

SUMMARY
Human skin is commonly colonized and infected by Staphylococcus aureus. Exactly how these organisms are sensed by keratinocytes has not been clearly delineated. Using a combination of metabolic and transcriptomic methodologies, we found that S. aureus infection is sensed as a metabolic stress by the hypoxic keratinocytes. This induces HIF1a signaling, which promotes IL-1 β production and stimulates aerobic glycolysis to meet the metabolic requirements of infection. We demonstrate that staphylococci capable of glycolysis, including WT and agr mutants, readily induce HIF1a responses. In contrast, Δpyk glycolytic mutants fail to compete with keratinocytes for their metabolic needs. Suppression of glycolysis using 2-DG blocked keratinocyte production of IL-1 β in vitro and significantly exacerbated the S. aureus cutaneous infection in a murine model. Our data suggest that S. aureus impose a metabolic stress on keratinocytes that initiates signaling necessary to promote both glycolysis and the proinflammatory response to infection.
Graphical Abstract
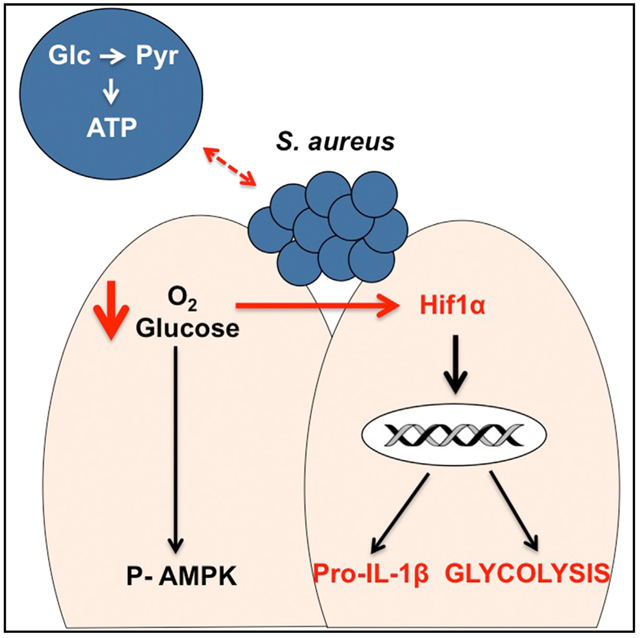
In Brief
Wickersham et al. show that human skin responds to the metabolic demands of S. aureus infection by increasing HIF1 α to promote aerobic glycolysis and IL-1 β production. Staphylococcal glycolysis is necessary to cause infection, while keratinocyte glycolysis is necessary for an effective host response to infection.
INTRODUCTION
Staphylococcus aureus are metabolically flexible pathogens that are highly adapted to persist on and within human tissues, with over 25% of individuals colonized with this organism (Jenkins et al., 2015). Colonization includes superficial residence of these bacteria on the skin as a component of the microbiome (Oh et al., 2016), intercalation within the layers of the epidermis (Nakatsuji et al., 2013), and intracellular persistence within keratinocytes (Soong et al., 2015). Skin and soft-tissue infections, as well as systemic S. aureus infection, are usually caused by colonizing strains, well documented in multiple whole-genome studies (Fitzgerald, 2014). Although typically considered to be extracellular pathogens, S. aureus can also persist within immune, endothelial, and epithelial cells that can serve as foci of persistent or recurrent infection. Often, these intracellular staphylococci harbor mutations in the agr locus affecting toxin expression (Shopsin et al., 2008; Smyth et al., 2012) or are small-colony variants (SCVs), typically metabolic auxotrophs (Kriegeskorte et al., 2011), reflecting adaptation to the host (Golubchik et al., 2013) (Proctor et al., 2006).
In response to S. aureus, keratinocytes initiate immune signaling. Like immune cells, keratinocytes undergo many fundamental changes following pathogen recognition. These include increasing the surface display of receptors, upregulating cytokine expression, and migrating to replace cells lost at the site of infection–all functions that require energy. To meet the increased metabolic demands of infection, many types of immune cells shift from oxidative phosphorylation to aerobic glycolysis, often referred to as metabolic reprogramming. This switch to glycolysis is critical for the successful host response to infection and has been documented in clinical settings using patient-derived data (Shalova et al., 2015; O’Neill and Pearce, 2016). Keratinocytes express pattern recognition and NOD (non-obese-diabetic)-like receptors, as well as the components of the inflammasome, providing multiple mechanisms to initiate proinflammatory signaling, but the relative importance of metabolic responses to infection in the skin is not established.
An important trigger for the induction of glycolysis is tissue hypoxia and the need to rapidly generate ATP in response to perceived infection. The human epidermis, which is oxygenated transdermally (O’Shaughnessy and Brown, 2015) (Ronquist et al., 2003), presents a relatively hypoxic environment that is further stressed by exposure to colonizing bacteria like S. aureus. Local hypoxia in immune cells drives inflammatory signaling through the activation of hypoxia-inducible factor 1a (HIF1α) signaling. This increases glucose uptake and facilitates glycolytic metabolism. HIF1α also stimulates the transcription of pro-interleukin (IL)-1 β, which contains an HRE (hypoxia-response element) in its promoter and is an important component of the inflammasome (Tannahill et al., 2013). As S. aureus colonization of the skin itself generates localized hypoxia (Lone et al., 2015), keratinocytes seem primed for the activation of the glycolytic pathway, even without substantial tissue invasion.
Staphylococci express several genes activated under conditions of hypoxia that facilitate skin and soft-tissue infections (Wilde et al., 2015). These include the srrAB locus, a two-component system that senses O2 (Pragman et al., 2007; Kinkel et al., 2013), and rex, a redox sensor that regulates the expression of genes necessary for metabolic activity in anaerobic conditions (Pagels et al., 2010). Loci important in bacterial glycolysis, like pyruvate kinase (pyk), have been found to mediate nitric oxide (NO) resistance required for virulence in skin and soft-tissue infection (Vitko et al., 2015). Moreover, S. aureus express four glucose transporters, suggesting that they are highly adapted to support a high level of glycolytic flux under situations when O2 is limited (Vitko et al., 2015).
As S. aureus is a member of the commensal flora and the major cause of human skin infection, both the organism and the host are highly adapted to each other. In the studies presented in this report, we postulated that infection causes metabolic stress sufficient to initiate HIF1α signaling and glycolysis, as the organisms and skin compete for the limited oxygen and glucose. We demonstrate not only that keratinocytes respond to the energy demands of infection by activating HIF1α and glycolysis, but also that S. aureus must generate ATP through glycolytic pathways in order to establish cutaneous infection.
