

Abstract
Introduction.
Macrophage migration inhibitory factor (MIF) is a pleiotropic inflammatory cytokine with upstream regulatory roles in innate and adaptive immunity and is implicated in the pathogenesis of autoimmune diseases including rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). Several classes of MIF inhibitors such as small molecule inhibitors and peptide inhibitors are in clinical development.
Areas Covered.
The role of MIF in the pathogenesis of RA and SLE is examined; the authors review the structure, physiology and signaling characteristics of MIF and the related cytokine D-DT/MIF-2. The preclinical and clinical trial data for MIF inhibitors are also reviewed; information was retrieved from PubMed and ClinicalTrials.gov using the keywords MIF, D-DT/MIF-2, CD74, CD44, CXCR2, CXCR4, Jab-1, rheumatoid arthritis, systemic lupus erythematosus, MIF inhibitor, small molecule, anti-MIF, anti-CD74, and peptide inhibitor.
Expert Opinion.
Studies in mice and genetic and clinical studies in humans demonstrate the therapeutic potential of MIF inhibition for RA and SLE. MIF- directed approaches could be particularly efficacious in patients with high expression MIF genetic polymorphisms. In patients with RA and SLE and high expression MIF alleles, targeted pharmacologic MIF inhibition could be part of a precision medicine approach. Anti-MIF pharmacotherapies could ultimately also be steroid-sparing in patients with chronic glucocorticoid dependence and/or refractory autoimmune disease.
Keywords: CD44, CD74, CXCR2, CXCR4, CXCR7, macrophage migration inhibitory factor (MIF), D-DT/MIF-2, precision medicine
1. Introduction.
Macrophage migration inhibitory factor (MIF) is a pleiotropic inflammatory cytokine important in the upstream regulation of both innate and adaptive immunological responses and has been implicated in the pathogenesis of autoimmune diseases including rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). MIF was the first “cytokine” function to be described in a report published in the Bulletin of the Johns Hopkins Hospital in 1932, which highlighted the ability of Mycobacteria-sensitized lymphocytes to arrest tissue macrophage migration in vitro.[1] While MIF has direct chemotactic effects similar to macrophage inflammatory protein-1α/β (MIP-1α/β) and monocyte chemoattractant protein 1 (MCP-1), its eponymic function ultimately was found to arise from its ability to desensitize the chemokine receptors CXCR2 and CXCR4.[2] The unambiguous molecular cloning of the human MIF gene was achieved and reported in 1993 following identification of its unique role in regulating the effects of endogenous cortisol.[3]
1.1. MIF Structure.
The MIF structure comprises a homo-trimer with a primary sequence that is well-conserved across evolutionary phylogeny ranging from mammals to invertebrates, plants, and protozoan species.[4] The human MIF monomer subunit is composed of 114 amino acids and has a mass of 12.5 kilo-Daltons.[5, 6] At the interface between monomer subunits, there is a tautomerase enzymatic activity. This enzymatic activity may be of vestigial function, as its physiologic substrate remains unknown.[7] The N-terminal proline residue within the tautomerase active site appears to interface with the region of the protein that binds to the MIF cognate receptor CD74.[8] (Figure 1) Mutation of this proline resulted in decreased MIF-CD74 binding in vitro and reduced MIF function in a genetic mouse model.[9]
Figure 1.
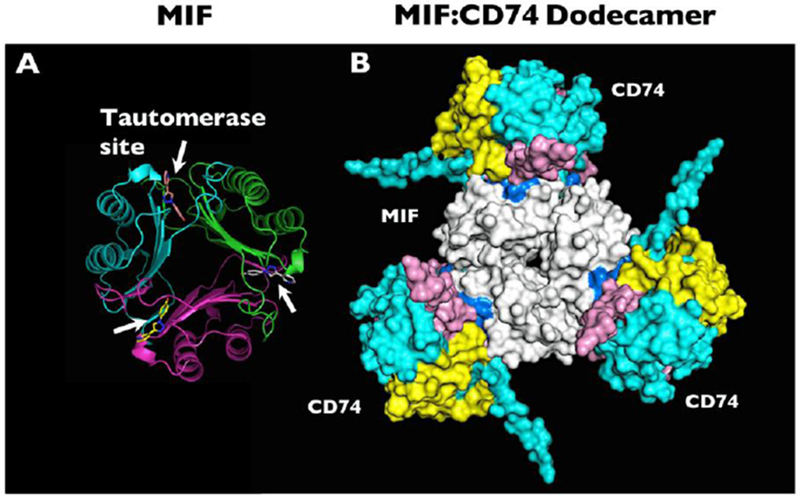
(a) Molecular structure of MIF based on x-ray crystallography, with white arrows indicating the locations of the tautomerase sites between adjacent monomers. The tautomerase sites are shown occupied by the small molecule MIF20. (b) Computational model representation of the MIF trimer (white, center) engaging with CD74 trimers (blue, yellow, and pink, outer). Many small molecule MIF inhibitors can occupy the MIF tautomerase sites that appear in close apposition to the CD74 receptor. Reprinted by permission from Springer: Metabolic brain disease. Predicted structure of MIF/CD74 and RTL1000/CD74 complexes, Meza Romero R., et al, COPYRIGHT 2016.
1.2. MIF Physiology.
Systemic MIF levels in humans follow a diurnal rhythm, peaking in the early morning hours in coordination with the levels of endogenous cortisol.[10] The normal MIF serum levels are in the 2–6 ng/mL range, but in the setting of acute stressors and/or inflammatory stimuli such as bacterial lipopolysaccharide (LPS), tumor necrosis factor (TNF), or interferon-γ (IFN- γ),[11] MIF levels increase several-fold subsequent to release from many cellular and tissue subtypes,[12] including monocytes/macrophages, B and T lymphocytes, granulocytes, platelets, dendritic cells, endothelial cells, and mesenchymal cells.[10, 13] Unlike most inflammatory cytokines, MIF is constitutively synthesized and stored in pre-formed intracellular pools. Following an acute stressor or inflammatory stimulus, MIF is then released from the cells through a process involving the Golgi-associated chaperone protein p115,[14] though in some cases export also may involve membrane blebbing and exosomes.[15]
Following its release, MIF acts in an autocrine and/or paracrine fashion to upregulate the synthesis of secondary inflammatory cytokines.[16] MIF also has an upstream role in regulating its own synthesis.[17] In a murine sepsis model, genetic Mif deletion resulted in decreased plasma levels of TNF, without an effect on IL-6 and IL-12 levels.[18] However, Mif deletion did result in decreased renal IL-6 levels in a mouse glomerulonephritis model,[19] and decreased serum IL-12 levels in response to systemic infection.[20] Overall studies have observed Mif knockout mice to have decreased macrophage synthesis of TNF, IL-1β, and prostaglandin E2,[21] and decreased lymphocyte production of IL-1β, IL-6, IL-17, and IL-23 in particular.[22] MIF-activated lymphocytes secrete IL-2, IL-17, and IFN-γ.[22, 23] MIF was shown to have a role in promoting neutrophil IL-1β release in a murine model of acute gout.[24] Though the mechanism for this remains loosely defined, MIF was found to co-localize with the NLRP3 and ASC proteins and regulated their expression and activation.[25] In human patients with SLE, immune complexes containing anti-U1-snRNP antibodies were shown to upregulate MIF expression in monocytes, leading to the increased activation of the NLRP3 inflammasome and resulting increased production of IL-1β.[26, 27]
In addition to these roles, MIF signaling is important for the appropriate surface expression of pattern recognition receptors in the innate immune response against infections. MIF stimulates increased activity of the PU.1 transcription factor, leading to upregulation of Toll-like receptor 4 (TLR4). MIF-deficient macrophages are less responsive to bacterial LPS due to downregulated surface expression of TLR4[28] and are similarly hypo-responsive to mycobacterial β-glucans due to downregulation of surface dectin-1.[29]
MIF signaling leads to enhanced cell survival and proliferation through several related pathways which differ between cell types. MIF signaling promotes cell survival through direct activation of the Akt pathway[30] and prevents apoptosis through inhibition of the tumor suppressor protein p53, a mechanism which is common between many cell types.[21] Given these roles in cell proliferation and survival, MIF also contributes to the inflammatory pathogenesis of different cancers.[31] High MIF expression has been reported in various tumors including glioblastoma multiforme, melanoma, and prostate, gastric, pancreatic, and lung cancers.[32–34]
In endothelial cells, MIF affects the expression of adhesion molecules necessary for leukocyte and lymphocyte migration and trafficking into inflamed tissues. Endogenous MIF signaling leads to endothelial cell expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin, as well as synthesis of the chemokines IL-8 and MCP-1. These effects facilitate leukocyte recruitment into inflamed tissues. Conversely, exogenous MIF signaling in the setting of concurrent TNF leads to endothelial cell expression of P-selectin, facilitating leukocyte rolling.[35]
1.3. D-DT/MIF-2.
A more recently characterized member of the MIF family that also binds to CD74 with high affinity is D-dopachrome tautomerase (D-DT) (also called MIF-2).[36] MIF-2 has many overlapping signaling functions as compared with MIF, as well as a described role in tissue protection from injury following ischemia-reperfusion.[37] (Figure 2) Like MIF, D-DT/MIF-2 is produced by many tissue and immune cell types, and its circulating levels in blood are similar to MIF.[38] D-DT/MIF-2 may also share a pro-oncogenic role with MIF, with signaling by both cytokines exerting a cooperative role in tumorigenesis.[39] D-DT/MIF-2 expression levels have been found to be increased in glioblastoma multiforme [40], pancreatic adenocarcinoma,[41] and renal cell carcinoma in particular.[39]
Figure 2.
