

Abstract
Protein kinase RNA-activated (PKR) is an interferon-inducible kinase that is potently activated by long double-stranded RNA (dsRNA). In a previous study, we found that snoRNAs exhibit increased association with PKR in response to metabolic stress. While it was unclear if snoRNAs also activated PKR in cells, activation in vitro was observed. snoRNAs do not exhibit the double-stranded character typically required for activation of PKR, but some studies suggest such RNAs can activate PKR if triphosphorylated at the 5′ terminus, or if they are able to form intermolecular dimers. To interrogate the mechanism of PKR activation by snoRNAs in vitro we focused on SNORD113. Using multiple methods for defining the 5′-phosphorylation state, we find that activation of PKR by SNORD113 does not require a 5′-triphosphate. Gel purification from a native gel followed by analysis using analytical ultracentrifugation showed that dimerization was also not responsible for activation. We isolated distinct conformers of SNORD113 from a native polyacrylamide gel and tracked the activating species to dsRNA formed from antisense RNA synthesized during in vitro transcription with T7 RNA polymerase. Similar studies with additional snoRNAs and small RNAs showed the generality of our results. Our studies suggest that a 5′ triphosphate is not an activating ligand for PKR, and emphasize the insidious nature of antisense contamination.
Keywords: PKR, snoRNA, dsRNA, antisense, T7 RNA polymerase, ss-dsRNA
INTRODUCTION
Protein kinase RNA-activated (PKR) is a ubiquitously expressed serine/threonine kinase that regulates translation in response to various cellular insults (Williams 1999; Garcia et al. 2006). Commonly described as a cellular RNA sensor for double-stranded RNA (dsRNA) produced during viral infection, PKR recognizes RNA with its amino-terminal dsRNA-binding domain (dsRBD) (Nanduri et al. 1998). The mechanism of PKR activation, in vitro and in vivo, has been predominantly interrogated by using the canonical PKR activator, dsRNA [e.g., poly(I:C)] (Tian and Mathews 2001; Cole 2007). Studies reveal that upon binding dsRNA, PKR is activated by a conformational change that promotes autophosphorylation via the carboxy-terminal kinase domain (Taylor et al. 1996; Romano et al. 1998). PKR and other dsRNA binding proteins bind dsRNA in a sequence-independent manner (Nanduri et al. 1998; Saunders and Barber 2003).
Several reports indicate that small RNAs containing single- and double-stranded regions (ss-dsRNA) can also activate PKR (Bevilacqua et al. 1998; Zheng and Bevilacqua 2004; Nallagatla et al. 2007; Mayo et al. 2016). Indeed, in previous studies we identified an interaction between PKR and small nucleolar RNAs (snoRNAs), in mouse embryonic fibroblasts, that is enhanced under conditions of metabolic stress (Youssef et al. 2015). We also observed that, at least in vitro, PKR is activated by both classes of snoRNA, box H/ACA and box C/D, and further, that all snoRNAs can activate PKR in vitro, even those that did not exhibit enhanced binding to PKR under conditions of metabolic stress. This led us to investigate the structural properties of snoRNAs that contribute to PKR activation in vitro.
In the absence of the conserved small nucleolar ribonucleoproteins that interact with snoRNAs in the nucleolus, predicted structures of snoRNAs show conformations that resemble imperfect hairpins (Youssef et al. 2015). Small hairpin RNAs are reported to activate PKR, and in some cases this activation requires the presence of a 5′-triphosphate (5′-ppp) (Nallagatla et al. 2007). Our initial investigation revealed that only some snoRNAs require a 5′-ppp to activate PKR, which we speculated reflected the specific secondary structure of the RNA. Recently, the dependence of PKR activation on the presence of a 5′-ppp for ss-dsRNAs has been called into question (Mayo et al. 2016).
In hopes of resolving existing controversies, and further defining what is necessary for an RNA to activate PKR, we performed detailed analyses of PKR activation in vitro. We focused on activation by SNORD113, the snoRNA previously identified as having the strongest dependence on the presence of a 5′-ppp (Youssef et al. 2015), but found that other RNAs showed similar properties. We examined the contribution of the 5′-ppp and RNA purification strategies to PKR activation. Our studies revealed that, for all tested RNAs, the 5′-ppp is dispensable for PKR activation. Most importantly, we determined that a minor conformer of SNORD113, which contains antisense SNORD113, is largely responsible for the activation of PKR. Our studies call into question the idea that PKR requires a 5′-ppp for activation by certain substrates.
RESULTS
PKR activation by SNORD113 does not require a 5′-triphosphate
We initially utilized two common methods to generate SNORD113 with and without a 5′-ppp. We used T7 RNA polymerase (RNAP) in in vitro transcription reactions to produce SNORD113 with a 5′-ppp. SNORD113 with a 5′-hydroxyl (5′-OH) was made by chemical synthesis. Each species of SNORD113 was gel purified after denaturing polyacrylamide gel electrophoresis (PAGE) prior to PKR activation assays. PKR is subject to substrate inhibition, displaying a bell-shaped curve of activation over a range of RNA concentrations. To make meaningful comparisons, we tested a range of RNA concentrations, allowing us to visualize at least part of the activation curve. In agreement with our previous experiments (Youssef et al. 2015), we saw strong activation of PKR with in vitro transcribed SNORD113 (5′-ppp), but not synthetic SNORD113 (5′-OH) (Fig. 1). To validate the importance of the 5'-ppp, we treated in vitro transcripts with three different phosphatases. For each enzyme treatment, a portion of SNORD113 transcript was set aside and left completely untreated (stock), incubated with buffer without phosphatase (mock treated), or incubated with the phosphatase. Somewhat surprisingly in light of prior studies (Nallagatla et al. 2007), treatment with calf intestinal phosphatase (CIP) and Antarctic phosphatase (AnP), which remove all three 5′-phosphates to leave a 5′-OH, as well as RNA 5′ pyrophosphohydrolase (RppH), which removes the pyrophosphate to leave a 5′-monophosphate (5′-p), did not alter the ability of the in vitro transcript to activate PKR (Fig. 2A,B). T4 polynucleotide kinase (T4 PNK) catalyzes the transfer of the ɣ-phosphate of ATP to a 5′-OH terminus of DNA or RNA (Richardson 1981). Control experiments showed that SNORD113 samples treated with phosphatase, but not those mock-treated, could be labeled with a 5′-32P using T4 PNK and [ɣ-32P]ATP, suggesting phosphatase treatments were effective at removing phosphates (Supplemental Fig. 1A). Additionally, stock- and mock-treated SNORD113 were not substrates for terminator 5′-phosphate-dependent exonuclease, which processively digests RNA only with a 5′-monophosphate (Supplemental Fig. 1B). Phosphatase-treated SNORD113 subsequently subjected to T4 PNK treatment with ATP to generate RNA species with a 5′-monophosphate was a substrate for Terminator (Supplemental Fig. 1B). Taken together, these results confirm that the majority of each of the RNA samples has the predicted 5′ termini after mock or phosphatase treatment and that a 5′-ppp is not required for activation of PKR by in vitro transcribed SNORD113.
