

Abstract
Aiming for glycolipid-based vesicles for targeted drug delivery, cationic Guerbet glycosides with spacered click functionality were designed and synthesized. The cationic charge promoted the distribution of the glycolipids during the formulation, thereby leading to homogeneously small vesicles. The positive surface charge of the vesicles stabilizes them against unwanted fusion and promotes interactions of the drug carriers with typical negative charge-dominated target cells. High bioconjugation potential of the functionalized glycolipids based on the copper-catalyzed azide alkyne cycloaddition makes them highly valuable components for targeted drug delivery systems.
Introduction
Despite the tremendous diversity of life, manifested in billions of species covering single and multicellular organisms, cellular processes are highly conserved through interspecies’ borders. This complicates the search for medical drugs, because the intended toxic effect on parasite cells is typically accompanied by unwanted side effects on host cells, constraining the application field and scope of medical drugs. Limited selectivity of biological active compounds has, therefore, created growing interest in a selective delivery to target cells.1,2 This concept is closely related to the immune response system and associated vaccinations, which also addresses hostile cells based on their surface topology rather than specific intracellular processes.
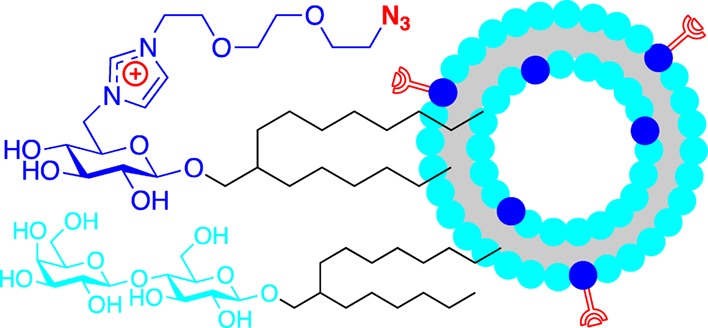
Selective drug delivery requires, on the one hand, an effective encapsulation of the biological active compound to avoid unwanted interactions with nontarget cells and on the other specific interactions of the carrier to ensure the delivery and subsequent action at the target.3,4 Vesicular assemblies of surfactants have been suggested as effective drug carriers. Their cell-like membrane provides a good barrier against unwanted exposure of the drug to nontarget cells. Moreover, they enable the encapsulation of both hydrophilic and hydrophobic compounds in the aqueous core and the bilayer membrane, respectively, thus making them universally applicable drug carriers. Incorporation of antigens for specific biological receptors provides an opportunity to direct a drug to target cells.5 In order to avoid a loss of the antigen, strong anchoring on the vesicle is required. Although this might be achieved by binding the antigen to an anchor lipid, which is subsequently applied as an additive in the vesicle formulation, such incorporation is expected to leave almost half of the antigens ineffective, because they are facing the inside of the drug carrier rather than the outside. Chemical coupling of functionalized antigens with complementary functional groups on vesicular assemblies, on the other side, enables a selective introduction of recognition domains on the extracellular surface,6,7 as shown in Figure 1.
Figure 1.

Conceptual design for targeted biofunctionalized vesicles.
Vesicles can be obtained with any type of surfactant, provided that the relative surface areas of the hydrophilic and hydrophobic region are balanced, hence promoting a lamellar assembly.8 Most commonly applied are anionic surfactants, like fatty acid salts.9 Besides, natural zwitterionic lipids, like DPPC and lecithin, have found attention as well.10,11 Due to the presence of ionic charges, the assembly behavior of both, anionic and zwitterionic, surfactants, is affected by external stimuli. Of particular relevance are the pH and certain electrolytes.12 While this stimuli response provides opportunities for easy vesicle formulation, e.g., in pH-induced micelle–vesicle transformations,13,14 it also constrains their stability. Higher assembly stability can be obtained with nonionic surfactants.15,16 Of particular interest here are glycolipids,17 which exhibit superior temperature tolerance compared to industrial ethylene oxide-based compounds. Moreover, their natural product-like structure ensures better biocompatibility.
Recently, we have developed vesicular drug carriers based on biantennary glycolipids.18 The double-chain region stabilizes lamellar assemblies while also providing access to bicontinuous cubic phases,19 which have been associated with membrane fusion processes.20,21 The bicontinuous cubic phase, hence, potentially provides an opportunity for the delivery of vesicle contents to a target cell.
Results and Discussion
Compound Design
In line with our previous work,7 we aimed for a click-functionalized glycolipid to be applied as a cosurfactant in lactose-based glycolipid vesicle formulations. The choice of the base surfactant reflected both economic and stability aspects: Although glucose is by far the least expensive carbohydrate, glycolipids based on monosaccharides exhibit unfavorably low HLB (hydrophilic–lipophilic balance) values if longer alkyl chains are applied. Since longer alkyl chains, however, ensure high assembly stability owing to a low critical assembly concentration,22,23 a more expensive disaccharide core provides advantages for the vesicle formulation. With this, background lactose became the resource of choice. Moreover, previous investigations have shown beneficial interactions of the axial hydroxyl group in galactosides on the stability of bilayer assemblies,24 thereby also favoring lactose over alternative disaccharides like maltose and isomaltose.
In view of the observation that application of ionic cosurfactants reduces the size for glycolipid vesicles while providing a narrow size distribution at the same time,18a we targeted to introduce a charge on the functionalized glycolipid. This way, the biofunctionalization anchor was expected to facilitate a favorable vesicle size as well, thereby avoiding the need of another cosurfactant. Moreover, the charge avoids clustering of the cosurfactants inside the vesicular assemblies due to repulsive interactions, thereby ensuring good distribution and accessibility of the anchor. Since typical target cells for drugs exhibit a negative surface charge, a cationic imidazolium was selected as an easily accessible building block. A flexible oligoethylene glycol spacer was chosen to mediate an effective bioconjugation without steric constraints by the vesicle surface, while a click chemistry approach, based on the copper-catalyzed Huisgen azide–alkyne cycloadditon,25−28 was selected for the coupling of vesicles with receptor antigens. As the latter can be easily functionalized with reactive propargyl reagents, the complementary azide coupling function is needed to be introduced on the anchor glycolipid. Considering the molecular size of the imidazolium ion, we chose to use a glucose carbohydrate for the anchor surfactant, as this lowers the synthetic costs. Hydrogen bonding interactions between the carbohydrate cores of the base glycolipid and the bioconjugation anchor aim to avoid a potential loss of targeting ligands on the drug carriers. The design of the functionalized glycolipid anchor is displayed in Figure 2.
Figure 2.

Structural design of the glycolipid core surfactant (A) and the functionalized bioconjugation anchor (B) for vesicular drug carriers.
Synthesis
The synthesis, as shown in Scheme 1, was based on previously reported Guerbet glucosides 3 with C12 and C16 hydrophobic domains.19a,19b These chain lengths were chosen to enable assembly studies without high material demand on the one hand and reasonably fast assembly kinetics on the other. Functionalization of the carbohydrate applied halogenation at C-6, following the established approach of Hanessian et al.29 The intermediates 4a and 4b were directly subjected to peracetylation to enable effective purification by chromatography and provided precursors 5a and 5b for the coupling with the imidazole component. The unsatisfactory yield of only 52% reflected obstacles for the removal of the triphenylphosphine oxide side product.
Scheme 1. Synthetic Scheme for the Functionalized Anchor Glycolipid.