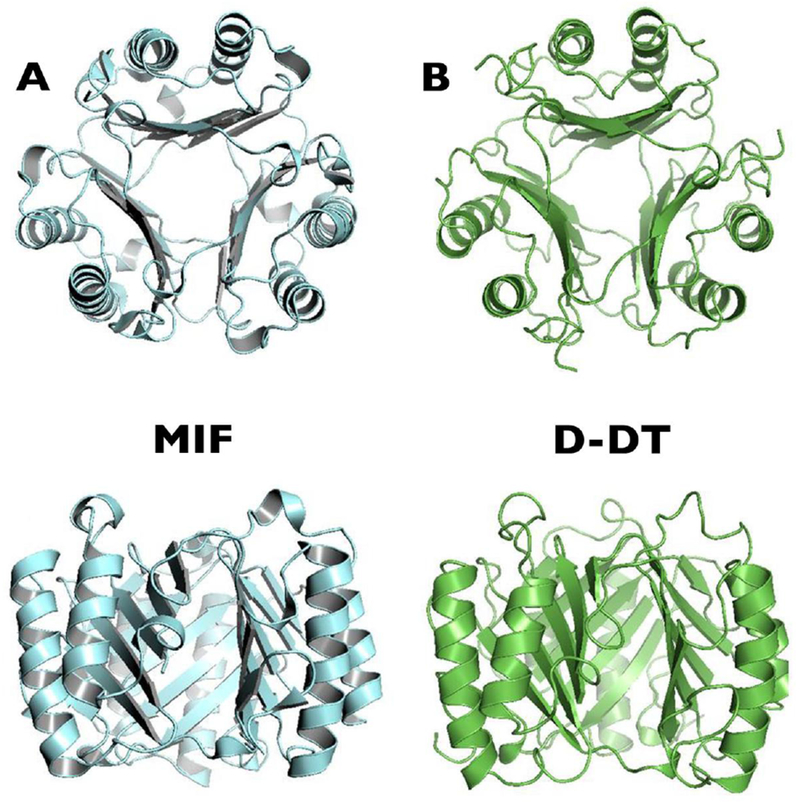
MIF family protein crystal structures. (A) Human MIF trimer. (B) Human D-DT/MIF-2 trimer. Reprinted with adaptations from Cytokine, 88, Meza-Romero R., et al, Modeling of both shared and distinct interactions between MIF and its homologue D-DT with their common receptor CD74, 62–70, Copyright (2016), with permission from Elsevier.
Despite having many overlapping functions, some studies report distinct roles for D-DT/MIF-2 in adipogenesis in particular.[42] D-DT/MIF-2 also lacks the pseudo-(E)LR motif present in MIF that is required for CXCR2 binding and activation.[43] As such, its role in inflammation and migration may be narrower than MIF. Another difference is that common genetic polymorphisms have not been described in the human D-DT/MIF-2 gene.
2. MIF Genetics.
During an investigation of RA patients, sequencing of the MIF locus revealed a variant four nucleotide microsatellite within the promoter region. This microsatellite was comprised of a C-A-T-T repeat present in 5–8 copies (e.g., −794 CATT5–8) (rs5844572); higher numbers of CATT repeats were found to be associated with higher baseline and stimulated MIF transcription.[44] These variant MIF alleles are common throughout different human populations. High expression MIF alleles in particular have been implicated in the pathogenesis of various autoimmune diseases, and the frequency of these alleles in human populations can exceed 20%.[45] The transcription factor ICBP90 was recently found to be the major protein regulating MIF mRNA transcription from this CATT5–8 promoter locus.[46] (Figure 3)
Figure 3.
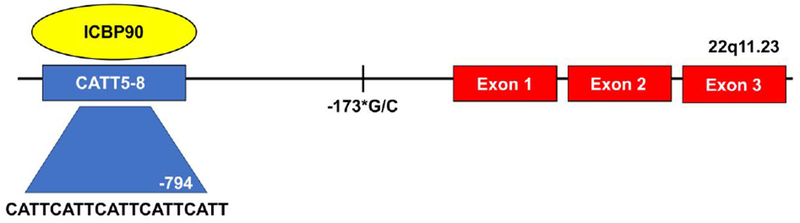
Diagram of the human MIF gene with 3 downstream exons, and the upstream microsatellite −794 CATT5–8 to which binds the transcription factor ICBP90. The number of CATT repeats correlates with basal and stimulated MIF expression. The single nucleotide polymorphism −173*G/C is in linkage disequilibrium with the CATT7 microsatellite repeat.
A second MIF promoter single nucleotide polymorphism (SNP) at −173*G/C (rs755622) has been identified, with the *C allele in linkage disequilibrium with the −794 CATT7 microsatellite.[47] Multiple studies have reported the −173*C allele and CATT7 in association with increased systemic MIF and an increased propensity for and/or severity of inflammatory rheumatological disease.[47–54]
3. MIF Receptors and Ligands.
3.1. CD74 and CD44.
The cognate MIF receptor comprises a signaling complex of ligand-binding CD74 coupled to signal-transducing CD44.[55, 56] CD74 is the cell surface expressed form of the class II invariant chain, its intracellular function to facilitate the loading of peptide fragments into MHC class II for antigen presentation. In the context of its function as the cognate MIF receptor, CD74 is expressed on the cell surface independent of MHC class II expression and can be found on virtually all nucleated cell types, either constitutively or after stimulatory induction.[57, 58]
CD44 is a broadly expressed cell adhesion molecule that mediates the activation of Src family kinases.[59] The gene for CD44 comprises 20 exons, 10 of which can undergo alternative splicing to generate various isoforms which differ in the length and structure of the protein ectodomain.[60] MIF signaling increases synthesis of the Tra2α splicing factor, which in turn leads to the transcription of CD44v3-v6 isoforms. These larger isoforms of CD44 are limited to expression in epithelium, proliferating cells, and in certain malignancies. The larger ectodomains facilitate extracellular matrix migration and provide binding sites for matrix metalloproteinases (MMPs) and growth factors such as fibroblast growth factor (FGF).[60]
Upon ligand binding with MIF, CD74 recruits CD44, whereupon both proteins become phosphorylated at their intracellular motifs and initiate downstream signal transduction.[61] (Figure 4) In monocytes and stromal cells such as fibroblasts, initial activation of CD44-associated Src tyrosine kinase and MEK leads to phosphorylation of ERK1/2 MAP kinases, activation of cytosolic phospholipase A2 (cPLA2), and the inhibition of p53 (blocking activation-induced apoptosis and contributing to MIF’s inflammatory and pro-survival effects).[62]
Figure 4.
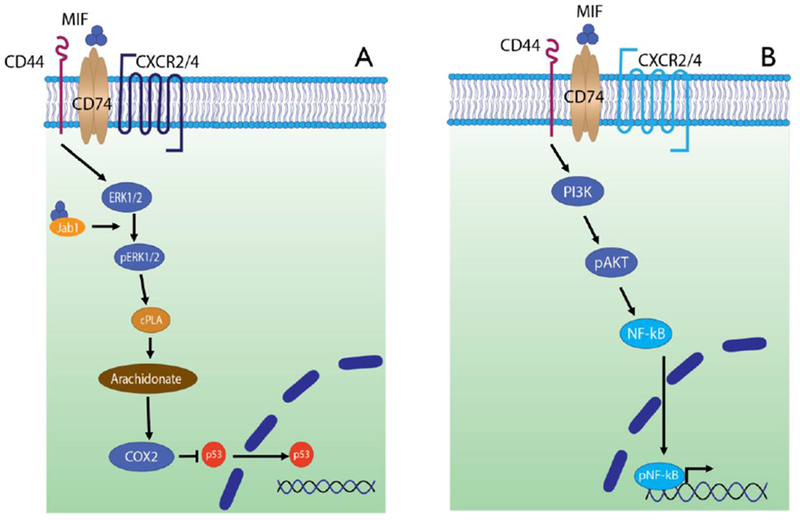
MIF signaling through the cognate surface receptor CD74. (A) CD74 and CD44 co-localize to mediate intracellular phosphorylation of ERK1/2, which in turn activates phospholipase A2 (cPLA2). This signaling cascade ultimately inhibits nuclear translocation of p53, promoting cell survival and proliferation. MIF interaction with intracellular Jab-1 may function to self-regulate these signaling pathways. CXCR2 is a non-cognate MIF ligand which can interact with CD74. (B) MIF signaling through CD74 leads to activation of phosphoinositide-3 kinase (PI3K), which then leads to phosphorylation and activation of Akt and then NFkB. The latter localizes to the nucleus and causes the transcription of inflammatory gene products. CXCR4 is a non-cognate MIF ligand which can interact with CD74.
