

Abstract
Neisseria gonorrhoeae, the sole causative agent of gonorrhea, constitutively undergoes diversification of the type IV pilus. Gene conversion occurs between one of several donor silent copies located in distinct loci and the recipient pilE gene, encoding the major pilin subunit of the pilus. A guanine quadruplex (G4) DNA structure and a cis-acting sRNA (G4-sRNA) are located upstream of the pilE gene and both are required for pilin antigenic variation (Av). We show that reduced sRNA transcription lowers pilin Av frequencies. Extended transcriptional elongation is not required for Av, since limiting the transcript to 32 nt allows for normal Av frequencies. Using chromatin immunoprecipitation assays (ChIP), we show that cellular G4s are less abundant when sRNA transcription is lower. In addition, using ChIP, we demonstrate that the G4-sRNA forms a stable RNA:DNA hybrid (R-loop) with its template strand. However, modulating R-loop levels by controlling RNase HI expression does not alter G4 abundance quantified through ChIP. Since pilin Av frequencies were not altered when modulating R-loop levels by controlling RNase HI expression, we conclude that transcription of the sRNA is necessary, but stable R-loops are not required to promote pilin Av.
Keywords: R-loop, guanine quadruplex, antigenic variation, recombination, gene diversification
Abbreviated Summary
Neisseria gonorrhoeae, the causative agent of gonorrhea, changes one of its surface exposed structures through DNA recombination, in a process known as antigenic variation. A small RNA is required for antigenic variation and we found that though it forms an RNA:DNA hybrid,hybrid stability is not important for antigenic variation. It is transcription of the small RNA that allows for the formation of an alternate DNA structure called a guanine quadruplex, that drives antigenic variation.
Graphical abstract
Introduction
Programmed genetic recombination allows for diversification of genes in many forms of life. Various bacterial, eukaryotic and viral pathogens utilize gene diversification to allow for immune avoidance and functional modification(s) (Palmer et al., 2016, Deitsch et al., 2009, Holland et al., 1982). These organisms alter surface structures through phase variation of genes, error prone replication, altered partitioning of genomic segments, and homologous recombination. In addition, mammalian immune cells use programmed genetic recombination to allow diversity in B cell responses (Maizels, 2005). Understanding how these species undergo high rates of recombination or mutation, while preserving genome function and viability over many generations is yet to be understood.
Neisseria gonorrhoeae (Gc) is a strict human pathogen and the causative agent of the sexually transmitted infection, gonorrhea (Quillin & Seifert, 2018). Symptomatic Gc infection is characterized by a robust innate immune response consisting of an influx of polymorphonuclear leukocytes to the site of infection. However, Gc can survive in and around these innate immune cells and possesses multiple mechanisms to avoid adaptive immune recognition, including antigenic variation (Av). The Type IV pilus of Gc and the closely-related pathogen Neisseria meningitidis (Mc), is an essential virulence factor. The Gc pilus promotes host cell adherence, biofilm formation, twitching motility, DNA transformation, and resistance to polymorphonuclear leukocytes (Rudel et al., 1992, Sparling, 1966, Wolfgang et al., 1998, Park et al., 2001, Stohl et al., 2013). Gc and Mc vary the gene encoding the major pilin subunit of the pilus, pilE, through changes in the coding sequence (Hagblom et al., 1985). Throughout the Gc and Mc genomes, distinct loci encode variant copies of the major pilin subunit that lack a promoter sequence, ribosome binding site, and signal sequence, known as silent copies (pilS). During pilin Av, a portion of a silent copy sequence replaces the analogous part of the pilE gene in a gene conversion process (Haas & Meyer, 1986, Meyer et al., 1984, Swanson et al., 1987). Any portion of the donating silent copy can be transferred into the recipient pilE locus, resulting in an altered pilE gene and thereby altering the pilin protein sequence. After gene conversion, the silent copy gene sequence, within its original locus, remains unchanged. Pilin Av requires many conserved recombination and repair factors that normally function in single strand gap and double strand break repair including RecA, RecOR, and RecG (Sechman et al., 2005, Mehr & Seifert, 1998, Mehr & Seifert, 1997). When other recombination and repair factors, RecQ, RecJ, and Rep, are mutated, pilin Av is lowered, but not totally abrogated, indicating these factors are involved in the gene conversion process, but may have redundant roles (Mehr & Seifert, 1998, Skaar et al., 2002, Kline & Seifert, 2005, Rotman et al., 2016, Xu & Seifert, 2018).
A conserved sequence located upstream of pilE can form an alternate DNA structure called a guanine quadruplex (G4) (Cahoon & Seifert, 2011, Cahoon & Seifert, 2009). G4 structures have been suggested to play specific roles in many molecular processes, including telomere maintenance, transcriptional regulation, DNA replication, viral packaging, and recombination (Rhodes & Lipps, 2015, Maizels & Gray, 2013). While historically the physical properties of G4 structures have been studied extensively in vitro, understanding the biological functions of G4 structures is an emerging field of research (Seifert, 2018). In vitro methods have allowed for the determination of biochemical parameters like folding, stability, and protein-binding partners for G4 structures found in many biological systems (Tarsounas & Tijsterman, 2013, Bhattacharyya et al., 2016, Asamitsu et al., 2019). Analysis of nucleotide mutations within the G4 sequence has shown that each individual G:C base pair is necessary for pilE G4 structure formation and pilin antigenic variation, but thatT:A base pairs within the sequence, which are not required to form the structure, are dispensable for pilin Av (Figure 1A) (Cahoon & Seifert, 2009).
Figure 1.

gar transcript levels influence pilin Av frequencies
A. Diagram of the G4 gar, pilE gene and G4 motif (blue box and highlighted in yellow). gar, the pilE G4-associated sRNA, is long and poorly expressed, as represented by the red dashed line indicating decreasing RNA transcription. In the mutants, the garP−10 sequence has been mutated to CCGTCC and the garP−35 sequence has been mutated to CCACTC. The G4 sRNA initiates within the G4 motif, using the C-rich side of the DNA as the template.
B. Alignment of the −35 promoter element to the G4 motif of common N. gonorrhoeae (Ng) and N. meningitidis (Nm) strains using CLC sequence viewer. The −35 and −10 promoter elements are denoted with a line along the top.
C. Transcript levels in the parent strain (recA6) and mutant strains garP−10 and garP−35 were measured using qRT-PCR. Strains were grown in liquid medium to obtain similar growth phase to maintain RNA quality. Primers were designed to only amplify within the sRNA region. The reported value represents the mean of three biological replicates +/− one standard deviation as indicated by the error bars. There is no significant difference between the transcript levels of garP−10 and garP−35 but the average garP−35 transcript levels (0.38) are 6x higher than the garP−10 (0.06).Unpaired student t-test **p<0.01
D. Cartoon showing the gar DNA sequence with a terminator sequence added (Green box) and the resulting RNA products. We constructed strains to create a84nt, 72 nucleotides and 32 nucleotides terminated gar . The 84 nucleotides sequence was used to determine the E. coli terminator efficiency. Pilin Av frequencies of the 72 and 32 nucleotides strains were determined by PacBio sequencing.
Pilin Av recombination initiation depends on a promoter located adjacent to the G4-forming sequence that initiates transcription of a cis-acting, noncoding RNA (sRNA) that starts within the G4 sequence (Figure 1A) (Cahoon & Seifert, 2013). Pilin Av does not occur if the sRNA is inverted or the DNA strand encoding the sRNA is switched (Cahoon & Seifert, 2009, Cahoon & Seifert, 2013). Pilin Av is also blocked when the G-rich DNA is switched to the template strand and the sRNA is transcribed from the normal strand (Cahoon & Seifert, 2013). The sRNA is transcribed on the leading strand of replication and is located approximately 280 kb away from the origin of replication (Tobiason & Seifert, 2006).
It is likely that the G-rich RNA transcript found opposite to G4 motif forms an RNA:DNA hybrid (R-loop) (Duquette et al., 2004). R-loops occur when a C-rich template DNA strand forms stable base pairs with a G-rich RNA transcript (Roy & Lieber, 2009).=While R-loops are formed during essential cellular processes like plasmid replication initiation and immunoglobulin class switch recombination (Dasgupta et al., 1987, Reaban & Griffin, 1990, Yu et al., 2003), they can cause genomic instability if not properly resolved. The RNase H enzyme family functions to degrade the RNA in R-loops in order to maintain genomic stability (Kogoma, 1986). If R-loops are not efficiently removed from the DNA by RNase H enzymes, they cause genomic instability by impeding transcription or replication (Santos-Pereira & Aguilera, 2015, Aguilera, 2002, Tuduri et al., 2009). DNA double strand breaks induced by R-loop formation are often correlated with chromosome translocations and cancers (Sollier & Cimprich, 2015, Helmrich et al., 2011). Mutation of R-loop resolving helicases, like RNase H, can result in increased R-loop formation and neurodegenerative disorders such as amyotrophic lateral sclerosis type 4 (Chen et al., 2004).
The pilE G4-associated sRNA (renamed gar here) is predicted to form an R-loop that functions to keep the G-rich DNA strand free, allowing the G4 structure to form, and/or may have a more direct role in the pilin Av initiation. There is evidence that RNase H controls R-loop formation, and thereby affecting Av in the eukaryotic parasite, Trypanosoma brucei. T. brucei is the causative agent of African sleeping sickness and utilizes Av to alter a variable surface glycoprotein (VSG) (McCulloch et al., 2015). R-loops form near VSG switch regions and upon deletion of the gene encoding RNase HI, R-loops near VSG are increased and VSG variation is increased (Briggs et al., 2018, Prister & Seifert, 2018).
