

Abstract
Methylglyoxal (MGO) is a reactive glycolytic metabolite associated with painful diabetic neuropathy at plasma concentration is between 500 nM and 5 μM. The mechanisms through which MGO causes neuropathic pain at these pathological concentrations are not known. Because MGO has been linked to diabetic neuropathic pain, which is prevalent and poorly treated, insight into this unsolved biomedical problem could lead to much needed therapeutics. Our experiments provide compelling evidence that ~1-μM concentrations of MGO activate the integrated stress response (ISR) in IB4-positive nociceptors in the dorsal root ganglion (DRG) of mice in vivo and in vitro. Blocking the integrated stress response with a specific inhibitor (ISRIB) strongly attenuates and reverses MGO-evoked pain. Moreover, ISRIB reduces neuropathic pain induced by diabetes in both mice and rats. Our work elucidates the mechanism of action of MGO in the production of pain at pathophysiologically relevant concentrations and suggests a new pharmacological avenue for the treatment of diabetic and other types of MGO-driven neuropathic pain.
Keywords: Integrated stress response, Methylglyoxal, ISRIB, TRPA1, IB4 neurons
1. Introduction
Diabetic neuropathic pain is a debilitating and poorly treated condition that has been linked to excessive production of methylglyoxal (MGO).5 Methylglyoxal is a reactive byproduct of several metabolic pathways in cells. It is degraded by enzymes called glyoxalases, which have also been linked to painful neuropathies.23,47 Previous studies have shown that MGO concentrations in the low nanomolar range (100-250 nM) are found in plasma of healthy subjects; however, MGO levels are elevated in the plasma of patients with painful diabetic neuropathy ranging between 500 and 5000 nM.1,5 Although this suggests that MGO promotes diabetic neuropathic pain, a mechanism that fully accounts for how these concentrations of MGO cause pain has yet to emerge. Multiple groups have demonstrated that MGO concentrations in the high micromolar and even millimolar range activate the voltage-gated sodium channel Nav1.8 and the transient receptor potential channel TRPA1.2,5,17,21 Although these findings might give some insight into clinical conditions where MGO levels can rise to exceptionally high levels, these concentrations are 30 to 100× higher than those observed in diabetic neuropathic pain patients.1,5 The first question we examine is if MGO produces pain at levels commonly observed in painful diabetic neuropathy.
The integrated stress response (ISR) pathway is activated by diverse cellular stressors that include metabolic toxins, double-stranded RNAs, misfolded protein accumulation, and starvation.39 These stressors activate 4 kinases, GCN2, PRK, PERK, and HRI, all of which phosphorylate the eukaryotic translation initiation factor eIF2α. eIF2α is a key regulatory factor for protein translation.43 When eIF2α is not phosphorylated by eIF2α kinases, it promotes translation initiation. When eIF2α is phosphorylated, it strongly inhibits canonical open reading frame translation reducing total cellular protein output. Interestingly, this results in increased translation from the 5′ untranslated region (UTR) of many mRNAs through a process called upstream open reading frame translation.3,19 It has recently been demonstrated that experimental diabetes activates the ISR in peripheral nerves and interfering the ISR signaling with chemical chaperones alleviate this effect.22 How the ISR is activated in DRG neurons and their axons under conditions leading to diabetic neuropathic pain is not known.
We hypothesized that MGO-induced activation of the ISR provides a mechanistic explanation for how metabolic disturbances (eg, diabetes) produce neuropathic pain. Our work shows that MGO, at concentrations that are found in patients with painful diabetic neuropathy, results in activation of the ISR in primarily IB4-positive DRG nociceptors. MGO-evoked pain is blocked by the ISR inhibitor, integrated stress response with a specific inhibitor (ISRIB),45 and this compound also reverses pain in animals with diabetic neuropathy. We conclude that MGO produces pain through the ISR and that inhibitors of the ISR, such as ISRIB, are promising potential therapeutics for the treatment of neuropathic pain.
2. Material and methods
2.1. Experimental animals
All animal procedures were approved by the Institutional Animal Care and Use Committees at The University of Texas at Dallas and by the Institutional Animal Care and Use Committee at Cinvestav (Protocol 0092-14). In addition, experiments were performed in accordance with the guidelines of the International Association for the Study of Pain. Behavioral experiments using ICR mice obtained from Harlan Laboratories were performed using mice between 4 and 8 weeks of age, weighing ~20 to 25 g at the start of the experiment. Diabetes modeling in rats was performed using male wistar rats between 200 to 230 g at the time of the streptozotocin (STZ) administration. Male eIF4ES209A mice on a C57BL/6 background were generated in the Sonenberg laboratory at McGill University, as described previously, 16 and bred at The University of Texas at Dallas to generate experimental animals. All animals were genotyped using DNA from ear clips taken at the time of weaning, and all animals were backcrossed to C57BL/6 background for at least 10 generations before experiments. Except for the experiments involving eIF4ES209A mice, all in vitro experiments were performed using ICR mice between the ages of 4 and 6 weeks at the start of the experiment. Behavioral experiments using eIF4ES209A and wild-type (WT) mice were performed using mice between the ages of 8 and 12 weeks, weighing ~20 to 25 g.
2.2. Antibodies and chemicals
Isolectin B4 (IB4) conjugated to Alexa Fluor 568 (cat # I21412, 1;1000). Guinea pig antitransient receptor potential V1 (TRPV1) antibody was procured from Neuromics (cat # GP14100, 1:1000). Mouse anti-NeuN antibody (cat # MAB377, 1:1000) was obtained from Millipore. Rabbit anti-p-PERKThr981 (cat # sc-32577, 1:500) antibody was purchased from Santa Cruz Biotechnology. Rabbit primary antibodies against p-eIF2αSer51 (cat # 9721, 1:1000; cat # 3398, 1:200), eIF2alpha (cat # 9722, 1:1000), BiP (cat # 3177, 1:1000), p-ERK (cat # 9101S, 1:3000), ERK (cat # 9102S, 1:3000), p-RS6 (cat # 2317, 1:1000), RS6(cat # 2317, 1:1000), and GAPDH (cat # 2118, 1:10000) were obtained from Cell Signaling Technology. Mouse Anti-Puromycin antibody was obtained from Millipore (cat # MABE343, 1:5000). Prostaglandin E2 (PGE2) (cat # 363-24-6) was purchased from Cayman Chemicals. Anisomycin (cat # A9789), A967079 (cat # SML0085), methylglyoxal (cat # 67028), ISRIB (cat # SML0843), 4-PBA (cat # SML0309), metformin (cat # 1396309), and STZ (cat # S0130) were purchased from Sigma. A769662 (cat # 3336) was obtained from Tocris. Alexa Fluor- and HRP-conjugated secondary antibodies were obtained from Life Technologies.
2.3. Dorsal root ganglion cell culture and Western blotting
Animals were killed by decapitation following anesthesia, and tissues were immediately frozen on dry ice. Frozen tissues were homogenized in lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1 mm EDTA, pH 8.0, and 1% Triton X-100) containing protease and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO) and homogenized using a pestle. Cultured primary DRG neurons were used to test the effects of MGO-induced ISR activation and the effects of the ISR inhibitors. For these experiments, mice (~20 g) were anesthetized with isoflurane and killed by decapitation. Dorsal root ganglions were dissected and placed in chilled HBSS (Invitrogen) until processed. Dorsal root ganglions were then digested in 1-mg/mL collagenase A (Roche, Mannheim, Germany) for 25 minutes at 37°C then subsequently digested in a 1:1 mixture of 1 mg/mL collagenase D and papain (Roche) for 20 minutes at 37°C. Dorsal root ganglions were then triturated in a 1:1 mixture of 1-mg/mL trypsin inhibitor (Roche) and bovine serum albumin (BioPharm Laboratories, Bluffdale, UT), then filtered through a 70-μm cell strainer (Corning, NY). Cells were pelleted then resuspended in DMEM/F12 with GlutaMAX (Thermo Fisher Scientific, MA) containing 10% fetal bovine serum (FBS; Thermo Fisher Scientific), 5-ng/mL NGF, 1% penicillin and streptomycin, and 3-μg/mL 5-fluorouridine with 7-μg/mL uridine to inhibit mitosis of non-neuronal cells and were distributed evenly in a 6-well plate coated with poly-D-lysine (Becton Dickinson, NJ). Dorsal root ganglion neurons were maintained in a 37°C incubator containing 5% CO2 with a media change every other day. On day 5, DRG neurons were treated, rinsed with chilled 1 × phosphate-buffered saline (PBS), harvested in lysis buffer containing protease and phosphatase inhibitors (Sigma-Aldrich), and then sonicated for 10 seconds. To clear debris, samples were centrifuged at 14,000 rpm for 15 minutes at 4°C. Ten to 15 μg of protein was loaded into each well and separated by a 10% SDS-PAGE gel. Proteins were transferred to a 0.45 polyvinylidene difluoride membrane (Millipore, MA) at 30 V overnight at 4°C. Subsequently, membranes were blocked with 5% nonfat dry milk in 1 × Tris buffer solution containing Tween 20 (TTBS) for at least 2 hours. Membranes were washed in 1 × TTBS 3 times for 5 minutes each then incubated with primary antibodies overnight at 4°C. The following day, membranes were washed 3 times in 1 × TTBS for 5 minutes each then incubated with the corresponding secondary antibodies at room temperature for 1 hour. After incubation, membranes were washed with 1 × TTBS 6 times for 5 minutes each. Signals were detected using Immobilon Western Chemiluminescent HRP Substrate (Millipore) and then visualized with Bio-Rad ChemiDoc Touch. Membranes were stripped using Restore Western Blot Stripping buffer (Thermo Fisher Scientific) and reprobed with another antibody. Analysis was performed using Image lab 6.0.1 software for Mac (Bio-Rad, Hercules, CA).
