

Abstract
Background
The objective of the current study was to study the molecular mechanism(s) underlying cardiac troponin I autoantibody (cTnIAAb) binding to cardiomyocyte and resultant myocardial damage/dysfunction.
Methods
cTnIAAb was purified from serum of 10 acute myocardial infarction (AMI) patients with left ventricular remodeling. Recombinant human cTnI was used to generate three mouse-derived monoclonal anti-cTnI antibodies (cTnImAb1, cTnImAb2, and cTnImAb3). The target proteins in cardiac myocyte membrane bound to cTnImAb and effect of cTnIAAb and cTnImAb on apoptosis and myocardial function were determined.
Findings
We found that cTnIAAb/cTnImAb1 directly bound to the cardiomyocyte membraneα-Enolase (ENO1) and triggered cell apoptosis via increased expression of ENO1 and Bax, decreased expression of Bcl2, subsequently activating Caspase8, Caspase 3, phosphatase and tensin homolog (PTEN) while inhibiting Akt activity. This cTnIAAb-ENO1-PTEN-Akt signaling axis contributed to increased myocardial apoptosis, myocardial collagen deposition, and impaired systolic dysfunction.
Interpretation
Results obtained in this study indicate that cTnIAAb is involved in the process of ventricular remodeling after myocardial injury.
Fund
The National Natural Science Foundation of China (Grant#: 81260026).
Keywords: Acute myocardial infarction, Cardiac troponin I autoantibody, Phosphatase and tensin homolog, Ventricular remodeling, α-Enolase
Abbreviations: cTnIAAb, cardiac troponin I autoantibody; AMI, acute myocardial infarction; PTEN, phosphatase and tensin homolog; ACS, acute coronary syndrome; DCM, dilated cardiomyopathy; HF, heart failure; VNC, ventricular noncompaction; PCI, percutaneous coronary intervention; CNBr, cyanogen bromide; SD, Sprague–Dawley; FITC, fluorescein isothiocyanate
Research in context.
Evidence before this study
Cardiac troponin I autoantibody (cTnIAAb) levels are elevated in patients with heart disease. cTnIAAb binds directly to cardiomyocytes, causing myocardial dysfunction.
Added value of the study
We examined the molecular mechanism by which cTnIAAb exerts its deleterious effects on the myocardium by purifying human cTnIAAb and creating recombinant mouse-derived antibodies. We identified α-Enolase (ENO1) as a novel cell surface-binding protein for cTnIAAb. We also demonstrated that ENO1 activates phosphatase and tensin homolog (PTEN) and suppresses AKT signaling, which induces myocardial apoptosis.
Implications of all available evidence
The cTnIAAb-ENO1-PTEN-Akt signaling axis increases myocardial apoptosis, myocardial collagen deposition, and impaired systolic dysfunction. Thus, cTnIAAb is not only an independent risk factor for ventricular remodeling after myocardial injury in heart disease patients, but it is also involved in ventricular remodeling.
Alt-text: Unlabelled Box
1. Introduction
cTnIAAb were found to be elevated in patients with heart diseases, including acute coronary syndrome (ACS), acute myocardial infarction (AMI), dilated cardiomyopathy (DCM), heart failure (HF), and ventricular noncompaction (VNC) [[1], [2], [3], [4], [5]]. Increased cTnIAAb significantly decreases the concentration of serum cTnI, which is a well-known cardiac biomarker [6,7], thus potentially causing false negative results on immunoassays for cTnI [1,8].
cTnIAAb, but not cardiac troponin T polyclonal autoantibody (cTnTAAb), was shown to directly bind to the myocardial cell membrane of mice and cause myocardial dysfunction [9,10]. In the mouse model of ischemia-reperfusion injury, cTnIAAb-positive mice exhibit more severe myocardial inflammation and worse prognosis [11]. Mechanistically, cTnIAAb induces direct myocardial damage and exaggerates the myocardial inflammatory response [12].
However, the exact mechanisms by which cTnIAAb renders its deleterious effects on the myocardium remain unclear. Studies have suggested that cTnIAAb binds to membrane cTnI of the myocardial cell and causes cardiac dysfunction through mediating the intracellular calcium flux [13]. However, given the presence of a large amount of endogenous cytosolic cTnI in myocardial cells, the potential contamination of cytosolic cTnI in the purified myocardial cell membrane proteins could not be ruled out [10].
In the present study we identified α-Enolase (ENO1) as a novel cell surface-binding protein for cTnIAAb using an unbiased approach. We also showed that ENO1 mediated cTnIAAb-induced myocardial cell apoptosis via activation of phosphatase and tensin homolog (PTEN) and suppression of AKT signaling. Our findings present a potential therapeutic target, ENO1, for treatment of cTnIAAb-positive patients with cardiac diseases.
2. Materials and methods
2.1. Patient selection and collection of serum samples
This study was approved by the Medical Ethics Committee at Changhai Hospital and Changcheng Hospital Affiliated to Nanchang University and patients enrolled provided signed informed consent. Patients admitted to the Emergency Department at Changhai Hospital and Changcheng Hospital Affiliated to Nanchang University from January 2013 to November 2014 were enrolled in this study. A total of 10 patients (8 males and 2 females) participated in the current study, and the mean age of patients was 73.7 ± 11.1 years (range from 49 to 86 years). These 10 patients were followed up for one year, and their left ventricular end-systolic volume was increased by >15% [14]. At admission, 6 were diagnosed as ST segment elevation myocardial infarction, and 4 as non-ST-segment elevation myocardial infarction. Besides, the blood cTnI concentration of all patients was higher than 0.1 μg/L. Those patients with idiopathic cardiomyopathy, congenital heart disease, rheumatic heart disease, valvular heart disease, autoimmune disease, kidney disease, liver disease, cancer, or other severe systemic illnesses were excluded from the current study. All patients were treated with either drug(s) or percutaneous coronary intervention (PCI) therapy within 12 h of symptom onset. Serum samples from ten cTnIAAb positive AMI patients with ventricular remodeling were collected, immediately aliquoted, and frozen at −80 °C for cTnIAAb affinity chromatography purification.
2.2. Purification of human cTnIAAb
Recombinant full-length human cTnI (50 mg) was coupled to 10 ml cyanogen bromide (CNBr)-activated Sepharose 4B (Instruction, Sweden) beads, and columns were packed according to manufacturer's guidelines. cTnIAAb-positive serum samples collected from patients were incubated with recombinant full-length human cTnI-coupled affinity column for 6 h at 4 °C, and subsequently washed with 10 bed volume of 1 × PBS (pH 7.2, 0.1 M). Thereafter, 0.1 M glycine (pH 3.0) was used to elute the target protein, and the eluate was immediately neutralized with 3 M Tris (pH 8.5). The eluate was dialyzed in a 10-kDa dialysis bag containing 1 × Phosphate Buffer solution (PBS, pH 7.2, 0.01 M), and the protein was concentrated by ultracentrifugation using an Amicon Μltra-15 Centrifugal Filter 10 K Device (Millipore, USA).
2.3. Cell culture, gene construction, and in vitro transfection
The animal protocol was planned in compliance with the animal protection, animal welfare, and ethical principles and was approved by the IACUC of the Second Military Medical University. Sprague–Dawley (SD) neonate rats were purchased from Experimental Animal Center of Second Military Medical University. Primary cardiomyocytes were isolated from neonatal 1-day-old SD rat offspring and cultured as described before. The gastric adenocarcinoma cell line BCG823 (Shanghai Sixth People's Hospital Central Laboratory) was maintained in RPMI-1640 medium (Gibco, USA) containing 10% fetal bovine serum (FBS; Gibco, USA). All cells were passaged using Accutase-Enzyme Cell Detachment Medium (eBioscience, USA). SiRNAs (Shanghai GenePharma) were transfected into cells using Lipofectamine 2000 (ThermoFisher Scientific) based on manufacturer's guidelines. The following siRNAs were used: Rat Eno1: 5′-GGCACAGAGAAUAAAUCUATT-3′; Negative control siRNA: 5′-UUCUCCGAACGUGUCACGUTT-3′.
2.4. Primary cardiomyocyte isolation, immunofluorescence staining and co-localization of cTnI and DiI cell membrane staining
Immunofluorescence staining of primary cultured neonatal rat cardiomyocytes was performed as previously described [15]. Immunofluorescence was observed by a fluorescence Olympus microscope (Japan). For cardiomyocyte membrane staining, cardiomyocytes were incubated with cTnIAAb, fluorescein isothiocyanate (FITC) and the live cell membrane dye Dil (working concentration, 5 μM, AAT Bioquest, USA) for additional 20 min at 37 °C, washed with pre-warmed high-glucose DMEM. The cardiomyocytes were then fixed with 4% paraformaldehyde at 4 °C for 30 min. Immunofluorescence was observed immediately using a Nikon ALR/AI confocal microscope (Japan).
2.5. Preparation of mouse anti-cTnI antibodies cTnImAb1, cTnImAb2 and cTnImAb3
cTnImAb1, cTnImAb2, and cTnImAb3 were generated in our laboratory. Recombinant full-length human cTnI was a gift from Shanghai Beyond Biological Diagnostics. The commercially available cTnImAb and cTnIpAb were purchased from Hytest (Finland). The four commercially available cTnImAb cloning numbers and their antigenic sites against human cTnI were 820 (a.a.r 26–35), 19C7 (a.a.r 41–49), 560 (a.a.r 83–93), and MF4 (a.a.r 190–196). Luminescent microspheres and biotinylated microspheres were gifts from Shanghai Beyond Biotechnology and were coated with individual commercially available cTnImAb 820, 19C7, 560, and MF4, separately (0.4 mg antibody/10 mg microspheres). The stock concentration of luminescent microspheres was 10 mg/ml, and the working concentration was 100 μg/ml. The stock concentration of biotinylated microspheres was 0.8 mg/ml, and the working concentration was 2 μg/ml. The molar ratio of biotinylated antibody to coated microspheres was 1:30 (1 mol/L antibody coated 30 mol/L biotin). The luminescence intensity was measured by homogeneous double antibody sandwich light-activated chemiluminescence (LICAHP and SP500 homogeneous light chemiluminescence analyzer, Shanghai Beyond Biotechnology). To determine the antigenic sites of cTnImAb1, cTnImAb2, and cTnImAb3, each of these three antibodies was incubated with two commercially available cTnImAbs, which had known antigenic sites on cTnI, and the recombinant full-length human cTnI to compete for binding sites. The same amount of mouse IgG was used as a control group. If the detected fluorescence signal compared with the control group decreased by ≥90%, it suggested the presence of a binding competition and the same antigenic sites were shared by the homemade antibody and the commercially available antibody. If the detected fluorescence signal was decreased between ≥50% and <90% compared with the control group, it suggested the presence of a partial competition and partial overlapping antigenic sites. If the detected fluorescence signal and that of the control group showed no significant difference, it suggested the absence of a binding competition and no similar antigenic sites were shared by the homemade antibody and the commercially available antibodies.
2.6. Flow cytometry
Briefly, cultured cells were incubated with one of the following primary antibodies: cTnImAb1, cTnImAb2, cTnImAb3, or cTnTmAb (Hytest, Finland, final concentration 100 μg/ml) at 37 °C for 2 h, then washed, trypsinized. After being washed with D-Hanks solution, the cells were incubated with anti-mouse IgG-FITC (eBioscience, USA) for 20 min, washed, and then immediately subjected to flow cytometric analysis.