The respective azido-terminated imidazole precursors 9, with varying spacer lengths from two to four ethylene oxide units, were obtained by nucleophilic substitution from the previously reported corresponding chlorides 82–84.30−32 The synthesis emphasizes the use of excess dichloride 7 in order to avoid spacer-linked diimidazoles, while application of low reaction temperatures avoided a bisalkylation of the imidazole.
Coupling of glycoside precursor 5 with imidazole 9 according to the Menschutkin reaction applied heating in a high boiling nonpolar solvent.33 The ionic nature of the imidazolium compounds 10 prevents an effective purification process. Therefore, equivalent reagent quantities were used, furnishing practically NMR-pure compounds. Final deprotection by transesterfication provided the functionalized glycolipids 11 in overall yields of about 43% based on the Guerbet glucosides 3.
Contrary to the halogenation of 3, both the Menschutkin coupling and the deprotection of 10 preceded in high yields without purification requirement. This is in line with previous investigations furnishing imidazolium-linked glycosides.14,34 Unexpected loss of compound during the deacetylation probably relates to adsorption of the cationic compound to the ion exchange resin, which was applied to remove the sodium methoxide catalyst. It may be avoided by aminolysis of the esters. However, the removal of the reaction side product would be more difficult.
Unlike the synthetic similarity, the compound design of 11 differs substantially from previously reported alkylimidazolium glycosides.14,34 While all compounds share the structural elements of sugar-based surfactants incorporating imidazolium cations, in 11, the imidazolium is not linking the surfactant antipodes. This difference has a tremendous impact on the surfactant biocompatibility, because it ensures easy degradability of the surfactant linkage.
Application Studies
Assembly studies with the functionalized glycolipids 11b by systematic surface tension measurements furnished critical aggregation concentrations (CACs) in the typical magnitude for the C16 chain, i.e., around 10–5 M. The CACs in Table 1 exceed the corresponding values for glycolipids incorporating equivalent hydrophobic domains by a factor of about 2.18a,35 This reflects the repulsive interactions of the surfactant head groups, destabilizing the assemblies. The minor difference between 11b3 and 11b4 is more likely related to slight differences in minor nonrecognized impurities, rather than reflecting the difference in the number of ethylene glycol units inside the spacer.
Table 1. Surface Properties of 11.
| surfactant | CMC (μM) | ΓCAC (mN m–1) | Ainterface (Å2) |
|---|---|---|---|
| 11b3 | 18 | 31 | 49 |
| 11b4 | 24 | 31 | 47 |
Lyotropic studies of the surfactants by contact penetration with water36 provided no visible textures under the optical polarizing microscope. In view of observed good water solubility, it was concluded that the surfactants only assemble in micelles; hence, the CAC was specified as critical micelle concentration (CMC). The absence of any anisotropic lyotropic phase is in contradiction to the behavior of the corresponding nonionic glucoside 3b.19c However, it is in line with the increased CAC; the repulsive interactions of the surfactant head groups do not only reduce the assembly stability but also increase the effective interfacial surface area of the head group. This is reflected in a moderate increase in the determined molecular surface area of about 10% with respect to nonionic alkyl glucosides.37,38 The imbalance of the hydrophilic and hydrophobic domains in terms of their respective surface areas leads to a conical shape of the surfactant, which promotes curved assembly geometries like micelles.39
While the pure cationic glycolipids 11 did not exhibit any anisotropic texture in contact with water, mixtures with dominating contents of the nonionic lactose-based Guerbet glycoside 12 showed high affinity for the aqueous lamellar phase, as indicated by extensive formation of myelin figures. Unlike for the solely β-anomeric 11, the base surfactant 12 was applied as a mixture of anomers.18a,35 This approach provides access to the synthetic product without chromatographic purification,40 thereby enhancing the economic viability.

Vesicles were prepared using the ethanolic injection method.41 Application of 5–10% of the cationic glycolipid 11b leads to the expected reduction of the vesicle size, as shown in Table 2. Moreover, the zeta potential of the vesicles confirmed a change in the surface charge, thereby proving the successful incorporation of the ionic cosurfactant. Regardless of the anionic or cationic character, the increased surface charge with respect to pure glycolipid vesicles enhances the stability of the vesicular assemblies owing to repulsive interactions of aggregates, constraining assembly fusions according to Oswald ripening.42 In fact, the vesicle dimensions remained practically unchanged upon storage for several days in a refrigerator.
Table 2. Vesicle Properties.
After storage for 1 week below 10 °C.
In order to evaluate the efficiency of the click coupling on vesicular assemblies incorporating the functionalized glycolipid 11, a model reaction on micellar assemblies of 11a4 was performed. Propargyl alcohol was used as a water-soluble alkyne component together with a copper(I) catalyst generated from copper sulfate and sodium ascorbate. The unusual probe enables the evaporative removal of the unreacted alkyne component after the coupling, which simplifies the analysis. The use of the micelle model instead of vesicles originated from unavailability of a suitable click-functionalized fluorescent or photometric quantifiable probe to evaluate the coupling process. Instead, the surfactant was extracted after the reaction, and the coupling process was investigated by spectroscopy. The low content of the functionalized glycolipid in vesicles prevents an effective monitoring of the coupling process, while such constrains do not apply for assemblies of the pure surfactant. However, since the latter are of micellar shape, as indicated previously, a different assembly was used as a model. The high surface density of the surfactants in the micelle was expected to prevent a complete conversion of the azide groups, as shown in Scheme 2. This, however, did not affect the monitoring. The IR analysis of the isolated surfactant after the micelle click coupling exhibited a drastic reduction of the azide peak, as depicted in Figure 3. This suggests a high yield for the click reaction despite the dense anchor packing. Moreover, the NMR analysis of the click product revealed an additional peak in the aromatic region, as shown in Figure 4. A comparison of this peak with 16, resembling a synthetic analogue of 14, confirmed the assignment of the triazole.
Scheme 2. Model Click Coupling on a Surfactant Micelle.

Figure 3.

IR spectra for model click coupling.
Figure 4.

1H NMR analysis of the aromatic region for model click coupling.
Conclusions
6-Halofunctionalization of Guerbet glucosides followed by subsequent Menschutkin reaction with azido-terminated oligo-ethoxylated imidazole provides easy access to cationic glycolipids with click functionality. The imidazolium cation increases the molecular solubility of the glycolipid due to repulsive interactions of the head groups and alters the assembly geometry of the surfactant. Its application as a cosurfactant on Guerbet lactosides furnished small vesicles with high size homogeneity and positive surface charge, reflecting the presence of the functionalized glycolipid. A micellar model study confirmed high bioconjugation potential for the functionalized glycolipids according to the CuAAC click reaction. Therefore, the cosurfactant has high potential for applications in targeted vesicular drug delivery.
Experimental Section
General Methods
Starting materials (synthesis grade) and solvents (AR-grade) were obtained from various commercial resources and used without prior purification. Reactions were followed by TLC, applying staining by dilute ethanolic H2SO4 followed by subsequent heating. Chromatographic purifications were performed on silica gel 60 (35–70 μm) using the flash technique. Optical rotations were measured on a JASCO P-1020 digital polarimeter using 10 cm cells. IR spectra were recorded on a Perkin Elmer ATR FTIR with 4 cm–1 resolution. NMR measurements were performed on 400 MHz spectrometers at room temperature (rt) using solvent peaks for calibration. 13C spectra applied the APT protocol. Assignments are based on the analyses of coupling patterns and HSQC correlations. Mass spectra were recorded in the ESI mode on an Agilent 6550 Q-TOF and a Jeol AccuTOF spectrometer.