2.4. Immunofluorescence
For primary neuronal cultures, DRG neurons were harvested and cultured for 5 days after the protocol described above with the exception that cells were distributed evenly on poly-D-lysine—coated coverslips (BD Falcon). After treatments, cells were fixed in ice-cold 10% formalin in 1 × PBS for 1 hour. Cells were then washed with 1 × PBS and permeabilized in PBS containing 10% heat-inactivated normal goat serum (NGS; Atlanta Biologicals, Atlanta, GA) and 0.02% Triton X-100 (Sigma) in 1 × PBS for 30 min and then blocked in 10% NGS in PBS for at least 1 hour. Primary antibodies were applied overnight at 4°C and the next day appropriate secondary antibodies (Alexa Fluor; Invitrogen) were applied for 1 hour. After additional PBS washes, coverslips were mounted on Superfrost plus slides with ProLong Gold antifade (Invitrogen). Images were taken using an Olympus FluoView 1200 confocal microscope and analyzed with ImageJ Version 1.48 (National Institutes of Health, Bethesda, MD) for Mac OS X (Apple). Images are representative of samples taken from 3 separate wells and presented as projections of z stacks. Using ImageJ, the corrected total cell fluorescence (CTCF) was calculated to determine the intensity of the signal between experimental groups. To do so, the integrated density and the area, as well as the background noise was measured and the CTCF calculated as equal to the integrated density – (area of selected cell × mean fluorescence of background readings). Corrected total cell fluorescence values from all groups were normalized to vehicle groups and expressed as %CTCF.
For tissues, animals were anesthetized with isoflurane and killed by decapitation, and tissues were frozen in O.C.T. on dry ice. Spinal cords were pressure ejected using chilled 1 × PBS. Sections of DRGs (20 mm) were mounted onto SuperFrost Plus slides (Thermo Fisher Scientific) and fixed in ice-cold 10% formalin in 1 × PBS for 1 hour then subsequently washed 3 times for 5 minutes each in 1 × PBS. Slides were permeabilized in 50% ethanol for 30 minutes. After 30 minutes, slides were washed 3 times for 5 minutes each in 1 × PBS. Tissues were blocked for at least 2 hours in 1× PBS and 10% NGS. Antibodies for IB4, TRPV1, NeuN, and p-eIF2αSer51 were applied and incubated with DRG sections on slides at 4°C overnight. Immunoreactivity was visualized after 1 hour incubation with Alexa Fluor secondary antibodies at room temperature. Images were taken using an Olympus FluoView 1200 confocal microscope, and colocalization analysis was performed as described previously.26 Images are presented as projections of z stacks, and they are representative of samples taken from 3 animals.
2.5. SUnSET assay
Dorsal root ganglion neurons were cultured for 5 days and then stimulated with MGO (1 μM) for 24 hours before the SUnSET assay.44 In the SUnSET approach, the structural analog of an aminoacyl-transfer RNA, puromycin, is used because it is readily incorporated into elongating polypeptides.37 This causes termination of peptide elongation and release of the nascent peptide. The levels of puromycin can be visualized using a highly specific monoclonal antibody. In our experiments, we used peripherin as a marker for nociceptive neurons. As a key negative control, we excluded the puromycin incubation. Homoharringtonine (50 μM, Sigma), an inhibitor of elongation, was used as a positive control and allowed to incubate at 37°C for 3 hours before the addition of puromycin (1 μM) for an additional 15 minutes. Immediately after the puromycin incubation, cells were washed in chilled HBSS containing 0.00036% digitonin (Sigma) for 2 min before fixation for the removal of background puromycin before immunofluorescence.
To calculate the puromycin incorporation, image analysis was performed using the ImageJ plug-in JACoP (Just Another Colocalization Plugin http://rsb.info.nih.gov/ij/plugins/track/jacop2.html).7 Manders’ overlap coefficient M1 (peripherin/puromycin; using thresholds) was calculated in images collected from all groups. The M1 coefficient varied from 0 to 1, the former corresponding to nonoverlapping images and the latter reflecting 100% colocalization between both images. The M1 overlap coefficient values obtained from all groups were normalized to vehicle + puromycin group values and expressed as % of normalized puromycin incorporation.
2.6. Ca2+ imaging
Dorsal root ganglion neurons were plated on glass-bottom poly-D-lysine-coated dishes (MatTek) and maintained in a 37°C incubator containing 5% CO2 with no media changes. One day after plating, cells were treated with MGO (1 μM) for 24 hours before the Ca2+ imaging experiments. Each dish was loaded with 10-μg/mL fura-2AM (Life Technologies, MA) in HBSS (Invitrogen, CA) supplemented with 0.25% wt/vol bovine serum albumin and 2-mm CaCl2 for 1 hour at 37°C. The cells were then changed to a bath solution (125-mM NaCl, 5-mM KCl, 10-mM HEPES, 1-mM CaCl2, 1-mM MgCl2, and 2-mM glucose, pH 7.4, adjusted with N-methyl glucamine to an osmolarity of ~300 mOsm) for 30 minutes in a volume of 2 mL for esterification. Dishes were then washed with 2 mL of bath solution before recordings. Only neurons were used in the analysis, and these were defined as cells with a ≥10% ratiometric change in intracellular Ca2+ in response to the 50-mM KCl perfusion. Maximum Ca2+ release was calculated by comparing the ratio value change by time compared with baseline. Experiments were conducted using the MetaFluor Fluorescence Ratio Imaging Software on an Olympus TH4–100 apparatus.
2.7. Behavior
Mice and rats were housed on 12-hour light/dark cycles with food and water available ad libitum. Mice were randomized to groups from multiple cages to avoid using mice from experimental groups that were cohabitating. Sample size was estimated by performing a power calculation using G*Power (version 3.1.9.2). With 80% power and an expectation of d = 2.2 effect size in behavioral experiments, and a set to 0.05, the sample size required was calculated as n = 6 per group. We therefore sought to have an n = 6 sample in all behavioral experiments. SD (set at 0.3) for the power calculation was based on previously published mechanical threshold data. The actual number of animals used in each experiment was based on available animals of the appropriate sex and weight but was at least n = 5 for behavior experiments. Animals were habituated for 1 hour to clear acrylic behavioral chambers before beginning the experiment. For intraplantar injections, drugs were injected in a total volume of 25 μL through a 30.5-gauge needle. For intrathecal injections, drugs were injected in a volume of 10 μL through a 30.5-gauge needle. For intraperitoneal injections, drugs were administered in a volume of 100 μL.
Mechanical paw withdrawal thresholds in mice and rats were measured using the up–down method with calibrated von Frey filaments (Stoelting Company, WoodDale, IL).11 To determine tactile allodynia in STZ-injected animals, von Frey filaments (Stoelting) were used to determine the 50% paw withdrawal threshold again using the up-down method. We calculated this threshold in mice and rats using the formula: 50% g threshold = 10(Xf + κδ)/10,000 where Xf = the value (in log units) of the final von Frey filament used, κ = the value from look up table for the pattern of positive and negative responses published previously,11 and ∂ = the mean difference (in log units) between stimuli. In normal rats (no pain), a value of 15 g was considered as the cutoff for stimulation. In rats injected with STZ, mechanical allodynia was considered to be present when paw withdrawal threshold below 4 g was observed, as previously described by Chaplan et al.11 Therefore, STZ rats that failed to achieve mechanical thresholds below 4 g were excluded from further experiments.
Thermal latency was measured using a Hargreaves device (IITC Life Science18) with heated glass Settings of 29°C glass, 40% active laser power, and 20 seconds cutoff were used.
The experimenter was blinded to the genotype of the mice and the drug condition in all experiments.
2.8. Streptozotocin-induced experimental diabetes in rodents
To produce experimental diabetes, male Wistar rats and C57BL/6 mice were intraperitoneally injected with a single dose of 50mg/kg and 150-mg/kg STZ, respectively. Control animals received an injection containing 0.9% saline. Diabetes was confirmed 1 week after injection by measurement of tail vein blood glucose levels. Only animals with a final blood glucose level of at least 15 mM were included in the study. Behavioral experiments with ISRIB were conducted at 6 weeks (mice) and 8 weeks (rats) after STZ administration.
2.9. Statistical analysis
All results are presented as the mean ± SEM. Statistical differences between 2 groups were determined by the Student t test. One- or 2-way analysis of variance, followed by Tukey, Dunnett, or Bonferroni test, was used to compare differences between more than 2 groups. Differences were considered to reach statistical significance when P < 0.05. Complete statistical analysis is detailed in figure legends.
