

Conspectus
A characteristic phenomenon of lanthanide–fullerene interactions is the transfer of metal valence electrons to the carbon cage. With early lanthanides such as La, a complete transfer of six valence electrons takes place for the metal dimers encapsulated in the fullerene cage. However, the low energy of the σ-type Ln–Ln bonding orbital in the second half of the lanthanide row limits the Ln2 → fullerene transfer to only five electrons. One electron remains in the Ln–Ln bonding orbital, whereas the fullerene cage with a formal charge of −5 is left electron-deficient. Such Ln2@C80 molecules are unstable in the neutral form but can be stabilized by substitution of one carbon atom by nitrogen to give azafullerenes Ln2@C79N or by quenching the unpaired electron on the fullerene cage by reacting it with a chemical such as benzyl bromide, transforming one sp2 carbon into an sp3 carbon and yielding the monoadduct Ln2@C80(CH2Ph). Because of the presence of the Ln–Ln bonding molecular orbital with one electron, the Ln2@C79N and Ln2@C80(R) molecules feature a unique single-electron Ln–Ln bond and an unconventional +2.5 oxidation state of the lanthanides.
In this Account, which brings together metallofullerenes, molecular magnets, and lanthanides in unconventional valence states, we review the progress in the studies of dimetallofullerenes with single-electron Ln–Ln bonds and highlight the consequences of the unpaired electron residing in the Ln–Ln bonding orbital for the magnetic interactions between Ln ions. Usually, Ln···Ln exchange coupling in polynuclear lanthanide compounds is weak because of the core nature of 4f electrons. However, when interactions between Ln centers are mediated by a radical bridge, stronger coupling may be achieved because of the diffuse nature of radical-based orbitals. Ultimately, when the role of a radical bridge is played by a single unpaired electron in the Ln–Ln bonding orbital, the strength of the exchange coupling is increased dramatically. Giant exchange coupling in endohedral Ln2 dimers is combined with a rather strong axial ligand field exerted on the lanthanide ions by the fullerene cage and the excess electron density localized between two Ln ions. As a result, Ln2@C79N and Ln2@C80(CH2Ph) compounds exhibit slow relaxation of magnetization and exceptionally high blocking temperatures for Ln = Dy and Tb. At low temperatures, the [Ln3+–e–Ln3+] fragment behaves as a single giant spin. Furthermore, the Ln–Ln bonding orbital in dimetallofullerenes is redox-active, which allows its population to be changed by electrochemical reactions, thus changing the magnetic properties because the change in the number of electrons residing in the Ln–Ln orbital affects the magnetic structure of the molecule.
Introduction
Increasing the single-ion magnetic anisotropy1−5 and engineering the intramolecular coupling between magnetic ions6−9 are the two main strategies for improving the performance of single-molecule magnets (SMMs). Realization of both approaches is possible in endohedral metallofullerenes (EMFs) with lanthanide (Ln) ions.10−13 High magnetic anisotropy has been achieved in clusterfullerenes,14−18 i.e., EMFs combining metals with nonmetal ions in the endohedral species,11 whereas dimetallofullerenes (di-EMFs) offer unprecedented possibilities for strong exchange coupling.19−23
Metal–metal bonding is a well-known phenomenon in the chemistry of transition metals,24 but lanthanide–lanthanide bonds in molecular compounds to date have been obtained only inside fullerene cages. Moreover, such Ln–Ln bonding interactions can be stabilized in di-EMFs in the form of a unique single-electron Ln–Ln bond, which is the main subject of this Account. The term single-electron Ln–Ln bond refers to the situation where the di-EMF molecule features a singly occupied Ln–Ln bonding molecular orbital (MO). The first di-EMFs featuring single-electron Ln–Ln bonds were the azafullerenes Ln2@C79N (Ln = Y, Tb, Gd), which were discovered in 2008 (it should be noted that because of the similarities in their electronic structures and oxidation states, Sc, Y, and La are considered on equal footing with 4f elements in this Account).25,26 Density functional theory (DFT) studies of di-EMFs in 2012–2014 showed that single-electron Ln–Ln bonds can be obtained in other di-EMFs.27,28 Stable La2@C80 monoadducts with single-electron Ln–Ln bonds were obtained in 2015 and 2016.29,30 Giant exchange coupling and promising SMM properties were predicted for Ln2@C79N (Ln = Gd, Dy) in 2015.31,32 These predictions for Gd2@C79N were confirmed experimentally in 201820,21 along with studies of the spin coherence properties of Gd2@C79N21 and its use for dynamical nuclear polarization at high fields.33 Ln2@C80(CH2Ph) monoadducts with a single-electron Ln–Ln bond were obtained for different lanthanides in 2017–2019, and excellent SMM performance was demonstrated for Dy and Tb di-EMFs.22,23 A high blocking temperature of magnetization and giant coercivity were also reported for Tb2@C79N in 2019.19 This short and incomplete chronicle highlights the fast progress achieved by concerted computational and experimental studies of di-EMFs with single-electron Ln–Ln bonds during the past few years. In this Account, we summarize the current knowledge on these unique molecules.
Valence State of Lanthanides in Metallofullerenes
A transfer of metal valence electrons to the fullerene results in zwitterionic EMF molecules with an endohedral cation and anionic cage. In monometallofullerenes, the oxidation state of the lanthanide varies: Sm, Eu, Tm, and Yb are divalent, but other lanthanides prefer a trivalent state. This difference correlates with the third ionization potentials (IP3) of lanthanide atoms (Figure 1a).36 The metals with IP3 exceeding 23 eV donate two electrons to the fullerene, whereas the IP3 values for trivalent La, Ce, Pr, Nd, Gd, Tb, Dy, Ho, Er, and Lu are below the threshold of 23 eV.
Figure 1.

(a) Third ionization potentials (IP3, blue dots) and energies of the 4fn5d16s2 → 4fn5d26s1 excitations (green dots) of lanthanides.34 The red dashed horizontal line marks the border between divalent and trivalent lanthanides in monometallofullerenes. (b) MO levels in C80-Ih and C82-C3v cages (black, occupied MOs; pink, vacant MOs) as well as La2 and Lu2 dimers. Reproduced with permission from ref (35). Copyright 2018 Elsevier. (c) Molecules of La2@C80-Ih with the La–La bonding LUMO and Lu2@C82-C3v with the Lu–Lu bonding HOMO.
Di-EMFs are well-known for the early lanthanides La and Ce and for heavier lanthanides such as Er and Lu. However, di-EMFs for metals in the middle of the lanthanide row could not be isolated until recently. Another puzzling phenomenon is the size and isomerism of fullerenes typical for di-EMFs with different lanthanides. The C80-Ih cage has enhanced stability in the 6– state and is therefore typical for the endohedral species that donate six electrons. The high abundances of La2@C80-Ih and Ce2@C80-Ih thus agree with the 3+ oxidation state of La and Ce. For Lu and Er, however, the most abundant di-EMF cages are C82-Cs(6) and C82-C3v(8), which are typical for EMFs with the fullerene charge of 4–,11 and the spectroscopic properties of Ln2@C82 and the clusterfullerenes Ln2S@C82 and Ln2C2@C82 are very similar.37,38 These facts suggest the fullerene charge of 4– in Er2@C82 and Lu2@C82, implying the 2+ oxidation state of Er and Lu. The different behaviors of La/Ce and Er/Lu in di-EMFs cannot be explained by the IP3 criterion and are rooted in the MO structure of the Ln2 dimers.
Figure 1b compares the MO levels of C80-Ih and C82-C3v(8) to those of La2 and Lu2.27,35 The fourfold-degenerate orbital occupied by two electrons makes C80-Ih unstable, but filling this MO with six more electrons produces a stable closed-shell electronic structure in C806–. La2 has a (6s)σg2(5d)πu4 configuration with six valence electrons,39 and since its occupied MO levels are considerably higher in energy than the LUMO in C80-Ih, six electrons are transferred from La2 to the fullerene in La2@C80-Ih, resulting in the charge distribution (La3+)2@C806–. La2@C80 has no valence electrons in the metal-localized orbitals, but its LUMO resembles the (6s)σg2 orbital of the La2 dimer (Figure 1c).
The orbitals of Lu2 with configuration (6s)σg2(6s)σu2(5d)πu2 are spread in a broader energy range than those of La2 (Figure 1b),39 and the (6s)σg2 level in Lu2 has a lower energy than the LUMO of C80-Ih. Therefore, the Lu2 dimer in Lu2@C80-Ih does not transfer six electrons as required for the closed-shell electron configuration. The fullerene C82-C3v(8) with two low-energy unoccupied MOs is a more suitable host for Lu2, as it is stabilized by the addition of four electrons.40 In Lu2@C82, four electrons from the (6s)σu2(5d)πu2 levels of Lu2 are transferred to the fullerene, whereas the low-energy (6s)σg2 orbital of Lu2 remains occupied. The formal charge distribution is (Lu2+)2@C824–, and the Lu–Lu bonding orbital resembling the (6s)σg2 MO of Lu2 is the HOMO of the di-EMF (Figure 1c).
In the middle of the lanthanide row, the energy of the (6s)σg2 MO in the Ln2 dimer is not low enough to stabilize the Ln2+ state but not high enough to ensure a transfer of six electrons in di-EMFs. Therefore, these lanthanides cannot produce di-EMFs with a closed-shell electronic structure. This seemingly simple conclusion was formulated only in 2014 in a computational study of Y2@C80-Ih,28 which showed that Y2@C80-Ih is a triplet with one electron populating the Y–Y bonding orbital and the formal charge distribution (Y2.5+)2@C805–. The same situation was also predicted for Gd and Lu.28,41 With only one electron occupying the (6s)σg2 Ln–Ln bonding orbital, such di-EMFs feature a single-electron Ln–Ln bond.
The valence states of lanthanides in di-EMFs are summarized in Figure 2a. Dimers of early lanthanides transfer six electrons to fullerenes, leading to a Ln3+ state and the Ln–Ln bonding orbital as the LUMO. Dimers of lanthanides at the end of the 4f row transfer only four electrons to fullerenes and keep the Ln–Ln bond in di-EMFs with a 2+ state of the Ln atoms. Finally, dimers of lanthanides in the middle of the row transfer five electrons to the hosting fullerene and form di-EMFs with a single-electron Ln–Ln bond and an oxidation state of Ln2.5+. The valence state of lanthanides in di-EMFs is thus governed by the energy matching between unoccupied MOs of the fullerene and the (6s)σg2 MO of the Ln2 dimer. Whereas the IP3 does not correlate with the (6s)σg2 orbital energy, the 4fn5d16s2 → 4fn5d26s1 excitation energy changes in accord with it (Figure 1a; see ref (27)). The state of the lanthanides in di-EMFs also depends on the fullerene host. For instance, Y and Er form both (Ln2+)2@C82 and (Ln2.5+)2@C80 di-EMFs (Figure 2a), and computations showed that the (La2.5+)2@C2n5– state may be preferable for C92–C96.42
Figure 2.