Detecting G4 structures within cells has been a barrier to demonstrating that these structures have distinct biological roles in cells (Seifert, 2018). We have used a previously described G4 recognizing monoclonal antibody (MAb) to specifically quantify the pilE G4 structure within Gc using chromatin immunoprecipitation (ChIP), and the R-loop specific S9.6 Mab in R-loop ChIP to quantify the pilE gar-mediated R-loop. We find that sRNA transcription initiation is a rate-limiting step for pilin Av, since reduced sRNA transcription reduces pilE G4 structure levels and pilin Av frequencies. While gar does form an R-loop, modulating R-loop stability by regulating RNase HI expression in Gc does not alter pilE G4 or pilin Av frequencies. We detected pilin Av frequencies using long read Pacific Biosciences amplicon sequencing and a custom analysis software (Ozer et al., 2019). These results show that the process of sRNA transcription is rate-limiting for G4 structure formation and pilin Av, while R-loop stability does not alter pilin Av.
Results
Altering the frequency of G4 sRNA transcription initiation alters pilin Av frequencies and G4 structure formation
It was previously reported that transcription of gar is required for pilin Av, since mutation of the −10 region of the gar promoter (garP−10) abrogated Av and mutation of the −35 promoter region (garP-35) lowered Av frequencies measured with a surrogate phase variation assay (Figure 1A) (Cahoon & Seifert, 2013). While it was reported that the G4 motif is conserved among Gc and Mc strainsthat undergo pilin Av (Cahoon & Seifert, 2009), we analyzed the conservation of the sRNA promoter sequence. We found that the −35 promoter element is completely conserved among the common lab strains FA1090, MS11, MC58 and 8013. There is one or two bases different in the middle of the −10 promoter element (Figure 1B). The first two and last base of the −10 element are the most conserved based on sigma70 promoter analysis in Escherichia coli, and since all the promoters conform to this consensus, we believe all will be functional to promote gar transcription (Rangel-Chavez et al., 2017). Some Mc strains (class II strains) possess a different non-variable pilE gene located in a different part of the genome, and the G4 motif and sRNA are not present in these strains (Cahoon & Seifert, 2009, Wormann et al., 2014). Attempts to visualize the sRNA by northern blot were unsuccessful, possibly due to low abundance of the sRNA and the lack of a transcriptional terminator leading to the production of many different sRNA species (Quillin et al., 2018). To determine gar abundance, we performed qRT-PCR with gar specific primers that amplify within the first 60 nucleotides of the sRNA transcript. The garP−10 mutation greatly reduced transcript levels near the background levels of the no reverse transcriptase controls. The garP−35 mutation had sRNA levels of about 30% of the parental strain (Figure 1C).
The garP−35 mutation was previously reported to lower pilin Av rates using the pilus dependent colony morphology change (PDCMC) assay in an inducible recA background (recA6) (Cahoon & Seifert, 2013). Pilin Av frequencies of garP−10 and garP−35 mutants without a regulatable recA(parental strain, FA1090) were determined by an novel method using PacBio sequencing (Ozer et al., 2019). All starting strains were confirmed to have the same, predominate 1–81–S2 pilE sequence by Sanger sequencing of each progenitor colony used to generate the progeny used for PacBio sequencing, although since pilin Av was unregulated each founder colony would start with different amounts of variant pilE subpopulations that were below the level of detection. At least 1000 colonies were isolated after ~19–20 generations of growth from two biological replicates, and total genomic DNA was extracted for each sample (Rotman et al., 2016). The pilE genes were amplified with pairs of primers, each with a different SMRT PacBio barcode for each strain. Individual amplicon sequences were determined by repetitive PacBio SMRTbell sequencing to reduce sequencing errors. The resulting sequences were de-multiplexed, filtered for quality, and analyzed to determine the pilin Av frequency for each strain (Ozer et al., 2019). The parental strain FA1090 had an average pilin Av frequency of 18.1%, the garP−35 strain had a frequency of 6.7%, and the garP−10 mutant strain had a frequency of 0.1% (Table 1 and Supplemental Table 1). These pilin Av frequency values ascertained by PacBio sequencing are the first to use an unregulated Gc strain and confirmed the effect of the gar promoter mutations to previous results using different sequencing technologies and an IPTG-regulated strain (Cahoon & Seifert, 2013, Rotman et al., 2016). These results confirm that that gar transcription initiation is rate-limiting for pilin Av.
Table 1.
Pilin Av Frequencies
| Strain | % Av |
|---|---|
| FA1090 | 18.1 |
| FA1090 G4 mutant | 0.2 |
| FA1090 garP−10 | 0.1 |
| FA1090 garP−35 | 6.4 |
| FA1090 gar 72 NT | 16.9 |
| FA1090 gar 32 NT | 17.6 |
| rnhA+nicsPlac::rnhA 0 mm IPTG | 10.7 |
| rnhA+nicsPlac::rnhA 0.005 mm IPTG | 13.9 |
| rnhA+nicsPlac::rnhA 1 mm IPTG | 13.6 |
| rnhA+nicsPlac::rnhA garP−10 0 IPTG | 0.2 |
This table shows the combined pilin av frequencies of each condition. All variable strains were tested in biological replicates. The individual pilin Av frequencies for each barcoded pool can be found in Supplemental Table 1 along with the number of reads analyzed for each pool.
An extended gar transcript is not required for pilin Av
The sRNA gar does not encode a defined transcription termination sequence and the transcript dissipates along the genome (Quillin et al., 2018). We introduced a Rho-independent terminator of trp from E. coli 84 bases downstream of the transcriptional start site to specifically halt transcription elongation (Reynolds et al., 1992). The efficiency of termination was determined by qRT-PCR using primers designed to amplify within the sRNA transcript and after the terminator sequence. The 84nt sRNA transcriptional terminator read through was 17% ± 6% (average ± SD), which is within the normal efficiency of Rho-independent termination (Reynolds et al., 1992). Two additional strains were constructed introducing the same terminator that would produce shorter transcripts to define the transcript necessary for Av (Figure 1D). These mutants were analyzed for pilin Av frequencies using PacBio sequencing and the 72nt and 32nt gar transcript strains produced pilin Av frequencies of 16.9% and 17.6%, respectively. Since reduced transcription in the garP−35 mutant resulted in reduced pilin Av frequencies, we conclude that the small amount of read through beyond the terminator sequence was not contributing to pilin Av. We conclude that terminating the majority of transcription 32 nucleotides after initiation is sufficient for the gar transcript to promote pilin Av, and that the extended gar transcript plays no role in the process.
RNase HI is encoded by NGO1162 and degrades RNA from R-loops
To determine whether the pilE G4 sRNA forms a stable R-loop with the template strand, we used ChIP with the S9.6 Mab that recognizes RNA:DNA hybrids (Hu et al., 2006, Chan et al., 2014). qPCR was used to quantify the immunoprecipitated DNA of the G4 sRNA region. As a negative control we also quantified the immunoprecipitated DNA ofan irrelevant ectopic site that does not have an abundant transcript and, thus, is predicted to have none or low levels of R-loops (Mehr & Seifert, 1998). As a positive control, we also conducted R-loop ChIP of the 16S RNA, as this is a very abundant transcript (Supplemental Figure 1). A polyclonal IgG mouse antibody was used to detect nonspecific, antibody-dependent Chip signals. Mab S9.6 ChIP demonstrated that stable R-loops exist at the G4 sRNA region. Importantly, the R-loop signal was reduced is garP−10 and –garP−35 mutants (Figure 2A and Supplement Figure 1).
Figure 2.

Transcription and modulation of rnhA modulates R-loop levels
A. R-loop levels of garP−10 and garP−35 strains relative to the parental strain (FA1090). Relative R-loop levels are calculated by determining the percent input after ChIP pulldown for each sample and the dividing by the percent input of FA1090. The reported value represents the mean of five biological replicates (individual dots) +/− one standard deviation as indicated by the error bars. There is no significant difference between garP−10and garP−35 . Unpaired student t-test **p<0.01 *p<0.05
B. The rnhA+nicsPlac::rnhA strain allows modulation of RNase HI. rnhA expression measured using qRT-PCR with Gc grown with different levels of IPTG in solid media. The average fold change compared to FA1090 (±standard deviation) for each IPTG level are: 0 IPTG-0.30 (0.09), 0.001 mM IPTG 0.61 (0.32), 0.005 mM IPTG 1.4 (0.5), 0.01 mM IPTG 4.3 (2.2), 1 mM IPTG 61.9 (33). The reported value represents the mean of five biological replicates (individual dots) +/− one standard deviation as indicated by the error bars. Unpaired student t-test . **p<0.01 *p<0.05
C. R-loop levels determined by R-loop ChIP when rnhA expression is modulated. The reported value represents the mean of six biological replicates (individual dots) +/− one standard deviation as indicated by the error bars. Unpaired student t-test **p<0.01
Since RNase H nucleases are the major means to remove R-loops, we asked whether modulating RNase H levels might influence gar R-loop stability. Gc encodes an RNase H1 ortholog (rnhA) at the FA1090 locus NGO1162 and an RNase HII ortholog (rnhB) at the locus NGO1789. An insertional mutant of rnhB showed no defects in growth, no other obvious phenotypes, and had parental levels of pilin Av when measured by the surrogate PDCMC assay (Supplemental Figure 2).