Physical Studies
All surfactants were dried in a desiccator over P2O5 for several hours prior to physical investigations. Distilled water with an electric conductivity of 1.1 ± 0.1 μS cm–1 at 25 °C was applied for all formulation studies. Lyotropic phase investigations were performed on a modified Amscope microscope with installed polarizers using the contact penetration technique.36 The assembly of surfactants was studied by systematic surface tension measurements on a Sigma 702 tensiometer using the Du Noüy ring method. Sample solutions were equilibrated for several hours before the measurement, while valid surface tension measurements required five replicates with a maximum standard deviation of 0.1 mN m–1. The critical aggregation concentration (CAC) was determined as the intersection of the regression lines for the concentration-dependent and the concentration-independent regions of the plot of the surface tension against the logarithmic surfactant concentration. The molecular surface area was calculated from the slope of the concentration-dependent region in the surface tension: log(c) plots based on the Gibbs adsorption isotherm.43 The formulation of vesicles applied the ethanolic injection method.41 An ethanolic surfactant solution was rapidly injected into a stirred 19th-fold volume of water through a 25 Ga bevel tip needle. The concentration was selected to obtain a final surfactant concentration of about 1 mM. Samples were left for several hours prior to analysis to ensure equilibration. Particle sizes were determined at rt by dynamic light scattering on a Malvern Zen 3600 Zetasizer, applying a measuring angle of 173°. Zeta potential measurements applied the same instrument.
2-Butyl-octyl 6-Bromo-6-deoxy-2,3,4-tri-O-acetyl-β-d-glucopyranoside (5a)
3a(19a,19b) (2.0 g, 5.7 mmol) and PPh3 (3.0 g, 11 mmol) were dissolved in DMF (20 mL), and about 2 mL of the solvent was evaporated to remove water inside the reaction mixture. The solution was cooled with ice and NBS (2.0 g, 11 mmol) was added followed by heating to 70 °C for 2 h under a N2 atmosphere. The excess reagent was destroyed by quenching with MeOH (10 mL), and the solvent was evaporated at reduced pressure. The crude mixture was subsequently peracetylated with acetic anhydride (4 mL) in pyridine (20 mL). The solvent and excess reagent were evaporated, and the residue distributed between CH2Cl2 and HCl (2 N). The organic phase was washed with water and dried over MgSO4. Purification of the crude product by chromatography (Hex/EtOAc, 7:1) furnished 5a (1.6 g, 52%) as a yellow syrup. [α]D25 – 5 (c 0.30, CHCl3). 1H NMR (400 MHz, CDCl3): δ 5.19 (dd∼t, H-3), 4.98 (dd∼t, H-4), 4.95 (dd, H-2), 4.47 (d, H-1), 3.83 (dd, α-CH2A), 3.69 (ddd, H-5), 3.45 (dd, H-6A), 3.38 (dd, H-6B), 3.34 (mc, α-CH2B), 2.04, 2.01, 1.99 (3 s, 3 × 3 H, Ac), 1.55 (mc, β-CH), 1.29–1.24 (16 H, mc, bulk-CH2), 0.87 (2 t, 6 H, CH3); 3J1,2 = 8.0, 3J2,3 = 10.0, 3J3,4 = 10.0, 3J4,5 = 10.0, 3J5,6A = 5.0, 3J5,6B = 3.0, 2J6 = 11.0 Hz; 13C NMR (100 MHz, CDCl3): δ 170.2, 169. 5, 169.1 (C=O), 100.8 (C-1), 73.3 (C-5), 72.9/ 72.8 (α), 72.6 (C-3), 71.4 (C-4), 71.2 (C-2), 37.9 (β), 31.8, 31.1, 30.9, 30.8, 30.7 (bulk-CH2), 30.6 (C-6), 29.64/29.63, 28.9, 28.8, 26.7, 26.5, 23.00/22.98 (bulk-CH2), 22.6 (ω-1), 20.61, 20.55, 20.52 (Ac), 14.0 (ω).
2-Hexyl-decyl 6-Bromo-6-deoxy-2,3,4-tri-O-acetyl-β-d-glucopyranoside (5b)
3b(19a,19b) (1.7 g, 4.2 mmol) and PPh3 (2.2 g, 8.4 mmol) were dissolved in DMF (12 mL), and about 2 mL of the solvent was evaporated to remove water inside the reaction mixture. The solution was cooled with ice and NBS (1.5 g, 8.4 mmol) was added followed by heating to 70 °C for 2 h under a N2 atmosphere. The excess reagent was destroyed by quenching with MeOH (10 mL), and the solvent was evaporated at reduced pressure. The crude mixture was subsequently peracetylated with acetic anhydride (4 mL) in pyridine (12 mL). The solvent and excess reagent were evaporated, and the residue distributed between CH2Cl2 and HCl (2 N). The organic phase was washed with water and dried over MgSO4. Purification of the crude product by chromatography (Hex/EtOAc, 9:1) furnished 5b (1.3 g, 52%) as a yellow syrup. [α]D25 – 4 (c 0.20, CHCl3). 1H NMR (400 MHz, CDCl3): δ 5.21 (dd∼t, H-3), 4.99 (dd∼t, H-4), 4.97 (dd, H-2), 4.49 (d, H-1), 3.84 (dd, α-CH2A), 3.69 (ddd, H-5), 3.46 (dd, H-6A), 3.39 (dd, H-6B), 3.35 (dd, α-CH2B), 2.06, 2.03, 2.01 (3 s, 3 × 3 H, Ac), 1.56 (mc, β-CH), 1.26 (24 H, mc, bulk-CH2), 0.88 (2 t, 6H, CH3); 3J1,2 = 8.0, 3J2,3 = 10.0, 3J3,4 = 10.0, 3J4,5 = 10.0, 3J5,6A = 3.0, 3J5,6B = 6.0, 2J6 = 11.0 Hz; 13C NMR (100 MHz, CDCl3): δ 170.3, 169.5, 169.1 (C=O), 100.8 (C-1), 73.3 (C-5), 72.9 (α), 72.7 (C-3), 71.4 (C-4), 71.2 (C-2), 37.9 (β), 31.9/31.8, 31.1 (bulk-CH2), 30.9 (C-6), 30.7, 30.04/30.02, 29.69/29.67, 29.6, 29.3, 26.8/26.7, 26.61/26.56 (bulk-CH2), 22.7 (ω-1), 20.7, 20.60, 20.57 (Ac), 14.1 (ω).
1-[2-(2-Azido-ethoxy)-ethyl]-1H-imidizaole (92)
A solution of 82(30) (0.42 g, 2.4 mmol) in DMF (10 mL) was treated with NaN3 (0.31 g, 4.8 mmol) and subsequently heated to 80 °C overnight. The solvent was evaporated at reduced pressure, and the remains were extracted with CH2Cl2 to provide 92 as a yellow liquid (0.40 g, 92%). IR (neat) ν (cm–1): 2931, 2873 (CH), 2102 (N3). 1H NMR (400 MHz, CDCl3): δ 7.53 (s, CHN2), 7.04–6.98 (m, 2 H, NCH), 4.13 (mc, 2 H, NCH2), 3.73, 3.59 (2 mc, 2 × 2 H, OCH2), 3.35 (mc, 2 H, CH2N3); 13C NMR (100 MHz, CDCl3): δ 137.4 (CHN2), 129.2, 119.4 (CHN), 70.5, 70.1 (OCH2), 50.6 (CH2N3), 47.1 (NCH2). HRMS (ESI) calcd for [M + H] [C7H12N5O]+ 182.1042; found, 182.1032.