Cell membrane binding competition experiments: cultured cells were first incubated with cTnImAb1, cTnImAb2, or cTnImAb3 (final concentration, 100 μg/ml) at 37 °C for 30 min, then incubated with cTnIAAb (final concentration, 100 μg/ml) at 37 °C for another 30 min, and next washed with D-Hanks solution. In the control group, only cTnIAAb was used. After being washed with D-Hanks solution, the cells were incubated with anti-human IgG-FITC (eBioscience, USA) at 37 °C for 20 min, washed, and then immediately subjected to flow cytometric analysis. Flow cytometry was carried out using a MACSQuant™ (Miltenyi biotec, Germany) or Aria II (Becton, Dickinson and Company, USA), and the data were analyzed using FlowJo software 7.6.2 (Tree Star Inc., Canada) or DIVA (Becton,Dickinson and Company, USA).
2.7. Purification and identification of cTnImAb membrane-bound protein
BCG823 cells (50 × 106) were lysed using NP-40 lysis buffer and the lysate was coupled with 10 mg of cTnImAb1 and CNBr-activated Sepharose 4B beads (2 ml) and processed as described above (Purification of human cTnIAAb). The sample was washed with glycine (pH 3.0, 0.1 M), and then the eluate was centrifuged using Amicon Mltra-15 Centrifugal Filter 10 K Device (Millipore, USA) to obtain concentrated protein. The concentrated protein was subjected to SDS-PAGE followed by Coomassie blue staining. The visualized protein band was cut, trypsin-digested, and analyzed by mass spectrometry (AB SCIEX TOF/TOFTM 5800, USA). The obtained peptide sequence was analyzed against human libraries and the peptide fingerprint database. Proteomic analysis was carried out at the Fudan University Protein Center (Shanghai, China).
2.8. Detection of apoptosis in cardiomyocytes and immunoblot analysis
Cell apoptosis was determined using Annexin V-FITC Apoptosis detection kit (eBioscience, USA). Caspase-3 activation was detected using the Cadp GLOW™ Fluorescein Active Caspase-3 Staining Kit (Biovision, USA). Flow cytometry was carried out using a MACSQuant™ (Miltenyi biotec, Germany) or Aria II(Becton, Dickinson and Company, USA), and the data were analyzed using FlowJo software 7.6.2 (Tree Star Inc., USA) or DIVA(Becton, Dickinson and Company, USA).
Apoptosis-related protein detection was performed using immunoblot analysis. Blots were probed using the following antibodies - rabbit anti-Bcl-2 monoclonal antibody, rabbit anti-Bax monoclonal antibody, rabbit anti-ENO1 monoclonal antibody, rabbit anti-Akt monoclonal antibody, rabbit anti-phospho-Ser380 PTEN monoclonal antibody, and rabbit anti-PTEN monoclonal antibody (all from Abcam, UK), mouse ENO1 polyclonal antibody (Abnova, USA), and rabbit anti-phospho-Thr308-Akt monoclonal antibody (Cell Signaling Technologies, USA). All blots were probed with mouse anti-β-actin monoclonal antibody (Proteintech, USA) to confirm equal loading across lanes.
2.9. Co-immunoprecipitation analysis
Co-IP was performed as previously described [16]. Briefly, cardiomyocytes (5 × 106) were lysed with the NP-40 protein lysis buffer (Pik Days, China) containing protease inhibitors (Selleckchem, USA). The whole cell lysates were incubated with 1 μg rabbit anti-ENO1 monoclonal antibody for 1.5 h followed by another incubation with 50 μl Protein A/G Plus-Agarose Immunoprecipitation Reagent (Santa Cruz Biotechnology, USA) on a rotator at 4 °C overnight. After centrifugation at 3000 g for 5 min at 4 °C, the precipitates were washed, boiled, and subjected to immunoblot analysis, with 30 μg of total protein lysates used as an input.
2.10. Quantitative real time polymerase chain reaction (qRT-PCR)
Briefly, total RNA was purified from cultured cardiomyocytes after 48 h of stimulation with vehicle (control), AngII (5 μM, Sigma-Aldrich, USA), cTnImAb1 or cTnTmAb using Trizol (Thermo Fisher Scientific, USA). RNA (100 ng) was used for reverse transcription using PrimeScript™ RT (Takara, Japan), and the obtained cDNA was used for qPCR using SYBRR PrimeScript™ RT-PCR (Takara, Japan) to detect the expression of cardiac disease markers, Nppa, Myh6, and Myh7. The expression of Actb (encoding β-actin) served as an internal control, and the qRT-PCR data were analyzed as previously reported [17]. The sequences of oligos used in qRT-PCR are shown below (5′ to 3′): Nppa_F: 5′-ATCACCAAGGGCTTCTTCCT-3′, Nppa_R: 5′-CCAGGTGGTCTA GCAGGTTC-3′; Myh6_F: 5′-GAGGCAAGAAGAAAGGCTCA-3′, Myh6_R: 5′-AAAGTGAGGATGGGTGGTC-3′; Myh7_F: 5′-TCAGCCTACCTCATGGGACT-3′, Myh7_R: 5′-GACATTCTGCCCTTTGGTGA-3′; Actb_F: 5′-CCAGATCATGTTTGAGACCTTCAA-3′, Actb_R: 5′-GTGGTACGACCAGAGGCATACA-3′.
2.11. Wild-type and mutant Eno1 expression plasmids, transfection, and analysis
The full length open reading frame of Eno1 was amplified by PCR with a high-fidelity DNA Polymerase (Takara, Japan) using the following primers: F: 5′-AGAGAATTC GGATCCATGTCTATTCTCAAGATCCATG-3′, and R: 5′-CTTCCATGGCTC GAGTTACTTGGCCAAGGGGTTTC-3′. The purified fragment was cloned into eukaryotic expression plasmid pCDNA3.1 N-Flag by In-Fusion (Clontech, China) and verified by Sanger sequencing. Site-Directed Mutagenesis (NEB, USA) was used to generate point mutations in pCDNA3.1-Eno1 (C119A, C339A, C357A, and C389A) using the following primers: C119A_F: 5′-CCTTGCCGTCgcCAAAGCTGGTGC-3′, C119A_R: 5′-GACACCCCCAGAATGGCG-3′; C339A_F: 5′-GTCCTGCAACgcCCTCCTGCTC-3′, C339A_R: 5′-TTCTCGTTCACGGCCTTG-3′; C357A_F: 5′-TCTTCAGGCGGCCAAGCTGGCCC-3′, C357A_R: 5′-GACTCGGTCACGGAGCCA-3′; C389A_F: 5′-TGTGGGGCTGGCCACTGGGCAG-3′, C389A_R: 5′-ACCAGGTCAGCGATGAAG-3′. The wild-type or mutant plasmids were transfected into 293 T cells using Lipofectamine 2000, and the expression of Flag-tagged proteins were analyzed by immunoblotting after 48 h.
2.12. Animal model
Four-week-old female BALB/C mice were purchased from the Animal Center at Second Military Medical University and housed at the small animal center in Changhai Hospital. Animals were randomly divided into three groups: the experimental group (n = 14), the antibody control group (n = 10), and the blank control group (n = 10). cTnImAb1 (200 μg/1000 μl), cTnTmAb (200 μg/1000 μl, Hytest, Finland) and mouse IgG (200 μg/1000 μl, Proteintech, USA) were intraperitoneally injected into the mice of these three groups, respectively, and maintained for 12 weeks. The antibody dosage and intraperitoneal injection were performed as previously reported [9,10].
2.13. Evaluation of cardiac function in mice
Cardiac function of mice was evaluated with the B-mode and M-mode of the VisualSonics © Vevo770 RMV707B operating system and high-frequency ultrasound probe (Canada). Briefly, mice were anesthetized by inhalation of 1% isoflurane (RWD, China) and fixed on a warm pad during the whole procedure of transthoracic measurements. Cardiac function indices and heart dimensions including left ventricular diameter, left ventricular function, left ventricular mass, left ventricular systolic, and diastolic posterior wall thickness were determined.
For the measurement of right ventricular pressure, mice were anesthetized and fixed on a warm pad as described for echocardiography. The left common carotid artery was isolated, and a cardiac catheter SPR-671 Millar was inserted through the left common carotid artery until it reached the left ventricle. Data were collected with MP150 data acquisition system and analyzed with Powerlab Chart5.0 software (AD Instruments, Australian). The right ventricular pressure and waveform were analyzed starting from the outside of the right jugular vein to the appearance of the ventricular waveform.
2.14. Evaluation of pathological changes in mice heart
The hearts were collected immediately after the sacrifice of mice, weighed, and photographed. Half of the ventricles was fixed at 4% paraformaldehyde, paraffin-embedded, and sectioned at 5 μm thickness for hematoxylin-eosin (H&E) staining, Trichrome Masson staining and immunohistochemistry against CD3 (sc-20047, Santa Cruz Biotechnology, USA), CD8 (sc-7188, Santa Cruz Biotechnology, USA), CD20 (sc-7735, Santa Cruz Biotechnology, USA), CD4 (NBP1–19371 SS, Novus Biological, USA), and CD68 (NB100-683SS, Novus Biological, USA) using DAB chromogenic method. The von Kossa staining (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) of calcified plague was carried out to analyze the calcification in the heart. The apoptosis of myocardium was observed by Transferase-mediated deoxyuridine triphosphate-biotin nick end labeling (TUNEL, Roche 11684795910, Switzerland). The other half of the ventricles was snap-frozen at −80 °C for subsequent immunoblot analysis of phosphorylation of the Akt signaling pathway and qRT-PCR for measurement of matrix metalloproteinase (MMPs) gene expression.
Mouse cardiac tissue total protein Akt signaling was screened using the PathScan® Akt Signaling Antibody Array Kit (Cell Signal, 9474, USA). The relative expression levels of mRNAs encoding matrix metalloproteinase (mmp1a, mmp3, mmp12) and metalloproteinase inhibitor (timp3) in myocardium were detected by qRT-PCR using the following primers (Sangon Biotechnology, China): mmp1a_F: 5′-TTCAAAGGCAGCAAAGTATGG-3′, mmp1a_R: 5′-TCCTCACAAACAGCAGCATC-3′; mmp3_F: 5′-TGGTGTTCCTGATGTTGGTG-3′, mmp3_R: 5′-TTTCAATGGCAGAATCCACA-3′; mmp12_F: 5′-GCAGCAGTTCTTTGGGCT A-3′, mmp12_R: 5′-CGCTTCATCCATCTTGACC-3′; timp3_F: 5′-CTTGCCTTGTTTTGTGACCTC-3′, timp3_R: 5′-TTGCTGATGCTCTTGTCTGG-3′; actb_F: 5′-ATATCGCTGCGCTGGTCGTC-3′, actb_R: 5′-AGGATGGCGTGA GGGAGAGC-3′.
2.15. Statistical analysis
Continuous variables are presented as mean ± standard deviation (SD) for data with a normal distribution, or as median ± standard deviation for the median with a non-normal distribution. The independent sample t-test or one-way analysis of variation (ANOVA) followed by a Tukey's post hoc test were used for determination of statistical significance of continuous variables.
The gray values of the signal of 16 phosphorylation sites obtained from the protein chip analysis was statistically compared with t-test and volcano chart.
A two-tailed test was used for all statistical analyses using statistical software SPSS version 23.0 (SPSS, Inc., USA) with p < 0.05 considered statistically significant.
3. Results
3.1. Human cTnIAAb binds myocardial cTnI of human, mouse, and rat
Recombinant full-length cTnI-coupled affinity column was used to purify and concentrate human cTnIAAb from 10 patients with left ventricular remodeling. On a sodium dodecyl sulfate (SDS) gel, two protein bands of approximately 28 kDa and 49 kDa molecular weights were clearly observed, in line with the sizes of the human IgG heavy and light chains, respectively (Fig. 1a). Purified cTnIAAb bound to cTnI present in human atrial tissues and mouse ventricular tissues, as well as to the recombinant full length cTnl (Fig. 1b, c). Also, protein hybridization analysis showed that cTnIAAb bound to other unknown protein(s) purified from cardiomyocytes, including a band at ~51 kDa (Fig. 1b). The human cTnIAAb also specifically bound to the primary cultured neonatal rat cardiomyocytes as revealed by immunofluorescence staining (Fig. 1d, e). Taken together, our results indicate that the human cTnIAAb purified from AMI patients with left ventricular remodeling is able to bind the myocardial cTnI of human, mouse, and rat origin as well as the recombinant full length cTnI.
Fig. 1.