3. Results
3.1. Methylglyoxal induces amplified pain behaviors in mice that require local protein synthesis
Plasma levels of MGO between 0.5 and 5 μM are correlated with painful neuropathy in humans.1,5,30 Substantially higher concentrations of MGO are associated with activation of ion channels including TRPA1.2,15,21 We sought to investigate the nociceptive responses produced by MGO in vivo across a broad range of concentrations [0.2 (0.1), 2 (1.1), 7.2 (4), and 720 (400) ng (μM) all in an injection volume of 25 μL]. Intraplantar administration of MGO, but not vehicle (PBS), produced a dose-dependent flinching behavior during the first 5 minutes after injection (Fig. 1A). Using the same dosing scheme, intraplantar administration of low doses of MGO (0.2, 2, and 7.2 ng) produced a short-term mechanical hypersensitivity to von Frey filament stimulation (Fig. 1B) lasting for approximately 1 day. A high dose of MGO (720 ng) produced a long-lasting mechanical hypersensitivity that lasted for ~2 weeks (Fig. 1C). We found that A967079 (30 μg, intraperitoneal [i.p.]), a TRPA1 channel antagonist, did not block the mechanical hypersensitivity produced by the administration of a low dose of MGO (Fig. 1D). A967079 did, however, produce a modest antinociceptive effect on mechanical hypersensitivity in response to a high dose of MGO (720 ng, Fig. 1E). This suggests that TRPA1 activation is not sufficient to fully account for MGO-evoked mechanical hypersensitivity.
Figure 1.
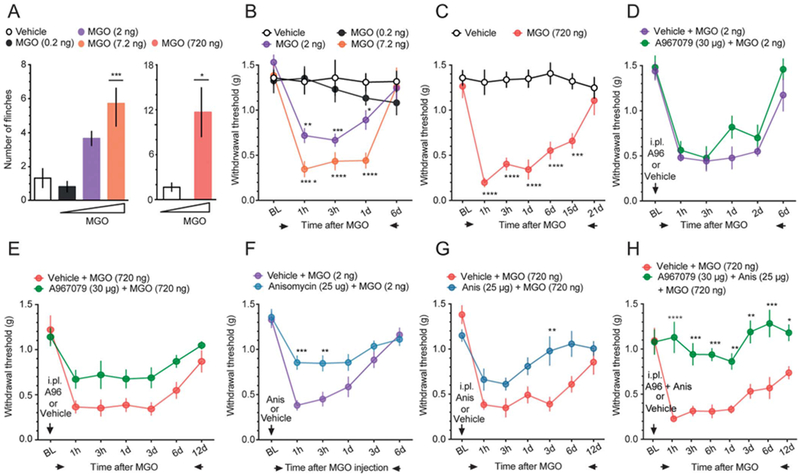
Intraplantar administration of methylglyoxal produces nociceptive responses: involvement of TRPA1 channels and nascent protein synthesis. (A) Intraplantar (i.p.) administration of MGO produces dose-dependent acute flinching behavior. One-way ANOVA: F(3, 20) = 10.22, P = 0.003; post-hoc Dunnett: ***P < 0.001, vehicle vs MGO (7.2 ng). Unpaired t test: t = 3.012; *P = 0.0131, vehicle vs MGO (720 ng). n = 6. (B) A low dose of MGO (0.2-7.2 ng/25 μL i.p.) induces a short-term mechanical hypersensitivity. Two-way ANOVA: F(12, 80) = 5.62, P < 0.001. Post-hoc Tukey; vehicle vs MGO (2 ng) at 1 hour: **P = 0.018, at 3 hours: ***P = 0.003, at 1 day: *P = 0.0398. vehicle vs MGO (7.2 ng) at 1 hour, 3 hours, 1 day: ****P = 0.0001. n = 6. (C) A high dose of MGO (720 ng/25 μL i.p.) produces long-lasting mechanical hypersensitivity. Two-way ANOVA: F(6, 60) = 8.163, P < 0.0001. Post-hoc Bonferroni; vehicle vs MGO (720 ng)at 1 hour, 3 hours, 1 day, and 6 days: ****P < 0.0001, at 15 days: ***P = 0.002. n = 6. (D) Methylglyoxal-induced short-term mechanical hypersensitivity is not blocked by A967079 (30 μg, i.p.), a TRPA1 channel antagonist. (E) A967079 (30 μg, i.p.) produced a minor antinociceptive effect after a high dose of MGO (720 ng/25 μL, i.p.). (F) Anisomycin (25 μg, i.p.), a nascent protein synthesis inhibitor, attenuates mechanical hypersensitivity produced by a low dose of MGO. Two-way ANOVA: F(5,132) = 3.343, P = 0.0071. Post-hoc Bonferroni; MGO + vehicle vs MGO + anisomycin at 1 hour: ****P = 0.0003, at 3 hours: **P = 0.0035. n = 12. (G) Mechanical hypersensitivity produced the high dose of MGO is prevented by anisomycin (25 μg, i.p.). Two-way ANOVA: F(6, 79) = 2.513, P = 0.0281. Post-hoc Bonferroni; MGO + vehicle vs MGO + anisomycin at 3 days: **P = 0.0035. (H) Coadministration of A967079 (30 μg, i.p.) and anisomycin (25 μg, i.p.) completely blocks the mechanical hypersensitivity produced by the high dose of MGO. Two-way ANOVA: F(7,70) = 3.866, P = 0.0013. Post-hoc Bonferroni; MGO + vehicle vs MGO + A967079 + Anisomycin at 1 hour: ****P = 0.0001, at 3 hours: *** P = 0.0004, at 6 hours: ***P = 0.0003, at 1 days: **P = 0.0038, at 3 days: ***P = 0.002, at 6 days: ****P < 0.0001, and at 12 days: *0.0254. n 5 6. ANOVA, analysis of variance.
Some studies have demonstrated that diabetic neuropathic pain is associated with activation of the ISR in peripheral nerves.22,31,32 Inspired by these observations, we hypothesized that MGO may be a causative factor in stimulating the ISR in diabetic neuropathic pain. Because the ISR is characterized by eIF2α phosphorylation, which can lead to translation from the 5′ UTR in a subset of mRNAs, we assessed whether a general protein synthesis inhibitor, anisomycin, would be capable to blocking MGO-evoked mechanical hypersensitivity. We injected low and high doses of intraplantar MGO in the presence of anisomycin (25 μg, i. p.), which partially blocked mechanical hypersensitivity produced by both low (Fig. 1F) and high (Fig. 1G) MGO doses. The long-lasting mechanical hypersensitivity produced by high-dose MGO was completely blocked by cotreatment with A967079 and anisomycin (Fig. 1H). These results indicate that MGO-induced pronociceptive responses require engagement of protein bio-synthetic pathways and, at high doses, TRPA1 channel activation.
3.2. Methylglyoxal activates the integrated stress response in mouse dorsal root ganglion neurons in vitro
To directly assess whether MGO induces the ISR (Fig. 2A) in DRG neurons, we cultured DRG neurons and stimulated them with 1-μM MGO, a low pathophysiological concentration that has been reported in the serum of humans suffering neuropathic pain.5,30 Methylglyoxal robustly increased the ISR marker immunoglobulin heavy-chainbinding protein (BiP) and produced the phosphorylation of the PKR-like endoplasmic reticulum-resident kinase (p-PERKThr981) (Fig. 2B), which in turn, increased the phosphorylation of the a subunit of the eukaryotic initiation factor 2 (eIF2) at serine 51 (p-eIF2αSer51) as measured by Western blot and immunocytochemistry (Figs. 2C and D). Activation of eIF2α is associated with a reduction in global translation.20 Using the SUnSET assay, we measured nascent protein synthesis levels in cultured DRG neurons. With vehicle treatment, we observed robust incorporation of puromycin indicative of robust levels of translation. However, introduction of either homoharringtonine, an inhibitor of elongation, or MGO significantly reduced nascent protein synthesis by 75% and 30%, respectively (Fig. 2E).
Figure 2.
