

Abstract
Herein we disclose the synthesis and full characterization of the first monocyclic aromatic 1,2,3,5-tetrazine, 4,6-diphenyl-1,2,3,5-tetrazine. Initial studies of its cycloaddition reactivity, mode, regioselectivity, and scope illustrate that it participates as the 4π-component of well-behaved inverse electron demand Diels–Alder reactions where it preferentially reacts with electron-rich or strained dienophiles. It was found to exhibit an intrinsic reactivity comparable to that of the isomeric 3,6-diphenyl-1,2,4,5-tetrazine, display a single mode of cycloaddition with reaction only across C4/N1 (no N2/N5 cycloaddition observed), proceed with a predictable regioselectivity (dienophile most electron-rich atom attaches to C4), and manifest additional reactivity complementary to the isomeric 1,2,4,5-tetrazines. It not only exhibits a remarkable cycloaddition reactivity, surprisingly good stability (e.g., stable to chromatography, long term storage, presence of H2O even as reaction co-solvent), and broad cycloaddition scope, but it also displays powerful orthogonal reactivity with the 1,2,4,5-tetrazines. Whereas the latter reacts at extraordinary cycloaddition rates with strained dienophiles (tetrazine ligation), the new and isomeric 1,2,3,5-tetrazine displays similarly remarkable cycloaddition rates and efficiencies with amidines (1,2,3,5-tetrazine/amidine ligation). The crossover reactivities (1,2,4,5-tetrazines with amidines vs 1,2,3,5-tetrazines with strained dienophiles) are sufficiently low to indicate they may be capable of use concurrently without competitive reactions.
Graphical Abstract

The first monocyclic aromatic 1,2,3,5-tetrazine:
Synthesis and full characterization
Reactive diene in inverse electron demand Diels-Alder reactions
Single mode of cycloaddition (C4/N1) and predictable regioselectivity
Orthogonal reactivity with 1,2,4,5-tetrazine without competitive reactions
INTRODUCTION
The inverse electron demand Diels–Alder cycloadditions of heterocyclic azadienes have provided powerful methodology for the synthesis of highly substituted and functionalized heterocycles1 widely used in organic synthesis and the pharmaceutical industry, in the divergent construction of screening libraries2 and in bioorthogonal conjugation.3,4 Our past efforts have provided systematic explorations of the reactions of 1,2,4,5-tetrazines,5 1,2,4-triazines,6 1,3,5-triazines,7 1,3,4-oxadiazoles,8 1,2-diazines9 and most recently 1,2,3-triazines.10 Each heterocyclic azadiene was found to possess a unique reactivity toward different classes of dienophiles, display now predictable modes of cycloaddition, and exhibit substantial substituent electronic effects impacting their intrinsic reactivity and cycloaddition regioselectivity. These insights permitted their use as key steps in more than 50 natural products total syntheses11 and helped inspire the development of the powerful 1,2,4,5-tetrazine based bioorthogonal conjugation and labeling technology (tetrazine ligation) widely used today. Consequently, it is remarkable in this day and age that no member of another fundamentally important heterocyclic azadiene ring system, the 1,2,3,5-tetrazines, has yet been reported (Figure 1A).
Figure 1.

(A) Structure of 1,2,3,5-tetrazine (1) and 4,6-diphenyl-1,2,3,5-tetrazine (2). (B) Reported structures bearing a nonaromatic 1,2,3,5-tetrazine core.
Limited attempts to prepare a 1,2,3,5-tetrazine have been described14 and no monocyclic aromatic 1,2,3,5-tetrazine as a discrete 6π heterocycle has been reported. Stability profiles established in computational studies suggest a decreased kinetic stability for 1,2,3,5-tetrazine compared with the well-known and isomeric 1,2,4,5-tetrazine (ΔEact 14 kcal/mol), but a greater thermodynamic stability than 1,2,4,5-tetrazine (7–8 kcal/mol).12 The principal challenge in the synthesis of 1,2,3,5-tetrazines is the construction of two consecutive N–N bonds, which are not present in 1,2,4,5-tetrazines but are found in the 1,2,3-triazines. Although no synthesis of a monocyclic 1,2,3,5-tetrazine as a 6π aromatic system has been reported, limited numbers of heterocyclic compounds bearing the 1,2,3,5-tetrazine core have been disclosed (Figure 1B).13–15 The nonaromatic 1,2,3,5-tetrazine core (3) was synthesized as part of fused heterocycles in the development of energetic materials,13 monocyclic tetrazinones have been described that were prepared by a 6π-electrocyclization of acyclic triazene isocyanate precursors14 and the synthesis of a library of ring-fused bicyclic tetrazinones has been reported of which a derivative (temozolomide, 4) is used clinically.15 The methods used in this work for the construction of the core ring system as well as those used for the preparation of 1,2,3-triazines10,16 provided valuable insights into the possible synthesis of aromatic 1,2,3,5-tetrazines. With even their stable existence in question, we explored several approaches for their synthesis. As a result of these efforts, herein we report the synthesis, characterization, properties, and cycloaddition reactivity of the first member of this new and previously unknown class of heterocycles.
RESULTS AND DISCUSSION
Synthesis and characterization of 4,6-diphenyl-1,2,3,5-tetrazine.