Multiple attempts to disrupt the rnhA gene were unsuccessful, even in a garP−10 mutant strain, showing that the lethality was not due to the presence of the gar sRNA. To overcome the potential lethality of the rnhA mutation, an IPTG-inducible copy of the rnhA gene driven by the lac promoter, was inserted into an intergenic region elsewhere on the Gc chromosome (nics) (Mehr & Seifert, 1998). The mutated rnhA gene was introduced into this strain at the NGO1162 locus with IPTG in the growth medium, to create the strain rnhA+nicsPlac::rnhA. The rnhA+nicsPlac::rnhA strain grew without IPTG in the growth medium, albeit more slowly than the parent strain. We reasoned that the ability to grow without rnhA induction was due to residual transcription from the repressed lac promoter. qPCR analysis confirmed that the rnhA+nicsPlac::rnhA strain produced ~30% of the rnhA transcript of the parental strain (Figure 2B). The rnhA+nicsPlac::rnhA strain showed a dose-dependent response to IPTG in the growth medium with 0.005 mM IPTG providing rnhA transcript levels equivalent to the parental FA1090 strain and up to a 50-fold overexpression of rnhA transcriptswith 1 mM IPTG (Figure 2B).
To determine whether rnhA levels affect R-loop stability, Gc was grown with and without IPTG to allow low and high levels of RNase HI expression. Mab S9.6 ChIP showed that when the rnhA+nicsPlac::rnhA strain was grown without IPTG, R-loop levels were 10-fold higher than parental levels (Figure 2). R-loop levels reached parental levels when rnhA+nicsPlac::rnhA was grown with 1 mM IPTG, which causes overexpression of rnhA (Figure 2C). The negative controls confirmed that the S9.6 ChIP was specific since S9.6 did not pull down the ectopic locus (Supplemental Figure 3A). Additionally, the R-loop signal was lost after treating ChIP samples with E. coli RNase HI in vitro after pulldown, showing that S9.6 was detecting R-loops and not non-RNA structures (Supplemental Figure 3B) (El Hage et al., 2014). rnhB modulation did not alter R-loop levels and was therefore not analyzed further (Supplemental figure 3C). These results show that RNase HI does process the pilE G4 R-loop, but that RNase HI overexpression did not reduce pilE G4 R-loop levels below that of the parental strain.
ChIP can detect and quantify the genomic pilE G4
It has been very difficult to detect or quantify G4 structures in cells. Many of the techniques used are limited in their inability to differentiate between G4 structures present within the cell and G4 structures that form after cell lysis and DNA extraction, particularly because the DNA is often denatured during extraction. To avoid this issue, we developed a G4 ChIP assay to detect and quantify G4 levels within the bacterial cell. After testing several MAbs and recombinant MAbs reactive to the G4 structure, we determined that the MAb, 1H6, which detects multiple G4 structures was the most sensitive antibody in our bacterial system (Henderson et al., 2014). To ensure that the 1H6-ChIP was detecting the presence of the pilE G4 within the bacteria, a G4 mutant in which 3 guanine tracts were disrupted, but the total number of G residues was retained, was used as a negative control. Additionally, a positive G4 ChIP control was performed using a mutant strain inactivated for two helicases, RecQ and Rep, active in unwinding G4s in vitro (Cahoon et al., 2013). The recQ rep double mutant had smaller colonies (Cahoon & Seifert, 2009). We detected a loss of the G4 ChIP signal in the G4 mutant and an increase in the recQ/rep mutant (Figure 3) demonstrating that this G4 ChIP assay is specific for the pilE G4 and detects and quantifies G4s existing within the bacterial cell.
Figure 3.

G4 ChIP detects intracellular G4 levels
The sequence of the G4 motif is 5’-GGGTGGGTTGGGTGGG and G4 mutant is 5’-GTGGGGGTGTGGTGGG with 3 of 4 G-tracts interrupted. G4 levels were determined from plate grown bacteria using G4 ChIP with the 1H6 Ab. The parental strain (recA6) and G4 mutant, and the double helicase mutant (recQ/rep) were tested for G4 levels. An alternate region of the genome (ectopic site) was amplified to test for non-specific DNA affinity by the Ab. IgG was used to determine general Ab binding. The reported value represents the mean of four biological replicates (individual dots) +/− one standard deviation as indicated by the error bars. Unpaired student t-test **p<0.01 *p<0.05
The gar ranscript is required for G4 formation and stability, but R-loop stability does not influence the pilE G4
The discovery of the requirement for transcription of the gar sRNA led to several hypotheses. First, transcription is necessary to open the DNA duplex allowing for the single stranded G-rich strand to fold into a G4 structure (Cahoon & Seifert, 2009). A second hypothesis, not mutually exclusive to the first hypothesis, is that the R-loop itself plays an active role in the process of pilin Av. To test the first hypothesis, we used the gar −10 and −35 promoter mutants. As expected, the garP−10 mutation that disrupts sRNA transcription and pilin Av also prevents the G4 ChIP signal (Figure 4A). The garP−35mutation, which produces an intermediate level of sRNA transcript (Figure 1B), reduced G4 levels (Figure 4A). Taken together these results confirm that transcriptional melting og the DNA duplex is the primary reason that pilin Av relies on the sRNA (Figure 4A and Supplemental Figure 4A).
Figure 4.

Transcription initiation, but not R-loop stability, influences G4 levels
A. G4 levels were determined using G4 ChIP on garP−10 and garP−35 with the G4 specific MAb, 1H6). Relative G4 levels are calculated by determining the % input after ChIP pulldown for each sample and the dividing by the % input of FA1090. Bar The reported value represents the mean of six biological replicates (individual dots) +/− one standard deviation as indicated by the error bars. Unpaired student t-test **p<0.01
B. G4 ChIP was used to determine G4 levels under low rnhA and high rnhA expression (rnhA+nicsPlac::rnhA 0 IPTG, and rnhA+nicsPlac::rnhA 1 mM IPTG). The reported value represents the mean of six biological replicates (individual dots) +/− one standard deviation as indicated by the error bars. Unpaired student t-test**p<0.01
Since down regulation of rnhA expression leads to an increase in R-loop levels, we asked whether the increased gar R-loop stability results in increased G4 formation. In the mutant strain where R-loop levels are high (rnhA+nicsPlac::rnhA in the absence of IPTG), G4 levels were similar to the FA1090 parent (Figure 4B and Supplemental Figure 4B). When rnhA+nicsPlac::rnhA was grown in the presence of 1mM IPTG, rnhA was over expressed, but R-loops levels were similar to parental levels, and G4 levels wereslightly reduced to 0.67 fold of parental levels. This result suggests that while transcription of the gar sRNA is necessary for G4 formation, that increased stability of the R-loop is not important.
R-loop stability does not influence pilin Av frequencies
While increased R-loop stability did not change G4 levels (Figure 4), it was possible that changes in R-loop levels or R-loop stability might still alter pilin Av frequencies. A PacBio sequencing assay was used to determine the pilin Av frequency of various strains (Ozer et al., 2019). Deep sequencing is the best method for determining Av frequency when the strains tested have different growth rates, a factor relevant to this work, as the rnhA+nicsPlac::rnhA strain exhibits slower growth in the absence of IPTG. Additionally, the longer reads generated during PacBio sequencing allows for the pilS silent copy donors to be quantified and defines the types of recombinants produced (Supplemental Figure 5 and 6) (Ozer et al., 2019).
Pilin Av frequencies were compared between the rnhA+nicsPlac::rnhA strain with varying RNase HI levels produced by growth on different amounts of IPTG (0.0, 0.005 or 1 mM IPTG). Pilin Av was abrogated when the G4 motif was mutated as reported previously (Cahoon & Seifert, 2009). In mutant strains where the sRNA promoter is altered, garP−10 and garP−35, pilin Av frequencies were similarly reduced, confirming result from the PDCMC assay (Cahoon & Seifert, 2013). However, pilin Av frequencies were not substantially affected by modulating the R-loop levels through the differential expression of RNase HI (Table 1 and Supplemental Tables 1 and 2). These results allow us to conclude that increasing R-loop stability does not affect pilin Av frequencies.
Discussion
We have determined that transcription of the G4 sRNA is the primary driver of pilin Av initiation. Mutation of the G4 sRNA promoter elements had previously been shown to lower or completely abrogate pilin Av, measured by the surrogate PDCMC assay (Cahoon & Seifert, 2013). In addition, a previously published 454 sequencing assay confirmed that a mutation in the −35 region of the sRNA promoter reduced pilin Av frequencies (Rotman et al., 2016). We have confirmed and extended these results to show that sRNA transcription initiation is rate limiting for pilin Av. We used a novel PacBio amplicon sequencing method to test for pilin Av frequencies in our different strains. A similar methodology could be used with Nanopore MinION sequencing which has lower rates of insertions and deletions, 4.9% and 7.8% respectively (Jain et al., 2015). However, PacBio CCS allows for multiple sequencing passes of the same DNA molecule, reducing the error rate over multiple reads. Moreover, while the sRNA transcript does not end at a specific terminator sequence, and transcript abundance appears to dissipate as downstream distance from the promoter sequence increases, only 32 nucleotides of the sRNA transcript is required for normal levels of pilin Av. The full activity of this shortened transcript in pilin Av suggests that the gar sRNA acts mainly opposite to the G4 motif and there is no function for extended sRNA transcription, at least for pilin Av. Since there is no gene downstream of the G4 sequence, there may not have been a reason to terminate this low abundance transcript. We have been unable to delete the A-rich sequence adjacent to the G4 forming motif (Figure 1) for unknown reasons, so we cannot rule out a role for this sequence within the sRNA in pilin Av. Since the G4 structure cannot form when DNA is double stranded, the DNA duplex needs to be opened by a helicase (Hardin et al., 1992, Li et al., 2002). The breaking of hydrogen bonds could be catalyzed by a dedicated helicase, or any of the helicase activities required for DNA replication, transcription, repair or recombination. Based on the phenotype of the −10 promoter mutant, we conclude that it is transcription by the house-keeping polymerase that opens the duplex to allow for G4 formation.
