

Summary
Many options now exist for constructing oral vaccines which, in experimental systems, have shown themselves to be able to generate highly effective immunity against infectious diseases. Their suitability for implementation in clinical practice, however, for prevention of outbreaks, particularly in low‐ and middle‐income countries (LMIC), is not always guaranteed, because of factors such as cost, logistics and cultural and environmental conditions. This brief overview provides a summary of the various approaches which can be adopted, and evaluates them from a pharmaceutical point, taking into account potential regulatory issues, expense, manufacturing complexity, etc., all of which can determine whether a vaccine approach will be successful in the late stages of development. Attention is also drawn to problems arising from inadequate diet, which impacts upon success in stimulating effective immunity, and identifies the use of lipid‐based carriers as a way to counteract the problem of nutritional deficiencies in vaccination campaigns.
Keywords: delivery, oral, Peyer’s patch, vaccine
Introduction
The conditions predisposing to the emergence of new infectious diseases – close contact with animals, opening up of new territory, population growth, etc. – are particularly prevalent in tropical and low‐ to‐middle‐income countries, where infrastructure is often lacking to provide rapid and widespread implementation of vaccine programmes. Under such circumstances, vaccines in an oral form can help to tackle problems of logistics, often faced by parenteral vaccines, in a very effective manner.
While injected vaccines necessitate administration in an aqueous form, which is usually not stable to extremes of temperature, the solid‐dose format of capsules or tablets lends itself to greater stability, which means that storage and transport can be effected without the need for cold‐chain procedures. Even injected vaccines in lyophilized form need to be reconstituted before use, which is not always a simple matter. Avoiding the use of needles also eliminates the possibility of contamination, and the need for rigorous containment measures, as well as supervised waste disposal, all of which are problematic for injected vaccines. Capsules take up little space, and are easily transported either in bulk or individually in blister packs. Administration does not need specialized medical skills, and the vaccine can even be taken home for self‐administration at a later date.
Efficacy of oral vaccines
In spite of widespread agreement regarding the desirability of oral vaccination, the approach has met with considerable resistance up to the present day. This seems to be because of doubts regarding the efficacy of orally administered vaccines, and concerns that the immune response mounted after oral vaccination does not match that achieved by injection. In reality, attempts to compare efficacy of injected and oral vaccines directly are not entirely valid, as the two routes initiate immunity in different parts of the body, and engender types of immunity different in qualitative rather than simply quantitative terms. Live attenuated vectors given orally have long been known to induce good mucosal and systemic responses 1. Non‐living oral vaccines can do the same, now that technologies have advanced to target antigens to the immune cells in the gut and appropriate immunostimulants are used 2, 3. In contrast, injected vaccines often afford poor levels of immunity in the intestine 4, so that their efficacy against intestinal pathogens is low in terms of providing a first line of defence, and the protection they offer only comes into play after the organism has crossed the gut wall and entered the body.
Under normal circumstances, antigens which enter the intestine are treated in the same way as dietary components, where any potential immune response is suppressed, in order to avoid allergic reactions 5. To generate a response in which the immune system is up‐regulated, it is necessary to administer the antigen in conjunction with an immunostimulant. In the case of live vectors, or whole‐killed organisms, this is provided by the organism itself – such as lipopolysaccharide 6 or cytosine–phosphate–guanine (CpG) 7. In some instances, the antigen transferred into the vector may be specially engineered to incorporate immunostimulants or cytokines, secreted at the same time as the antigen, a strategy being explored extensively in cancer immunotherapy 8. In non‐living vaccine constructs, antigens can be combined with immunostimulants possessing properties specifically adapted to the gut environment, of which the best known is cholera toxin B fragment (CTB), which binds specifically to GM1 gangliosides on the cell surface, prior to ingestion of cholera toxin by the cell 9.
There is a general perception that mucosal immune responses are short‐lived. This may, in part, be the result of observations on systemic immunity generated after oral administration, where cells produced by the gut appear in the bloodstream initially but migrate to other mucosal sites and focus their attention locally. In fact, antibody‐secreting cells in the gut mucosa have been demonstrated to persist for decades 10, and long‐lived plasma cells provide long‐lived mucosal immunity 11, 12. It has been noted that, in certain cases, cytotoxic T lymphocyte 13 and long‐lived immunoglobulin (Ig)A mucosal memory 14 is more readily achieved via oral immunization than via systemic administration.
In addition, up‐regulation of the immune response leading to significant long‐term mucosal and systemic immunity can be readily achieved by supplementing with an additional oral boost. While there is an understandable reluctance to rely on boosters with injected vaccines, because of poor patient acceptability, it is important to rid the scientific community of this prejudice with regard to oral vaccination. Boosting is a natural mechanism whereby the body gradually increases its baseline immunity as a result of repeated exposure to antigens. In the case of oral administration, the objection of patients to repeat administration is removed, as a tablet or capsule is completely innocuous compared with an injection and can even be self‐administered at the convenience of the patients themselves. For many conditions, even as simple as headaches, patients are happy to take pills several times a day, or once a day for extended periods of time, so the prospect of receiving a restricted number of booster administrations of an oral vaccine is in no way an impediment to the success of the oral vaccination approach. Provided that an immuno‐upregulator is always combined with antigen, administration of a booster dose should not induce tolerance towards that antigen (see further discussion later in this review).
The intestine is the largest immune organ in the body, acting as host to 70–80% of the body’s immune cells 15. These cells can be located in one of several different environments to which vaccine carriers and their antigens can gain access: (i) the Peyer’s patches 16, highly structured lymphoid follicles specialized for initiation of immune responses through extensive antigen processing and presentation on dendritic cells; (ii) the lamina propria, where lymphocytes (lamina propria lymphocytes, LPL) and dendritic cells are distributed throughout the lymphatic drainage channels; and (iii) mesenteric lymph nodes 17, into which lymphatics from the lamina propria flow; the same lymphatics can also feed ultimately into the peripheral systemic blood circulation via the thoracic duct. See reference 18 for an overview of the components of the gut immune system. An additional compartment, the intestinal epithelial layer itself, contains a large number of T cells (intraepithelial lymphocytes, IEL 19) which perform important functions in maintaining the intestinal cell barrier intact 20, 21. Both IEL and LPL comprise a large proportion of unusual T cell subsets, Tγδ cells, although the γδ populations in the two sites are functionally different, those in the lamina propria capable of secreting significant amounts of inflammatory cytokines such as interleukin (IL)‐17 22, while intestinal IEL T cell receptor (TCR)‐γδ cells (and TCR‐αβCD8+) can secrete anti‐inflammatory cytokines such as IL‐10 and transforming growth factor (TGF)‐β 23, 24. It is likely, therefore, that IEL play a role in down‐regulation of unwanted responses to non‐pathogen‐related antigens.
Oral vaccine carrier vehicles can access one or all of these sites (Fig. 1). Particulate materials (including viruses and bacteria) can be taken up by the M cells covering the Peyer’s patches, and such uptake can be enhanced by making the particles lipidic in nature, or by placing on their surface ligands which can be recognized by M cells. Antigenic materials can also cross the intestinal cell barrier, either by passing through the cells (transcytosis) or in between them (paracellular transport though opening of tight junctions). Recently, studies have indicated that antigens can be taken up by enterocytes via a phagocytic process 25, 26. It is also worth noting that the initial stage in the lipid‐processing pathway of intestinal cells involves uptake of bile salt/oil micelles of approximately 0·4 mm diameter – the same size as particulate carriers discussed later. Antigen taken up via this route should survive intact, as the level of proteases is low. After exocytosis from the enterocytes, the antigens and/or their carriers have opportunity to interact directly with itinerant memory T cells and phagocytes located in the lamina propia. The physicochemical nature of the antigen can determine the route in the body subsequently taken – small water‐soluble molecules can diffuse across the semi‐permeable membranes of blood microcapillaries and pass via the hepatic portal vein through the liver to the peripheral bloodstream, while large particulate and lipidic materials will be retained in the lymphatics (maximizing contact with intestinal lymphocytes) and then drain into the mesenteric lymph nodes, or eventually pass into the bloodstream via the thoracic duct.
Figure 1.
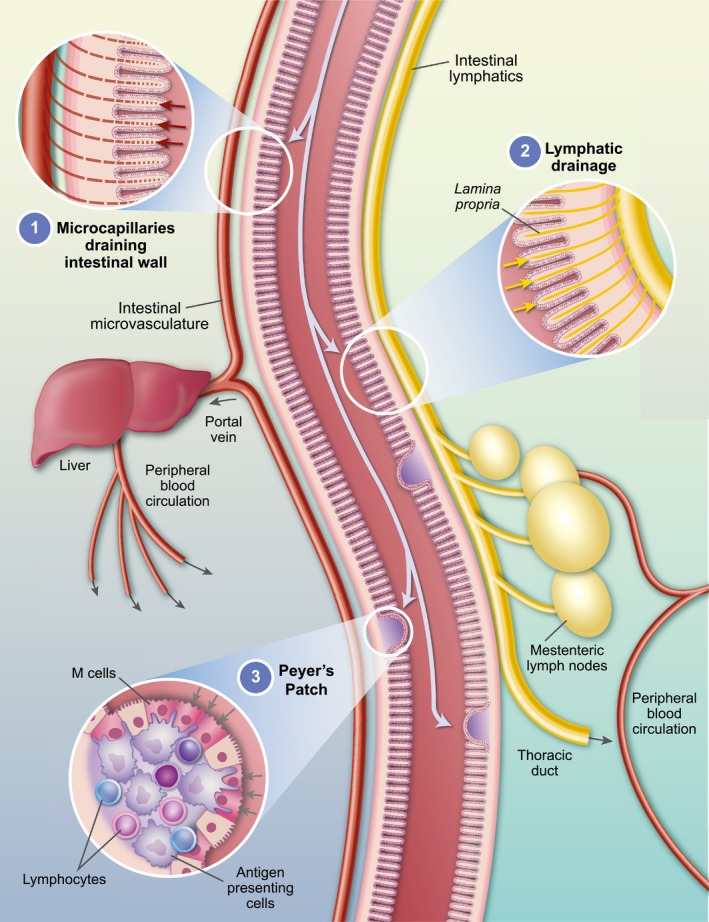
Routes of oral vaccine‐delivered antigens through the intestine, accessing different parts of the immune system of the body. (1) Via microcapillaries into the local and peripheral blood circulation; (2) via the lamina propria and intestinal lymphatics into the mesenteric lymph nodes; (3) direct uptake though M cells into the Peyer’s patches.