The widely utilized syntheses of the isomeric 1,2,4,5-tetrazines employ an oxidative aromatization of dihydro-1,2,4,5-tetrazines as an effective final step,17 suggesting that the preparation of the 1,2,3,5-tetrazine system from a non-aromatic dihydro precursor likely would be similarly successful. In a series of studies focused on the reactivity of 1,2,3-triazolium amides, Butler et al. reported a beautiful reaction cascade to furnish N2,N5-diaryl substituted 2,5-dihydro-1,2,3,5-tetrazines (Scheme 1A).18 We envisioned that subsequent removal of the N-aryl substituents by either oxidation or SNAr reactions, followed by a subsequent oxidation of the resulting dihydrotetrazine would be a promising approach to prepare 4,6-disubstituted 1,2,3,5-tetrazines.
Scheme 1.
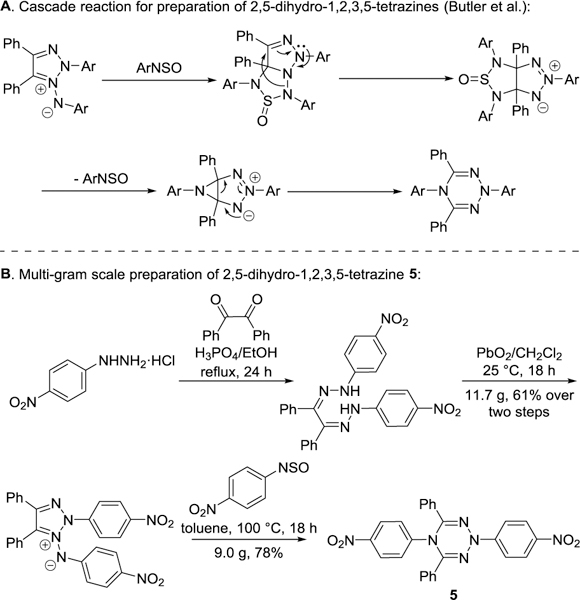
For initial studies, we chose to target the preparation of the substituted tetrazine 2, 4,6-diphenyl-1,2,3,5-tetrazine. We began our study with development of a multigram scale preparation of 5,18 a reported dihydro-1,2,3,5-tetrazine bearing p-nitrophenyl groups as the N-substituents (Scheme 1B). In order to investigate the oxidative dearylation effected by reagents like cerium ammonium nitrate (CAN), hydrogenation of 5 was conducted to yield 2,5-dihydrotetrazine 7 in which the N-substituents were reduced to p-anilinyl as an appropriate reaction precursor (Scheme 2). The alternative SNAr reaction of 5, using thiophenol as the nucleophile, proceeded smoothly to yield the stable N5-dearylated product 6. However, the second dearylation of the remaining N2-substituent was found to be difficult and has not yet been achieved even under harsher reaction conditions.
Scheme 2.
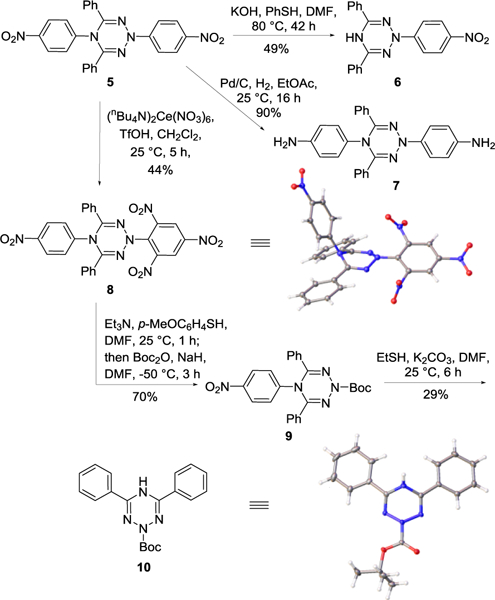
A serendipitous finding was observed when the oxidative dearylations of 5, 6, and 7 were examined. CAN or 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) mediated oxidative dearylations of 6 and most notably 7 were not successful where oxidation and decomposition of the dihydrotetrazine system were observed prior to the desired N-dearylations. With 5, however, treatment with (nBu4N)2Ce(NO3)6, a CAN equivalent that is soluble in organic solvents, resulted in selective dinitration of the N2-p-nitrophenyl substituent, affording 8 in 44% yield. The formation and structure of 8 were established by NMR and HRMS and unambiguously confirmed with a single crystal X-ray structure determination (Scheme 2).19 CAN-mediated nitrations have been reported,20 and the presence of stoichiometric acid was needed to effectively promote the reaction that provided 8.21 Notably, use of CAN instead of (nBu4N)2Ce(NO3)6 either in acetonitrile or methanol resulted in little or no reaction product and CH2Cl2 was found to be the most effective solvent for the reaction of those examined (Figure 2). Although the attempted oxidation of 5 did not remove the aryl substituents, we viewed this transformation as an activation of the unreactive N2-aryl moiety, permitting subsequent SNAr dearylation.
Figure 2.

Discovery and optimization of the nitration reaction of 5.