Characterization of purified human cTnIAAb.
a. Purified human cTnIAAb was subjected to sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) followed by Coomassie blue staining, which revealed two protein bands with sizes of ~51 kDa and ~28 kDa in line with the characteristic molecular weights of immunoglobulins.
b. Human cTnIAAb bound to protein lysates purified from human atria (lane 1 from left), mouse ventricles (lane 2 from left), and recombinant full-length cTnI (lane 3 from left) at the position of ~24 kDa as revealed by Western blotting. Binding to protein lysates purified from mouse liver tissue was used as a negative control (lane 4 from left). Left panel, non-reduced protein sample, anti-cTnIAAb blot; middle panel, reduced protein sample, anti-cTnIAAb blot; right panel, reduced protein sample, anti-cTnImAb blot.
c. Representative images of immunohistochemical staining showing positive staining of cTnIAAb in tissue sections prepared from human atria and mouse ventricles. Human IgG was used as a negative control. Bar = 50 μm.
d&e. Representative images showing fluorescence immunostaining of primary cultured neonatal Sprague-Dawley rat cardiomyocyte using human cTnIAAb. d, bar = 50 μm. e, bar = 20 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. Human cTnIAAb can directly bind to the myocardial cell membrane and induce cardiomyocyte apoptosis
Previous studies from our group and others have shown that the high circulating levels of cTnIAAb are closely correlated with the severity of myocardial injury and poor prognosis of AMI patients [5,[9], [10], [11], [12]], indicating that cTnIAAb has direct or indirect detrimental effects on the myocardium. We cultured neonatal rat cardiomyocytes in the absence or presence of human cTnIAAb(100 μg/ml) and found that cTnIAAb did not increase the expression levels of cardiac hypertrophy-related genes (Supplementary Fig. 1). However, cTnIAAb triggered cardiomyocyte apoptosis (Fig. 2a, b), accompanied by significant activation of caspase 3 (Fig. 2c, d), increased expression of the pro-apoptotic marker Bax, and decreased expression of the pro-survival marker Bcl2 (Fig. 2e, f).
Supplementary Fig. 1.