In all types of cells, R-loops are often found opposite G4 forming sequences, since G-rich RNA preferentially binds to C-rich DNA (Gyi et al., 1996). We confirmed an R-loop was forming in the G4 sRNA region by R-loop ChIP and confirmed that R-loop formation depends on G4 sRNA, gar, transcription. Understandably, in the −10 mutant defective for transcription initiation, there is no R-loop formation. Similarly, when transcription efficiency is low in the −35 mutant, R-loop levels are reduced. To understand how changes in R-loop levels or R-loop stability may influence pilin Av, we created the rnhA+nicsPlac::rnhA strain, allowing for modulation of rnhA expression with IPTG. However, in strain rnhA+nicsPlac::rnhA in the absence of IPTG, wherein R-loop levels were increased 10-fold above parental levels, there was no increase in G4 levels, showing that increased R-loop abundance does not influence G4 formation or stability. Most importantly, in this same strain under the same conditions of increased R-loop stability, Av frequencies were unaffected. Without IPTG, this strain grows slowly, and we presume that there is an increase in R-loops throughout the genome. An increase in the stability of all R-loops may be create genomic instability influence the pilin Av process by recruiting essential recombination and repair factors. Surprisingly, when rnhA was overexpressed, R-loop levels were not significantly different from the parental strain. This result shows that, even though lowering RNase HI increases sRNA R-loop abundance, there exist R-loops that remain unaffected by parental and overexpressed levels of RNase HI. The most likely explanation for this effect is that RNase HI is not acting on its own to control R-loop stability, and that activity on the sRNA is linked to other processes such as DNA replication. It is also possible that a small portion of the sRNA can form a structure that is resistant to digestion, such as triplex DNA or dsRNA (Hartono et al., 2018). Interestingly, when rnhA was overexpressed, there was a slight decrease in G4 levels, even though overall R-loop levels remained unchanged. This effect may be due to an independent effect on the bacterial cell of expressing the RNase HI 50-fold above normal expression levels. As this small decrease in G4 levels during rnhA overexpression does not affect pilin Av frequencies, this discrepancy may reflect the fact that ChIP is not a perfectly quantitative assay.
Gc also encodes the RNase HII family enzyme rnhB (NGO1789). A rnhB mutant and complement had similar growth phenotypes and no effect on pilin Av as measured by PDCMC. While RNase HII family enzymes can act on R-loops, they are more often involved in removing ribonucleotides during replication (Itaya, 1990, McDonald et al., 2012). The lack of an rnhB phenotype suggests that misincorporated ribonucleotides are not involved in pilin Av. Since low rnhA expression leads to a large increase in R-loop levels, we conclude that in Gc, RNase HI is the primary enzyme functioning to remove R-loops.
Though critical for pilin Av and other microbial cellular processes (Seifert, 2018), G4 levels within a bacterial cell have not been previously reported. Therefore, we developed a G4 ChIP assay to understand how the G4 functions during pilin Av. Using G4 ChIP, and G4 and helicase mutants as negative and positive controls, respectively, we were able to quantify a cellular G4-specific signal. Since the 1H6 Mab also has affinity to T-rich (thiamine-rich) DNA in addition to G4 structures (Kazemier et al., 2017), and there is a T-rich sequence near the G4 motif (Figure 1), it was possible that we were precipitating this T-rich sequence and not the G4 structure. However, the T-rich region is present in both the G4 mutant and parent strain, yet the G4 signal is lost in the G4 mutant (Figure 3). The 1H6 MAb also has affinity to Ts contained within the loops of a G4 structure, which are present in the pilE G4 (Kazemier et al., 2017). Whether the 1H6 MAb detects the G4 structure itself or the specific structure of the G4 loop Ts is immaterial since the MAb only pulls down G4 DNA and not G4 mutant DNA. Additionally, G4 ChIP signals increased when two G4 interacting helicases (RecQ and Rep) were inactivated. If G4 folding was only occurring during the nucleic acid extraction or the ChIP protocol, we would expect the G4 signal to be similar between the double helicase mutant and the parent strain. Therefore, G4 ChIP with MAb 1H6 reliably detects and quantifies G4s within the cell.
The critical role for the gar sRNA in Av appears to be opening duplex DNA through transcription, although there are a few other plausible roles for the sRNA in pilin Av. In all of the strains that allow sRNA transcription there is some level of R-loop formation and the R-loop would still prevent the C-rich strand from reannealing to the G-rich strand. Thus, R-loop formation and limited stability may still be important for G4 folding. The sRNA could have an additional role in pilin Av by directly engaging with the G4 DNA structure. The gar transcript contains three G-tracts and one or more of the tracts could participate in an intermolecular G4 structure creating a DNA/RNA G4 (Xu et al., 2012). We consider this hypothesis unlikely since individual mutation of 11 of the 12 of the G4 G:C base pairs abrogates Av, and mutation of the 12th lowers pilin Av since a 13th G can compensate for its loss (Cahoon & Seifert, 2009). If the sRNA participated in an alternative G4 structure, all four G-tracts may not be required for pilin Av, particularly since the first tract is not present in the sRNA. The sRNA or R-loop formation could also be recruiting factors to pilE to enhance recombination, but this idea remains to be tested. The exact sequence requirement of the G4 transcript has not been determined. The G4 motif cannot be mutated without altering the G4 DNA structure. The A-rich region of the sRNA could also play an additional role in pilin Av. It should be noted that since transcription is required for R-loop formation, we have not directly tested for a role of the R-loop in pilin Av without transcription opening the duplex and allowing formation of the G4 structure.
We conclude that sRNA transcription is rate limiting for G4 formation and although R-loops do form at this location, increased R-loop stability is dispensable for pilin Av. We hypothesize pilin Av is initiated through a DNA break caused by stalled chromosomalreplication (Figure 5). Either the G4 or R-loop upstream of pilE could participate in creating a DNA break. However based on these results; it is more likely that the G4 blocks replication, because G4 levels and Av occur similarly when R-loop levels are high or low. If R-loops were participating in genomic instability, we would predict that increased R-loops in the Rnase HI under expressing strain would cause an increase in Av frequency through additional DNA breaks. However, the pilin Av frequency when R-loop levels are increased is slightly lower compared to normal levels of R-loops. R-loops have been implicated in many pathologies including oncogenesis by way of chromosomal translocations and neurological syndromes (Bhatia et al., 2014, Groh et al., 2014, Haeusler et al., 2014). Since most stable R-loops are G-rich, the role of G4s and other alternative DNA and DNA/RNA structures has not always been investigated. The identification of roles for R-loops in mediating positive biological processes is less well established than detrimental roles. R-loops are necessary for the initiation of replication in some plasmids and mitochondrial DNA (Xu & Clayton, 1996, Fukuoh et al., 1997). R-loops are also important for Trypanosoma brucei VSG Av since the loss of RNase HI influenced Av frequencies (Briggs et al., 2018). R-loops are also implicated in Ig class switch, where the RNA G4 aids recruitment of the cytidine deaminase, which leads to recombination (Zhang et al., 2014, Zheng et al., 2015). We predict that other diversity generation systems, many that have G-rich segments associated with them (Walia & Chaconas, 2013, Giacani et al., 2012, Glover et al., 2013, Smargiasso et al., 2009, Briggs et al., 2018, Beaume et al., 2013), may also depend on R-loops and associated G4s to induce programmed recombination.
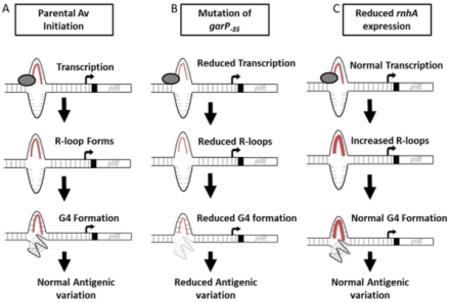
Figure 5.

Summary of results
Transcription of the G4 sRNA is the first step in Av. In A, RNA polymerase (grey oval) opens the DNA duplex. The sRNA transcript (red line) remains bound to the C rich strand of DNA, while the other DNA strand can fold into a G4 structure (grey structure with three square planes and zig zag loops). In B, reduced transcription causes reduced G4 formation and subsequent Av. In C, RNase H levels are low so R-loops are high, however, R-loop stability is dispensable for G4 levels and pilin Av.