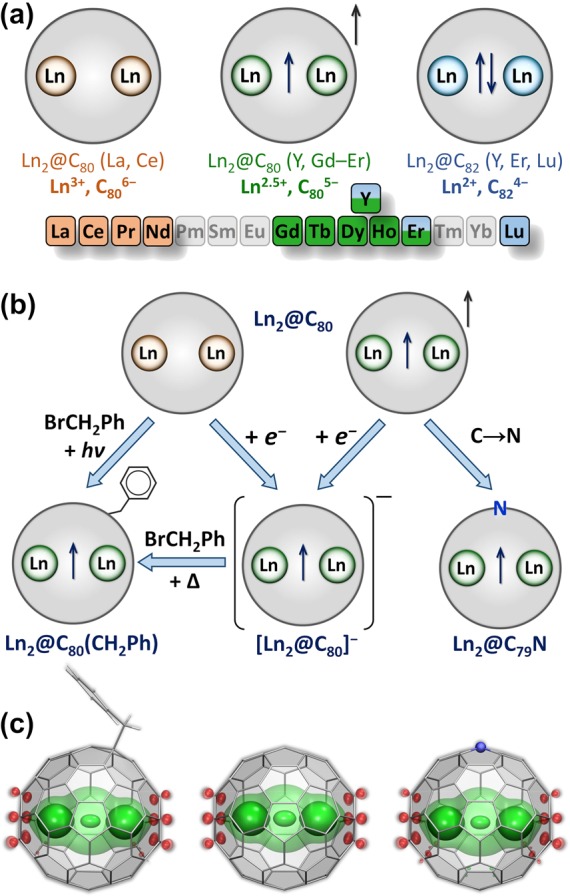
(a) Oxidation states of lanthanides in dimetallofullerenes; the color code for different states (light brown for Ln3+, light green for Ln2.5+, and light blue for Ln2+) is used in subsequent figures. (b) Stabilization of di-EMFs with a single-electron Ln–Ln bond in the form of Ln2@C80(CH2Ph), [Ln2@C80]−, and Ln2@C79N. (c) Spin density distributions in Gd2@C80(CH2Ph), [Gd2@C80]−, and Gd2@C79N (“+”, green; “–”, red; transparent and solid isosurfaces have isovalues of 0.0012 and 0.014, respectively).
Stabilization of Single-Electron Ln–Ln Bonds in Dimetallofullerenes
Ions of Dimetallofullerenes
Depending on the nature of the frontier orbitals of Ln2@C2n, different strategies are required to stabilize single-electron Ln–Ln bonds (Figure 2b). For (Ln3+)2@C2n6–, a single-electron Ln–Ln bond is created by addition of one electron to the Ln–Ln bonding LUMO. Indeed, reduction of La2@C80 leads to the radical anion [La2@C80]− with a single-electron Ln–Ln bond.43,44 In (Ln2.5+)2@C2n5–, the single-electron Ln–Ln bond is already present, but the fullerene has an unpaired electron, and addition of one electron produces an anion with a closed-shell structure of the cage, (Ln2.5+)2@C2n6–.23,41,42 Formation of such anions is crucial for the extraction of (Ln2.5+)2@C2n5– from the carbon soot after arc-discharge synthesis. The radical nature results in high reactivity and polymerization of such di-EMFs, making them insoluble in CS2 or aromatic solvents. However, dimethylformamide (DMF) extracts EMFs in the form of anions.45−47 Thus, single-electron reduction of both (Ln3+)2@C2n6– and (Ln2.5+)2@C2n5– di-EMFs yields (Ln2.5+)2@C2n6– anions with a closed-shell electronic structure of the fullerene.
For (Ln2+)2@C2n4– with a two-electron Ln–Ln bond, an obvious route to attain a single-electron Ln–Ln bond is a one-electron oxidation. Electrochemical studies of Ln2@C82 (Ln = Sc, Y, Er, Lu) proved that oxidation is a metal-based process forming (Ln2.5+)2@C2n4– cations.37,48,49 Chemical oxidation of Sc2@C82 and Er2@C82 with [(4-BrC6H4)3N]SbCl6 gave corresponding cation radicals,37 and an electron paramagnetic resonance (EPR) study of [Sc2@C82]+ revealed a giant 45Sc hyperfine coupling constant of 19.9 mT, in agreement with the formation of a singly occupied Sc–Sc bonding MO. For the Er analogue, superconducting quantum interference device (SQUID) magnetometry showed considerable differences in the magnetization behaviors of Er2@C82 and its salt [Er2@C82]+SbCl6–.37
Dimetallofullerene Derivatives
(Ln2.5+)2@C2n6– anions react with benzyl bromide to give noncharged benzyl monoadducts Ln2@C2n(CH2Ph). A series of Ln2@C80(CH2Ph) compounds ({Ln2} hereafter) with Ln2 = Y2, Gd2, Tb2, Dy2, Ho2, Er2, TbGd, and TbY were obtained by this approach.22,23 A single-crystal X-ray diffraction study of {Dy2} proved selective addition of the benzyl group to pentagon/hexagon/hexagon ([5,6,6]) junctions.23 The reaction of La2@C80 with benzyl bromide under UV irradiation proceeds as a radical addition and also yields a [5,6,6] {La2} monoadduct.29 The unpaired electron formed when the benzyl radical is attached to the fullerene is transferred to the La–La bonding orbital and yields a single-electron La–La bond. Another monoadduct with single-electron La–La bond, La2@C80(C3N3Ph2) (Figure 3b), was obtained by thermal reaction of La2@C80 with 3-chloro-5,6-diphenyltriazine.30 Radical trifluoromethylation has been used to functionalize EMF mixtures, which among other products gave Ln2@C80(CF3) monoadducts (Ln = Y,28,50 Gd51).
Figure 3.
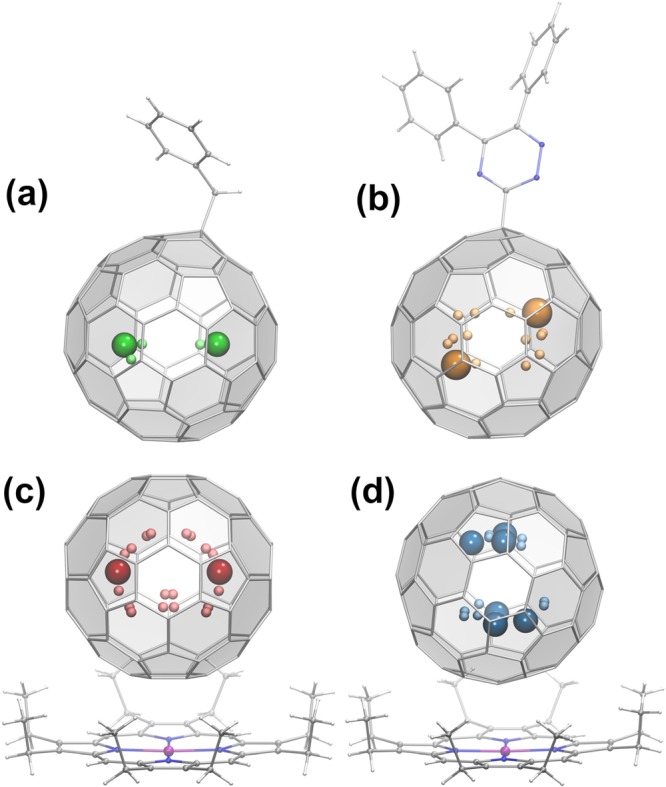
Molecular structures of di-EMFs with a single-electron Ln–Ln bond from single-crystal X-ray diffraction: (a) Dy2@C80(CH2Ph);23 (b) La2@C80(C3N3Ph2);30 (c) Tb2@C79N·Ni(OEP);26 (d) Gd2@C79N·Ni(OEP).21 Large colored spheres show the Ln sites with the highest occupancies. The Ln···Ln distances are 3.896(1) Å (Dy), 3.784(2) Å (La), 3.902(1) Å (Tb), and 3.835(9) Å (Gd).
Azafullerenes Ln2@C79N
Since C79N5– is isoelectronic with C806–, substitution of one carbon with nitrogen stabilizes (Ln2.5+)2@C805– in the form of the azafullerene (Ln2.5+)2@C79N5–. Y2@C79N, Gd2@C79N, and Tb2@C79N were obtained by arc-discharge synthesis in the presence of nitrogen.25,26 Single-crystal XRD studies of Tb2@C79N and Gd2@C79N cocrystallized with Ni(OEP) (Figure 3c,d)21,26 proved that their fullerene cages are based on C80-Ih, but the exact position of the nitrogen could not be determined. DFT computations showed that substitution of a [5,6,6] carbon in C80-Ih by nitrogen gives the most stable Ln2@C79N structures.19,26,31
Role of the Fullerene Cage
Despite the +2.5 oxidation state of the lanthanides, both Ln2@C80(CH2Ph) and Ln2@C79N compounds are air-stable, which is not typical for compounds with lanthanides in unconventional oxidation states. Thus, the fullerene cage provides sufficient protection to the (Ln2.5+)2 species with a single-electron Ln–Ln bond. Another important aspect is the spatial confinement of Ln2 dimers. Coulomb repulsion between lanthanide ions in di-EMFs is much stronger than the stabilization energy of the bonding interactions.27 Therefore, Ln ions tend to maximize the Ln···Ln distance, but since the extent of the Ln2 dimer is limited by the fullerene, metal ions remain at distances allowing bond formation. The bonding between strongly repulsive metal ions in di-EMFs has been identified as “oxymoron”27 or “unwilling” bonding.52
Magnetic Resonance Studies of Di-EMFs with Single-Electron Ln–Ln Bonds
Figure 2c shows that {Gd2}, [Gd2@C80]−, and Gd2@C79N have almost identical distributions of the spin density, with three maxima. Two of these are due to Gd 4f7 electrons, whereas the third one is associated with the unpaired electron in the Gd–Gd bonding orbital. A similar distribution is expected for other lanthanides with partially filled 4f shells, whereas for Y, La, and Lu only the spatial distribution of the unpaired valence electron in the Ln–Ln bonding MO will be seen. The experimental proof of such spin distributions is provided by magnetic resonance techniques.
