bulavin D. V., Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol. Cell 23, 757–764 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Bulavin D. V., Demidov O. N., Saito S., Kauraniemi P., Phillips C., Amundson S. A., Ambrosino C., Sauter G., Nebreda A. R., Anderson C. W., Kallioniemi A., Fornace A. J. Jr., Appella E., Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat. Genet. 31, 210–215 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Belova G. I., Demidov O. N., Fornace A. J. Jr., Bulavin D. V., Chemical inhibition of Wip1 phosphatase contributes to suppression of tumorigenesis. Cancer Biol. Ther. 4, 1154–1158 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Nannenga B., Lu X., Dumble M., Van Maanen M., Nguyen T.-A., Sutton R., Kumar T. R., Donehower L. A., Augmented cancer resistance and DNA damage response phenotypes in PPM1D null mice. Mol. Carcinog. 45, 594–604 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Shreeram S., Hee W. K., Demidov O. N., Kek C., Yamaguchi H., Fornace A. J. Jr., Anderson C. W., Appella E., Bulavin D. V., Regulation of ATM/p53-dependent suppression of myc-induced lymphomas by Wip1 phosphatase. J. Exp. Med. 203, 2793–2799 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levine A. J., Oren M., The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 9, 749–758 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y., Kulesz-Martin M., p53 protein at the hub of cellular DNA damage response pathways through sequence-specific and non-sequence-specific DNA binding. Carcinogenesis 22, 851–860 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Vousden K. H., Prives C., Blinded by the light: The growing complexity of p53. Cell 137, 413–431 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Brooks C. L., Gu W., Ubiquitination, phosphorylation and acetylation: The molecular basis for p53 regulation. Curr. Opin. Cell Biol. 15, 164–171 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Vousden K. H., Lane D. P., p53 in health and disease. Nat. Rev. Mol. Cell Biol. 8, 275–283 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Brooks C. L., Gu W., The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2, 456–462 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito A., Lai C.-H., Zhao X., Saito S., Hamilton M. H., Appella E., Yao T.-P., p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 20, 1331–1340 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donehower L. A., Harvey M., Slagle B. L., McArthur M. J., Montgomery C. A. Jr., Butel J. S., Bradley A., Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356, 215–221 (1992). [DOI] [PubMed] [Google Scholar]
- 18.Wu K., Yu X., Huang Z., Zhu D., Yi X., Wu Y.-L., Hao Q., Kemp K. T. II, Elshimali Y., Iyer R., Nguyen K. T., Zheng S., Chen G., Chen Q.-H., Wang G., Vadgama J. V., Wu Y., Targeting of PP2Cδ by a small molecule c23 inhibits high glucose-induced breast cancer progression in vivo. Antioxid. Redox Signal. 30, 1983–1998 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu L., Scolnick D. M., Trievel R. C., Zhang H. B., Marmorstein R., Halazonetis T. D., Berger S. L., p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol. 19, 1202–1209 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pao G. M., Janknecht R., Ruffner H., Hunter T., Verma I. M., CBP/p300 interact with and function as transcriptional coactivators of BRCA1. Proc. Natl. Acad. Sci. U.S.A. 97, 1020–1025 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang H., Somasundaram K., Peng Y., Tian H., Zhang H., Bi D., Weber B. L., El-Deiry W. S., BRCA1 physically associates with p53 and stimulates its transcriptional activity. Oncogene 16, 1713–1721 (1998). [DOI] [PubMed] [Google Scholar]
- 22.Hickson I., Zhao Y., Richardson C. J., Green S. J., Martin N. M. B., Orr A. I., Reaper P. M., Jackson S. P., Curtin N. J., Smith G. C. M., Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 64, 9152–9159 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Gatei M., Scott S. P., Filippovitch I., Soronika N., Lavin M. F., Weber B., Khanna K. K., Role for ATM in DNA damage-induced phosphorylation of BRCA1. Cancer Res. 60, 3299–3304 (2000). [PubMed] [Google Scholar]
