

Globoside has long been considered the primary receptor of B19V. However, its expression does not correlate well with B19V binding and uptake and cannot explain the pathogenesis or the remarkable narrow tissue tropism of the virus. By using a knockout cell line, we demonstrate that globoside does not have the expected function as a cell surface receptor required for B19V entry, but it has an essential role at a postentry step for productive infection. This finding explains the natural resistance to infection associated with individuals lacking globoside, contributes to a better understanding of B19V restricted tropism, and offers novel strategies for the development of antiviral therapies.
KEYWORDS: B3GalNT1, Gb4, P antigen, parvovirus B19, globoside, globoside synthase, receptor, virus entry, virus tropism
ABSTRACT
Globoside (Gb4) is considered the primary receptor of parvovirus B19 (B19V); however, its expression does not correlate well with the attachment and restricted tropism of the virus. The N terminus of VP1 (VP1u) of B19V interacts with an as-yet-unknown receptor required for virus internalization. In contrast to Gb4, the VP1u cognate receptor is expressed exclusively in cells that B19V can internalize. With the aim of clarifying the role of Gb4 as a B19V receptor, we knocked out the gene B3GalNT1 coding for the enzyme globoside synthase in UT7/Epo cells. Consequently, B3GalNT1 transcripts and Gb4 became undetectable in the knockout (KO) cells without affecting cell viability and proliferation. Unexpectedly, virus attachment, internalization, and nuclear targeting were not disturbed in the KO cells. However, NS1 transcription failed, and consequently, genome replication and capsid protein expression were abrogated. The block could be circumvented by transfection with a B19V infectious clone, indicating that Gb4 is not required after the generation of viral double-stranded DNA with resolved inverted terminal repeats. While in wild-type (WT) cells, occupation of the VP1u cognate receptor with recombinant VP1u disturbed virus binding and blocked the infection, antibodies against Gb4 had no significant effect. In a mixed population of WT and KO cells, B19V selectively infected WT cells. This study demonstrates that Gb4 does not have the expected receptor function, as it is dispensable for virus entry; however, it is essential for productive infection, explaining the resistance of the rare individuals lacking Gb4 to B19V infection.
IMPORTANCE Globoside has long been considered the primary receptor of B19V. However, its expression does not correlate well with B19V binding and uptake and cannot explain the pathogenesis or the remarkable narrow tissue tropism of the virus. By using a knockout cell line, we demonstrate that globoside does not have the expected function as a cell surface receptor required for B19V entry, but it has an essential role at a postentry step for productive infection. This finding explains the natural resistance to infection associated with individuals lacking globoside, contributes to a better understanding of B19V restricted tropism, and offers novel strategies for the development of antiviral therapies.
INTRODUCTION
Human parvovirus B19 (B19V) is a prominent human pathogen which is typically associated with erythema infectiosum, or fifth disease, a worldwide disease affecting mostly school-aged children (1). B19V infection may cause arthropathies in adults and hydrops fetalis in pregnant women. In individuals with underlying immune or hematologic disorders, B19V may cause severe cytopenia, myocarditis, vasculitis, glomerulonephritis, or encephalitis (2). The 5.6-kb linear single-stranded DNA genome of B19V is encapsidated into a small nonenveloped icosahedral capsid and encodes three nonstructural proteins (NS1, 11 kDa and 7.5 kDa) and two capsid proteins (VP1 and VP2). The capsid consists of 60 structural subunits, of which approximately 95% are VP2 (58 kDa) and 5% are VP1 (83 kDa) (3). VP1 is identical to VP2 except for an additional N-terminal region of 227 amino acids, the “VP1 unique region” (VP1u). Although VP2 proteins are the main component of the capsid, VP1u is critical to elicit an efficient immune response (4). The N terminus of VP1u is rich in neutralizing epitopes (5, 6), denoting the critical role of this region in the infection process.
B19V has an exceptionally narrow tissue tropism almost exclusively infecting human erythroid progenitor cells in the bone marrow. During natural infection, B19V can replicate in cells from the erythroid lineage at the BFU-E and CFU-E stages of differentiation, which accounts for the hematological disorders observed during the infection (7). The neutral glycosphingolipid globoside (Gb4), also known as P antigen, is considered the primary cellular receptor of B19V (8). A large body of evidence suggests that B19V recognizes Gb4 and that its expression is important for infection. Gb4 is expressed in various types of cells, but it is particularly abundant in human erythroid progenitor cells in the bone marrow, which are also the natural host cells of the virus (7). B19V exhibits hemagglutinating activity, which can be inhibited by soluble or lipid-associated Gb4 (9, 10). Binding of B19V to Gb4 was demonstrated by thin-layer chromatography (8). Cryo-electron microscopy (cryoEM) image reconstructions suggested that B19V binds Gb4 in the depressions on the 3-fold axes of the capsid (11). Probably the most convincing finding suggesting the essential role of Gb4 in B19V infection is the fact that the rare individuals lacking Gb4 are not susceptible to the infection and, accordingly, have no detectable B19V antibodies (12). The reason for the resistance to B19V infection has been attributed to the lack of the primary receptor required for virus internalization. However, attempts to demonstrate the specific role of Gb4 as the primary receptor required for virus entry into permissive cells have not been undertaken.
The pathogenicity and extreme narrow tropism of B19V do not correlate with the wide-ranging Gb4 expression. It was shown that different expression levels of Gb4 in cells do not correlate with B19V binding to the cells and that its expression is required but not sufficient for productive infection (13). Attempts to demonstrate B19V binding to membrane-associated Gb4 in vitro failed. No binding signals above background controls were observed in sensitive assays employing fluorescence-labeled liposomes, radiolabeled B19 protein capsids, surface plasmon resonance, and isothermal titration microcalorimetry (10). In this study, cryoEM image reconstruction at high resolution also failed to confirm B19V binding to Gb4. In another study, binding of B19 virus-like particles (VLPs) to Gb4 in supported lipid bilayers was reported (14). These contradictory results may be explained by a complex interaction in which glycosphingolipid clustering, accessibility, and other plasma membrane molecules may influence the binding to Gb4. Besides Gb4, other glycosphingolipids have been shown to interact with B19V (15).
Although under certain conditions, the interaction of B19V with Gb4 seems undeniable, its role as the primary receptor required for virus entry remains uncertain. Despite Gb4 expression, some cell lines cannot be infected because the virus cannot be internalized, thus suggesting that other receptor molecules are critical for the uptake of the virus into susceptible cells. α5β1 integrin (16) and Ku80 autoantigen (17) have been proposed as potential coreceptors for B19V infection. However, the restricted uptake of B19V does not correspond with their expression profiles. In an earlier study, we showed that VP1u contains a receptor-binding domain (RBD), which mediates the uptake of the virus (18, 19). The receptor that binds the VP1u-RBD has not yet been identified, but its expression profile is far more restricted than that of Gb4, limiting B19V internalization and infection exclusively in cells at erythropoietin-dependent erythroid differentiation stages (20). Although VP1u is not accessible in native capsids, interaction with surface receptors in susceptible cells can render VP1u accessible (21, 22). This process could be partially reproduced by incubation of native capsids with soluble Gb4 (23).
Nevertheless, despite substantial efforts, the unequivocal interplay of B19V with Gb4 in the context of a capsid-receptor interaction required for virus entry has not yet been demonstrated. To clarify the role of Gb4 as the primary virus receptor, the B3GalNT1 gene, coding for globoside synthase, was knocked out. The loss of this enzyme, which catalyzes the transition of globotriaosylceramide (Gb3) to Gb4 (24), leads to the elimination of Gb4 and downstream glycosphingolipids. The B3GalNT1 knockout (KO) cell line was used to investigate the contribution of Gb4 to virus entry. The results revealed an unexpected essential role of Gb4 at a postentry step.