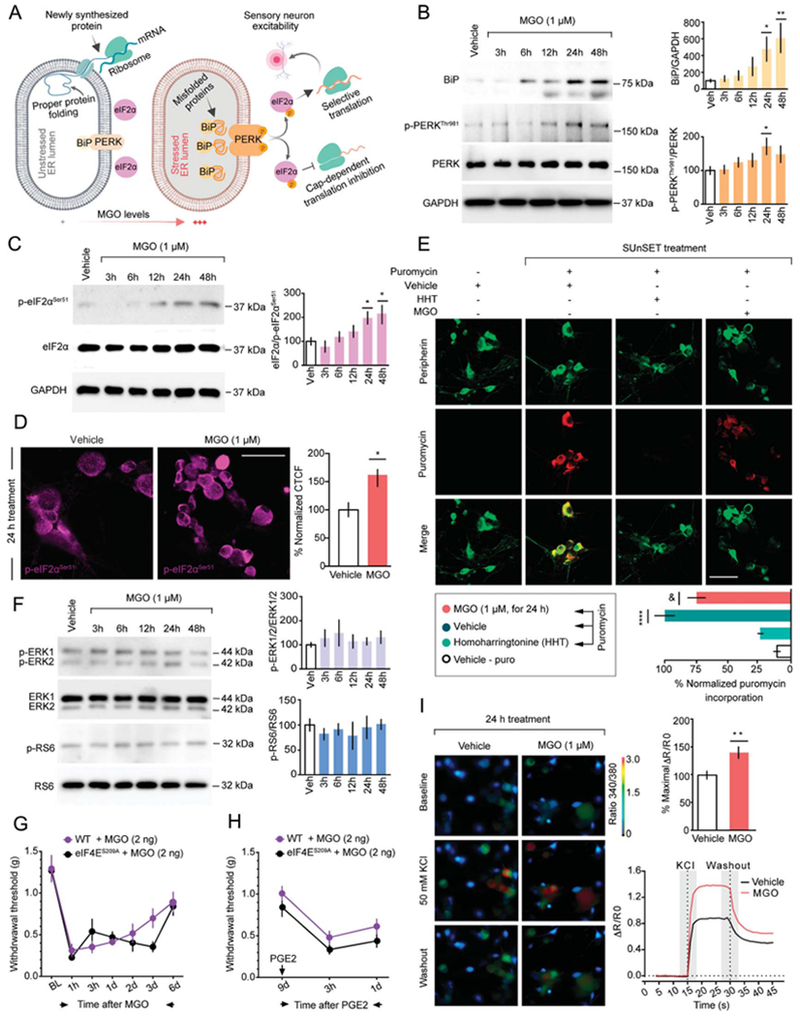
Methylglyoxal induces the ISR in cultured mouse DRG neurons. (A) Schematic representation of ISR signaling in DRG neurons under normal and pathophysiological MGO concentrations. (B) Incubation of DRG neurons at day 6 in vitro with MGO (1 μM) up-regulates the expression of the immunoglobulin heavy-chain-binding protein (BiP) and phosphorylates the PKR-like ER-localized eIF2α kinase (PERKThr981). For BiP: 1-way ANOVA, F(5,26) = 4.339, P = 0.0053; post-hoc Dunnett: vehicle (veh) + MGO at 24 hours: *P = 0.0472 and at 48 hours: **P = 0.0050. n = 5 to 6. For p-PERKThr981: 1-way ANOVA, F(5,30) = 2.161, P = 0.0851; post-hoc Dunnett: vehicle + MGO at 24 hours: *P = 0.0427. n = 6. (C and D) BiP activation and PERKThr981 phosphorylation converge on the subsequent phosphorylation of the eukaryotic initiation factor 2α at serine 51 (eIF2αSer51). For Western blot: 1-way ANOVA, F(5, 30) = 4.52, P = 0.0034; post-hoc Dunnett: vehicle + MGO at 24 hours: *P = 0.0475, at 48 hours: 0.0144. n = 6. For immunofluorescence: Unpaired t test: t = 2.396; *P = 0.0224, vehicle (n = 10) vs MGO (n = 25). (E) In the surface sensing of translation (SUnSET) method, cultured DRG neurons are incubated with MGO (1 μM) for 24 hours after addition of puromycin (1 μM) for an additional 15 minutes. Incubation with either MGO (1 μM for 24 hours) or the elongation inhibitor homoharringtonine (HHT) (50 μM for 1 hour), but not vehicle, significantly reduces nascent protein synthesis in DRG neurons. Staining is shown from top to bottom for puromycin (green), peripherin (red), or a merge. 1-way ANOVA, F(3, 21) = 28.83, P < 0.0001; post-hoc Tukey: ****P < 0.0001, veh-puro vs vehicle + puro; &P = 0.0445, vehicle + puro vs MGO + puro. (F) Treatment with MGO (1 μM) represses the phosphorylation of proteins mainly associated with cap-dependent translation such as the extracellular signal-regulated kinases 1 and 2 (ERK1/2) and the ribosomal protein S6. (G) Methylglyoxal (2 ng, i.p.) administration produces mechanical hypersensitivity in mice lacking phosphorylation of the cap-binding protein eIF4E (eIF4ES209A) and (H) precipitates mechanical hypersensitivity after an intraplantar injection of prostaglandin E2 (PGE2) at day 9 after i.p. MGO. (I) Activation of the ISR by MGO (1 μM, for 24 hours), but not vehicle, drives neuronal hyperexcitability in cultured DRG neurons revealed by an increase in Ca2+ signaling when they are stimulated with 50 mM KCl. Unpaired t test: = 3.13; *P = 0.0022, vehicle (n = 57) vs MGO (n = 76). Scale bar, 50 μm. ANOVA, analysis of variance; ISR, integrated stress response.
Because MGO induced eIF2α phosphorylation, we also assessed whether MGO influenced pathways that control protein synthesis through other mechanisms. Extracellular signal-regulated kinases 1 and 2 (ERK) and mechanistic target of rapamycin complex 1 (mTORC1) activity modulate translation through signaling to eIF4E, an initiation factor involved the regulation of cap-dependent translation.33,36 We found that MGO had no detectable influence on activation of ERK1/2 or the phosphorylation of the mTORC1 effector, ribosomal protein S6 (p-RS6, Fig. 2F). To support our in vitro findings, we assessed whether MGO engages eIF4E signaling in vivo. We found that mice lacking eIF4E phosphorylation at serine 209 (eIF4ES209A) show similar mechanical hypersensitivity compared with WT mice after intraplantar MGO administration (Fig. 2G). We also examined hyperalgesic priming in these mice because our previous work suggests that eIF4E signaling in nociceptors is critical for the establishment of hyperalgesic priming.36 No differences in mechanical hypersensitivity between WT and eIF4ES209A were observed when the animals were challenged with a 100-ng dose of prostaglandin E2 (PGE2) at day 9 (Fig. 2H). Therefore, as opposed to a variety of other stimuli that cause hyperalgesic priming, MGO produces priming but it does not depend on eIF4E signaling.
It has been shown that at a concentration of 150 μM in vitro, MGO increases DRG neuronal excitability while higher concentrations (250-750 μM) are cytotoxic in a dose-dependent manner.42 Using a similar time course to our data showing that MGO causes activation of the ISR, we assessed whether MGO is capable of sensitizing DRG neurons at lower concentrations. To do so, we exposed DRG neurons to 1-μM MGO for 24 hours and then assessed their intracellular Ca2+ levels in response to a depolarizing concentration of K+ ions. Dorsal root ganglion neurons treated with MGO showed an increase in Ca2+ responses to 50-mM KCl suggesting an increase in neuronal excitability (Fig. 2I). Together, our findings demonstrate that low μM concentrations of MGO activate the ISR, engage eIF2αSer51 phosphorylation, and promote sensitization of nociceptor excitability.
3.3. Methylglyoxal activates the integrated stress response primarily in IB4-positive dorsal root ganglion neurons in vivo
Our previous observations in vitro support the notion that MGO triggers the ISR and stimulates eIF2αSer51 phosphorylation in DRG neurons. We next characterized the profile of eIF2αSer51 phosphorylation in lumbar DRGs with sustained, systemic MGO administration in mice. We gave daily intraperitoneal (i.p.) MGO injections for 6 days using an MGO dose described to produce pain hypersensitivity in rodents.30 We found that i.p. administration of MGO, but not vehicle, produced a robust mechanical hypersensitivity in mice tested daily at 3 hours after the MGO injection (Fig. 3A). Interestingly, thermal hyperalgesia was only transiently induced during the early phase (day 1-2) of MGO treatment (Fig. 3B). Using the same experimental protocol, we found that MGO caused a sustained increase in eIF2αSer51 phosphorylation in lumbar DRGs (Fig. 3C). In addition, MGO also produced a robust increase on eIF2αSer51 phosphorylation in the sciatic nerve, but this effect was only observed at day 3 (Fig. 3C). This may be due to differential regulation of glyoxalase activity in the sciatic nerve vs in the DRG. We examined patterns of eIF2α phosphorylation within the DRG after MGO treatment. Methylglyoxal caused an increase of eIF2αSer51 phosphorylation in small–medium diameter neurons in the DRG (Fig. 3D).
Figure 3.
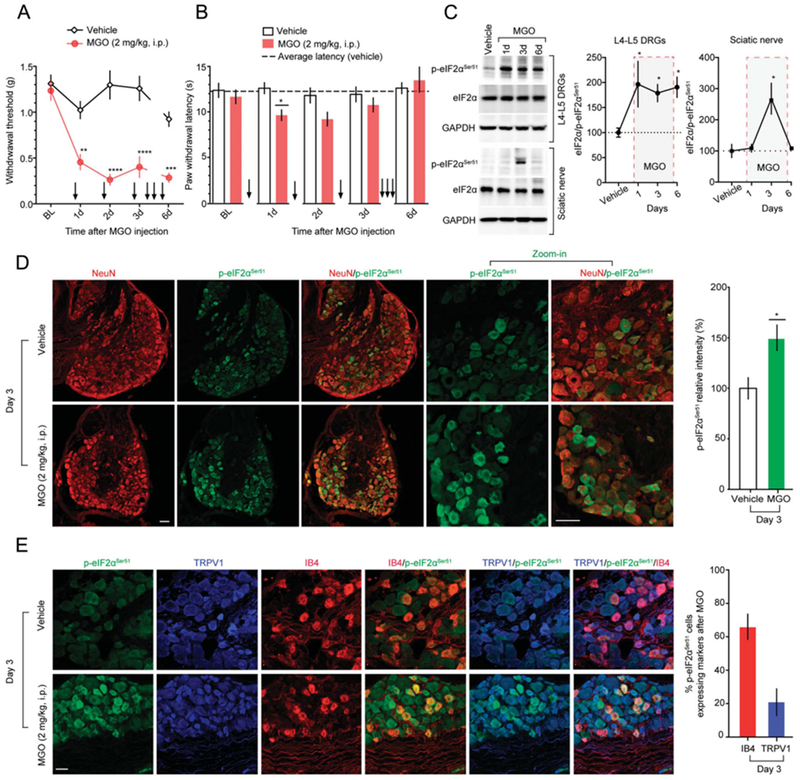
Methylglyoxal produces pain hypersensitivity and triggers the ISR in DRG neurons in vivo. (A) Daily administration of MGO (2 mg/kg, i.p; for 6 days) markedly increases mechanical sensitivity in mice tested at 3 hours after MGO injection. Two-way ANOVA, F(4, 40) = 7.156, P = 0.0002. Post-hoc Bonferroni; vehicle vs MGO at 1 days: **P = 0.0012, at 2 days: ****P < 0.0001, at 3 days: ****P = 0.0001, and at 6 days: ***P = 0.002. n = 6. (B) Methylglyoxal (2 mg/kg, i.p) produces a transient thermal hypersensitivity. Two-way ANOVA, F(4,107) = 1.771, P = 0.1400. Post-hoc Bonferroni; vehicle vs MGO at 1 day: *P = 0.0329. n = 12. (C) Methylglyoxal (2 mg/kg, i.p) produces a sustained increase on eIF2αSer51 phosphorylation, from day 1 to 6, in L4-L5 DRGs. One-way ANOVA, F(3,9) = 3.742, P = 0.05; post-hoc Dunnett: veh vs MGO at 1 day: *P = 0.0479, at 3 days: *P = 0.0456, at 6 days: *P = 0.0259. n = 4. Methylglyoxal also produces a transient significant increase on p-eIF2αSer51 at day 3 in the sciatic nerve of mice. One-way ANOVA, F(3,11) = 7.35, P = 0.0056. Post-hoc Dunnett: *P = 0.0055, vehicle vs MGO at day 3. n = 4. (D) The increase on eIF2αSer51 phosphorylation in the DRGs at day 3 is mainly present in neurons as shown by the colocalization with the neuronal marker NeuN. Unpaired t test: t = 2.967; *P = 0.0048, vehicle vs MGO. n = 24. (E) After i.p. MGO administration, 65.96% of p-eIF2α-positive cells in the DRG express IB4 and 20.82% express TRPV1. Scale bar, 50 μm. ANOVA, analysis of variance; ISR, integrated stress response.