1-(8-Azido-3,6-dioxa-octyl)-1H-imidizaole (93)
A solution of 83(31) (0.54 g, 2.5 mmol) in DMF (10 mL) was treated with NaN3 (0.40 g, 6.2 mmol) and subsequently heated to 80 °C overnight. The solvent was evaporated at reduced pressure, and the remains were extracted with CH2Cl2 to provide 93 as a yellow liquid (0.51 g, 91%). IR (neat) ν (cm–1): 2872 (CH), 2102 (N3). 1H NMR (400 MHz, CDCl3): δ 7.46 (s, CHN2), 6.95–6.92 (m, 2 H, CHN), 4.04 (mc, 2 H, NCH2), 3.68 (mc, 2 H, OCH2), 3.56–3.53 (m, 6 H, OCH2), 3.29 (mc, 2 H, CH2N3); 13C NMR (100 MHz, CDCl3): δ 137.4 (CHN2), 129.0, 119.3 (CHN), 70.6, 70.54, 70.50, 70.0 (OCH2), 50.6 (CH2N3), 47.0 (NCH2). HRMS (ESI) calcd for [M + H] [C9H16N5O2]+ 226.1304; found, 226.1304.
1-(11-Azido-3,6,9-trioxa-undecyl)-1H-imidizaole (94)
A solution of 84(32) (0.30 g, 1.1 mmol) in DMF (20 mL) was treated with NaN3 (0.15 g, 2.3 mmol) and subsequently heated to 80 °C overnight. The solvent was evaporated at reduced pressure, and the remains were extracted with CH2Cl2 to provide 94 as a yellow liquid (0.27 g, 91%). IR (neat) ν (cm–1): 2873 (CH), 2102 (N3). 1H NMR (400 MHz, CDCl3): δ 7.52 (s, CHN2), 7.01–6.97 (m, 2 H, CHN), 4.09 (t, 2 H, NCH2), 3.72 (t, 2 H, OCH2), 3.66–3.56 (m, 12 H, OCH2), 3.36 (t, 2 H, CH2N3); 13C NMR (100 MHz, CDCl3): δ 137.4 (CHN2), 129.0, 119.3 (CHN), 70.6, 70.51, 70.50, 70.4, 69.9 (OCH2), 50.6 (CH2N3), 47.0 (NCH2). HRMS (ESI) calcd for [M + H] [C11H20N5O3]+ 270.1566; found, 270.1563.
2-Butyl-octyl 6-[1-(5-Azido-3-oxa-pentyl)-3H-imidazolium-3-yl]-6-deoxy-β-d-glucopyranoside Bromide (11a2)
A solution of 5a (0.18 g, 0.33 mmol) and 92 (60 mg, 0.33 mmol) in xylene (4 mL) was heated to 130 °C. When TLC indicated the absence of the starting material, the solvent was evaporated to provide 10a2 (0.23 g, 97%) as a yellow syrup. The intermediate 10a2 (0.11 g, 0.15 mmol) was subjected to Zemplén deacetylation in MeOH (5 mL) using a catalytic amount of NaOMe. After stirring at rt overnight, the catalyst was removed by treatment with Amberlite IR120 (H+), and the solvent was evaporated to furnish 11a2 (85 mg, 93%) as a yellow syrup.
10a2
[α]D25 – 18 (c 0.28, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 10.05 (bs, imidazole), 7.42, 7.36 (2 mc, 2 H, imidazole), 5.17 (dd∼t, H-3), 4.85 (dd∼mc, H-2), 4.72 (dd∼bd, H-6A), 4.68 (dd∼t, H-4), 4.62–4.44 (m, 3 H, H-6B, CH2Nimidazole), 4.52 (d, H-1), 4.04 (mc, H-5), 3.91 (mc, 2 H, CH2O), 3.76–3.52 (m, 3 H, EG-CH2, α-CH2-A), 3.35 (mc, 2 H, CH2N3), 3.26 (mc, α-CH2-B), 2.18, 1.95, 1.91 (3 s, 3 × 3 H, Ac), 1.48 (mc, β-CH), 1.18 (mc, 24 H, bulk-CH2), 0.82 (t, 6 H, CH3); 3J1,2 = 8, 3J2,3 = 9, 3J3,4 = 9, 3J4,5 = 9, 2J6 = 11.5 Hz. 13C NMR (100 MHz, CDCl3): δ = 170.6, 169.7, 169.2 (CO), 122.8, 122.5 (imidazole), 101.0 (C-1), 73.4 (α), 72.1 (C-3), 71.4 (C-5), 71.0 (C-2), 70.1 (EG-CH2), 68.8 (CH2O), 68.3 (C-4), 50.5 (CH2N3), 50.2 (CH2Nimidazole), 49.6 (C-6), 37.9 (β), 31.8 (ω-2), 31.0, 30.8, 30.7, 30.4, 29.60/29.58, 28.9/28.8 (bulk-CH2), 26.7/26.6 (γ), 23.0, 22.6 (ω-1), 21.4, 20.45, 20.42 (Ac), 14.0 (ω).
11a2
[α]D25 – 14 (c 0.23, MeOH). IR (neat) ν (cm–1): 3376 (OH), 2955, 2925, 2858 (CH), 2107 (N3). 1H NMR (400 MHz, CD3OD): δ 8.55 (s, <1 H, imidazole), 7.72, 7.62 (2 s, 2 H, imidazole), 4.66 (dd∼d, H-6A), 4.48–4.41 (m, 3 H, CH2N, H-6B), 4.26 (d, H-1), 3.93 (t, 2 H, CH2O), 3.73–3.61 (m, 4 H, CH2O, α-OCH2-A, H-5), 3.42–3.36 (m, 4 H, CH2N3, H-3, α-CH2), 3.17 (dd, H-2), 3.08 (dd∼t, H-3), 1.61 (mc, β-CH), 1.40–1.32 (m, 16 H, bulk-CH2), 0.93 (m, 6 H, CH3); 3J1,2 = 8.0, 3J2,3 = 9.5, 3J3,4 = 9.5, 3J4,5 = 9.5, 3J5,6A < 2, 3J5,6B = 7.0, 2J6 = 14.0 Hz; 13C NMR (100 MHz, CD3OD): δ 138.7 (imidazole-CHN2), 125.0, 123.9 (imidazole), 105.0 (C-1), 77.8 (C-3), 75.1 (C-2), 74.9 (C-5), 74.32/ 74.30 (α), 72.5 (C-4), 71.4, 69.8 (EG-CH2), 51.9 (C-6), 51.8 (CH2N3), 51.1 (CH2N), 39.7 (β), 33.2, 32.41/32.35, 32.1/32.0, 31.00, 30.33/30.28, 28.04/27.98, 24.3 (bulk-CH2), 23.9 (ω-1), 14.6 (ω). HRMS (ESI) calcd for [M – Br] [C25H46N5O6]+ 512.3448, 513.3482 (28%); found, 512.3437, 513.3470 (32%).
2-Butyl-octyl 6-[1-(11-Azido-3,6,9-trioxa-undecyl)-3H-imidazolium-3-yl]-6-deoxy-β-d-glucopyranoside Bromide (11a4)
A solution of 5a (0.19 g, 0.35 mmol) and 94 (96 mg, 0.35 mmol) in xylene (3 mL) was heated to 130 °C. When TLC indicated the absence of the starting material, the solvent was evaporated to provide 10a4 (0.25 g, 85%) as a yellow syrup. The intermediate 10a4 (92 mg, 0.11 mmol) was subjected to Zemplén deacetylation in MeOH (5 mL) using a catalytic amount of NaOMe. After stirring at rt overnight, the catalyst was removed by treatment with Amberlite IR120 (H+), and the solvent was evaporated to furnish 11a4 (70 mg, 90%) as a yellow syrup.