For Y and La di-EMFs, spin properties are straightforwardly analyzed with EPR spectroscopy. The EPR spectrum of {Y2} in toluene at room temperature (Figure 4a) shows a triplet isotropic hyperfine structure due to the two equivalent 89Y nuclei (IY = 1/2). Very similar solution spectra were reported for Y2@C79N,26 Y2@C80(CF3),28 and a mixture of [Y2@C2n]− anions.23 At low temperature, the spectra change to an axial hyperfine pattern (Figure 4b).23 The EPR parameters (Table 1) prove the localization of the unpaired spin on the Y2 dimer: The large aiso(89Y) values show that the s electrons of Y make a large contribution to the spin density, in agreement with DFT-calculated spin populations of 0.26 (s), 0.08 (p), and 0.16 (d) of each Y atom in {Y2}. The A tensor anisotropy is caused by contributions from p and d orbitals of Y to the spin density. Finally, deviations of giso from the free electron value of 2.0023 and a considerable anisotropy of the g tensor also point to localization of the unpaired spin on the metal atoms. Likewise, localization of the spin density on the La2 dimer in di-EMFs with single-electron La–La bonds also follows from the EPR studies of [La2@C80]−,28,41 La2@C80(C3N3Ph2),30 and {La2}29 (Table 1).
Figure 4.

(a, b) X-band EPR spectra of {Y2} in toluene at (a) room temperature and (b) 150 K. (c, d) Q-band (34 GHz) and X-band (9.4 GHz) EPR spectra of {Gd2} in frozen toluene. The inset in (d) shows the room-temperature spectrum. (e) 1H NMR spectra of {Ln2} compounds in CS2 at room temperature. Asterisks in the spectrum of {TbGd} denote the signals of {Tb2}. Adapted with permission from (a, b) ref (23) and (c–e) ref (22). Copyright 2017 and 2019, respectively, Springer Nature.
Table 1. EPR Parameters of Y and La Di-EMFs with a Single-Electron Ln–Ln Bond.
EPR studies of 4f di-EMFs are complicated by fast relaxation and strong magnetic anisotropy, and successful measurements have been reported only for Gd2@C79N21,25,33 and {Gd2} to date.22 Coupling of the Gd 4f spins to the unpaired electron spin gives a large-spin ground state with a characteristic zero-field splitting (ZFS) pattern, which can be resolved in the EPR spectra of Gd2@C79N and {Gd2} at low temperatures (Figure 4c,d). The ZFS can be ascribed to the spin S = 15/2 originating from ferromagnetic coupling of the two 4f7 spins (S = 7/2) and the unpaired electron spin (S = 1/2). The spin Hamiltonian takes the form
| 1 |
where the first two terms describe the ZFS and the last term represents the Zeeman effect. The X- and Q-band EPR spectra of {Gd2} near 100 K are reproduced by the parameters D = 1.00(2) GHz, E = 0.22(4) GHz, and giso = 1.987 (Figure 4c,d).22 For Gd2@C79N, the analogous parameters are D = 0.96(6) GHz, E = 0.14(1) GHz, and giso = 1.99.54 The spin coherence properties of Gd2@C79N solutions were studied by pulsed EPR experiments.21 Phase memory times (Tm) of up to 1.6 μs (5 μs with dynamic decoupling) were found at 5 K, and the possibility of coherent spin manipulation was confirmed by Rabi oscillations in echo-detected nutation experiments. The use of Gd2@C79N for improving dynamic nuclear polarization (DNP) at high fields was also demonstrated.33
For lanthanides other than Gd, the localization of an unpaired spin on the metal dimers in {Ln2} has been verified by NMR spectroscopy.22 Localization of the spin on the carbon cage would make the detection of 13C or 1H NMR signals problematic. However, solution 1H NMR spectra of {Ln2} compounds exhibit well-defined resonances of benzyl protons (Figure 4e) strongly shifted from their positions in diamagnetic compounds (3–7 ppm). These paramagnetic 1H shifts are caused by the dipolar magnetic field of the endohedral Ln2 dimer. Since molecules in solution rotate, the isotropic contributions of the dipolar field average out, and paramagnetic shifts (δpara) reflect the magnetic anisotropy of the endohedral Ln2 dimer. In a point-dipole approximation, dipolar (or pseudocontact) paramagnetic shifts take the form55
| 2 |
where Ri and θi are polar coordinates of the ith proton in the coordinate system centered on the Ln2 dimer with the polar axis along the Ln–Ln bond, and χ∥ (χ⊥) is the longitudinal (transverse) magnetic susceptibility of the Ln2 dimer. Since the first factor in eq 2 (the geometrical factor) is very similar for all {Ln2} molecules, variation of the 1H NMR spectra along the {Ln2} series is mainly caused by the difference of χ∥ and χ⊥. Thus, 1H chemical shifts reveal the sign and relative magnitude of the magnetic anisotropy. From a comparison of the 1H NMR spectra in Figure 4e, we conclude that {Dy2} and {Tb2} have similar magnetic properties since their 1H chemical shifts are almost identical. The {Ho2} values are a factor of ca. 2 smaller, indicating a lower magnetic anisotropy. Substitution of one Tb in {Tb2} by Y to obtain {TbY} expectedly results in a considerable decrease in the paramagnetic shift. For {TbGd}, a close similarity to the 1H NMR spectra of {TbY} might be expected because isotropic spins do not produce dipolar paramagnetic shifts. However, the 1H shifts in {TbGd} are 2 times larger than those in {TbY} (Figure 4e), which shows that the isotropic Gd3+ spin is locked to the anisotropic Tb3+ spin by strong exchange interactions. Finally, the 1H resonances in {Er2} are shifted in the opposite direction in comparison to other {Ln2} molecules, revealing the opposite sign of the magnetic anisotropy.
Moving Electrons into and out of Ln–Ln Bonds
Since the Ln–Ln bonding MO in di-EMFs is either a LUMO, HOMO, or singly occupied MO, redox reactions of di-EMFs may populate or depopulate this orbital. According to their redox behavior, di-EMFs with Ln–Ln bonding MOs can be divided into four classes (Figure 5a). Ln2@C2n with La and Ce represent class I. They have a metal-based LUMO, and their reduction proceeds via the formation of a single-electron Ln–Ln bond. Oxidation of these di-EMFs is a cage-based process, and the corresponding redox potentials do not depend on the encapsulated metal. Class II is represented by di-EMFs of Y, Er, and Lu.37,48,49,56 They have a doubly occupied metal-based HOMO, which loses one electron upon oxidation to form a single-electron Ln–Ln bond in the cation. The first oxidation potential is thus strongly metal-dependent. At the same time, reduction of these di-EMFs is a cage-based process with a metal-independent potential.
Figure 5.

(a) Four types of di-EMF redox behavior. (b) Cyclic voltammograms of {Ln2} (Ln = Er, Ho, Dy) in o-dichlorobenzene (o-DCB). Vertical dashed lines denote the reduction (E1/2(0/−)) and oxidation (E1/2(+/0)) potentials of {Ho2}. (c) Redox potentials in the {Ln2} series. Horizontal dotted lines denote potentials of the fullerene-based redox processes, and dashed green lines show the variation of the potential of the redox-active Ln–Ln orbital. (d) Schematic description of the MO levels in {Er2}, {Gd2}, and {La2}. Cage MOs are shown in gray and metal-based MOs in green.
Ln2@C80(CH2Ph) adducts belong to class III or IV, and their redox behavior is illustrated in Figure 5b with {Er2}, {Ho2}, and {Dy2};22,23 the first redox potentials of the {Ln2} series are summarized in Figure 5c. The reduction potential shifts from −0.4 to −0.9 V in going from Er to Gd, whereas the oxidation potential remains virtually constant at +0.5 V (potentials are referenced versus Fe(Cp)2+/0). A further increase in the Ln3+ ionic radius leads to a drop in the oxidation potential to +0.15 V in {La2},29 but the reduction potential of {La2} is shifted from the {Gd2} value by only −0.06 V. These changes in redox potentials can be rationalized using the MO scheme in Figure 5d. In {Y2} and {Er2}–{Gd2}, assigned to class III, the LUMO is the vacant Ln–Ln bonding orbital, which is populated in the first reduction step, giving a two-electron Ln–Ln bond (Figure 5a). The reduction potential is therefore metal-dependent. At the same time, the HOMO of these {Ln2} molecules is a cage-based orbital, whereas the singly occupied Ln–Ln bonding MO has lower energy and remains unaffected by oxidation. Both the occupied and vacant components of the Ln–Ln bonding MO increase in energy upon moving from heavier to lighter lanthanides. At a certain threshold, the order of the metal- and fullerene-cage-based orbitals changes (Figure 5d). Then the cage-based MO becomes the LUMO with a metal-independent reduction potential, whereas the metal-based MO becomes the HOMO, and the oxidation potential becomes metal-dependent and starts to shift negatively with increasing metal size. This is the situation in {La2}, the only member of class IV to date.
The reduction mechanism of di-EMFs in class III has been confirmed by 1H NMR spectroscopy.22 Population of the fullerene-based MO by the surplus electron in {Ln2}− would lead to delocalization of the unpaired spin over the fullerene cage, and 1H NMR spectra of such anions would not be detectable. However, single-electron reduction of {Ln2} with cobaltocene gave {Ln2}− anions with well-defined 1H NMR resonances (Figure 6), proving the formation of a two-electron Ln–Ln bond.
Figure 6.