To further classify the subset of DRG neurons that display eIF2α phosphorylation responsiveness, we performed immunostaining for transient receptor potential cation channel subfamily V member 1 (TRPV1) and isolectin (IB4), 2 well-known markers for peptidergic and nonpeptidergic nociceptive neurons, respectively. In naive mice, it has been recently reported that 36% of p-eIF2α–positive cells express IB4 and 20.8% express TRPV1.26 After 3 days of MGO treatment, we found that 65.96% of p-eIF2α–positive cells express IB4 and 20.82% express TRPV1, indicating that MGO induces the ISR primarily in nonpeptidergic neurons (Fig. 3E). This parallels our behavioral observations because the IB4 class of nociceptors plays a key role in mechanical but not thermal pain,10 whereas the TRPV1 population contributes primarily to thermal pain in the mouse.9
3.4. Targeting the integrated stress response with the small molecule inhibitor, integrated stress response with a specific inhibitor, blocks and reverses methylglyoxal-evoked pain
Integrated stress response with a specific inhibitor is a small molecule inhibitor of the ISR that interacts specifically with eIF2B to counteract the effect of eIF2α phosphorylation on cap-dependent translation.45,46,50,54 Integrated stress response with a specific inhibitor does not block eIF2α phosphorylation but long-term treatment with ISRIB may reverse eIF2α phosphorylation through an unknown mechanism.46 Intraplantar or intraperitoneal treatment with ISRIB prevented low-dose MGO-induced mechanical hypersensitivity (Figs. 4A and B). Similarly, intraperitoneal ISRIB on day 6 after high-dose MGO treatment transiently reversed long-lasting mechanical hypersensitivity produced by MGO (Fig. 4C). In cultured DRG neurons treated with MGO(1 μM) for 24 hours, ISRIB (200 nM) significantly reduced MGO-induced eIF2αSer51 phosphorylation (Figs. 4D and E). Incubation with either vehicle or ISRIB alone failed to yield significant changes on eIF2αSer51 phosphorylation. Moreover, mice treated contemporaneously with i.p. MGO and i.p. ISRIB for 3 days did not develop mechanical (Fig. 4F) or thermal (Fig. 4G) hypersensitivity. ISRIB also prevented increased eIF2αSer51 phosphorylation in L4-L5 DRGs taken from these mice as measured by Western blot (Fig. 4H) and immunohistochemistry (Fig. 4I). Finally, we assessed whether ISRIB could reverse mechanical hypersensitivity in mice or rats with diabetic neuropathic pain induced by STZ treatment 6 or 8 weeks previously, respectively. For these experiments, we used intrathecal doses of ISRIB because of the large amount of drug needed to dose rats with systemic injections and because of potential differences in pharmacokinetics between species with systemic dosing. Importantly, intrathecal drug delivery bathes DRGs with the administered drug. A single intrathecal administration of ISRIB (3-300 ng), but not vehicle, significantly reversed mechanical hypersensitivity, in a dose-dependent fashion, for about 6 hours in diabetic mice (Fig. 4J) and rats (Fig. 4K). Together, these results show that ISRIB both blocks and reverses MGO-induced pain in mice and rats, and that targeting the ISR with ISRIB may be an efficacious treatment in a model of diabetic neuropathic pain.
Figure 4.
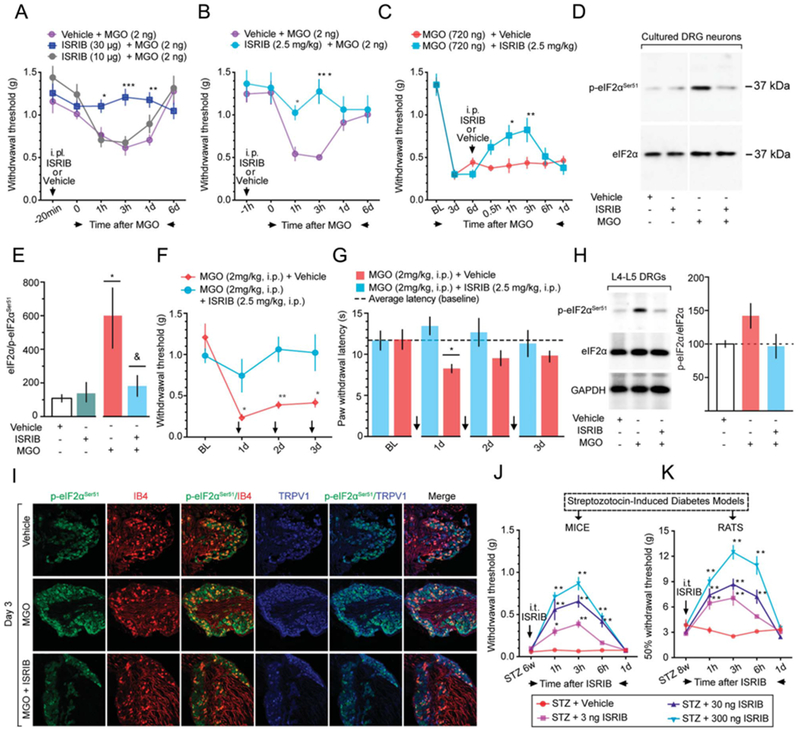
Integrated stress response with a specific inhibitor reverses the effects of eIF2αSer51 phosphorylation and attenuates mechanical hypersensitivity in a model of experimental diabetes. (A) i.p. (3,10 μg) or (B) i.p. (2.5 mg/kg) administration of ISRIB prevents the mechanical hypersensitivity produced by a low-dose MGO (2 ng, i.p.). For i.p., ISRIB: 2-way ANOVA, F(10,75) = 4.093, P = 0.002. Post-hoc Bonferroni: vehicle + MGO vs MGO + ISRIB (10 μg) at 1 hour: *P = 0.0386, at 3 hours: ***P = 0.001, at 24 hours: **P = 0.0028. For i.p. ISRIB: 2-way ANOVA, F(5,50) = 3.921, P = 0.0045. Post-hoc Bonferroni: vehicle + MGO vs MGO + ISRIB (2.5 mg/kg) at 1 hour: *P = 0.0396, and at 3 hours: ***P = 0.0002. n = 6. (C) Integrated stress response with a specific inhibitor (2.5 mg/kg, i.p.) partially reverses the long-lasting mechanical hypersensitivity produced by a high-dose MGO (720 ng, i.p.). Two-way ANOVA, F(7,70) = 3.037, P = 0.0076. Post-hoc Bonferroni; vehicle + MGO vs ISRIB + MGO at 1 hour: *P = 0.0219 and at 3 hours: **P = 0.0097. n = 6. (D and E) In cultured DRG neurons, ISRIB (200 nM, 4-hour treatment) reverses the eIF2αSer51 phosphorylation produced by MGO (1 μM, 24-hour treatment). One-way ANOVA, F(3,8) = 7.854, P = 0.0091. Post-hoc Dunnett: *P = 0.0178, vehicle vs MGO; &P = 0.405, MGO + vehicle vs MGO + ISRIB. n = 3. (F) Integrated stress response with a specific inhibitor (2.5 mg/kg, i.p.) for 3 consecutive days, prevents the mechanical and (G) thermal hypersensitivity, and normalizes the MGO-induced eIF2αSer51 phosphorylation in L4-L5 DRGs by Western blot (H) and IHC (I). For mechanical hypersensitivity: 2-way ANOVA, F(3, 30) = 6.528, P = 0.0016. Post-hoc Bonferroni; MGO + vehicle vs MGO + ISRIB at 1 day: *P = 0.0458, at 2 days: *P = 0.0043, and at 3 days: 0.0115. n = 6. Forthermal hypersensitivity: 2-way ANOVA, F(3,30) = 2.575, P = 0.0724. Post-hoc Bonferroni; MGO + vehicle vs MGO + ISRIB at 1 day: *P = 0.0116. n = 6. (J) In mice and (K) rats, ISRIB (3-300 ng, intrathecal), but not vehicle, reverses the mechanical hypersensitivity produced by streptozotocin-induced experimental diabetes. For mice (J): Two-way ANOVA, F(12,130) = 10.64, P < 0.0001. Post-hoc Bonferroni; STZ + vehicle vs STZ + ISRIB (3 ng) at 1h: *P = 0.0141, at 3 h: ****P < 0.0001. STZ + vehicle vs STZ + ISRIB (30 ng) at 1,3,6h: ****P < 0.0001. STZ + vehicle vs STZ + ISRIB (300 ng) at 1,3,6 h: ****P < 0.0001. n = 6. For rats (K): Two-way ANOVA, F(12,88) = 11.92, P < 0.0001. Post-hoc Bonferroni; STZ + vehicle vs STZ + ISRIB (3 ng) at 1 h: **P = 0.0017, at 3 h: ****P<0.0001. STZ + vehicle vs STZ + ISRIB (30 ng) at 1,3,6 h: ****P < 0.0001. STZ + vehicle vs STZ + ISRIB (300 ng) at 1,3,6 h: ****P < 0.0001. n = 6. Arrows in the figure indicate the time points of drug administration. Scale bar, 50 μm. ANOVA, analysis of variance.