With 8 in hand, two sequential SNAr dearylations were conducted. As expected, the N2-picryl group is now more vulnerable to nucleophilic attack by an aryl thiol (p-MeOC6H4SH, Et3N) under mild conditions (25 °C, 1 h), leaving the N5-substituent intact. The released free amine was found to be unstable at 25 °C, which is in sharp contrast to its isomer 6 (stable at 80 °C). As a result, a subsequent in situ Boc-protection was performed to provide 9 in 70% overall yield. Interestingly, normal protection conditions using a weak base (Et3N) were not effective, and a strong base (NaH) was required. Conducting the reaction at low temperature (–50 °C) was also found to be important in this protection step to avoid unproductive consumption of the free amine. The second nucleophilic substitution reaction was more challenging due to the instability of 9 (compared to 5) in the presence of nucleophiles. Use of an alkyl thiol as a stronger nucleophile was found to promote the remaining SNAr reaction under mild conditions (EtSH, K2CO3, DMF, 25 °C, 6 h), providing the N2-Boc protected dihydrotetrazine 10 (29%, see Table S2 in Supporting Information for conditions surveyed). Because limited structural information was provided by NMR, the structure and solid-state conformation of 10 were unambiguously determined by X-ray crystallography (Scheme 2).19 The final oxidation of 10 proceeded smoothly with a range of oxidants under mild reaction conditions, affording the desired 4,6-diphenyl-1,2,3,5-tetrazine (2). A spontaneous loss of the Boc group was observed to accompany the oxidation. Compared with other oxidants, including CAN or (nBu4N)2Ce(NO3)6, MnO2 was found to be the most effective and convenient reagent of those examined, providing 2 in high yield (80%) with simple filtration removal of the oxidant following completion of the reaction (Figure 3). Tetrazine 2 is semi-stable on a silica gel column, although a rapid column chromatography purification was important to provide the product in high yield.
Figure 3.

Oxidation of dihydrotetrazine 10 to tetrazine 2.
4,6-Diphenyl-1,2,3,5 tetrazine 2 is a yellow crystalline solid at room temperature, and crystalline needles suitable for X-ray single crystal diffraction analysis were obtained by recrystallization from CH2Cl2, CHCl3, or MeCN.19 The crystal structure is presented in Figure 4, alongside the structure of 2,5-dihydro-1,2,3,5-tetrazine 10 for comparison. All atoms in 2 were found to be nearly coplanar and equalization of the bond lengths in the central 1,2,3,5-tetrazine core was observed, with C–N bond lengths of 1.338–1.356 Å and N–N bond lengths of 1.326–1.328 Å, depicting a clear aromatic character for the delocalized 6π-aromatic 1,2,3,5-tetrazine ring (typical bond lengths: C–N: 1.416 Å, C=N: 1.279 Å, N–N: 1.420 Å, N=N: 1.240 Å22). This is in sharp contrast to the dihydrotetrazine derivatives 8 and 10, where the dihydro-1,2,3,5-tetrazine core adopt boat conformations, and bond lengths of 1.392–1.393 Å, 1.284–1.286 Å and 1.424–1.425 Å were found for the C–N bonds, C=N bonds and N–N bonds in 10, respectively. The 1H NMR spectrum of tetrazine 2 exhibited a single set of phenyl peaks, indicative of free rotation of the phenyl ring in the solution. Notably, the ortho-protons of the phenyl substituent exhibit a downfield chemical shift of 8.8 ppm (same set of protons in 10 has a chemical shift of 7.8 ppm), resulting from a combined inductive and deshielding effect of the aromatic tetrazine. The tetrazine was found to display a characteristic downfield carbon peak (163 ppm) in the 13C NMR, like that of the carbon found in 3,6-diphenyl-1,2,4,5-tetrazine (164 ppm).
Figure 4.

Top and side view of single crystal X-ray structures of 10 (left) and 2 (right). The Boc-group in 10 is omitted for clarity.
Tetrazine 2 was found to be stable at 25 °C in the crystalline state, in a chloroform solution, and even in a protic solvent (CD3CN/D2O, V/V = 7:3), all for at least 5 days after which its monitoring was discontinued. It was also found to be stable for at least 24 h at 25 °C in 70% H2O/CH3CN, 50% and 80% H2O/DMSO, CH3CN/PBS buffer (1:1), and hexafluoroisopropanol (HFIP). It can be stored at 4 °C for more than 50 days without observable decomposition. To obtain insights into its thermal stability, the kinetics of thermal consumption of tetrazine 2 were monitored by 1H NMR at 80 °C, the lowest temperature at which measurable conversion was readily observed. A clean transformation of tetrazine 2 to benzonitrile and N2 was found, with no observation of other byproducts or intermediates. First-order kinetics for the transformation were observed, with a rate constant of k = 0.0293 h−1 (8.15 × 10−6 s−1) and half-life of t1/2 = 23.6 h (8.50 × 104 s) (Supporting Information Figure S2). Although it is kinetically less stable than the corresponding 1,2,4,5-tetrazine in agreement with earlier computational studies,12 the observed stability of tetrazine 2 permits easy handling under normal conditions and it can be warmed to elevated temperatures in solvent to promote cycloadditions with predictable limitations on the conditions used.21
We also examined the photophysical and electrochemical properties of 2. The UV-Vis spectrum of 2 showed a strong absorption (molar extinction coefficient ε = 2.52 × 104 L·mol−1·cm−1) peak at 275 nm, corresponding to the π-π* transition of the phenyl rings and tetrazine core (Figure 5). A much weaker absorption (ε = 4.90 × 102 L·mol−1·cm−1) was observed at 396 nm, corresponding to the n-π* transition of the tetrazine. It displayed no fluorescence, being distinct from the isomeric and weakly fluorescent 3,6-diphenyl-1,2,4,5-tetrazine (ϕ = 1.5 × 10−4).23 Tetrazine 2 was found to undergo a quasi-reversible single-electron reduction at –1.37 V (vs Fc+/Fc, –0.78 V vs N. H. E.) in cyclic voltammetry (CV), revealing its electron-deficient nature (Figure 5). In contrast to its reduction potential, no observable oxidation was observed when a positive scan was performed on 2. The reduction potential of 2 is comparable but slightly lower than 3,6-diphenyl-1,2,4,5-tetrazine (–1.21 V vs Fc+/Fc),24 indicating a similar electron deficiency and likely similar reactivity in inverse electron demand Diels–Alder cycloadditions.