Effect of cTnIAAb treatment on the expression of hypertrophic genes in vitro. Neonatal rat cardiomyocytes were treated for 48h with cTnIAAb (100 μg/L), cTnTmAb (100 μg/L), mouse IgG (100μg/L) and AngII (1μM, Sigma), respectively. AngII was used as positive control. Thereafter, total RNA was isolated using TRIzol reagent, and the hypertrophic markers, Nppa, Nppb and myh7/myh6 were quantified using RT-PCR and normalized to β-actin housekeeping gene. *p<0.05 compared to control (t-test, n=3).
Fig. 2.

Human cTnIAAb binds membrane of cultured cardiomyocytes and causes cell apoptosis.
a&b. Flow cytometric analysis results showing that cTnIAAb treatment for 48 h triggered apoptosis in neonatal rat cardiomyocytes. a, Annexin V-PI staining was used for flow cytometric analysis. Representative images are shown. b, Quantification of AnnexinV/PI staining from 3 independent experiments. *p = .000 (ANOVA analysis).
c & d. Flow cytometric analysis results showing that cTnIAAb treatment for 48 h triggered apoptosis in neonatal rat cardiomyocytes. c, Active Caspase-3 staining was used for flow cytometric analysis. Representative images are shown. d, Quantification of Annexin-V/PI staining from 3 independent experiments. *p = .000 (ANOVA analysis).
e. cTnIAAb treatment for 48 h significantly increased expression of Bax and decreased expression of Bcl2 as revealed by immune blot analysis. β-actin was used as a loading control. Shown are representative blots of 3 independent experiments.
f. Quantification of immunoblot analysis using ImageJ software shown in e. n = 3, *p = .000 (ANOVA analysis).
g. Human cTnIAAb specifically bound to the primary cultured myocardial cell membrane as revealed by cell membrane staining and flow cytometric analysis. Shown are representative images of 3 independent experiments.
Our findings suggest that cTnIAAb can bind directly to the myocardial cell membrane and elicit subsequent pathophysiological changes in cells. Indeed, further experiments of live cardiomyocytes using flow cytometry and laser scanning confocal microscopy confirmed the presence of binding site(s) on the myocardial cell membrane for human cTnIAAb (Fig. 2g, Supplementary Fig. 2). Collectively, these findings indicate that human cTnIAAb directly binds to the myocardial cell membrane and causes cell apoptosis, but not hypertrophy.
Supplementary Fig. 2.

Representative immunofluorescence images showing co-localization of cTnIAAb and cell membrane marker as revealed by immunofluorescence confocal microscopy. Green, cTnlAAb; red, Dil, a membrane marker; blue, nuclei. Bar = 10 μm.
3.3. cTnIAAb binds to α-enolase (ENO1) on the myocardial cell surface
Due to the limited availability of purified human cTnIAAb, we used recombinant human cTnI to generate three mouse-derived monoclonal anti-cTnI antibodies (cTnImAb1, cTnImAb2, and cTnImAb3) for use in place of human cTnIAAb in subsequent experiments. Both cTnImAb1 and cTnImAb3 bound to the rat myocardial cell surface, while cTnImAb2 showed weaker binding (Fig. 3a). Moreover, both cTnImAb1 and cTnImAb3, but not cTnImAb2, significantly inhibited the binding of cTnIAAb to myocardial cells (Fig. 3b, c), suggesting that the binding to myocardial cells between cTnIAAb and cTnImAb1/ cTnImAb3 was competitive. It was earlier shown that cTnI mAbs were bond to the epitope-dependent gastric adenocarcinoma cell line [18]. Our results showed that cTnImAb1 and cTnImAb3 also bound to BCG823 (Fig. 3d), consistent with previous study using clone 2F6.6, a.a.r 13–36 [19]. Similarly, purified human cTnIAAb bound to BCG823 cells as well, and this binding was substantially blocked by cTnImAb1 and cTnImAb3 (Fig. 3e, f).
Fig. 3.

cTnImAbs as bioactive alternative for cTnIAAb.
a. Differential binding affinities of cTnImAb1, −2, and −3 for myocardial membrane as revealed by flow cytometry. cTnImAb1 exhibited the highest binding force.
b & c. Flow cytometry showed that cTnImAb1 was a potent competitor against human cTnIAAb in binding to the myocardial cell membrane. Shown are representative images (b) and quantification (c). *p = .000 (ANOVA analysis).
d. Differential binding affinities of cTnImAb1, −2 and −3 for the membranes of BCG823 cells as revealed by flow cytometry. cTnImAb1 exhibited the highest affinity.
e & f. Flow cytometry showed that cTnImAb1 was a potent competitor against human cTnIAAb in binding to the membrane of BCG823 cells. Shown are representative images (e) and quantification (f). *p = .000 (ANOVA analysis).
Further experiments suggested that the binding sites for cTnImAb1 were located on the N-terminal amino acid residues 26–49 of cTnI (Supplemental Tables 1 & 2). This range overlapped the previously reported antigenic sites (a.a.r 13–36) for cTnImAb (2F6.6) [18] and the stable mid-fragment of the cTnI molecule (a.a.r 30–110) [19]. Taken together, the above findings indicate that human cTnIAAb and cTnImAb1/cTnImAb3 share the same or similar antigenic sites on cTnI and that cTnImAb1 could be used in place of human cTnIAAb in the subsequent studies to identify the potential myocardial cell surface binding partner(s) for cTnIAAb and the potential biological effects of this binding.
The BCG823 cell surface showed a high abundance of cTnIAAb binding protein(s), but intracellular cTnI was not detected at mRNA or protein level (Beckman Access2, <0.04 μg / L), in contrast with previous finding; of note though, Chen et al. also found that cTnI detection was epitope-dependent and mRNA expression assayed by semi-quantitative PCR was ten thousand times less than in cardiomyocytes [18]. Hence, we lysed BCG823 cells and performed affinity chromatography with cTnImAb1 to identify cTnImAb1 binding candidate(s). A clear protein band near 51 kDa was observed (Fig. 4a), which was determined to be ENO1 by mass spectrometry and a Mascot search (Supplementary Fig. 3). Further in vitro binding analysis showed that the purified binding protein specifically bound to anti-ENO1 monoclonal antibody, but not to cTnImAb1 (Fig. 4b, left).
Fig. 4.

cTnIAAb binds to α-enolase (ENO1) in myocardial cell membrane.
a. The concentrated purified protein from BCG823 cell lysates through cTnImAb1-dextran affinity chromatography was subjected to SDS-PAGE followed by Coomassie blue staining, which revealed one single band.
b. The purified protein from BCG823 cell lysates specifically bound to ENO1 mAb, but could not bind to cTnImAb1 again.
Left: Western blotting showing that the purified protein from BCG823 cell lysates through cTnImAb1-dextran affinity chromatography could not interact with cTnImAb1 again.
Middle: Western blotting showing that the purified protein from BCG823 cell lysates through cTnImAb1-dextran affinity chromatography could specifically bind to ENO1 mAb (51 KDa). Right: Western blotting showing that the non-reduced protein lysates from mouse cardiomyocytes interacted with cTnImAb1 at both 28 kDa and 51 kDa, while the reduced protein lysates interacted with cTnImAb1 only at 28 kDa.
c. Immunoblot analysis showed that cTnImAb1 and anti-ENO1 pAb bound to the immunoprecipitation-purified endogenous dimeric ENO1 from cardiomyocytes.
Far Left: Mouse cTnImAb1 strongly interacted with the unreduced ENO1 dimer (line 6, ~98 kDa) immunoprecipitated by rabbit ENO1 mAb155102, while it weakly interacted with the reduced endogenous ENO1 (line 3, ~47 kDa). Mouse cTnImAb1 interacted with myocardial cTnI (lines 2 and 5, ~28 kDa) as well as the unreduced input protein sample (line 2, ~98 kDa).
Left: Mouse ENO1 pAb strongly interacted with the unreduced ENO1 dimer (line 6, ~98 kDa) immunoprecipitated by rabbit ENO1 mAb155102, while it weakly interacted with the reduced endogenous ENO1 (line 3, ~47 kDa). Mouse ENO1 pAb interacted with myocardial ENOI (lines 2 and 5, ~47 kDa) as well as the unreduced input protein sample (line 2, ~98 kDa).
Right: Rabbit ENO1 mAb155955 interacted with ENO1 in the input protein sample (lines 2 and 5) as well as the immunoprecipitated endogenous ENO1 by rabbit ENO1 mAb155102 (lines 3 and 6). Rabbit ENO1 mAb155102 also bound to anti rabbit IgG (lines 4 and 7).
Far Right: Mouse actin mAb interacted with the actin in the input protein sample (lines 2 and 5). All images are representative of at least 3 independent iterations.
d. Identification of FLAG-tagged recombinant wild-type and C389A, C357A, C339A, C119A ENO1 protein expressed in 293 T cells.
e. The reaction of recombinant ENO1 protein and 4 point-mutated ENO1 proteins with cTnImAb1 and Anti-ENO1 (non-reducing sample) respectively. Results shown are representative immunoblot analysis of three independent co-immunoprecipitation analysis. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Supplementary Fig. 3.