FIGURE 1.
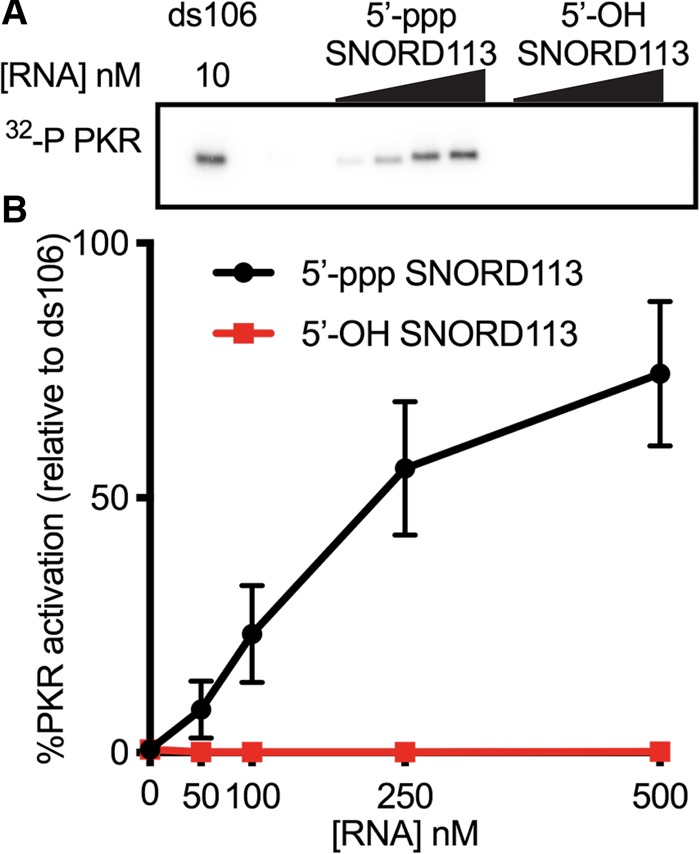
PKR is activated by in vitro transcribed SNORD113, but not chemically synthesized SNORD113. Each SNORD113 species was initially purified by denaturing PAGE prior to the PKR autophosphorylation assay. (A) Representative PhosphorImage of an autophosphorylation assay with in vitro transcribed SNORD113 (5′-ppp) and chemically synthesized SNORD113 (5′-OH). SNORD113 concentrations were 50, 100, 250, and 500 nM; perfectly duplexed RNA 106 bp in length (ds106) was 10 nM. (B) Activation data as assayed in A were normalized to ds106 and averaged values were plotted. Error bars ± SD; n = 3 with at least two biological replicates per sample.
FIGURE 2.

PKR is activated by in vitro transcribed SNORD113 after phosphatase treatment. SNORD113 was initially purified by denaturing PAGE followed by the indicated phosphatase treatments. (A) Representative PhosphorImage of an autophosphorylation assay with in vitro transcribed SNORD113 treated without (mock) or with the indicated phosphatase; (CIP) calf intestinal phosphatase, (AnP) antarctic phosphatase, (RppH) RNA 5′ pyrophosphohydrolase. SNORD113, 100 nM, 500 nM, 1000 nM; ds106, 10 nM. (B) Activation data as assayed in A were normalized to ds106 and averaged values were plotted. Error bars ± SD; n = 3 with at least two biological replicates.
PKR activation is affected by gel purification of SNORD113
After treatment with CIP or Antarctic phosphatase, SNORD113 should have a 5′-hydroxyl, the same terminus found on the chemically synthesized SNORD113. Yet, only the phosphatase-treated SNORD113 was able to activate PKR. Thus, we next focused on understanding why phosphatase-treated SNORD113 activated PKR while chemically synthesized SNORD113 did not, even though both RNAs had a 5′-OH. Interestingly, studies from other laboratories indicate that two RNAs, similar in structure, show different PKR activation after CIP treatment, calling into question the importance of the 5′-ppp (Nallagatla et al. 2007; Mayo et al. 2016). Comparison of the CIP protocols used in these studies revealed two main differences: (i) quenching CIP activity with EDTA, and (ii) gel purification after CIP treatment (Nallagatla et al. 2007; Mayo et al. 2016). We tested each variable to understand the downstream effect on PKR activation.
In vitro transcribed SNORD113 was purified from a denaturing gel and then mock or CIP treated followed by addition of EDTA or water. SNORD113 samples were then gel purified a second time after denaturing or native PAGE (Fig. 3A,B). SNORD113 from each treatment ran as one band after denaturing PAGE (referred to as Den), and two bands after native PAGE (referred to as N-Top and N-Bot), as visualized by UV shadowing. Each of these bands was extracted, and used for downstream experiments.
FIGURE 3.

Activity of in vitro transcribed SNORD113 purified from different PAGE systems. As shown in panel A, SNORD113 was initially purified by denaturing PAGE (8% polyacrylamide, 8 M urea; see Materials and Methods) and then subjected to additional treatments (EDTA, CIP) and subsequent gel purification (B). SNORD113 adopted two primary conformers visible by UV shadow of the native gel, referred to as N-Top (top band), or N-Bot (bottom band); Den indicates RNA purified from a denaturing gel. Each band imaged was extracted from the gel and rerun on either a denaturing or native PAGE stained with Sybr Gold to visualize the RNA (C), or subjected to an autophosphorylation assay (D). Autophosphorylation activity was normalized to activation by ds106. Average activation values ± SD are shown below. n = 3 with at least two biological replicates. SNORD113, 500 nM; ds106, 10 nM; PKR, 100 nM.
The first aspect we wanted to examine was RNA size and conformer distribution after gel purification. During subsequent electrophoresis, each SNORD113 sample again ran as one band of the same size by denaturing PAGE, indicating that CIP treatment and gel purification did not affect the stability or purity of SNORD113 (Fig. 3C). Native PAGE analysis after either denaturing or native gel purification revealed that SNORD113 conformers are extremely dynamic (Fig. 3C). SNORD113 conformers reequilibrated to N-Top and N-Bot bands, regardless of which band was initially purified.
Each SNORD113 sample was then tested for PKR activation. We observed no significant difference in PKR activation when comparing the mock to CIP-treated RNAs (Fig. 3D), consistent with earlier results. However, we observed a striking difference when comparing the activation between RNAs purified from the denaturing gel and those purified from the native gel. RNA purified from a denaturing gel (Den) retained potent PKR activation, while RNA purified from a native gel showed decreased activation, for both the N-Top and N-Bot. These results confirmed that at the tested concentration, the presence or absence of the 5′-ppp generated by mock/CIP treatment has little to no effect on PKR activation, but isolation of particular RNA conformers does.