Diabetes is commonly treated with the AMP-activated protein kinase (AMPK) activator, metformin, and other, more specific AMPK activators are in development for treatment of diabetes.13 Metformin and other AMPK activators seem to reduce diabetic neuropathic pain independently of glucose control.40 AMPK activation reduces eIF2α phosphorylation in several cell types in vitro and in vivo.8,14,29 4-PBA is a chemical chaperone that has been shown to reduce diabetic complications including neuropathic pain in rodents,22 and this compound also inhibits eIF2α phosphorylation.12,22,38 The specific AMPK activator A769662 and the chemical chaperone 4-PBA both attenuated low-dose MGO-evoked mechanical hypersensitivity (Figs. 5A and B). High-dose MGO-evoked mechanical hypersensitivity was also significantly reversed by systemic treatment with 4-PBA and metformin (Figs. 5C and D). We used DRG neurons in culture to assess whether these compounds could attenuate MGO-stimulated eIF2αSer51 phosphorylation. A-769662 and 4-PBA reversed the effect of MGO on eIF2αSer51 phosphorylation and metformin produced a trend toward reduced phosphorylation (Figs. 6A–E). These results further support the hypothesis that MGO induces the ISR to cause mechanical hypersensitivity.
Figure 5.
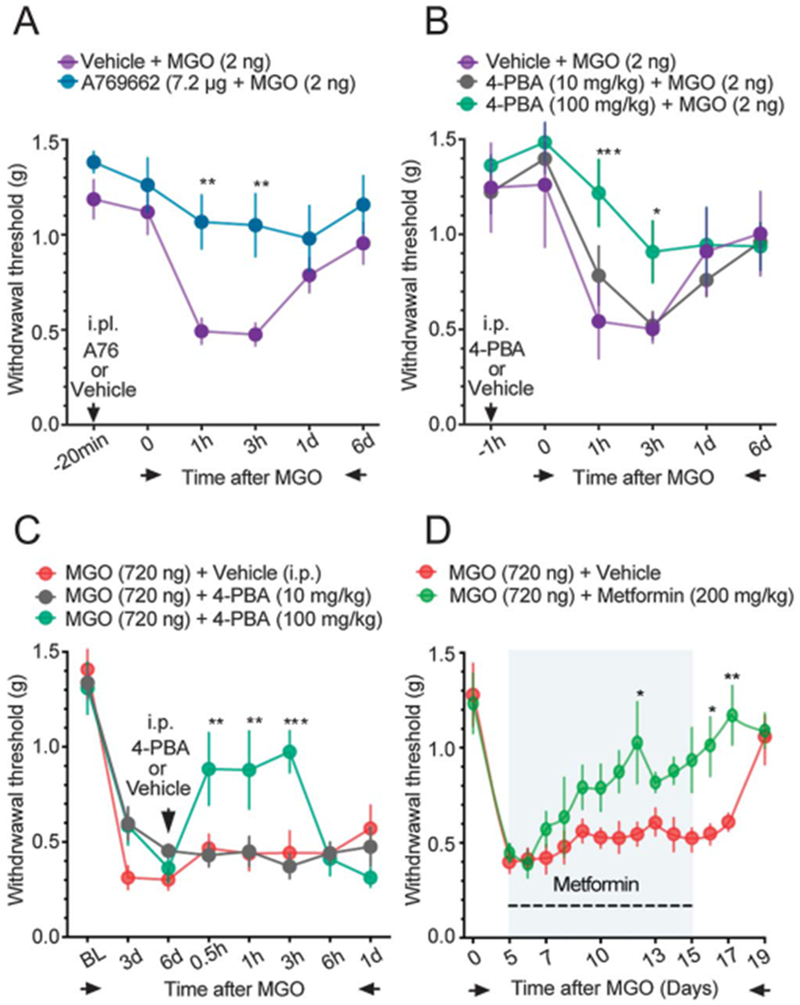
Effects of metformin, A769662, and 4-PBA on MGO-induced mechanical hypersensitivity. (A) A769662 (7.2 μg, i.p.) prevents the mechanical hypersensitivity produced by a low dose of MGO (2 μg, i.p.). (B) 4-PBA (100 mg/kg, i.p.) attenuates mechanical hypersensitivity produced by the low dose of MGO, whereas the low dose of 4-PBA (10 mg/kg) has no significant antinociceptive effect. For A769662: 2-way ANOVA, F(5, 60) = 1.424, P = 0.2287. Post-hoc Bonferroni; vehicle + MGO vs a769662 + MGO at 1 hour: **P = 0.0078, at 3 hours: **P = 0.0079. n = 6. For 4-PBA: 2-way ANOVA, F(10,90) = 1.591, P = 0.1221. Post-hoc Bonferroni; vehicle + MGO vs 4-PBA (100 mg/kg) + MGO at 1 hour: ***P = 0.0001, at 3 hours: **P = 0.0273. n = 6. (C) 4-PBA (10-100 mg/kg, i.p.) administration at day 6 reverses the mechanical hypersensitivity produced by a high-dose of MGO (720 ng, i.p.). Two-way ANOVA, F(14,105) = 3.857, P < 0.001. Post-hoc Bonferroni; MGO + vehicle vs MGO+ 4-PBA at 0.5 hours: **P = 0.0074, at 1 hour: **P = 0.0045, at 3 hours: ***P = 0.0005. n = 6. (D) Metformin (200 mg/kg, per os) administration for 10 days, starting at day 5, reverses the mechanical hypersensitivity and accelerates the recovery to baseline mechanical thresholds in mice injected with a high dose of MGO (720 ng, i.p.). Two-way ANOVA, F(13,130) = 1.872, P < 0.001. Post-hoc Bonferroni; MGO + vehicle vs MGO+metformin at 12 days: *P = 0.0143, at 15 days: P = 0.0762, at 16 days: **P = 0.0316, at 17 days: **P = 0.0029. n = 6. ANOVA, analysis of variance.
Figure 6.
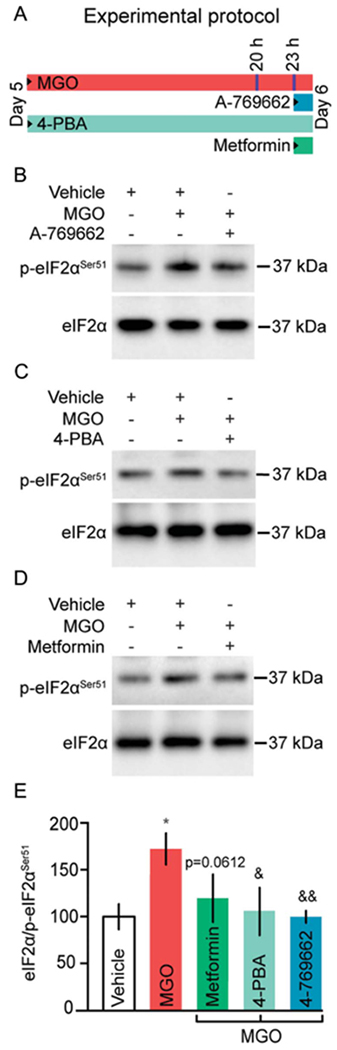
In vitro effects of A769662, 4-PBA, and metformin on MGO-induced eIF2αSer51 phosphorylation. (A) Schematic representation of the experimental protocol used for MGO or drug treatments. Cultured DRG neurons were stimulated at day 5 in vitro with MGO (1 μM) for 24 hours in the presence of vehicle, A769662 (200 μM, for 1 hour), 4-PBA (1 μM, for 24 hours), or metformin (20 mM, for 1 hour). (B) A-769662 and (C) attenuate eIF2αSer51 phosphorylation in cultured DRG neurons treated with MGO. (D) Treatment with metformin for 1 hour produces a trend towards the reduction of eIF2αSer51 phosphorylation. One-way ANOVA, F(4, 27) = 3.981, P = 0.0115. Post-hoc Bonferroni: *P = 0.0023, vehicle vs MGO; &P = 0.0612, MGO vs metformin; *P = 0.0023, MGO vs 4-PBA; &&P = 0.0072, vehicle vs A769662. n = 4. (E) Quantification of B–D. ANOVA, analysis of variance.