Figure 5.
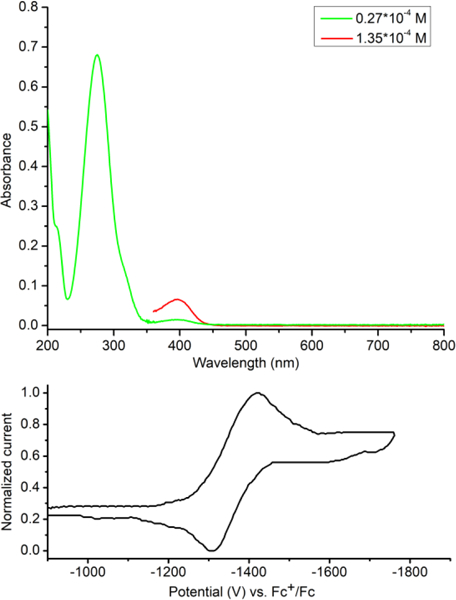
UV-Vis spectrum (top) and cyclic voltammetry data (bottom) of tetrazine 2. UV-Vis spectrum was measured in CH3CN. CV (scan rate, 40 mV/s) measurement was carried out in a solution of nBu4NBF4 (100 mM) in CH3CN.
Cycloaddition reactivity.
We previously reported the discovery of the unusually rapid inverse electron demand Diels–Alder cycloadditions of a series of 1,2,3-triazines with amidines as heterodienophiles.10 As a 5-aza-1,2,3-triazine, 1,2,3,5-tetrazines were anticipated to react with amidines to provide analogous cycloaddition products. When tetrazine 2 in acetonitrile (40 mM) was treated with benzamidine 11a, complete conversion to 12a was found after 2 h at 25 °C even at the dilute reaction concentrations. An exclusive C4/N1 versus N2/N5 mode of cycloaddition and a single reaction regioselectivity were observed, affording triphenyl-1,3,5-triazine 12a in near quantitative yield (97%). The scope of this cycloaddition of tetrazine 2 with amidines was defined (Figure 6). All amidines, including aryl amidines (11a–f), heteroaryl amidines (11g–j), and aliphatic amidines (11k–n), provided the corresponding fully substituted 1,3,5-triazines (12a–n) in excellent yields (87–99%) under these remarkably mild reaction conditions (40 mM, 25 °C, 2 h). Notably, the reaction proceeded effectively even with electron-deficient (hetero)arylamidines (11f–h, 11j). To examine the compatibility of this cycloaddition with protic solvents including water, the cycloaddition of tetrazine 2 with amidine 11l was also conducted in CH3CN/H2O (V/V = 7:3, 2 h, 89% and 3:7, 24 h, 94%), where the reaction proceeded smoothly even in 70% H2O/CH3CN and provided triazine 12l in the same yield observed in CH3CN alone. In order to gain a deeper insight into the reaction mechanism and kinetics, 1H NMR was used to monitor the reaction between tetrazine 2 (10 mM, CD3CN) and amidine 11k (2.0 equiv) at 25 °C. Fast consumption of tetrazine 2 and accumulation of product 12k were observed with appreciable product formation within 5 min, 90% conversion within 1 h, and full (>95%) conversion after 2 h. It is worth noting that 1H NMR signals from only tetrazine 2, amidine 11k, and triazine 12k were observed throughout the course of the reaction, without detection of any reaction intermediates or byproducts (Figure 7). Therefore, it is the cycloaddition reaction or addition-cyclization reaction between tetrazine and amidine that is the rate-determining step, which is followed by rapid sequential elimination of N2 and NH3 from the cycloadduct. From the NMR studies, the second-order rate constant of this reaction was determined to be k = (5.87±0.10)*10−2 M−1·s−1 (Supporting Information Figure S3).
Figure 6.
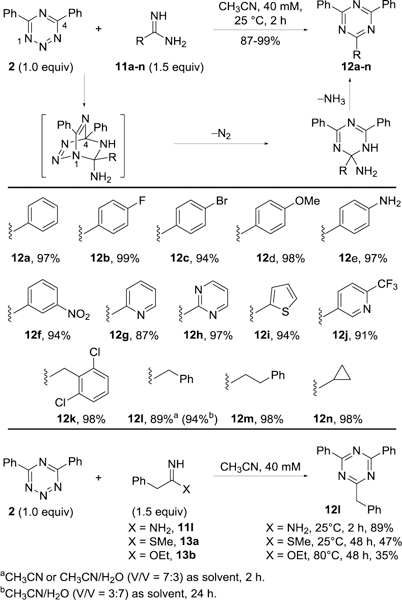
Scope of the cycloaddition between 2 and amidines, imidates and thioimidates.
Figure 7.

1H NMR study of the reaction between tetrazine 2 and amidine 11k (2 equiv) over 120 min (10 mM, 25 °C, CD3CN).