RESULTS
S. aureus Impose Metabolic Stress upon Keratinocytes
Keratinocytes are poised to rapidly respond to the presence of pathogenic bacteria. We postulated that S. aureus infection forces keratinocytes to compete with the organisms for available glucose and O2. Since staphylococci can reside within keratinocytes as well as cause superficial infection, we compared several mutant strains with differing levels of intracellular accumulation and metabolic capabilities. These included wild-type (WT) USA300 LAC MRSA, a frequent cause of human skin and soft-tissue infection; an agr mutant, lacking toxin production and capable of intracellular accumulation (Soong et al., 2015); a Δpyk glycolytic mutant, lacking pyruvate kinase; and a DpckA gluconeogenesis mutant, lacking phosphoenolpyruvate carboxykinase and making it unable to regenerate glucose for energy under stressed conditions (Vitko et al., 2015). Strains with mutations in the srrA/B locus (White et al., 2014), a two-component system important in O2 sensing and the coordinate response to hypoxia (Kinkel et al., 2013; Yarwood et al., 2001), and rex, a regulator of anaerobic metabolism and responses to NO, were also studied (Pagels et al., 2010). Growth curves of the WT and mutant strains under the experimental conditions used and in anaerobiasis were analyzed to compare their relative abilities to proliferate (Figure S1A). Screening studies that were performed with transposon mutants are interpreted with caution, and constructed deletions were used for most of the studies.
The metabolic response of keratinocytes under control conditions or infected with various S. aureus strains, as well as the bacterial strains themselves, was studied using a Seahorse analyzer (Figures 1A-1D). This enabled us to compare infected and uninfected keratinocytes by following O2 consumption rate (OCR), which reflects oxidative phosphorylation and generation of ATP through the tricarboxylic acid (TCA) cycle, and glycolytic activity measured as extracellular acidification rate (ECAR), which reflects lactate production as the endpoint of aerobic glycolysis. The Seahorse assay involved following the addition of glucose to stimulate metabolism, oligomycin to suppress oxidative phosphorylation, and 2-DG (deoxyglucose) to shut off glycolysis.
Figure 1. S. aureus Infection Induces O2 Consumption and Glycolysis in Human Keratinocytes.

(A–D) A Seahorse analyzer was used to monitor the metabolic activity via oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) of uninfected primary keratinocytes (HEKn) and those exposed to S. aureus metabolic (A), oxygen-sensing (B), and toxin-deficient (C) strains for 3 hr (MOI, 20:1) or to heat-killed staphylococci. The metabolic activity of staphylococci at the same concentration alone was also measured (D). The additions of glucose, oligomycin, and 2-DG (deoxyglucose) are indicated as vertical lines.
(E–H) In (E) and (G), intracellular uptake (2 hr) and persistence (24 hr) of S. aureus strains were quantified by gentamicin exclusion assay comparing WT (LAC USA300), LAC agr null mutants, Tn mutants in the Je2 background, and Δpyk and ΔpckA deletion mutants in the LAC background. (F and H) Cell viability assessed by trypan blue exclusion.
(I and J) Immunoblots showing phospho-AMPK of human keratinocytes exposed to PBS, WT, or agr S. aureus (I), as well as Δpyk and ΔpckA (J), after 4 or 24 hr.
Densitometry normalized to actin is shown for comparison.
Representative experiments from at least two independent assays are shown. For all graphs, each data point is the mean value ± SEM (n = 3). *p < 0.05; ****p < 0.0001, by one-way ANOVA. SeaHorse statistical significances are compared to PBS alone.
Following a 3-hr incubation with S. aureus (MOI, 20:1), the substantial oxidative activity of the keratinocytes infected with WT USA300 S. aureus strain LAC was evident as a significantly increased OCR was observed throughout the course of the assay in comparison to the lower OCR associated with the uninfected keratinocyte control. The limited effect of oligomycin on the infected keratinocytes, which inhibits oxidative phosphorylation, suggests that the metabolic activity is primarily due to glycolysis. The Δpyk glycolytic mutant, in contrast to the complemented strain, did not contribute to OCR or to the glycolytic activity of the infected cultures, despite persisting in the media used for the assays (Figure S1B and S1C). Assays measuring the proton production rate (PPR), a different assessment of glycolytic activity, confirmed the Δpyk phenotype (Figure S1D). The ΔpckA mutant at baseline minimally increased the OCR and was very close to the WT strain in stimulating glycolytic activity in response to added glucose (Figure 1A). We similarly compared strains with mutations affecting O2 sensing using rex and srrA/B mutants (Figure 1B). The srrA/B and rex mutants were comparable to WT strains in O2 consumption and exhibited slightly less (srrA/B) or slightly more (rex) glycolytic activity than the parental strain (Figure 1B). The metabolic impact of an agr mutant lacking toxin expression, but capable of increased intracellular accumulation, was similarly examined in keratinocytes (Figure 1C). In response to glucose, the agr strain modestly increased OCR and especially glycolytic activity in parallel with the WT parent.
The overall contribution of bacterial metabolism to the net changes in OCR and ECAR was negligible, indicating that the changes in metabolism were driven primarily by the keratinocyte response to infection (Figure 1D). Of note, heat-killed bacteria failed to elicit metabolic changes in the keratinocytes, suggesting that bacterial metabolism was affecting the keratinocytes, not simply the presence of pathogen-associated molecular patterns (PAMPs).
The metabolic response did not correlate with intracellular uptake and persistence of the staphylococcal strains as assessed by a gentamicin protection assay (Figure 1E). The agr mutant accumulated to significantly greater amounts within the keratinocytes than the other strains but did not differ significantly in its induction of glycolytic activity in the infected cells (Figure 1C). There was slightly greater intracellular persistence of the ΔpckA mutant than the WT parental strain and significantly less intracellular recovery of the Δpyk strain (Figure 1G). The rex and srrA/B mutants were recovered from within the infected keratinocytes to similar amounts at 24 hr, indicating that the relative amounts of intracellular accumulation of the different mutants did not correlate with their effects on the overall metabolic activity of the infected keratinocytes. Rates of keratinocyte viability over the course of the experiment did not differ substantially among the various strains (Figures 1F and 1H).