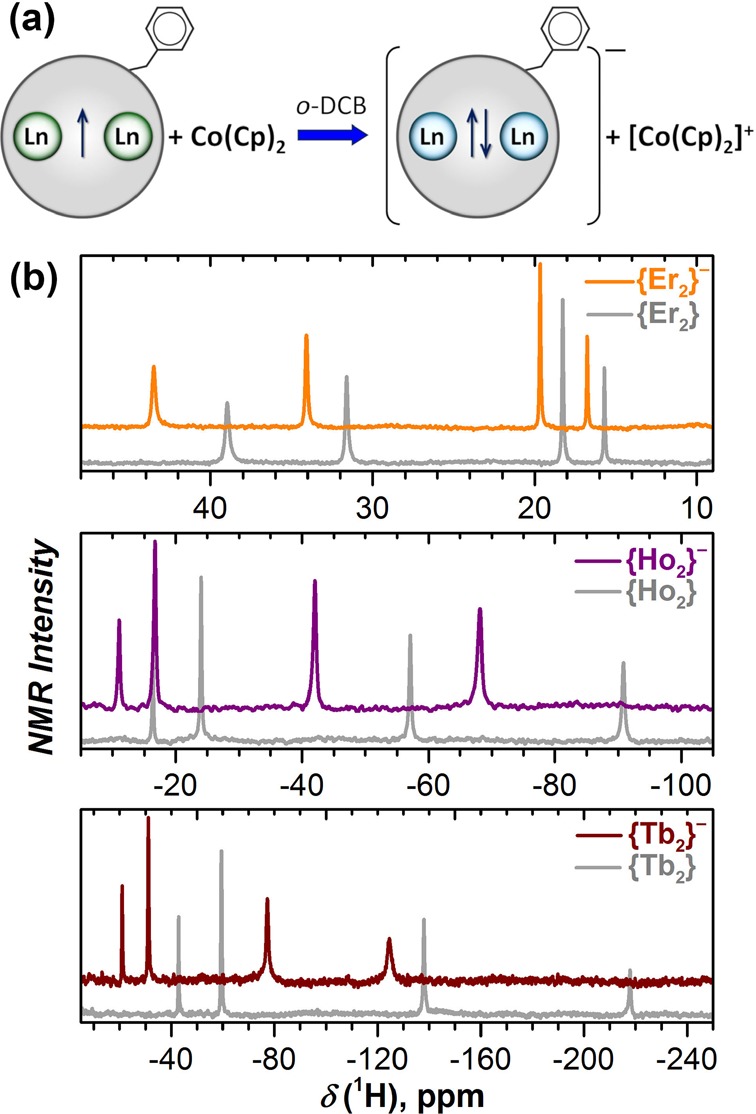
(a) Reaction of {Ln2} with cobaltocene. (b) Room-temperature 1H NMR spectra of {Er2}−, {Tb2}−, and {Ho2}− anions in o-DCB-d4 solution (colored lines) compared with the spectra of pristine {Ln2} compounds (gray lines). Reproduced with permission from ref (22). Copyright 2019 Springer Nature.
Single-Molecule Magnetism in Di-EMFs with Single-Electron Ln–Ln Bonds
The core property of SMMs is slow relaxation of the magnetization, leading to blocking of the magnetization and magnetic hysteresis. Below the blocking temperature (TB), the magnetic properties depend on the way that the current state of the sample was obtained, whereas above TB the sample reaches thermodynamic equilibrium faster than the controlled parameter (temperature, magnetic field, etc.) is changed. With TB values of 22, 29, and 28 K (Figure 7a–c), respectively, {Dy2},23 {Tb2},22 and Tb2@C79N19 are the strongest SMMs among all EMF-SMMs.13 For comparison, the highest TB values in nitride, carbide, sulfide, and oxide clusterfullerenes are 7–8 K (Dy2ScN@C80-Ih16 and DySc2N@C80-Ih14), 7 K (DyYTiC@C80-Ih15), 4 K (Dy2S@C82-C3v17), and 7 K (Dy2O@C82-C3v18). {Ho2} and {Er2} do not show blocking of the magnetization above 2 K.22
Figure 7.

(a–c) Magnetization blocking temperatures of (a) {Dy2} (b), {Tb2}, and (c) Tb2@C79N. The magnetic field was 0.2–0.3 T, and the temperature sweep rate was 5 K min–1. (d–f) Magnetic hysteresis of (d) {Dy2}, (e) {Tb2}, and (f) Tb2@C79N. The magnetic field sweep rate was 3 mT s–1 in (d) and (f) and 9.5 mT s–1 in (e). (g–i) Magnetization relaxation times of (g) {Dy2}, (h) {Tb2}, and (i) Tb2@C79N. Adapted with permission from (a, d, g) ref (23), (b, e, h) ref (22), and (c, f, i) ref (19). Copyright 2017 and 2019 Springer Nature and 2019 Wiley-VCH, respectively.
Since TB depends on the temperature sweep rate, it was suggested to use TB100, the temperature at which the magnetization relaxation time is 100 s. The TB100 values for {Dy2} (18.2 K), {Tb2} (25.2 K), and Tb2@C79N (24.1 K) are second only to those of Dy metallocenium cations, which hold the absolute records among all SMMs with TB100 up to 53–65 K,3−5 and recently discovered TbII(CpiPr5)2 with TB100 = 52 K.57 We are not aware of other SMMs with TB100 values higher than those of {Tb2} and Tb2@C79N; at this moment, the closest follower with TB100 of 20 K is the dinuclear Tb metallocene complex with a N23– radical bridge.6
{Dy2}, {Tb2}, and Tb2@C79N exhibit magnetic hysteresis with large remanence below TB (Figure 7d–f). The magnetic hysteresis of {Tb2} is uniquely broad with giant coercive fields of 8 T at 5–10 K (Figure 7e).22 Similarly large coercivity was reported only in the aforementioned dinuclear-radical-bridged Tb complex6 and has no further analogues among molecular magnets. For Tb2@C79N, a 2-fold smaller but still large coercive field of 3.8 T is found between 2 and 10 K (Figure 7f).19
In order to identify the relaxation mechanisms of SMMs, magnetization relaxation times τM are measured at various temperatures and magnetic fields. The total relaxation rate (τM–1) is a sum of the rates of contributing processes:
| 3 |
The first term in eq 3 describes relaxation of the magnetization via quantum tunneling of magnetization (QTM), which occurs when spin-up and spin-down levels are quasi-degenerate. Since QTM is not thermally activated, the relaxation rate τQTM–1 is temperature-independent. However, QTM depends on the magnetic field and can be quenched completely when Zeeman splitting lifts the degeneracy of spin-up and spin-down levels. In the Orbach mechanism (the second term in eq 3), spin reversal involves overcoming the energy barrier Ueff, which corresponds to a certain excited state of the system, and hence follows the Arrhenius law. Finally, two power-law temperature terms describe the single-phonon direct mechanism (n1 = 1) and the two-phonon Raman relaxation mechanism (n2 = 4–9).
The magnetization relaxation times of {Dy2}, {Tb2}, and Tb2@C79N are plotted in Figure 7g–i in Arrhenius coordinates (log(τM) vs T–1).19,22,23 For {Ho2} and {Er2}, the relaxation times are orders of magnitude shorter and will not be discussed here.22 {Dy2}, {Tb2}, and Tb2@C79N show similar temperature dependences of τM. At low temperatures in zero field, the relaxation times are temperature-independent, which points to relaxation via QTM. In this regime, the combined spin of the [Ln3+–e–Ln3+] system flips as a single entity with rather long τQTM: 0.9 h in {Dy2}, 18 h in {Tb2}, and 4.5 h in Tb2@C79N. A magnetic field of 0.3–0.4 T quenches the QTM and slows the relaxation down to months or even years (Figure 7g,h). For {Dy2}, dilution of the sample in a polystyrene matrix also resulted in a significant increase in the relaxation time,23 showing that intermolecular dipolar fields play an important role in facilitating the QTM mechanism of EMF-SMMs.14,23
Above 20 K, all three di-EMFs show a linear regime in Arrhenius coordinates, which points to the Orbach mechanism with the barriers Ueff of 613 ± 8 K in {Dy2}, 799 ± 2 K in {Tb2}, and 757 ± 4 K in Tb2@C79N. These energies are assigned to the pure exchange-excited states, in which the spin of one of the lanthanide ions is flipped (Figure 7h). In many SMMs showing zero-field QTM at the lowest temperatures, the temperature increase first activates the Raman mechanism, which then in turn gives way to the Orbach regime at higher temperatures. As a result, the Raman mechanism is often the main limiting factor reducing the blocking temperature of magnetization of SMMs with large Orbach barriers.58 In {Tb2} and Tb2@C79N, the strong coupling between the two Ln ions greatly suppresses the Raman mechanism, so that the QTM is superseded directly by the Orbach mechanism as the dominant mode of relaxation. As a result, although the Orbach barriers of {Tb2} and Tb2@C79N are smaller than in a handful of SMMs with Ueff > 1000 K discovered during the past few years (including Dy2ScN@C80 with Ueff = 1735 K16), the TB and TB100 values of {Tb2} and Tb2@C79N are higher than for many of those.
Coupling of Spins in Dimetallofullerenes with Single-Electron Ln–Ln Bonds
In designing a spin Hamiltonian describing di-EMFs with a single-electron Ln–Ln bond, the molecule is considered to be a three-center spin system [Ln3+–e–Ln3+] (Figure 8a). The lanthanides are treated as Ln3+ ions with corresponding ligand-field (LF) Hamiltonians and spin operators ĴLn, whereas the exchange Hamiltonian includes the direct Ln–Ln interaction with coupling constant j12 and exchange interactions between the lanthanide spins and the unpaired electron spin ŝ with exchange constants K′ and K″:
| 4 |
Equation 4 implies the general case of two different lanthanides in one molecule (Ln′ and Ln″) and hence, two different Ln–e coupling constants (K′ and K″) are needed. For homometallic systems with nonsymmetric molecules, a small difference between K′ and K″ values can be ignored in the first approximation.
Figure 8.

(a) Schematic representation of di-EMFs with a single-electron Ln–Ln bond as a three-center [Ln3+–e–Ln3+] spin system. (b) Alignment of the Tb spins (green arrows) and the unpaired electron spin (red arrow) in Tb2@C79N. (c) Alignment of the Ln and unpaired electron spins (red arrows) in {Ln2} molecules. The spins of Er ions are visualized as ellipsoids built upon the g tensors of their single-ion ground states. (d) Low-energy spectrum of the spin Hamiltonian (eq 6) for {Tb2} with Keff = 55 cm–1. Red lines visualize transition probabilities. Tb and unpaired spins are shown with green and red arrows, respectively. Dashed arrows show the QTM and Orbach mechanisms.