To further probe whether the reaction of amidines with 1,2,3,5-tetrazine 2 represents a single-step cycloaddition or a stepwise addition-cyclization, we conducted the reaction of 2 with amidine 11l that was doubly labeled with 15N (Figure 8). The reaction produced the 1,3,5-triazine product 121 in superb yield (94%) with incorporation of a single 15N label (1.07 ±0.01) as established by mass spectrometry (Supporting Information Table S3) and NMR experiments (Supporting Information Figure S4). This is consistent with a single-step cycloaddition and, while not ruling out a stepwise addition-cyclization mechanism, the latter would have been expected to provide a near equal mixture of singly and doubly 15N labeled triazine products (ca. 1.5 vs 1 15N). If the cycloaddition does proceed in a non-concerted fashion with generation of a zwitterionic intermediate,25 it would appear to require tight contact ion pair association following the initial addition with collapse to the [4+2] cycloadduct (vs proton transfer) prior to loss of nitrogen.
Figure 8.
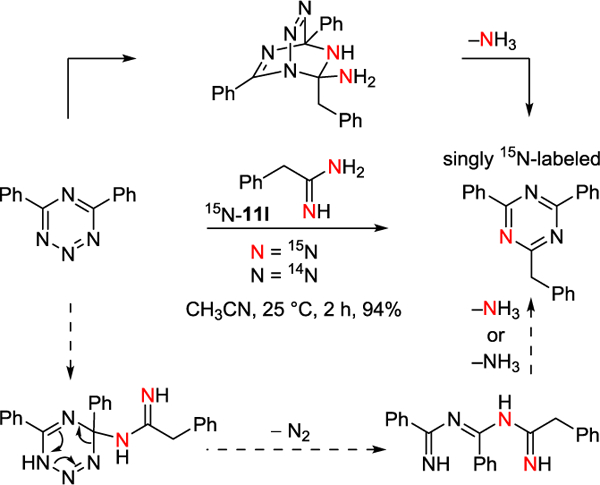
Reaction of 2 with doubly 15N labeled 11l.
Imidates and thioimidates are related heterodienophiles reported to react with some heterocyclic azadienes, but which typically display a lower reactivity than the corresponding amidines and a similar reactivity difference was observed with tetrazine 2 (Figure 6). Whereas the reaction of 2 with amidine 11l proceeds smoothly under mild conditions (2 h, 25 °C, 89%), a longer reaction time was required for the corresponding thioimidate 13a (48 h, 25 °C) and even then the formation of the same triazine 12l was observed in a lower yield (47%). With an increase in the amount of dienophile (5 equiv) and reaction concentration (400 mM), requiring CHCl3 as solvent for solubility purposes, the reaction provided 12l in good yield (68%, 48 h, 25 °C). The corresponding imidate 13b exhibited an even lower reactivity and the reaction between 2 and 13b required an elevated temperature (35%, 48 h, 80 °C) or higher concentration (43%, 400 mM in CHCl3, 5 equiv 13b, 48 h, 25 °C) and longer reaction times, providing more modest yields of the triazine 12l.
Additional dienophiles classes were examined, including electron-rich olefins and alkynes (Figure 9). The initial cycloaddition reaction between 4,6-diphenyl-1,2,3,5-tetrazine (2) with enamines 14a–d in acetonitrile is fast even at room temperature and is accompanied by loss of N2. This is followed by a slower amine elimination with rearomatization, requiring the longer reaction times and subsequent elevated temperatures. The same exclusive C4/N1 (vs N2/N5) cycloaddition mode was observed and occurs with a predictable exclusive regioselectivity (dienophile most electron-rich atom attaches to C4), affording substituted pyrimidines (15a–d) in good yields. In the case of 14b, the use of CHCl3 (41%) versus CH3CN (43%) as an alternative solvent provided similar yields of 15b, although the impact of solvent on the slow and occasionally problematic final aromatization was not investigated in detail. A similar reactivity was observed for the cycloaddition of tetrazine 2 with ynamines (16a–b), where the reaction proceeded smoothly under mild conditions. Complete conversion of tetrazine 2 to pyrimidine 17a (90% yield) was observed after 1 h (25 °C), whereas the reaction of the sterically more hindered ynamine 16b was found to be slower, providing pyrimidine 17b (92% yield) after 24 h.
Figure 9.
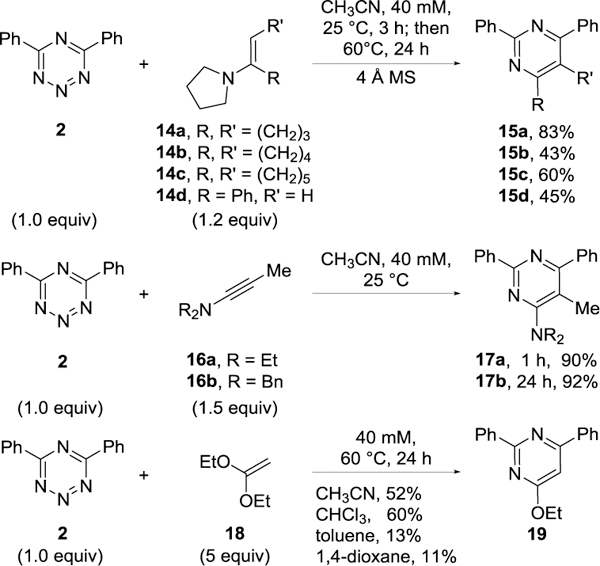
Reaction of 2 with enamines, ynamines and a ketene acetal.
Treatment of tetrazine 2 with ketene acetal 18 in warm acetonitrile (60 °C) provided the expected pyrimidine product 19 in good yield (52%) after 24 h (Figure 9). An interesting solvent effect was observed in this cycloaddition where the reaction in CHCl3 gave a slightly improved yield (60%), but a slower reaction and incomplete conversion of the tetrazine 2 was observed when the reaction was conducted in toluene (13%) or 1,4-dioxane (11%) under otherwise identical conditions.