Another way to assess the metabolic impact of S. aureus infection on the keratinocytes is to follow the phosphorylation of AMPK, the major cellular sensor of ATP (Mihaylova and Shaw, 2011). In response to infection, there was induction of phosphorylated AMPK (P-AMPK) (Figures 1I and 1J). Of note, the Δpyk mutant, unable to initiate glycolysis, did not induce P-AMPK. These results are consistent with the metabolic data, indicating that the addition of S. aureus imposes a metabolic stress upon the infected keratinocytes.
S. aureus Induce HIF1α Signaling in Keratinocytes
Under hypoxic conditions, as often present in settings of infection, HIF1α signaling is induced to rapidly meet the metabolic requirements for host defenses (Weichhart et al., 2015; O’Neill et al., 2016; O’Neill and Pearce, 2016). Under normoxyic conditions, mammalian target of rapamycin (mTOR) induces the translation of HIF1α1 and HIF1α2, which are modified by prolyl hydroxylases that subsequently interact with the ubiquitin E3 ligase of the von Hippel-Landau complex to target the proteins for proteasomal degradation (Ward and Thompson, 2012). In hypoxic conditions, prolyl hydroxylase activity is suppressed, and HIF1a accumulates. HIF1α-dependent signaling promotes the expression of over 1,000 genes, many of which orchestrate the uptake of glucose and expression of the components necessary for glycolysis (Semenza, 2013), as well as transcription of pro-IL-1β (Tannahill et al., 2013).
We analyzed RNA sequencing (RNA-seq) data using Ingenuity Pathway Analysis (IPA) software (QIAGEN) comparing gene expression in control and S. aureus USA300-infected human keratinocytes in primary culture (HEKn cells) and in HaCaT cell lines after 1 hr of exposure (MOI, 100:1). HIF1α and numerous HIF1α-regulated genes that contribute to the transport and utilization of glucose necessary for glycolysis were induced, and several were confirmed by qRT-PCR (Figures 2A and 2B). There was a 22-fold increase in HIF1α expression and similar levels of induction of EGLN3, which catalyzes the formation of 4-hydroxyl proline on HIF; PFK1 (phosphofrucktokinase 1), the enzyme necessary to commit to glycolysis; and HK2 (hexokinase 2), which phosphorylates glucose to initiate metabolism. The induction of known targets of HIF1α signaling was completely blocked in cells treated with a HIF1α inhibitor, whereas tumor necrosis factor (TNF) expression was not significantly altered (Figure 2C). IL-1β, a component of the inflammasome and major cytokine in skin defenses (Cho et al., 2012; Kitur et al., 2016), was also significantly upregulated. The agr mutant did not significantly increase HIF1α transcription (Figure 2D), but both the WT and agr mutant increased the amounts of HIF1α protein recovered in the infected keratinocytes, consistent with the major regulation of HIF1α abundance through stabilization that is dependent upon the activity of the prolyl hydroxylases (Figure 2E). Activation of HIF1α in vivo was confirmed in S. aureus-infected skin grafts maintained on severe combined immunodeficiency (SCID) mice (Soong et al., 2015) (Figure 2F). Sections of the infected skin grafts demonstrated neovascularization (indicated by an arrow), consistent with the upregulation of VEGFA by HIF1α (Figure 2G). These results confirm that staphylococcal infection contributes to keratinocyte hypoxia. The keratinocytes respond to infection by upregulating the HIF1α cascade to increase glucose uptake and promote glycolysis to generate energy.
Figure 2. Staphylococcal Induction of HIF1α in Keratinocytes.

(A) An Ingenuity Pathway Analysis (IPA) was performed on RNA-seq data obtained from both human keratinocytes in primary culture (HEKn) and HaCaTs exposed to S. aureus (WT) USA300 LAC for 1 hr (MOI, 100:1). Heatmap shows the relative activation of genes responsive to HIF1α.
(B and C) qRT-PCR (B) and ELISAs (C) were performed on keratinocytes in primary culture exposed to PBS or WT S. aureus, with and without a small-molecule HIF1α inhibitor.
(D and E) qRT-PCR (D) and immunoblot (E) of HIF1α in primary keratinocytes after 24 hr of WT or agr infection. Densitometry normalized to actin is included for comparison.
(F) Immunofluorescence staining of infected and control human skin grafts with α-keratin (red) and α-HIF1α (green) demonstrating co-localization (yellow) in the infected grafts.
(G) H&E staining of sections from infected skin grafts. Arrow indicates areas of neovascularization.
Representative data from at least two independent experiments are shown. For all graphs, each data point is the mean value ± SEM (n = 4). *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001, by one-way ANOVA.
S. aureus Glycolytic Activity Is Required to Activate HIF1α Responses
Having observed that the S. aureus glycolytic mutant Δpyk did not induce keratinocyte metabolic activity (Figure 1A), we predicted that only S. aureus strains able to participate in glycolysis would induce HIF1α accumulation (Figure 3). At the level of transcription, only the WT strain significantly induced HIF1α expression and that of the other HIF1α-associated genes (EGLN3, HK2, and PFK1) (Figure 3A). More importantly, the post-translational modification of HIF1α regulates the accumulation of the transcription factor. We observed substantially more HIF1α protein accumulation as detected by western blot in keratinocyte lysates from the WT and ΔpckA-infected keratinocytes relative to the glycolytic mutant Δpyk (Figure 3B). We noted that the phosphorylation of S6, a marker for proliferative activity, was suppressed over the course of infection, but only by the bacterial strains capable of glycolysis (Figure 3B). The induction of HIF1α also correlated with the upregulation of the inflammasome-associated cytokines IL-1β and IL-18 (Figure 3C). The Δpyk mutant failed to induce expression of these cytokines, but it did significantly induce IL-17A, IL-12p40, IL-12p70, TNF, IL-8, and interferon γ (IFNγ) which are not HIF1α dependent (Figure 3C).
Figure 3. HIF1α Induction of Cytokines.

(A) qRT-PCR showing induction of HIF1α and HIF1α-dependent genes in keratinocytes exposed to PBS or WT USA300, Δpyk, and ΔpckA S. aureus for 24 hr.
(B) Immunoblot showing induction of HIF1α and P-S6 in primary keratinocytes exposed to PBS or WT USA300S. aureus for 4 or 24 hr. Densitometry normalized to actin is included for comparison.