Oral vaccines on the market
A number of oral vaccines are, or have been, on the market, details of which are listed in Table 1. The majority of these are live attenuated organisms, the first and most well‐known being the polio vaccine 27, an oral version of which, developed by Sabin 28, was first made available commercially in 1961. The first version of this vaccine comprised three separate strains (serotypes) attenuated by sequential passage through a range of different monkey cell types derived from tissues such as skin, testicle and kidney 29. This oral version was introduced after the majority of cases were already dramatically reduced by use of the original Salk inactivated polio vaccine, administered by injection, but the oral vaccine has played a major role in extending that decline as a result of the easier logistics of distribution and boosting, despite the issues with cold chains. Although polio has been eradicated in all but three countries worldwide 30, a combination of injected and oral vaccines have continued to be employed in populations at potential risk to maintain herd immunity or to protect travellers.
Table 1.
Oral vaccines in clinical use.
| Infection | Vaccine type | Manufacturer | Trade name |
|---|---|---|---|
| Poliomyelitis | Live attenuated polio vaccine | Many discontinued | |
| Rotavirus | Live attenuated monovalent human rotavirus strain | GlaxoSmithKline | Rotarix® |
| Live attenuated monovalent human rotavirus strain | Bharat Biotech Int. Ltd | Rotavac® | |
| Live attenuated monovalent human rotavirus strain | CRPVB, Vietnam | Rotavin‐M1® | |
| Pentavalent live vaccine | Merck & Co., Inc. | RotaTeq® | |
| Pentavalent live vaccine (lyophilized) | Serum Institute (India) | Rotasiil® | |
| Typhoid | Ty21a live attenuated vaccine | Previously PaxVax Berna GmbH now Emergent Biosolutions | Vivotif® |
| Cholera | Cholera toxin B subunit and inactivated V. cholerae 01 whole cells | Valneva | Dukoral® |
| Live attenuated V. cholerae 01 strain (CVD 103.HgR) | PaxVax | Vaxchora® |
Additionally, oral vaccines against adenovirus types 4 (Ad4) and 7 (Ad7) for prevention of febrile respiratory illness (FRI) are being produced by the US military for treatment of trainees. The vaccines are live attenuated viruses administered orally 109. This table is not intended to be a comprehensive list.
Being the first live attenuated vaccine ever employed commercially, polio vaccine and its precursors ran into many teething troubles, such as contamination and reversion, and even now vaccine‐induced polio has been reported 31, 32, although cases are far fewer than would be expected in an unvaccinated population. What has never been in doubt, however, is that the oral vaccine is highly efficacious in both adults and children, particularly in generating mucosal antibodies in the gut.
The polio virus does not specifically target immune tissues in the gut, but the attenuation process knocks out its ability to infect neural cells 33 while retaining its capacity for reproduction in intestinal epithelium 34. Consequently, small doses of the live vaccine can result in rapid multiplication and secretion of large amounts of antigen, which are sufficient to trigger the intestinal immune system into action.
A second live attenuated viral vaccine has recently come onto the market: oral rotavirus vaccine, introduced first in the United States in 2006, and now used globally 35. The protection rate varies markedly from study to study and country to country 36, but significant reductions in mortality are observed, and the vaccine can be given to children as young as 6 weeks of age.
Two other marketed oral vaccines are both directed at bacterial targets: Salmonella enterica serovar typhi and Vibrio cholera. The live attenuated oral typhoid vaccine, Ty21a (Paxvax, introduced in 2014) appears to have similar efficacy to the injected Vi polysaccharide vaccine: ~50% protection after 3 years 37.
There are several different oral cholera vaccines on the market, some of which are live attenuated, and some are whole‐killed bacteria. In the case of the whole‐killed bateria, manufactured by Valneva, the whole organism is supplemented with additional CTB, and large quantities are contained within each dose (Dukoral package leaflet information). CTB is a strong mucosal adjuvant 38, 39, 40, and this probably explains the efficacy of the vaccine even in the absence of an antigen‐amplifying effect due to replication of live organisms in the gut. In the case of all the other vaccines (both bacterial and viral), however, even in the absence of a strong immunostimulants such as CTB, other immune triggers are present in the form of CpG (bacterial), single‐stranded RNA or double‐stranded DNA, which bind to Toll‐like receptors (TLR)‐9, ‐7/8 and ‐3, respectively. One interesting aspect of the use of the cholera vaccines is the proposal that they can be employed reactively (i.e. after an outbreak has been initiated), rather than just in a prophylactic mode 41.
Experimental vaccines
Clearly, on the basis of the vaccines that are already being used in humans in large numbers for long periods of time, immunization via the oral route is a viable, efficacious and economic method of protecting against lethal diseases. Because of their more recent appearance, oral vaccines against emerging diseases are still in the experimental stage, and many different approaches are being explored. However, all these approaches share many features in common.
At the outset, it is important for several myths about challenges confronting oral vaccines to be dispelled. One is the barrier presented by the stomach. In fact, technologies for allowing agents to pass unharmed across the stomach have existed for decades, and are employed in a number of widely used pharmaceuticals. This involves filling the vaccine, in solid form, into capsules, which are coated with a polymer film (e.g. cellulose acetate phthalate, or Eudragit) which is insoluble at the low pH of the human stomach but dissolves readily at pH values above 5–5·5, after having passed unaltered into the small intestine. The common practice of presenting conventional vaccines in liquid form is one imposed by the need to inject into the body using a syringe. For oral vaccines, where the ingested antigen becomes dispersed in the liquid found in the lumen of the gut, presentation in solid form is more practical and promises higher levels of stability. Orally targeted vaccines are thus ideally suited for capsule or tablet administration.
The second myth is that the mucus layer covering the intestinal wall is a barrier to uptake. While this is true for absorption and uptake of materials via the enterocytes, the mucus layer covering the M cells of the Peyer’s patches is sparse or non‐existent as the dome of the Peyer’s patches contains few, if any, goblet cells, so mucin secretion is greatly reduced. This is indeed to be expected, as it is the job of M cells to take up material of high molecular weight, which cannot easily diffuse across a thick mucus layer.
One generalization which holds for essentially all oral vaccine carriers and targeting vehicles is that the carrier should be particulate. Reasons for this are as follows:
M cells in the Peyer’s patches have a high propensity for uptake of particulates, the optimal size range centering approximately 0·1–0·2 mm in diameter;
where ligands are employed to target the vaccine to structures on the surface of M cells, presentation of ligands as a multivalent array on the surface of a particle encourages internalization, compared with binding of ligands in a monomeric form;
inclusion of both antigens and immunostimulants/potentiators in the same particle ensures that these agents can reach the same cell and act in concert, rather than becoming physically separated during passage along the gut; and
encapsulation within particles can protect protein antigens from attack and breakdown by intestinal proteases.
Live vectors
Attenuated bacteria
One type of particulate carrier which has received a great deal of attention is the live attenuated vector. This has the advantage that the quantity of antigen payload delivered can be amplified as a result of growth and limited replication inside the host, and that immunostimulants are co‐expressed together with the antigens and targeting agents. The most successful ones to date are derived from pathogenic strains, as these have recognition structures which strongly stimulate the host immune system. Indeed, it is their pathogenicity which has caused the host to evolve specifically to recognize these structures. One vector which has been widely researched is the S. enterica serova typhi Ty21a attenuated strain, as this already has a long history of use in humans as a vaccine for typhoid fever.
Although good results have been obtained in animal experiments, studies in humans using strains expressing heterologous antigens (i.e. antigens not native to the organism being used as vector) show that their efficacy leaves something to be desired 42. One problem is that, because the vectors are originally pathogenic, great efforts are made to attenuate them, and in the process their immunogenicity may be reduced. A compounding issue is that introduction of foreign antigen into the vector, for in‐situ production in a large quantity, takes its toll on the organism in terms of metabolic stress, so that the vectors are much less viable (particularly in an essentially hostile environment) when loaded with the additional burden of antigen production 43. As a result, invasion and colonization, etc. are less effective. An ingenious way to overcome this is to manipulate the vector so that the antigen only starts being produced after the vector has penetrated the host tissues, enabling it to concentrate its energy on invasion, before a promoter kicks in (e.g. at low oxygen concentration) to start producing antigen after the organism has become well established. This and other promoters employed are summarized in da Silva et al 44. Other issues needing attention are the locations in the bacterium of the genetic sequence. Larger amounts of antigen can be produced from plasmid‐expressed DNA than chromosomal, but more care needs to be taken to ensure that plasmids are maintained from generation to generation and distributed evenly among daughter cells. This can be achieved by designing the plasmids so that they contain a gene essential for the survival of the whole organism (see review by Kotton & Hohmann 45 for a summary of approaches). An operator‐repressor system (ORT) has also been developed for this purpose 46. Finally, the protein may be engineered so that it either stays in the cytoplasm, becomes transferred to the periplasmic space, is expressed on the outer membrane or is secreted. Each of these may result in a different type of immunity being generated 47. In addition, the magnitude of the immune response is not always linearly related to the dose of organism administered 48.
Encouraging results with bacterial vectors have been obtained using strong immunogens (e.g. diphtheria toxoid, Escherichia coli heat‐labile toxin or tetanus toxoid fragment C), each of which have their own adjuvanting capacity, but responses in human clinical trials against ‘weak’ immunogens have so far been disappointing (see Galen et al. 38). Further work on the issues described above may well change the situation.