Mass spectrometric analysis and Mascot search results determined that ENO1 was the protein purified from BCG823 cell lysates through cTnImAb1–dextran affinity chromatography. A. Mass spectrum from mass spectrometric analysis. B. Secondary mass spectrum. C. Mascot search results showed that the only protein with a statistical significance was ENO1.
BLASTP analysis revealed no sequence homology or similar linear epitope between cTnI and ENO1 (data not shown) while ElliPro prediction software [20] revealed the existence of similar conformational epitopes, which were located on a.a.r 41.42.43.44 of cTnI and on a.a.r 6.27.74.79 of ENO1 (data not shown). Therefore, ENO1 obtained by affinity chromatography may cause ENO1 conformational epitope loss during acid elution, and thus cannot bind to cTnImAb1. cTnImAb1 specifically bound to ENO1 (near 49KD and 97KD) and cTnI (28KD) when we performed immunoblot analysis on unreduced samples of primary rat myocardial tissue lysate (Fig. 4b, right). We then co-immunoprecipitated endogenous ENO1 from primary SD rat neonatal cardiomyocyte lysate and analyzed by immunoblotting using mouse Anti-ENO1 pAb (Fig. 4c, left) and mouse cTnImAb1 (Fig. 4c, far left), respectively, along with another rabbit anti-ENO1 mAb (Fig. 4c, right), and mouse β-actin mAb (Fig. 4c, far right). Both cTnImAb1 and anti-ENO1pAb bound to the immunoprecipitation-purified endogenous dimeric ENO1 from cardiomyocytes.
To provide more direct evidence of the binding of cTnImAb1 to ENO1 conformational epitopes, we generated FLAG-tagged expression plasmids for wild-type and four ENO1 cysteines to alanine point mutations, C119A, C339A, C357A, C389A (Fig. 4d). Immunoprecipitation showed that exogenously expressed ENO1-FLAG can specifically bind to both ENO1 mAb and cTnI mAb1, whereas the point mutation C357A binding to cTnI mAb1 was significantly suppressed (Fig. 4e). Taken together our data indicates that cTnI mAb1 binds to ENO1 on cardiac myocyte surface, recognizing conformational epitopes similar to ENO1 and cTnI proteins.
3.4. cTnImAb1 induces cardiomyocyte apoptosis by up-regulating ENO1 expression
We next examined whether ENO1 plays a role in cTnIAAb-triggered cell apoptosis. As expected, cTnImAb1 also induced apoptosis of the primary cultured neonatal rat cardiomyocytes in a dose- (Fig. 5a–c) and time- (Fig. 5d–f) dependent manner, as assessed by Annexin V/PI staining (Fig. 5a, b, d, e), or increase in Bax expression and decrease in Bcl2 expression (Fig. 5c, f), while mouse IgG and cTnTmAb did not significantly affect cardiomyocyte death (Supplementary Fig. 4a-d). We also found that cTnImAb1 treatment significantly increased expression of ENO1 at both protein and mRNA levels in cardiomyocytes (Fig. 5g-k and Supplementary Fig. 5), and silencing Eno1 in myocardial cells attenuated the apoptotic effects of cTnImAb1 on myocardial cells (Fig. 5l, m) as well as reversed the cTnImAb1–induced changes in the expression levels of Bax and Bcl-2 (Fig. 5n, o). Taken together, these results suggest that cTnImAb1 induces cardiomyocyte apoptosis at least in part through binding to membrane ENO1 and enhancing the expression of ENO1.
Fig. 5.

cTnImAb1 upregulates the expression of ENO1 and induces cardiomyocyte apoptosis, while silencing ENO1 attenuates cTnImAb1–induced cardiomyocyte apoptosis.
a & b. cTnImAb1 treatment (0, 25, 50, or 100 μg/ml) for 48 h induced cardiomyocyte apoptosis in a dose-dependent manner as revealed by flow cytometry. a, representative images. b, quantification of 3 independent experiments. *p = .000 (ANOVA analysis).
c. cTnImAb1 treatment for 48 h at indicated doses increased Bax, cleaved Caspase3, and caspase8/p18 expression, and decreased Bcl2 expression in a dose-dependent manner as revealed by immunoblot analysis. β-actin served as a loading control. Shown are representative images of 3 independent experiments.
d&e. cTnImAb1 treatment (50 μg/ml) for indicated times induced cardiomyocyte apoptosis in a time-dependent manner as revealed by flow cytometry. d, representative images. e, quantification of 3 independent experiments, *p = .000 (ANOVA analysis).
f. cTnImAb1 treatment (50 μg/ml) for indicated times increased Bax, cleaved Caspase3, and caspase8/p18 expression, and decreased Bcl2 expression in a dose-dependent manner as revealed by immunoblot analysis. β-actin served as a loading control. Shown are representative images of 3 independent experiments.
g-i. cTnImAb1 treatment (50 μg/ml) for 48 h increased ENO1 expression of primary cultured cardiomyocytes (i) compared with mouse IgG (g) and cTnTmAb (h) treatments as revealed by immunohistochemistry. Bar = 50 μm.
j&k. cTnImAb1 treatment (50 μg/ml) for 48 h increased ENO1 expression compared with mouse IgG treatments as revealed by immunoblot analysis. β-actin served as a loading control. Shown are representative images (j) and quantification by ImageJ-mediated densitometry analysis (k) of 3 independent experiments. *p = .000 (t-test).
l&m. Silencing ENO1 attenuated cTnImAb1 (50 μg/ml)-induced cardiomyocyte apoptosis compared with mock-siRNA group. l, Annexin V-PI staining was used for flow cytometric analysis. Representative images are shown. m, Quantification of Annexin-V/PI staining from 3 independent experiments. 24 h *p = .008, 48 h *p = .000 (t-test).
n&o. RNAi-mediated silencing of Eno1 in primary cultured cardiomyocytes significantly reduced the cTnImAb1 (50 μg/ml)-induced cardiomyocyte apoptosis by increasing Bax expression and decreasing Bcl2 expression compared with mock-siRNA group. Representative immunoblot images are shown in n. ImageJ-based quantification of ENO1 (*ENO1-siRNA p = .000, Mock-siRNA p = .000), Bax (*ENO1-siRNA p = .000, Mock-siRNA p = .000), and Bcl2 (*ENO1-siRNA p = .003, Mock-siRNA p = .020) protein expression is shown in o. (ANOVA analysis).
Supplementary Fig. 4.

cTnTmAb and mouse-IgG did not induce apoptosis in primary cultured cardiomyocytes. A. Treatment with mouse-IgG at different concentrations (0, 25μg/ml, 50μg/ml, 100μg/ml) or 50μg/ml mouse-IgG for 48h did not induce myocardial apoptosis as revealed by FACS. B. Treatment with cTnTmAb at different concentrations (0, 25μg/ml, 50μg/ml, 100μg/ml) or 50μg/ml cTnTmAb for 48h did not induce myocardial apoptosis as revealed by FACS. C. Treatment with mouse-IgG at different concentrations (0, 25μg/ml, 50μg/ml, 100μg/ml) or 50μg/ml mouse-IgG for 48h did not alter the expression of Bax, Bcl2 and cleaved Caspase3 as revealed by immunoblot analysis. β-actin served as a loading control. D. Treatment with cTnTmAb at different concentrations (0, 25μg/ml, 50μg/ml, 100μg/ml) or 50μg/ml cTnTmAb for 48h did not alter the expression of Bax, Bcl2 and cleaved Caspase3 as revealed by immunoblot analysis. β-actin served as a loading control.
Supplementary Fig. 5.

cTnImAb1 significantly up-regulated Eno1 mRNA expression in primary cultured cardiomyocytes compared with mouse-IgG and cTnTmAb groups.
3.5. cTnImAb1 induces cardiac pathological changes and dysfunction
We next established a myocardial injury mouse model as previously reported [10] and explored the effects of cTnImAb1 on myocardial function. cTnImAb1 treatment impaired left ventricular systolic function of mice compared with treatment with either cTnTmAb or mouse IgG (Fig. 6a, b, and Supplemental Tables 3). Mmp3 mRNA expression was significantly up-regulated in cTnImAb1-injected mice compared with cTnTmAb-injected mice and mouse IgG-injected mice, suggesting that cTnImAb1-injected mice developed heart failure (Supplementary Fig. 6).
Fig. 6.