(C) ELISA performed using supernatants from primary keratinocytes exposed to PBS or WT USA300 S. aureus background strains for 24 hr.
(D) qRT-PCR showing induction of HIF1α and HIF1α-dependent gene (EGLN3) in keratinocytes exposed to PBS and Je2 mutant rex or srrA/B S. aureus for 24 hr.
(E) Immunoblot showing induction of HIF1α and P-S6 in cells exposed to PBS or Je2 mutant rex or srrA/B for 4 and 24 hr. Densitometry normalized to actin is included for comparison.
(F) ELISAs performed using supernatants from primary keratinocytes exposed to PBS or Je2 WT, rex, or srrA/B S. aureus for 24 hr.
(G) P-AKT was detected by immunoblot from primary keratinocytes infected with WT and agr S. aureus for 2 and 24 hr (bar indicates deletion of irrelevant lanes). Densitometry normalized to actin is included for comparison.
(H) Sections of biopsied human skin grafts maintained on SCID mice stained for P-AKT. Arrows indicate P-AKT at basal and superficial layers of the epidermis.
(I) ELISA performed using supernatants from HaCaTs treated with inhibitors of AKT (triciribine) or TBK1/IKKε (BX795) with and without WT S. aureus for 24 hr.
Representative results from at least two independent experiments are shown. For all graphs, each data point is the mean value ± SEM (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001, by one-way ANOVA.
The O2 and redox sensing mutants, srrA/B and rex, were somewhat impaired in their induction of HIF1α and EGLN3 gene expression (Figure 3D). However, these mutants that are capable of glycolysis stimulated HIF1α protein accumulation to levels similar to those achieved by infection with the WT strain (Figure 3E). Neither the rex nor the srrA/B mutants were significantly impaired in their induction of IL-1β and IL-18, in comparison to the WT strain (Figure 3F). Thus, once adapted to the ambient conditions, the bacteria also activate similar keratinocyte responses.
Our data suggest that S. aureus stimulate both glycolysis and IL-1β production through the accumulation of HIF1α as a consequence of local hypoxia. However, as noted in Figure 3B, S. aureus also appeared to be stimulating the transcription of HIF1α. Several upstream signaling pathways converge at the central regulators of metabolism mTOR/AKT, which increase HIF1α translation by 4E-BP1 (Weichhart et al., 2015). The induction of P-AKT/mTOR could occur via a staphylococcal activation of a G protein-coupled receptor (GPCR) at the cell surface, through phosphatidylinositol 3-kinase (PI3K) or through Toll-like receptor (TLR) signaling via TBK1/IKKε (Everts et al., 2014). We found that both WT and agr mutant S. aureus stimulate P-AKT (Figure 3G) in vitro and in infected human skin grafts maintained on SCID mice (Figure 3H). However, treatment of keratinocytes with wortmannin (10 mM) or rapamycin (10 mM) to inhibit PI3K and mTOR, respectively, had no significant effects on cytokine production, despite potently suppressing P-AMPK (Figure S2). Similarly, inhibition of TBK1 and AKT with BX795 or triciribine, respectively, increased keratinocyte IL-1β expression induced by WT S. aureus (Figure 3I). These observations suggest that the major impact of S. aureus on the keratinocytes is metabolic and that TBK1 or GPCR signaling is not as important in the induction of keratinocyte signaling as the metabolic impact of infection.
Glycolytic Metabolism Is a Key Component of Host Defense to Cutaneous Infection
To determine the in vivo significance of keratinocyte glycolysis in S. aureus infection, we treated mice with 2-DG to inhibit glycolysis 1 day prior to and during the course infection with WT or the Δpyk mutant. Although the 2-DG-treated mice had equivalent bacterial loads to the PBS-treated controls in the 5-mm punch biopsy (Figure 4A), the extent of the associated dermonecrosis was significantly greater in the 2-DG-treated mice, reflecting an overall greater bacterial load (Figures 4B and 4D). Bacterial accumulation was dependent on the glycolytic capabilities of S. aureus, as there was a significantly lower bacterial burden in Δpyk-infected mice (Figure 4C) and no obvious dermonecrotic lesions (Figure 4D). Over the course of infection, 2-DG-treated mice had significantly less TNF produced after 1 day and less IL-1β after 5 days, while the Δpyk failed to elicit any proinflammatory response (Figure 4E). The histopathological changes observed in the 2-DG-treated mice included substantially increased vascularity and adiposity (a marker of inflammation) in the 2-DG-treated mice (Figure 4F). Although there were no differences in neutrophil or natural killer (NK) cell recruitment in response to 2-DG treatment, there were significantly more interstitial macrophages in fluorescence-activated cell sorting (FACS) analysis of the infected skin lesions (Figure 4G).
Figure 4. Participation of Glycolysis in Host Response to S. aureus Skin Infection.

(A and B) Bacterial load after 5 days (A) and dermonecrosis (B) in mice treated with PBS or 2-DG (n = 14).
(C) Bacterial load after 5 days in mice infected with WT or Δpyk S. aureus (n = 7 and 9).
(D) Images of mouse lesions after 5 days of WT or Δpyk infection with and without 2-DG.
(E) Cytokine measurements by ELISA from skin biopsies of the infected sites after 1 day and 5 days (n = 5).
(F) H&E staining of skin sections taken from WT infected mice with and without 2-DG treatment. Insets indicate areas of adipogenesis, and arrow indicates blood vessels.
(G) Cell populations recruited in response to cutaneous WT infection in PBS- and 2-DG-treated mice after 5 days of exposure to WT S. aureus.
(H and I) ELISA (H) and lactate dehydrogenase (LDH) (I) assays performed on supernatants obtained from primary keratinocytes treated with 2-DG and WT, mutant, or heat-killed S. aureus strains for 24 hr (n = 4).
Representative results from at least two independent experiments are shown. For all graphs, each data point is the mean value ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001, by one-way ANOVA.
In vitro studies with 2-DG-treated keratinocytes demonstrated significantly decreased induction of IL-1β as compared to PBS-treated controls (Figure 4H). Under control conditions, there was increased cytotoxicity associated with 2-DG treatment that was further increased by infection with organisms capable of glycolysis, including the complemented but not the Δpyk mutant (Figure 4I). These studies further indicate a central role for the induction of keratinocyte glycolysis as part of an effective host response to staphylococcal skin infection as well as demonstrating the importance of staphylococcal glycolytic activity in establishing infection.