Non‐pathogenic bacterial vectors
The use of vectors which are commensal in the human gut, for which attenuation procedures are unnecessary, is an alternative approach which holds promise. As these organisms are adapted to the host intestine, they need to expend much less energy to survive and can devote all their efforts to production of antigens. The archetypal vectors of the class are members of the group of lactic acid bacteria, including lactobacilli and lactococcus species, as well as streptococci 49. Although they are essentially non‐pathogenic and can survive for long periods in the human gut, they can interact with the gut immune system and induce inflammatory reactions in dendritic cells, such as cytokine secretion and up‐regulation of major histocompatibility complex (MHC) surface presentation molecules 50. Lactobacilli can adhere to enterocyte membranes, thus preventing them from being cleared rapidly from the intestinal tract 51. Their surface coat of poly teichoic acid (PTA) appears to be immunostimulatory, as mutants lacking PTA, but expressing foreign proteins, induce a suppressive response against that protein in the same way as does the gut immune system for food antigens 52. Different strains of lactobacilli (e.g. casei, acidophilus, gassneri) stimulate the immune system in different ways, leading to different types of immune response, so there is the possibility of matching the immunity generated to the organism being targeted 53, 54.
Other lactic acid bacteria which have been the subject of much study are lactococci (e.g. lactococcus lacti), which do not have as long a residence time in the gut as lactobacilli, or are immunostimulatory to the same extent, but seem to be able to produce large quantities of antigen and are easy to manipulate genetically. Some strains have been engineered so that they can be internalized by mammalian cells, and as such can act as vectors for DNA vaccines 55.
Viral vectors
Viruses are also good candidates as vectors for vaccine antigens. Adenoviruses in particular can withstand the environment of the gut, and can infect intestinal cells 56. Their replication mechanisms can be employed to insert antigens onto cell membranes, and this generates strong T cell immune responses, which can be enhanced 57. Because space within a virus capsid is limited, insertion of large amounts of genetic material may not always be easy. Another concern is that pre‐existing immunity to the viral carrier may impair their ability to function as vaccines, particularly if a booster is required 58. Although experiments in rodents suggested that pre‐existing immunity (acquired in the form of maternal antibody) was not a problem 59, a dose‐ranging study in humans showed that those subjects with antibodies against the carrier developed significantly reduced responses against the foreign antigen at lower doses of virus, but that as the dose was increased, existing immunity no longer influenced the outcome 60. Whether or not dosing at the high levels reported is routinely feasible is an issue which needs careful examination.
One solution to the problem of pre‐existing immunity – or more specifically, the avoidance of generating immunity against the carrier in a priming administration which then inhibits a booster response – is to use different carriers for the prime and booster. This approach provides two shots at generating specific types of immunity adapted to the target, and was employed successfully in rodents, generating responses T and B cell responses against HIV antigen through the use of attenuated oral Listeria monocytogenes, followed‐up with the AD4 administered intra‐vaginally 61. The complexity of the system, however, will be a significant obstacle to implementation in clinical practice.
An alternative method of circumventing the effects of pre‐existing immunity is to mask the surface of the virus so that antibodies are unable to bind. This has been achieved by coating the virus covalently with liposomes 62. Under these circumstances, the normal method of entry of the virus is probably blocked, but this can be overcome by making the liposomes cationic, which provides a mechanism for targeting to the cell surface and aids internalization. Thereafter, replication is able to proceed normally, although subsequent cell‐to‐cell transfer would be inhibited, which may be a good safety mechanism.
The underlying concern with all approaches involving live vectors is the lack of control over the replication process once ingested and the possibility of reverting to a pathogenic form, either by spontaneous mutation or acquisition of new genes from environmental flora. In the case of polio, the incidence of reversion was low, but finite, and the possibility that similar events can occur with other, less well‐researched vectors, cannot be ruled out. Consequently, in many countries, regulatory authorities are still very reluctant to approve introduction of vaccines based on live vectors, and their perception as methodologies still in the experimental phase is a reason for resistance to their introduction from the West into low‐ and middle‐income countries (LMIC) for emerging diseases. The absence of such safety concerns is one reason for continued focus on non‐living carriers for antigen delivery in vaccines for emerging diseases. Here, the antigenic material can either be whole‐killed organisms or subunits thereof, and the need for containment procedures and avoidance of contamination is markedly reduced.
Non‐living vaccines
Foodstuffs
The recognition that foodstuffs are materials which are highly compatible with the intestinal tract, and that plants can be employed to synthesize proteins easily in large quantities, has led to the suggestion that certain foods could be used as agents for delivering antigens orally as vaccines 63. A number of studies showed success using CTB or E. coli toxin fragment as antigen 64, 65, but these cannot be considered as demonstrations of enhanced delivery to the immune system, as CTB/ETB are immunostimulatory in their own right in the absence of any carrier system, provided that a sufficiently large amount of proteins is ingested. Using the same approach with other antigens could risk inducing suppression of immune responses to the antigen rather than stimulation, unless something such as CTB were administered at the same time. An extension of this approach might be to work with fusion proteins of antigen and CTB together. In both cases, however, immunity against bystander molecules could be generated as a result of unanticipated physical association with the CTB molecule, as no provision is made for encapsulation to restrict such non‐specific interactions. Even if antigen and immunostimulant were to be co‐encapsulated with a food granule (e.g. yeast‐derived beta‐glucan particles 66) a mechanism for targeting to Peyer’s patches would still be required. Work directed towards use of foodstuff‐based vaccines as a general principle is now focusing on plants as a method of antigen production, rather than in‐vivo delivery of antigen.
Encapsulation vehicles
The advantage of building a carrier using pharmaceutical principles ‘from the ground up’ is that one has freedom to incorporate any number of specific agents (immunostimulants, immunopotentiators, etc. – see Table 2) permitting one to induce an immune response tailored specifically to the infectious organism being targeted. As time progresses, more and more such agents will be identified, which can lead to ever more sophisticated vehicles targeting only the right arms of the immune response at the right time. Even with the current range of stimulators available (see Table 2) strong and specific responses can be brought about in a number of ways. In addition, encapsulation within a carrier prevents the antigen being broken down by gut digestive processes before reaching the immune system, and permits targeting ligands to be attached to the outside of the particle to enhance uptake by the gut immune system, particularly the Peyer’s patches (see Table 3).
Table 2.
Targets of immunostimulants employed in vaccines.
| Target | Agent | References |
|---|---|---|
| TLR‐2 | Mannan, zymosan, beta‐glucan, muramyl dipeptide (MDP) | 110, 111, 112 |
| TLR‐2/1 | Bacterial lipopeptide; Pam3CSK4 | 113, 114, 115 |
| TLR‐2/6 | Mycoplasma derived macrophage‐activating lipopeptide (MALP‐2) | 116 |
| TLR‐3 | dsRNA | 117, 118, 119 |
| TLR‐4 | LPS, glycoinositol phospholipids, mannan; monophosphoryl lipid A (MPLA) | 120, 121, 122 |
| TLR‐5 | Flagellin | 123 |
| TLR‐6 | Zymosan, beta‐glucan | 124 |
| TLR‐7, TLR‐8 | ssRNA; imiquimod | 125, 126, 127 |
| TLR‐9 | CpG DNA | 128 |
| GM1 | Cholera toxin B fragment (CTB), E. coli toxin B fragment (ETB) | Jobling 9, Stratmann 36 |
| NOD1 NOD2 | Muramyl dipeptide (MDP) | 129 |
| STING | cGAMP | 130 |
| IL‐1 receptor | IL‐1 | 131 |
| IL‐7, IL‐12 and IL‐15 receptors | IL‐7, IL‐12, IL‐15 | 132 |
| Mincle | Trehalose dibehenate (TDB) | 133 |
See Hug et al. 134 for a general review on Toll‐like receptors (TLR) in relation to the human gut.
CpG = cytosine–phosphate–guanine; IL = interleukin; LPS = lipopolysaccharide; NOD = non‐obese diabetic.
Table 3.
Ligands targeting to Peyer’s patch M cells.
| Target | Ligand | Ref |
|---|---|---|
| Lectin targets | ||
| lectin α1,2 fucose | UEA‐1 | 135, 136 |
| α‐L‐fucose | Aleuria auranitia (AAL) | 137 |
| (D‐glcNAc)2,sialic acid | Triticum vulgaris (wheat germ) WGA | 138 |
| N‐glycans/repeated oligosaccharide | Galectin‐9 | 139 |
| Microbial targets | ||
| β1 integrin | Invasin (Yersinia) | 140, 141 |
| Glycoprotein 2 IgA immunogloblin receptors | FimH (E. coli, Salmonella) | 142, 143, 144 |
| C5aR | OmpH (Yersinia) | 145 |
| Cellular prion protein | Hsp60 of Brucella abortus | 146 |
| ANXA5 | Lipid A domain of LPS (gram‐negative bacteria) | 147 |
| PGLRP‐1 | Bacterial peptidoglycan | 148 |
| Claudin 4 | C‐term domain of enterotoxin (Clostridium perfringens) | 149 |
| α2,3 sialic acid | σ1 protein (reovirus) | 150 |
| PFAR | Phosphorylcholine moiety of LPS | 151 |
| C5aR | Peptide Co1 (SFHQLPARSPLP) | 152 |
| Other | ||
| P2X7 receptor, formyl peptide receptor 2 | Cathelicidin LL‐37 | 153 |
As mentioned above, inclusion of a strong immunostimulant is key to ensuring that suppression of the immune response to administered antigen does not occur. To date, the most successful and widespread stimulant employed for this purpose is CTB 67. This is present in the oral cholera vaccine Dukoral™, providing a history of safe oral use in humans. Immune responses and protection in cholera models have been demonstrated with recombinantly produced CTB 68, 69, and strong responses to other antigens are generated when co‐administered with recombinant CTB 70, 71, confirming that the effect is due to the presence of CTB alone, and not the presence of low levels of cholera toxin A fragment (CTA).