Analytical ultracentrifugation analysis of SNORD113
Previous studies report an RNA conformer-dependent activation of PKR after gel purification following native PAGE (Heinicke et al. 2009; Heinicke and Bevilacqua 2012; Nallagatla et al. 2013). These studies concluded that RNA dimerization, by increasing intermolecular dsRNA, promotes PKR activation, whereas monomeric RNA does not activate PKR. To test whether this is the case for SNORD113, we used sedimentation velocity analyses to determine the association state of each RNA. Since we observed activation did not depend on a 5′-ppp, we used our stock SNORD113. Untreated SNORD113 was gel purified after denaturing PAGE (Den), or native PAGE (isolating the N-Top and N-Bot bands), resuspended in AU200 buffer (see Materials and Methods), and analyzed via sedimentation velocity AUC (SV-AUC). The data from three independent SNORD113 gel purifications were each fit to a continuous sedimentation coefficient distribution [c(s)] using the program SEDFIT. Each RNA sample (Den, N-Top, and N-Bot) had a predominant species with a sedimentation coefficient of 5.0 corresponding to a dimer, although its abundance differed slightly between samples (83% ± 6%) (Table 1; Fig. 4A; Supplemental Tables S1, S2). Two minor species (<10% on average) were also detected with sedimentation coefficients of 3.5 (monomer) and ∼7.5 (>Dimer, approximately tetramer). RNA samples were recovered after SV-AUC and analyzed by native PAGE. As previously observed, SNORD113 N-Top and N-Bot reequilibrated after PAGE purification. The major species observed by SV-AUC was a dimer, and since N-Top was the most abundant conformer on a native PAGE (Fig. 4B), we predicted this band is likely a dimer of SNORD113. The distribution of the monomeric species in each sample correlated with the observed abundance of the N-Bot, making this a good candidate for monomeric SNORD113. Comparing our AUC results to our activation data of PKR after gel purification (Figs. 3, 4B), it is of note that we see no correlation between the presence of a SNORD113 dimer and PKR activation, in contrast to prior studies of some other RNAs (Heinicke et al. 2009; Heinicke and Bevilacqua 2012; Nallagatla et al. 2013).
TABLE 1.
Hydrodynamic properties and molecular weights of SNORD113 conformers
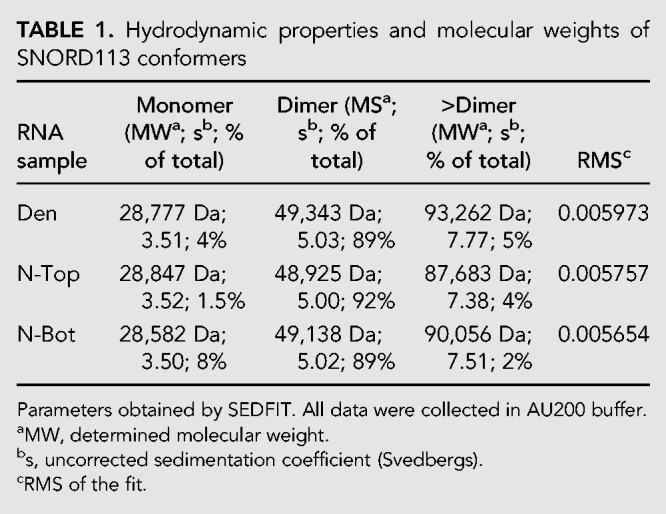
FIGURE 4.

Sedimentation velocity analytical ultracentrifugation (SV-AUC) analyses of SNORD113. (A) Sedimentation coefficient c(s) distribution analysis of SNORD113 conformers. [RNA] was 0.5 µM in AU200 buffer. Peaks corresponding to monomer, dimer, and >dimer species of SNORD113 are indicated. (B) Native PAGE analysis of SNORD113 recovered after SV-AUC. RNA was visualized with SYBR Gold. Below the gel are the amounts of SNORD113 monomer, dimer, and >dimer as determined by SV-AUC; the average PKR activation by each RNA sample is indicated in the bottom row. The SV-AUC results from two additional SNORD113 purifications are shown in Supplemental Tables S1 and S2.
Isolation of a potent PKR activator from SNORD113
We routinely relied on isolating abundant RNA species that were visible by UV shadowing (Fig. 3B). However, there was one species of SNORD113 that ran between N-Top and N-Bot, not always visible by UV shadowing, but sometimes apparent when staining with SYBR Gold (Fig. 4B). In an attempt to fully understand SNORD113 conformers, we repeated the mock and CIP treatments, and native gel purification as indicated in Figure 3, including the isolation of anything that ran in between N-Top and N-Bot. The newly isolated N-Mid was an extremely potent PKR activator (Fig. 5; Supplemental Fig. S2). Importantly, this activity was also independent of mock or CIP treatment, and therefore the presence of a 5′-ppp or 5′-OH. SNORD113 N-Mid required significantly lower concentrations to maximally activate PKR than stock SNORD113 or SNORD113 purified from a denaturing gel after mock/CIP treatments (∼10–250 nM for the N-Mid, and 500–1000 nM for SNORD113 Den). We observed variability in the concentration of SNORD113 N-Mid required for maximum activation, and this likely reflected the variable composition of the middle band we isolated after native gel purification. Isolation of synthetic SNORD113 RNA from the same “middle” region after native PAGE did not enrich for a similar RNA conformer, nor did it result in any PKR activation (Supplemental Fig. S3).
FIGURE 5.