DISCUSSION
Keratinocytes have a major role in host defense, providing both a physical and immunological barrier against infection. The factors that initiate keratinocyte signaling in the presence of a substantial skin microbiome consisting of both commensal and pathogenic flora are not completely understood. Our studies suggest that S. aureus infection induces considerable metabolic stress in keratinocytes as directly measured by changes in OCR and induction of glycolysis in infected cells. Bacterial competition for glucose and O2 stimulates the keratinocyte response, and bacteria unable to participate in glycolysis fail to activate the skin cells. The limited availability of O2 in the human epidermis increases susceptibility to metabolic stress and reliance upon glycolysis to rapidly generate energy in response to perceived infection (O’Shaughnessy and Brown, 2015).
The keratinocyte response to S. aureus is mediated by the induction of HIF1α signaling. Numerous studies have identified HIF1α as a major transcription factor activated in the setting of infection to rapidly meet increased metabolic requirements (Ward and Thompson, 2012). This is especially relevant in the setting of the relative hypoxia of human skin. Staphylococcal induction of HIF1α directly correlated with the organism’s ability to use glycolysis to generate ATP, and only the glycolytic mutant Δpyk that did not impose a metabolic stress upon the keratinocytes completely failed to stimulate keratinocyte metabolism or HIF1α activity. WT and toxin-deficient agr mutant strains, despite substantially different abilities to persist within keratinocytes, both induced HIF1α accumulation. A growing literature documents the central role of HIF1α in coordinating metabolic and inflammatory responses, as well as participating in epithelial barrier function in the setting of infection (Rezvani et al., 2011). HIF1α has been shown to be induced and stabilized in the setting of low O2 as present in the skin (Boutin et al., 2008; Ronquist et al., 2003), contributing to local production of antimicrobial peptides (Peyssonnaux et al., 2008) and supporting barrier function (Wong et al., 2015). Our studies suggest a more fundamental role for HIF1α in coordinating the metabolic and immune functions of the skin in the setting of acute infection.
A major consequence of HIF1α signaling is the generation of proinflammatory cytokines—particularly, mature IL-1β and IL- 18 associated with the inflammasome. The presence of staphylococcal PAMPS that are known to be immunostimulatory was not sufficient to induce an IL-1β response in the absence of glycolytic activity. Numerous studies have identified IL-1β as being especially important in the effective clearance of S. aureus skin infection (Cho et al., 2012; Soong et al., 2012). Various toxins expressed by the USA300 MRSA strains used in these experiments, including Hla, LukAB, and PVL, have each been shown to be capable of stimulating the NLRP3 inflammasome and IL-1β (Craven et al., 2009; Holzinger et al., 2012; Melehani et al., 2015). However, in the presence of an HIF1α inhibitor, the IL-1β response was virtually eliminated. These observations indicate that the induction of HIF1α and the generation of pro-IL-1β are likely to be critical steps to prime the keratinocyte for subsequent activation of the inflammasome by S. aureus toxins. The stimulation of glycolysis and IL-1β signaling appear to be generic responses of keratinocytes to S. aureus and not solely dependent upon the expression of specific virulence factors or toxins.
We observed that staphylococcal genes involved in sensing hypoxia (srrA/B), but not oxidative or nitrosative stress (rex), only modestly affected the ability of S. aureus to stimulate a keratinocyte HIF1α response. These mutants confer a somewhat diminished metabolic stress upon the keratinocytes, but once adapted to the milieu, they are capable of using glycolysis to generate energy. Previous studies indicated an impaired ability of the srrA/B mutants to cause osteomyelitis, which may be due to limited ability to initiate glycolysis or increased susceptibility to the reactive oxygen species (ROS) generated by recruited immune cells (Wilde et al., 2015). A more conclusive phenotype has been ascribed to the Δpyk glycolytic mutant, which is significantly attenuated in models of skin and soft-tissue infection (Vitko et al., 2016), as was a S. aureus mutant lacking all four glucose transporters similarly interfering with glycolytic activity (Vitko et al., 2015). Although the Δpyk mutant stimulated an IL-17A response, it did not activate metabolic stress or stimulate HIF1α accumulation, nor did it suppress proliferative signaling mediated by phosphorylation of S6.
The systemic inhibition of glycolysis with 2-DG resulted in significantly larger S. aureus-induced skin lesions with delayed healing and decreased IL-1β production, which is a key factor in the resolution of skin infection (Cho et al., 2012). The increased abundance of dermal adipocytes in the 2-DG-treated mice is consistent with recent studies documenting the importance of adipocytes in protection from S. aureus infection (Zhang et al., 2015). In contrast, 2-DG was found to have protective effects in a model of systemic L. monocytogenes infection. Thus, there are clearly important differences in the metabolic responses to specific pathogens and sites of infection (Wang et al., 2016). There is a growing literature linking the induction of specific metabolic pathways and immune signaling in macrophages, dendritic cells, and the many different types of T cells. For keratinocytes, this association between metabolic activity and keratinocyte function in host defense is especially important. The relative hypoxia of the skin lowers their threshold for activation of HIF1α and glycolysis and provides a common mechanism to provide both the metabolic and proinflammatory response to cutaneous colonization/infection. Overall, our findings confirm the clinical importance of glycolysis in promoting host defenses—specifically, in the context of S. aureus skin infection.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Growth Curves
The USA300 LAC strain and an agr deletion mutant have been previously described (Soong et al., 2015). The Δpyk and ΔpckA mutants and a complemented Δpyk mutant also in the LAC background were compared to their USA300 LAC parent (Vitko et al., 2015). Tn mutants from the Je2 NARSA (Network on Antimicrobial Resistance in Staphylococcus aureus) library were used for screening studies, and the rex and the srrA and srrB mutants were compared with the Je2 parental control; the srrA/B null mutant in the LAC background and parent strains were obtained from W. Nauseef (White et al., 2014). For growth curves, starter cultures were grown overnight in Luria broth (LB) media and then diluted to an optical density at 600 nm (OD600) of 0.1 in LB, DMEM, or Dermalife media with and without 0.5% sodium pyruvate (n = 4). Cultures were grown in either an anaerobic chamber or a standard 37°C incubator, and the OD600 was taken at 30-min intervals in a 96-well plate using an automated Tecan reader.