One should be aware that a great deal of work has been performed using CTB as an agent to induce antigen‐specific tolerance 72. Almost invariably, however, this is observed when the antigen is linked covalently (or fused) to CTB, rather than simply co‐administered. Why the CTB behaves in this altered fashion is not clear, although it is to be expected that attaching exogenous molecules at certain sites may prevent polymerization which may change its interaction with lipid rafts, even though binding to GM1 is maintained 73. It may also be that other sites on the surface of the molecule are masked by the antigen, interfering with its stimulatory effect. Occasions have been reported where free CTB administered orally can tolerize, although this is when the CTB comes into contact with the gut milieu and the proteases therein, so it is likely that structural modifications have taken place. This is an additional argument for administering CTB in encapsulated form, where contact with extracellular protease will be avoided. It is worth mentioning in passing that conjugation of antigens to CTB or E. coli EtxB does not always lead to tolerance. Polysaccharides conjugated chemically to EtxB gave a construct which stimulated generation of antibodies to polysaccharide in both serum and stools of mice after oral administration 74.
Three types of synthetic vaccine carrier suitable for the gut environment have been studied, their physical characteristics being illustrated in Fig. 2. Membrane vesicles such as liposomes have been employed since the 1970s, following observation that these carriers worked well after parenteral administration. In parallel, polymeric particle matrices have been extensively researched, including PLGA and chitosan nanoparticles. Finally, oil droplet‐based vehicles have shown promise, and for reasons described later may come to be the method of choice for synthetic carriers. All these vehicles are constructed from generally‐recognized‐as‐safe (GRAS)‐listed materials, with good safety profiles, so regulatory requirements can easily be met and toxicity is unlikely to be an issue. Methods of manufacture on a commercial scale have been established for many years, and use standard, non‐specialized equipment, so technology transfer and production plants can be set up anywhere in the world.
Figure 2.
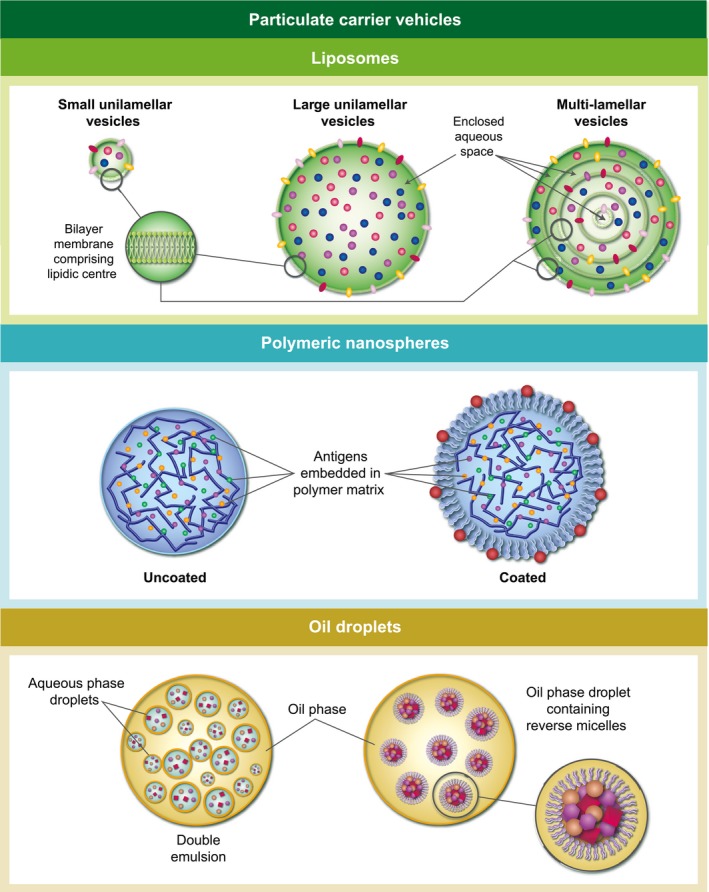
Different types of particulate carrier employed as oral vaccine vehicles. (1) Membrane vesicles (liposomes); (2) polymeric micro/nanoparticles; (3) oil droplets and emulsions.
Liposomes
Liposomes are well established as pharmaceutical carriers, and have been on the market for parenteral administration in applications such as cancer chemotherapy and treatment of candidiasis 75. They are membrane vesicles in which the membrane is generally made up of components similar to those in natural cell membranes (principally phospholipids) and, for this reason, they are considered to be highly compatible with biological systems. The amphiphilic nature of phospholipids means that they are suitable for incorporating a wide range of hydrophilic and hydrophobic components. Thus, water‐soluble antigens can be encapsulated within the aqueous space enclosed by the membrane, while membrane‐associated antigens (e.g. viral) and lipidic immunostimulants such as cord factor, muramyl dipeptide or lipopolysaccharide can be held within the membrane itself. To fulfil such requirements, multi‐lamellar vesicles are the most appropriate to employ as they have roughly equal proportions of hydrophilic and lipophilic space within the vesicles. Their size can be tailored by extrusion through controlled‐pore membrane filters, and they are physically more stable to lyophilization than are liposomes composed of just one single outer membrane.
Liposomes composed of commonly available phospholipids (phosphatidyl choline from egg or soya) are unstable in the gut environment, being prone to breakdown by bile salts and phopholipases. Consequently, alternative lipids are often used, which give more robust membranes resistant to enzymic attack – for example, synthetic distearoyl PC, sphingomyelin or archebacterial lipids, where the amphiphile is double‐headed, and spans both sides of the ‘bilayer’ membrane. Use of such lipids complicates the manufacture process, however, and incorporation of encapsulants is not 100%, so purification procedures need to be conducted, followed by extensive characterization to make sure that consistency is maintained from batch to batch. Successful generation of oral immune responses has been reported in a number of disease models 76. See Corthésy et al. for a general review of lipid‐based particles as mucosal vaccine adjuvants, including the oil droplet approaches described here later 77.
A variant of liposomes, ISCOMs (Immune Stimulating COMplexes), in which phospholipids are co‐formulated with an immunostimulatory saponin amphiphile, has shown promise as a vaccine vehicle, inducing strong humoral and T cell responses after parenteral administration 78, 79. The vesicles are small and contain little internal aqueous space, so antigens incorporated into ISCOMs are usually inserted into the outer membrane. As such, the vehicle was originally considered best suited for presentation of viral and other membrane‐bound antigens. In fact, however, it emerges that good immune responses are generated even if antigens are not physically incorporated into the ISCOMS 80 so, under some circumstances, use with a wider range of antigens can be envisaged.
A small number of studies have been conducted 81, 82, 83, with some success, looking at ISCOMs administered orally, although the authors indicated that high doses of antigen were administered 84. It should be noted that intrinsic properties of the vehicle may limit its applicability via the oral route – namely, that the vesicles are labile and readily disassembled by detergents in the gut, that antigens which are not incorporated physically into ISCOMs can be easily separated from the immunostimulant by dilution in intestinal fluid, that antigens presented on the surface are exposed to protease attack, and that the antigens incorporated into ISCOMs are restricted to the subset which is membrane‐bound. Finally, the saponins in particulate form display toxicity to many cancer cells and, to a lesser extent, to normal cells 85, so there may be regulatory issues which need to be addressed.
Polymeric particulates
Recent studies conducted with thiol organosilica nanoparticles have shown that these can be taken up spontaneously by Peyer’s patches via transcytosis through M cells, and that optimal uptake was dependent on their size 86. A diameter of 0·1 μm seems to be preferred, although larger particles could also be taken up to lesser extents, probably via a different mechanism. Other workers looked at uptake of hydrophobic latex particles by Peyer’s patches, and also found that sub‐micron particles were taken up preferentially 87. Coating the particles with block co‐polymers to confer a hydrophilic surface to the particles inhibited uptake, showing that M cells have a natural preference for uptake of hydrophobic particles. Uptake of hydrophilic particles bearing charge, however, either positive or negative, has also been observed, implicating non‐specific interactions with certain surface‐bound receptors. It is worth mentioning as an aside that, although liposomes are composed of lipids (particularly phospholipids) they cannot be considered to be hydrophobic, as it is their highly polar phosphoryl choline headgroups which are outermost on the vesicle, presenting a very hydrophilic surface. Inclusion of specific targeting ligands on the outside of liposomes is therefore to be recommended in order to achieve uptake by Peyer’s patches.
Under the right circumstances, polymeric particles seem to be able to access Peyer’s patches unaided, which makes their cost lower and facilitates manufacture. They are also stable in the dry form, and can easily be reconstituted in aqueous media. A potential issue with polymeric particles, however, is their disintegration kinetics. They need to be able to break down in order to release their contents when inside the cell. Conversely, their disintegration should not be so rapid that they come apart in the intestinal lumen before reaching the Peyer’s patches. A fine balance needs to be struck, therefore. Other concerns are that large amounts of either hydrophilic or hydrophobic agents encapsulated within the body of the particle may disrupt the matrix and alter the disintegration kinetics, or that the matrix may disrupt the antigen itself and cause deleterious structural changes. A final problem is that the particles may not be so versatile in the nature and number of encapsulants they can hold; small molecules may leak out of the matrix while large proteins are held back. This is not a concern when used as drug delivery vehicles, where incorporation of a single agent is all that is required. For vaccines, however, inclusion of a large number of different components is desirable, as these can synergize to stimulate the immune system to best effect, and it may not be possible, in polymeric particles, to control the behaviour of all these agents optimally at the same time.