Methods
Gc Strains and Growth Conditions
Bacterial strains used in this study were derivatives of the FA1090 clinical isolate 1–81–S2 that was re-isolated from a symptomatic, human-volunteer infection (Seifert et al., 1994). All gonococcal strains carried the 1–81–S2 pilus variant sequence as determined by Sanger sequencing (Seifert et al., 1994). The Escherichia coli strain DH5α was used for expression of mutant constructs in plasmid cloning vectors and grown on Luria-Bertani Broth. N. gonorrhoeae strains were maintained on Gc medium base (Difco) plus Kellogg supplement I (22.2 mM glucose, 0.68 mM glutamine, 0.45 mM cocarboxylase) and Kellogg supplement II (1.23 mM Fe(NO3)3) (Kellogg et al., 1968). When noted, strains contain the modified recA gene, recA6, which places the endogenous gene under the control of the lac promoter, allowing for IPTG-dependent regulation of pilin Av (Seifert, 1997). When stated, Gc were propagated in liquid culture using a previously described method (Stohl et al., 2005) to obtain a uniform population in mid-exponential growth phase.
Mutant Strain Construction
KOD polymerase (Novagen) was used to amplify the upstream and downstream regions of rnhA, encoding Rnase HI, (FA1090 locus, NGO1162, Genbank ID) with phosphorylated primers rnhA-usF and rnhAusR-NotI, and rnhA-dsR and rnhAdsF-NotI (Supplemental Table 3 and 4). The 3′ primer of the upstream region (rnhAusR-NotI) and 5′ primer of the downstream region (rnhAdsF-NotI) contained complementary sequences to introduce a NotI restriction site. Splicing overlap extension PCR (SOE‐PCR) was used to combine the upstream and downstream regions, creating a mutant construct containing a NotI restriction site where the rnhA coding sequence had been. rnhB (FA1090 locus, NGO1789, Genbank ID) was deleted and complemented similarly with primers r2usF and r2usRkpnI, and r2dsR and r2dsFkpnI deleting rnhB and inserting a KpnI site. These constructs were cloned into pSMART LCAmp (Lucigen 40300–2) following the manufacturer’s instructions and introduced into E. coli DH5α by electroporation. The rnhA and rnhB mutant constructs-containing pSMART LCAmp sequence, pSMART-ΔrnhA and pSMART-ΔrnhB, were confirmed by Sanger sequencing. Both plasmids were isolated using the QIAprep Spin Miniprep Kit (Qiagen 27104) according to the manufacturer’s instructions, digested with NotI or KpnI (NEB R0189S), and gel-purified using the QIAquick gel extraction kit according to the manufacturer’s instructions (Qiagen 28115). The chloramphenicol resistance (CmR) gene was digested from plasmid pHSS6-Cm2–9 using NotI (Seifert et al., 1990), gel purified using the QIAquick gel extraction kit, introduced into the NotI site of pSMART-ΔrnhA, and ligated using T4 DNA Ligase (ThermoFisher EL0014). The KanR gene from pBSL86 (Alexeyev, 1995) was digested with KpnI and cloned into pSMART-ΔrnhB. The pSMART-ΔrnhA-CmR and pSMART-ΔrnhB-KanR ligation products were electroporated into E. coli DH5α selecting for chloramphenicol resistance at (50 ug/ml) and Kanamycin resistance at (50 ug/ml) respectively.
The RNase HI complementation construct was made using primers rnhAFPacI and rnhARPmeI, containing PmeI and PacI cut sites, to amplify the rnhA gene in strain FA1090. The amplified PCR product and recipient plasmid pGCC4 (Mehr & Seifert, 1998) were both cut with PmeI and PacI, then ligated together with T4 DNA ligase. The pGCC4-rnhA complementation plasmid was isolated using the QIAprep Spin Miniprep Kit and the rnhA insertion sequence was confirmed by Sanger sequencing using primers Lctpout and LacPFor. rnhB was complemented similarly with primers R2FPacI and R2RFseI and inserted into pGCC4. Plasmid constructs pSMART-ΔrnhA-CmR and pGCC4-rnhA, and pSMART-ΔrnhB-KanR and pGCC4-rnhB were introduced into the Gc chromosome by natural transformation as described previously (Stein 1991) and confirmed by PCR and Sanger sequencing.
Rho-independent terminator mutants of the G4 sRNA, G4 mutation, and garP−10 were synthesized as gBlocks (IDT) (Reynolds et al., 1992) . All terminator mutant gBlocks were cut with PacI and inserted into the PacI and EcoRV cut site of the pilE plasmid containing the region between USS2-pilArev2 (Cahoon & Seifert, 2009) and transformed into DH5α. The transformants were confirmed by Sanger sequencing and then transformed into the chromosome of FA1090 recA6 and FA1090. Kanamycin resistant (KanR) colonies were selected and transformants were verified by PCR and Sanger sequencing with KanFor and RT-G43R. The G4 mutant was cloned similarly in the pilE plasmid containing the region between USS2-pilArev2 and was transformed into FA1090.
Mutation of the −10 box of the G4 sRNA promoter was synthesized as a gBlock (IDT). The gBlock was cloned into vector pBlunt-TOPOII (Thermo Fisher). The plasmid was then extracted and transformed into the parental FA1090, the rnhA mutant, and rnhB mutant strains. Transformants that no longer produced nonpiliated blebs over extended growth on solid medium (Seifert, 1997) were screened by PCR and subsequent sequencing to identify G4 sRNA −10 mutants.
Mutation of the −35 promoter element in FA1090 was performed by isolating genomic DNA from the recA6 −35mutant (Cahoon & Seifert, 2013). The genomic DNA was transformed into FA1090 by spot transformation, and Kan resistant colonies isolated and sequenced.
qRT-PCR to detect sRNA and mRNA Expression
For transcriptional terminator experiments, strains were grown in liquid medium to mid log phase (ODA550~0.5), back-diluted to an OD A550 of 0.07 and grown for four hours. Since low expression of RNase HI causes a growth defect, RNA was collected from all strains after overnight growth on solid media instead of liquid growth. For all Gc strains for which rnhA transcript expression would be tested by quantitative reverse transcription PCR (qRT-PCR), all strains were grown on GC agar medium overnight and lawns were swabbed for qRT-PCR. After growth on either liquid or solid GCB medium + Kellogg’s supplements I and II, bacteria were treated with RNA-Protect reagent (Qiagen 76526) and pelleted by centrifugation. Total RNA was purified using the RNeasy kit (Qiagen 74104) according to the manufacturer’s instructions. Purified RNA was treated with RQ1 DNase (Promega M6101) to remove contaminating genomic DNA, then further purified with the RNeasy kit. RNA was reverse transcribed using Superscript III polymerase (Invitrogen 18080093). To assure there was no genomic DNA contamination in the samples undergoing RT-qPCR, no reverse transcriptase controls were included with all samples tested. cDNA was amplified using the iQ SYBR Green Supermix (BioRad 170–8882) according to the manufacturer’s instructions with the following cycling parameters: one cycle, 95°C 3 min.; 40 cycles, 95°C 10 s, 58°C 30 s (Anderson & Seifert, 2013). Relative transcript abundance was determined using the ΔΔCt method of Schmittgen and Livak (Schmittgen & Livak, 2008). Housekeeping gene omp3 was used as the reference gene internal control (Stohl et al., 2005). All amplifications yielded a single product based on melting temperature analysis, all primer sets were optimized to assure uniform primer efficiency (Schmittgen & Livak, 2008). Expression results for mutant strains are expressed as the mean fold change in transcript abundance ± one standard deviation compared to the FA1090 recA6 or FA1090 parental strain (Anderson & Seifert, 2013).
The efficiency of the E. coli Rho-independent terminator sequence in halting sRNA transcription was measured by qRT-PCR using internal primers (RT-G4start and RT-midfor) within the first part of the sRNA and external primers designed to detect the sRNA sequence after the terminator sequence (RT-G4start and RT-termfor). The ratio of read-through sRNA was compared to total transcript levels by comparing the external primer set to the internal primer set. (RT-G4start and RT-midfor, RT-G4start an RT-termFor). rnhA transcript levels were determined using primers RT-rnhAF2 and RT-rnhAR2.
PDCMC Assay
The surrogate Av assay was performed as previously described (Sechman et al., 2005), with exception that the strains did not contain a regulatable recA gene. Therefore, the pilE sequence of each seeding colony was determined by sanger sequencing to ensure each assay started with the same pilE sequence (Seifert et al., 1994). A single colony (confirmed to be 1–81–S2)was spread on GCB agar plates and colony morphology tracked starting at 22 hours. Ten fully piliated colonies were selected and followed for 6 hours with scoring every two hours. Each bleb accounted for one point and four or more blebs was scored as a four.
RNA:DNA Hybrid ChIP Assay
ChIP was performed using confluent lawns of Gc grown overnight at 37°C with 5% CO2 on GCB agar + Kellogg supplements I and II with or without 1 mM IPTG. Total nucleic acids were extracted using phenolchloroform without exposure to heat (Wilson, 2001). 100 ug of DNA was then digested with 1 ul Mung Bean Nuclease (Promega M4311) for 30 min at 37 degrees C to stabilize R-loops (Wahba et al., 2016, Mehta & Laimins, 2018). DNA/RNA was ethanol precipitated in 50 ul 7.5M ammonium acetate and 250 ul ethanol at −80°C for 1 hour and centrifuged for 15 min at 15000xg at 4°C. The pellet was washed in 70% ethanol and the desiccated pellet was dissolved in 100 ul ddH2O. DNA was sonicated with a Diagnode Bioruptor with the settings: 30 seconds on and 30 seconds off, for 30 min at 4°C.