In B cells, CD44-associated Syk tyrosine kinase leads to Akt phosphorylation and downstream NFκB activation.[63] At least among B cells, MIF binding to CD74 also results in the intra-membrane cleavage of CD74 via the SPPL2a protease to produce a 42 amino acid intracellular domain which has a role in the positive regulation of B cell maturation.[64] This fragment translocates to the nucleus to activate p65-NFκB, upregulate TAp63, and stimulate Bcl-2 leading to enhanced cell survival.[65] Another signaling pathway in B cells involves the engagement of the tyrosine kinase receptor c-Met with CD74/CD44, which permits sensitization of c-Met to autocrine hepatocyte growth factor/scatter factor (HGF). Signaling through the latter supplements the proliferation and survival mechanisms in B cells.[66]
A soluble form of the CD74 ectodomain (sCD74) formed by apparent membrane proteolysis also has been described, initially in the context of patients with autoimmune hepatitis and primary biliary cholangitis.[67] This sCD74 receptor binds to extra-cellular MIF, neutralizes its activity, and may function in regulating systemic MIF signaling.[68] There is evidence that SPPL2a protease cleavage of CD74 may account for the formation of sCD74.[64]
3.2. CXCR2, CXCR4, and CXCR7.
The CXCR2, CXCR4, and CXCR7 chemokine receptors associate with CD74, and MIF is a non-cognate ligand for these receptors.[69] MIF signaling through the CD74-CXCR2 complex elicits monocyte/macrophage chemotaxis, and migration arrest results from CXCR2 desensitization.[2] The binding of MIF to CXCR2 requires the presence of the pseudo-(E)LR motif (Asp-44-X-Arg-11) formed by the folding of non-adjacent aspartate and arginine residues in the MIF protein.[43] The interaction between MIF and CXCR4 requires an arginine-leucine-arginine (RLR) structural motif that binds to the N-terminus of CXCR4.[70] MIF signaling through the CD74-CXCR4-CXCR7 complex has been demonstrated to have a role in lymphocyte chemotaxis particularly in B cells.[71] These chemokine receptors are variably expressed on different stromal and leukocyte populations, conferring tissue specificity to MIF action in different physiologic and pathologic contexts.[2, 72]
3.3. Jab-1.
In the cytosol, endogenous MIF can bind to Jun-c activation domain-binding protein 1 (Jab-1) and inhibits its function, resulting in the downregulation of Jab-1 signaling pathways. The MIF and Jab-1 interaction leads to the reduced transcription of AP-1 specific pathways, while the NFκB pathway remains unaffected. In addition, MIF interaction with Jab-1 leads to the inhibition of JNK signaling pathways, which are otherwise activated by unbound Jab-1. Another activity of Jab-1 involves the degradation of the cyclin dependent kinase inhibitor p27Kip1, which promotes cell proliferation. The MIF-Jab-1 interaction thus opposes this latter effect, causes persistence of p27Kip1 rather than degradation, and results in cell cycle growth arrest.[73] Overall, the MIF-Jab-1 interaction appears to counter-regulate the inflammatory effects of MIF signaling through the cognate CD74 receptor.
4. MIF and Steroids.
MIF antagonizes and regulates the immunosuppressive effects of endogenous cortisol. MIF suppresses the cortisol-induced expression of the NFκB inhibitor IκB, resulting in increased NFκB-associated transcription of inflammatory genes.[74] MIF also suppresses the glucocorticoid-induced expression of MAP kinase phosphatase-1, which normally dephosphorylates and inactivates the inflammatory ERK1/2, JNK, and p38 MAP kinase signaling pathways.[75, 76] MIF activation of ERK1/2 MAP kinase reverses the corticosteroid-mediated suppression of phospholipase A2 activity, resulting in the enhanced synthesis of arachidonic acid, which promotes the activation of JNK-mediated inflammatory signaling pathways.[62]
Wang and colleagues studied human patients with SLE and reported higher MIF levels in serum and in peripheral blood mononuclear cells from patients with “steroid-resistant” disease, as well as increased NFκB and decreased IκB levels in this group.[77] Among SLE patients, systemic MIF levels have been considered as a potential biomarker for steroid-resistant disease.[78] Griga and colleagues studied patients with Crohn’s Disease and determined that those with the −173*C MIF promoter SNP, which is in linkage disequilibrium with the high expression, functional CATT7 microsatellite, had greater overall glucocorticoid requirements for disease management compared to those without this variant.[79]
5. MIF and Rheumatoid Arthritis (RA).
Collagen-induced arthritis (CIA) is a mouse model for RA that follows immunization with a mixture of complete Freund’s adjuvant and type II collagen. Affected mice develop inflammatory arthritis in the context of anti-type II collagen auto-antibodies and a primarily T cell-driven response. Genetic Mif deficiency reduces arthritis severity in this model and in the related adjuvant-induced arthritis (AIA) animal model, revealing MIF to be a critical mediator of inflammation and joint destruction in these diseases.[80–82]
Patients with RA who carry the CATT5 allele have been reported to have milder, non-erosive disease, while patients with CATT6–8 alleles have more severe and erosive phenotypes.[44] These results were subsequently confirmed by Radstake and colleagues, in which the high expression CATT7 allele was correlated with increased circulating MIF levels and more severe erosive radiologic joint damage.[49]
Patients with RA have elevated serum and synovial tissue MIF levels compared to healthy controls,[83, 84] and patients with high expression MIF alleles have increased serum levels compared to those with low expression alleles.[83] MIF is present at increased concentrations within synovial macrophages and fibroblast-like synoviocytes (FLS) of patients with RA, as well as within the synovial fluid of inflamed joints.[85]
MIF has numerous roles in the maintenance of inflammation within the synovial microenvironment of the RA joint. MIF induces expression of phospholipase A2 and cyclooxygenase-2 (COX-2) in FLS cells, leading to increased expression of IL-1, IL-6, IL-8, prostaglandin E2, and MMPs, the latter directly contributing to cartilage destruction and bone erosions. MIF signaling leads to FLS cell proliferation and inhibits apoptosis, resulting in synovial hyperplasia and pannus formation.[86]
MIF signaling also leads to upregulated expression of MMP-9 and MMP-13 from murine osteoblasts,[87] a mechanism that may be involved in the development of bone erosions in human patients with RA. Another mechanism may involve T cell-derived MIF upregulating RANKL expression, which leads to osteoclastogenesis.[88]
Treatment of cultured FLS from patients with RA with anti-MIF results in decreased IL-1β-mediated transcription of phospholipase A2 and COX-2.[89] The role of MIF signaling in FLS contributes to synovial hyperplasia and the pannus formation that is one of the hallmarks of RA. Rheumatoid pannus demonstrates features of a locally-invasive tumor.[90] MIF signaling resulting in cell proliferation, angiogenesis,[91] and inhibition of apoptosis contributes to this invasive pannus formation, and these pathophysiological mechanisms may be similar to MIF implicated malignant transformation.
6. MIF and Systemic Lupus Erythematosus (SLE).
In two different mouse models of SLE (MRL/lpr and NZB/NZW F1), treatment with either an anti-MIF antibody or a small molecule MIF antagonist resulted in a similar degree of decreased leukocyte recruitment to the kidneys, improved renal function and histological glomerulonephritis, and reduced systemic inflammatory cytokine and chemokine production, in particular TNF and the monocyte chemoattractant CCL2. However, MIF inhibition did not affect T and B cell activation, anti-dsDNA antibody levels, or glomerular IgG deposition.[92] Genetic Mif deficiency in MRL/lpr mice resulted in a similar amelioration of inflammation as compared with pharmacologic MIF inhibition.[19, 92]
A role for MIF in the pathophysiology of human SLE was supported by the finding of a correlation between increased systemic MIF levels and Systemic Lupus International Collaborating Clinics/American College of Rheumatology (SLICC/ACR) disease activity index scores.[93] In a genetic study by Sreih and colleagues, a cohort of 1,369 patients with SLE were assessed for MIF genotype and circulating MIF levels. Among patients with established disease, high expression MIF alleles (CATT6–8) and increased circulating MIF levels correlated with serositis, nephritis, and central nervous system involvement.[94] Elevated MIF levels in the serum,[95, 96] kidneys,[97] and urine[98] have been correlated with active systemic disease as well.
However, among patients with high expression MIF alleles (CATT7, −173*C haplotype), there was a lower rate of SLE and antinuclear antibody (ANA) positivity compared to healthy controls.[94] Dysregulated clearance of apoptotic debris has been implicated in the pathogenesis of the loss of self-tolerance and autoantibody formation against nuclear components in SLE.[99] It has been hypothesized that among patients without SLE, high systemic MIF expression facilitates the clearance of apoptotic debris and immune complexes leading to a reduced risk of developing pathologic autoantibodies and loss of immune tolerance.[94] Conversely, low systemic MIF expression may lead to the persistence of apoptotic and immune complex debris, resulting in B cell expansion and the development of antinuclear antibodies.
A particular role for MIF in mediating the inflammation of glomerulonephritis has been suggested by studies correlating immune-mediated renal damage with local tissue MIF levels. In human patients with various forms of proliferative glomerulonephritis, Lan and colleagues associated up-regulated tissue MIF expression with renal insufficiency, cellular damage, and leukocyte infiltrates.[97] These mechanisms may similarly account for the development of glomerulonephritis in the context of SLE.
7. MIF Directed Therapeutics.
MIF as a therapeutic target could be particularly advantageous for patients with higher background and inducible MIF expression, such as among those with CATT6–8 alleles or the −173*C/CATT7 haplotype. Furthermore, MIF inhibition might be particularly efficacious for patients with autoimmune inflammatory conditions and concurrent steroid refractoriness or long-standing glucocorticoid dependence.[100] (Table 1)
Table 1.