RESULTS
Generation of B3GalNT1 KO UT7/Epo cell line.
To determine the role of Gb4 in B19V infection, we sought to generate a UT7/Epo cell line devoid of Gb4. To this end, the B3GalNT1 gene, coding for globoside synthase, was knocked out. Globoside synthase is responsible for the biosynthesis of Gb4 from its precursor Gb3 (24). The knockout of the B3GalNT1 gene would abolish the synthesis of Gb4 and its downstream glycosphingolipids (Fig. 1A). The strategy of the knockout is depicted in Fig. 1B. UT7/Epo cells were cotransfected with two plasmids, one coding for Cas9 endonuclease and one of three genomic RNAs (gRNAs) targeting the B3GalNT1 gene, and a second plasmid containing homologous arms for homology-directed repair and a cassette containing red fluorescence protein (RFP) and a puromycin coding sequence. Fluorescence-activated cell sorting (FACS) was used to concentrate RFP-expressing cells by two bulk cell sortings before performing a final single-cell sort (Fig. 1C).
FIG 1.

Generation of B3GalNT1 KO UT7/Epo cell line. (A) Schematic depiction of the biosynthetic pathways of the (neo)lacto- and globo-series. Globoside (Gb4-ceramide) is synthesized out of Gb3-ceramide with the help of globoside synthase (B3GalNT1). (B) A set of plasmids was employed to disrupt the B3GalNT1 gene. The CRISPR/Cas9 knockout plasmid was used to generate double-strand breaks at the target site using Cas9 endonuclease. A gRNA specific for the B3GalNT1 gene was used as a guide for Cas9. The homology-directed repair (HDR) plasmid provided homologous 5′ and 3′ arms of the cleavage site and could be used as a template for the double-strand break, leading to the disruption of the gene and introducing a puromycin resistance and RFP marker. (C) Cells cotransfected with the two plasmids showed a yellow fluorescence from the GFP and RFP markers (3 days posttransfection). Cells were sorted in consecutive FACS (13, 42, and 62 days posttransfection) and a single-cell sort to select RFP-expressing cells.
Knockout cells lack B3GalNT1 transcripts, do not express Gb4, and proliferate normally.
The presence of B3GalNT1 transcripts was tested in the WT and transfected cells. Total poly(A) mRNA was isolated and used to detect B3GalNT1 mRNA by reverse transcription-PCR (RT-PCR). While in WT cells, an amplicon of the expected size was detected, no detectable signal was observed from two independent single cell-derived RFP-expressing clones. A nested RT-PCR allowed the detection of B3GalNT1 mRNA from 1% of WT cells. Despite the increased sensitivity, B3GalNT1 transcripts remained undetectable in the transfected cells (Fig. 2A). The expression of Gb4 was examined by confocal immunofluorescence microscopy with a specific antibody (25). Gb4 was abundantly expressed in WT cells; however, no specific signal was detectable in the transfected cells (Fig. 2B). A common feature of parvoviruses is their dependence on host cell factors present during cell replication. Hence, it was important to verify that the removal of B3GalNT1 did not alter the replication rate of the cells compared to the WT cells. The two cell types showed no visible morphological differences (Fig. 2C) and exhibited similar growth curves (Fig. 2D).
FIG 2.

B3GalNT1 KO UT7/Epo cells lack B3GalNT1 transcripts, do not express Gb4, and proliferate normally. (A) Detection of B3GalNT1 mRNA. Total mRNA was isolated from WT cells and from two single cell-derived RFP-expressing clones (KO1 and KO2) and used to detect B3GalNT1 transcripts by RT-qPCR. The amplicons were used in a nested PCR to ensure sufficient sensitivity. Dilutions (1% and 10%) of the WT amplicons were loaded as a reference. GAPDH mRNA was used as a loading control. (B) Detection of Gb4 by immunofluorescence. WT and KO cells were stained with anti-Gb4 antibody, fixed, and visualized by confocal microscopy. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). (C) Phase-contrast images of WT and KO cells showing no morphological differences. (D) Cell proliferation of WT and KO cells. Cells were incubated at 37°C and counted at the indicated days.
Gb4 is dispensable for B19V cell attachment and internalization.
WT and Gb4 KO UT7/Epo cells were used to compare B19V binding and internalization by confocal immunofluorescence microscopy. KG1a cells were used as controls. These cells derived from bone marrow acute myelogenous leukemia and lack the VP1u cognate receptor required for virus internalization (18). B19V was incubated with the cells for 1 h at 4°C. After several washing steps, the cells were fixed and stained with antibody 860-55D against intact capsids. As shown in Fig. 3A, B19V was able to bind WT and KO cells without noticeable differences. Binding to KG1a cells was less efficient. To verify the capacity of B19V to internalize, the cells were incubated at 37°C for 1 h, trypsinized to remove noninternalized viruses, fixed, and examined by confocal microscopy. Similar to the binding assay, no significant difference was observed in cells with or without Gb4. The virus internalized and displayed the characteristic endocytic distribution (Fig. 3B). As expected, B19V was unable to internalize KG1a cells, which lack the VP1u cognate receptor required for virus uptake. The capacity of B19V to bind and internalize WT and Gb4 KO cells was examined by quantitative PCR (qPCR). B19V was incubated with the cells for 1 h at 4°C for binding or at 37°C for internalization. Cells incubated at 37°C for 1 h were trypsinized to remove noninternalized virus. Cells incubated at 4°C served as controls (no internalization). Total DNA was extracted, and the amount of viral DNA was quantified by qPCR. Regardless of the presence or absence of Gb4, B19V was able to bind (Fig. 3C) and internalize (Fig. 3D) UT7/Epo cells without significant differences, confirming the results obtained by confocal immunofluorescence microscopy. Similar results were obtained from two independent single cell-derived knockout clones.
FIG 3.

Gb4 is dispensable for B19V cell attachment, internalization, and VP1u exposure. (A) Detection of B19V attachment by immunofluorescence. B19V was incubated with cells at 4°C for 1 h, followed by four washes with cold PBS. Cells were fixed, stained with antibody 860-55D against capsids, and visualized by confocal microscopy. (B) Detection of B19V internalization by immunofluorescence. B19V was incubated with cells at 37°C for 1 h, washed four times with PBS, and trypsinized to remove noninternalized viruses. Cells were fixed, stained with antibody 860-55D, and visualized by confocal microscopy. (C) Quantification of B19V attachment. B19V was incubated with cells at 4°C for 1 h, followed by four washes with cold PBS. The number of virions bound to the cells was quantified by PCR. (D) Quantification of B19V internalization. B19V was incubated with cells at 37°C for 1 h, washed four times with PBS, trypsinized to remove noninternalized viruses, and quantified by PCR. WT cells incubated at 4°C serve as negative controls (no internalization). (E) Quantification of VP1u exposure from free virus or bound to cells. Virions were immunoprecipitated with antibody 860-55D against capsids (total capsids) and a rabbit antibody against the PLA2 region (α-VP1u), followed by qPCR. Normal rabbit IgG was used as a negative control. P values were calculated according to Student’s t test. *, P < 0.05; **, P < 0.01; ns, no significant difference.
Gb4 is dispensable for VP1u externalization.