cTnImAb1 induces cardiac dysfunction and pathological changes in mouse heart.
a & b. Representative images of echocardiography (a) and screen shots of catheterization (b) showing decreased cardiac function in mice of the cTnImAb1 group compared with the mouse IgG and cTnTmAb groups.
c. Representative images of hematoxylin and eosin (H&E) staining showing epicardial calcified plaque formation in the mouse hearts of the cTnImAb1 group, which were not seen in those of the mouse IgG and cTnTmAb groups. Bar = 50 μm.
d. Representative images of Masson's Trichrome staining showing interstitial collagen deposition in the mouse hearts of cTnImAb1 group, which were not seen in those of the mouse IgG and cTnTmAb groups. Bar = 50 μm.
e & f. Representative images of TUNEL staining (e) and quantification (f) in the mouse hearts of the cTnImAb1 group compared with that in the mouse IgG and cTnTmAb groups. Bar = 10 μm. *p = .000 (ANOVA analysis).
Supplementary Fig. 6.
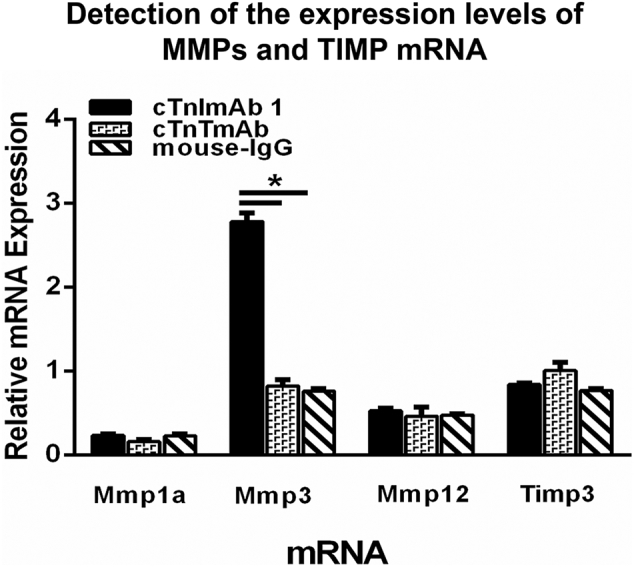
Effects of cTnImAb1, cTnTmAb, and mouse-IgG on the expression of mRNAs encoding matrix metalloproteinase and matrix metalloproteinase inhibitors in mouse hearts. cTnImAb1 treatment significantly increased the expression of matrix metalloproteinase-3 (Mmp3) but not Mmp12 and metal matrix metalloproteinase inhibitor of matrix protein 3 (Timp3) in the hearts compared with treatment with either cTnTmAb or mouse IgG. *p<0.05 vs. cTnTmAb and mouse IgG(t-test, n=3).
Although the global view of mouse hearts from these three groups did not show any obvious differences (Supplementary Fig. 7), pathological examination showed that the hearts of cTnImAb1–injected mice exhibited increased collagen deposition (Fig. 6c-d), and myocardial apoptosis (Fig. 6e, f). There was no obvious increase in infiltration of immune cells, as revealed by immunostaining against CD3, CD4, CD8, CD20, and CD68 (Supplementary Fig. 8).
Supplementary Fig. 7.

Representative global view of the harvested hearts comparing the cTnImAb1, cTnTmAb and mouse IgG treatment groups (Group cTnImAb1 n=21, Group cTnTmAb n=13, Group mouse IgG n=14).
Supplementary Fig. 8.

cTnImAb1 treatment did not increase inflammatory cell infiltration in the mouse heart compared with the cTnTmAb and mouse IgG groups. Immunohistochemical staining against CD4, CD8, CD20, and CD68 was performed on heart tissue sections. Representative images are shown (Group cTnImAb1 n=21, Group cTnTmAb n=13, Group mouse IgG n=14).
3.6. cTnImAb1 causes activation of PTEN / Akt pathway and increased cardiomyocyte apoptosis
To further clarify the signaling pathways involved in cTnImAb1–induced myocardial apoptosis, we used PathScan® Akt Signaling Antibody Array analysis to investigate the changes in phosphorylation of 16 proteins related to the Akt signaling pathway in mouse myocardial tissues (Fig. 7a-c) and found that PTEN Ser380 phosphorylation was significantly upregulated and phosphorylation of Akt Thr308 was significantly downregulated (Fig. 7a-e). In vitro experiments further confirmed that cTnImAb1 stimulation of the primary cardiomyocytes induced PTEN Ser380 phosphorylation at 8 h, which was decreased after 24 h of treatment, while the expression of Akt Thr308 in 8–12 h was significantly reduced and then recovered after 48 h. In contrast, mouse IgG had no significant effects on PTEN Ser380 phosphorylation, but enhanced the expression of Akt Thr308 from 24 to 36 h (Fig. 7f-h). In addition, knockdown of eno1 expression by specific siRNA significantly reduced the PTEN Ser380/total PTEN ratio within 4–12 h, while significantly increasing the Akt Thr308/total Akt ratio within 4–12 h compared with the mock-siRNA group (Fig. 7i-l). These findings collectively suggest that cTnImAb1 activates PTEN/Akt signaling in myocardial cells, which is, at least in part if not all, mediated by ENO1.
Fig. 7.

cTnImAb1 treatment increases ENO1 expression followed by activation of PTEN and suppression of Akt signaling in mouse hearts.
a. Heat map from PathScan® Akt Signaling Antibody Array kit analysis showing changes in 16 phosphorylated proteins.
b & c. Statistical analysis of heat map data in a and volcano plot showing that cTnImAb1 treatment significantly increased PTEN Ser380 phosphorylation in the mouse heart. 1. PTEN Ser380, *p = .023, (t-test). 2. GSK-3b Ser9, *p = .038, (t-test). 3. PTEN Ser380, *p = .031 (t-test).
d&e. Immunoblot analysis showed that cTnImAb1 treatment significantly increased ENO1 expression and PTEN Ser380 phosphorylation but suppressed Akt Thr308 phosphorylation. β-actin served as a loading control. e is the statistical analysis of d. ENO1 *p = .002, pPTEN/PTEN/Actin, *p = .003, pAkt/Akt/Actin, *p = .003. (ANOVA analysis).
f-h. cTnImAb1 treatment (50 μg/ml) induced changes in ENO1 expression and phosphorylation of PTEN Ser380 and Akt Thr308 at different time points in cultured cardiomyoyctes. cTnImAb1 increased the p-Ser380-PTEN/total PTEN ratio at 8 h but decreased the pThr308-AKT/total Akt ratio at 8–12 h. Mouse IgG treatment did not alter pSer380 PTEN but significantly increased the pThr308-Akt/total Akt ratio at 24–36 h. f shows representative blot images of three independent experiments. g and h represent ImageJ-based densitometry analysis of relative changes in p-Ser380-PTEN/total PTEN ratio (G, 8 h *p = .020, 12 h *p = .024, t-test) and pThr308-AKT/total Akt (H, 12 h *p = .018, 24 h *p = .001, 36 h *p = .001, t-test).
i-l. Knockdown of ENO1 significantly suppressed cTnImAb1–induced alteration in PTEN/Akt signaling. Cultured cardiomyocytes were transfected with siRNA targeting Eno1 and then subjected to cTnImAb1 treatment (50 μg/ml) for 4–48 h as indicated. Mock-siRNA was used as a control. Depletion of Eno1 suppressed the p-PTEN Ser380/total PTEN ratio and increased the pAkt-Thr308/total Akt ratio at 4, 8, and 12 h, respectively. i shows representative blot images of three independent experiments. j represents ImageJ-based densitometry analysis of relative changes in ENO1 protein levels (J,48 h *p = .000, t-test). k and l represent ImageJ-based densitometry analysis of relative changes in p-Ser380-PTEN/total PTEN ratio (k, 48 h *p = .000, t-test) and pThr308-AKT/total Akt (l, 6 h *p = .000, 12 h *p = .000, t-test).
4. Discussion
The results of the current study cumulatively showed that human cTnIAAb interacts with ENO1 present in the myocardial membrane and induces apoptosis of cultured cardiomyocytes in in vitro and in vivo models. The cTnImAb1, which possessed bioactivity similar to that of human cTnIAAb, triggered cardiomyocyte apoptosis via binding membrane ENO1, increasing ENO1 expression, promoting phosphorylation of PTEN Ser380, and suppressing Akt activity in the mouse model of cTnImAb-induced myocardial injury (Fig. 8).
Fig. 8.