MIF Directed Therapeutics
| Small molecule inhibitors. | |||
|---|---|---|---|
| Name: | Description: | Target: | Reference: |
| N-Acetyl-P-benzoquinone (NAPQI) | Metabolite of acetaminophen/paracetamol | MIF tautomerase site | 105 |
| ISO-1 | 4,5-dihydro-3-(4-hydroxyphenyl)-5-isoxazoleacetic acid methyl ester | MIF tautomerase site | 32, 106, 107 |
| 4-IPP | 4-Iodo-6-Phenylpyrimidine | MIF tautomerase site | 108–112 |
| SCD-19 | Isocoumarin | MIF tautomerase site | 113, 114 |
| Ibudilast | 3-isobutyryl-2-isopropylpyrazolo-(1,5-a) pyridine | MIF, proximal to the tautomerase site | 112, 115, 116 |
| Ebselen | Synthetic organo-selenium | MIF trimer | 117 |
| p425 | Azo sulfonated organic acid | MIF trimer | 118 |
| Monoclonal antibodies. | |||
| Name: | Description: | Target: | Reference: |
| imalumab | Human monoclonal | MIF | 121 |
| milatuzumab | Humanized monoclonal | CD74 | 122–128 |
| Peptide inhibitors. | |||
| Name: | Description: | Target: | Reference: |
| DRα1-MOG-35–55 | HLA-DRα1 domain and myelin oligodendrocyte glycoprotein 35–55 peptide | CD74 | 131 |
| RTL1000 | HLA-DRα1β1 and myelin oligodendrocyte glycoprotein 35–55 peptide | CD74 | 132, 133 |
| C36L1 peptide | Immunoglobulin complementarity-determining region (CDR) peptide | CD74 | 135 |
| MIF-(40-49), MIF-(47-56) | Resemble the N-like loop of MIF that binds to CXCR2 | CXCR2 | 136 |
MIF: macrophage migration inhibitory factor protein;
HLA: human leukocyte antigen;
MOG: myelin oligodendrocyte glycoprotein
7.1. Small Molecule Inhibitors.
Small molecule MIF inhibitors have been identified through various screening strategies,[101–103] and there has been a particular focus upon exploiting the MIF tautomerase site, which is involved in the interaction with CD74, in the design of such inhibitors.[104]
The first small molecule inhibitor of MIF that was identified was the acetaminophen metabolite N-acetyl-p-benzoquinone (NAPQI), which was reported to irreversibly bind to the proline-1 residue in the MIF tautomerase active site and decrease its cell-binding activity.[105] This finding opened the way for further investigations of small molecule inhibitors involving the tautomerase active site.
Subsequently, the first synthetically designed small molecule MIF inhibitor to be described was the isoxazoline compound ISO-1. ISO-1 binds in the MIF tautomerase active site and inhibits downstream signaling effects.[106] In the experimental autoimmune encephalomyelitis (EAE) animal model of multiple sclerosis (MS), ISO-1 treatment reduced disease severity and duration. Further, ISO-1 decreased synthesis of TNF, IFN-γ, IL-4, and IL-17 from murine splenocytes of mice with EAE.[107] ISO-1 treatment also has been shown to reduce proliferation and mitogenic signaling in glioblastoma cells.[32]
The small molecule antagonist 4-iodo-6-phenylpyrimidine (4-IPP) covalently binds to the N-terminal proline within both the MIF and D-DT/MIF-2 tautomerase sites and arrests their signaling functions.[108] 4-IPP was found to have 5–10 times greater specificity for MIF than for D-DT/MIF-2 when compared to ISO-1, and resulted in decreased migration and growth of human lung adenocarcinoma cells.[109] Treatment of human melanoma cells in vitro with 4-IPP suppresses their endogenous expression of programmed death ligand 1 (PDL-1)[110], and this effect could account for the decreased tumor growth and angiogenesis in response to 4-IPP treatment in a mouse model of human melanoma.[111] Combined inhibition of both MIF and D-DT/MIF-2 has the potential for greater therapeutic efficacy in conditions driven by the overlapping signaling functions of these cytokines.[112]
SCD-19 is a small molecule inhibitor that binds in the MIF tautomerase active site and blocks its extracellular signaling functions. SCD-19 treatment resulted in decreased tumor volumes in a mouse model of lung cancer,[113] and ameliorated the inflammatory response in a model of chronic Pseudomonas aeruginosa lung disease.[114]
The small molecule inhibitor ibudilast is furthest in clinical evaluation. Ibudilast is a non-selective but principally phosphodiesterase 4 inhibitor being investigated for the treatment of MS,[115] and is a non-competitive inhibitor of the MIF tautomerase enzymatic activity, binding not at the N-terminal proline but adjacent to the active site.[116] Ibudilast binds MIF with micromolar affinity, and decreases mononuclear cell chemotaxis and downstream synthesis of MIF-dependent cytokines including IL-1β, IL-6, and TNF.[112]
Ebselen is a synthetic organo-selenium compound being investigated for use in various inflammatory disorders. Ebselen has a unique mechanism of causing MIF trimer disassociation into monomer subunits. Ouertatani-Sakouhi and colleagues determined that ebselen-mediated MIF disassociation disrupts MIF-CD74 signaling interactions, and results in hyperresponsiveness of signaling through the non-cognate receptors CXCR2 and CXCR4.[117]
The inhibitor p425 is an azo compound sulfonated organic acid that has a unique mechanism of action. It binds allosterically to the MIF trimer surface, not within the tautomerase site, through hydrophobic bonds and blocks the interaction between MIF and its cognate receptor CD74.[118]
7.2. Monoclonal antibodies.
Initial studies involving treatment of mice with CIA with an anti-MIF neutralizing antibody led to delayed onset and reduced frequency of disease.[119] Similar findings were observed in a related anti-type II collagen/LPS (anti-CII Ab/LPS) inflammatory arthritis mouse model, with decreased inflammatory cytokine levels and synovial inflammation in those animals treated with anti-MIF antibodies.[82] Lan and colleagues pursued experiments with a neutralizing murine anti-MIF monoclonal antibody for the treatment of experimentally-induced anti-glomerular basement membrane disease (anti-GBM) in a rat model. Anti-MIF treatment resulted in decreased proteinuria, maintained renal function, and decreased histological inflammatory changes.[120]
The fully human anti-MIF monoclonal antibody imalumab is the only candidate that has advanced in clinical testing, initially in patients with lupus nephritis (, discontinued with limited enrollment). Imalumab has been more extensively studied for the treatment of cancer ( for malignant solid tumors, for ovarian cancer, and for metastatic colorectal cancer). Outcomes from these early phase trials in heavily-pretreated patients with metastatic cancer reported that imalumab was well-tolerated by the patients.[121]
The anti-CD74 monoclonal antibody milatuzumab is a humanized form of the mouse anti-CD74 LL1.[122] Milatuzumab was evaluated in a 24-week phase 1b clinical trial for the treatment of SLE (), with patients demonstrating improvement per the British Isles Lupus Assessment Group (BILAG) index and SLE Disease Activity Index (SLEDAI) scoring systems, and only mild-moderate adverse effects.[123] Further safety and efficacy trials have been pursued in patients with graft-versus-host disease (), multiple myeloma ( and ), chronic lymphocytic leukemia ( and ), and B-cell non-Hodgkin lymphoma (). With these early phase clinical trials having been completed and milatuzumab reportedly having been well-tolerated by patients, further development has nonetheless not been continued.[124] Early outcomes of the combination therapy of milatuzumab with the anti-CD20 monoclonal antibody rituximab were also promising for the treatment of mantle cell lymphoma.[125] Milatuzumab has been granted orphan designation by the United States Food and Drug Administration (FDA) and European Medicines Agency (EMA) for multiple myeloma and chronic lymphocytic leukemia.[126–128]
Nanobodies, which are single domain antigen-binding fragments, are emerging as a new class of biologic therapeutics. Advantages of nanobodies include their high solubility, stability, and their greater tissue penetration compared to conventional antibodies.[129] Anti-MIF nanobodies with nanomolar affinity for both murine and human MIF have been developed and may prove beneficial for the treatment of inflammatory conditions.[130]
7.3. Peptide inhibitors.
A newer class of emerging therapeutics are small peptides that have shown potential in blocking MIF and/or its receptors.