Tetrazine 2 was also found to react with the electron-rich styrene 20 at an elevated temperature (80 °C), where cycloaddition is followed by an in situ oxidation to afford the aromatic pyrimidine product 21 (Figure 10). A slow conversion and low yield (9%) were found when the reaction was conducted in acetonitrile, whereas an improved yield (28%) was observed when 2 was warmed with neat 20. Although the latter results might appear modest, they do highlight the exceptional reactivity that can be expected of the 1,2,3,5-tetrazines, rivaling that of the 1,2,4,5-tetrazines and exceeding that of the respective triazines.
Figure 10.

Reaction of 2 with an electron-rich styrene and a strained alkyne.
Strained alkenes and alkynes are an especially interesting and important dienophile class. They serve as an excellent group of dienophiles and their reactions with 1,2,4,5-tetrazines proceed at remarkable rates under extraordinarily mild conditions even at dilute reaction concentrations.4 Consequently, variations on these reactions have been explored and adopted for powerful bioorthogonal conjugation and labeling techniques.3,4 In order to investigate whether 1,2,3,5-tetrazine 2 possesses similar or a distinctive reactivity, the reaction of 1,2,3,5-tetrazine 2 with the strained alkyne 22 was examined. Efficient cycloaddition with formation of the fused pyrimidine 23 in excellent yield (97%) was observed under mild (25 °C) and modestly concentrated reaction conditions (400 mM 2, 4 equiv dienophile) and progressively slower conversions and decreased yields were observed under increasingly more dilute reaction concentrations (40 mM 2, 28%; 5 mM 2, no reaction) with the balance of the materials being recovered starting tetrazine 2 and alkyne 22 (Figure 10). Again, only a single mode of cycloaddition was observed (C4/N1 vs N2/N5).
Reactivity comparison between two isomeric tetrazines: 4,6-diphenyl-1,2,3,5-tetrazine (2) versus 3,6-diphenyl-1,2,4,5-tetrazine (24).
In order to compare what became evident as distinct cycloaddition behaviors, the reactivity of 4,6-diphenyl-1,2,3,5-tetrazine (2) and the well-known and similarly substituted 3,6-diphenyl-1,2,4,5-tetrazines (24) was compared side-by-side and in competition studies with representative members of the different dienophile classes. Notably, these would not be the most reactive members of either tetrazine class, but the comparisons do provide the first opportunity to define cycloaddition distinctions. Three dienophiles were chosen for study, amidine 11l, ynamine 16a, and cyclooctyne 22, representing examples of a heterodienophile, an electron-rich dienophile, and a strained dienophile, respectively. Separate reactions between each of the tetrazines and dienophiles were conducted, followed by competition reactions where 1 equivalent of dienophile was added to an equimolar mixture of 1,2,3,5-tetrazine 2 and 1,2,4,5-tetrazine 24 (1 equiv each) (Figure 11).
Figure 11.

Comparison of the reactivity of 1,2,3,5-tetrazine 2 and 1,2,4,5-tetrazine 24 with different dienophile classes.
Three different outcomes were observed for the three dienophiles. While amidine 11l rapidly reacted with 1,2,3,5-tetrazine 2 under mild conditions (25 °C, 40 mM, CH3CN, 2 h, 89%), no reaction between the same amidine 11l and 1,2,4,5-tetrazine 24 was observed under the same reaction conditions, consistent with reports of the modest reactivity of 1,2,4,5-tetrazines toward amidines.1a Perhaps even more remarkable, 1,3,5-triazine 12l was observed as the only product (92%) when an equimolar mixture of the two tetrazines was treated with amidine 11l, reflecting the much greater reactivity of 2. A completely reversed cycloaddition preference was observed when cyclooctyne was used as the dienophile. Consistent with prior reports, 3,6-diphenyl-1,2,4,5-tetrazine (24) possessed an outstanding reactivity toward cyclooctyne 22, rapidly forming the pyridazine 25 (25 °C, 5 mM, CH3CN, 10 min, 95%). By contrast, the reaction of 1,2,3,5-tetrazine 2 with the strained cyclooctyne 22 did not produce any pyrimidine 23 under the same reaction conditions even after prolonged reaction times (48 h). Expectedly, only pyridazine 25 was isolated (98%) when an equimolar of mixture of the two tetrazines 2 and 24 were treated with cyclooctyne 22 (1 equiv), reflecting now the greater reactivity of 24. Ynamine 16a was found to react with either tetrazine individually (25 °C, 40 mM, 1 h), affording the corresponding pyrimidine 17a (90%) or pyridazine 26 (95%) as products. The treatment of ynamine 16a (1 equiv) with an equimolar mixture of the two tetrazines gave both 17a (15%) and 26 (53%) as reaction products, revealing a competitive but slightly lower reactivity of 1,2,3,5-tetrazine 2 than 1,2,4,5-tetrazine 24 toward the ynamine dienophile (1:3.5).