20 ul per sample of Protein-A Dynabeads (Invitrogen) were incubated with Herring testes DNA and BSA and incubated overnight at 4C. Beads were incubated with 20 μg/ml of anti-DNA/RNA hybrid S9.6 MAb (generously provided by Dr. Nicholas Proudfoot, Oxford University) or a normal mouse polyclonal IgG control antibody (sc2025 Santa Cruz Biotechnology) at 4°C for 8 hours. Supernatant was removed and 1 ml IP buffer (1% Triton X-100, 0.1% sodium deoxycholate, 10 mM EDTA, 50 mM Tris-HCL pH 8.0) and 100 ul sheared DNA were added to each tube and mixed overnight at 4°C. Bead complexes were washed eight times with 1 mL of RIPA buffer (50 mM Hepes pH 8.0, 1 mM EDTA pH 8.0, 1% NP40, 0.7% sodium Deoxycholate, 0.5 M LiCl, and Protease inhibitor). Complexes were suspended in 1 mL of 1X TE buffer and centrifuged at 3000 rpm for 3 minutes. The complexes were treated with 50μl of elution buffer (10 mM Tris pH 8.0, 1 mM EDTA, 10% SDS) at 65°C for 10 minutes, vortexing periodically during incubation. The eluted complexes were spun for 1 minute at 15000 rpm and then transferred to fresh tubes. This process was repeated with an additional 50 ul elution buffer. The precipitated DNA was purified by using a Qiagen DNA purification kit following the manufacturer’s protocol. (Mehta & Laimins, 2018, Wong et al., 2010). Primers RT-G4start and RT-midFor were used to amplify the G4 region, USaspcF1 and USaspcR1 were used to amplify the ectopic site, and 16sF1 and 16sF2 were used to amplify the 16s region. The ectopic site is used as a negative control, since transcription at this site is low, the pull down of this region of the genome should be low and represent non-specific binding of the S9.6 antibody (Mehr & Seifert, 1998). 16S RNA-specific primers were used in as a positive control, as transcription in this region is consistently high enough that R-loops should always be present. All primers were determined to have similar levels of efficiency based on the procedure of Livak et al. (Schmittgen & Livak, 2008). On bead treatment with E. coli Rnase H (NEB) was performed twice (El Hage et al., 2014).
G4 ChIP Assay
When multiple G4 specific antibodies (including BG4 (Biffi et al., 2013) and an unpublished phage display G4 MAbs) were tested for G4 ChIP signal (Hansel-Hertsch et al., 2018) and low background binding to DNA, only the 1H6 MAb produced a G4-specific signal. G4 ChIP was performed similarly to the previously described R-loop ChIP protocol, with the exception of not conducting the mung bean nuclease treatment. After extraction, 100 ug of DNA was digested with 10 ul BfaI (NEB). The extracted DNA was ethanol precipitated as described above. The digested DNA was added to beads blocked with either 2ug of MAb IH6 (MABE1126 Millipore) (Henderson et al., 2014) or a control mouse IgG (Santa Crux Technologies) in IP buffer, and incubated room overnight at 4C. Beads were washed eight times in RIPA buffer and complexes were suspended in 1 mL of 1X TE buffer and spun down at 3000 rpm for 3 minutes. The complexes were treated with 50 μl of elution buffer twice at 65 C. The precipitated DNA was quantified by qPCR with the primers RT-G43F and RT-G4R. The primer set used to quantify R-loop levels could be used in G4 ChIP as well, however, PCR efficiency across a G4 structure can affect the quantification. Since, we wanted to compare the parent and G4 mutant, we designed a primer set outside the G4 motif to avoid this complication.
PacBio Sequencing to determine Pilin Av Frequency
Strains were revived from frozen glycerol stocks at −80 C by streaking for overnight growth for 18 hours on GCB agar + Kellogg supplements I and II. A single colony was collected using a sterile 6 mm filter disk and dispersed in 500 ul GCBL with vigorous vortex mixing and dilutions were plated on GCB plates to obtain 200–400 colonies per plate. The remainder of the cell suspension was pelleted, washed with 1X PBS, and the bacteria were lysed in cell lysis buffer. The lysed bacterial suspension was used as a template for PCR and subsequent sequencing with primers PilRBS and Sp3A. This step ensured that all strains sequenced began with the same variant pilE sequence (1–81–S2) at the beginning of the experiment (Seifert et al., 1994).
rnhA+nicsPlac::rnhA without IPTG grows slower than the parent strain. To match the number of generations of the mutant strain to the parent strain during growth preceding extraction for PacBio sequencing, the strains were grown for different amounts of time that would amount to the same colony forming units (CFU) per single colony ratio (CFU/colony) for both strains, normalizing the growth state of all strains sequenced (Rotman et al., 2016). The rnhA mutant grown for 26 hours matched the CFU/colony value of the parental FA1090 strain at 22 hours, so these respective time points were used for these strains. At least 1000 total colonies of each strain were pooled in liquid GCB medium + Kellogg Supplements I and II, from which genomic DNA was isolated using the QiaAmp kit (Qiagen 51304). The pilE gene was amplified using the following reaction: 1 ng genomic DNA, 20 uM dNTPs, 1X Phusion reaction buffer, 0.5 uM Primer 1, 0.5 uM Primer 2, 3% DMSO, 1 unit Phusion Hot Start Flex (NEB) Polymerase and water. The reaction was run under the following conditions: 98°C for 30 s for initial denaturation and Polymerase activation, 98°C for 10 s, 65°C for 30 s then 0.3°C reduced in each cycle, 72°C 1min, repeat cycles 30 times, final extension for 5 min at 72°C.
A unique 16 base barcode identifier for each sample was encoded by including the PacBio barcode sequence (Verhey et al., 2018) on both the Forward and Reverse primers that amplified the pilE locus amplicon sequenced in this study (Supplemental Table 5). Amplification with the primers PilRBS-TTTCCCCTTTCAATTAGGAG and OpaERev- GGGTTCCGGGCGGTGTTTC yielded 788 bp of pilE sequence within the 820 bp total amplicon containing the unique barcodes. PCR products were gel extracted without Ethidium bromide exposure. Gel extraction was performed with the QiaQuick gel extraction kit (Qiagen) according to the manufacturer’s instructions, except the gel slices were dissolved at room temperature to maintain DNA integrity. The DNA was eluted with TE buffer and pooled to obtain 300 ng of DNA per sample. Samples were submitted to the University of Maryland Genomic Resource Center, where the amplicons were cleaned with SPRI clean up, quantified and pooled into two runs. The pools underwent SMRTbell library prep and subsequent sequencing on the Pacific Biology Sequel machine SMRT cell with v3 reagents (Verhey et al., 2018). Frequencies were analyzed using a custom program (https://github.com/egonozer/switchAmp) (Ozer et al., 2019). Reads can be accessed under the SRA project SRP214219 with the bioproject accession number PRJNA553228 (Supplemental Table 2).
The silent copy choice for each variation event was also analyzed using the SwitchAmp program. There were four common silent copy choices among all the strains tested. The tail sequence of silent copies 2c1 and 6c1 (2c1:6c1 HVT) was one of the common events, similar to previously published results (Criss et al., 2005, Rotman et al., 2016). We also observed a mosaic sequence containing both 1–81–S2 and the 2c1:6c1 tail sequence that was not observed in the negative control strains. Additionally, the use of silent copy 3c1 was common among all the strains. The forth major category of variants was multivariant, in which the read contained silent copy sequence shared among multiple copies, so a single donor could not be identified (Supplemental Figure 5 and 6) (Ozer et al., 2019).
Supplementary Material
Author Summary.
Neisseria gonorrhoeae, the causative agent of gonorrhea, changes one of its surface exposed structures to avoid immune detection. This process is known as antigenic variation and involves a complex process of DNA recombination. There are two alternate DNA and RNA structures that may be involved in DNA recombination adjacent to the gene that undergoes antigenic variation, a guanine quadruplex and an RNA:DNA hybrid made from a small RNA. Both of these structures can cause nicks or breaks in DNA, and this work investigates if the RNA:DNA hybrid influences the guanine quadruplex or antigenic variation. We found that reducing transcription lowers both guanine quadruplex and antigenic variation, but increasing the RNA:DNA hybrids does not change either one.
Acknowledgements
We thank Sarah Quillin and Linda Hu for editorial assistance and Kavi Mehta for experimental advice. We are grateful to Nicholas Proudfoot at the Sir William Dunn School of Pathology at University of Oxford for the generous gift of the S9.6 antibody. We acknowledge the University of Maryland Genomic Resource Center for completing the PacBio sequencing. This work was supported by NIH grant R37 AI033493. This work was also supported by the Northwestern University NUSeq Core Facility.