In previous studies, we showed that B19V VP1u is not accessible on the capsid surface but becomes accessible upon interaction with susceptible UT7/Epo cells and erythrocytes at 4°C (21, 22). This conformational change was partially reproduced upon incubation of B19V with soluble Gb4 (23). Later, we demonstrated that this rearrangement is essential to allow the interaction of VP1u with its cognate receptor required for virus internalization (18–20). Since B19V is able to internalize in KO cells, it can be assumed that receptor molecules other than Gb4 can mediate the conformational change. To test this hypothesis, B19V was incubated with the cells at 4°C to allow attachment. After several washing steps and cell lysis, the virions were immunoprecipitated with antibody 860-55D against capsids (total capsids) and an antibody against VP1u, followed by qPCR. While in native free virions VP1u was mostly not accessible, it became largely exposed following attachment to WT or to KO cells without significant differences (Fig. 3E). This result confirms that Gb4 is not required for VP1u exposure and explains the capacity of the virus to internalize KO cells.
The absence of Gb4 does not affect either the accumulation of the incoming B19V in the nuclear fraction or the externalization of the viral DNA.
To verify whether the internalized virus is able to follow the infectious route and traffic to the nucleus in the absence of Gb4, nuclei from infected cells were isolated and tested for the presence of B19V DNA. The integrity of the isolated nuclei was judged by light microscopy after trypan blue staining. The purity of the nuclear fraction and the absence of cytoplasmic contamination were verified using antibodies against lamin A/C (nuclear inner membrane marker), glyceraldehyde 3-phosphate dehydrogenase (GAPDH; cytosolic marker), and sarco/endoplasmic reticulum Ca2+-ATPase (SERCA; endoplasmic reticulum marker) (Fig. 4A). Nuclei isolated after 10 min of infection served as negative controls. Viral DNA was quantified from the purified nuclei by qPCR, and the results were normalized by the quantification of the β-actin gene. At 3 h postinfection (pi), similar amounts of viral DNA accumulated in the nuclear fraction from WT and KO cells (Fig. 4B). In a recent study, we showed that a large proportion of B19V particles accumulating in the nuclear fraction of the infected cells have the viral DNA accessible and therefore are sensitive to nuclease digestion (26). As shown in Fig. 4B, the amount of nuclease-sensitive virus was also similar between WT and KO cells. These results indicate that in the Gb4 KO cells, the internalized virus can reach the nuclear fraction and can externalize the DNA with the same efficiency as in wild-type cells.
FIG 4.

Nuclear targeting and viral DNA accessibility are not disturbed in Gb4 KO cells. (A) Absence of cytoplasmic contamination from two nuclear preparations (Nuc1 and Nuc2). Lamin A/C (nuclear inner membrane), SERCA2 ATPase (endoplasmic reticulum), and GAPDH (cytoplasmic proteins). WC, whole cells. (B) Nuclei from infected cells were isolated at 10 min and 3 h postinfection and were treated or not with DNase I. Total DNA was extracted and the viral DNA was quantified by PCR. *, P < 0.05; ***, P < 0.001; ns, no significant difference.
Gb4 is essential for NS1 expression.
In UT7/Epo cells, the expression of NS1 occurs early after nuclear entry (27) and is required for DNA replication and productive infection (28, 29). The presence of NS1 mRNA in WT and KO cells was tested at increasing times pi by an NS1-specific quantitative RT-PCR. While increasing amounts of NS1 transcripts were detected in the WT cells, no NS1 transcripts above the background level were detected in Gb4 KO cells (Fig. 5A). The presence of NS1 proteins was examined by Western blotting with a monoclonal antibody (MAb) against the NS1 protein (30). In WT cells, the NS1 protein was detectable from day 1 pi. However, in KO cells, NS1 proteins remained undetectable up to day 4 pi (Fig. 5B). The absence of NS1 expression in KO cells was further confirmed by immunofluorescence microscopy with the antibody against NS1 (Fig. 5C). Similar results were obtained from two independent single-cell-derived knockout clones.
FIG 5.
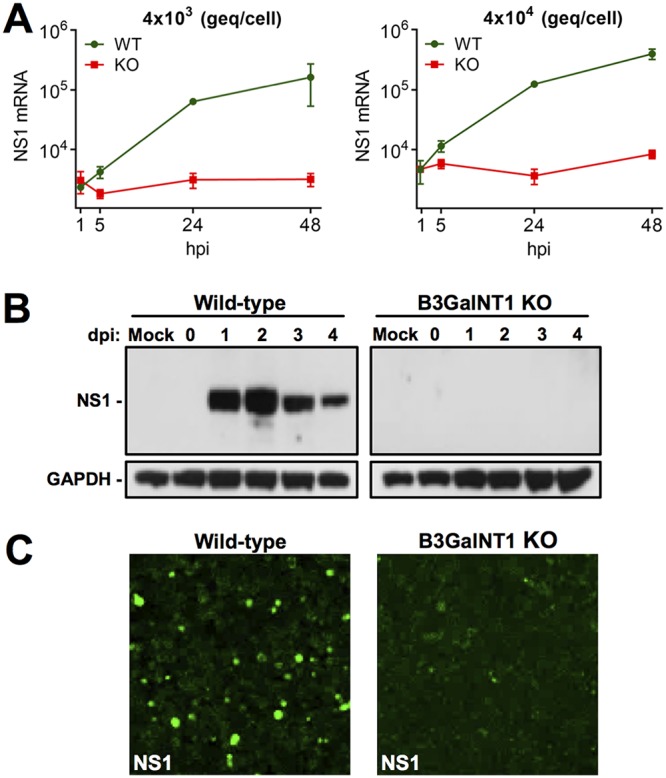
NS1 expression is blocked in Gb4 KO cells. (A) Detection of NS1 transcripts in WT and KO cells. Cells were infected with the indicated genome eqivalents per cell. Total mRNA was extracted at increasing hours postinfection (hpi), and the NS1 mRNA was quantified by RT-qPCR. (B) Detection of NS1 protein by Western blotting at increasing days postinfection (dpi). Cells were infected with 4 × 104 geq/cell. GAPDH was used as a loading control. Mock-infected samples were used as a negative control. (C) Detection of NS1 by immunofluorescence confocal microscopy. Cells were infected with a high dose of B19V (4 × 105 geq/cell). At 4 days postinfection, the cells were fixed and stained with a human NS1 antibody and a goat anti-human Alexa Fluor 488.
B19V DNA replication and capsid proteins are not detectable in Gb4 KO cells.
Following second-strand synthesis, NS1 is required to initiate and maintain virus replication through a rolling hairpin mechanism, which involves the resolution of the inverted terminal repeats (ITRs) (29). Accordingly, the lack of NS1 in KO cells should prohibit virus replication and subsequent steps of the infection, such as capsid protein expression. To verify the capacity of B19V to replicate, WT and KO cells were infected and at 1 h and 3 days pi, low-molecular-weight DNA was extracted, and B19V DNA species were examined by Southern blotting. Incoming single-stranded DNA (ssDNA) species were detected in both cell types. In infected WT cells, monomer and dimer replicative forms (mRF and dRF, respectively) were detected, confirming the existence of an active replication process. However, only a small amount of DNA with a molecular weight corresponding to mRF was observed in KO cells, probably corresponding to unprocessed covalently closed double-stranded DNA (dsDNA) or self-hybridized incoming ssDNA of opposite polarity (Fig. 6A). KpnI digestion confirmed the presence of viral dsDNA species in WT and KO cells (Fig. 6B).
FIG 6.