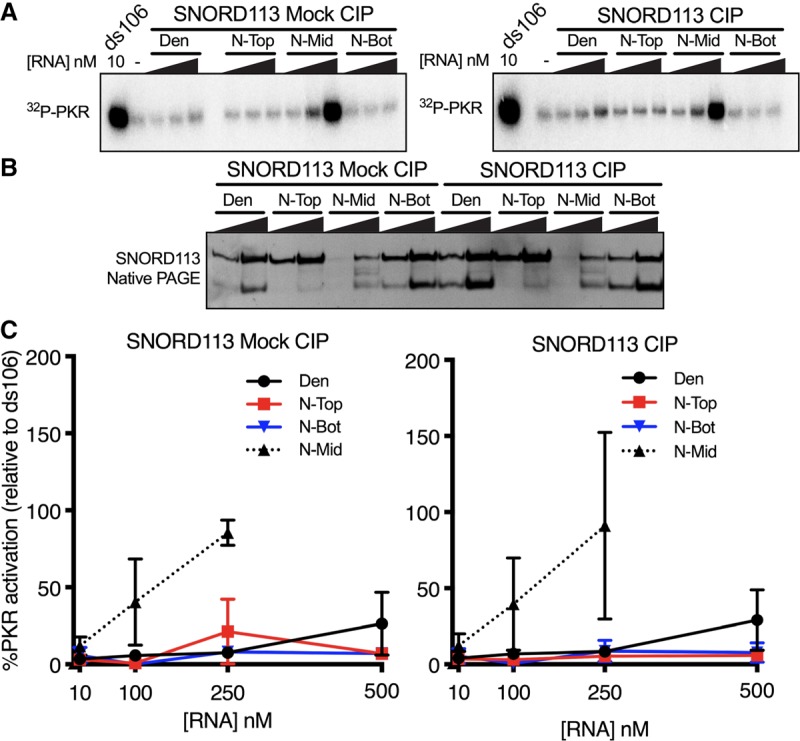
Minor SNORD113 species is a potent PKR activator. SNORD113 species were gel purified after 8% denaturing-PAGE or 8% native-PAGE prior to the following experiments. (A) Representative PhosphorImage of an autophosphorylation assay with SNORD113 Den, N-Top, N-Bot at 10 nM, 100 nM, 500 nM; N-Mid 10 nM, 100 nM, 250 nM; ds106, 10 nM. (B) 8% native-PAGE analysis of SNORD113 species. (C) Averaged activation data as assayed in A. Error bars ± SD; n = 3 with at least two biological replicates. We believe the large error bars reflect differing amounts of contaminating antisense RNA produced by T7 RNAP in different in vitro transcription reactions. At present we do not know the parameters that affect this variable.
We also observed that PKR activation by another snoRNA, SNORA71, as well as a previously reported ss-dsRNA, ss-dsRNA(9,11) (Nallagatla et al. 2007), did not require the presence of a 5′-ppp (Supplemental Fig. 4A,B). Rather, native gel purification of each RNA revealed that, like SNORD113, a minor species was responsible for PKR activation (Supplemental Fig. 4C–E).
Characterization of the activating SNORD113 conformer
The presence of an activating species in the in vitro transcribed SNORD113 RNA but not in the chemically synthesized SNORD113 RNA led us to carefully examine the conditions used for the preparation of RNA in these studies (Supplemental Fig. S5). All of our in vitro transcription reactions were performed with T7 RNAP under high yield conditions. These conditions occasionally lead to aberrant side products such as untemplated 3′-extensions (Triana-Alonso et al. 1995; Gholamalipour et al. 2018), longer than expected RNAs (Cazenave and Uhlenbeck 1994; Rong et al. 1998), and generation of antisense RNA (Konarska and Sharp 1989; Biebricher and Luce 1996; Mu et al. 2018). A comparison of the isolated N-Mid band of in vitro transcribed SNORD113 to perfectly duplexed sense and antisense synthetic SNORD113 revealed that these species comigrate in native PAGE (Fig. 6A). To test if N-Mid contains antisense SNORD113 we performed a northern analysis on synthetic SNORD113 RNAs and in vitro transcribed SNORD113. We detected a strong and specific signal with a probe designed to hybridize with antisense SNORD113 using synthetic antisense SNORD113 and synthetic dsSNORD113 as positive controls (Fig 6B, lanes 3,4). Importantly, this probe for antisense RNA did not hybridize with synthetic sense SNORD113 (Fig. 6B, lane 2). However, we did detect antisense SNORD113 in N-Mid (Fig. 6B, lane 6, red asterisk).
FIGURE 6.
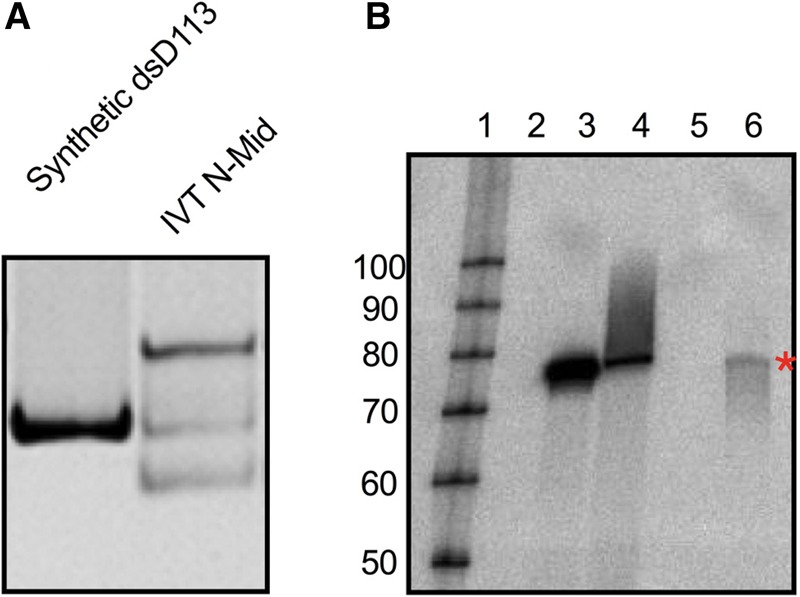
SNORD113 N-Mid contains antisense RNA. (A) Native PAGE comparison of dsSNORD113 (generated by annealing synthetic sense and synthetic antisense RNA) to in vitro transcribed middle band (IVT N-Mid). A total of 200 nM of each RNA was subjected to 8% native PAGE and visualized with SYBR Gold. (B) RNA samples (500 ng) were separated by denaturing PAGE and subjected to a northern blot analysis with a 32-P-labeled probe designed to detect antisense SNORD113. A representative blot (n = 3, two biological replicates with an additional technical replicate) is shown. (Lane 1) Labeled RNA decade marker (Thermo Fisher); (lane 2) synthetic sense SNORD113 RNA; (lane 3) synthetic antisense SNORD113 RNA; (lane 4) dsSNORD113; (lane 5) empty; (lane 6) in vitro transcribed SNORD113 N-Mid. Red asterisk indicates detection of antisense SNORD113 in the N-Mid lane.
Antisense RNA can be generated during in vitro transcription with T7 RNAP from either the sense RNA, or the nontemplate DNA strand (Konarska and Sharp 1989; Mu et al. 2018). We incubated synthetic sense SNORD113 RNA in our reaction conditions for high yield in vitro transcription with and without T7 RNAP to determine if the RNA itself could be acting as a template for antisense transcription. No PKR activation was observed with untreated synthetic sense SNORD113, or synthetic sense SNORD113 after incubation without T7 RNAP (mock). However, synthetic sense SNORD113 gained the ability to activate PKR after incubation in the presence of T7 RNAP (Supplemental Fig. S6A) suggesting that antisense RNA was generated from the sense RNA strand during in vitro transcription.