10a4
[α]D25 – 24 (c 0.30, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 10.15 (bs, imidazole), 7.52, 7.39 (2 mc, 2 H, imidazole), 5.21 (dd∼t, H-3), 4.87 (dd, H-2), 4.76–4.63 (m, 2 H, H-6), 4.67 (dd∼t, H-4), 4.55 (d, H-1), 4.58–4.39 (m, 2 H, CH2Nimidazole), 4.07 (ddd, H-5), 3.88 (mc, 2 H, CH2O), 3.74–3.57 (m, 11 H, α-CH2-A, EG-CH2), 3.36 (t∼bs, 2 H, CH2N3), 3.29 (mc∼bs, α-CH2-B), 2.22, 1.98, 1.94 (3 s, 3 × 3 H, Ac), 1.48 (mc, β-CH), 1.21 (mc, 16 H, bulk-CH2), 0.84 (t, 6 H, CH3); 3J1,2 = 8.0, 3J2,3 = 9.5, 3J3,4 = 9.5, 3J4,5 = 10.0, 3J5,6A = 3.5, 3J5,6B = 5.0, 2J6 = 14.0 Hz. 13C NMR (100 MHz, CDCl3): δ = 170.6, 169.7, 169.2 (CO), 138.4 (imidazole-CHN2), 122.72, 122.67 (imidazole), 101.0 (C-1), 73.35/73.31 (α), 72.1 (C-3), 71.3 (C-5), 71.0 (C-2), 70.5, 70.3 (3), 69.9 (EG-CH2), 68.9 (CH2O), 68.2 (C-4), 50.6 (CH2N3), 49.9 (CH2Nimidazole), 49.4 (C-6), 37.9 (β), 31.7 (ω-2), 31.0, 30.7, 30.6, 30.4, 29.56/29.54, 28.9/28.7 (bulk-CH2), 26.7/26.5 (γ), 22.9, 22.5 (ω-1), 21.3, 20.41, 20.37 (Ac), 14.0 (ω).
11a4
[α]D25 – 9 (c 0.10, MeOH). IR (neat) ν (cm–1): 3372 (OH), 2925, 2859 (CH), 2105 (N3). 1H NMR (400 MHz, CD3OD): δ 9.02 (s, <1 H, imidazole), 7.73, 7.61 (2 s, 2 H, imidazole), 4.67 (dd, H-6A), 4.49–4.44 (m, 3 H, CH2N, H-6B), 4.27 (d, H-1), 3.89 (t, 2 H, CH2O), 3.71–3.68 (m, 12 H, EG-CH2, α-CH2-A, H-5), 3.43–3.35 (m, 4 H, CH2N3, H-3, αCH2-B), 3.16 (dd, H-2), 3.08 (dd∼t, H-4), 1.60 (β-CH), 1.32 (mc, 16 H, bulk-CH2), 0.93 (m, 6 H, CH3); 3J1,2 = 8.0, 3J2,3 = 10.0, 3J3,4 = 10.0, 3J4,5 = 10.0, 3J5,6A = 2.5; 2J6 = 14.5 Hz; 13C NMR (100 MHz, CD3OD): δ 138.8 (imidazole-CHN2), 124.7, 124.1 (imidazole), 105.0 (C-1), 77.8 (C-3), 75.1 (C-2), 74.9 (C-5), 74.30/74.28 (α), 72.5 (C-4), 71.68, 71.65, 71.50, 71.51, 71.2, 69.9 (EG), 51.9 (C-6), 51.1 (CH2N3), 51.0 (CH2N), 39.6 (β), 33.2, 32.4/32.3, 32.1/32.0, 31.0, 30.31/30.25, 28.02/27.95, 24.3 (bulk-CH2), 23.9 (ω-1), 14.6 (ω). HRMS (ESI) calcd for [M – Br] [C29H54N5O8]+ 600.3974, 601.4006 (32%); found, 600.3958, 601.3986 (38%).
2-Hexyl-decyl 6-Deoxy-6-[1-(8-azido-3,6-dioxa-octyl)-3H-imidazolium-3-yl]-β-d-glucopyranoside Bromide (11b3)
A solution of 5b (0.22 g, 0.37 mmol) and 93 (83 mg, 0.37 mmol) in xylene (1 mL) was heated to 130 °C. When TLC indicated the absence of the starting material, the solvent was evaporated to provide 10b3 (0.27 g, 89%) as a yellow syrup. The intermediate 10b3 (0.27 g, 0.33 mmol) was subjected to Zemplén deacetylation in MeOH (3 mL) using a catalytic amount of NaOMe. After stirring at rt overnight, the catalyst was removed by treatment with Amberlite IR120 (H+), and the solvent was evaporated to furnish 11b3 (0.22 g, 97%) as a yellow syrup.
10b3
[α]D25 – 16 (c 0.56, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 10.16 (s, imidazole), 7.46, 7.35 (2 mc, 2 H, imidazole), 5.18 (dd∼t, H-3), 4.85 (dd, H-2), 4.73 (dd, H-6A), 4.65 (dd∼t, H-4), 4.65 (dd, H-6B), 4.52 (d, H-1), 4.52 (ddd∼mc, CH2Nimidazole-A), 4.41 (ddd∼mc, CH2Nimidazole-B), 4.05 (ddd, H-5), 3.87 (mc, 2 H, CH2O), 3.73–3.55 (m, 6 H, EG-CH2), 3.68 (dd, -CH2-A), 3.34 (t, 2 H, CH2N3), 3.27 (dd, α-CH2-B), 2.20, 1.95, 1.91 (3 s, 3 × 3 H, Ac), 1.46 (mc, β-CH), 1.19 (mc, 24 H, bulk-CH2), 0.82 (t, 6 H, CH3); 3J1,2 = 8.0, 3J2,3 = 9.5, 3J3,4 = 9.5, 3J4,5 = 9.5, 3J5,6A = 2.5, 3J5,6B = 6.0, 2J6 = 14.5, 3JαA,β = 5.0, 3JαB,β = 6.0, 2Jα = 9.5, 3JCH2N-B,CH2O = 6.0/3.5 , 2JCH2N = 14.0 Hz. 13C NMR (100 MHz, CDCl3): δ = 170.5, 169.6, 169.1 (CO), 138.4 (imidazole-CHN2), 122.7, 122.6 (imidazole), 100.9 (C-1), 73.25/73.28 (α), 72.0 (C-3), 71.2 (C-5), 70.9 (C-2), 70.20, 70.15, 69.8 (EG-CH2), 68.7 (CH2O), 68.2 (C-4), 50.5 (CH2N3), 49.9 (CH2Nimidazole), 49.4 (C-6), 37.9 (β), 31.7 (ω-2), 30.9, 30.6, 29.9/29.8, 29.50/29.48, 29.4, 29.2 (bulk-CH2), 26.7/26.6/26.50/26.45 (γ), 22.5 (ω-1), 21.3, 20.4, 20.3 (Ac), 13.9 (ω).