The material of choice for construction of microparticles for use in vivo is poly lactic glycolic acid (PLGA) 88, as this is biodegradable and has a long history of use in clinical practice. The hydrophilic/hydrophobic balance of these particles can be controlled by varying the ratio of lactic/glycolic acids employed. In addition, PLGA particles have been constructed using coatings such as polymethyl methacrylate 89 or containing TLR agonists such as monophosphoryl lipid A (MPLA), CpG and Pam3‐Cys‐Ser‐Lys4 (Pam3CSK4) 90. Microparticles constructed of chitosan have also been investigated, as chitosan has immunostimulating properties in its own right, because of its polycationic nature 91. For comprehensive reviews of the performance of polymeric particles in disease models see references 92, 93.
Oil droplets
The recognition that Peyer’s patch M cells have a predilection for hydrophobic particulates leads to the realization that the archetypal hydrophobic particle – an oil droplet – may also have potential as an oral vaccine carrier. This particularly makes sense bearing in mind that oils have been used as the basis for parenteral adjuvant formulations for decades, at least in experimental studies, in the form of Freund’s complete and incomplete adjuvants, where mineral oil is taken up by phagocytic cells, and appears to have a stimulating effect in its own right. A number of oil‐containing injectable vaccines are currently on the market, but in these formulations the antigen is located outside the oil droplets. For formulations administered orally, where the antigen needs to be protected from digestive enzymes and carried by the droplets to Peyer’s patches, the antigen must be encapsulated within the oil phase itself, a requirement which presents a challenge, as protein antigens are generally hydrophilic while the oil is hydrophobic. Two strategies have been employed to overcome this fundamental incompatibility.
The standard approach is production of what are known as double emulsions, where an aqueous phase is dispersed as droplets in an oil medium which is then itself dispersed in an aqueous phase 94. Different amphiphiles stabilizing the two types of droplet are required to facilitate their formation, as the relative size of head group and lipid tail varies in each case. Much energy needs to be expended to make the water‐in‐oil droplets small enough to fit into the final oil droplet of 0·2 mm diameter or less. Success has been achieved in generating immune responses after oral administration 95. However, use of these formulations in commercial practice may be dogged by stability issues, as there is the possibility of leakage of antigens from the core droplets, either during manufacture or storage; in addition, the presence of free water in the aqueous droplets raises the possibility of hydrolytic degradation of antigen, meaning that a cold chain is required during storage and distribution.
These problems are overcome by a more recent formulation methodology in which the antigens are incorporated into the oil phase not inside water droplets but within reverse micelles 96. Indeed, the technique employed is to form the reverse micelles first, and then dissolve them in the oil to form a clear solution. The end result is an antigen‐containing oil phase which can self‐emulsify in water (for example, in the lumen of the gut) to form droplets of the right size. Minimal leakage occurs during the self‐emulsification process and, interestingly, the same amphiphiles which emulsify the oil into droplets are also suitable for the formation of reverse micelles. No free water is present in the reverse micelles (this is taken off during lyophilization) so a cold chain should not be needed, as the threat of hydrolytic degradation is minimized. The absence of water also means that the bulk oil phase can be filled into a capsule for ease of administration. Unlike all the other particulate formulations described here, the method of manufacture involving reverse micelles dictates that incorporation is 100%, thus avoiding wastage and simplifying purification and characterization procedures. These formulations have been employed successfully in a number of preclinical disease models 68, 97, including challenge studies 98, and are ready for testing in the clinic.
One big advantage that the oil‐based formulations have over other types of carrier is that they can stably incorporate a large variety of different immunopotentiators in large quantities. These include the fat‐soluble vitamins A, D and E, all of which are known to play important roles in support of responses generated in the intestinal mucosa. While liposomes can, in, theory incorporate these agents into their membranes, large amounts will destabilize the membrane so their ability to deliver them is limited. Vitamin E, in oil‐based vaccine formulations, increases antibody responses and modulates expression of interleukins such as IL‐6, and chemokines belonging to CC and CXC classes, as well as aiding recruitment of granulocytes 99. Vitamin A, specifically retinoic acid, enhances the ability of dendritic cells to present antigen 100, encourages the production of cytokines promoting a T helper type 1 (Th1) response 101 and influences the homing 102 response of cells – in this case the ability of lymphoid cells to return to mucosal tissues after having migrated from Peyer’s patches or mesenteric lymph nodes 103. Vitamin D plays an important role in controlling T cell development in the later stages of immune stimulation 104, and in particular increases the pool of memory T cells in infectious disease 105. An oral lipid formulation containing all those vitamins in combination has been reported in Moore et al. 106, demonstrating successfully the boosting of an immune response against plague (Yersinia pestis antigens F1 and V). This work has also pioneered a combination of injected prime and oral booster, as a rapid way of maximizing responses to the disease.
The role played by these (and other) dietary components in the proper working of the immune system makes it clear that adequate nutrition is an important prerequisite for generating strong immune responses. Unfortunately, in LMIC, malnutrition of populations at risk is a serious problem 107, 108, and the correlation between famine and rise of epidemic disease is well accepted. Clearly, a vaccine which is able to supply these micronutrients at the right place, and at the right time (i.e. during initiation of an immune response), is a very desirable objective, and this is an extremely significant advantage of oil‐based oral vaccines for protection against emerging diseases.
Conclusion
There is no reason to believe that new infectious diseases will not continue to emerge in the future, and the likelihood is that these will appear in their severest form in LMICs, where climate, living standards, public health issues and poor nutrition combine to increase transmission, and weaken natural immune resistance. The new generations of vaccine developed for combatting these diseases need to be ones which can perform well in such an environment, i.e. can be stored and distributed easily, are readily acceptable by the local population, can easily be handled by non‐medical personnel with risk of contamination and show clear evidence of efficacy. These criteria are all fulfilled by oral vaccines, and future initiatives designed to tackle the problems of emerging diseases, particularly in LMIC, must make oral vaccination a high priority.
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Vaccines for emerging pathogens: from research to the clinic. Part two. Clinical and Experimental Immunology 2019, 198: 141–142.
A novel vaccine platform using glucan particles for induction of protective responses against Francisella tularensis and other pathogens. Clinical and Experimental Immunology 2019, 198: 143–152.
Vaccines for emerging pathogens: prospects for licensure. Clinical and Experimental Immunology 2019, 198: 170–183.
References
- 1. Lin IYC, Van TTH, Smooker PM. Live‐attenuated bacterial vectors: tools for vaccine and therapeutic agent delivery. Vaccines 2015; 3:940–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pandit J, Sultana Y, Kalam MA. Newer technologies in oral vaccine delivery. Current Drug Therapy 2014; 9:173–87. [Google Scholar]
- 3. Kim SH, Jang YS. The development of mucosal vaccines for both mucosal and systemic immune induction and the roles played by adjuvants. Clin Exp Vaccine Res 2017; 6:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Belyakov IM, Ahlers JD. What role does the route of immunization play in the generation of protective immunity against mucosal pathogens? J Immunol 2009; 183:6883–92. [DOI] [PubMed] [Google Scholar]
- 5. Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol 2003; 3:331–41. [DOI] [PubMed] [Google Scholar]
- 6. Arenas J. The role of bacterial lipopolysaccharides as immune modulator in vaccine and drug development. Endocr Metab Immune Disord Drug Targets 2012; 12:221–35. [DOI] [PubMed] [Google Scholar]
- 7. Pisetsky D. Immune activation by bacterial DNA: a new genetic code. Immunity 1996; 5:303–10. [DOI] [PubMed] [Google Scholar]
- 8. Young PA, Morrison SL, Timmerman JM. Antibody‐cytokine fusion proteins for treatment of cancer: engineering cytokines for improved efficacy and safety. Semin Oncol 2014; 41:623–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jobling MG, Yang Z, Kam WR, Lencer WI, Holmes RK. A single native ganglioside GM1‐binding site is sufficient for cholera toxin to bind to cells and complete the intoxication pathway. mBio 2012; 3:e00401–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Landsverk OJB, Snir O, Casado RB et al Antibody‐secreting plasma cells persist for decades in human intestine. J Exp Med 2017; 214:309–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baumgarth N, Rothaeusler K. Extrafollicular foci‐derived B cells provide long‐lived mucosal immunity. J immunol 2009; 182(Suppl 1):84.16.19109138 [Google Scholar]