4. Discussion
Nerve injury is accompanied by changes in translational signaling that are associated with the pathogenesis of neuropathic pain. Although alterations in cap-dependent translation signaling have been widely observed, increased eIF2α phosphorylation has also been found repeatedly.22,26,31,32,34,52 The origin of eIF2α phosphorylation and the resulting consequences in DRG neurons have not been described previously. We find that MGO, which is associated with diabetic neuropathy5 as well as discogenic neuropathies,30 induces eIF2α phosphorylation in DRG neurons in vitro and in vivo at concentrations that are consistent with those observed in patients with diabetic neuropathic pain. Moreover, several studies have shown that MGO activates TRPA1 with an EC50 between 100 and 300 μM,15,21 concentrations that are 30 to 100 times higher than those found in patients. Although the exact mechanisms by which MGO enters the peripheral nerves are not entirely clear, it has been shown that MGO produces the redistribution of claudin-5 and β-catenin, 2 junctional proteins important for the regulation of the blood-brain barrier permeability.49 Specifically, MGO produces a redistribution of β-catenin in brain endothelial cells resulting in fragmentation and loss of the continuous cortical pattern of β-catenin. This phenomenon may produce damage to the basal lamina weakening microvascular wall structures resulting in an increase of brain vessel permeability to MGO.28,51 These same mechanisms are likely to be engaged at the nerve—blood barrier. Moreover, it is important to emphasize that, unlike the central nervous system, the cell body—rich area of the DRG possess a high density of platelet-endothelial cell adhesion molecule (CD31)-positive cells, and this anatomical feature is coupled with relative lack of a functional nerve—blood barrier in these capillaries.24 Therefore, we may expect that DRG neurons might be particularly likely to accumulate methylglyoxal specially because glyoxalase I (GLO1) expression and activity are constitutively very low in the peripheral nervous system.5,6 Our findings suggest that clinically relevant concentrations of MGO stimulate the ISR. Importantly, MGO-induced nociceptive plasticity requires protein synthesis and is robustly attenuated by the specific ISR inhibitor ISRIB. Because eIF2α phosphorylation and ISR induction stimulates translation from the 5′ UTR,19,48 we speculate that this form of translation leads to the production of proteins that sensitize the excitability of DRG neurons.
Taken together with the existing literature, our data suggest a new paradigm for understanding translation regulation and pain plasticity. Translation regulation is fundamentally required for the development of plasticity in nociceptors, which underlies chronic pain; however, the type of translation that is engaged differs substantially depending on the underlying cause of the plasticity. Unlike proinflammatory cytokines, growth factors and traumatic nerve injury, which all stimulate signaling to eIF4E through mTOR and ERK,25,35 MGO turns on ISR, engages eIF2α phosphorylation, and suppresses general, cap-dependent translation. However, MGO-evoked pain still requires active protein synthesis, indicating that translation events from the 5′ UTR48 likely underlie this effect. These translation regulation signaling events have important implications for understanding neuropathic pain mechanisms and developing therapeutics. Our work suggests that plasma levels of MGO could be developed as a biomarker for ISR-targeting therapies for neuropathic pain. Although more work is obviously needed to develop this idea, it would be one of the first biomarker-based patient stratification strategies for the treatment of neuropathic pain driven by specific mechanistic insight.
An intriguing aspect of our work is the relatively specific induction of the ISR by MGO in IB4-positive neurons. Methylglyoxal produces more robust mechanical than thermal hypersensitivity, and this effect is consistent with the known function of IB4-positive neurons in the generation of mechanical pain.10 An important question is why does MGO act with relative specificity on these neurons. TRPA1 is primarily expressed in these IB4-positive neurons but, again, the concentrations of MGO used in our study to activate the ISR are substantially less than is needed to activate the TRPA1 ion channel, suggesting that this may not be the cause. Other receptor-operated mechanisms or differential glyoxalase activity and expression may be altered in this subpopulation of sensory neurons to transduce this MGO signal to result in eIF2α phosphorylation. Our identification of this subset of nociceptor as a key target for MGO can enable discovery in this area. A key neuroanatomical feature of diabetic neuropathic pain is die-back of C-fiber innervation of the epidermis, an effect that occurs in mice and humans.4 The neurons that innervate the epidermis in mice are almost entirely IB4-positive. A key physiological feature of diabetic neuropathic pain is ongoing burning pain in affected extremities. This type of pain is believed to be driven primarily by TRPV1-expressing nociceptors. In mice, IB4-positve neurons mostly do not express TRPV1, and these IB4-positive neurons in mice do not contribute to heat pain.10,53 However, rat DRG sensory neurons show considerable overlap between TRPV1 and IB4,41 an overlap that is very likely consistent with what is observed in humans given that topical capsaicin clearly activates epidermal nerve fibers in man, and it has been suggested that most human epidermal nerve fibers show TRPV1 immunoreactivity,27 although this is an area of active debate. Interestingly, these TRPV1 immunoreactive epidermal nerve fibers seem to show die-back in painful neuropathies in humans.27 Therefore, although some neurochemical properties of these epidermally projecting neurons may differ between mice and humans, a selective effect of MGO on these neurons is supported by our mouse findings and consistent with the etiology of diabetic neuropathic pain in humans.
Our work demonstrates that pathophysiological concentrations of MGO activate the ISR in IB4-positive DRG neurons. This results in mechanical hypersensitivity that is attenuated by treatments that block the ISR. Our findings provide new insight into MGO-induced pain and provide a rationale for development of ISRIB as a neuropathic pain therapeutic.
Acknowledgments
The authors also thank members of the Price, Dussor, and Campbell laboratories for useful discussions that benefited the project.
This work was supported by NIH Grants R01NS065926 (T.J.P.), R01NS102161 (T.J.P.), and R01NS100788 (Z.T.C.); and Conacyt grants CB-2012/179294 (V.G.-S.) and CB-2013/222720 (V.G.-S.).
Footnotes
Conflict of interest statement
The authors have no conflict of interest to declare.
P. Barragan-Iglesias received a Postdoctoral Conacyt Fellowship (274414). A.B. Salinas-Abarca is a graduate Conacyt fellowship (277977).
References
- [1].Andersen ST, Witte DR, Dalsgaard EM, Andersen H, Nawroth P, Fleming T, Jensen TM, Finnerup NB, Jensen TS, Lauritzen T, Feldman EL, Callaghan BC, Charles M. Risk factors for incident diabetic polyneuropathy in a cohort with screen-detected type 2 diabetes followed for 13 years: ADDITION-Denmark. Diabetes Care 2018;41:1068–75. [DOI] [PubMed] [Google Scholar]
- [2].Andersson DA, Gentry C, Light E, Vastani N, Vallortigara J, Bierhaus A, Fleming T, Bevan S. Methylglyoxal evokes pain by stimulating TRPA1. PLoS One 2013;8:e77986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Barbosa C, Peixeiro I, Romao L. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet 2013;9: e1003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Beiswenger KK, Calcutt NA, Mizisin AP. Epidermal nerve fiber quantification in the assessment of diabetic neuropathy. Acta Histochem 2008;110:351–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bierhaus A, Fleming T, Stoyanov S, Leffler A, Babes A, Neacsu C, Sauer SK, Eberhardt M, Schnolzer M, Lasitschka F, Neuhuber WL, Kichko TI, Konrade I, Elvert R, Mier W, Pirags V, Lukic IK, Morcos M, Dehmer T, Rabbani N, Thornalley PJ, Edelstein D, Nau C, Forbes J, Humpert PM, Schwaninger M, Ziegler D, Stern DM, Cooper ME, Haberkorn U, Brownlee M, Reeh PW, Nawroth PP. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat Med 2012;18:926–33. [DOI] [PubMed] [Google Scholar]
- [6].Bierhaus A, Nawroth PP. Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia 2009;52:2251–63. [DOI] [PubMed] [Google Scholar]
- [7].Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 2006;224:213–32. [DOI] [PubMed] [Google Scholar]
- [8].Boss M, Newbatt Y, Gupta S, Collins I, Brune B, Namgaladze D. AMPK-independent inhibition of human macrophage ER stress response by AICAR. Sci Rep 2016;6:32111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 1997;389:816–24. [DOI] [PubMed] [Google Scholar]
- [10].Cavanaugh DJ, Lee H, Lo L, Shields SD, Zylka MJ, Basbaum AI, Anderson DJ. Distinct subsets of unmyelinated primary sensory fibers mediate behavioral responses to noxious thermal and mechanical stimuli. Proc Natl Acad Sci U S A 2009;106:9075–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994; 53:55–63. [DOI] [PubMed] [Google Scholar]