11b3
[α]D25 – 13 (c 0.14, MeOH). IR (ATR, neat) ν (cm–1): 3354 (OH), 2824, 2856 (CH), 2104 (N3). 1H NMR (400 MHz, CD3OD): δ = 8.52 (bs, <1 H, imidazole), 7.70, 7.59 (2 bs, 2 H, imidazole), 4.65 (dd∼bd, H-6A), 4.44 (mc, 3 H, H-6B, CH2Nimidazole), 4.25 (d, H-1), 3.88 (t, 2 H, CH2O), 3.70–3.59 (m, 8 H, EG-CH2, α-CH2-A, H-5), 3.44–3.31 (m, 4 H, H-3, CH2N3, β-CH2-B), 3.14 (dd, H-2), 3.06 (dd∼t, H-4), 1.59 (mc, β-CH), 1.30 (mc, 24 H, bulk-CH2), 0.91 (t, 6 H, CH3); 3J1,2 = 8.0, 3J2,3 = 9.0, 3J3,4 = 9.0, 3J4,5 = 9.0, 2J6 = 14.5, 3JCH2N,CH2O = 4.5 Hz. 13C NMR (100 MHz, CD3OD): δ = 124.7, 124.1 (imidazole), 105.0 (C-1), 77.8 (C-3), 75.1 (C-2), 74.9 (C-5), 74.3 (α), 72.5 (C-4), 71.59, 71.58, 71.2 (EG-CH2), 70.0 (CH2O), 51.94 (CH2N3), 51.87 (C-6), 51.1 (CH2Nimidazole), 39.6 (β), 33.2 (ω-2), 32.34/32.33, 32.28/32.26, 31.30/31.29, 31.0, 30.88/30.86, 30.6 (bulk-CH2), 28.02/28.01/27.96/27.95 (γ), 23.9 (ω-1), 14.6 (ω). HRMS (ESI) calcd for [M – Br] [C31H58N5O7]+ 612.4336, 613.4370 (34%), 614.4404 (5%); found, 612.4337, 613.4354 (40%), 614.4410 (12%).
2-Hexyl-decyl 6-[1-(11-Azido-3,6,9-trioxa-undecyl)-3H-imidazolium-3-yl-]-6-deoxy-β-d-glucopyranoside Bromide (11b4)
A solution of 5b (0.14 g, 0.24 mmol) and 94 (64 mg, 0.24 mmol) in xylene (2 mL) was heated to 130 °C. When TLC indicated the absence of the starting material, the solvent was evaporated to provide 10b4 (0.18 g, 88%) as a yellow syrup. The intermediate 10b4 (0.18 g, 0.21 mmol) was subjected to Zemplén deacetylation in MeOH (3 mL) using a catalytic amount of NaOMe. After stirring at rt overnight, the catalyst was removed by treatment with Amberlite IR120 (H+), and the solvent was evaporated to furnish 11b4 (0.14 g, 90%) as a yellow syrup.
10b4
[α]D25 – 17 (c 0.20, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 10.44 (s, imidazole), 7.70, 7.38 (2 mc, 2 H, imidazole), 5.23 (dd∼t, H-3), 4.90 (dd, H-2), 4.80 (dd, H-6A), 4.70 (dd, H-6B), 4.68 (dd∼t, H-4), 4.56 (d, H-1), 4.53 (ddd∼mc, CH2Nimidazole-A), 4.43 (ddd∼mc, CH2Nimidazole-B), 4.06 (ddd, H-5), 3.91 (mc, 2 H, CH2O), 3.74 (dd, α-CH2-A), 3.71–3.58 (m, 10 H, EG-CH2), 3.39 (t, 2 H, CH2N3), 3.33 (dd, α-CH2-B), 2.27, 2.01, 1.96 (3 s, 3 × 3 H, Ac), 1.51 (mc, β-CH), 1.24 (mc, 24 H, bulk-CH2), 0.87 (t, 6 H, CH3); 3J1,2 = 8.0, 3J2,3 = 9.5, 3J3,4 = 9.5, 3J4,5 = 10.0, 3J5,6A = 5.5, 3J5,6B = 2.5, 2J6 = 15.0, 3JαA,β = 5.0, 3JαB,β = 6.0, 2Jα = 9.5, 3JCH2N-B,CH2O = 6.0/5.5, 2JCH2N = 14.5 Hz. 13C NMR (100 MHz, CDCl3): δ = 170.6, 169.7, 169.2 (CO), 138.8 (imidazole-CHN2), 122.8, 122.7 (imidazole), 101.2 (C-1), 73.50/73.47 (α), 72.1 (C-3), 71.4 (C-5), 71.0 (C-2), 70.6, 70.4 (3), 70.0 (EG-CH2), 68.9 (CH2O), 68.1 (C-4), 50.6 (CH2N3), 49.9 (CH2Nimidazole), 49.4 (C-6), 38.0 (β), 31.8 (ω-2), 31.0, 30.8, 30.00/29.98, 29.64/29.62, 29.57, 29.3 (bulk-CH2), 26.8/26.7/26.63/26.59 (γ), 22.6 (ω-1), 21.4, 20.5, 20.4 (Ac), 14.0 (ω).
11b4
[α]D25 – 12 (c 0.49, MeOH). IR (ATR, neat) ν (cm–1): 3355 (OH), 2923, 2855 (CH), 2104 (N3). 1H NMR (400 MHz, CD3OD): δ = 9.02 (bs, <1 H, imidazole), 7.72, 7.59 (2 d, 2 H, imidazole), 4.66 (dd, H-6A), 4.45 (dd, H-6B), 4.44 (t, 2 H, CH2Nimidazole), 4.26 (d, H-1), 3.88 (t, 2 H, CH2O), 3.69–3.60 (m, 12 H, EG-CH2, α-CH2-A, H-5), 3.40 (dd∼t, H-3), 3.40–3.31 (m, 3 H, CH2N3, β-CH2-B), 3.15 (dd, H-2), 3.07 (dd∼t, H-4), 1.59 (mc, β-CH), 1.30 (mc, 24 H, bulk-CH2), 0.90 (t, 6 H, CH3); 3J1,2 = 8.0, 3J2,3 = 9.0, 3J3,4 = 9.5, 3J4,5 = 9.0, 3J5,6A = 2.5, 3J5,6B = 7.5, 2J6 = 14.5, 3JCH2N,CH2O = 4.5 Hz. 13C NMR (100 MHz, CD3OD) δ = 138.4 (very weak, imidazole-CHN2), 124.6, 124.1 (imidazole), 105.0 (C-1), 77.7 (C-3), 75.1 (C-2), 74.9 (C-5), 74.3 (α), 72.5 (C-4), 71.66, 71.64, 71.58, 71.49, 71.1 (EG-CH2), 69.9 (CH2O), 51.9 (CH2N3), 51.9 (C-6), 51.1 (CH2Nimidazole), 39.6 (β), 33.18/33.17 (ω-2), 32.3, 32.2, 31.3, 31.0, 30.86/30.84, 30.6 (bulk-CH2), 28.00/27.98/27.93/27.92 (γ), 23.9 (ω-1), 14.6 (ω). HRMS (ESI) calcd for [M – Br] [C33H62N5O8]+ 656.4598, 657.4632 (37%); found, 656.4587, 657.4616 (42%).
Micelle Click Coupling (14)
A micellar dispersion of 11a4 (14 mg, 20 μmol) in water (1 mL) was treated with propargyl alcohol (10 μL, 0.17 mmol), sodium ascorbate (2 mg, 0.01 mmol), and CuSO4×5aq (1 mg, 4 μmol). The mixture was stirred at rt overnight before it was diluted with water (5 mL) and extracted with nBuOH (3 × 5 mL). The organic phases were combined and evaporated to provide the click-coupled surfactant for analysis.
Acknowledgments
We gratefully acknowledge assistance of Professor Dr. Kam Toh Seok and Dr. Low Yun Yee for recording mass spectra. This work was supported by the University of Malaya in the form of a student scholarship for Mr. Ean Wai Goh and under research grants RG383-17AFR and PG149-2015A. The Malaysian Ministry of Education provided additional support under research grant FP032-2013B.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b02809.