Gd Dimetallofullerenes
As the LF terms for isotropic Gd spins can be neglected, the effective spin Hamiltonian of {Gd2} or Gd2@C79N takes the form
| 5 |
DFT computations on {Gd2},23 Gd2@C79N,31,32 and [Gd2@C80]−41,59 showed that direct Gd–Gd interactions are antiferromagnetic with j12 < −1 cm–1, whereas the Gd–electron exchange is ferromagnetic with K′ and K″ values of 180–200 cm–1. Taking into account the small difference between K′ and K″ and the small value of j12 allows further simplification of the spin Hamiltonian to obtain the last equality in eq 5 with a single effective exchange constant Keff, whose value can be estimated from a comparison of experimental and simulated χT curves. For Gd2@C79N20,21 (Figure 9a) and {Gd2},22 good agreement is achieved for Keff = 170 ± 10 cm–1 and 160 ± 10 cm–1, respectively. These values are unprecedentedly large for lanthanide molecular magnets. The largest Gd–radical coupling constants in other compounds are 6 cm–1 for Gd–nitroxide,60,61 −10 cm–1 for Gd–bipyrimidyl,62 and −27 cm–1 for Gd–N23– radical bridges.8,63 The mechanism of the strong exchange interactions in the [Gd3+–e–Gd3+] system has been discussed in refs (31), (32), and (59) but is not fully understood at present.
Figure 9.
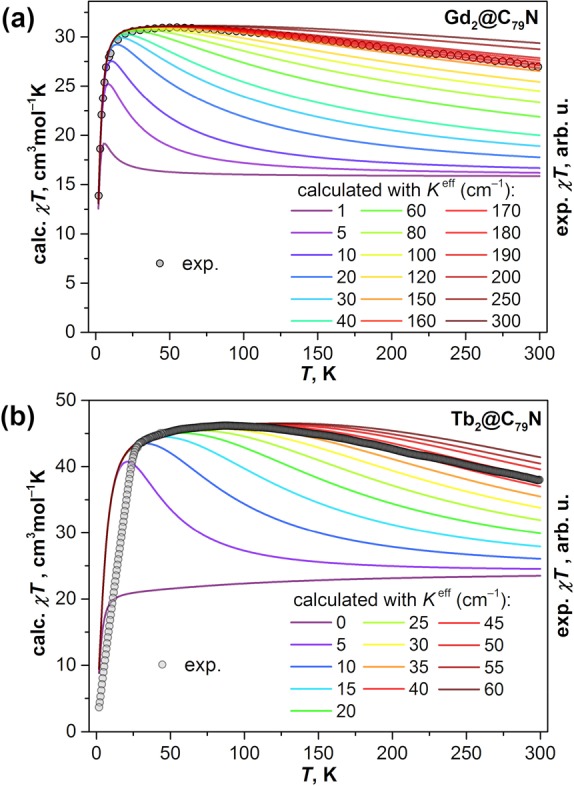
Comparison of experimental χT values (dots, arbitrary units) measured for (a) Gd2@C79N and (b) Tb2@C79N in a field of 1 T to the results of calculations for different values of the coupling constant Keff. Adapted with permission from (a) ref (20) and (b) ref (19). Copyright 2018 Royal Society of Chemistry and 2019 Wiley-VCH, respectively.
Di-EMFs with Magnetically Anisotropic Lanthanides
The low-energy states of [Ln3+–e–Ln3+] systems with anisotropic lanthanides are described by the same Hamiltonian as in eq 5 but with the addition of ligand field terms:
| 6 |
Ab initio calculations at the CASSCF/RASSI-SO level revealed strong axiality of the ligand field for Tb3+ and Dy3+ ions in {Ln2} and Ln2@C79N molecules (Figure 8b,c).22,23 Dy3+ and Tb3+ have large-spin ground states with collinear quantization axes aligned parallel to the Ln–Ln bond (Figure 8c) and LF splittings of 900–1000 cm–1. In the |J, mJ⟩ basis, the ground state Kramers doublet (KD) of Dy3+ is a pure state with |mJ| = 15/2, but higher energy KDs have a more mixed character. In Tb3+, the four lowest-energy pseudo-Kramers doublets are essentially pure mJ states with 99–100% contributions of |mJ| = 6, 5, 4, and 3.22 Similar energies and mJ state compositions are found for Tb3+ in Tb2@C79N,19 but the magnetic axes of the Tb ions are tilted from the Tb–Tb bond by 7° (Figure 8b). A high-spin ground state is also predicted for Ho3+, but with a considerably smaller LF splitting, stronger tilting of the easy axes of the Ho ions (Figure 8c), and mixed mJ character with the leading term of 64% |±8⟩. For Er3+, an easy-plane ground state is predicted in agreement with NMR data (Figure 8c).22
With ĤLF parameters from ab initio calculations, the Keff value can be estimated in a similar fashion as described for Gd2@C79N. Figure 9b shows that for Tb2@C79N the best match to the experimental χT curve is obtained for Keff = 40–45 cm–1.19 The Keff values estimated from the χT curves are 48–53 cm–1 for {Tb2}, 30–35 cm–1 for {Dy2}, ca. 40 cm–1 for {Ho2}, and ca. 20 cm–1 for {Er2}.22,23
Relaxation of the magnetization in di-EMFs can be understood using the spectrum of the spin Hamiltonian (eq 6), as shown in Figure 8d for {Tb2}. [Ln3+–e–Ln3+] is a Kramers system for any lanthanide and has a rigorous twofold degeneracy of the spin states in zero field. In the ground-state doublet of {Tb2}, all three spins are aligned along the Tb–Tb axis (Figure 8c). Negligible transverse components of the g tensor and the large total spin result in a low efficiency of QTM within this doublet, as transverse fields (e.g., from intermolecular interactions) cannot couple to increase the tunneling gap. Next follow the states with LF excitations in one or both lanthanides, forming branches with a gradual decrease of the moment at higher energies. These states do not provide efficient relaxation pathways. The first pure exchange-excited states, in which one of the Tb spins is flipped, appear at energies comparable to twofold LF excitations. In these states, Tb spins cancel each other out, leading to a small total moment with an almost free unpaired spin. As these states are very efficient for relaxation of the magnetization, the Orbach regimes found in the temperature dependence (Figure 7g–i) correspond to relaxation via these states. An assumption that Ueff corresponds to the energy of the first pure exchange-excited state (Figure 8d) allows an independent estimation of Keff, which gives values of 55 cm–1 for {Tb2} and 32 cm–1 for {Dy2}, in good agreement with Keff estimated from the χT curves.22,23 For comparison, in dinuclear-radical-bridged Tb and Dy complexes, the largest Ln–radical coupling constants are −23.1 cm–1 (Tb) and −7.2 cm–1 (Dy).6 It should be noted that if the ground states of the lanthanide spins are of the Ising type with Jz = ±J, the energy of the pure exchange-excited state with the exchange-only Hamiltonian is 2JKeff, which allows a very simple but less precise estimation of Keff as Ueff/2J.
At the end of this section, we would like to note that comparison
of the coupling constants between the compounds should be done cautiously
because the values will depend on the definition of the exchange Hamiltonian.
Whereas Ĥexchange in eq 6 is defined through the whole momentum ĴLn, another popular approach is based
on the use of pseudospin (s̃Ln = 1/2) to describe the ground state of lanthanides.31,64,65 The same energy difference between
ferromagnetically and antiferromagnetically coupled states in the
−2Keffŝ·ĴLn and  exchange formalisms would require
a keff that is 2J times
larger
than Keff, where J is
the total momentum of a lanthanide. Another option may be to use only
the spin ŜLn without the angular momentum
and to define the exchange term as −2jeffŝ·ŜLn. Likewise, jeff can be obtained by scaling Keff with the factor J/S. For Dy3+ with J = 15/2 and S = 5/2 as
an example, the Keff value of 32 cm–1 for {Dy2} is
equivalent to keff = 960 cm–1 or jeff = 96 cm–1.
This substantial dependence of the coupling constant on the form of
the exchange Hamiltonian should be taken into account carefully when
comparing the values from different works.
exchange formalisms would require
a keff that is 2J times
larger
than Keff, where J is
the total momentum of a lanthanide. Another option may be to use only
the spin ŜLn without the angular momentum
and to define the exchange term as −2jeffŝ·ŜLn. Likewise, jeff can be obtained by scaling Keff with the factor J/S. For Dy3+ with J = 15/2 and S = 5/2 as
an example, the Keff value of 32 cm–1 for {Dy2} is
equivalent to keff = 960 cm–1 or jeff = 96 cm–1.
This substantial dependence of the coupling constant on the form of
the exchange Hamiltonian should be taken into account carefully when
comparing the values from different works.
Mixed-Metal Di-EMFs
Di-EMFs synthesized using two lanthanides at once comprise a mixture of Ln′2, Ln″2, and Ln′Ln″ species, and some such mixtures can be separated into individual components by recycling HPLC. In particular, separation of the Tb–Y system afforded pure {TbY}.22 Replacement of one Tb ion in {Tb2} by Y dramatically increases the relaxation rates and changes the relaxation mechanism and hysteretic behavior (Figure 10a). {TbY} shows a narrow magnetic hysteresis below 5 K with an opening in the field of 0.1–1.0 T (Figure 10a). At 2 K in a magnetic field of 0.3 T, the relaxation time of {TbY} is 2.9 s (compared with ca. 6 years estimated for {Tb2} at 3 K), and in zero field τM drops to only a few ms.22 Thus, the presence of two lanthanide spins with uniaxial anisotropy in {Ln2} is essential for a good di-EMF SMM.
Figure 10.
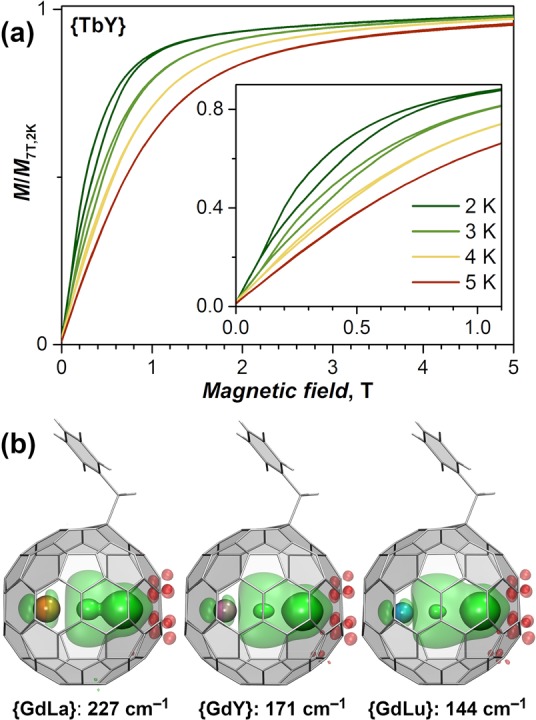
(a) Magnetization curves for {TbY} below 5 K. Reproduced with permission from ref (22). Copyright 2019 Springer Nature. (b) DFT-computed spin density distribution and Gd electron exchange coupling constants in {GdLa}, {GdY}, and {GdLu} (PBE0/DKH-TZVP level of theory; the isosurface visualization parameters are the same as in Figure 2).