Contamination of an RNA of interest with any level of antisense transcript is undesirable because it will hybridize with the sense strand, forming dsRNA, which potently activates PKR. We therefore sought to identify conditions that would prevent antisense transcription of SNORD113. T7 RNAP has become the polymerase of choice to produce large amounts of RNA, and various methods have been described to either prevent aberrant T7 RNAP activity during in vitro transcription, or to remove contaminating dsRNA after in vitro transcription (Mellits et al. 1990; Pe'ery and Mathews 1997; Gholamalipour et al. 2018; Mu et al. 2018). Two other RNA polymerases, T3 and SP6, are commercially available and can also be used to generate RNA. We compared SNORD113 generated from our standard high yield in vitro transcription conditions to SNORD113 generated from modified T7 RNAP transcription conditions (reduced [Mg2+] or [UTP] concentrations) and SNORD113 produced by T3 and SP6 RNAPs. Each RNA was gel purified after denaturing PAGE by visualization with UV shadowing as in previous experiments. SNORD113 generated under our standard in vitro transcription conditions potently activated PKR and contained an appreciable amount of N-Mid (Supplemental Fig. S6B). SNORD113 generated by T7 RNAP with reduced [UTP] and [Mg2+] activated PKR at levels above background, albeit at lower levels than standard SNORD113, and with less N-Mid visible. SNORD113 produced by T3 or SP6 activated PKR just above background levels, with no N-Mid detectable by SYBR Gold staining. We observed slower migrating species in the SNORD113 samples generated by T3 and SP6. These possibly represent higher order oligomers or alternate SNORD113 conformers, and, importantly, did not activate PKR. We attempted to remove contaminating antisense RNA after standard in vitro transcription by annealing SNORD113 with full-length DNA oligos and treating the samples with RNaseH (Supplemental Fig. S6C). We were not able to identify any conditions that led to antisense removal by RNaseH, perhaps because the sense and antisense duplex was resistant to the refolding conditions tested.
DISCUSSION
We previously reported that PKR bound snoRNAs in vitro and in vivo (Youssef et al. 2015). In this previously published study we were unable to definitively show that snoRNAs could activate PKR in vivo, but for certain snoRNAs we observed activation in vitro. Here we report follow-up studies designed to understand the mechanistic basis of this in vitro activation. snoRNAs do not fold into the rod-like dsRNA shape typically associated with PKR activation, so we considered features reported to allow activation of RNAs with single-stranded and double-stranded character, such as a 5′-ppp (Nallagatla et al. 2007) or dimerization between two RNAs (Heinicke et al. 2009; Heinicke and Bevilacqua 2012; Nallagatla et al. 2013). We compared synthetic transcripts with 5′-OH, to those synthesized with T7 RNAP and exposed to various phosphatases to remove the 5′-ppp. Our studies largely focused on a representative box C/D snoRNA, SNORD113, but were extended to multiple snoRNAs as well as a previously reported ss-dsRNA (Supplemental Fig. S4). In all cases we observed that activation did not require a 5′-ppp (Figs. 1, 2; Supplemental Fig. S4). Using native gel purification followed by analytical ultracentrifugation, we found that dimerization was also not required for activation (Fig. 4). However, using successive gel purifications of the in vitro transcribed RNA, we were able to isolate a potent activator and found that this sample contained antisense RNA (Figs. 5, 6). Our studies provide yet another cautionary tale to the myriad examples of dsRNA binding proteins reported to have an expanded substrate repertoire, only to find that in the end the results could be explained by contaminating dsRNA.
Do snoRNAs bind PKR in vivo?
Using RNA immunoprecipitation, as well as UV-crosslinking in cells, we previously concluded that snoRNAs bind PKR in vivo. At present we have no reason to question this conclusion, although the conditions that allow PKR to bind snoRNAs, and whether the binding has a biological function, is unclear. Indeed, our previously reported studies included gel-shift analyses showing that SNORD113 binds PKR in vitro with a relatively high affinity (∼16 nM), and this binding is dependent on functional dsRNA binding motifs. Notably, in the absence of purification of the activating species, a much higher concentration of SNORD113 (∼1 µM) is required for maximal activation. Taken together these observations suggest that snoRNAs can indeed bind PKR, but they cannot activate PKR. Rather, our studies described here indicate the observed PKR activation by in vitro transcribed snoRNAs is entirely due to contaminating dsRNA.
snoRNAs are abundant noncoding RNAs that reside in the nucleolus. Based on mfold predictions, snoRNAs adopt hairpin-like structures in the absence of proteins, albeit with some helical imperfections (Youssef et al. 2015). Many of these predicted hairpins include 11–16 contiguous base pairs of dsRNA, the minimum length required to bind the dsRBD of PKR (Manche et al. 1992; Bevilacqua and Cech 1996). The ability of PKR to accommodate helical imperfections (Zheng and Bevilacqua 2000), and the localization of PKR to the nucleus in mouse embryonic fibroblasts (Youssef et al. 2015), may account for the association we observe between snoRNAs and PKR in cells. However, how PKR would effectively compete for binding snoRNAs in the presence of their cognate snoRNA binding proteins is entirely unclear (Bachellerie et al. 2002).
Do RNA dimers promote PKR activation?
RNA is structurally dynamic, adopting different secondary and tertiary conformations in solution based on the buffer conditions. Indeed, several prior studies conclude that particular conformers of RNA display different potentials for activating PKR. The HDV ribozyme, HIV-TAR RNA, and unmodified tRNAs (Heinicke et al. 2009; Heinicke and Bevilacqua 2012; Nallagatla et al. 2013), have all been shown to adopt monomers and homodimers in solution. In these studies, PKR activation assays following purification from a native gel allowed the authors to conclude that dimeric conformers are responsible for PKR activation, while monomeric species are inhibitory to PKR activation. For these RNAs, dimerization allows the length of dsRNA to approach 33 bp, the minimum dsRNA length for PKR activation, thus facilitating PKR dimerization and autophosphorylation. While RNA produced by T7 RNAP potentially contains contaminating antisense RNA, our studies indicate that at least for some RNAs, gel purification after native PAGE effectively separates sense-antisense duplexes from sense homodimers. This observation, and the fact that the prior studies included gel purification from a native gel, substantiates that in some cases homodimerization of an RNA can promote PKR activation. However, while our SV-AUC analyses indicated that the N-Top conformer of SNORD113 was a homodimer (Fig. 4), we found that this SNORD113 species does not activate PKR. This difference may indicate that the SNORD113 homodimer does not adopt a structure that increases the amount of dsRNA available to PKR above 33 bp. Importantly, the presence of a 5′-ppp on the monomer or homodimer of SNORD113 is not enough to trigger PKR activation.
What is the best way to ensure RNA samples do not contain antisense RNA?