Graphical mechanism summary: Human cTnIAAb interacts with ENO1 present in the myocardial membrane and induces apoptosis of cultured cardiomyocytes. Human cTnIAAb can trigger cardiomyocyte apoptosis via binding membrane ENO1, increasing ENO1 expression, promoting phosphorylation of PTEN Ser380, and suppressing Akt activity in the mouse model of cTnImAb-induced myocardial injury. This induces the pro-apoptotic Bax and suppresses the anti-apoptotic Bcl2 expression resulting in cardiomyocyte apoptosis.
To the best of our knowledge, we identified for the first time that human cTnIAAb bound to ENO1 in the membrane of cardiomyocytes and subsequently caused cardiomyocyte apoptosis. ENO1, which was discovered as a glycolytic enzyme as well as a plasminogen receptor [[21], [22], [23]], is present in the cytoplasm but also on the cell surface as a dimer [[24], [25], [26]]. Involvement of ENO1 in tumorigenesis has been investigated [[27], [28], [29]]; however, studies of its direct role in mediating myocardial function and dysfunction have been limited. Previous studies showed that ENO1 expression was upregulated in the hypertrophic hearts of spontaneously hypertensive rats and in the diabetic hearts of a rat model [25,26]. We revealed here that ENO1 mediated the pro-apoptotic activity of cTnIAAb in cardiomyocytes. In addition, ENO1 exhibits non-enzymatic activities and is considered as a multifunctional protein [23]. For example, on the tumor cell surface, ENO1, as a fibrin lysosomal receptor, regulates the proliferation and migration of cell [30,31]. A more recent study has shown that ENO1 expression was different in different tissues and was relatively higher in the heart, and ENO1 promotes cardiomyocyte apoptosis independent of its enzymatic activity [32]. Thus, the non-enzymatic activity of ENO1 is expected to play important roles in cellular activity as well. Indeed, ENO1 has been shown to function as autoantigens in various diseases including Hashimoto's encephalopathy, lupus nephritis, retinopathy, rheumatic arthritis, systemic sclerosis, autoimmune ovarian failure, relapsing polychondritis and lung cancer [33]. Although we have provided evidence that ENO1 mediated cTnImAb1–induced cardiomyocyte apoptosis, whether this function involves its enzymatic activity remains to be elucidated.
Although the non-enzymatic activity is involved in a variety of cellular events, the ENO1 downstream effectors have not been fully deciphered. Overexpression of Myc promoter-binding protein-1 (MBP-1), a short version of ENO1, induces cell stress through the AKT/PERK/eIF2α axis [32]. Also, ENO1 was shown to be involved in the differentiation of alveolar epithelial cells through Wnt-β-catenin signaling [34]. In the heart, ENO1 contributes to doxorubicin-induced cardiomyocyte injury through suppression of AMPK [35]. In the present study, we found that ENO1, once bound by cTnlmAb1, triggered apoptosis at least partially via the PTEN signaling axis. PTEN is a negative regulator of Akt, and the implication of PTEN signaling in mediating cardiac physiology and pathophysiology has been well documented [36,37]. PTEN has therefore been proposed as a potential therapeutic target for the treatment of ischemic cardiac injury [38]. In both in vitro and vivo experiments, we found that cTnIAAb induced cell apoptosis, which coincided with increased phosphorylation of PTEN Ser380 and decreased phosphorylation of Akt Thr308. Therefore, we believe that the PTEN signaling axis was the downstream effector of the functional interaction between cTnIAAb and ENO1, at least in cardiomyocytes.
One limitation of the current study is that we were unable to obtain a sufficient amount of human cTnIAAb to replicate all in vitro and in vivo studies performed with cTnImAb. Also, given that human cTnIAAb is a polyclonal antibody, whether other proteins in cardiomyocyte membrane are also involved in cTnIAAb-triggered cell death or other pathophysiological activities remains to be elucidated. In addition, Eno1 knockout mice should be used in the future to further confirm the role of ENO1 in cTnImAb1–induced cardiac injury in vivo.
Of note, protein components in cardiomyocytes, such as cTnI, rarely come into contact with the immune system during embryonic development, and often referred to as masked antigens. However, when myocardial cells are damaged, intracellular cTnI is released into the blood circulation, which may stimulate immunity that results in production of cTnIAAb. In the present study, injection of cTnImAb1 significantly accentuated cardiac dysfunction in mice with no evidence of inflammatory infiltration. In addition, in our in vitro experiment, cTnIAAb stimulation failed to induce the cardiomyocyte hypertrophic response. Thus, we believe that the heart failure and pathological changes in the hearts induced by cTnIAAb within 12 weeks were mainly associated with apoptosis and not with inflammation.Mechanistically, ENO1 is the cTnIAAb binding partner on the cardiomyocyte cell surface and mediates cTnIAAb-induced cell death through potentiation of PTEN phosphorylation and suppression of Akt phosphorylation. Hence, the cTnIAAb-ENO1-PTEN-Akt signaling axis plays an important role in cTnIAAb-linked pathophysiological events, particularly apoptosis. Our findings reveal a novel potential therapeutic target, i.e., ENO1, for treatment of the cTnIAAb-positive AMI patients and in ventricular remodeling.
The following are the supplementary data related to this article.
This video shows the animation for 3D reconstruction of primary myocardial cells in Supplement Figure 2A. In this video, the primary myocardial cells co-stained with cTnIAAb-FITC and DiI are scanned in layers using a laser confocal microscope to obtain a set of optical sections, and then two-color image processing was carried out by 3D reconstruction method to obtain real 3D structure of cells. The surface morphology and the internal structure of the cell were observed by rotating to different angles.
Supplementary material
Declaration of Competing Interests
None of the authors have any relationships/conditions/circumstances that present a potential conflict of interest.
Author contributions
QS and GST designed the whole experiments and guided the entire experiments, and are responsible for all experiment data; YW and YHQ carried out the experiments; YW prepared the manuscript; YL provided human cardiac tissue samples; LZ helped with animal modeling; XXZ were in charge of follow-up of cardiologic disease patients, and provided patient blood samples; YYL helped with the flow cytometry experiment; and SWL designed the guided the molecular biological experiments. All authors have read the manuscript and approved the submission.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (Grant#: 81260026) and the Doctoral Innovation Fund from Second Military Medical University of China. The funders had no role in study design, data collection, data analysis, interpretation, writing of the manuscript. We are grateful for the help from the Dr. Yalin Sun (swinel@hotmail.com) from Department of Statistics at Second Military Medical University, Dr. Junbo Ge (jbge@zs-hospital.sh.cn) from the Cardiovascular Laboratory, Zhongshan Hospital, Fudan University, Dr. Xin Hongbo (hongboxin@yahoo.com) from Nanchang University, and Dr. Jun Zhang (156088435@qq.com) from Shanghai Delta Health Hospital.
Contributor Information
Gu-sheng Tang, Email: drake015@163.com.
Qian Shen, Email: shenq611@163.com.
References
- 1.Tang G., Wu Y., Zhao W. Multiple immunoassay systems are negatively interfered by circulating cardiac troponin I autoantibodies. Clin Exp Med. 2012;12:47–53. doi: 10.1007/s10238-011-0141-x. [DOI] [PubMed] [Google Scholar]
- 2.Shmilovich H., Danon A., Binah O. Autoantibodies to cardiac troponin I in patients with idiopathic dilated and ischemic cardiomyopathy. Int J Cardiol. 2007;117:198–203. doi: 10.1016/j.ijcard.2006.04.077. [DOI] [PubMed] [Google Scholar]
- 3.Eriksson S., Hellman J., Pettersson K. Autoantibodies against cardiac troponins. N Engl J Med. 2005;352:98–100. doi: 10.1056/NEJM200501063520123. [DOI] [PubMed] [Google Scholar]