Literature Cited
- Aguilera A (2002) The connection between transcription and genomic instability. The EMBO journal 21: 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexeyev MF (1995) Three kanamycin resistance gene cassettes with different polylinkers. BioTechniques 18: 52, 54,, 56. [PubMed] [Google Scholar]
- Anderson MT, and Seifert HS (2013) Phase variation leads to the misidentification of a Neisseria gonorrhoeae virulence gene. PloS one 8: e72183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asamitsu S, Obata S, Yu Z, Bando T, and Sugiyama H (2019) Recent Progress of Targeted G-Quadruplex-Preferred Ligands Toward Cancer Therapy. Molecules 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaume N, Pathak R, Yadav VK, Kota S, Misra HS, Gautam HK, and Chowdhury S (2013) Genome-wide study predicts promoter-G4 DNA motifs regulate selective functions in bacteria: radioresistance of D. radiodurans involves G4 DNA-mediated regulation. Nucleic acids research 41: 76–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia V, Barroso SI, Garcia-Rubio ML, Tumini E, Herrera-Moyano E, and Aguilera A (2014) BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 511: 362–365. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya D, Mirihana Arachchilage G, and Basu S (2016) Metal Cations in G-Quadruplex Folding and Stability. Front Chem 4: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G, Tannahill D, McCafferty J, and Balasubramanian S (2013) Quantitative visualization of DNA G-quadruplex structures in human cells. Nat Chem 5: 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs E, Crouch K, Lemgruber L, Lapsley C, and McCulloch R (2018) Ribonuclease H1-targeted R-loops in surface antigen gene expression sites can direct trypanosome immune evasion. PLoS genetics 14: e1007729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoon LA, Manthei KA, Rotman E, Keck JL, and Seifert HS (2013) Neisseria gonorrhoeae RecQ helicase HRDC domains are essential for efficient binding and unwinding of the pilE guanine quartet structure required for pilin antigenic variation. J Bacteriol 195: 2255–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoon LA, and Seifert HS (2009) An alternative DNA structure is necessary for pilin antigenic variation in Neisseria gonorrhoeae. Science 325: 764–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoon LA, and Seifert HS (2011) Focusing homologous recombination: pilin antigenic variation in the pathogenic Neisseria. Molecular microbiology 81: 1136–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoon LA, and Seifert HS (2013) Transcription of a cis-acting, noncoding, small RNA is required for pilin antigenic variation in Neisseria gonorrhoeae. PLoS pathogens 9: e1003074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YA, Aristizabal MJ, Lu PY, Luo Z, Hamza A, Kobor MS, Stirling PC, and Hieter P (2014) Genome-wide profiling of yeast DNA:RNA hybrid prone sites with DRIP-chip. PLoS genetics 10: e1004288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, Dierick I, Abel A, Kennerson ML, Rabin BA, Nicholson GA, Auer-Grumbach M, Wagner K, De Jonghe P, Griffin JW, Fischbeck KH, Timmerman V, Cornblath DR, and Chance PF (2004) DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 74: 1128–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criss AK, Kline KA, and Seifert HS (2005) The frequency and rate of pilin antigenic variation in Neisseria gonorrhoeae. Molecular microbiology 58: 510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S, Masukata H, and Tomizawa J (1987) Multiple mechanisms for initiation of ColE1 DNA replication: DNA synthesis in the presence and absence of ribonuclease H. Cell 51: 1113–1122. [DOI] [PubMed] [Google Scholar]
- Deitsch KW, Lukehart SA, and Stringer JR (2009) Common strategies for antigenic variation by bacterial, fungal and protozoan pathogens. Nature reviews. Microbiology 7: 493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquette ML, Handa P, Vincent JA, Taylor AF, and Maizels N (2004) Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev 18: 1618–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hage A, Webb S, Kerr A, and Tollervey D (2014) Genome-wide distribution of RNA-DNA hybrids identifies RNase H targets in tRNA genes, retrotransposons and mitochondria. PLoS genetics 10: e1004716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoh A, Iwasaki H, Ishioka K, and Shinagawa H (1997) ATP-dependent resolution of R-loops at the ColE1 replication origin by Escherichia coli RecG protein, a Holliday junction-specific helicase. The EMBO journal 16: 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacani L, Brandt SL, Puray-Chavez M, Reid TB, Godornes C, Molini BJ, Benzler M, Hartig JS, Lukehart SA, and Centurion-Lara A (2012) Comparative investigation of the genomic regions involved in antigenic variation of the TprK antigen among treponemal species, subspecies, and strains. J Bacteriol 194: 4208–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover L, Alsford S, and Horn D (2013) DNA break site at fragile subtelomeres determines probability and mechanism of antigenic variation in African trypanosomes. PLoS pathogens 9: e1003260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh M, Lufino MM, Wade-Martins R, and Gromak N (2014) R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS genetics 10: e1004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyi JI, Conn GL, Lane AN, and Brown T (1996) Comparison of the thermodynamic stabilities and solution conformations of DNA.RNA hybrids containing purine-rich and pyrimidine-rich strands with DNA and RNA duplexes. Biochemistry 35: 12538–12548. [DOI] [PubMed] [Google Scholar]
- Haas R, and Meyer TF (1986) The repertoire of silent pilus genes in Neisseria gonorrhoeae: evidence for gene conversion. Cell 44: 107–115. [DOI] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, Rothstein JD, and Wang J (2014) C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507: 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagblom P, Segal E, Billyard E, and So M (1985) Intragenic recombination leads to pilus antigenic variation in Neisseria gonorrhoeae. Nature 315: 156–158. [DOI] [PubMed] [Google Scholar]
- Hansel-Hertsch R, Spiegel J, Marsico G, Tannahill D, and Balasubramanian S (2018) Genome-wide mapping of endogenous G-quadruplex DNA structures by chromatin immunoprecipitation and high-throughput sequencing. Nat Protoc 13: 551–564. [DOI] [PubMed] [Google Scholar]
- Hardin CC, Watson T, Corregan M, and Bailey C (1992) Cation-dependent transition between the quadruplex and Watson-Crick hairpin forms of d(CGCG3GCG). Biochemistry 31: 833–841. [DOI] [PubMed] [Google Scholar]
- Hartono SR, Malapert A, Legros P, Bernard P, Chedin F, and Vanoosthuyse V (2018) The Affinity of the S9.6 Antibody for Double-Stranded RNAs Impacts the Accurate Mapping of R-Loops in Fission Yeast. Journal of molecular biology 430: 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmrich A, Ballarino M, and Tora L (2011) Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Molecular cell 44: 966–977. [DOI] [PubMed] [Google Scholar]
- Henderson A, Wu Y, Huang YC, Chavez EA, Platt J, Johnson FB, Brosh RM Jr., Sen D, and Lansdorp PM (2014) Detection of G-quadruplex DNA in mammalian cells. Nucleic acids research 42: 860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland J, Spindler K, Horodyski F, Grabau E, Nichol S, and VandePol S (1982) Rapid evolution of RNA genomes. Science 215: 1577–1585. [DOI] [PubMed] [Google Scholar]
- Hu Z, Zhang A, Storz G, Gottesman S, and Leppla SH (2006) An antibody-based microarray assay for small RNA detection. Nucleic acids research 34: e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itaya M (1990) Isolation and characterization of a second RNase H (RNase HII) of Escherichia coli K-12 encoded by the rnhB gene. Proceedings of the National Academy of Sciences of the United States of America 87: 8587–8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Fiddes IT, Miga KH, Olsen HE, Paten B, and Akeson M (2015) Improved data analysis for the MinION nanopore sequencer. Nature methods 12: 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemier HG, Paeschke K, and Lansdorp PM (2017) Guanine quadruplex monoclonal antibody 1H6 cross-reacts with restrained thymidine-rich single stranded DNA. Nucleic acids research 45: 5913–5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg DS Jr., Cohen IR, Norins LC, Schroeter AL, and Reising G (1968) Neisseria gonorrhoeae. II. Colonial variation and pathogenicity during 35 months in vitro. Journal of Bacteriology 96: 596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline KA, and Seifert HS (2005) Role of the Rep helicase gene in homologous recombination in Neisseria gonorrhoeae. J Bacteriol 187: 2903–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogoma T (1986) RNase H-defective mutants of Escherichia coli. J Bacteriol 166: 361–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Wu P, Ohmichi T, and Sugimoto N (2002) Characterization and thermodynamic properties of quadruplex/duplex competition. FEBS Lett 526: 77–81. [DOI] [PubMed] [Google Scholar]
- Maizels N (2005) Immunoglobulin gene diversification. Annual review of genetics 39: 23–46. [DOI] [PubMed] [Google Scholar]
- Maizels N, and Gray LT (2013) The G4 genome. PLoS genetics 9: e1003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch R, Morrison LJ, and Hall JPJ (2015) DNA Recombination Strategies During Antigenic Variation in the African Trypanosome. Microbiol Spectr 3: MDNA3–0016-2014. [DOI] [PubMed] [Google Scholar]
- McDonald JP, Vaisman A, Kuban W, Goodman MF, and Woodgate R (2012) Mechanisms employed by Escherichia coli to prevent ribonucleotide incorporation into genomic DNA by Pol V. PLoS genetics 8: e1003030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehr IJ, and Seifert HS (1997) Random shuttle mutagenesis: gonococcal mutants deficient in pilin antigenic variation. Molecular microbiology 23: 1121–1131. [DOI] [PubMed] [Google Scholar]