B19V DNA replication and capsid proteins are not detectable in Gb4 KO cells. (A) Southern blot analysis of low-molecular-weight DNA extracted from infected cells at the indicated times postinfection. Linearized pM20 infectious clone was used as a marker. dRF, dimer replicative form; mRF, monomer replicative form. (B) Southern blot analysis of low-molecular-weight DNA extracted from infected cells at 3 days postinfection and digested or not with KpnI. (C) Detection of VP1/VP2 transcripts in WT and KO cells. Cells were infected with the indicated genome equivalents per cell. mRNA was extracted at increasing hours postinfection (hpi), and the VP1/VP2 mRNA was quantified by RT-qPCR. (D) Detection of viral capsid proteins by Western blotting with antibody 3113-81C at increasing days postinfection (dpi). GAPDH was used as a loading control. (E) Detection of viral capsid proteins by immunofluorescence confocal microscopy. Cells were infected with a high dose of B19V (4 × 105 viruses per cell). At 4 days postinfection, the cells were fixed and stained with mouse antibody R9283 and goat anti-mouse Alexa Fluor 488.
The active replication of the viral DNA enhances readthrough of the proximal poly(A) and the polyadenylation of transcripts at the distal poly(A), which encode for the capsid proteins (31). Therefore, the absence of NS1 and virus replication in the KO cells should prohibit the generation of transcripts encoding structural proteins. As expected and regardless of the quantities of virions used, capsid protein mRNA was undetectable in KO cells (Fig. 6C). While capsid proteins accumulated at increasing days pi in WT cells, no detectable proteins were observed in KO cells (Fig. 6D). The lack of capsid protein expression was further confirmed by immunofluorescence microscopy with an antibody against viral structural proteins (Fig. 6E).
Transfection with a B19V infectious clone can circumvent the Gb4 block.
Transfection with a B19V infectious clone was performed to evaluate the permissiveness of KO cells to B19V infection when the full-length dsDNA with resolved ITRs is directly transferred to the nucleus by nucleofection. The B19V infectious clone was digested with SalI to release the full-length genomes and used to transfect WT and KO cells using a Nucleofector device. As shown in Fig. 7A and B, similar amounts of NS1 and VP1/2 mRNAs were detected in the two cell types. At 4 days posttransfection, the cells were examined by immunofluorescence microscopy with an antibody against viral capsid proteins. Although the efficiency of transfection is typically low in UT7/Epo cells, similar amounts of positively transfected cells were observed in the two cell types (Fig. 7C). This result demonstrates that Gb4 is dispensable for B19V infection when viral dsDNA with resolved ITRs are already present in the nucleus.
FIG 7.
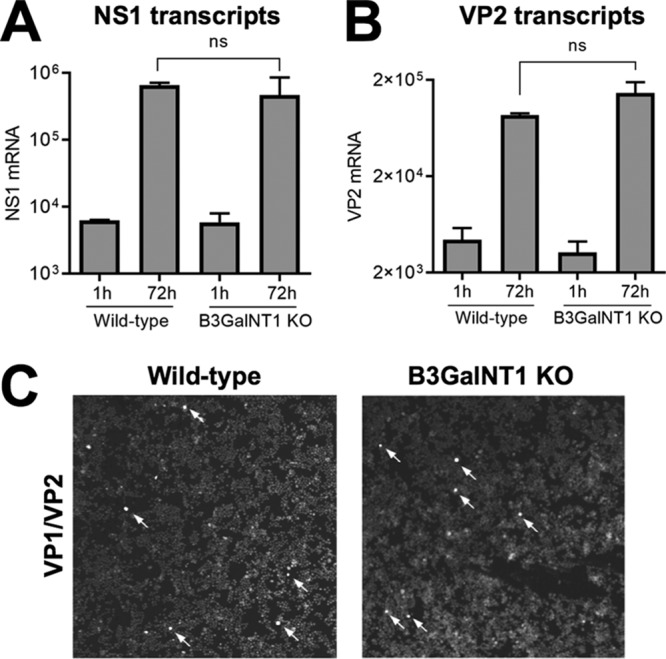
Transfection with a B19V infectious clone can circumvent the Gb4 block. Transfection of WT and KO cells with pM20. (A and B) mRNA was extracted 1 h or 72 h posttransfection and quantified by RT-qPCR with primers for NS1 mRNA (A) or VP1/VP2 mRNA (B). (C) At 3 days posttransfection, cells were fixed and stained with antibody R9283 against viral capsid proteins. Arrows indicate cells with positive staining. ns, no significant difference.
Interaction of B19V with the VP1u cognate receptor but not with Gb4 is essential for virus internalization.
Although B19V does not require Gb4 for cell attachment, uptake, and nuclear targeting, a transient interaction with Gb4 at the plasma membrane may still be important for the infection at a postentry step. In order to test this hypothesis, WT cells were incubated with the antibody against Gb4, and subsequently, B19V was added at 4°C to allow virus attachment. In parallel, cells were preincubated with a ΔC128 recombinant VP1u (recVP1u) construct, to block the interaction of B19V with the VP1u cognate receptor, which is required for virus uptake (18). ΔC128 lacks the C-terminal 128 amino acids of VP1u and has an intact receptor-binding domain (RBD). As a control, cells were incubated with a truncated ΔN29 recVP1u construct. This construct lacks the N-terminal 29 amino acids of VP1u, which disable the RBD. The expression and purification of the VP1u constructs have been described elsewhere (18). While the antibody against Gb4 had no effect on virus binding (Fig. 8A), approximately 50% binding inhibition was observed in the presence of the ΔC128 recVP1u (Fig. 8B). Similar cell-binding inhibition was observed when the cells were incubated with ΔC128 recVP1u and the Gb4 antibody together (Fig. 8C). Cells incubated with the truncated ΔN29 recVP1u or normal chicken IgY had no effect on virus binding.
FIG 8.

Effect of recVP1u and Gb4 antibody in virus binding and NS1 expression in wild-type cells. (A to C) Quantification of B19V binding to wild-type cells in the presence of chicken anti-Gb4 (A), recVP1u (ΔC128 mutant; 70 ng) (B), or both (C). (D and E) NS1 mRNA expression was quantified 24 h postinfection in the presence of anti-Gb4 (D) or recVP1u (ΔC128 mutant; 70 ng) (E). NS1 mRNA was quantified after 1 h as background control. Normal chicken IgY and truncated recVP1 (ΔN29 mutant; 70 ng), lacking the receptor-binding domain, were used as controls. *, P < 0.05; ns, no significant difference.
We next analyzed the effect of the Gb4 antibody and the ΔC128 recVP1u on B19V NS1 expression. The presence of the antibody did not affect NS1 expression (Fig. 8D). In sharp contrast, in the presence of the ΔC128 recVP1u, NS1 expression was completely inhibited. As a control, cells incubated with the truncated ΔN29 recVP1u or normal chicken IgY had no effect on the infection (Fig. 8E).
B19V selectively infects WT cells in a mixed population of WT and KO cells.