Our use of successive gel purifications allowed us to isolate a potent PKR activator, the N-Mid conformer of SNORD113. Multiple observations indicate this conformer is dsRNA formed from contaminating antisense RNA produced during in vitro transcription. For example, N-mid comigrates during native PAGE with dsRNA formed between synthetically prepared sense and antisense SNORD113, and hybridizes to sense oligos used as a probe in northern analyses (Fig. 6). The presence of SNORD113 dsRNA due to contaminating antisense SNORD113 explains the increased stability of the N-mid conformer, which is resistant to hybridization with DNA in RNaseH assays (Supplemental Fig. S6). Additionally, N-Mid potently activated PKR at concentrations close to those required for activation by ds106 (10–50 nM), a perfectly base-paired dsRNA. The characterization of the N-mid conformer of SNORD113 as dsRNA makes our studies completely consistent with previous studies showing that PKR activation by perfectly duplexed RNA is unaffected by the presence or absence of a 5′-ppp (Nallagatla et al. 2007).
Our results appear broadly applicable to RNA made by T7 RNAP. For two additional snoRNAs and ss-dsRNA(9,11) (Nallagatla et al. 2007), we were able to isolate a minor species after native PAGE that strongly activated PKR at lower concentrations than the same RNA purified after denaturing PAGE (Supplemental Fig. S4C–E). This suggests that contaminating antisense RNA produced by T7 RNAP is a confounding factor when analyzing RNA in vitro, as emphasized in a previous study focused on the immunogenicity of in vitro transcribed RNAs in cells (Mu et al. 2018).
T7 RNAP is commonly used by researchers to generate large amounts of RNA, despite numerous reports of unwanted side products. Indeed, after incubation with T7 RNAP under high-yield transcription conditions, synthetic sense SNORD113 RNA activated PKR (Supplemental Fig. S6A), indicating T7 RNAP can use ssRNA as a template to make antisense RNA. This suggests that during T7 transcription the desired sense RNA accumulates and can serve as a template for antisense transcription. While denaturing gel purification will remove longer and shorter RNAs generated by template switching and early abortive cycling of T7 RNAP, respectively, it will not remove an antisense transcript that is identical in length to the sense strand (Milligan and Uhlenbeck 1989; Cazenave and Uhlenbeck 1994). Numerous methods have been proposed to reduce or abolish production of antisense RNA that is not removed by purification after denaturing PAGE, ranging from attention to template DNA structure, modified transcription conditions, and enzymatic removal of dsRNA. Our studies indicate that gel purification after native PAGE can allow for separation of sense-antisense duplexes from sense homodimers and monomers.
Our findings add to numerous reports over many years documenting examples where aberrant production of antisense RNA confounded interpretation of experimental results, emphasizing the insidious nature of this problem. Indeed, unexpected activation of PKR with a known RNA inhibitor, VAI, led to the identification of contaminating antisense RNA as a product of T7 transcription (Mellits et al. 1990; Pe'ery and Mathews 1997). RIG-I, a cytosolic protein involved in innate immune sensing of viral infections was initially thought to be activated by ssRNA containing a 5′-ppp (Hornung et al. 2006; Pichlmair et al. 2006). The RNAs used were generated by T7 RNAP; analysis of the purity and identity of the activating RNAs revealed that the RNA was actually double-stranded with a 5′-ppp on a blunt terminus (Schlee et al. 2009; Wang et al. 2010). The discovery of dsRNA in transcription reactions eventually led to understanding in the RNA interference field of why both sense and antisense preparations could induce mRNA silencing (Fire et al. 1998). In future studies it will be essential to completely remove contaminating antisense RNA from PKR-activating RNAs, such as ss-dsRNAs. While these side products can account for <1% of a transcription reaction, our data indicate this is enough to robustly activate PKR.
MATERIALS AND METHODS
PKR expression and purification
For PKR expression, pPET-PKR/PPase plasmid (Addgene #42934, Lemaire et al. 2005) was transformed into Escherichia coli BL21(DE3) Rosetta cells. Cells were grown in LB medium at 37°C until OD600 ∼ 0.6. PKR expression was induced with IPTG (0.5 mM) for 16 h at 19°C. Cells were lysed by sonication, and the lysate was centrifuged for 30 min at 17,000g. PKR was purified from clarified lysates as previously described (Lemaire et al. 2005; Anderson et al. 2011). Peak fractions were pooled, glycerol was added to a final concentration of 20%, and samples were flash frozen and stored at −80°C. Aliquots were thawed and subjected to gel filtration chromatography prior to use. PKR was eluted from the gel filtration column in buffer A (20 mM HEPES buffer pH 7.5, 150 mM NaCl, 0.1 mM EDTA, 10% glycerol, and 10 mM β-mercaptoethanol [BME]). Peak fractions from the gel filtration column were stored in buffer A supplemented with glycerol to a final concentration of 20%, at 4°C, for up to 2 wk.
PKR autophosphorylation assay
PKR (100 nM) was incubated with specified concentrations of RNA for 10 min at 30°C in 20 mM HEPES (pH 7.5), 100 mM NaCl, 4 mM MgCl2, 1.5 mM DTT, 0.1 mM ATP and 1.5 µCi [ɣ-32P]ATP (PerkinElmer). Reactions were quenched by addition of SDS-sample buffer (188 mM Tris-Cl pH 6.8, 3% SDS, 30% glycerol, 0.01% bromophenol blue, 15% BME) and resolved on 4%–15% PAGE (BioRad). After electrophoresis, gels were fixed in 7% acetic acid for 15 min and then dried. Gels were exposed to a PhosphorImage screen and quantified on a Typhoon Trio PhosphorImager or an Amersham Typhoon (GE Healthcare). Values were normalized to autophosphorylation reactions performed with perfectly duplexed RNA of 106 bp at 10 nM. Autophosphorylation assays were repeated at least three times for each RNA.
RNA synthesis and purification
RNAs were in vitro transcribed under high yield conditions (4 mM of each NTP) with T7 RNA polymerase using PCR templates (Phusion, NEB). Our standard T7 transcription buffer was 40 mM Tris pH 8.1, 2 mM Spermidine, 20 mM DTT, 20 mM MgCl2, 0.1% Triton X-100. In all cases, RNA was subsequently gel-purified after denaturing PAGE (8% polyacrylamide, 8 M Urea) prior to additional treatments. RNA bands were identified by UV shadowing, excised from the gel, and eluted by crushing and soaking overnight at 25°C in 0.5 M NH4OAc, 0.1 M EDTA, 0.1% SDS. Acrylamide was removed using Spin-x columns, and RNA was ethanol precipitated. Chemically synthesized sense SNORD113 containing a 5′-OH was prepared by the University of Utah DNA/peptide synthesis core. Chemically synthesized antisense SNORD113 containing a 5′-OH was obtained from Integrated DNA Technologies (IDT). Synthetic RNAs were gel-purified after denaturing PAGE as described for in vitro transcribed RNA.