Combining different lanthanides in one di-EMF is promising for the design of molecular magnets with tunable spin properties.66 One of the unique aspects of this approach is the possibility to tune the Ln–electron exchange interactions by varying the relative size of the lanthanides, as illustrated in Figure 10b for {GdLa}, {GdY}, and {GdLu}. With the increase in the lanthanide size from Lu to Y and further to La, the maximum of the spin density due to the unpaired valence electron shifts closer and closer to Gd. Broken-symmetry DFT calculations show that this shift is accompanied by a strong variation of the Gd–electron exchange coupling constant from 144 cm–1 in {GdLu} to 227 cm–1 in {GdLa}.
Outlook
Although dedicated studies of dimetallofullerenes with single-electron lanthanide–lanthanide bonds with the focus on redox and magnetic properties have been pursued for only a few years, impressive advantages of dimetallofullerenes as molecular magnets are already clear. However, the field is still in its infancy, and further studies required to fully exploit the potential of these molecules are outlined below:
-
1.
Only a few Ln′Ln″ combinations have been studied to date in di-EMFs. Further exploration of mono- and heterobimetallic systems may give exceptional control over the spin properties of dimetallofullerenes, such as spin–spin interactions, frontier orbital energies, and single-ion magnetic anisotropy.
-
2.
Most of the studied di-EMFs with a single-electron Ln–Ln bond are based on the fullerene C80-Ih. Comparison of the EPR and SMM properties of Ln2@C79N and Ln2@C80(CH2Ph) shows a noticeable influence of the fullerene cage on the magnetic properties of di-EMFs,19,20,22 which is not fully understood and should be studied further.
-
3.
Endohedral fullerenes are able to undergo multiple derivatization reactions,67 which affect the fullerene π system. The mutual influence of Ln–Ln bonds and exohedral chemistry and the variation of the spin properties of di-EMFs with functionalization have not been studied to date.
-
4.
Ln–Ln bonds in fullerenes are redox-active,35 and the electron transfer can dramatically change the magnetic properties of di-EMFs. Electron transfer can also be realized intramolecularly by creating donor–acceptor dyads with photoexcited electron transfer,68 which may result in optically controlled magnetism.
-
5.
Spins encapsulated within carbon cages are isolated from the environment but still can be manipulated externally, which has rich implications for quantum information processing.69 The first experiments in this direction21 demonstrate the great potential of single-electron Ln–Ln bonds.
-
6.
The chemical and thermal stability of fullerenes allow the design of EMF-based supramolecular architectures. One- and two-dimensional arrays of some EMF-SMMs have already been reported,13 and this should be feasible for di-EMFs as well. The strong interaction of core 4f electrons with the frontier Ln–Ln bonding orbital may give unique spin-transport properties. Thus, di-EMFs with a single-electron Ln–Ln bond have excellent perspectives in molecular spintronics.
-
7.
The single-electron lanthanide–lanthanide bond in di-EMFs is challenging with regard to the development of a theoretical framework required for interpretation of experimental data. Although treating a single-electron Ln–Ln bond as a three-spin [Ln3+–e–Ln3+] system gives a reasonable description of the magnetic properties, the validity of this approach remains questionable. Implementation of more robust ab initio approaches and severe testing of model spin Hamiltonians are required for better modeling of the magnetic properties of di-EMFs.
Acknowledgments
The authors acknowledge funding from the EU’s Horizon 2020 Programme, European Research Council (Grant 648295 to A.A.P.) and Marie Skłodowska-Curie Action (Grant 748635 to S.M.A.), and the Deutsche Forschungsgemeinschaft (Grants PO 1602/4-1 and 1602/5-1 to A.A.P.). We thank H. Dorn (Virginia Tech) and S. Stevenson (Purdue University Fort Wayne) for providing some dimetallofullerene samples, M. Zalibera (Slovak University of Technology) for help with EPR studies, T. Greber and A. Kostanyan (University of Zürich) for help with magnetometry, and many colleagues at IFW Dresden, including A. Beger, B. Büchner, C.-H. Chen, V. Dubrovin, S. Gaß, L. Hozoi, K. Nenkov, U. Nitzsche, R. Ray, M. Richter, M. Rosenkranz, N. Samoylova, S. Schiemenz, A. U. B. Wolter, and F. Ziegs, for experimental, theoretical, and technical support of the studies of di-EMFs.
Biographies
Fupin Liu received his Ph.D. in 2014 from the University of Science and Technology of China in Hefei, where he worked with Prof. S. Yang. In 2015 he joined the Fullerene group at IFW Dresden in Germany and is currently supervising synthetic and crystallographic studies of metallofullerenes.
Lukas Spree obtained his Master’s degree in applied natural science from Freiberg University in 2016 and then joined the Fullerene group at IFW Dresden to study the synthesis, chemical functionalization, and magnetic properties of metallofullerenes with F. Liu and A. A. Popov.
Denis S. Krylov obtained his Master’s degree in nanobiophysics in 2013 from TU Dresden. In 2014 he joined the Popov group at IFW Dresden to study the magnetic properties of EMFs by SQUID magnetometry, XMCD, and STM techniques. After his Ph.D. defense in 2018, he joined the Center for Quantum Nanoscience at Ewha Womans University (Seoul, Korea) to study spin properties of atoms and molecules on the nanoscale.
Georgios Velkos obtained his Master’s degree in nanosciences and nanotechnologies in 2017 from Aristotle University of Thessaloniki in Greece with an internship at Oxford University. Then he joined the Popov group at IFW Dresden to study the magnetic properties of metallofullerenes.
Stanislav M. Avdoshenko received M.S. (2007) and Ph.D. (2009) on computational studies of fullerene derivatives from Moscow State University. In 2009–2012 he was an Erasmus Fellow at TU Dresden, studying transport phenomena in complex systems. He then moved to the United States, first to the School of Materials Engineering at Purdue University (2012–2014) and then to the Institute for Computational Engineering and Sciences at UT Austin (2014–2016). In 2017 he received the prestigious Marie Skłodowska-Curie Fellowship to support his independent research at IFW Dresden. His group is now working on the theory of spin properties of lanthanide and transition metal systems, including those of metallofullerenes, and on quantum spin and charge dynamics in molecular materials.
Alexey A. Popov received M.S. (1999) and Ph.D. (2003) in chemistry from Moscow State University (MSU) in Russia. He was a Senior Researcher at MSU until 2008, when he received an Alexander von Humboldt Fellowship to work on endohedral metallofullerenes at IFW Dresden, where he now heads the Fullerene group. His research interests include experimental and computational studies of EMFs, including their synthesis, derivatization, magnetism, spectroelectrochemistry, and photophysics. He is a recipient of an ERC Consolidator Grant for the studies of bulk and surface magnetism of EMF-SMMs.
The authors declare no competing financial interest.
References
- Liu J.-L.; Chen Y.-C.; Tong M.-L. Symmetry strategies for high performance lanthanide-based single-molecule magnets. Chem. Soc. Rev. 2018, 47, 2431–2453. 10.1039/C7CS00266A. [DOI] [PubMed] [Google Scholar]
- Harriman K. L. M.; Errulat D.; Murugesu M. Magnetic Axiality: Design Principles from Molecules to Materials. Trends in Chemistry 2019, 1, 425–439. 10.1016/j.trechm.2019.04.005. [DOI] [Google Scholar]
- Goodwin C. A. P.; Ortu F.; Reta D.; Chilton N. F.; Mills D. P. Molecular magnetic hysteresis at 60 K in dysprosocenium. Nature 2017, 548, 439–442. 10.1038/nature23447. [DOI] [PubMed] [Google Scholar]
- Guo F.-S.; Day B. M.; Chen Y.-C.; Tong M.-L.; Mansikkamäki A.; Layfield R. A. Magnetic hysteresis up to 80 K in a dysprosium metallocene single-molecule magnet. Science 2018, 362, 1400–1403. 10.1126/science.aav0652. [DOI] [PubMed] [Google Scholar]