In earlier studies, we showed that not all B19V bound to UT7/Epo cells at 4°C can internalize the cells after raising the temperature to 37°C. Instead, some viruses detach from the cells. The detached viruses have VP1u exposed, remain infectious, and are able to bind cells more efficiently than are viruses exposed for the first time to the cells (23). The numbers of bound viruses becoming detached from WT or KO cells were similar (Fig. 9A). We then sought to test whether the virus detached from the Gb4-expressing cells gains the ability to infect cells without Gb4. To this end, WT and KO cells were mixed (1:1) and incubated with a high dose of B19V (4 × 105 genome equivalents [geq]/cell) at 37°C. This approach was also intended to explore a possible complementing effect of Gb4 via juxtacrine signaling. As shown in Fig. 9B, a large proportion of WT cells became infected, as judged by the expression of viral capsid proteins. However, none of the KO cells, which are recognized by the expression of red fluorescent protein, were productively infected. The fact that viruses exposed to Gb4-expressing cells remain unable to infect KO cells further confirms that the interaction with Gb4 at the plasma membrane, if occurring, is not critical for infection.
FIG 9.

B19V selectively infects WT cells in a mixed population of WT and KO cells. (A) Detachment of B19V from WT and KO cells. B19V was incubated with the cells at 4°C. Following five washes to remove unbound virus, the cells were transferred to 37°C to allow internalization and detachment. At 0 min (background control) and 30 min, the cells were pelleted. The numbers of viruses bound to the cells and in the supernatant (detached) were quantified by PCR. (B) B19V infection in a mixed population of UT7/Epo WT and KO cells. WT and KO cells were mixed (1:1) and infected with a high dose of B19V (4 × 105 viruses per cell). At 4 days postinfection, the cells were washed, fixed in 4% formaldehyde to preserve the RFP signal, and stained with a mouse antibody R9283 and a goat anti-mouse Alexa Fluor 488. KO cells are recognized by the expression of RFP. ns, no significant difference.
DISCUSSION
The cell receptor of a virus plays a central role in the infection, as it represents a major determinant of the host range, tissue tropism, and viral pathogenesis. The glycosphingolipid globoside or P antigen (Gb4) has long been considered the primary cell receptor of B19V (8). Individuals with a rare mutation in the B3GalNT1 gene do not express Gb4 and are naturally resistant to the infection (12). However, the role of Gb4 as a B19V receptor remains controversial, as its expression does not correlate well with virus attachment, internalization, or the remarkable narrow tissue tropism of B19V. To clarify the role of Gb4 and to explore its function as the primary receptor of B19V, the B3GalNT1 gene, coding for globoside synthase, was knocked out in UT7/Epo cells, leading to the elimination of Gb4 and downstream glycosphingolipids (24). The glycosphingolipid PX2, which is also generated by globoside synthase (32), would also be lost in the B3GalNT1 KO cells. Systematic and quantitative analyses of the progression of B19V infection in WT and KO cells revealed that Gb4 does not have the expected function as a cell surface receptor required for B19V entry. Instead, Gb4 has an essential role at a postentry step for the expression of the incoming viral genomes and productive infection.
Although Gb4 is dispensable for virus entry, the interaction of B19V with Gb4 cannot be excluded. There is solid experimental evidence that the interaction may occur under certain conditions. For example, the interaction should be possible on the surface of erythrocytes since soluble Gb4 can inhibit B19V-mediated hemagglutination (9, 10). In an earlier study, we observed that incubation of B19V with erythrocytes or with UT7/Epo cells triggered conformational changes in the capsid leading to VP1u externalization (21, 22). This conformational change could be partially mimicked by incubation of capsids with soluble Gb4 (23). Since VP1u also became exposed following attachment to KO cells (Fig. 3E), other structures besides Gb4 should be recognized by B19V at the plasma membrane, which can trigger VP1u exposure. In this respect, it has been suggested that B19V recognizes several glycosphingolipids (15). A stable interaction of B19V with Gb4 could not be demonstrated in a systematic study using various sensitive assays (10). In another study, the interaction of B19 virus-like particles with Gb4 was documented (14). These apparently contradictory results may reflect the existence of an interaction exclusively under specific conditions, such as receptor clustering, the surrounding molecular environment, or structural conformations. However, the fact that B19V may recognize Gb4 under certain conditions should not imply that the interaction occurs at the plasma membrane of host cells and in the context of a virus-receptor interaction required for entry.
Glycosphingolipids are mainly associated with the plasma membrane; however, they are also found in intracellular compartments. Gb4 has been found associated with secretory granules (33) and intermediate filaments (34, 35). Accordingly, the relevant interaction with Gb4 may not occur at the plasma membrane but after virus entry. In line with this concept, we could not block virus attachment and the infection by targeting plasma membrane Gb4 with specific antibodies. The specificity of the Gb4 antibody used in this study has been thoroughly documented in previous publications (25) and further confirmed in our experiments where no signal was visible in KO cells (Fig. 2B). In sharp contrast to the Gb4 antibody, masking the VP1u cognate receptor with recVP1u disturbed the binding of the virus and completely abrogated the infection (Fig. 8). Furthermore, in a cell mixture containing equal amounts of WT and KO cells, B19V infected exclusively the cells expressing Gb4 (Fig. 9). Of note, Gb4 is a relatively small molecule, and its accessibility at the plasma membrane can be compromised by surrounding molecules. Pretreatment of cells with pronase or neuraminidase has been shown to significantly increase the accessibility of the cryptic Gb4 to antibodies (33).
Although B19V capsids may interact with Gb4 intracellularly, the essential role of Gb4 may not be related to direct physical interaction with incoming B19V capsids. Instead, the glycosphingolipid may provide a permissive intracellular environment for B19V replication. Erythropoietin (EPO) signaling is not required for virus entry and nuclear targeting, but it is essential for virus DNA replication (36). Similar to EPO, Gb4 may also promote signal transduction required for B19V infection. Clustered glycosphingolipids at the cell surface membrane interact with functional membrane proteins, such as integrins, growth factor receptors, and tetraspanins. Gb4 is involved in a variety of cellular processes, including cell adhesion, growth, motility, and cell differentiation (37, 38). Gb4 was found to promote activation of extracellular signal-related kinase (ERK), to induce the enhanced activity of specific transcription factors, and to accelerate cell differentiation (39–41). Accordingly, Gb4 may provide the required intracellular conditions for the expression of the incoming genomes.
Our data suggest that Gb4 is essential at a postentry step between nuclear entry and before the generation of dsDNA with resolved ITRs. Accumulation of the incoming capsids in the nuclear fraction and the accessibility of their DNA were similar in WT and KO cells, suggesting that Gb4 is not required for these steps. However, it cannot be excluded that Gb4 is required for the translocation of the perinuclear cytosolic capsids into the nucleus. Following virus entry into the host nucleus, the incoming ssDNA genome is converted to the double-stranded monomer replicative form (mRF), which serves as the template for virus replication and transcription. In KO cells, dsDNA corresponding in size to mRF was barely detectable in KO cells, and the dimer RF (dRF) typically observed during active DNA replication was not observed (Fig. 6A and B). The presence of viral dsDNA in KO cells suggests that Gb4 is not essential for second-strand synthesis from the incoming ssDNA genomes. However, in the absence of Gb4, the mRF remains transcriptionally inactive (Fig. 6C to E). Without NS1, the viral DNA cannot be processed further at the terminal resolution sites to initiate active DNA replication by the rolling hairpin mechanism, explaining the lack of virus replication and protein expression in the KO cells. It cannot be excluded, however, that all or a proportion of dsDNA species observed in KO cells originate from hybridized incoming ssDNA of opposite polarity. Introduction of full-length viral dsDNA with resolved ITRs by transfection can circumvent the block (Fig. 7), suggesting that Gb4 is required at a postentry step between nuclear entry and the generation of full-length viral dsDNA.