Experimental data for additional products 11a and 11b as well as for the reference compound 16, graphs for surface tension measurements, OPM texture for contact penetration of vesicle mixed surfactant with water, example of vesicle size distribution by dynamic light scattering measurement, example of vesicle zeta potential distribution, and complete NMR spectra of all compounds (PDF)
Author Present Address
‡ Present address: College of Pathological Analysis Technologies, Al-Bayan University, Baghdad, Iraq
The authors declare no competing financial interest.
Supplementary Material
References
- Charman W. N.; Chan H.-K.; Finnin B. C.; Charman S. A. Drug delivery: a key factor in realising the full therapeutic potential of drugs. Drug Dev. Res. 1999, 46, 316–327. . [DOI] [Google Scholar]
- Odiba A.; Ukebu C.; Anoubi O.; Chukwunomyelum I. Making drugs safer: improving drug delivery and reducing the side effect of drugs on the human biochemical system. Nanotechnol. Rev. 2016, 5, 183–194. 10.1515/ntrev-2015-0055. [DOI] [Google Scholar]
- Kamaly N.; Xiao Z.; Valencia P. M.; Radovic-Moreno A. F.; Farokhzad O. C. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem. Soc. Rev. 2012, 41, 2971–3010. 10.1039/c2cs15344k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty C.; Das M.; Kanwar J. R.; Sahoo S. K. Receptor mediated tumor targeting: an emerging approach for cancer therapy. Curr. Drug Delivery 2011, 8, 45–58. 10.2174/156720111793663606. [DOI] [PubMed] [Google Scholar]
- Jones M. N. Carbohydrate-mediated liposomal targeting and drug delivery. Adv. Drug Delivery Rev. 1994, 13, 215–249. 10.1016/0169-409X(94)90013-2. [DOI] [Google Scholar]
- Ito H.; Kamachi T.; Yashima E. Specific surface modification of the acetylene-linked glycolipid vesicle by click chemistry. Chem. Commun. 2012, 48, 5650–5652. 10.1039/c2cc31666h. [DOI] [PubMed] [Google Scholar]
- Tabandeh M.; Goh E. W.; Salman A. A.; Heidelberg T.; Hussen R. S. D. Functionalized glycolipids for potential bioconjugation of vesicles. Carbohydr. Res. 2018, 469, 14–22. 10.1016/j.carres.2018.08.016. [DOI] [PubMed] [Google Scholar]
- Soussan E.; Cassel S.; Blanzat M.; Rico-Lattes I. Drug delivery by soft matter: matrix and vesicular carriers. Angew. Chem., Int. Ed. 2009, 48, 274–288. 10.1002/anie.200802453. [DOI] [PubMed] [Google Scholar]
- Morigaki K.; Walde P. Fatty acid vesicles. Curr. Opin. Colloid Interface Sci. 2007, 12, 75–80. 10.1016/j.cocis.2007.05.005. [DOI] [Google Scholar]
- Lohner K.; Sevcsik E.; Pabst G. Chapter Five Liposome-based biomembrane mimetic systems: implications for lipid-peptide interactions. Adv. Planar Lipid Bilayers Liposomes 2008, 6, 103–137. 10.1016/S1554-4516(07)06005-X. [DOI] [Google Scholar]
- Eibl H.; Kaufmann-Kolle P. Medical Application of synthetic phospholipids as liposomes and Drugs. J. Liposome Res. 1995, 5, 131–148. 10.3109/08982109509039914. [DOI] [Google Scholar]
- Li W.; Yang Y.; Liu L.; Tan X.; Luo T.; Shen J. Dual stimuli-responsive self-assembly transition in zwitterionic/anionic surfactant systems. Soft Matter 2015, 11, 4283–4289. 10.1039/C5SM00627A. [DOI] [PubMed] [Google Scholar]
- Jiang Z.; Liu J.; Sun K.; Dong J.; Li X.; Mao S.; Du Y.; Liu M. pH- and concentration-induced micelle-to-vesicle transitions in pyrrolidone-based Gemini surfactants. Colloid Polym. Sci. 2014, 292, 739–747. 10.1007/s00396-013-3145-0. [DOI] [Google Scholar]
- Salman A. A.; Goh E. W.; Heidelberg T.; Hussen R. S. D.; Ali H. M. Bis-(alkylimidazolium)-glycosides – promising materials for easy vesicle preparation. J. Mol. Liq. 2016, 222, 609–613. 10.1016/j.molliq.2016.07.089. [DOI] [Google Scholar]
- Zhang L.; Somasundaran P.; Maltesh C. Electrolyte effects on the surface tension and micellization of n-Dodecyl β-D-maltoside solutions. Langmuir 1996, 12, 2371–2373. 10.1021/la950670w. [DOI] [Google Scholar]
- Mahale N. B.; Thakkar P. D.; Mali R. G.; Walunj D. R.; Chaudhari S. R. Niosomes: Novel sustained release nonionic stable vesicular systems — An overview. Adv. Colloid Interface Sci. 2012, 183-184, 46–54. 10.1016/j.cis.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Faivre V.; Rosilio V. Interest of glycolipids in drug delivery: from physiochemical properties to drug targeting. Expert Opin. Drug Delivery 2010, 7, 1013–1048. 10.1517/17425247.2010.511172. [DOI] [PubMed] [Google Scholar]
- a Mak O. W.; Seman N.; Harun N. H.; Hussen R. S. D.; Rodzi N. M.; Heidelberg T. Nanovesicles based on mixtures of a biantennary glycolipid with ionic cosurfactants. J. Surfactants Deterg. 2015, 18, 973–980. 10.1007/s11743-015-1746-y. [DOI] [Google Scholar]; b Tabandeh M.; Salman A. A.; Goh E. W.; Heidelberg T.; Hussen R. S. D. Renewable resources-based approach to biantennary glycolipids. Chem. Phys. Lipids 2018, 212, 111–119. 10.1016/j.chemphyslip.2018.01.011. [DOI] [PubMed] [Google Scholar]
- (a) Hashim R.; Hassan H.; Hamzah A. S.; Vill V.; Wulf M.. Synthesis of branched-chain alkyl glucosides and their liquid crystal behaviour. e-Liq. Cryst. Commun., Feb. 10, (2003); http://www.e-lc.org/tmp/Rauzah__Hashim_2003_02_05_07_19_24.pdf. [Google Scholar]; b Hashim R.; Hashim H. H. A.; Rodzi N. Z. M.; Hussen R. S. D.; Heidelberg T. Branched chain glycosides: enhanced diversity for phase behavior of easily accessible synthetic glycolipids. Thin Solid Films 2006, 509, 27–35. 10.1016/j.tsf.2005.09.009. [DOI] [Google Scholar]; c Brooks N. J.; Hamid H. A. A.; Hashim R.; Heidelberg T.; Seddon J. M.; Conn C. E.; Husseini S. M. M.; Zahid N. I.; Hussen R. S. D. Thermotropic and lyotropic liquid crystalline phases of Guerbet branched-chain -D-glucosides. Liq. Cryst. 2011, 38, 1725–1734. 10.1080/02678292.2011.625689. [DOI] [Google Scholar]; d Zahid N. I.; Abou-Zied O. K.; Hashim R.; Heidelberg T. Fluorescence probing of the temperature-induced phase transition in a glycolipid self-assembly: Hexagonal ↔ micellar and cubic ↔ lamellar. Langmuir 2012, 28, 4989–4995. 10.1021/la3001976. [DOI] [PubMed] [Google Scholar]