- [12].Chiang CK, Wang CC, Lu TF, Huang KH, Sheu ML, Liu SH, Hung KY. Involvement of endoplasmic reticulum stress, autophagy, and apoptosis in advanced glycation end products-induced glomerular mesangial cell injury. Sci Rep 2016;6:34167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Coughlan KA, Valentine RJ, Ruderman NB, Saha AK. AMPK activation: a therapeutic target for type 2 diabetes? Diabetes Metab Syndr Obes 2014;7:241–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dong Y, Zhang M, Wang S, Liang B, Zhao Z, Liu C, Wu M, Choi HC, Lyons TJ, Zou MH. Activation of AMP-activated protein kinase inhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes 2010;59: 1386–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Eberhardt MJ, Filipovic MR, Leffler A, de la Roche J, Kistner K, Fischer MJ, Fleming T, Zimmermann K, Ivanovic-Burmazovic I, Nawroth PP, Bierhaus A, Reeh PW, Sauer SK. Methylglyoxal activates nociceptors through transient receptor potential channel A1 (TRPA1): a possible mechanism of metabolic neuropathies. J Biol Chem 2012;287:28291–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Furic L, Rong L, Larsson O, Koumakpayi IH, Yoshida K, Brueschke A, Petroulakis E, Robichaud N, Pollak M, Gaboury LA, Pandolfi PP, Saad F, Sonenberg N. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc Natl Acad Sci U S A 2010;107:14134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Griggs RB, Laird DE, Donahue RR, Fu W, Taylor BK. Methylglyoxal requires AC1 and TRPA1 to produce pain and spinal neuron activation. Front Neurosci 2017;11:679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. PAIN 1988;32:77–88. [DOI] [PubMed] [Google Scholar]
- [19].Hinnebusch AG, Ivanov IP, Sonenberg N. Translational control by 5’-untranslated regions of eukaryotic mRNAs. Science 2016;352:1413–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hinnebusch AG, Lorsch JR. The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb Perspect Biol 2012;4: a011544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Huang Q, Chen Y, Gong N, Wang YX. Methylglyoxal mediates streptozotocin-induced diabetic neuropathic pain via activation of the peripheral TRPA1 and Nav1.8 channels. Metabolism 2016;65:463–74. [DOI] [PubMed] [Google Scholar]
- [22].Inceoglu B, Bettaieb A, Trindade da Silva CA, Lee KS, Haj FG, Hammock BD. Endoplasmic reticulum stress in the peripheral nervous system is a significant driver of neuropathic pain. Proc Natl Acad Sci U S A 2015; 112:9082–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jack MM, Ryals JM, Wright DE. Protection from diabetes-induced peripheral sensory neuropathy-a role for elevated glyoxalase I? Exp Neurol 2012;234:62–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jimenez-Andrade JM, Herrera MB, Ghilardi JR, Vardanyan M, Melemedjian OK, Mantyh PW. Vascularization of the dorsal root ganglia and peripheral nerve of the mouse: implications for chemical-induced peripheral sensory neuropathies. Mol Pain 2008;4:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Khoutorsky A, Price TJ. Translational control mechanisms in persistent pain. Trends Neurosci 2018;41:100–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Khoutorsky A, Sorge RE, Prager-Khoutorsky M, Pawlowski SA, Longo G, Jafarnejad SM, Tahmasebi S, Martin LJ, Pitcher MH, Gkogkas CG, Sharif-Naeini R, Ribeiro-da-Silva A, Bourque CW, Cervero F, Mogil JS, Sonenberg N. eIF2alpha phosphorylation controls thermal nociception. Proc Natl Acad Sci U S A 2016;113:11949–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lauria G, Morbin M, Lombardi R, Capobianco R, Camozzi F, Pareyson D, Manconi M, Geppetti P. Expression of capsaicin receptor immunoreactivity in human peripheral nervous system and in painful neuropathies. J Peripher Nerv Syst 2006;11:262–71. [DOI] [PubMed] [Google Scholar]
- [28].Li W, Maloney RE, Circu ML, Alexander JS, Aw TY. Acute carbonyl stress induces occludin glycation and brain microvascular endothelial barrier dysfunction: role for glutathione-dependent metabolism of methylglyoxal. Free Radic Biol Med 2013;54:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Liang B, Wang S, Wang Q, Zhang W, Viollet B, Zhu Y, Zou MH. Aberrant endoplasmic reticulum stress in vascular smooth muscle increases vascular contractility and blood pressure in mice deficient of AMP-activated protein kinase-alpha2 in vivo. Arterioscler Thromb Vasc Biol 2013;33:595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu CC, Zhang XS, Ruan YT, Huang ZX, Zhang SB, Liu M, Luo HJ, Wu SL, Ma C. Accumulation of methylglyoxal increases the advanced glycation end-product levels in DRG and contributes to lumbar disk herniation-induced persistent pain. J Neurophysiol 2017;118:1321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lupachyk S, Watcho P, Obrosov AA, Stavniichuk R, Obrosova IG. Endoplasmic reticulum stress contributes to prediabetic peripheral neuropathy. Exp Neurol 2013;247:342–8. [DOI] [PubMed] [Google Scholar]
- [32].Lupachyk S, Watcho P, Stavniichuk R, Shevalye H, Obrosova IG. Endoplasmic reticulum stress plays a key role in the pathogenesis of diabetic peripheral neuropathy. Diabetes 2013;62:944–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Melemedjian OK, Asiedu MN, Tillu DV, Peebles KA, Yan J, Ertz N, Dussor GO, Price TJ. IL-6- and NGF-induced rapid control of protein synthesis and nociceptive plasticity via convergent signaling to the eIF4F complex. J Neurosci 2010;30:15113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Melemedjian OK, Asiedu MN, Tillu DV, Sanoja R, Yan J, Lark A, Khoutorsky A, Johnson J, Peebles KA, Lepow T, Sonenberg N, Dussor G, Price TJ. Targeting adenosine monophosphate-activated protein kinase (AMPK) in preclinical models reveals a potential mechanism for the treatment of neuropathic pain. Mol Pain 2011;7:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Melemedjian OK, Khoutorsky A. Translational control of chronic pain. Prog Mol Biol Transl Sci 2015;131:185–213. [DOI] [PubMed] [Google Scholar]
- [36].Moy JK, Khoutorsky A, Asiedu MN, Black BJ, Kuhn JL, Barragan-Iglesias P, Megat S, Burton MD, Burgos-Vega CC, Melemedjian OK, Boitano S, Vagner J, Gkogkas CG, Pancrazio JJ, Mogil JS, Dussor G, Sonenberg N, Price TJ. The MNK-eIF4E signaling axis contributes to injury-induced nociceptive plasticity and the development of chronic pain. J Neurosci 2017;37:7481–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nathans D Puromycin inhibition of protein synthesis: incorporation of puromycin into peptide chains. Proc Natl Acad Sci U S A 1964;51:585–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006;313:1137–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep 2016;17:1374–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Price TJ, Das V, Dussor G. Adenosine monophosphate-activated protein kinase (AMPK) activators for the prevention, treatment and potential reversal of pathological pain. Curr Drug Targets 2016;17:908–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Price TJ, Flores CM. Critical evaluation of the colocalization between calcitonin gene-related peptide, substance P, transient receptor potential vanilloid subfamily type 1 immunoreactivities, and isolectin B4 binding in primary afferent neurons of the rat and mouse. J Pain 2007;8:263–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Radu BM, Dumitrescu DI, Mustaciosu CC, Radu M. Dual effect of methylglyoxal on the intracellular Ca2+ signaling and neurite outgrowth in mouse sensory neurons. Cell Mol Neurobiol 2012;32:1047–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ron D Translational control in the endoplasmic reticulum stress response. J Clin Invest 2002;110:1383–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 2009;6:275–7. [DOI] [PubMed] [Google Scholar]
- [45].Sidrauski C, McGeachy AM, Ingolia NT, Walter P. The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife 2015;4:e05033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sidrauski C, Tsai JC, Kampmann M, Hearn BR, Vedantham P, Jaishankar P, Sokabe M, Mendez AS, Newton BW, Tang EL, Verschueren E, Johnson JR, Krogan NJ, Fraser CS, Weissman JS, Renslo AR, Walter P. Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the integrated stress response. Elife 2015;4:e07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Skapare E, Konrade I, Liepinsh E, Strele I, Makrecka M, Bierhaus A, Lejnieks A, Pirags V, Dambrova M. Association of reduced glyoxalase 1 activity and painful peripheral diabetic neuropathy in type 1 and 2 diabetes mellitus patients. J Diabetes Complications 2013;27:262–7. [DOI] [PubMed] [Google Scholar]
- [48].Starck SR,Tsai jC , Chen K, Shodiya M,Wang L,Yahiro K, Martins-Green M, Shastri N, Walter P. Translation from the 5’ untranslated region shapes the integrated stress response. Science 2016;351:aad3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Toth AE, Walter FR, Bocsik A, Santha P, Veszelka S, Nagy L, Puskas LG, Couraud PO,Takata F, Dohsgu S, Kataoka Y, Deli MA. Edaravone protects against methylglyoxal-induced barrier damage in human brain endothelial cells. PLoS One 2014;9:e100152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Tsai JC, Miller-Vedam LE, Anand AA, Jaishankar P, Nguyen HC, Renslo AR, Frost A, Walter P. Structure of the nucleotide exchange factor eIF2B reveals mechanism of memory-enhancing molecule. Science 2018;359: eaaq0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wang CX, Shuaib A. Critical role of microvasculature basal lamina in ischemic brain injury. Prog Neurobiol 2007;83:140–8. [DOI] [PubMed] [Google Scholar]
- [52].Yang ES, Bae JY, Kim TH, Kim YS, Suk K, Bae YC. Involvement of endoplasmic reticulum stress response in orofacial inflammatory pain. Exp Neurobiol 2014;23:372–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].ZwickM Davis BM, Woodbury CJ Burkett JN, Koerber HR Simpson JF, Albers KM. Glial cell line-derived neurotrophic factor is a survival factor for isolectin B4-positive, but not vanilloid receptor 1-positive, neurons in the mouse. J Neurosci 2002;22:4057–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zyryanova AF, Weis F, Faille A, Alard AA, Crespillo-Casado A, Sekine Y, Harding HP, Allen F, Parts L, Fromont C, Fischer PM, Warren AJ, Ron D. Binding of ISRIB reveals a regulatory site in the nucleotide exchange factor eIF2B. Science 2018;359:1533–6. [DOI] [PMC free article] [PubMed] [Google Scholar]