In summary, this study reveals that Gb4 does not have the expected receptor function in B19V infection, as it is dispensable for virus entry and trafficking. However, Gb4 is required at a postentry step for productive infection, either through direct interaction with incoming viruses or by means of signal transduction. The failure of B19V to infect productively Gb4 KO cells, even at a very high multiplicity of infection or in the presence of WT cells, explains the natural resistance of the rare individuals lacking Gb4 to B19V infection. Further studies will aim to investigate the postentry step that is defective in Gb4 KO cells, and the information obtained will provide novel opportunities for the development of selective antiviral therapies.
MATERIALS AND METHODS
Cells and viruses.
The human megakaryoblastoid UT7/Epo cells were maintained in Eagle’s minimal essential medium (MEM) containing 5% fetal calf serum (FCS) and 2 U/ml recombinant erythropoietin (EPO). The KG1a human erythroleukemia cells were cultured in Iscove’s modified Dulbecco’s medium (IMDM) containing 10% FCS. B19V-infected plasma (4 × 109 genome equivalents [geq]/μl) was obtained from a donation center (CSL Behring AG, Charlotte, NC). The plasma IgGs were removed by using HiTrap protein G high-performance (HP) columns (GE Healthcare, Chicago, IL).
Antibodies.
The human monoclonal antibody (MAb) 860-55D was purchased from Mikrogen (Neuried, Germany). The antibody recognizes a conformational epitope and does not react with disassembled capsid proteins (30). The human MAb against NS1 (24) was kindly provided by G. Gallinella. The mouse antibodies 3113-81C (United States Biological, Boston, MA) and R9283 (Merck, Burlington, MA) were used in Western blotting and immunofluorescence analyses, respectively. The antibody against the viral PLA2 region was obtained as previously described (22). The polyclonal chicken anti-Gb4 IgY antibody JM07/164-4 was kindly provided by J. Müthing. The preparation and specificity of the antibody against Gb4 have been reported in previous publications (25). Rabbit anti-GAPDH and mouse anti-SERCA were purchased from Abcam (Cambridge, MA). Rabbit anti-lamin A/C was purchased from Cell Signaling Technologies (Danvers, MA).
Generation of B3GalNT1 knockout UT7/Epo cells.
One day prior to transfection, 2 × 106 UT7/Epo cells were seeded in a 6-well plate with 3 ml MEM containing 5% FCS and 2 U/ml EPO. A plasmid to generate a double-strand break in the target gene (β-1,3-Gal-T3 CRISPR/Cas9 KO green fluorescence protein [GFP]) and a plasmid for homology-directed repair (β-1,3-Gal-T3 HDR RFP) (Santa Cruz Biotechnology, TX) were used for transfection using Lipofectamine 3000 (Invitrogen, CA), according to the manufacturer’s instructions. Doubly transfected cells were sorted by FACS (FACSAria; BD Biosciences, NJ). Serial bulk cell sorting was performed to isolate and concentrate RFP-expressing cells, followed by a final single-cell sorting.
Analysis of B3GalNT1 KO cells.
mRNA was isolated using the Dynabeads mRNA Direct kit (Invitrogen, CA), according to the manufacturer’s instructions. For detection of the B3GalNT1 mRNA, the Luna Universal one-step reverse transcription-quantitative PCR (RT-qPCR) kit (New England BioLabs, Ipswich, MA) was used. The forward primer was 5′-CTCCTGAGTTTCTTTGTGATGTGG-3′, and the reverse primer was 5′-CATTACGTACTTGGCATTGGGG-3′. The nested-PCR forward primer was 5′-CCCCACTACAATGTGATAGAACGC-3′, and the reverse primer was 5′-GGCAAAACTCAGTTACCCACC-3′. For the detection of Gb4, cells were incubated in phosphate-buffered saline (PBS) containing 2% bovine serum albumin at 4°C for 20 min. Subsequently, the cells were resuspended in 50 μl PBS containing 2% goat serum and incubated with the chicken antibody against Gb4 (0.5 μl) at 4°C for 1 h. After several washing steps with ice-cold PBS, the cells were fixed with methanol-acetone (1:1) at –20°C for 4 min, blocked with 10% goat serum in PBS, and incubated with a polyclonal antibody against chicken IgY Alexa Fluor 594 (Abcam). The proliferation rates of wild-type and transfected cells were examined. For each measurement, 105 cells were harvested, resuspended in fresh medium, and seeded in a 12-well plate. At different time points, cells were counted using a Moxi Z automated cell counter (Orflo Technologies, Ketchum, ID).
Virus binding and internalization.
Cells (3 × 105) were resuspended in 50 μl MEM without FCS. For binding, B19V was added (104 virions per cell) and incubated at 4°C for 1 h. For internalization, the samples were incubated at 37°C for 1 h, washed 4 times with PBS at 4°C, and trypsinized at 37°C for 4 min to remove noninternalized viruses. Subsequently, the samples were examined by confocal immunofluorescence microscopy and by qPCR. Cells were fixed with methanol-acetone (1:1) at –20°C for 4 min, blocked with 10% goat serum in PBS, and incubated with antibody 860-55D against capsids. Anti-human IgG Alexa Fluor 488 (Invitrogen, Carlsbad, CA) was used as the secondary antibody. For qPCR, DNA was extracted using the DNeasy blood and tissue kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions, and used for qPCR using iTaq polymerase (iTaq Universal SYBR green supermix; Bio-Rad, CA) with the following primers: forward, 5′-GGGCAGCCATTTTAAGTGTTT-3′; and reverse, 5′-CCAGGAAAAAGCAGCCCAG-3′.
Nuclear targeting.
Cells (6 × 105) were resuspended in 100 μl MEM and incubated with B19V (104 virions per cell) at 4°C for 1 h to allow virus binding. After three washes with PBS, the cells were resuspended in 500 μl MEM containing 5% FCS and 2 U/ml EPO and incubated at 37°C. After 3 h, cells were pelleted and washed thrice with PBS, and the nuclei were extracted using the Nuclei EZ Prep kit (Sigma-Aldrich, MO), as previously described (26). Trypan blue staining was used to evaluate the integrity of the isolated nuclei. The absence of cytoplasmic contamination was examined with antibodies against lamin A/C (nuclear inner membrane), SERCA (endoplasmic reticulum), and GAPDH (cytoplasmic proteins). Total DNA was extracted from the isolated nuclei, and B19V DNA was quantified as specified above.
NS1 and capsid protein expression.
The expression of the incoming viral genome in infected WT and KO cells was examined. NS1 and VP1/VP2 mRNA were detected by quantitative RT-PCR and proteins by Western blotting and immunofluorescence. UT7/Epo WT and KO cells were incubated with B19V for up to 4 days. The cells were washed four times with PBS, and mRNA was extracted as described above. For the detection of NS1 mRNA, the following primers were used: forward primer, 5′-GGGGCAGCATGTGTTAA-3′; and reverse primer, 5′-AGTGTTCCAGTATATGGCATGG-3′. For the detection of VP1/VP2 mRNA, the following primers were used: forward primer, 5′-CATGCACACCTACTTTCCCAA-3′; and reverse primer, 5′-GGAGGATGGGGTTTGCATCA-3′. The presence of NS1 and VP1/VP2 proteins was analyzed by Western blotting and by immunofluorescence using specific antibodies. For immunofluorescence, cells were fixed with methanol-acetone (1:1), blocked with 10% goat serum in PBS, and incubated with the antibody against NS1 or viral structural proteins. Secondary antibodies with conjugated Alexa Fluor 488 (Invitrogen) were used against the primary antibodies. The samples were visualized using confocal microscopy (Zeiss LSM 880).
Southern blot.