- 12. Lemke A, Kraft M, Roth K, Riedel R, Lammerding D, Hauser AE. Long‐lived plasma cells are generated in mucosal responses and contribute to the bone marrow plasma cell pool in mice. Immunology 2016; 9:83–97. [DOI] [PubMed] [Google Scholar]
- 13. Gallichan WS, Rosenthal KL. Long‐lived cytotoxic lymphocyte memory in mucosal tissues after mucosal but not systemic immunization. J Exp Med 1996; 184:1879–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bemark M, Hazanov H, Stromberg A et al Limited clonal relatedness between gut IgA plasma cells and memory B cells after oral immunisation. Nat Commun 2016; 7:12698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vighi G, Marcucci F, Sensi L, Di Cara G, Frati F. Allergy and the gastrointestinal system. Clin Exp Immunol 2008; 153:3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jung C, Hugot J‐P, Barreau F. Peyer’s patches: the immune sensors of the intestine. Int J Inflamm 2010; 2010: 823710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Macpherson AJ, Smith K. Mesenteric lymph nodes at the center of immune anatomy. J Exp Med 2006; 203:497–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chassaing B, Kumar M, Baker MT, Singh V, Vijay‐Kumar M. Mammalian gut immunity. Biomed J 2014; 37:246–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sheridan S, Lefrancois L. Intraepithelial lymphocytes: to serve and protect. Curr Gastroenterol Rep 2010; 12:513–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qiu Y, Yang H. Effects of intraepithelial lymphocyte‐derived cytokines on intestinal mucosal barrier function. J Interferon Cytokine Res 2013; 33:551–62. [DOI] [PubMed] [Google Scholar]
- 21. Vitale S, Picascia S, Gianfrani C. The cross‐talk between enterocytes and intraepithelial lymphocytes. Mol Cell Pediatr 2016; 3:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hayday A, Deban L. Mucosal T cell receptor γδ intraepithelial T cells. Mucosal Immunol 2015; 1:765–76. [Google Scholar]
- 23. Saurer L, Mueller C. T cell‐mediated immunoregulation in the gastrointestinal tract. Allergy 2009; 64:505–19. [DOI] [PubMed] [Google Scholar]
- 24. Ruberti M, Fernandes LGR, Simioni PU, Gabriel DL, Yamada AT, Tamashiro WMC. Phenotypical and functional analysis of intraepithelial lymphocytes from small intestine of mice in oral tolerance. Clin Dev Immunol 2012; article ID 208054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reineke JJ, Daniel Y, Cho DY et al Unique insights into the intestinal absorption, transit, and subsequent biodistribution of polymer‐derived microspheres. PNAS 2013; 110:13803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Howe SE, Lickteig DJ, Plunkett KN. Ryerse JS, Konjufca V. The uptake of soluble and particulate antigens by epithelial cells in the mouse small intestine. PLOS ONE 2014; 9:e86656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baicus A. History of polio vaccination. World J Virol 2012; 1:108–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sabin AB, Ramos‐Alvarez M, Alvarez‐Amezquita J et al Live, orally given poliovirus vaccine. Effects of rapid mass immunization on population under conditions of massive enteric infection with other viruses. JAMA 1960; 173:1521–6. [DOI] [PubMed] [Google Scholar]
- 29. Sabin AB, Boulger LR. History of Sabin attenuated poliovirus oral live vaccine strains. J Biol Standard 1973; 1:115–8. [Google Scholar]
- 30. World Health Organization (WHO) . Polio vaccines: WHO position paper, March, 2016. Wkly Epidemiol Rec 2016; 91:145–168.27039410 [Google Scholar]
- 31. Kew O, Morris‐Glasgow V, Landaverde M et al Outbreak of poliomyelitis in hispaniola associated with circulating type 1 vaccine‐derived poliovirus. Science 2002; 296:356–9. [DOI] [PubMed] [Google Scholar]
- 32. Shimizu H, Thorley B, Paladin FJ et al Circulation of type 1 vaccine‐derived poliovirus in the Philippines in 2001. J Virol 2001; 2004:13512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gromeier M, Bossert B, Arita M, Nomoto A, Wimmer E. Dual stem loops within the poliovirus internal ribosomal entry site control neurovirulence. J Virol 1999; 73:958–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ochs K, Zeller A, Saleh L et al Impaired binding of standard initiation factors mediates poliovirus translation attenuation. J Virol 2003; 77:115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. World Health Organization (WHO) . Rotavirus vaccines: WHO position paper, January, 2013. Wkly Epidemiol Rec 2013; 88:49–64. [PubMed] [Google Scholar]
- 36. Tate J, Parashar UD. Rotavirus vaccines in routine use. Clin Infect Dis 2014; 59:1291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. World Health Organization (WHO) . Typhoid vaccines: WHO position paper, March, 2018. Wkly Epidemiol Rec 2018; 93:153–172. [Google Scholar]
- 38. Wang M, Bregenholt S, Petersen JS. The cholera toxin B subunit directly costimulates antigen‐primed CD4� T cells ex vivo . Scand J Immunol 2003; 58:342–9. [DOI] [PubMed] [Google Scholar]
- 39. Gloudemans AK, Plantinga M, Guilliams M et al The mucosal adjuvant cholera toxin B instructs non‐mucosal dendritic cells to promote IgA production via retinoic acid and TGF‐beta. PLOS ONE 2013; 8:e59822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stratmann T. Cholera toxin subunit B as adjuvant – an accelerator in protective immunity and a break in autoimmunity. Vaccines 2015; 3:579–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. World Health Organization (WHO) . Oral cholera vaccines in mass immunization campaigns: guidance for planning and use. World Health Organization 2010; ISBN 9789241500432. [Google Scholar]
- 42. Galen JE, Pasetti MF, Tennant S, Olveira‐Ruiz P, Sztein MB, Levine MM. Salmonella enterica serovar typhi live vector vaccines finally come of age. Immunol Cell Biol 2009; 87:400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Galen JE, Levine MM. Can a ‘flawless’ live vector vaccine strain be engineered? Trends Microbiol 2001; 9:372–6. [DOI] [PubMed] [Google Scholar]
- 44. da Silva AJ, Zangirolami TC, Novo‐Mansur MTM, Giordano RC, Martins EAL. Live bacterial vaccine vectors: an overview. Braz J Microbiol 2014; 45:1117–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kotton C, Hohmann EL. Enteric pathogens as vaccine vectors for foreign antigen delivery. Infect Immun 2004; 72:5535–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cranenburgh RM, Lewis KS, Hanak JAJ. Effect of plasmid copy number and lac operator sequence on antibiotic‐free plasmid selection by operator‐repressor titration in Escherichia coli . J Mol Microbiol Biotechnol 2004; 7:197–203. [DOI] [PubMed] [Google Scholar]
- 47. Martinoli C, Chiavelli A, Rescigno M. Entry route of Salmonella typhimurium directs the type of induced immune response. Immunity 2007; 27:975–84. [DOI] [PubMed] [Google Scholar]
- 48. Tacket CO, Pasetti MF, Sztein MB, Livio S, Levine MM. Immune responses to an oral typhoid vaccine strain that is modified to constitutively express Vi capsular polysaccharide. J Infect Dis 2004; 190:565–70. [DOI] [PubMed] [Google Scholar]
- 49. Wells J, Mercenier A. Mucosal delivery of therapeutic and prophylactic molecules using lactic acid bacteria. Nat Rev Microbiol 2008; 6:349–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mohamadzadeh M, Duong T, Sandwick SJ, Hoover T, Klaenhammer TR. Dendritic cell targeting of Bacillus anthracis protective antigen expressed by Lactobacillus acidophilus protects mice from lethal challenge. Proc Natl Acad Sci 2009; 106:4331–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Coconnier M‐H, Klaenhammer TR, Kerneis S, Bernet M‐F, Servini AL. Protein‐mediated adhesion of lactobacillus acidophilus BG2FO4 on human enterocyte and mucus‐secreting cell lines in culture. Appl Environ Microbiol 1992; 58:2034–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lightfoot YL, Mohamadzadeh M. Tailoring gut immune responses with lipoteichoic acid‐deficient Lactobacillus acidophilus . Front Immunol 2013; 4:2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stoeker L, Nordone S, Gunderson S et al Assessment of Lactobacillus gasseri as a candidate oral vaccine vector. Clin Vaccine Immunol 2011; 18:1834–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mohamadzadeh M, Klaenhammer TR. Specific Lactobacillus species differentially activate Toll‐like receptors and downstream signals in dendritic cells. Exp Rev Vaccines 2008; 7:1155–64. [DOI] [PubMed] [Google Scholar]
- 55. de Azevedo M, Meijerink M, Taverne N, Pereira VB, LeBlanc JG, Azevedo V. Recombinant invasive Lactococcus lactis can transfer DNA vaccines either directly to dendritic cells or across an epithelial cell monolayer. Vaccine 2015; 33:4807–12. [DOI] [PubMed] [Google Scholar]
- 56. Chen RF, Lee CY. Adenoviruses types, cell receptors and local innate cytokines in adenovirus infection. Int Rev Immunol 2014; 33:45–53. [DOI] [PubMed] [Google Scholar]
- 57. Forbes EK, de Cassan SC, Llewellyn D et al Cell responses induced by adenoviral vectored vaccines can be adjuvanted by fusion of antigen to the oligomerization domain of C4b‐binding protein. PLOS ONE 2012; 7:e44943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Saxena M, Van TTH, Baird FJ, Coloe PJ, Smooker PM. Pre‐existing immunity against vaccine vectors – friend or foe? Microbiology 2013; 159:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Xiang ZQ, Gao GP, Reyes‐Sandoval A, Li Y, Wilson M, Ertl HCJ. Oral Vaccination of mice with adenoviral vectors is not impaired by pre‐existing immunity to the vaccine carrier. J Virol 2003; 77:10780–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gurwith M, Lock M, Taylor EM et al Safety and immunogenicity of an oral, replicating adenovirus serotype 4 vector vaccine for H5N1 influenza: a randomised, double‐blind, placebo‐controlled, phase 1 study. Lancet Infect Dis 2013; 13:238–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li ZX, Zhang MX, Zhou CH, Zhao XY, Iijima N, Frankel FR. Novel vaccination protocol with two live mucosal vectors elicits strong cell‐mediated immunity in the vagina and protects against vaginal virus challenge. J Immunol 2008; 180:2504–13. [DOI] [PubMed] [Google Scholar]