The novel peptide inhibitor DRα1-MOG-35–55 is a chimeric molecule comprising the HLA-DRα1 domain and the myelin oligodendrocyte glycoprotein 35–55 (MOG) peptide. This peptide binds to CD74 and downregulates signaling on CD74+ monocytes, showing efficacy in reducing axonal damage and histological inflammation in a murine model of MS (EAE).[131]
The related inhibitor RTL1000 combines the DRβ1 domain (DRα1β1-MOG-35–55) with the DRα1 domain and MOG moiety and blocks MIF and MIF-2 binding to CD74.[132] A phase 1 clinical trial of RTL1000 in human patients with MS was well-tolerated without significant safety signals being observed.[133] Notably, the expression of MIF and D-DT/MIF-2 are increased in male patients with secondary forms of MS, suggesting the possibility of a precision medicine approach to the treatment of these patients.[134]
Figueiredo and colleagues reported on the development of C36L1, a 17 amino acid peptide that binds to CD74 on monocytes and dendritic cells. Peptide binding leads to decreased MIF-CD74 signaling and results in the decreased synthesis of immunosuppressive factors including TGF-β, IL-10, and PDL-1 with resulting increased antitumoral immunity from CD8+ cytotoxic T cells in a melanoma model.[135]
Inhibition of MIF binding to its non-cognate receptors with peptide therapeutics also has been explored. The synthetic peptides MIF-(40–49) and MIF-(47–56) resemble the N-like loop of parent MIF responsible for secondary site binding to CXCR2 and compete for this binding site. In vitro experiments with MIF-CXCR2 peptide inhibition resulted in decreased monocyte arrest upon aortic endothelial cells.[136]
8. Conclusions.
MIF is a pleiotropic inflammatory cytokine with upstream immunoregulatory effects that is induced as part of the innate and adaptive immune responses. MIF is constitutively expressed among a broad distribution of cell types, and in the setting of an appropriate stimulus is released from intracellular pools as well as synthesized de novo. Its effects include stimulation of synthesis of downstream inflammatory cytokines (including IL-1β, IL-6, TNF, and IFN-γ), promotion of cell proliferation, and arrest of apoptotic pathways. MIF signaling through non-cognate cell receptors mediates leukocyte trafficking and arrest at sites of inflammation.
MIF genetic polymorphisms are common across global populations and microsatellite numbers may have expanded to increase MIF expression in response to lethal infections such as invasive pneumococcus.[137, 138] High expression MIF alleles have been correlated with severity and joint erosions in RA, and with end-organ manifestations in established SLE disease.
The development of MIF inhibitors is being pursued for the treatment of various malignancies and autoimmune conditions. Small molecule inhibitors binding within the MIF tautomerase site negatively affect protein binding with CD74. Small molecules with unique mechanisms of action include ebselen, which causes MIF trimer disassociation, and p425, which binds the MIF trimer surface to cause disrupted CD74 signaling. Monoclonal antibodies including imalumab (anti-MIF) and milatuzumab (anti-CD74) have been evaluated in early phase clinical trials, with reports of efficacy in the treatment of SLE and cancer. The development of peptide inhibitors as a third therapeutic class may expand the potential armamentarium of anti-MIF clinical therapeutics even further.
9. Expert Opinion.
Significant experimental, genetic, and clinical research has implicated MIF in the pathogenesis of inflammation and autoimmunity, in particular of RA and SLE. Due to the upstream regulatory role of MIF in the pathogenesis of these inflammatory diseases, it has been considered to be a good candidate cytokine for targeted inhibition, and MIF inhibitors may have a broader spectrum of clinical effects compared to blocking more downstream inflammatory cytokines such as TNF or IL-6.
Precision medicine is an approach to patient care that allows physicians to choose treatments based on a genetic understanding of an individual patient’s disease, and tailoring therapeutics to the personal level. A precision medicine approach could be advantageous in patients with RA or SLE who have high expression MIF alleles. Such patients might be optimally treated with MIF inhibitors with the goal to reduce systemic MIF levels to those typical of low genotypic MIF expressers (e.g., those patients with CATT5 alleles). This approach, rather than complete MIF blockade, would likely be better tolerated by patients with fewer adverse effects, as other important physiologic functions of MIF could otherwise be preserved.
Despite research advances and the recent development of biologic and small molecule tyrosine kinase inhibitor treatments for autoimmune diseases, glucocorticoids continue to be commonly used by clinicians for the treatment of autoimmunity and inflammation. Advantages of glucocorticoids include their broad immunosuppressive effect and rapid onset of action. However, many patients can become dependent on high doses of glucocorticoids to maintain disease control and/or remission status. MIF has a unique role among cytokines through its counter regulation of glucocorticoid action. As such, the pharmacologic approach of MIF inhibition could increase the potency of endogenous or exogenous steroids. This approach could reduce steroid dependence and ultimately be steroid-sparing for patients across the spectrum of autoimmune diseases. This could also have the consequence of decreasing long-term glucocorticoid morbidities in such patients, such as secondary diabetes mellitus, weight gain and obesity, and metabolic bone disease.
The development of pharmacological MIF inhibitors for indications including autoimmunity and cancer will continue. Clinical trials thus far have been limited to phase 1 or II studies, and the various therapeutics tested have in general been well tolerated with a mild-to-moderate adverse effect profile. Despite the development of small molecule and peptide inhibitors, trials involving the monoclonal antibodies imalumab and milatuzumab comprise much of the human safety and efficacy data thus far. As such, the prospect for human patient treatment with the small molecule and peptide inhibitors remains more uncertain. Ibudilast, while inhibiting MIF, also has other relevant anti-inflammatory actions related to its inhibition of phosphodiesterases.
While most of the clinical trial research with pharmacologic MIF inhibitors has been directed at cancer or MS, there is significant potential that antagonism of MIF or D-DT/MIF-2 will be effective for the treatment of RA and SLE. Genetic Mif deficiency in animal models of these diseases results in phenotypic effects comparable with pharmacologic MIF inhibition. In human patients, systemic and focal MIF upregulation at sites of inflammation such as the synovium in RA and the kidneys in SLE has direct correlations with disease pathogenesis. Ibudilast and milatuzumab are furthest along in clinical development, with milatuzumab showing evidence of efficacy in a phase 1 tolerability study in human SLE. Further exploration of treatment with small molecule, antibody, and peptide MIF inhibitors for RA and SLE are warranted.
A precision medicine approach could also streamline the clinical development of MIF inhibitors. Clinical trials would become more cost effective with recruitment limited to high genotypic MIF expressers. The utilization of patient-specific MIF genotyping may offer the possibility of directing more effective and less toxic clinical care for autoimmune diseases such as RA and SLE. Among patients with high expression MIF alleles and more severe disease phenotypes, and/or chronic glucocorticoid dependence or steroid resistance, the use of MIF-directed pharmacological treatment might be especially warranted.
Article Highlights.
MIF is a pleotropic inflammatory cytokine with upstream immunoregulatory roles and its activity counter-regulates the effects of endogenous corticosteroids.
Commonly occurring high expression MIF alleles, defined by the presence of greater than 5 CATT repeats at the −794 MIF promoter site or the −173*G/C single nucleotide polymorphism in linkage disequilibrium with CATT7, are associated with greater severity and erosive rheumatoid arthritis, and greater severity of systemic lupus erythematosus in patients with established disease.
MIF signals through the CD74/CD44 receptor complex to activate intracellular kinases and NFκB transcriptional pathways. MIF signaling through the non-cognate ligands CXCR2, CXCR4, and CXCR7 has been implicated in mediating leukocyte trafficking and migration arrest at sites of inflammation.
Experimental animal studies of Mif gene deletion and anti-MIF treatment in models of rheumatoid arthritis and systemic lupus erythematosus ameliorate disease activity.
MIF directed pharmacologic therapeutics in development for treatment of human patients include small molecule inhibitors, monoclonal antibodies and nanobodies, and peptide inhibitors.
Acknowledgments
Funding.
This work was supported by funding from the National Institutes of Health (N.I.H.) to R Bucala. JB Bilsborrow receives salary support from the N.I.H. T32 training grant program. E Doherty and PV Tilstam are supported by the Arthritis Foundation Award #548970.
Footnotes
Declaration of Interest.
R Bucala is a co-inventor on patents describing the potential clinical applications of MIF antagonists and MIF genotyping. In addition, he is a co-founder of MIFCOR, Inc., which has licensed MIF modulators from Yale University and is endeavoring to develop MIF agonists.
The authors have no other relevant affiliations or financial involvements with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
References
Publications of special note have been highlighted as either of interest (*) or of considerable interest (**) to readers.
- 1.Rich AR, L. MR, The nature of allergy in tuberculosis as revealed by tissue culture studies. Bulletin of the Johns Hopkins Hospital, 1932. 50: p. 115–31. [Google Scholar]
- 2.Bernhagen J, et al. , MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med, 2007. 13(5): p. 587–96. [DOI] [PubMed] [Google Scholar]
- 3.*.Bernhagen J, et al. , MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature, 1993. 365(6448): p. 756–9. [DOI] [PubMed] [Google Scholar]; Report on the successful cloning of the human MIF gene.
- 4.Sparkes A, et al. , The non-mammalian MIF superfamily. Immunobiology, 2017. 222(3): p. 473–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suzuki M, et al. , Crystal structure of the macrophage migration inhibitory factor from rat liver. Nat Struct Biol, 1996. 3(3): p. 259–66. [DOI] [PubMed] [Google Scholar]
- 6.*.Sun HW, et al. , Crystal structure at 2.6-A resolution of human macrophage migration inhibitory factor. Proc Natl Acad Sci U S A, 1996. 93(11): p. 5191–6. [DOI] [PMC free article] [PubMed] [Google Scholar]; First report of the human MIF protein crystal structure.