These results, along with the observations on the reactivities of tetrazines with other dienophiles, indicates the dependence of inverse electron demand Diels–Alder reactivity on both the core structure of the tetrazine as well as the dienophile itself. Compared to the 1,2,4,5-tetrazine 24, 1,2,3,5-tetrazine 2 exhibited an outstanding reactivity toward heterodienophiles (amidines), a comparable reactivity toward electron-rich dienophiles (ynamines, enamines, ketene acetal), and a lower reactivity toward strained olefins and alkynes (cyclooctyne). Houk et al. recently reported a comprehensive computational study of the reaction of the full and systematic series of heterocyclic azadienes with olefinic dienophiles.26 A selectivity for C–C bond formation over C–N bond formation within a given azadiene was predicted, defining a preferred intrinsic mode of cycloaddition, and the cycloaddition reactivity was found to follow a general trend of two C–C bond > C–C and C–N bond > two C–N bond formations. Thus, in addition to the relative energies of the reacting LUMO energies (slightly lower LUMO for 1,2,4,5-tetrazine vs 1,2,3,5-tetrazine, ΔE = 0.03–0.04 eV, both of which are much lower than any of the isomeric triazines, ΔE = 0.5–0.8 eV; MNDO/AM1), this correspondence between cycloaddition reactivity and nature of the bond formation at the diene termini was attributed to both a lower distortion energy of the dienes/dienophiles and productive interaction energy (orbital overlap efficiency) in the reaction transition state, where both are more favorable for the cycloaddition of olefinic dienophiles (ethylene) with 1,2,4,5-tetrazine versus 1,2,3,5-tetrazine (ΔEact = 9.4 kcal/mol). Consequently, a 1,2,4,5-tetrazine was predicted to be more reactive than a 1,2,3,5-tetrazine toward olefin/alkyne dienophiles. Whereas we found that the expected distinction was modest with an ynamine (electron-rich alkyne), we found that it was remarkably pronounced with a strained alkyne. However, the origin of the remarkably efficient and high reactivity of amidines with a 1,2,3,5-tetrazine, especially compared with a 1,2,4,5-tetrazine, is unclear. It is possible that the polarized nature of both the diene and dienophile and the polarity match of the reacting partners might result in a change in reaction mechanism, with the reaction proceeding now through a non-concerted cycloaddition or asynchronous cycloaddition rather than a synchronous concerted cycloaddition. However, we find no evidence to support a simple addition-cyclization reaction in direct reaction monitoring (Figure 7), 15N-labeling studies (Figure 8), or through intercepted reaction byproducts. Alternatively, it is plausible that the interaction energy of an amidine with a 1,2,3,5-tetrazine is much more favorable than with a 1,2,4,5-tetrazine, where the latter but not the former would suffer destabilizing electrostatic interactions of its ring nitrogens with the amidine amine substituent in the [4+2] cycloaddition transition state. Regardless of the origin but as a result of the remarkable distinctions in cycloaddition reactivity, a new orthogonal reaction pair was discovered, the 1,2,3,5-tetrazine/amidine pair, complementing the powerful 1,2,4,5-tetrazine/strained alkyne pair. Whereas 1,2,4,5-tetrazine displays extraordinary cycloaddition rates with strained dienophiles (tetrazine ligation), the new and isomeric 1,2,3,5-tetrazine displays similarly remarkable cycloaddition rates and efficiencies with amidines (1,2,3,5-tetrazine/amidine ligation). When following the reactions of 4,6-diphenyl-1,2,3,5-tetrazine with amidines (1H NMR), only starting material and product are observed and no intermediate(s) or byproducts are detected (see Figure 7). The crossover reactivities (1,2,4,5-tetrazines with amidines vs 1,2,3,5-tetrazines with strained dienophiles) are sufficiently low to indicate they may be used concurrently without competitive reactions. Both reactions proceed at low reaction concentrations under mild conditions and are compatible with H2O as the reaction co-solvent, providing potential new applications in orthogonal or sequential ligation reactions. Such studies are presently under investigation.
CONCLUSIONS
Herein we disclosed a novel synthesis and the full characterization of the first member of a monocyclic aromatic 1,2,3,5-tetrazine, 4,6-diphenyl-1,2,3,5-tetrazine. It is remarkable that no member of such a fundamental and important heterocyclic ring system had been previously described. Its synthesis and full characterization, including 1H/13C NMR, X-ray structure, intrinsic chemical stability, UV spectrum, and quasi reversible single-electron reduction (–1.37 V vs Fc+/Fc) are reported. Although we continue to define its cycloaddition reactivity, mode, regioselectivity, and scope, we have found that: (1) its reactivity is comparable to that of the isomeric 3,6-diphenyl-1,2,4,5-tetrazine, (2) it displays a single mode of cycloaddition with reaction only across C4/N1 (no N2/N5 cycloaddition observed), (3) its cycloaddition occurs with a predictable regioselectivity (dienophile most electron-rich atom attaches to C4), and (4) it participates as the 4π-component of well-behaved inverse electron demand Diels–Alder reactions preferentially reacting with electron-rich or strained dienophiles. Although we are only now beginning to explore the full cycloaddition scope, we have already found that it displays additional reactivity complementary to the isomeric 1,2,4,5-tetrazines. It not only displays the expected remarkable cycloaddition reactivity, a surprisingly good stability (e.g., stable to chromatography, long term storage, presence of H2O even as reaction co-solvent), and broad cycloaddition scope, but it also exhibits a powerful orthogonal reactivity with the 1,2,4,5-tetrazines that may be especially useful, including in the fields of click ligation and bioconjugation. Whereas the latter displays extraordinary cycloaddition rates with strained dienophiles (tetrazine ligation), the new and isomeric 1,2,3,5-tetrazine displays similarly remarkable cycloaddition rates and efficiencies with amidines (1,2,3,5-tetrazine/amidine ligation). The crossover reactivities (1,2,4,5-tetrazines with amidines vs 1,2,3,5-tetrazines with strained dienophiles) are sufficiently low to indicate they may be capable of concurrent use without competitive reactions. Extensions of the work to the study of additional substituted 1,2,3,5-tetrazines, the development of additional and needed synthetic approaches to this previously unknown class of heterocyclic azadienes, and their applications are in progress and will be reported in due time.