- McClain K. R.; Gould C. A.; Chakarawet K.; Teat S.; Groshens T. J.; Long J. R.; Harvey B. G. High-temperature magnetic blocking and magneto-structural correlations in a series of dysprosium(III) metallocenium single-molecule magnets. Chem. Sci. 2018, 9, 8492–8503. 10.1039/C8SC03907K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demir S.; Gonzalez M. I.; Darago L. E.; Evans W. J.; Long J. R. Giant coercivity and high magnetic blocking temperatures for N23- radical-bridged dilanthanide complexes upon ligand dissociation. Nat. Commun. 2017, 8, 2144. 10.1038/s41467-017-01553-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demir S.; Jeon I.-R.; Long J. R.; Harris T. D. Radical ligand-containing single-molecule magnets. Coord. Chem. Rev. 2015, 289–290, 149–176. 10.1016/j.ccr.2014.10.012. [DOI] [Google Scholar]
- Rinehart J. D.; Fang M.; Evans W. J.; Long J. R. Strong exchange and magnetic blocking in N23--radical-bridged lanthanide complexes. Nat. Chem. 2011, 3, 538–542. 10.1038/nchem.1063. [DOI] [PubMed] [Google Scholar]
- Rinehart J. D.; Fang M.; Evans W. J.; Long J. R. A N23– Radical-Bridged Terbium Complex Exhibiting Magnetic Hysteresis at 14 K. J. Am. Chem. Soc. 2011, 133, 14236–14239. 10.1021/ja206286h. [DOI] [PubMed] [Google Scholar]
- Popov A. A.; Yang S.; Dunsch L. Endohedral Fullerenes. Chem. Rev. 2013, 113, 5989–6113. 10.1021/cr300297r. [DOI] [PubMed] [Google Scholar]
- Yang S.; Wei T.; Jin F. When metal clusters meet carbon cages: endohedral clusterfullerenes. Chem. Soc. Rev. 2017, 46, 5005–5058. 10.1039/C6CS00498A. [DOI] [PubMed] [Google Scholar]
- Popov A. A.Endohedral Fullerenes: Electron Transfer and Spin; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar]
- Spree L.; Popov A. A. Recent advances in single molecule magnetism of dysprosium-metallofullerenes. Dalton Trans 2019, 48, 2861–2871. 10.1039/C8DT05153D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krylov D.; Liu F.; Brandenburg A.; Spree L.; Bon V.; Kaskel S.; Wolter A.; Buchner B.; Avdoshenko S.; Popov A. A. Magnetization relaxation in the single-ion magnet DySc2N@C80: quantum tunneling, magnetic dilution, and unconventional temperature dependence. Phys. Chem. Chem. Phys. 2018, 20, 11656–11672. 10.1039/C8CP01608A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandenburg A.; Krylov D. S.; Beger A.; Wolter A. U. B.; Büchner B.; Popov A. A. Carbide clusterfullerene DyYTiC@C80 featuring three different metals in the endohedral cluster and its single-ion magnetism. Chem. Commun. 2018, 54, 10683–10686. 10.1039/C8CC04736G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krylov D. S.; Liu F.; Avdoshenko S. M.; Spree L.; Weise B.; Waske A.; Wolter A. U. B.; Büchner B.; Popov A. A. Record-high thermal barrier of the relaxation of magnetization in the nitride clusterfullerene Dy2ScN@C80-Ih. Chem. Commun. 2017, 53, 7901–7904. 10.1039/C7CC03580B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-H.; Krylov D. S.; Avdoshenko S. M.; Liu F.; Spree L.; Yadav R.; Alvertis A.; Hozoi L.; Nenkov K.; Kostanyan A.; Greber T.; Wolter A. U. B.; Popov A. A. Selective arc-discharge synthesis of Dy2S-clusterfullerenes and their isomer-dependent single molecule magnetism. Chem. Sci. 2017, 8, 6451–6465. 10.1039/C7SC02395B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.; Velkos G.; Liu F.; Sudarkova S. M.; Wang Y.; Zhuang J.; Zhang H.; Li X.; Zhang X.; Büchner B.; Avdoshenko S. M.; Popov A. A.; Chen N. Single Molecule Magnetism with Strong Magnetic Anisotropy and Enhanced Dy···Dy Coupling in Three Isomers of Dy-Oxide Clusterfullerene Dy2O@C82. Advanced Science 2019, 1901352. 10.1002/advs.201901352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velkos G.; Krylov D.; Kirkpatrick K.; Spree L.; Dubrovin V.; Büchner B.; Avdoshenko S.; Bezmelnitsyn V.; Davis S.; Faust P.; Duchamp J.; Dorn H.; Popov A. A. High blocking temperature of magnetization and giant coercivity in the azafullerene Tb2@C79N with a single-electron Tb–Tb bond. Angew. Chem., Int. Ed. 2019, 58, 5891–5896. 10.1002/anie.201900943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velkos G.; Krylov D. S.; Kirkpatrick K.; Liu X.; Spree L.; Wolter A. U. B.; Buchner B.; Dorn H. C.; Popov A. A. Giant exchange coupling and field-induced slow relaxation of magnetization in Gd2@C79N with a single-electron Gd-Gd bond. Chem. Commun. 2018, 54, 2902–2905. 10.1039/C8CC00112J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z.; Dong B.-W.; Liu Z.; Liu J.-J.; Su J.; Yu C.; Xiong J.; Shi D.-E.; Wang Y.; Wang B.-W.; Ardavan A.; Shi Z.; Jiang S.-D.; Gao S. Endohedral Metallofullerene as Molecular High Spin Qubit: Diverse Rabi Cycles in Gd2@C79N. J. Am. Chem. Soc. 2018, 140, 1123–1130. 10.1021/jacs.7b12170. [DOI] [PubMed] [Google Scholar]
- Liu F.; Velkos G.; Krylov D. S.; Spree L.; Zalibera M.; Ray R.; Samoylova N. A.; Chen C.-H.; Rosenkranz M.; Schiemenz S.; Ziegs F.; Nenkov K.; Kostanyan A.; Greber T.; Wolter A. U. B.; Richter M.; Büchner B.; Avdoshenko S. M.; Popov A. A. Air-stable redox-active nanomagnets with lanthanide spins radical-bridged by a metal–metal bond. Nat. Commun. 2019, 10, 571. 10.1038/s41467-019-08513-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Krylov D. S.; Spree L.; Avdoshenko S. M.; Samoylova N. A.; Rosenkranz M.; Kostanyan A.; Greber T.; Wolter A. U. B.; Büchner B.; Popov A. A. Single molecule magnet with an unpaired electron trapped between two lanthanide ions inside a fullerene. Nat. Commun. 2017, 8, 16098. 10.1038/ncomms16098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddle S. T.Molecular Metal–Metal Bonds; Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Fu W.; Zhang J.; Fuhrer T.; Champion H.; Furukawa K.; Kato T.; Mahaney J. E.; Burke B. G.; Williams K. A.; Walker K.; Dixon C.; Ge J.; Shu C.; Harich K.; Dorn H. C. Gd2@C79N: Isolation, Characterization, and Monoadduct Formation of a Very Stable Heterofullerene with a Magnetic Spin State of S = 15/2. J. Am. Chem. Soc. 2011, 133, 9741–9750. 10.1021/ja202011u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo T.; Xu L.; Beavers C. M.; Olmstead M. M.; Fu W.; Crawford T. D.; Balch A. L.; Dorn H. C. M2@C79N (M = Y, Tb): Isolation and Characterization of Stable Endohedral Metallofullerenes Exhibiting M···M Bonding Interactions inside Aza[80]fullerene Cages. J. Am. Chem. Soc. 2008, 130, 12992–12997. 10.1021/ja802417d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov A. A.; Avdoshenko S. M.; Pendás A. M.; Dunsch L. Bonding between strongly repulsive metal atoms: an oxymoron made real in a confined space of endohedral metallofullerenes. Chem. Commun. 2012, 48, 8031–8050. 10.1039/c2cc32568c. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Kitaura R.; Shinohara H. Metal-Dependent Stability of Pristine and Functionalized Unconventional Dimetallofullerene M2@Ih-C80. J. Phys. Chem. C 2014, 118, 13953–13958. 10.1021/jp5028492. [DOI] [Google Scholar]
- Bao L.; Chen M.; Pan C.; Yamaguchi T.; Kato T.; Olmstead M. M.; Balch A. L.; Akasaka T.; Lu X. Crystallographic Evidence for Direct Metal–Metal Bonding in a Stable Open-Shell La2@Ih-C80 Derivative. Angew. Chem., Int. Ed. 2016, 55, 4242–4246. 10.1002/anie.201511930. [DOI] [PubMed] [Google Scholar]
- Yamada M.; Kurihara H.; Suzuki M.; Saito M.; Slanina Z.; Uhlik F.; Aizawa T.; Kato T.; Olmstead M. M.; Balch A. L.; Maeda Y.; Nagase S.; Lu X.; Akasaka T. Hiding and Recovering Electrons in a Dimetallic Endohedral Fullerene: Air-Stable Products from Radical Additions. J. Am. Chem. Soc. 2015, 137, 232–238. 10.1021/ja509956y. [DOI] [PubMed] [Google Scholar]
- Rajaraman G.; Singh M. K.; Yadav N. Record High Magnetic Exchange and Magnetization Blockade in Ln2@C79N (Ln = Gd(III) and Dy(III)) Molecules: A Theoretical Perspective. Chem. Commun. 2015, 51, 17732–17735. 10.1039/C5CC06642E. [DOI] [PubMed] [Google Scholar]
- Cimpoesu F.; Frecus B.; Oprea C. I.; Ramanantoanina H.; Urland W.; Daul C. On exchange coupling and bonding in the Gd2@C80 and Gd2@C79N endohedral dimetallo-fullerenes. Mol. Phys. 2015, 113, 1712–1727. 10.1080/00268976.2015.1007107. [DOI] [Google Scholar]
- Wang X.; McKay J. E.; Lama B.; van Tol J.; Li T.; Kirkpatrick K.; Gan Z.; Hill S.; Long J. R.; Dorn H. C. Gadolinium based endohedral metallofullerene Gd2@C79N as a relaxation boosting agent for dissolution DNP at high fields. Chem. Commun. 2018, 54, 2425–2428. 10.1039/C7CC09765D. [DOI] [PubMed] [Google Scholar]
- Kramida A. E.; Ralchenko Y.; Reader J.. NIST Atomic Spectra Database, version 5.6.1 [online]; National Institute of Standards and Technology: Gaithersburg, MD, 2019; available at https://physics.nist.gov/asd.