Tissue Culture, Infections, and Cytokine Panels
The human HaCaT keratinocyte cell line was grown in DMEM with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. HEKn cells were obtained from GIBCO and grown in Dermalife (Lifeline Cell Technologies) with 1% FBS and 1% penicillin/streptomycin. Primary cells were only used up to passage 7. For infections, media were changed to DMEM or Dermalife without penicillin/ streptomycin the night before infection. Bacteria were grown in LB to an OD600 of 1 and added to cell media 1 hr after fibronectin was added at 10 mg/mL. Inhibitors rapamycin (10 μM), wortmannin (10 μM), compound C (20 μM), Hif1α inhibitor (10 μM), 2-DG (100 μM), triciribine (20 μM), and BX795 (5 μM) were added 1 hr before infection. For gentamicin exclusion assays, media was changed to DMEM or Dermalife with 500 mg/mL gentamicin 2 hr after infection. Cells were lifted from the tissue culture plate with Tryple Express (Life Technologies), serially diluted, and plated on LB agar 2 and 24 hr after gentamicin was added. Cellular viability was assessed by trypan blue staining and measured using a “Countess” Illuminator. For cytokine panels, 500 mg/mL gentamicin was added to the infected keratinocytes after 2 hr of incubation, and supernatants were collected at 24 hr and sent to Eve Technologies for cytokine analysis, or analysis was performed using commercially available kits (Biolegend).
SCID:hu Mice and Confocal Staining
SCID:hu mouse graft methods were performed as previously described (Soong et al., 2015). In brief, NOD-scid IL2Rγ (NSG) mice (the Jackson Laboratory) were grafted with human neonatal foreskin, per Columbia University Institutional Review Board (IRB) protocol AAAN6306. After maturation (3–6 weeks), a 10-μL aliquot of 108 colony-forming units (CFUs) of USA300 or PBS were applied dropwise. Skin grafts were excised after 72 hr. Skin biopsies were fixed in 4% paraformaldehyde for 24 hr and then 70% ethanol for 24 hr. Sections were then preserved in paraffin blocks and mounted on immune slides in 6-mm sections. Sections were deparaffinized using SafeClear xylene substitute and ethanol and then microwaved in sodium citrate buffer (sodium citrate, 10 mM; 0.05% Tween 20, pH 6) for three 5-min rounds for antigen presentation. Slides were washed in Tween 20 0.5% for 10 min to permeabilize the sections and then washed in Triton X-100. Sections were then blocked in 10% normal donkey serum in 1% BSA for 1 hr, and primary antibodies were applied overnight in 1% BSA at 4°C. Slides were washed in Triton X-100, and secondary antibodies (Molecular Probes Alexafluores) were applied in 1% BSA for 1 hr. Coverslips were then mounted using Vectashield. Controls were performed using only secondary antibody.
Western Blots and Antibodies
Cell lysates were frozen in RIPA + HALT. Blots were run using Bolt mini gels (Lifetech) and transferred using the iBlot machine (Lifetech). Antibodies for Hif1a (Cayman Chemical), P-S6 (Cell Signaling), S6 (Cell Signaling), P-AKT (Cell Signaling), AKT (Cell Signaling), P-AMPK (Cell Signaling), AMPK (Cell Signaling), and β-actin (Santa Cruz Biotechnology) were used.
RNA Analysis and siRNA
ON-TARGETplus SMARTpool Human Hif1α siRNA and ON-TARGETplus non-targeting scrambled control pool siRNAs were purchased from Dharmacon and transfected into HEKn cells using Lipofectamine RNAiMAX (Life Technologies) per the manufacturer’s instructions. Cell lysates were saved in TRK Lysis Buffer, and RNA was isolated using the EZNA RNA Kit (Omega Biotech). cDNA was synthesized from RNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and analyzed using the StepOne qPCR machine and Power SYBR Green PCR Master Mix (Applied Bio-systems). The following target genes and primers are listed as follows: Hif1α (forward: CCCTAACGTGTTATCTGTCGCT; reverse: AGTAGCTGCATGATCG TCTGG); EGLN3 (forward: GCGTCTCCAAGCGACAC; reverse: TTTCCCGGA TAGCAAGCCAC); HK2 (forward: CAAAGTGACAGTGGGTGTGG; reverse: GCCAGGTCCTTCACTGTCTC); PFK1 (forward: AAGCATCATCGAAACGC TCTC; reverse: GGTGCCCGTGTCTTCTTTGT); and β-actin (forward: AGAGC TACGAGCTGCCTGAC; reverse: AGCACTGTGTTGGCGTACAG).
Lactate Dehydrogenase Assay
Lactate dehydrogenase assay was performed as per manufacturer’s instructions (Roche) and as previously described (Kitur et al., 2016).
Seahorse Glycolysis Stress Test
HaCaTs or HEKn cells were infected at 60,000 cells per well according to Seahorse protocol. Fn (fibronectin) was added 1 hr before infection, and bacteria were added (MOI, 20:1) for 3 hr. Seahorse analysis was performed according to the XF Glycolysis Stress Test Kit User Guide (wells were washed in XF Base Medium + 200 mM L-glutamine, incubated at 37°C without CO2 for 45 min, then placed in the Seahorse analyzer). The analysis consisted of a 20-min observation period followed by the addition of glucose to stimulate glycolysis, the addition of oligomycin to suppress oxidative phosphorylation, and the addition of 2-DG to block glycolysis, each at 20-min intervals. The metabolic activity of the bacteria was determined by adding the same amount of bacteria in other Seahorse assays without any HaCaTs or HEKn cells.
RNA-Seq Analysis
HaCaTs and HEKn cells were infected with WT USA300 S. aureus (MOI, 100:1) for 1 hr before cell lysates were prepared. Poly(A) pull-down was used to enrich mRNAs from total RNA samples (200 ng-1 μg per sample, RIN > 8 required) and to proceed to library preparation by using the Illumina TruSeq RNA Prep Kit. Libraries were then sequenced using Illumina HiSeq 2500 at the Columbia Genome Center. The samples were multiplexed in each lane, which yields the targeted number of single-end/paired-end 100-bp reads for each sample as a fraction of 180 million reads for the whole lane.