- 24.Cortez D., Wang Y., Qin J., Elledge S. J., Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 286, 1162–1166 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Lu X., Bocangel D., Nannenga B., Yamaguchi H., Appella E., Donehower L. A., The p53-induced oncogenic phosphatase PPM1D interacts with uracil DNA glycosylase and suppresses base excision repair. Mol. Cell 15, 621–634 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Parrales A., Iwakuma T., Targeting oncogenic mutant p53 for cancer therapy. Front. Oncol. 5, 288 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reinhardt H. C., Schumacher B., The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 28, 128–136 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pei D., Zhang Y., Zheng J., Regulation of p53: A collaboration between Mdm2 and Mdmx. Oncotarget 3, 228–235 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ventura A., Kirsch D. G., McLaughlin M. E., Tuveson D. A., Grimm J., Lintault L., Newman J., Reczek E. E., Weissleder R., Jacks T., Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661–665 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Xue W., Zender L., Miething C., Dickins R. A., Hernando E., Krizhanovsky V., Cordon-Cardo C., Lowe S. W., Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosen E. M., BRCA1 in the DNA damage response and at telomeres. Front. Genet. 4, 85 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scata K. A., El-Deiry W. S., p53, BRCA1 and breast cancer chemoresistance. Adv. Exp. Med. Biol. 608, 70–86 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Xu X., Qiao W., Linke S. P., Cao L., Li W.-M., Furth P. A., Harris C. C., Deng C.-X., Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat. Genet. 28, 266–271 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Kastan M. B., Bartek J., Cell-cycle checkpoints and cancer. Nature 432, 316–323 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Bartkova J., Hořejší Z., Koed K., Krämer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J. M., Lukas C., Ørntoft T., Lukas J., Bartek J., DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Wu Y., Sarkissyan M., Mcghee E., Lee S., Vadgama J. V., Combined inhibition of glycolysis and AMPK induces synergistic breast cancer cell killing. Breast Cancer Res. Treat. 151, 529–539 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu Y., Yu X., Yi X., Wu K., Dwabe S., Atefi M., Elshimali Y., Kemp K. T. II, Bhat K., Haro J., Sarkissyan M., Vadgama J. V., Aberrant phosphorylation of SMAD4 Thr277-mediated USP9x-SMAD4 interaction by free fatty acids promotes breast cancer metastasis. Cancer Res. 77, 1383–1394 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rahman A. A., Makpol S., Jamal R., Harun R., Mokhtar N., Ngah W. Z. W., Tocotrienol-rich fraction, [6]-gingerol and epigallocatechin gallate inhibit proliferation and induce apoptosis of glioma cancer cells. Molecules 19, 14528–14541 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bauer D. E., Canver M. C., Orkin S. H., Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. J. Vis. Exp. 2015, e52118 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Granja A. G., Nogal M. L., Hurtado C., Salas J., Salas M. L., Carrascosa A. L., Revilla Y., Modulation of p53 cellular function and cell death by African swine fever virus. J. Virol. 78, 7165–7174 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang N., Wei P., Gong A., Chiu W.-T., Lee H.-T., Colman H., Huang H., Xue J., Liu M., Wang Y., Sawaya R., Xie K., Yung W. K. A., Medema R. H., He X., Huang S., FoxM1 promotes beta-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell 20, 427–442 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dang E., Yang S., Song C., Jiang D., Li Z., Fan W., Sun Y., Tao L., Wang J., Liu T., Zhang C., Jin B., Wang J., Yang K., BAP31, a newly defined cancer/testis antigen, regulates proliferation, migration, and invasion to promote cervical cancer progression. Cell Death Dis. 9, 791 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaaw8417/DC1
Fig. S1. Effect of PP2Cδ inhibition on apoptosis, cell cycle progression, and senescence in MCF-7 cells.
Fig. S2. p53 acetylation was decreased upon BRCA1 knockdown.
Fig. S3. Phosphorylation of BRCA1 on Ser1423/1524 mediates apoptosis triggered by PP2Cδ inhibition.
Fig. S4. C23 markedly increases basal and UV-induced ATM phosphorylation in MCF-7 cells.
Table S1. Sequences for sgRNA oligos.