- Popov A. A. Redox-active metal–metal bonds between lanthanides in dimetallofullerenes. Curr. Opin. Electrochem. 2018, 8, 73–80. 10.1016/j.coelec.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki T.; Suenaga K.; Lian Y. F.; Gu Z. N.; Shinohara H. Intrafullerene electron transfers in Sm-containing metallofullerenes: Sm@C2n (74 ≤ 2n ≤ 84). J. Mol. Graphics Modell. 2001, 19, 244–251. 10.1016/S1093-3263(00)00093-0. [DOI] [PubMed] [Google Scholar]
- Samoylova N. A.; Avdoshenko S. M.; Krylov D. S.; Thompson H. R.; Kirkhorn A.; Rosenkranz M.; Schiemenz S.; Ziegs F.; Wolter A. U. B.; Yang S.; Stevenson S.; Popov A. A. Confining the spin between two metal atoms within the carbon cage: Redox-active metal-metal bonds in dimetallofullerenes and their stable cation radicals. Nanoscale 2017, 9, 7977–7990. 10.1039/C7NR02288C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y.; Okazaki T.; Okubo S.; Akachi M.; Ohno Y.; Mizutani T.; Nakamura T.; Kitaura R.; Sugai T.; Shinohara H. Enhanced 1520 nm photoluminescence from Er3+ ions in di-erbium-carbide metallofullerenes (Er2C2)@C82 (isomers I, II, and III). ACS Nano 2007, 1, 456–462. 10.1021/nn700235z. [DOI] [PubMed] [Google Scholar]
- Cao X.; Dolg M. Pseudopotential study of lanthanum and lutetium dimers. Theor. Chem. Acc. 2002, 108, 143–149. 10.1007/s00214-002-0372-8. [DOI] [Google Scholar]
- Valencia R.; Rodríguez-Fortea A.; Poblet J. M. Understanding the Stabilization of Metal Carbide Endohedral Fullerenes M2C2@C82 and Related Systems. J. Phys. Chem. A 2008, 112, 4550–4555. 10.1021/jp800419d. [DOI] [PubMed] [Google Scholar]
- Velloth A.; Imamura Y.; Kodama T.; Hada M. Theoretical Insights into the Electronic Structures and Stability of Dimetallofullerenes M2@Ih-C80. J. Phys. Chem. C 2017, 121, 18169–18177. 10.1021/acs.jpcc.7b03533. [DOI] [Google Scholar]
- Li Q.-Z.; Zheng J.-J.; He L.; Nagase S.; Zhao X. La–La bonded dimetallofullerenes [La2@C2n]−: species for stabilizing C2n (2n = 92–96) besides La2C2@C2n. Phys. Chem. Chem. Phys. 2018, 20, 14671–14678. 10.1039/C8CP01371C. [DOI] [PubMed] [Google Scholar]
- Kato T. Metal dimer and trimer within spherical carbon cage. J. Mol. Struct. 2007, 838, 84–88. 10.1016/j.molstruc.2007.01.027. [DOI] [Google Scholar]
- Tsuchiya T.; Wielopolski M.; Sakuma N.; Mizorogi N.; Akasaka T.; Kato T.; Guldi D. M.; Nagase S. Stable Radical Anions Inside Fullerene Cages: Formation of Reversible Electron Transfer Systems. J. Am. Chem. Soc. 2011, 133, 13280–13283. 10.1021/ja205391v. [DOI] [PubMed] [Google Scholar]
- Bubnov V. P.; Laukhina E. E.; Kareev I. E.; Koltover V. K.; Prokhorova T. G.; Yagubskii E. B.; Kozmin Y. P. Endohedral metallofullerenes: A convenient gram-scale preparation. Chem. Mater. 2002, 14, 1004–1008. 10.1021/cm011106b. [DOI] [Google Scholar]
- Kareev I. E.; Bubnov V. P.; Laukhina E. E.; Dodonov A. F.; Kozlovski V. I.; Yagubskii E. B. Experimental evidence in support of the formation of anionic endohedral metallofullerenes during their extraction with N,N-dimethylformamide. Fullerenes, Nanotubes, Carbon Nanostruct. 2005, 12, 65–69. 10.1081/FST-120027135. [DOI] [Google Scholar]
- Tsuchiya T.; Wakahara T.; Lian Y. F.; Maeda Y.; Akasaka T.; Kato T.; Mizorogi N.; Nagase S. Selective extraction and purification of endohedral metallofullerene from carbon soot. J. Phys. Chem. B 2006, 110, 22517–22520. 10.1021/jp0650679. [DOI] [PubMed] [Google Scholar]
- Pan C.; Shen W.; Yang L.; Bao L.; Wei Z.; Jin P.; Fang H.; Xie Y.-P.; Akasaka T.; Lu X. Crystallographic Characterization of Y2C2n (2n = 82, 88–94): Direct Y-Y Bonding and Cage-Dependent Cluster Evolution. Chem. Sci. 2019, 10, 4707–4713. 10.1039/C9SC00941H. [DOI] [Google Scholar]
- Shen W.; Bao L.; Wu Y.; Pan C.; Zhao S.; Fang H.; Xie Y.; Jin P.; Peng P.; Li F.-F.; Lu X. Lu2@C2n (2n = 82, 84, 86): Crystallographic Evidence of Direct Lu–Lu Bonding between Two Divalent Lutetium Ions Inside Fullerene Cages. J. Am. Chem. Soc. 2017, 139, 9979–9984. 10.1021/jacs.7b04421. [DOI] [PubMed] [Google Scholar]
- Kareev I. E.; Lebedkin S. F.; Bubnov V. P.; Yagubskii E. B.; Ioffe I. N.; Khavrel P. A.; Kuvychko I. V.; Strauss S. H.; Boltalina O. V. Trifluoromethylated endohedral metallofullerenes: Synthesis and characterization of Y@C82(CF3)5. Angew. Chem., Int. Ed. 2005, 44, 1846–1849. 10.1002/anie.200461497. [DOI] [PubMed] [Google Scholar]
- Kareev I. E.; Bubnov V. P.; Yagubskii E. B. Endohedral gadolinium-containing metallofullerenes in the trifluoromethylation reaction. Russ. Chem. Bull. 2008, 57, 1486–1491. 10.1007/s11172-008-0192-7. [DOI] [Google Scholar]
- Foroutan-Nejad C.; Vicha J.; Marek R.; Patzschke M.; Straka M. Unwilling U-U bonding in U2@C80: cage-driven metal-metal bonds in di-uranium fullerenes. Phys. Chem. Chem. Phys. 2015, 17, 24182–24192. 10.1039/C5CP04280A. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Wang T.; Li Y.; Li J.; Wu J.; Wu B.; Jiang L.; Wang C. Steering Metallofullerene Electron Spin in Porous Metal–Organic Framework. J. Am. Chem. Soc. 2015, 137, 15055–15060. 10.1021/jacs.5b10796. [DOI] [PubMed] [Google Scholar]
- Hu Z.; Dong B.-W.; Liu Z.; Liu J.-J.; Su J.; Yu C.; Xiong J.; Shi D.-E.; Wang Y.; Wang B.-W.; Ardavan A.; Shi Z.; Jiang S.-D.; Gao S. Correction to “Endohedral Metallofullerene as Molecular High Spin Qubit: Diverse Rabi Cycles in Gd2@C79N. J. Am. Chem. Soc. 2018, 140, 6183–6183. 10.1021/jacs.8b04311. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Luchinat C.; Parigi G.. Solution NMR of Paramagnetic Molecules. Application to Metallobiomolecules and Models; Elsevier Science B.V.: Amsterdam, 2001. [Google Scholar]
- Lu X.; Hu S.; Shen W.; Duan G.; Yang L.; Jin P.; Xie Y.; Akasaka T. Crystallographic and Theoretical Investigations of Er2@C2n (2n = 82, 84, 86): Indication of Distance-Dependent Metal-Metal Bonding Nature. Chem. - Eur. J. 2019, 25, 11538–11544. 10.1002/chem.201902321. [DOI] [PubMed] [Google Scholar]
- Gould C.; McClain K. R.; Yu J.; Groshens T. J.; Furche F.; Harvey B. G.; Long J. R. Synthesis and Magnetism of Neutral, Linear Metallocene Complexes of Terbium(II) and Dysprosium(II). J. Am. Chem. Soc. 2019, 141, 12967–12973. 10.1021/jacs.9b05816. [DOI] [PubMed] [Google Scholar]
- Winpenny R. E. P.; Chilton N. F.; Giansiracusa M.; Collison D.; Kostopoulos A. Correlating Blocking Temperatures with Relaxation Mechanisms in Monometallic Single-Molecule Magnets with High Energy Barriers (Ueff> 600 K). Chem. Commun. 2019, 55, 7025–7028. 10.1039/C9CC02421B. [DOI] [PubMed] [Google Scholar]
- Mansikkamäki A.; Popov A. A.; Deng Q.; Iwahara N.; Chibotaru L. F. Interplay of spin-dependent delocalization and magnetic anisotropy in the ground and excited states of [Gd2@C78]− and [Gd2@C80]−. J. Chem. Phys. 2017, 147, 124305. 10.1063/1.5004183. [DOI] [PubMed] [Google Scholar]
- Kanetomo T.; Yoshitake T.; Ishida T. Strongest Ferromagnetic Coupling in Designed Gadolinium(III)–Nitroxide Coordination Compounds. Inorg. Chem. 2016, 55, 8140–8146. 10.1021/acs.inorgchem.6b01072. [DOI] [PubMed] [Google Scholar]
- Kanetomo T.; Kihara T.; Miyake A.; Matsuo A.; Tokunaga M.; Kindo K.; Nojiri H.; Ishida T. Giant Exchange Coupling Evidenced with a Magnetization Jump at 52 T for a Gadolinium–Nitroxide Chelate. Inorg. Chem. 2017, 56, 3310–3314. 10.1021/acs.inorgchem.6b02685. [DOI] [PubMed] [Google Scholar]
- Demir S.; Zadrozny J. M.; Nippe M.; Long J. R. Exchange Coupling and Magnetic Blocking in Bipyrimidyl Radical-Bridged Dilanthanide Complexes. J. Am. Chem. Soc. 2012, 134, 18546–18549. 10.1021/ja308945d. [DOI] [PubMed] [Google Scholar]
- Meihaus K. R.; Corbey J. F.; Fang M.; Ziller J. W.; Long J. R.; Evans W. J. Influence of an Inner-Sphere K+ Ion on the Magnetic Behavior of N23– Radical-Bridged Dilanthanide Complexes Isolated Using an External Magnetic Field. Inorg. Chem. 2014, 53, 3099–3107. 10.1021/ic4030102. [DOI] [PubMed] [Google Scholar]
- Chibotaru L. F.; Ungur L. Ab initio calculation of anisotropic magnetic properties of complexes. I. Unique definition of pseudospin Hamiltonians and their derivation. J. Chem. Phys. 2012, 137, 064112. 10.1063/1.4739763. [DOI] [PubMed] [Google Scholar]
- Velloth A.; Imamura Y.; Hada M. Functionalization of Endohedral Metallofullerenes toward Improving Barrier Height for the Relaxation of Magnetization for Dy2@C80–X (X = CF3, C3N3Ph2). Inorg. Chem. 2019, 58, 1208–1215. 10.1021/acs.inorgchem.8b02652. [DOI] [PubMed] [Google Scholar]
- Velloth A.; Imamura Y.; Kodama T.; Hada M. Quantum Chemical Study on Endohedral Heteronuclear Dimetallofullerene M1M2@Ih-C80 Toward Molecular Design. J. Phys. Chem. C 2017, 121, 27700–27708. 10.1021/acs.jpcc.7b08302. [DOI] [Google Scholar]
- Jin P.; Li Y.; Magagula S.; Chen Z. Exohedral functionalization of endohedral metallofullerenes: Interplay between inside and outside. Coord. Chem. Rev. 2019, 388, 406–439. 10.1016/j.ccr.2019.02.028. [DOI] [Google Scholar]
- Rudolf M.; Kirner S. V.; Guldi D. M. A multicomponent molecular approach to artificial photosynthesis - the role of fullerenes and endohedral metallofullerenes. Chem. Soc. Rev. 2016, 45, 612–630. 10.1039/C5CS00774G. [DOI] [PubMed] [Google Scholar]
- Atzori M.; Sessoli R. The Second Quantum Revolution: Role and Challenges of Molecular Chemistry. J. Am. Chem. Soc. 2019, 141, 11339–11352. 10.1021/jacs.9b00984. [DOI] [PubMed] [Google Scholar]