- 7.Rosengren E, et al. , The immunoregulatory mediator macrophage migration inhibitory factor (MIF) catalyzes a tautomerization reaction. Mol Med, 1996. 2(1): p. 143–9. [PMC free article] [PubMed] [Google Scholar]
- 8.Pantouris G, et al. , An Analysis of MIF Structural Features that Control Functional Activation of CD74. Chem Biol, 2015. 22(9): p. 1197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fingerle-Rowson G, et al. , A tautomerase-null macrophage migration-inhibitory factor (MIF) gene knock-in mouse model reveals that protein interactions and not enzymatic activity mediate MIF-dependent growth regulation. Mol Cell Biol, 2009. 29(7): p. 1922–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petrovsky N, et al. , Macrophage migration inhibitory factor exhibits a pronounced circadian rhythm relevant to its role as a glucocorticoid counter-regulator. Immunol Cell Biol, 2003. 81(2): p. 137–43. [DOI] [PubMed] [Google Scholar]
- 11.Calandra T, et al. , The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J Exp Med, 1994. 179(6): p. 1895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehmann LE, et al. , Plasma levels of macrophage migration inhibitory factor are elevated in patients with severe sepsis. Intensive Care Med, 2001. 27(8): p. 1412–5. [DOI] [PubMed] [Google Scholar]
- 13.Calandra T and Roger T, Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol, 2003. 3(10): p. 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merk M, et al. , The Golgi-associated protein p115 mediates the secretion of macrophage migration inhibitory factor. J Immunol, 2009. 182(11): p. 6896–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Costa-Silva B, et al. , Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol, 2015. 17(6): p. 816–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greven D, Leng L, and Bucala R, Autoimmune diseases: MIF as a therapeutic target. Expert Opin Ther Targets, 2010. 14(3): p. 253–64. [DOI] [PubMed] [Google Scholar]
- 17.Santos LL and Morand EF, Macrophage migration inhibitory factor: a key cytokine in RA, SLE and atherosclerosis. Clin Chim Acta, 2009. 399(1–2): p. 1–7. [DOI] [PubMed] [Google Scholar]
- 18.Bozza M, et al. , Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J Exp Med, 1999. 189(2): p. 341–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoi AY, et al. , Macrophage migration inhibitory factor deficiency attenuates macrophage recruitment, glomerulonephritis, and lethality in MRL/lpr mice. J Immunol, 2006. 177(8): p. 5687–96. [DOI] [PubMed] [Google Scholar]
- 20.Flores M, et al. , Macrophage migration inhibitory factor (MIF) is critical for the host resistance against Toxoplasma gondii. FASEB J, 2008. 22(10): p. 3661–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitchell RA, et al. , Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci U S A, 2002. 99(1): p. 345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stojanovic I, et al. , Macrophage migration inhibitory factor stimulates interleukin-17 expression and production in lymph node cells. Immunology, 2009. 126(1): p. 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bacher M, et al. , An essential regulatory role for macrophage migration inhibitory factor in T-cell activation. Proc Natl Acad Sci U S A, 1996. 93(15): p. 7849–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galvao I, et al. , Macrophage migration inhibitory factor drives neutrophil accumulation by facilitating IL-1beta production in a murine model of acute gout. J Leukoc Biol, 2016. 99(6): p. 1035–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lang T, et al. , Macrophage migration inhibitory factor is required for NLRP3 inflammasome activation. Nat Commun, 2018. 9(1): p. 2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin MS, et al. , U1-small nuclear ribonucleoprotein activates the NLRP3 inflammasome in human monocytes. J Immunol, 2012. 188(10): p. 4769–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shin MS, et al. , Macrophage Migration Inhibitory Factor Regulates U1 Small Nuclear RNP Immune Complex-Mediated Activation of the NLRP3 Inflammasome. Arthritis Rheumatol, 2019. 71(1): p. 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roger T, et al. , MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature, 2001. 414(6866): p. 920–4. [DOI] [PubMed] [Google Scholar]
- 29.Das R, et al. , Macrophage migration inhibitory factor (MIF) is a critical mediator of the innate immune response to Mycobacterium tuberculosis. Proc Natl Acad Sci U S A, 2013. 110(32): p. E2997–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lue H, et al. , Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the Akt pathway and role for CSN5/JAB1 in the control of autocrine MIF activity. Oncogene, 2007. 26(35): p. 5046–59. [DOI] [PubMed] [Google Scholar]
- 31.Bucala R and Donnelly SC, Macrophage migration inhibitory factor: a probable link between inflammation and cancer. Immunity, 2007. 26(3): p. 281–5. [DOI] [PubMed] [Google Scholar]
- 32.Mangano K, et al. , Pathogenic role for macrophage migration inhibitory factor in glioblastoma and its targeting with specific inhibitors as novel tailored therapeutic approach. Oncotarget, 2018. 9(25): p. 17951–17970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otvos B, et al. , Cancer Stem Cell-Secreted Macrophage Migration Inhibitory Factor Stimulates Myeloid Derived Suppressor Cell Function and Facilitates Glioblastoma Immune Evasion. Stem Cells, 2016. 34(8): p. 2026–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meyer-Siegler KL, et al. , Macrophage migration inhibitory factor (MIF) gene polymorphisms are associated with increased prostate cancer incidence. Genes Immun, 2007. 8(8): p. 646–52. [DOI] [PubMed] [Google Scholar]
- 35.Cheng Q, et al. , Macrophage migration inhibitory factor increases leukocyte-endothelial interactions in human endothelial cells via promotion of expression of adhesion molecules. J Immunol, 2010. 185(2): p. 1238–47. [DOI] [PubMed] [Google Scholar]
- 36.*.Merk M, et al. , The D-dopachrome tautomerase (DDT) gene product is a cytokine and functional homolog of macrophage migration inhibitory factor (MIF). Proc Natl Acad Sci U S A, 2011. 108(34): p. E577–85. [DOI] [PMC free article] [PubMed] [Google Scholar]; First description of the D-DT/MIF-2 protein and comparison of its differences and similarities with MIF.
- 37.Qi D, et al. , The vestigial enzyme D-dopachrome tautomerase protects the heart against ischemic injury. J Clin Invest, 2014. 124(8): p. 3540–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merk M, et al. , D-dopachrome tautomerase (D-DT or MIF-2): doubling the MIF cytokine family. Cytokine, 2012. 59(1): p. 10–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Reilly C, et al. , Targeting MIF in Cancer: Therapeutic Strategies, Current Developments, and Future Opportunities. Med Res Rev, 2016. 36(3): p. 440–60. [DOI] [PubMed] [Google Scholar]
- 40.Presti M, et al. , Overexpression of macrophage migration inhibitory factor and functionally-related genes, D-DT, CD74, CD44, CXCR2 and CXCR4, in glioblastoma. Oncol Lett, 2018. 16(3): p. 2881–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo D, et al. , D-dopachrome tautomerase is over-expressed in pancreatic ductal adenocarcinoma and acts cooperatively with macrophage migration inhibitory factor to promote cancer growth. Int J Cancer, 2016. 139(9): p. 2056–67. [DOI] [PubMed] [Google Scholar]
- 42.Kim BS, et al. , The macrophage migration inhibitory factor protein superfamily in obesity and wound repair. Exp Mol Med, 2015. 47: p. e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weber C, et al. , Structural determinants of MIF functions in CXCR2-mediated inflammatory and atherogenic leukocyte recruitment. Proc Natl Acad Sci U S A, 2008. 105(42): p. 16278–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.**.Baugh JA, et al. , A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun, 2002. 3(3): p. 170–6. [DOI] [PubMed] [Google Scholar]; Initial description of the MIF promoter CATT microsatellite, and its association with rheumatoid arthritis phenotypes.
- 45.Zhong XB, et al. , Simultaneous detection of microsatellite repeats and SNPs in the macrophage migration inhibitory factor (MIF) gene by thin-film biosensor chips and application to rural field studies. Nucleic Acids Res, 2005. 33(13): p. e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.*.Yao J, et al. , Transcription factor ICBP90 regulates the MIF promoter and immune susceptibility locus. J Clin Invest, 2016. 126(2): p. 732–44. [DOI] [PMC free article] [PubMed] [Google Scholar]; Report of the ICBP90 transcription factor which regulates MIF expression through binding to the CATT microsatellite region.
- 47.Donn RP, et al. , A novel 5’-flanking region polymorphism of macrophage migration inhibitory factor is associated with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum, 2001. 44(8): p. 1782–5. [DOI] [PubMed] [Google Scholar]
- 48.Donn R, et al. , A functional promoter haplotype of macrophage migration inhibitory factor is linked and associated with juvenile idiopathic arthritis. Arthritis Rheum, 2004. 50(5): p. 1604–10. [DOI] [PubMed] [Google Scholar]
- 49.Radstake TR, et al. , Correlation of rheumatoid arthritis severity with the genetic functional variants and circulating levels of macrophage migration inhibitory factor. Arthritis Rheum, 2005. 52(10): p. 3020–9. [DOI] [PubMed] [Google Scholar]
- 50.Llamas-Covarrubias MA, et al. , Macrophage migration inhibitory factor (MIF): genetic evidence for participation in early onset and early stage rheumatoid arthritis. Cytokine, 2013. 61(3): p. 759–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Donn RP, et al. , Macrophage migration inhibitory factor gene polymorphism is associated with psoriasis. J Invest Dermatol, 2004. 123(3): p. 484–7. [DOI] [PubMed] [Google Scholar]
- 52.Morales-Zambrano R, et al. , Macrophage migration inhibitory factor (MIF) promoter polymorphisms (−794 CATT5–8 and −173 G>C): association with MIF and TNFalpha in psoriatic arthritis. Int J Clin Exp Med, 2014. 7(9): p. 2605–14. [PMC free article] [PubMed] [Google Scholar]
- 53.Amoli MM, et al. , Macrophage migration inhibitory factor gene polymorphism is associated with sarcoidosis in biopsy proven erythema nodosum. J Rheumatol, 2002. 29(8): p. 1671–3. [PubMed] [Google Scholar]
- 54.Wu SP, et al. , Macrophage migration inhibitory factor promoter polymorphisms and the clinical expression of scleroderma. Arthritis Rheum, 2006. 54(11): p. 3661–9. [DOI] [PubMed] [Google Scholar]
- 55.**.Leng L, et al. , MIF signal transduction initiated by binding to CD74. J Exp Med, 2003. 197(11): p. 1467–76. [DOI] [PMC free article] [PubMed] [Google Scholar]; First report of CD74 as the cell surface receptor for MIF.