- (a) Siegel D. P.The relation between bicontinuous inverted cubic phases and membrane fusion. In Bicontinuous Liquid Crystals; Surfactant Science Series127. Lynch M. L., Spicer P. T., Eds.; CRC-Press: Boca Raton, 2005; pp. 58–98; [Google Scholar]; b Tenchov B. G.; MacDonald R. C.; Siegel D. P. Cubic Phases in Phosphatidylcholine-Cholesterol Mixtures: Cholesterol as Membrane “Fusogen”. Biophys. J. 2006, 91, 2508–2516. 10.1529/biophysj.106.083766. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tenchov B. G.; MacDonald R. C.; Lentz B. R. Fusion peptides promote formation of bilayer cubic phases in lipid dispersions. An X-ray diffraction study. Biophys. J. 2013, 104, 1029–1037. 10.1016/j.bpj.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappolt M. Formation of curved membranes and membrane fusion Processes studied by synchrotron X-ray-scattering techniques. Adv. Planar Bilayers Liposomes 2013, 17, 29–54. 10.1016/B978-0-12-411516-3.00002-4. [DOI] [Google Scholar]
- Tanford C. Hydrophobic free energy, micelle formation and the association of proteins with amphiphiles. J. Mol. Biol. 1972, 67, 59–74. 10.1016/0022-2836(72)90386-5. [DOI] [PubMed] [Google Scholar]
- Budin I.; Prywes N.; Zhang N.; Szostak J. W. Chain-length heterogeneity allows for the assembly of fatty acid vesicles in dilute solutions. Biophys. J. 2014, 107, 1582–1590. 10.1016/j.bpj.2014.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Teoh T. C.; Heidelberg T.; Hashim R.; Gary S. Computer modeling and simulations of thermotropic and lyotropic alkyl glycoside bilayers. Liq. Cryst. 2007, 34, 267–281. 10.1080/02678290601130192. [DOI] [Google Scholar]; b Hashim R.; Mirzadeh S. M.; Heidelberg T.; Minamikawa H.; Yoshiaki T.; Sugimura A. A reevaluation of the epimeric and anomeric relationship of glucosides and galactosides in thermotropic liquid crystal self-assemblies. Carbohydr. Res. 2011, 346, 2948–2956. 10.1016/j.carres.2011.10.032. [DOI] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Click chemistry: diverse chemical function from a few good reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. . [DOI] [PubMed] [Google Scholar]
- Moses J. E.; Moorhouse A. D. The growing applications of click chemistry. Chem. Soc. Rev. 2007, 36, 1249–1262. 10.1039/B613014N. [DOI] [PubMed] [Google Scholar]
- Lallana E.; Riguera R.; Fernandez-Megia E. Reliable and efficient procedures for the conjugation of biomolecules through Huisgen azide-alkyne Cycloadditions. Angew. Chem., Int. Ed. 2011, 50, 8794–8804. 10.1002/anie.201101019. [DOI] [PubMed] [Google Scholar]
- Liang L.; Astruc A. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) ‘click’ reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. 10.1016/j.ccr.2011.06.028. [DOI] [Google Scholar]
- Hanessian S.; Ponpipom M. M.; Lavallee P. Procedures for the direct replacement of primary hydroxyl groups in carbohydrates by halogen. Carbohydr. Res. 1972, 24, 45–56. 10.1016/S0008-6215(00)82258-2. [DOI] [Google Scholar]
- Yan J. M.; Jiang Z. L.; Zhang L.; Xie R. G. A new synthesis of 1-(−haloalkyl)-imidazole. Chin. Chem. Lett. 1998, 9, 519–520. [Google Scholar]
- Akiyama T.; Ibata C.; Fujihara H. Water soluble palladium and gold nanoparticles functionalized by a new phosphine with zwitterionic liquid based on imidazolium sulfonate linked ethylene glycol moiety. Heterocycles 2010, 80, 925–931. [Google Scholar]
- Ishida Y.; Sasaki D.; Miyauchi H.; Saigo K. Design and synthesis of novel imidazolium-based ionic liquids with a pseudo crown-ether moiety: diastereomeric interaction of a racemic ionic liquid with enantiopure europium complexes. Tetrahedron Lett. 2004, 45, 9455–9459. 10.1016/j.tetlet.2004.10.073. [DOI] [Google Scholar]
- Abboud J. M.; Notario R.; Bertan J.; Sola M.. One century of physical chemistry: the Menshutkin reaction. In Progress in Physical Organic Chemistry , vol 19; Taft R. W., Ed.; Wiley: New York, 1993; pp 1–182. [Google Scholar]
- Salman A. A.; Tabandeh M.; Heidelberg T.; Hussen R. S. D.; Ali H. M. Alkyl-imidazolium glycosides: non-ionic—cationic hybrid surfactants from renewable resources. Carbohydr. Res. 2015, 412, 28–33. 10.1016/j.carres.2015.04.022. [DOI] [PubMed] [Google Scholar]
- Hussen R. S. D.Niosomes From Chain-Branched Glycosides for Drug Delivery. Synthesis, Formulation and Evaluation, Lambert Academic Publishing, 2012. [Google Scholar]
- von Minden H. M.; Brandenburg K.; Seydel U.; Koch M. H. J.; Garamus V.; Willumeit R.; Vill V. Thermotropic and lyotropic properties of long chain alkyl glycopyranosides. Part II. Disaccharide headgroups. Chem. Phys. Lipids 2000, 106, 157–179. 10.1016/S0009-3084(00)00151-1. [DOI] [PubMed] [Google Scholar]
- Nguan H. S.; Heidelberg T.; Hashim R.; Tiddy G. J. T. Quantitative analysis of the packing of alkyl glycosides: a comparison of linear and branched alkyl chains. Liq. Cryst. 2010, 37, 1205–1213. 10.1080/02678292.2010.492245. [DOI] [Google Scholar]
- Molina-Bolivar J. A.; Ruiz C. C.. Self-assembly and micellar structures of sugar-based surfactants: Effect of temperature and salt addition. In Sugar-Based Surfactants; Surfactant Science Series 143, Ruiz C. C., Ed.; CRC Press: Boca Raton, 2008; pp. 61–104. [Google Scholar]
- (a) Israelachvili J.Intermolecular and Surface Forces. Academic Press: London, 1992. [Google Scholar]; (b) Israelachvili J. N.Soft and biological structures. In Intermolecular and Surface Forces, 3rd ed.; Israelachvili J. N., Ed.; Academic Press: Boston, 2011; pp. 535–576. [Google Scholar]
- Heidelberg T.; Chuan R.; Chie N. C.; Anwar S. A.; Hashim R. Synthesis and surfactant study on isomeric octyl glucosides. Mal. J. Sci. 2009, 28, 105–113. [Google Scholar]
- Pons M.; Foradada M.; Estelrich J. Liposomes obtained by the ethanol injection method. Int. J. Chem. 1993, 95, 51–56. 10.1016/0378-5173(93)90389-W. [DOI] [Google Scholar]
- Sun D.; Kang S.; Liu C.; Lu Q.; Cui L.; Hu B. Effect of Zeta Potential and particle size on the stability of SiO2 nanospheres as carrier for ultrasound imaging contrast agents. Int. J. Electrochem. Sci. 2016, 11, 8520–8529. 10.20964/2016.10.30. [DOI] [Google Scholar]
- Rosen M. J., Kunjappu J. T.. Surfactants and Interfacial Phenomena, 4th ed.; Wiley Interscience: Hoboken, 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.