- 4.Erer H.B., Guvenc T.S., Kemik A.S. Troponin and anti-troponin autoantibody levels in patients with ventricular noncompaction. PLoS One. 2013;8 doi: 10.1371/journal.pone.0057648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pettersson K., Eriksson S., Wittfooth S. Autoantibodies to cardiac troponin associate with higher initial concentrations and longer release of troponin I in acute coronary syndrome patients. Clin Chem. 2009;55:938–945. doi: 10.1373/clinchem.2008.115469. [DOI] [PubMed] [Google Scholar]
- 6.Wu A.H., Apple F.S., Gibler W.B. National academy of clinical biochemistry standards of laboratory practice: recommendations for the use of cardiac markers in coronary artery diseases. Clin Chem. 1999;45:1104–1121. [PubMed] [Google Scholar]
- 7.Adams J.E., 3rd, Bodor G.S., Davila-Roman V.G. Cardiac troponin I. A marker with high specificity for cardiac injury. Circulation. 1993;88:101–106. doi: 10.1161/01.cir.88.1.101. [DOI] [PubMed] [Google Scholar]
- 8.Bohner J., von Pape K.W., Hannes W., Stegmann T. False-negative immunoassay results for cardiac troponin I probably due to circulating troponin I autoantibodies. Clin Chem. 1996;42:2046. [PubMed] [Google Scholar]
- 9.Nishimura H., Okazaki T., Tanaka Y. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 10.Okazaki T., Tanaka Y., Nishio R. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat Med. 2003;9:1477–1483. doi: 10.1038/nm955. [DOI] [PubMed] [Google Scholar]
- 11.Volz H.C., Buss S.J., Li J. Autoimmunity against cardiac troponin I in ischaemia reperfusion injury. Eur J Heart Fail. 2011;13:1052–1059. doi: 10.1093/eurjhf/hfr098. [DOI] [PubMed] [Google Scholar]
- 12.Goser S., Andrassy M., Buss S.J. Cardiac troponin I but not cardiac troponin T induces severe autoimmune inflammation in the myocardium. Circulation. 2006;114:1693–1702. doi: 10.1161/CIRCULATIONAHA.106.635664. [DOI] [PubMed] [Google Scholar]
- 13.Miettinen K.H., Eriksson S., Magga J. Clinical significance of troponin I efflux and troponin autoantibodies in patients with dilated cardiomyopathy. J Card Fail. 2008;14:481–488. doi: 10.1016/j.cardfail.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 14.Zhou S., Wang H.R., Xue M. Relationship between cardiac troponin I autoantibody and left ventricular remodeling in patients with acute myocardial infarction. Chin Circ J. 2018;33:322–326. [Google Scholar]
- 15.LaFramboise W.A., Scalise D., Stoodley P. Cardiac fibroblasts influence cardiomyocyte phenotype in vitro. Am J Physiol Cell Physiol. 2007;292:C1799–C1808. doi: 10.1152/ajpcell.00166.2006. [DOI] [PubMed] [Google Scholar]
- 16.Kan A., Mohamedali A., Tan S.H. An improved method for the detection and enrichment of low-abundant membrane and lipid raft-residing proteins. J Proteomics. 2013;79:299–304. doi: 10.1016/j.jprot.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 17.Maayah Z.H., Ansari M.A., El Gendy M.A., Al-Arifi M.N., Korashy H.M. Development of cardiac hypertrophy by sunitinib in vivo and in vitro rat cardiomyocytes is influenced by the aryl hydrocarbon receptor signaling pathway. Arch Toxicol. 2014;88:725–738. doi: 10.1007/s00204-013-1159-5. [DOI] [PubMed] [Google Scholar]
- 18.Chen C., Liu J.B., Bian Z.P. Cardiac troponin I is abnormally expressed in non-small cell lung cancer tissues and human cancer cells. Int J Clin Exp Pathol. 2014;7:1314–1324. [PMC free article] [PubMed] [Google Scholar]
- 19.Savukoski T., Twarda A., Hellberg S. Epitope specificity and IgG subclass distribution of autoantibodies to cardiac troponin. Clin Chem. 2013;59:512–518. doi: 10.1373/clinchem.2012.194860. [DOI] [PubMed] [Google Scholar]
- 20.Ponomarenko J., Bui H.H., Li W. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008;9:514. doi: 10.1186/1471-2105-9-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cappello P., Principe M., Bulfamante S., Novelli F. Alpha-Enolase (ENO1), a potential target in novel immunotherapies. Front Biosci (Landmark Ed) 2017;22:944–959. doi: 10.2741/4526. [DOI] [PubMed] [Google Scholar]
- 22.Mori Y., Yamaguchi M., Terao Y., Hamada S., Ooshima T., Kawabata S. alpha-Enolase of Streptococcus pneumoniae induces formation of neutrophil extracellular traps. J Biol Chem. 2012;287:10472–10481. doi: 10.1074/jbc.M111.280321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diaz-Ramos A., Roig-Borrellas A., Garcia-Melero A., Lopez-Alemany R. alpha-Enolase, a multifunctional protein: its role on pathophysiological situations. J Biomed Biotechnol. 2012;2012 doi: 10.1155/2012/156795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piast M., Kustrzeba-Wojcicka I., Matusiewicz M., Banas T. Molecular evolution of enolase. Acta Biochim Pol. 2005;52:507–513. [PubMed] [Google Scholar]
- 25.Zhu L.A., Fang N.Y., Gao P.J., Jin X., Wang H.Y. Differential expression of alpha-enolase in the normal and pathological cardiac growth. Exp Mol Pathol. 2009;87:27–31. doi: 10.1016/j.yexmp.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 26.Lu N., Zhang Y., Li H., Gao Z. Oxidative and nitrative modifications of alpha-enolase in cardiac proteins from diabetic rats. Free Radic Biol Med. 2010;48:873–8781. doi: 10.1016/j.freeradbiomed.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 27.Hsiao K.C., Shih N.Y., Chu P.Y. Anti-alpha-enolase is a prognostic marker in postoperative lung cancer patients. Oncotarget. 2015;6:35073–35086. doi: 10.18632/oncotarget.5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murtas C., Bruschi M., Candiano G., Bonanni A., Ghiggeri G.M. Anti-alpha-enolase antibodies in membranous nephropathy: isotype matters. Clin Exp Nephrol. 2017;21:171–172. doi: 10.1007/s10157-016-1263-y. [DOI] [PubMed] [Google Scholar]
- 29.Principe M., Ceruti P., Shih N.Y. Targeting of surface alpha-enolase inhibits the invasiveness of pancreatic cancer cells. Oncotarget. 2015;6:11098–11113. doi: 10.18632/oncotarget.3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.White-Al Habeeb N.M., Di Meo A., Scorilas A. Alpha-enolase is a potential prognostic marker in clear cell renal cell carcinoma. Clin Exp Metastasis. 2015;32:531–541. doi: 10.1007/s10585-015-9725-2. [DOI] [PubMed] [Google Scholar]
- 31.Maranto C., Perconti G., Contino F., Rubino P., Feo S., Giallongo A. Cellular stress induces cap-independent alpha-enolase/MBP-1 translation. FEBS Lett. 2015;589:2110–2116. doi: 10.1016/j.febslet.2015.06.030. [DOI] [PubMed] [Google Scholar]
- 32.Gao S., Li H., Feng X.J. alpha-Enolase plays a catalytically independent role in doxorubicin-induced cardiomyocyte apoptosis and mitochondrial dysfunction. J Mol Cell Cardiol. 2015;79:92–103. doi: 10.1016/j.yjmcc.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 33.Ji H., Wang J., Guo J. Progress in the biological function of alpha-enolase. Anim Nutr. 2016;2:12–17. doi: 10.1016/j.aninu.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mutze K., Vierkotten S., Milosevic J., Eickelberg O., Konigshoff M. Enolase 1 (ENO1) and protein disulfide-isomerase associated 3 (PDIA3) regulate Wnt/beta-catenin-driven trans-differentiation of murine alveolar epithelial cells. Dis Model Mech. 2015;8:877–890. doi: 10.1242/dmm.019117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhan P., Zhao S., Yan H. alpha-Enolase promotes tumorigenesis and metastasis via regulating AMPK/mTOR pathway in colorectal cancer. Mol Carcinog. 2017;56:1427–1437. doi: 10.1002/mc.22603. [DOI] [PubMed] [Google Scholar]
- 36.Wang J., Chen H., Zhou Y. Atorvastatin inhibits myocardial apoptosis in a swine model of coronary microembolization by regulating PTEN/PI3K/Akt signaling pathway. Cell Physiol Biochem. 2016;38:207–219. doi: 10.1159/000438622. [DOI] [PubMed] [Google Scholar]
- 37.Sussman M.A., Volkers M., Fischer K. Myocardial AKT: the omnipresent nexus. Physiol Rev. 2011;91:1023–1070. doi: 10.1152/physrev.00024.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roe N.D., Xu X., Kandadi M.R. Targeted deletion of PTEN in cardiomyocytes renders cardiac contractile dysfunction through interruption of Pink1-AMPK signaling and autophagy. Biochim Biophys Acta. 2015;1852:290–298. doi: 10.1016/j.bbadis.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This video shows the animation for 3D reconstruction of primary myocardial cells in Supplement Figure 2A. In this video, the primary myocardial cells co-stained with cTnIAAb-FITC and DiI are scanned in layers using a laser confocal microscope to obtain a set of optical sections, and then two-color image processing was carried out by 3D reconstruction method to obtain real 3D structure of cells. The surface morphology and the internal structure of the cell were observed by rotating to different angles.
Supplementary material