- 56.Shi X, et al. , CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity, 2006. 25(4): p. 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Henne C, et al. , Surface expression of the invariant chain (CD74) is independent of concomitant expression of major histocompatibility complex class II antigens. Immunology, 1995. 84(2): p. 177–82. [PMC free article] [PubMed] [Google Scholar]
- 58.Bucala R and Shachar I, The integral role of CD74 in antigen presentation, MIF signal transduction, and B cell survival and homeostasis. Mini Rev Med Chem, 2014. 14(14): p. 1132–8. [DOI] [PubMed] [Google Scholar]
- 59.Lesley J, Hyman R, and Kincade PW, CD44 and its interaction with extracellular matrix. Adv Immunol, 1993. 54: p. 271–335. [DOI] [PubMed] [Google Scholar]
- 60.Ponta H, Sherman L, and Herrlich PA, CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol, 2003. 4(1): p. 33–45. [DOI] [PubMed] [Google Scholar]
- 61.Yoo SA, et al. , MIF allele-dependent regulation of the MIF coreceptor CD44 and role in rheumatoid arthritis. Proc Natl Acad Sci U S A, 2016. 113(49): p. E7917–E7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mitchell RA, et al. , Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J Biol Chem, 1999. 274(25): p. 18100–6. [DOI] [PubMed] [Google Scholar]
- 63.Gore Y, et al. , Macrophage migration inhibitory factor induces B cell survival by activation of a CD74-CD44 receptor complex. J Biol Chem, 2008. 283(5): p. 2784–92. [DOI] [PubMed] [Google Scholar]
- 64.Schneppenheim J, et al. , The intramembrane protease SPPL2a promotes B cell development and controls endosomal traffic by cleavage of the invariant chain. J Exp Med, 2013. 210(1): p. 41–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lantner F, et al. , CD74 induces TAp63 expression leading to B-cell survival. Blood, 2007. 110(13): p. 4303–11. [DOI] [PubMed] [Google Scholar]
- 66.Gordin M, et al. , c-Met and its ligand hepatocyte growth factor/scatter factor regulate mature B cell survival in a pathway induced by CD74. J Immunol, 2010. 185(4): p. 2020–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Assis DN, et al. , The role of macrophage migration inhibitory factor in autoimmune liver disease. Hepatology, 2014. 59(2): p. 580–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Soppert J, et al. , Soluble CD74 Reroutes MIF/CXCR4/AKT-Mediated Survival of Cardiac Myofibroblasts to Necroptosis. J Am Heart Assoc, 2018. 7(17): p. e009384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schwartz V, et al. , A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett, 2009. 583(17): p. 2749–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lacy M, et al. , Identification of an Arg-Leu-Arg tripeptide that contributes to the binding interface between the cytokine MIF and the chemokine receptor CXCR4. Sci Rep, 2018. 8(1): p. 5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alampour-Rajabi S, et al. , MIF interacts with CXCR7 to promote receptor internalization, ERK1/2 and ZAP-70 signaling, and lymphocyte chemotaxis. FASEB J, 2015. 29(11): p. 4497–511. [DOI] [PubMed] [Google Scholar]
- 72.Kapurniotu A, Gokce O, and Bernhagen J, The Multitasking Potential of Alarmins and Atypical Chemokines. Front Med (Lausanne), 2019. 6: p. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kleemann R, et al. , Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature, 2000. 408(6809): p. 211–6. [DOI] [PubMed] [Google Scholar]
- 74.Daun JM and Cannon JG, Macrophage migration inhibitory factor antagonizes hydrocortisone-induced increases in cytosolic IkappaBalpha. Am J Physiol Regul Integr Comp Physiol, 2000. 279(3): p. R1043–9. [DOI] [PubMed] [Google Scholar]
- 75.Roger T, et al. , Macrophage migration inhibitory factor promotes innate immune responses by suppressing glucocorticoid-induced expression of mitogen-activated protein kinase phosphatase-1. Eur J Immunol, 2005. 35(12): p. 3405–13. [DOI] [PubMed] [Google Scholar]
- 76.Aeberli D, et al. , Endogenous macrophage migration inhibitory factor modulates glucocorticoid sensitivity in macrophages via effects on MAP kinase phosphatase-1 and p38 MAP kinase. FEBS Lett, 2006. 580(3): p. 974–81. [DOI] [PubMed] [Google Scholar]
- 77.Wang FF, et al. , New insights into the role and mechanism of macrophage migration inhibitory factor in steroid-resistant patients with systemic lupus erythematosus. Arthritis Res Ther, 2012. 14(3): p. R103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bucala R, Approaching the immunophysiology of steroid resistance. Arthritis Res Ther, 2012. 14(3): p. 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Griga T, et al. , A polymorphism in the macrophage migration inhibitory factor gene is involved in the genetic predisposition of Crohn’s disease and associated with cumulative steroid doses. Hepatogastroenterology, 2007. 54(75): p. 784–6. [PubMed] [Google Scholar]
- 80.Santos LL, et al. , Reduced arthritis in MIF deficient mice is associated with reduced T cell activation: down-regulation of ERK MAP kinase phosphorylation. Clin Exp Immunol, 2008. 152(2): p. 372–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leech M, et al. , Regulation of p53 by macrophage migration inhibitory factor in inflammatory arthritis. Arthritis Rheum, 2003. 48(7): p. 1881–9. [DOI] [PubMed] [Google Scholar]
- 82.Ichiyama H, et al. , Inhibition of joint inflammation and destruction induced by anti-type II collagen antibody/lipopolysaccharide (LPS)-induced arthritis in mice due to deletion of macrophage migration inhibitory factor (MIF). Cytokine, 2004. 26(5): p. 187–94. [DOI] [PubMed] [Google Scholar]
- 83.Bae SC and Lee YH, Associations between circulating macrophage migration inhibitory factor (MIF) levels and rheumatoid arthritis, and between MIF gene polymorphisms and disease susceptibility: a meta-analysis. Postgrad Med J, 2018. 94(1108): p. 109–115. [DOI] [PubMed] [Google Scholar]
- 84.Onodera S, et al. , High expression of macrophage migration inhibitory factor in the synovial tissues of rheumatoid joints. Cytokine, 1999. 11(2): p. 163–7. [DOI] [PubMed] [Google Scholar]
- 85.Leech M, et al. , Macrophage migration inhibitory factor in rheumatoid arthritis: evidence of proinflammatory function and regulation by glucocorticoids. Arthritis Rheum, 1999. 42(8): p. 1601–8. [DOI] [PubMed] [Google Scholar]
- 86.Morand EF, New therapeutic target in inflammatory disease: macrophage migration inhibitory factor. Intern Med J, 2005. 35(7): p. 419–26. [DOI] [PubMed] [Google Scholar]
- 87.Onodera S, et al. , Macrophage migration inhibitory factor up-regulates matrix metalloproteinase-9 and −13 in rat osteoblasts. Relevance to intracellular signaling pathways. J Biol Chem, 2002. 277(10): p. 7865–74. [DOI] [PubMed] [Google Scholar]
- 88.Gu R, et al. , Macrophage migration inhibitory factor is essential for osteoclastogenic mechanisms in vitro and in vivo mouse model of arthritis. Cytokine, 2015. 72(2): p. 135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sampey AV, et al. , Regulation of synoviocyte phospholipase A2 and cyclooxygenase 2 by macrophage migration inhibitory factor. Arthritis Rheum, 2001. 44(6): p. 1273–80. [DOI] [PubMed] [Google Scholar]
- 90.You S, et al. , The Tumor-Like Phenotype of Rheumatoid Synovium: Molecular Profiling and Prospects for Precision Medicine. Arthritis Rheumatol, 2018. 70(5): p. 637–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Amin MA, et al. , Migration inhibitory factor mediates angiogenesis via mitogen-activated protein kinase and phosphatidylinositol kinase. Circ Res, 2003. 93(4): p. 321–9. [DOI] [PubMed] [Google Scholar]
- 92.Leng L, et al. , A small-molecule macrophage migration inhibitory factor antagonist protects against glomerulonephritis in lupus-prone NZB/NZW F1 and MRL/lpr mice. J Immunol, 2011. 186(1): p. 527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Foote A, et al. , Macrophage migration inhibitory factor in Systemic Lupus Erythematosus. J Rheumatol, 2004. 31: p. 268–273. [PubMed] [Google Scholar]
- 94.Sreih A, et al. , Dual effect of the macrophage migration inhibitory factor gene on the development and severity of human systemic lupus erythematosus. Arthritis Rheum, 2011. 63(12): p. 3942–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Connelly KL, et al. , Association of MIF, but not type I interferon-induced chemokines, with increased disease activity in Asian patients with systemic lupus erythematosus. Sci Rep, 2016. 6: p. 29909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mizue Y, et al. , Quantitation of macrophage migration inhibitory factor (MIF) using the one-step sandwich enzyme immunosorbent assay: elevated serum MIF concentrations in patients with autoimmune diseases and identification of MIF in erythrocytes. Int J Mol Med, 2000. 5(4): p. 397–403. [DOI] [PubMed] [Google Scholar]
- 97.Lan HY, et al. , Expression of macrophage migration inhibitory factor in human glomerulonephritis. Kidney Int, 2000. 57(2): p. 499–509. [DOI] [PubMed] [Google Scholar]
- 98.Vincent FB, et al. , Analysis of urinary macrophage migration inhibitory factor in systemic lupus erythematosus. Lupus Sci Med, 2018. 5(1): p. e000277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Colonna L, Lood C, and Elkon KB, Beyond apoptosis in lupus. Curr Opin Rheumatol, 2014. 26(5): p. 459–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Flaster H, et al. , The macrophage migration inhibitory factor-glucocorticoid dyad: regulation of inflammation and immunity. Mol Endocrinol, 2007. 21(6): p. 1267–80. [DOI] [PubMed] [Google Scholar]
- 101.Ouertatani-Sakouhi H, et al. , Kinetic-based high-throughput screening assay to discover novel classes of macrophage migration inhibitory factor inhibitors. J Biomol Screen, 2010. 15(4): p. 347–58. [DOI] [PubMed] [Google Scholar]
- 102.Cournia Z, et al. , Discovery of human macrophage migration inhibitory factor (MIF)-CD74 antagonists via virtual screening. J Med Chem, 2009. 52(2): p. 416–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jorgensen WL, et al. , Benzisothiazolones as modulators of macrophage migration inhibitory factor. Bioorg Med Chem Lett, 2011. 21(15): p. 4545–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Orita M, et al. , Macrophage migration inhibitory factor and the discovery of tautomerase inhibitors. Curr Pharm Des, 2002. 8(14): p. 1297–317. [DOI] [PubMed] [Google Scholar]
- 105.**.Senter PD, et al. , Inhibition of macrophage migration inhibitory factor (MIF) tautomerase and biological activities by acetaminophen metabolites. Proc Natl Acad Sci U S A, 2002. 99(1): p. 144–9. [DOI] [PMC free article] [PubMed] [Google Scholar]; Report on the first small molecule inhibitor of the MIF tautomerase site.