Real-Time Analysis (RTA; Illumina) was used for base calling, and bcl2fastq (version 1.8.4; Illumina) was used for converting BCL to fastq format, coupled with adaptor trimming. The reads were mapped to a reference genome (human: NCBI/build37.2; mouse: UCSC [University of California, Santa Cruz]/mm9) using TopHat (Trapnell et al., 2009), with four mismatches (-read-mismatches = 4) and ten maximum multiple hits (-max-multihits = 10). To tackle the mapping issue of reads that are from exon-exon junctions, TopHat infers novel exon-exon junctions ab initio and combines them with junctions from known mRNA sequences (refgenes) as the reference annotation. The relative abundance (a.k.a. expression level) of genes and splice isoforms was estimated using Cufflinks (Trapnell et al., 2010) with default settings. Differentially expressed genes were tested under various conditions using DESeq (Anders and Huber, 2010). It is an R package based on a negative binomial distribution that models the number reads from RNA-seq experiments and tests for differential expression. Expression values were analyzed using IPA software, as previously described (Ahn et al., 2016).
Model of Skin Infection
Six- to 8-week-old sex-matched C57BL/6J mice (the Jackson Laboratory) were inoculated subcutaneously on the back with 2 × 106 CFUs of WT USA300 or the Δpyk mutant in 100 μL PBS or PBS alone as a control, and lesions were measured daily and followed for 5 days as previously described (Kitur et al., 2016). 2-DG (1 g/kg) treatment was initiated the day prior to infection and followed over the course of the experiment. Punch biopsies (5 mm) were taken at the borders of the infected lesion and normal-appearing skin. The biopsied tissue was homogenized through 40-mm filters prior to the hypotonic lysis of red blood cells. Recovered bacteria were enumerated. Keratinocytes and immune cells were spun down, and a homogenate was used for cytokine ELISA. Immune cells were stained with fluorescently conjugated markers and analyzed by FACS, as described previously (Ahn et al., 2016). Skin specimens were also fixed in 70% ethanol for histology, performed by the Columbia University Medical Center. Trichrome staining was done, and images were examined on a Zeiss Axiocm MRc. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) protocol (AAAR0406 and AAAR5412).
Ethics Statement
Animal experiments were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the NIH, the Animal Welfare Act, and U.S. federal law. The Institutional Animal Care and Use Committee of Columbia University approved the protocol (AC-AAAH2350 and AC-AAAG7408).
Statistical Analysis
Multiple comparisons were analyzed using one-way or two-way ANOVA with Dunnett’s posttest. Normal distributions were analyzed by Student’s t test. Mouse samples were compared using the nonparametric Mann-Whitney test. RNA-seq analysis was conducted using the Benjamini-Hochberg multiple comparison test. Statistical analyses were performed with GraphPad Prism software, with significance defined as p < 0.05.
Supplementary Material
Highlights.
S. aureus use glycolysis to infect skin
Infection imposes a metabolic stress on keratinocytes
HIF1α is increased in infected keratinocytes
Keratinocytes use glycolysis to meet the demands of infection
ACKNOWLEDGMENTS
We thank William Nauseef, University of Iowa, for providing srrA/B mutants. This work was supported by NIH grants 5R01HL079395 and 5R01AI103854 to A.P. Research reported in this publication was performed in the CCTI Flow Cytometry Core, supported in part by the Office of the Director, NIH, under award S10RR027050.The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The proteomics core facility is funded by NIH P30 CA013696-39S3, and the CCTI Flow Cytometry Core is funded by NIH S10 RR027050.
Footnotes
ACCESSION NUMBERS
The accession number for the RNA-seq data reported in this paper is GEO: GSE95080.
SUPPLEMENTAL INFORMATION
Supplemental Information includes two figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.02.055.
REFERENCES
- Ahn D, Perñaloza H, Wang Z, Wickersham M, Parker D, Patel P, Koller A, Chen EI, Bueno SM, Uhlemann AC, and Prince A (2016). Acquired resistance to innate immune clearance promotes Klebsiella pneumoniae ST258 pulmonary infection. JCI Insight 1, e89704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutin AT, Weidemann A, Fu Z, Mesropian L, Gradin K, Jamora C, Wiesener M, Eckardt KU, Koch CJ, Ellies LG, et al. (2008). Epidermal sensing of oxygen is essential for systemic hypoxic response. Cell 133, 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JS, Guo Y, Ramos RI, Hebroni F, Plaisier SB, Xuan C, Granick JL, Matsushima H, Takashima A, Iwakura Y, et al. (2012). Neutrophil-derived IL-1β is sufficient for abscess formation in immunity against Staphylococcus aureus in mice. PLoS Pathog. 8, e1003047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E, Ting JP, and Duncan JA (2009). Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE 4, e7446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJ, et al. (2014). TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat. Immunol 15, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald JR (2014). Evolution of Staphylococcus aureus during human colonization and infection. Infect Genet Evol. 21, 542–547. [DOI] [PubMed] [Google Scholar]
- Golubchik T, Batty EM, Miller RR, Farr H, Young BC, Larner-Svensson H, Fung R, Godwin H, Knox K, Votintseva A, et al. (2013). Within-host evolution of Staphylococcus aureus during asymptomatic carriage. PLoS ONE 8, e61319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzinger D, Gieldon L, Mysore V, Nippe N, Taxman DJ, Duncan JA, Broglie PM, Marketon K, Austermann J, Vogl T, et al. (2012). Staphylococcus aureus Panton-Valentine leukocidin induces an inflammatory response in human phagocytes via the NLRP3 inflammasome. J. Leukoc. Biol 92, 1069–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins A, Diep BA, Mai TT, Vo NH, Warrener P, Suzich J, Stover CK, and Sellman BR (2015). Differential expression and roles of Staphylococcus aureus virulence determinants during colonization and disease. MBio 6, e02272–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkel TL, Roux CM, Dunman PM, and Fang FC (2013). The Staphylococcus aureus SrrAB two-component system promotes resistance to nitrosative stress and hypoxia. MBio 4, e00696–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitur K, Wachtel S, Brown A, Wickersham M, Paulino F, Peñaloza HF, Soong G, Bueno S, Parker D, and Prince A (2016). Necroptosis promotes Staphylococcus aureus clearance by inhibiting excessive inflammatory signaling. Cell Rep. 16, 2219–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegeskorte A, König S, Sander G, Pirkl A, Mahabir E, Proctor RA, von Eiff C, Peters G, and Becker K (2011). Small colony variants of Staphylococcus aureus reveal distinct protein profiles. Proteomics 11, 2476–2490. [DOI] [PubMed] [Google Scholar]