- Mehr IJ, and Seifert HS (1998) Differential roles of homologous recombination pathways in Neisseria gonorrhoeae pilin antigenic variation, DNA transformation and DNA repair. Molecular microbiology 30: 697–710. [DOI] [PubMed] [Google Scholar]
- Mehta K, and Laimins L (2018) Human Papillomaviruses Preferentially Recruit DNA Repair Factors to Viral Genomes for Rapid Repair and Amplification. mBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer TF, Billyard E, Haas R, Storzbach S, and So M (1984) Pilus genes of Neisseria gonorrheae: chromosomal organization and DNA sequence. Proceedings of the National Academy of Sciences of the United States of America 81: 6110–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozer EA, Prister LL, Yin SY, Ward BH, Ivanov SS, and Seifert HS (2019) PacBio amplicon sequencing method to measure pilin antigenic variation frequencies of Neisseria gonorrhoeae. Biorxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer GH, Bankhead T, and Seifert HS (2016) Antigenic Variation in Bacterial Pathogens. Microbiol Spectr 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HS, Wolfgang M, van Putten JP, Dorward D, Hayes SF, and Koomey M (2001) Structural alterations in a type IV pilus subunit protein result in concurrent defects in multicellular behaviour and adherence to host tissue. Molecular microbiology 42: 293–307. [DOI] [PubMed] [Google Scholar]
- Prister LL, and Seifert HS (2018) R-loops modulate Trypanosome antigenic variation. PLoS genetics 14: e1007809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quillin SJ, Hockenberry AJ, Jewett MC, and Seifert HS (2018) Neisseria gonorrhoeae Exposed to Sublethal Levels of Hydrogen Peroxide Mounts a Complex Transcriptional Response. mSystems 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quillin SJ, and Seifert HS (2018) Neisseria gonorrhoeae host adaptation and pathogenesis. Nature reviews. Microbiology 16: 226–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel-Chavez C, Galan-Vasquez E, and Martinez-Antonio A (2017) Consensus architecture of promoters and transcription units in Escherichia coli: design principles for synthetic biology. Mol Biosyst 13: 665–676. [DOI] [PubMed] [Google Scholar]
- Reaban ME, and Griffin JA (1990) Induction of RNA-stabilized DNA conformers by transcription of an immunoglobulin switch region. Nature 348: 342–344. [DOI] [PubMed] [Google Scholar]
- Reynolds R, Bermudez-Cruz RM, and Chamberlin MJ (1992) Parameters affecting transcription termination by Escherichia coli RNA polymerase. I. Analysis of 13 rho-independent terminators. Journal of molecular biology 224: 31–51. [DOI] [PubMed] [Google Scholar]
- Rhodes D, and Lipps HJ (2015) G-quadruplexes and their regulatory roles in biology. Nucleic acids research 43: 8627–8637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotman E, Webber DM, and Seifert HS (2016) Analyzing Neisseria gonorrhoeae pilin antigenic variation using 454 sequencing technology. J Bacteriol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy D, and Lieber MR (2009) G clustering is important for the initiation of transcription-induced R-loops in vitro, whereas high G density without clustering is sufficient thereafter. Mol Cell Biol 29: 3124–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudel T, van Putten JP, Gibbs CP, Haas R, and Meyer TF (1992) Interaction of two variable proteins (PilE and PilC) required for pilus-mediated adherence of Neisseria gonorrhoeae to human epithelial cells. Molecular microbiology 6: 3439–3450. [DOI] [PubMed] [Google Scholar]
- Santos-Pereira JM, and Aguilera A (2015) R loops: new modulators of genome dynamics and function. Nat Rev Genet 16: 583–597. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, and Livak KJ (2008) Analyzing real-time PCR data by the comparative CT method. Nature Protocols 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
- Sechman EV, Rohrer MS, and Seifert HS (2005) A genetic screen identifies genes and sites involved in pilin antigenic variation inNeisseria gonorrhoeae. Molecular microbiology 57: 468–483.15978078 [Google Scholar]
- Seifert HS (1997) Insertionally inactivated and inducible recA alleles for use in Neisseria. Gene 188: 215–220. [DOI] [PubMed] [Google Scholar]
- Seifert HS (2018) Above and Beyond Watson and Crick: Guanine Quadruplex Structures and Microbes. Annu Rev Microbiol 72: 49–69. [DOI] [PubMed] [Google Scholar]
- Seifert HS, Ajioka RS, Paruchuri D, Heffron F, and So M (1990) Shuttle mutagenesis of Neisseria gonorrhoeae: pilin null mutations lower DNA transformation competence. J Bacteriol 172: 40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert HS, Wright CJ, Jerse AE, Cohen MS, and Cannon JG (1994) Multiple gonococcal pilin antigenic variants are produced during experimental human infections. The Journal of clinical investigation 93: 2744–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar EP, Lazio MP, and Seifert HS (2002) Roles of the recJ and recN genes in homologous recombination and DNA repair pathways of Neisseria gonorrhoeae. J Bacteriol 184: 919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smargiasso N, Gabelica V, Damblon C, Rosu F, De Pauw E, Teulade-Fichou MP, Rowe JA, and Claessens A (2009) Putative DNA G-quadruplex formation within the promoters of Plasmodium falciparum var genes. BMC Genomics 10: 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollier J, and Cimprich KA (2015) Breaking bad: R-loops and genome integrity. Trends Cell Biol 25: 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparling PF (1966) Genetic transformation of Neisseria gonorrhoeae to streptomycin resistance. J Bacteriol 92: 1364–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohl EA, Criss AK, and Seifert HS (2005) The transcriptome response of Neisseria gonorrhoeae to hydrogen peroxide reveals genes with previously uncharacterized roles in oxidative damage protection. Molecular microbiology 58: 520–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohl EA, Dale EM, Criss AK, and Seifert HS (2013) Neisseria gonorrhoeae metalloprotease NGO1686 is required for full piliation, and piliation is required for resistance to H2O2- and neutrophil-mediated killing. mBio 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson J, Bergstrom S, Boslego J, and Koomey M (1987) Gene conversion accounts for pilin structural changes and for reversible piliation “phase” changes in gonococci. Antonie Van Leeuwenhoek 53: 441–446. [DOI] [PubMed] [Google Scholar]
- Tarsounas M, and Tijsterman M (2013) Genomes and G-quadruplexes: for better or for worse. Journal of molecular biology 425: 4782–4789. [DOI] [PubMed] [Google Scholar]
- Tobiason DM, and Seifert HS (2006) The obligate human pathogen, Neisseria gonorrhoeae, is polyploid. PLoS biology 4: e185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuduri S, Crabbe L, Conti C, Tourriere H, Holtgreve-Grez H, Jauch A, Pantesco V, De Vos J, Thomas A, Theillet C, Pommier Y, Tazi J, Coquelle A, and Pasero P (2009) Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat Cell Biol 11: 1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey TB, Castellanos M, and Chaconas G (2018) Analysis of recombinational switching at the antigenic variation locus of the Lyme spirochete using a novel PacBio sequencing pipeline. Molecular microbiology 107: 104–115. [DOI] [PubMed] [Google Scholar]
- Wahba L, Costantino L, Tan FJ, Zimmer A, and Koshland D (2016) S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes & Development 30: 1327–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walia R, and Chaconas G (2013) Suggested role for G4 DNA in recombinational switching at the antigenic variation locus of the Lyme disease spirochete. PloS one 8: e57792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson K (2001) Preparation of genomic DNA from bacteria Current protocols in molecular biology / edited by Frederick M Ausubel … [et al. ] Chapter 2: Unit 2 4. [DOI] [PubMed] [Google Scholar]
- Wolfgang M, Lauer P, Park HS, Brossay L, Hebert J, and Koomey M (1998) PilT mutations lead to simultaneous defects in competence for natural transformation and twitching motility in piliated Neisseria gonorrhoeae. Molecular microbiology 29: 321–330. [DOI] [PubMed] [Google Scholar]
- Wong PP, Pickard A, and McCance DJ (2010) p300 alters keratinocyte cell growth and differentiation through regulation of p21(Waf1/CIP1). PloS one 5: e8369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wormann ME, Horien CL, Bennett JS, Jolley KA, Maiden MC, Tang CM, Aho EL, and Exley RM (2014) Sequence, distribution and chromosomal context of class I and class II pilin genes of Neisseria meningitidis identified in whole genome sequences. BMC Genomics 15: 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, and Clayton DA (1996) RNA-DNA hybrid formation at the human mitochondrial heavy-strand origin ceases at replication start sites: an implication for RNA-DNA hybrids serving as primers. The EMBO journal 15: 3135–3143. [PMC free article] [PubMed] [Google Scholar]
- Xu J, and Seifert HS (2018) Analysis of Pilin Antigenic Variation in Neisseria meningitidis by Next-Generation Sequencing. J Bacteriol 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Ishizuka T, Yang J, Ito K, Katada H, Komiyama M, and Hayashi T (2012) Oligonucleotide models of telomeric DNA and RNA form a Hybrid G-quadruplex structure as a potential component of telomeres. The Journal of biological chemistry 287: 41787–41796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Chedin F, Hsieh CL, Wilson TE, and Lieber MR (2003) R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nature immunology 4: 442–451. [DOI] [PubMed] [Google Scholar]
- Zhang ZZ, Pannunzio NR, Han L, Hsieh CL, Yu K, and Lieber MR (2014) The strength of an Ig switch region is determined by its ability to drive R loop formation and its number of WGCW sites. Cell Rep 8: 557–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Vuong BQ, Vaidyanathan B, Lin JY, Huang FT, and Chaudhuri J (2015) Non-coding RNA Generated following Lariat Debranching Mediates Targeting of AID to DNA. Cell 161: 762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.