- 106.Lubetsky JB, et al. , The tautomerase active site of macrophage migration inhibitory factor is a potential target for discovery of novel anti-inflammatory agents. J Biol Chem, 2002. 277(28): p. 24976–82. [DOI] [PubMed] [Google Scholar]
- 107.Fagone P, et al. , Contribution of the macrophage migration inhibitory factor superfamily of cytokines in the pathogenesis of preclinical and human multiple sclerosis: In silico and in vivo evidences. J Neuroimmunol, 2018. 322: p. 46–56. [DOI] [PubMed] [Google Scholar]
- 108.Rajasekaran D, et al. , Targeting distinct tautomerase sites of D-DT and MIF with a single molecule for inhibition of neutrophil lung recruitment. FASEB J, 2014. 28(11): p. 4961–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Winner M, et al. , A novel, macrophage migration inhibitory factor suicide substrate inhibits motility and growth of lung cancer cells. Cancer Res, 2008. 68(18): p. 7253–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Imaoka M, et al. , Macrophage migration inhibitory factor-CD74 interaction regulates the expression of programmed cell death ligand 1 in melanoma cells. Cancer Sci, 2019. 110(7): p. 2273–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Soumoy L, et al. , Role of Macrophage Migration Inhibitory Factor (MIF) in Melanoma. Cancers (Basel), 2019. 11(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gunther S, et al. , Role of MIF and D-DT in immune-inflammatory, autoimmune, and chronic respiratory diseases: from pathogenic factors to therapeutic targets. Drug Discov Today, 2019. 24(2): p. 428–439. [DOI] [PubMed] [Google Scholar]
- 113.Mawhinney L, et al. , Macrophage migration inhibitory factor (MIF) enzymatic activity and lung cancer. Mol Med, 2015. 20: p. 729–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tynan A, et al. , Macrophage migration inhibitory factor enhances Pseudomonas aeruginosa biofilm formation, potentially contributing to cystic fibrosis pathogenesis. FASEB J, 2017. 31(11): p. 5102–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fox RJ, et al. , Phase 2 Trial of Ibudilast in Progressive Multiple Sclerosis. N Engl J Med, 2018. 379(9): p. 846–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cho Y, et al. , Allosteric inhibition of macrophage migration inhibitory factor revealed by ibudilast. Proc Natl Acad Sci U S A, 2010. 107(25): p. 11313–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ouertatani-Sakouhi H, et al. , Identification and characterization of novel classes of macrophage migration inhibitory factor (MIF) inhibitors with distinct mechanisms of action. J Biol Chem, 2010. 285(34): p. 26581–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bai F, et al. , A novel allosteric inhibitor of macrophage migration inhibitory factor (MIF). J Biol Chem, 2012. 287(36): p. 30653–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mikulowska A, et al. , Macrophage migration inhibitory factor is involved in the pathogenesis of collagen type II-induced arthritis in mice. J Immunol, 1997. 158(11): p. 5514–7. [PubMed] [Google Scholar]
- 120.Lan HY, et al. , The pathogenic role of macrophage migration inhibitory factor in immunologically induced kidney disease in the rat. J Exp Med, 1997. 185(8): p. 1455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mahalingam D, et al. , Safety and efficacy analysis of imalumab, an anti-oxidized macrophage migration inhibitory factor (oxMIF) antibody, alone or in combination with 5-fluorouracil/leucovorin (5-FU/LV) or panitumumab, in patients with metastatic colorectal cancer (mCRC). Annals of Oncology, 2016. 27(Supplement 2): p. ii102–ii117. [Google Scholar]
- 122.Stein R, et al. , Antiproliferative activity of a humanized anti-CD74 monoclonal antibody, hLL1, on B-cell malignancies. Blood, 2004. 104(12): p. 3705–11. [DOI] [PubMed] [Google Scholar]
- 123.Wallace DJ, et al. , CT-01 Phase IB study of IMMU-115 (humanised ANTI-CD74 antibody) targeting antigen presenting cells in patients with systemic lupus erythematosus (SLE). Lupus Science & Medicine, 2016. 3(Suppl 1): p. A37. [Google Scholar]
- 124.Harris J, et al. , Rediscovering MIF: New Tricks for an Old Cytokine. Trends Immunol, 2019. [DOI] [PubMed] [Google Scholar]
- 125.Alinari L, et al. , Combination anti-CD74 (milatuzumab) and anti-CD20 (rituximab) monoclonal antibody therapy has in vitro and in vivo activity in mantle cell lymphoma. Blood, 2011. 117(17): p. 4530–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Food and Drug Administration. Cumulative List of all Products that have received Orphan Designation. 2009, May 5; Available from: https://www.fda.gov/media/76409/download.
- 127.European Medicines Agency, EU/3/08/601. 2009, June 29.
- 128.European Medicines Agency. EU/3/08/602. 2009, June 29; Available from: https://www.ema.europa.edu/en/medicines/human/orphan-designations/eu308602.
- 129.Harmsen MM and De Haard HJ, Properties, production, and applications of camelid single-domain antibody fragments. Appl Microbiol Biotechnol, 2007. 77(1): p. 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sparkes A, et al. , Novel half-life extended anti-MIF nanobodies protect against endotoxic shock. FASEB J, 2018. 32(6): p. 3411–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Meza-Romero R, et al. , HLA-DRalpha1 constructs block CD74 expression and MIF effects in experimental autoimmune encephalomyelitis. J Immunol, 2014. 192(9): p. 4164–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Meza-Romero R, et al. , Predicted structure of MIF/CD74 and RTL1000/CD74 complexes. Metab Brain Dis, 2016. 31(2): p. 249–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Offner H, et al. , RTL therapy for multiple sclerosis: a Phase I clinical study. J Neuroimmunol, 2011. 231(1–2): p. 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Benedek G, et al. , MIF and D-DT are potential disease severity modifiers in male MS subjects. Proc Natl Acad Sci U S A, 2017. 114(40): p. E8421–E8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Figueiredo CR, et al. , Blockade of MIF-CD74 Signalling on Macrophages and Dendritic Cells Restores the Antitumour Immune Response Against Metastatic Melanoma. Front Immunol, 2018. 9: p. 1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kraemer S, et al. , MIF-chemokine receptor interactions in atherogenesis are dependent on an N-loop-based 2-site binding mechanism. FASEB J, 2011. 25(3): p. 894–906. [DOI] [PubMed] [Google Scholar]
- 137.Yende S, et al. , The influence of macrophage migration inhibitory factor gene polymorphisms on outcome from community-acquired pneumonia. FASEB J, 2009. 23(8): p. 2403–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Savva A, et al. , Functional polymorphisms of macrophage migration inhibitory factor as predictors of morbidity and mortality of pneumococcal meningitis. Proc Natl Acad Sci U S A, 2016. 113(13): p. 3597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]