Cells were infected with B19V (4 × 104 virions per cell), harvested at 1 h or 3 days postinfection, washed with PBS, resuspended in 50 μl Tris-buffered saline (TBS), and lysed with 750 μl lysis buffer (10 mM Tris [pH 8], 10 mM EDTA, 0.6% SDS, and 200 μg proteinase K per ml) at room temperature (RT) for 1 h. NaCl (200 μl, 1 M final concentration) was added and incubated overnight at 4°C. The samples were then spun at 16,000 × g for 30 min to pellet precipitated chromosomal DNA. Total DNA was extracted from the supernatant and quantified as specified above. The viral DNA species were separated on a 0.8% agarose gel, depurinated, denatured, and neutralized before being transferred onto a positively charged nylon membrane (Amersham Hybond-N+; GE Healthcare) by capillary transfer using 20× saline-sodium citrate (SSC) buffer. The DNA was fixed to the membrane by baking. The membrane was incubated for 2 h in hybridization buffer (7% [wt/vol] SDS, 0.125 M sodium phosphate buffer [pH 7.2], 0.25 M NaCl, 1 mM EDTA, 45% [vol/vol] formamide) at 42°C. DNA was detected by overnight hybridization with a 32P-labeled probe of 944 nucleotides (nt) (B19V J35 isolate, nt 773 to 1716). The membrane was washed four times at 42°C with 2× SSC and 0.1% SDS for 5 min and twice with 0.1× SSC and 0.1% SDS for 30 min before detection with a phosphorimager (Typhoon FLA 9500; GE Healthcare).
Transfection.
The B19V infectious clone pM20 (0.5 μg) (42) was digested with SalI to release the full-length genome from the plasmid backbone. The linearized genome was transfected into the cells (105) using Lipofectamine 3000 reagent (Invitrogen), as described above. B19V NS1 and capsid protein mRNAs were examined 3 days posttransfection by RT-qPCR, and capsid proteins were analyzed by immunofluorescence microscopy, as specified above.
Quantification of VP1u exposure from free and cell-bound virus.
Virus binding to UT7/Epo WT or KO cells was carried out at 4°C (2 × 104 viruses per cell) for 1 h. Cells were washed 4× with 1% bovine serum albumin in PBS (PBSA 1%) and once with PBS. Cells were lysed in lysis buffer (1% NP40, 150 mM NaCl, 50 mM Tris-HCl [pH 8], 5 mM EDTA), and the cell debris was removed by centrifugation at 10,000 × g for 10 min at 4°C. The supernatant was transferred to a new tube and incubated with antibody 860-55D against the viral capsid (total capsids) or with an antibody against the PLA2-region (α-VP1u) at 4°C for 1 h. Protein G Plus-agarose (20 μl; Santa Cruz Biotechnology) was added and incubated overnight at 4°C. The beads were washed 4× with PBSA 1% and once with PBS, followed by DNA extraction and qPCR.
ACKNOWLEDGMENTS
This study was supported by a grant from the Swiss National Science Foundation (SNSF grant 31003A_179384 to J.B.).
We are grateful to J. Müthing (University of Münster, Germany) for providing the antibody against globoside and to G. Gallinella (University of Bologna, Italy) for providing the antibody against NS1.
REFERENCES
- 1.Young NS, Brown KE. 2004. Parvovirus B19. N Engl J Med 350:586–597. doi: 10.1056/NEJMra030840. [DOI] [PubMed] [Google Scholar]
- 2.Servey JT, Reamy BV, Hodge J. 2007. Clinical presentations of parvovirus B19 infection. Am Fam Physician 75:373–376. [PubMed] [Google Scholar]
- 3.Cotmore SF, McKie VC, Anderson LJ, Astell CR, Tattersall P. 1986. Identification of the major structural and nonstructural proteins encoded by human parvovirus B19 and mapping of their genes by prokaryotic expression of isolated genomic fragments. J Virol 60:548–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zuffi E, Manaresi E, Gallinella G, Gentilomi GA, Venturoli S, Zerbini M, Musiani M. 2001. Identification of an immunodominant peptide in the parvovirus B19 VP1 unique region able to elicit a long-lasting immune response in humans. Viral Immunol 14:151–158. doi: 10.1089/088282401750234529. [DOI] [PubMed] [Google Scholar]
- 5.Anderson S, Momoeda M, Kawase M, Kajigaya S, Young NS. 1995. Peptides derived from the unique region of B19 parvovirus minor capsid protein elicit neutralizing antibodies in rabbits. Virology 206:626–632. doi: 10.1016/S0042-6822(95)80079-4. [DOI] [PubMed] [Google Scholar]
- 6.Saikawa T, Anderson S, Momoeda M, Kajigaya S, Young NS. 1993. Neutralizing linear epitopes of B19 parvovirus cluster in the VP1 unique and VP1-VP2 junction regions. J Virol 67:3004–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi T, Ozawa K, Takahashi K, Asano S, Takaku F. 1990. Susceptibility of human erythropoietic cells to B19 parvovirus in vitro increases with differentiation. Blood 75:603–610. [PubMed] [Google Scholar]
- 8.Brown KE, Anderson SM, Young NS. 1993. Erythrocyte P antigen: cellular receptor for B19 parvovirus. Science 262:114–117. doi: 10.1126/science.8211117. [DOI] [PubMed] [Google Scholar]
- 9.Brown KE, Cohen BJ. 1992. Haemagglutination by parvovirus B19. J Gen Virol 73:2147–2149. doi: 10.1099/0022-1317-73-8-2147. [DOI] [PubMed] [Google Scholar]
- 10.Kaufmann B, Baxa U, Chipman PR, Rossmann MG, Modrow S, Seckler R. 2005. Parvovirus B19 does not bind to membrane-associated globoside in vitro. Virology 332:189–198. doi: 10.1016/j.virol.2004.11.037. [DOI] [PubMed] [Google Scholar]
- 11.Chipman PR, Agbandje-McKenna M, Kajigaya S, Brown KE, Young NS, Baker TS, Rossmann MG. 1996. Cryo-electron microscopy studies of empty capsids of human parvovirus B19 complexed with its cellular receptor. Proc Natl Acad Sci U S A 93:7502–7506. doi: 10.1073/pnas.93.15.7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown KE, Hibbs JR, Gallinella G, Anderson SM, Lehman ED, McCarthy P, Young NS. 1994. Resistance to parvovirus B19 infection due to lack of virus receptor (erythrocyte P antigen). N Engl J Med 330:1192–1196. doi: 10.1056/NEJM199404283301704. [DOI] [PubMed] [Google Scholar]
- 13.Weigel-Kelley KA, Yoder MC, Srivastava A. 2001. Recombinant human parvovirus B19 vectors: erythrocyte P antigen is necessary but not sufficient for successful transduction of human hematopoietic cells. J Virol 75:4110–4116. doi: 10.1128/JVI.75.9.4110-4116.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nasir W, Nilsson J, Olofsson S, Bally M, Rydell GE. 2014. Parvovirus B19 VLP recognizes globoside in supported lipid bilayers. Virology 456-457:364–369. doi: 10.1016/j.virol.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 15.Cooling LL, Koerner TA, Naides SJ. 1995. Multiple glycosphingolipids determine the tissue tropism of parvovirus B19. J Infect Dis 172:1198–1205. doi: 10.1093/infdis/172.5.1198. [DOI] [PubMed] [Google Scholar]
- 16.Weigel-Kelley KA, Yoder MC, Srivastava A. 2003. Alpha5betaI integrin as a cellular coreceptor for human parvovirus B19: requirement of functional activation of beta1 integrin for viral entry. Blood 102:3927–3933. doi: 10.1182/blood-2003-05-1522. [DOI] [PubMed] [Google Scholar]