Modified T7 in vitro transcription reactions had [Mg2+] reduced to 5 mM, or [UTP] reduced to 0.5 mM. All other reagent concentrations were as described above for high yield. We designed primers which modified the promoter sequence to generate PCR templates for in vitro transcription with either SP6 or T3. T3 and SP6 enzymes were purchased from Thermo Fisher Scientific. SNORD113 in vitro transcription by T3 and SP6 was carried out using the provided 5× transcription buffer and [NTP] of 4 mM.
Phosphatase treatment
To remove the 5′-ppp generated during in vitro transcription, a final concentration of 2 µM RNA in 100 µL was incubated with 10U of phosphatase (CIP, AnP, or RppH) for 1 h at 37°C. The RNA was purified by phenol–chloroform extraction, ethanol precipitation, and PAGE purification as indicated. To confirm the removal of the 5′-ppp, RNA samples treated with and without phosphatase were subsequently treated with T4 PNK in the presence of [ɣ-32P]ATP. RNAs were electrophoresed on native and denaturing gels, which were dried and exposed to PhosphorImage screens and imaged on a Typhoon Trio PhosphorImager (GE Healthcare). RNA samples were additionally treated with T4 PNK in the presence of only unlabeled ATP, followed by treatment with terminator exonuclease. RNAs were separated on native and denaturing gels, and visualized with SYBR gold stain.
RNase H treatment
DNA oligos corresponding to the full-length sequence of SNORD113 were ordered from IDT. RNA and DNA were mixed in 10 µL at a 1:4 pmol ratio (RNA:DNA) in 20 mM Tris pH 7.5, 100 mM NaCl, 0.5 mM EDTA, and refolded by boiling at 95°C for 5 min before placing on ice. RNA/DNA hybrids were mock or RNaseH treated for 1 h at 37°C. The RNaseH reaction was terminated by phenol–chloroform extraction followed by ethanol precipitation. Samples were resuspended in 20 µL H2O and either mock or DNAse I treated for 30 min at 37°C; RNA was purified using Zymo RNA clean and concentrate columns.
Analytical ultracentrifugation
SNORD113 oligomeric state was determined by sedimentation velocity analytical ultracentrifugation (Beckman Coulter Optima XL-I) after purification on either denaturing or native PAGE in AU200 buffer (20 mM HEPES pH 7.5, 200 mM NaCl, 0.1 mM EDTA, 0.1 mM TCEP). Samples (420 µL) and matching AU200 buffer (440 µL) were centrifuged at 50,000 RPM in carbon epon 2-sector centerpieces loaded in an An60-Ti rotor. To preserve RNA integrity, centerpieces and quartz windows were first rinsed with RNaseZap and then sterile deionized water. Radial distributions were recorded via absorbance at 260 nm (0.003 radial step size, one replicate) at 20°C. The c(s) continuous sedimentation distribution function of the data were determined via analysis by SEDFIT (Schuck 2000). Density (ρ) of AU200 was calculated with SEDNTERP (1.008 g/mL) (Laue et al. 1992). The partial specific volume (vbar) for SNORD113 (0.5 mL/g) was calculated after identifying the buoyant molecular weight (M*) of the majority species (dimer) from the sedimentation velocity data using the formula: M* = M (1 − vbar*ρ).
Northern blot
SNORD113 samples were subjected to denaturing PAGE and transferred to a nylon membrane. RNAs were crosslinked to the membrane with UV using a Stratagene Stratalinker set to 120,000 µJ. DNA probes were chemically synthesized at the University of Utah DNA/peptide synthesis core, purified after denaturing PAGE prior to 5′-end labeling with γ-[32P]-ATP and T4 PNK. Membranes were prehybridized at 43°C in ExpressHyb hybridization solution (Clontech) for 30 min. Hybridization of 32P-labeled probes to SNORD113 was performed in ExpressHyb hybridization solution overnight at 43°C. Membranes were washed once in 2× saline sodium citrate (SSC), 0.1% SDS buffer for 15 min at room temperature followed by one wash in 0.5× SSC, 0.1% SDS buffer for 15 min at room temperature.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Bass and Cazalla laboratories for reagents and discussions. We thank Dr. Mark Metzstein for reminding us once again of the antisense RNA problem. Oligonucleotides were synthesized by the DNA/Peptide Facility, part of the Health Science Center Cores at the University of Utah. This work was supported by funds to B.L.B. from the National Institutes of Aging of the National Institutes of Health (8DP1AG044162) and the H.A. and Edna Benning Presidential Endowed Chair.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.071910.119.
REFERENCES
- Anderson E, Pierre-Louis WS, Wong CJ, Lary JW, Cole JL. 2011. Heparin activates PKR by inducing dimerization. J Mol Biol 413: 973–984. 10.1016/j.jmb.2011.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachellerie JP, Cavaillé J, Hüttenhofer A. 2002. The expanding snoRNA world. Biochimie 84: 775–790. 10.1016/S0300-9084(02)01402-5 [DOI] [PubMed] [Google Scholar]
- Bevilacqua PC, Cech TR. 1996. Minor-groove recognition of double-stranded RNA by the double-stranded RNA-binding domain from the RNA-activated protein kinase PKR. Biochemistry 35: 9983–9994. 10.1021/bi9607259 [DOI] [PubMed] [Google Scholar]
- Bevilacqua PC, George CX, Samuel CE, Cech TR. 1998. Binding of the protein kinase PKR to RNAs with secondary structure defects: role of the tandem A-G mismatch and noncontiguous helixes. Biochemistry 37: 6303–6316. 10.1021/bi980113j [DOI] [PubMed] [Google Scholar]
- Biebricher CK, Luce R. 1996. Template-free generation of RNA species that replicate with bacteriophage T7 RNA polymerase. EMBO J 15: 3458–3465. 10.1002/j.1460-2075.1996.tb00712.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazenave C, Uhlenbeck OC. 1994. RNA template-directed RNA synthesis by T7 RNA polymerase. Proc Natl Acad Sci 91: 6972–6976. 10.1073/pnas.91.15.6972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JL. 2007. Activation of PKR: an open and shut case? Trends Biochem Sci 32: 57–62. 10.1016/j.tibs.2006.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. 1998. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391: 806–811. 10.1038/35888 [DOI] [PubMed] [Google Scholar]
- Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. 2006. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 70: 1032–1060. 10.1128/MMBR.00027-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholamalipour Y, Karunanayake Mudiyanselage A, Martin CT. 2018. 3′ end additions by T7 RNA polymerase are RNA self-templated, distributive and diverse in character—RNA-seq analyses. Nucleic Acids Res 46: 9253–9263. 10.1093/nar/gky796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinicke LA, Bevilacqua PC. 2012. Activation of PKR by RNA misfolding: HDV ribozyme dimers activate PKR. RNA 18: 2157–2165. 10.1261/rna.034744.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinicke LA, Wong CJ, Lary J, Nallagatla SR, Diegelman-Parente A, Zheng X, Cole JL, Bevilacqua PC. 2009. RNA dimerization promotes PKR dimerization and activation. J Mol Biol 390: 319–338. 10.1016/j.jmb.2009.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzózka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann K-K, Schlee M, et al. 2006. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314: 994–997. 10.1126/science.1132505 [DOI] [PubMed] [Google Scholar]
- Konarska MM, Sharp PA. 1989. Replication of RNA by the DNA-dependent RNA polymerase of phage T7. Cell 57: 423–431. 10.1016/0092-8674(89)90917-3 [DOI] [PubMed] [Google Scholar]
- Laue TM, Shah B, Ridgeway T, Peletier S. 1992. Computer-aided interpretation of analytical sedimentation data for proteins. Royal Society of Chemistry, Cambridge. [Google Scholar]
- Lemaire PA, Lary J, Cole JL. 2005. Mechanism of PKR activation: dimerization and kinase activation in the absence of double-stranded RNA. J Mol Biol 345: 81–90. 10.1016/j.jmb.2004.10.031 [DOI] [PubMed] [Google Scholar]
- Manche L, Green SR, Schmedt C, Mathews MB. 1992. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol Cell Biol 12: 5238–5248. 10.1128/MCB.12.11.5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo CB, Wong CJ, Lopez PE, Lary JW, Cole JL. 2016. Activation of PKR by short stem–loop RNAs containing single-stranded arms. RNA 22: 1065–1075. 10.1261/rna.053348.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellits KH, Pe'ery T, Manche L, Robertson HD, Mathews MB. 1990. Removal of double-stranded contaminants from RNA transcripts: synthesis of adenovirus VA RNAI from a T7 vector. Nucleic Acids Res 18: 5401–5406. 10.1093/nar/18.18.5401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan JF, Uhlenbeck OC. 1989. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol 180: 51–62. 10.1016/0076-6879(89)80091-6 [DOI] [PubMed] [Google Scholar]
- Mu X, Greenwald E, Ahmad S, Hur S. 2018. An origin of the immunogenicity of in vitro transcribed RNA. Nucleic Acids Res 46: 5239–5249. 10.1093/nar/gky177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nallagatla SR, Hwang J, Toroney R, Zheng X, Cameron CE, Bevilacqua PC. 2007. 5′-triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science 318: 1455–1458. 10.1126/science.1147347 [DOI] [PubMed] [Google Scholar]
- Nallagatla SR, Jones CN, Ghosh SKB, Sharma SD, Cameron CE, Spremulli LL, Bevilacqua PC. 2013. Native tertiary structure and nucleoside modifications suppress tRNA's intrinsic ability to activate the innate immune sensor PKR. PLoS One 8: e57905 10.1371/journal.pone.0057905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanduri S, Carpick BW, Yang Y, Williams BR, Qin J. 1998. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J 17: 5458–5465. 10.1093/emboj/17.18.5458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pe'ery T, Mathews MB. 1997. Synthesis and purification of single-stranded RNA for use in experiments with PKR and in cell-free translation systems. Methods 11: 371–381. 10.1006/meth.1996.0435 [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan C-P, Näslund TI, Liljeström P, Weber F, Reis e Sousa C. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314: 997–1001. 10.1126/science.1132998 [DOI] [PubMed] [Google Scholar]
- Richardson CC. 1981. 16 Bacteriophage T4 polynucleotide kinase. Enzymes 14: 299–314. 10.1016/S1874-6047(08)60342-X [DOI] [Google Scholar]
- Romano PR, Garcia-Barrio MT, Zhang X, Wang Q, Taylor DR, Zhang F, Herring C, Mathews MB, Qin J, Hinnebusch AG. 1998. Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2α kinases PKR and GCN2. Mol Cell Biol 18: 2282–2297. 10.1128/MCB.18.4.2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong M, Durbin RK, McAllister WT. 1998. Template strand switching by T7 RNA polymerase. J Biol Chem 273: 10253–10260. 10.1074/jbc.273.17.10253 [DOI] [PubMed] [Google Scholar]
- Saunders LR, Barber GN. 2003. The dsRNA binding protein family: critical roles, diverse cellular functions. FASEB J 17: 961–983. 10.1096/fj.02-0958rev [DOI] [PubMed] [Google Scholar]
- Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W, Coch C, Janke M, Mihailovic A, Wardle G, et al. 2009. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31: 25–34. 10.1016/j.immuni.2009.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78: 1606–1619. 10.1016/S0006-3495(00)76713-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DR, Lee SB, Romano PR, Marshak DR, Hinnebusch AG, Esteban M, Mathews MB. 1996. Autophosphorylation sites participate in the activation of the double-stranded-RNA-activated protein kinase PKR. Mol Cell Biol 16: 6295–6302. 10.1128/MCB.16.11.6295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Mathews MB. 2001. Functional characterization of and cooperation between the double-stranded RNA-binding motifs of the protein kinase PKR. J Biol Chem 276: 9936–9944. 10.1074/jbc.M007328200 [DOI] [PubMed] [Google Scholar]
- Triana-Alonso FJ, Dabrowski M, Wadzack J, Nierhaus KH. 1995. Self-coded 3′-extension of run-off transcripts produces aberrant products during in vitro transcription with T7 RNA polymerase. J Biol Chem 270: 6298–6307. 10.1074/jbc.270.11.6298 [DOI] [PubMed] [Google Scholar]
- Wang Y, Ludwig J, Schuberth C, Goldeck M, Schlee M, Li H, Juranek S, Sheng G, Micura R, Tuschl T, et al. 2010. Structural and functional insights into 5′-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat Struct Mol Biol 17: 781–787. 10.1038/nsmb.1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BR. 1999. PKR; a sentinel kinase for cellular stress. Oncogene 18: 6112–6120. 10.1038/sj.onc.1203127 [DOI] [PubMed] [Google Scholar]
- Youssef OA, Safran SA, Nakamura T, Nix DA, Hotamisligil GS, Bass BL. 2015. Potential role for snoRNAs in PKR activation during metabolic stress. Proc Natl Acad Sci 112: 5023–5028. 10.1073/pnas.1424044112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Bevilacqua PC. 2000. Straightening of bulged RNA by the double-stranded RNA-binding domain from the protein kinase PKR. Proc Natl Acad Sci 97: 14162–14167. 10.1073/pnas.011355798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Bevilacqua PC. 2004. Activation of the protein kinase PKR by short double-stranded RNAs with single-stranded tails. RNA 10: 1934–1945. 10.1261/rna.7150804 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.