- 62. Naito T, Kaneko Y, Kozbor D. Oral vaccination with modified vaccinia virus Ankara attached covalently to TMPEG‐modified cationic liposomes overcomes pre‐existing poxvirus immunity from recombinant vaccinia immunization. J Gen Virol 2007; 88:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Arntzen CJ. Pharmaceutical foodstuffs – oral immunization with transgenic plants. Nat Med Vaccine Suppl 1998; 4:502–3. [DOI] [PubMed] [Google Scholar]
- 64. Nochi T, Takagi H, Yuki Y et al Rice‐based mucosal vaccine as a global strategy for cold‐chain‐ and needle‐free vaccination. Proc Natl Acad Sci 2007; 104:10986–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Soh HS, Chung HY, Lee HH et al Expression and functional validation of heat‐labile enterotoxin B (LTB) and cholera toxin B (CTB) subunits in transgenic rice (Oryza sativa). Springerplus 2015; 4:148–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. De Smet R, Allais L, Cuvelier CA. Recent advances in oral vaccine development. Yeast‐derived beta‐glucan particles. Hum Vaccines Immunother 2014; 10:1309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Holmgren J, Lycke N, Czerkinsky C. Cholera toxin and cholera B subunit as oral‐mucosal adjuvant and antigen vector systems. Vaccine 1993; 11:1179–84. [DOI] [PubMed] [Google Scholar]
- 68. Okuno T, Kashige N, Satho T, Irie K. Hiramatsu Y, Sharmin T. Expression and secretion of cholera toxin B subunit in Lactobacilli. Biol Pharm Bull 2013; 36:952–8. [DOI] [PubMed] [Google Scholar]
- 69. Price GA, McFann K, Holmes RK. Immunization with cholera toxin B subunit induces high‐level protection in the suckling mouse model of cholera. PLOS ONE 2013; 2013:e57269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Park SJ, Chun SK, Kim PH. Intraperitoneal delivery of cholera toxin B subunit enhances systemic and mucosal antibody responses. Mol Cell 2003; 16:106–12. [PubMed] [Google Scholar]
- 71. Kubota E, Joh T, Tanida S et al Vaccination against helicobacter pylori with recombinant cholera toxin B‐subunit. Helicobacter 2005; 10:345–52. [DOI] [PubMed] [Google Scholar]
- 72. Sun JB, Czerkinsky C, Holmgren J. Mucosally induced immunological tolerance, regulatory T cells and the adjuvant effect by cholera toxin B subunit. Scand J Immunol 2010; 71:1–11. [DOI] [PubMed] [Google Scholar]
- 73. Moreno‐Altamirano MMB, Aguilar‐Carmona I, Sanchez‐Garcia FJ. Expression of GM1, a marker of lipid rafts, defines two subsets of human monocytes with differential endocytic capacity and lipopolysaccharide responsiveness. Immunology 2007; 120:536–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Andrade G, New RRC, Sant’Anna OA et al A universal polysaccharide conjugated vaccine against O111 E. coli . Hum Vaccine Immunother 2014; 10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bulbake U, Doppalapudi S, Kommineni N, Khan W. Liposomal formulations in clinical use: an updated review. Pharmaceutics 2017; 9:12–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schwendener RA. Liposomes as vaccine delivery systems: a review of the recent advances. Ther Adv Vaccines 2014; 2:159–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Corthésy B, Bioley G. Lipid‐based particles: versatile delivery systems for mucosal vaccination against infection. Front Immunol 2018; 9:431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lövgren Bengtsson K, Morein B, Osterhaus ADME. ISCOM technology‐based Matrix M™ adjuvant: success in future vaccines relies on formulation. Exp Rev Vaccine 2011; 10:401–3. [DOI] [PubMed] [Google Scholar]
- 79. Reimer JM, Karlsson KH, Lovgren‐Bengtsson K, Magnusson SE, Fuentes A, Stertman L. Matrix‐M™ adjuvant induces local recruitment, activation and maturation of central immune cells in absence of antigen. PLOS ONE 2012; 7:e41451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lovgren K, Morein B. The requirement of lipids for the formulation of immunostimulating complexes (ISCOMS). Biotechnol Appl Biochem 1988; 10:161–72. [PubMed] [Google Scholar]
- 81. Sjolander A, Cox JC. Uptake and adjuvant activity of orally delivered saponin and ISCOM™ vaccines. Adv Drug Deliv Rev 1999; 34:321–38. [DOI] [PubMed] [Google Scholar]
- 82. Mowat AMcl, Maloy K, Smith RE, Donachie A. Iscoms as Mucosal Vaccine Vectors InGregoriadis G, McCormack B, Allison AC, eds. Vaccine Design. NATO ASI Series (Series A: Life Sciences). Boston, MA: Springer, 1997: 293. [Google Scholar]
- 83. Van Pinxteren LAH, Bruce MG, Campbell I et al Effect of oral rotavirus/iscom vaccines on immune responses in gnotobiotic lambs. Vet Immunol Immunopathol 1999; 7:53–67. [DOI] [PubMed] [Google Scholar]
- 84. Azevedo MSP, Gonzalez AM, Yuan LJ et al Oral versus intranasal prime/boost regimen using attenuated human rotavirus or VP2 and VP6 virus‐like particles with immunostimulating complexes influences protection and antibody‐secreting cell responses to rotavirus in a neonatal gnotobiotic pig model. Clin Vaccine Immunol 2010; 17:420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hu KF, Berenjian S, Larsson R et al Nanoparticulate Quillaja saponin induces apoptosis in human leukemia cell lines with a high therapeutic index. Int J Nanomed 2010; 5:51–62. [PMC free article] [PubMed] [Google Scholar]
- 86. Awaad A, Nakamura M, Ishimura K. Imaging of size‐dependent uptake and identification of novel pathways in mouse Peyer’s patches using fluorescent organosilica particles. Nanomedicine 2012; 8:627–36. [DOI] [PubMed] [Google Scholar]
- 87. Florence AT, Hillery AM, Hussain N, Oral Janu PU. Uptake and translocation of nanoparticles: a real but useful phenomenon? In Gregoriadis G, McCormack P, Poste G, eds. Targeting of Drugs 4. NATO ASI Series (Series A: Life Sciences). Boston, MA: Springer, 1994: 273:173–81. [Google Scholar]
- 88. O’Hagan DT, Singh M. Poly (lactide‐coglycolide) microparticles as vaccine adjuvants. Exp Rev Vaccines 2000; 42:91–103. [Google Scholar]
- 89. Zhang L, Yang WD, Hu CH, Wang QC, Wu YK. Properties and applications of nanoparticle/microparticle conveyors with adjuvant characteristics suitable for oral vaccination. Int J Nanomed 2018; 13:2973–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Madan‐Lala R, Pradhan P, Roy K. Combinatorial delivery of dual and triple agonists via polymeric pathogen‐like particles synergistically enhances innate and adaptive immune responses. Nat SciRep 2017; 7:2530–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. van der Lubben IM, Verhoef JC, Borchard G, Junginger HE. Chitosan for mucosal vaccination. Adv Drug Deliv Rev 2001; 52:139–44. [DOI] [PubMed] [Google Scholar]
- 92. Soares EFP, Borges OMF. Oral vaccination through Peyer’s patches: update on particle uptake. Curr Drug Deliv 2018; 15:321–30. [DOI] [PubMed] [Google Scholar]
- 93. Oyewumi MO, Kumar A, Cui ZR. Nano‐microparticles as immune adjuvants: correlating particle size and the resultant immune responses. Exp Rev Vaccines 2010; 9:1095–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Muschiolik G, Dickinson E. Double emulsions relevant to food systems: preparation, stability, and applications. Comp Rev Food Sci Food Safety 2017; 16:532–55. [DOI] [PubMed] [Google Scholar]
- 95. Liau JJ, Prestidge CA, Hook S, Barnes TJ. Development of a multi‐compartmental oral vaccine delivery system. Drug Delivery Lett 2016; 6:57–62. [Google Scholar]
- 96. New RRC, Kirby CK. Solubilisation of hydrophilic drugs in oily formulations. Adv Drug Deliv Rev 1997; 25:59–69. [Google Scholar]
- 97. Domingos MO, Lewis DJ, Jansen T, Zimmerman DH, Williamson ED, New RRC. A new oil‐based antigen delivery formulation for both oral and parenteral vaccination. Open Drug Deliv J 2008; 2:52–60. [Google Scholar]
- 98. Prabakaran M, Madhan S, Prabhu N, Geng YH, New R, Kwang J. Reverse micelle‐encapsulated recombinant baculovirus as an oral vaccine against H5N1 infection in mice. Antiviral Res 2010; 86:180–7. [DOI] [PubMed] [Google Scholar]
- 99. Morel S, Didierlaurent A, Bourgignon P et al Adjuvant system AS03 containing alpha‐tocopherol modulates innate immune response and leads to improved adaptive immunity. Vaccine 2011; 29:2461–73. [DOI] [PubMed] [Google Scholar]
- 100. Pino‐Lagos K, Benson MJ, Noelle RJ. Retinoic acid in the immune system. Ann N Y Acad Sci 2008; 1143:170–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hall JA, Grainger JR, Spencer SP, Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity 2011; 35:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mora JR, von Andrian UH. Differentiation and homing of IgA‐secreting cells. Immunology 2008; 1:96–109. [DOI] [PubMed] [Google Scholar]
- 103. Mora JR, Iwata M, von Andrian UH. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol 2008; 8:685–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Looman KIM, Jansen MAE, Voortman T et al The role of vitamin D on circulating memory T cells in children: the Generation R study. Pediatr Allergy Immunol 2017; 28:579–87. [DOI] [PubMed] [Google Scholar]
- 105. Chun RF, Liu PT, Modlin RL, Adams JS, Hewison M. Impact of vitamin D on immune function: lessons learned from genome‐wide analysis. Front Physiol 2014; 5:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Moore BD, New RRC, Butcher W et al Dual Route Vaccination for Plague with Emergency Use Applications. Vaccine 2018; 36:5210–7. [DOI] [PubMed] [Google Scholar]