- 17.Munakata Y, Saito-Ito T, Kumura-Ishii K, Huang J, Kodera T, Ishii T, Hirabayashi Y, Koyanagi Y, Sasaki T. 2005. Ku80 autoantigen as a cellular coreceptor for human parvovirus B19 infection. Blood 106:3449–3456. doi: 10.1182/blood-2005-02-0536. [DOI] [PubMed] [Google Scholar]
- 18.Leisi R, Ruprecht N, Kempf C, Ros C. 2013. Parvovirus B19 uptake is a highly selective process controlled by VP1u, a novel determinant of viral tropism. J Virol 87:13161–13167. doi: 10.1128/JVI.02548-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leisi R, Di Tommaso C, Kempf C, Ros C. 2016. The receptor-binding domain in the VP1u region of parvovirus B19. Viruses 8:61. doi: 10.3390/v8030061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leisi R, Von Nordheim M, Ros C, Kempf C. 2016. The VP1u receptor restricts parvovirus B19 uptake to permissive erythroid cells. Viruses 8:265. doi: 10.3390/v8100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ros C, Gerber M, Kempf C. 2006. Conformational changes in the VP1-unique region of native human parvovirus B19 lead to exposure of internal sequences that play a role in virus neutralization and infectivity. J Virol 80:12017–12024. doi: 10.1128/JVI.01435-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bönsch C, Kempf C, Ros C. 2008. Interaction of parvovirus B19 with human erythrocytes alters virus structure and cell membrane integrity. J Virol 82:11784–11791. doi: 10.1128/JVI.01399-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bönsch C, Zuercher C, Lieby P, Kempf C, Ros C. 2010. The globoside receptor triggers structural changes in the B19 virus capsid that facilitate virus internalization. J Virol 84:11737–11746. doi: 10.1128/JVI.01143-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chien JL, Williams T, Basu S. 1973. Biosynthesis of a globoside-type glycosphingolipid by a -N-acetylgalactosaminyltransferase from embryonic chicken brain. J Biol Chem 248:1778–1785. [PubMed] [Google Scholar]
- 25.Legros N, Dusny S, Humpf HU, Pohlentz G, Karch H, Müthing J. 2017. Shiga toxin glycosphingolipid receptors and their lipid membrane ensemble in primary human blood-brain barrier endothelial cells. Glycobiology 27:99–109. doi: 10.1093/glycob/cww090. [DOI] [PubMed] [Google Scholar]
- 26.Caliaro O, Marti A, Ruprecht N, Leisi R, Subramanian S, Hafenstein S, Ros C. 2019. Parvovirus B19 uncoating occurs in the cytoplasm without capsid disassembly and it is facilitated by depletion of capsid-associated divalent cations. Viruses 11:430. doi: 10.3390/v11050430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolfisberg R, Ruprecht N, Kempf C, Ros C. 2013. Impaired genome encapsidation restricts the in vitro propagation of human parvovirus B19. J Virol Methods 193:215–225. doi: 10.1016/j.jviromet.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 28.Zhi N, Mills IP, Lu J, Wong S, Filippone C, Brown KE. 2006. Molecular and functional analyses of a human parvovirus B19 infectious clone demonstrates essential roles for NS1, VP1, and the 11-kilodalton protein in virus replication and infectivity. J Virol 80:5941–5950. doi: 10.1128/JVI.02430-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ganaie SS, Qiu J. 2018. Recent advances in replication and infection of human parvovirus B19. Front Cell Infect Microbiol 8:166. doi: 10.3389/fcimb.2018.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gigler A, Dorsch S, Hemauer A, Williams C, Kim S, Young NS, Zolla-Pazner S, Wolf H, Gorny MK, Modrow S. 1999. Generation of neutralizing human monoclonal antibodies against parvovirus B19 proteins. J Virol 73:1974–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guan W, Cheng F, Yoto Y, Kleiboeker S, Wong S, Zhi N, Pintel DJ, Qiu J. 2008. Block to the production of full-length B19 virus transcripts by internal polyadenylation is overcome by replication of the viral genome. J Virol 82:9951–9963. doi: 10.1128/JVI.01162-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Westman JS, Benktander J, Storry JR, Peyrard T, Hult AK, Hellberg Å, Teneberg S, Olsson ML. 2015. Identification of the molecular and genetic basis of PX2, a glycosphingolipid blood group antigen lacking on globoside-deficient erythrocytes. J Biol Chem 290:18505–18518. doi: 10.1074/jbc.M115.655308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katz HR, Austen KF. 1986. Plasma membrane and intracellular expression of globotetraosylceramide (globoside) in mouse bone marrow-derived mast cells. J Immunol 136:3819–3824. [PubMed] [Google Scholar]
- 34.Gillard BK, Heath JP, Thurmon LT, Marcus DM. 1991. Association of glycosphingolipids with intermediate filaments of human umbilical vein endothelial cells. Exp Cell Res 192:433–444. doi: 10.1016/0014-4827(91)90062-y. [DOI] [PubMed] [Google Scholar]
- 35.Gillard BK, Thurmon LT, Marcus DM. 1992. Association of glycosphingolipids with intermediate filaments of mesenchymal, epithelial, glial, and muscle cells. Cell Motil Cytoskeleton 21:255–271. doi: 10.1002/cm.970210402. [DOI] [PubMed] [Google Scholar]
- 36.Chen AY, Guan W, Lou S, Liu Z, Kleiboeker S, Qiu J. 2010. Role of erythropoietin receptor signaling in parvovirus B19 replication in human erythroid progenitor cells. J Virol 84:12385–12396. doi: 10.1128/JVI.01229-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kojima N, Hakomori S. 1991. Synergistic effect of two cell recognition systems: glycosphingolipid-glycosphingolipid interaction and integrin receptor interaction with pericellular matrix protein. Glycobiology 1:623–630. doi: 10.1093/glycob/1.6.623. [DOI] [PubMed] [Google Scholar]
- 38.D’Angelo G, Capasso S, Sticco L, Russo D. 2013. Glycosphingolipids: synthesis and functions. FEBS J 280:6338–6353. doi: 10.1111/febs.12559. [DOI] [PubMed] [Google Scholar]
- 39.Song Y, Withers DA, Hakomori S. 1998. Globoside-dependent adhesion of human embryonal carcinoma cells, based on carbohydrate-carbohydrate interaction, initiates signal transduction and induces enhanced activity of transcription factors AP1 and CREB. J Biol Chem 273:2517–2525. doi: 10.1074/jbc.273.5.2517. [DOI] [PubMed] [Google Scholar]
- 40.Park SY, Kwak CY, Shayman JA, Kim JH. 2012. Globoside promotes activation of ERK by interaction with the epidermal growth factor receptor. Biochim Biophys Acta 1820:1141–1148. doi: 10.1016/j.bbagen.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakamura T, Chiba Y, Naruse M, Saito K, Harada H, Fukumoto S. 2016. Globoside accelerates the differentiation of dental epithelial cells into ameloblasts. Int J Oral Sci 8:205–212. doi: 10.1038/ijos.2016.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhi N, Zádori Z, Brown KE, Tijssen P. 2004. Construction and sequencing of an infectious clone of the human parvovirus B19. Virology 318:142–152. doi: 10.1016/j.virol.2003.09.011. [DOI] [PubMed] [Google Scholar]