- Lone AG, Atci E, Renslow R, Beyenal H, Noh S, Fransson B, Abu-Lail N, Park JJ, Gang DR, and Call DR (2015). Colonization of epidermal tissue by Staphylococcus aureus produces localized hypoxia and stimulates secretion of antioxidant and caspase-14 proteins. Infect. Immun. 83, 3026–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melehani JH, James DB, DuMont AL, Torres VJ, and Duncan JA (2015). Staphylococcus aureus Leukocidin A/B (LukAB) kills human monocytes via host NLRP3 and ASC when extracellular, but not intracellular. PLoS Pathog. 11, e1004970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, and Shaw RJ (2011). The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 13, 1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuji T, Chiang HI, Jiang SB, Nagarajan H, Zengler K, and Gallo RL (2013). The microbiome extends to subepidermal compartments of normal skin. Nat. Commun 4, 1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA, and Pearce EJ (2016). Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med 213, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA, Kishton RJ, and Rathmell J (2016). A guide to immunometabolism for immunologists. Nat. Rev. Immunol 16, 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shaughnessy RF, and Brown SJ (2015). Insight from the air-skin interface. J. Invest. Dermatol 135, 331–333. [DOI] [PubMed] [Google Scholar]
- Oh J, Byrd AL, Park M, Kong HH, and Segre JA; NISC Comparative Sequencing Program (2016). Temporal stability of the human skin microbiome. Cell 165, 854–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagels M, Fuchs S, Pané-Farré J, Kohler C, Menschner L, Hecker M, McNamarra PJ, Bauer MC, von Wachenfeldt C, Liebeke M, et al. (2010). Redox sensing by a Rex-family repressor is involved in the regulation of anaerobic gene expression in Staphylococcus aureus. Mol. Microbiol 76, 1142–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyssonnaux C, Boutin AT, Zinkernagel AS, Datta V, Nizet V, and Johnson RS (2008). Critical role of HIF-1alpha in keratinocyte defense against bacterial infection. J. Invest. Dermatol 128, 1964–1968. [DOI] [PubMed] [Google Scholar]
- Pragman AA, Herron-Olson L, Case LC, Vetter SM, Henke EE, Kapur V, and Schlievert PM (2007). Sequence analysis of the Staphylococcus aureus srrAB loci reveals that truncation of srrA affects growth and virulence factor expression. J. Bacteriol 189, 7515–7519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor RA, von Eiff C, Kahl BC, Becker K, McNamara P, Herrmann M, and Peters G (2006). Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat. Rev. Microbiol 4, 295–305. [DOI] [PubMed] [Google Scholar]
- Rezvani HR, Ali N, Serrano-Sanchez M, Dubus P, Varon C, Ged C, Pain C, Cario-André M, Seneschal J, Taïeb A, et al. (2011). Loss of epidermal hypoxia-inducible factor-1α accelerates epidermal aging and affects re-epithelialization in human and mouse. J. Cell Sci 124, 4172–4183. [DOI] [PubMed] [Google Scholar]
- Ronquist G, Andersson A, Bendsoe N, and Falck B (2003). Human epidermal energy metabolism is functionally anaerobic. Exp. Dermatol 12, 572–579. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2013). HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Invest 123, 3664–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalova IN, Lim JY, Chittezhath M, Zinkernagel AS, Beasley F, Hernández-Jiménez E, Toledano V, Cubillos-Zapata C, Rapisarda A, Chen J, et al. (2015). Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1α. Immunity 42, 484–498. [DOI] [PubMed] [Google Scholar]
- Shopsin B, Drlica-Wagner A, Mathema B, Adhikari RP, Kreiswirth BN, and Novick RP (2008). Prevalence of agr dysfunction among colonizing Staphylococcus aureus strains. J. Infect. Dis 198, 1171–1174. [DOI] [PubMed] [Google Scholar]
- Smyth DS, Kafer JM, Wasserman GA, Velickovic L, Mathema B, Holzman RS, Knipe TA, Becker K, von Eiff C, Peters G, et al. (2012). Nasal carriage as a source of agr-defective Staphylococcus aureus bacteremia. J. Infect. Dis. 206, 1168–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong G, Chun J, Parker D, and Prince A (2012). Staphylococcus aureus activation of caspase 1/calpain signaling mediates invasion through human keratinocytes. J. Infect. Dis 205, 1571–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong G, Paulino F, Wachtel S, Parker D, Wickersham M, Zhang D, Brown A, Lauren C, Dowd M, West E, et al. (2015). Methicillin-resistant Staphylococcus aureus adaptation to human keratinocytes. MBio 6, e00289–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. (2013). Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitko NP, Spahich NA, and Richardson AR (2015). Glycolytic dependency of high-level nitric oxide resistance and virulence in Staphylococcus aureus. MBio 6, e00045–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitko NP, Grosser MR, Khatri D, Lance TR, and Richardson AR (2016). Expanded glucose import capability affords Staphylococcus aureus optimized glycolytic flux during infection. MBio 7, e00296–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Huen SC, Luan HH, Yu S, Zhang C, Gallezot JD, Booth CJ, and Medzhitov R (2016). Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation. Cell 166, 1512–1525.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, and Thompson CB (2012). Signaling in control of cell growth and metabolism. Cold Spring Harb. Perspect. Biol 4, a006783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichhart T, Hengstschläger M, and Linke M (2015). Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol 15, 599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MJ, Boyd JM, Horswill AR, and Nauseef WM (2014). Phosphatidylinositol-specific phospholipase C contributes to survival of Staphylococcus aureus USA300 in human blood and neutrophils. Infect. Immun 82, 1559–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde AD, Snyder DJ, Putnam NE, Valentino MD, Hammer ND, Lonergan ZR, Hinger SA, Aysanoa EE, Blanchard C, Dunman PM, et al. (2015). Bacterial hypoxic responses revealed as critical determinants of the host-pathogen outcome byTnSeq analysis of Staphylococcus aureus invasive infection. PLoS Pathog. 11, e1005341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WJ, Richardson T, Seykora JT, Cotsarelis G, and Simon MC (2015). Hypoxia-inducible factors regulate filaggrin expression and epidermal barrier function. J. Invest. Dermatol 135, 454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarwood JM, McCormick JK, and Schlievert PM (2001). Identification of a novel two-component regulatory system that acts in global regulation of virulence factors of Staphylococcus aureus. J. Bacteriol 183, 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L-J, Guerrero-Juarez CF, Hata T, Bapat SP, Ramos R, Plikus MV, and Gallo RL (2015). Innate immunity. Dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science 347, 67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.