- 107. Sack DA, Qadri F, Svennerholm AM. Determinants of responses to oral vaccines in developing countries. Ann Nestlé (UK) 2008; 66:71–9. [Google Scholar]
- 108. Levine MM. Immunogenicity and efficacy of oral vaccines in developing countries: lessons from a live cholera vaccine. BMC Biol 2010; 8:129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Radin JM, Hawkesworth AW, Blair PJ et al Dramatic decline of respiratory illness among US military recruits after the renewed use of adenovirus vaccines. Clin Inf Diseases 2014; 59:962–8. [DOI] [PubMed] [Google Scholar]
- 110. Fournier B, Philpott DJ. Recognition of Staphylococcus aureus by the Innate immune system. Clin Microbiol Rev 2005; 18:521–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Schwandner R, Dziarske R, Wesche H, Rother M, Kirschning CJ. Peptidoglycan and lipoteichoic acid‐induced cell activation is mediated by toll‐like receptor 2. J Biol Chem 1999; 274:17406–9. [DOI] [PubMed] [Google Scholar]
- 112. Kakutani R, Adachi Y, Takata H, Kuriki T, Ohno N. Essential role of Toll‐like receptor 2 in macrophage activation by glycogen. Glycobiology 2012; 22:146–59. [DOI] [PubMed] [Google Scholar]
- 113. Farhat K, Riekenberg S, Heine H et al Heterodimerisation of TLR2 with TLR1 or TLR6 expands the ligand spectrum but does not lead to differential signalling. J Leukocyte Biol 2008; 83:692–701. [DOI] [PubMed] [Google Scholar]
- 114. Takeuchi A, Sato S, Horiuchi T et al Role of Toll‐like receptor 1 in mediating immune response to microbial lipoproteins. J Immunobiol 2002; 169:1–14. [DOI] [PubMed] [Google Scholar]
- 115. Funderburg NT, Jadlowsky JK, Lederman MM, Feng ZN, Wienberg W, Sieg SF. The Toll‐like receptor 1/2 agonists Pam3CSK4 and human β‐defensin‐3 differentially induce interleukin‐10 and nuclear factor‐κB signalling patterns in human monocytes. Immunology 2011; 134:151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Takeuchi O, Kaufmann A, Grote K et al Preferentially the R‐stereoisomer of the mycoplasmal lipopeptide macrophage‐activating lipopeptide‐2 activates immune cells through a Toll‐like receptor 2‐ and MyD88‐dependent signalling pathway. J Immunol 2000; 164:554–7. [DOI] [PubMed] [Google Scholar]
- 117. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double‐stranded RNA and activation of NF‐κB by Toll‐like receptor 3. Nature 2001; 413:732–8. [DOI] [PubMed] [Google Scholar]
- 118. Liu L, Botos I, Wang Y et al Structural basis of Toll‐like receptor 3 Signalling with double‐stranded RNA. Science 2008; 320:379–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Tatematsu M, Seya T, Matsumoto M. Beyond dsRNA: Toll‐like receptor 3 signalling in RNA‐induced immune responses. Biochem J 2014; 458:195–201. [DOI] [PubMed] [Google Scholar]
- 120. Cui WG, Joshi NS, Liu Y, Meng HL, Kleinstein S, Kaech SM. TLR4 ligands lipopolysaccharide and monophosphoryl lipid A differentially regulate effector and memory CD8+ T cell differentiation. J Immunol 2014; 192:4221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Romero CD, Varma TK, Hobbs JB, Reyes A, Driver B, Sherwood ER. The Toll‐like receptor 4 agonist monophosphoryl lipid A augments innate host resistance to systemic bacterial infection. Infect Immun 2011; 79:3576–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Netea MG, Gow NAR, Munro CA et al Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll‐like receptors. J Clin Invest 2006; 116:1642–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Yoon SL, Kurnasov O, Natarajan V et al Structural basis of TLR5‐flagellin recognition and signalling. Science 2012; 17:859–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Takeuchi O, Kawai T, Sanjo H et al TLR6: a novel member of an expanding Toll‐like receptor family. Gene 1999; 231:65. [DOI] [PubMed] [Google Scholar]
- 125. Dowling DJ. Recent advances in the discovery and delivery of TLR7/8 agonists as vaccine adjuvants. ImmunoHorizons 2018; 2:185–97. [DOI] [PubMed] [Google Scholar]
- 126. Colak E, Leslie A, Zausmer K et al RNA and imidazoquinolines are sensed by distinct TLR7/8 ectodomain sites resulting in functionally disparate signalling events. J Immunol 2014; 192:5963–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Heil F, Hemmi H, Hochrein H et al Species‐specific recognition of single‐stranded RNA via Toll‐like receptor 7 and 8. Science 2004; 303:1526–9. [DOI] [PubMed] [Google Scholar]
- 128. Cornelie S, Hoebeke J, Schacht A‐M et al Direct evidence that Toll‐like receptor 9 (TLR9) functionally binds plasmid DNA by specific cytosine‐phosphate‐guanine motif recognition. J Biol Chem 2004; 279:15124–9. [DOI] [PubMed] [Google Scholar]
- 129. Caruso R, Warner N, Inohara N, Núñez G. NOD1 and NOD2: signalling, host defense, and inflammatory disease. Immunity 2014; 41:898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Cai X, Chiu YH, Chen ZJ. The cGAS‐cGAMP‐STING pathway of cytosolic DNA sensing and signaling. Mol Cell 2014; 54:289–96. [DOI] [PubMed] [Google Scholar]
- 131. Kayamuro H, Yoshioka Y, Abe Y et al Interleukin‐1 family cytokines as mucosal adjuvants for induction of protective immunity against influenza virus. J Virology 2010; 84:12703–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Stevceva L, Moniuszko M, Hitchens K, Manzanero S, Werninghaus K, Ferrari MG. Utilizing IL‐12, IL‐15 and IL‐7 as mucosal vaccine adjuvants. Lett Drug Des Discov 2006; 3:586–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Schoenen H, Bodendorfer B, Hitchens K et al Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose‐dibehenate. J Immunol 2010; 184:2756–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Hug H, Mohajeri MH, La Fata G. Toll‐like receptors: regulators of the immune response in the human gut. Nutrients 2018; 10:203–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Foster N, Clark MA, Jepson MA, Hirst BH. Ulex europaeus 1 lectin targets microspheres to mouse Peyer’s patch M‐cells in vivo . Vaccine 1998; 16:536–41. [DOI] [PubMed] [Google Scholar]
- 136. Chen H, Torchilin V, Langer R. Lectin‐bearing polymerized liposomes as potential oral vaccine carriers. Pharm Res 1996; 13:1378–83. [DOI] [PubMed] [Google Scholar]
- 137. Clark MA, Jepson MA, Simmons NI, Hirst BH. Differential surface characteristics of M cells from mouse intestinal Peyer’s and caecal patches. Histochem J 1994; 26:271–80. [PubMed] [Google Scholar]
- 138. Gebert A, Hach G. Differential binding of lectins to M cells and enterocytes in the rabbit cecum. Gastroenterology 1993; 105:1350–61. [DOI] [PubMed] [Google Scholar]
- 139. Hirabayashi J, Hashidate T, Arata Y et al Oligosaccharide specificity of galectins: a search by frontal affinity chromatography. Biochem Biophys Acta 2002; 1572:232–54. [DOI] [PubMed] [Google Scholar]
- 140. Clark MA, Hirst BH, Jepson MA. M‐cell surface beta1 integrin expression and invasion‐medicated targeting of Yersinia pseudotuberculosis to mouse Peyer’s patch M cells. Infect Immun 1998; 66:1237–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Critchley‐Thorne RJ, Stagg AK, Vassaux G. Recombinant Escherichia coli expressing invasion targets the Peyer’s patches: the basis for a bacterial formulation for oral vaccination. Mol Ther 2006; 14:183–91. [DOI] [PubMed] [Google Scholar]
- 142. Sato S, Kaneto A, Shibata N et al Transcription factor Spi‐B‐dependent and –independent pathways for the development of Peyer’s patch M cells. Mucosal Immunol 2013; 6:838–46. [DOI] [PubMed] [Google Scholar]
- 143. Hase K, Kawano K, Nochi T et al Uptake through glycoprotein 2 of FimH(+) bacteria by M cells initiates mucosal immune response. Nature 2009; 462:226–30. [DOI] [PubMed] [Google Scholar]
- 144. Ohno H, Hase K. Glycoprotein 2 (GP2): grabbing the FimH bacteria into M cells for mucosal immunity. Gut Microbes 2010; 1:407–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Kim SH, Jung DI, Yang IY et al M cells expressing the complement C5a receptor are efficient targets for mucosal vaccine delivery. Eur J Immunol 2011; 41:3219–29. [DOI] [PubMed] [Google Scholar]
- 146. Nakato G, Hase K, Suzuki M, Kimura M, Ato M, Hanazato M. Brucella abortus exploits a cellular prion protein on intestinal M cells as an invasive receptor. J Immunol 2012; 189:1540–4. [DOI] [PubMed] [Google Scholar]
- 147. Rand JH, Wu XX, Lin EY, Griffel A, Gialanella P, McKitrick JC. Annexin A5 binds to lipopolysaccharide and reduces its endotoxin activity. MBio 2012; 3:e00292–e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Osani A, Sashinami H, Asano K, Li SJ, Hu DL, Nakane A. Mouse peptidoglycan recognition protein PGLYRP‐1 plays a role in the host innate immune response against Listeria monocytogenes infection. Infect Immun 2011; 79:858–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Lo DD, Ling J, Eckelhoefer AH. M cell targeting peptide can enhance mucosal IgA responses. BMC Biotechnol 2012; 12:7m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Wolf JL, Kauffman RS, Finberg R, Dambrauskas R, Fields BN, Trier JS. Determinants of reovirus interaction with the intestinal |M cells and absorptive cells of murine intestine. Gastroenterology 1983; 85:291–300. [PubMed] [Google Scholar]
- 151. Tyrer PC, Foxwell AR, Kyd JM, Otczyk DC, Cripps AW. Receptor mediated targeting of M‐cells. Vaccine 2007; 25:3204–9. [DOI] [PubMed] [Google Scholar]
- 152. Kim SH, Kim YN, Kim J, Jang YS. C5a receptor targeting of partial non‐structural protein 3 of dengue virus promotes antigen‐specific IFN‐γ‐producing T‐cell responses in a mucosal dengue vaccine model. Cell Immunol 2018; 325:41–7. [DOI] [PubMed] [Google Scholar]
- 153. Kim SH, Lee HY, Jang YS. Expression of the ATP‐gated P2X7 receptor on M cells and its modulating role in the mucosal immune environment. Immune Netw 2015; 15:1–44. [DOI] [PMC free article] [PubMed] [Google Scholar]