Supplementary Material
ACKNOWLEGEMENTS
We are especially grateful to the National Institutes of Health for financial support of the studies (CA042056, DLB) and a JITRI Fellowship (ZCW). We thank Dr. Laura Pasternack and Dr. Jason Chen for the analysis of labeling experiments, Dr. Milan Gembicky and Dr. Arnold Rheingold of the Crystallography Facility at the University of California, San Diego for the X-ray structure determinations of 2, 8 and 10 and Dr. Paul Richardson (Pfizer) for the differential scanning calorimetry data on 2 and 8.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:10.1021/jacs.xxxxxxx. Full experimental details and characterization data (PDF)
The authors declare no competing financial interests.
REFERENCES
- 1.(a) Boger DL Diels–Alder Reactions of Azadienes. Tetrahedron 1983, 39, 2869–2939. [Google Scholar]; (b) Boger DL Diels–Alder Reactions of Heterocyclic Azadienes. Scope and Applications. Chem. Rev. 1986, 86, 781–793. [Google Scholar]
- 2.(a) Moisan L; Odermatt S; Gombosuren N; Carella A; Rebek J Jr.. Synthesis of an Oxazole–Pyrrole–Piperazine Scaffold as an α‐Helix Mimetic. Eur. J. Org. Chem. 2008, 10, 1673–1676. [Google Scholar]; (b) Volonterio A; Moisan L; Rebek J Jr.. Synthesis of Pyridazine-Based Scaffolds as α-Helix Mimetics. Org. Lett. 2007, 9, 3733–3736. [DOI] [PubMed] [Google Scholar]; (c) Biros SM; Moisan L; Mann E; Carella A; Zhai D; Reed J; Rebek J Jr. Heterocyclic α-Helix Mimetics for Targeting Protein–Protein Interactions. Bioorg. Med. Chem. Lett. 2007, 17, 4641–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lahue BR; Lo S-M; Wan Z-K; Woo GHC; Snyder JK Intramolecular Inverse-Electron-Demand Diels−Alder Reactions of Imidazoles with 1,2,4-Triazines: A New Route to 1,2,3,4-Tetrahydro-1,5-Naphthyridines and Related Heterocycles. J. Org. Chem. 2004, 69, 7171–7182. [DOI] [PubMed] [Google Scholar]
- 3.(a) Devaraj NK; Weissleder; Hilderbrand SA Tetrazine-Based Cycloadditions: Application to Pretargeted Live Cell Imaging. Bioconjugate Chem. 2008, 19, 2297–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Blackman ML; Royzen M; Fox JM Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels−Alder Reactivity. J. Am. Chem. Soc. 2008, 130, 13518–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Devaraj NK; Upadhyay R; Haun JB; Hilderbrand SA; Weissleder R. Fast and Sensitive Pretargeted Labeling of Cancer Cells through a Tetrazine/trans‐Cyclooctene Cycloaddition. Angew. Chem., Int. Ed. 2009, 48, 7013–7016. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li Z; Cai H; Hassink M; Blackman ML; Brown RCD; Conti PS; Fox JM Tetrazine–trans-Cyclooctene Ligation for the Rapid Construction of 18F Labeled Probes. Chem. Commun. 2010, 46, 8043–8045. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Rossin R; Verkerk PR; van den Bosch SM; Vulders RCM; Verel I; Lub J; Robillard MS In vivo Chemistry for Pretargeted Tumor Imaging in Live Mice. Angew. Chem., Int. Ed. 2010, 49, 3375–3378. [DOI] [PubMed] [Google Scholar]; (f) Haun JB; Devaraj NK; Hilderbrand SA; Lee H; Weissleder R. Bioorthogonal Chemistry Amplifies Nanoparticle Binding and Enhances the Sensitivity of Cell Detection. Nat. Nanotechnol. 2010, 5, 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) McKay CS; Finn MG Click Chemistry in Complex Mixtures: Bioorthogonal Bioconjugation. Chem. Biol. 2014, 21, 1075–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Denk C; Svatunek D; Filip T; Wanek T; Lumpi D; Fröhlich J; Kuntner C; Mikula H. Development of a 18F‐Labeled Tetrazine with Favorable Pharmacokinetics for Bioorthogonal PET Imaging. Angew. Chem., Int. Ed. 2014, 53, 9655–9659. [DOI] [PubMed] [Google Scholar]; (i) Wu H; Devaraj NK Advances in Tetrazine Bioorthogonal Chemistry Driven by the Synthesis of Novel Tetrazines and Dienophiles. Acc. Chem. Res. 2018, 51, 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Han H; Devaraj NK; Lee J; Hilderbrand SA; Weissleder R; Bawendi MG Development of a Bioorthogonal and Highly Efficient Conjugation Method for Quantum Dots Using Tetrazine− Norbornene Cycloaddition. J. Am. Chem. Soc. 2010, 132, 7838–7839. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schoch J; Weissler M; Jäschke A. Post-Synthetic Modification of DNA by Inverse-Electron-Demand Diels−Alder Reaction. J. Am. Chem. Soc. 2010, 132, 8846–8847. [DOI] [PubMed] [Google Scholar]; (c) Pipkorn R; Waldemar W; Didinger B; Koch M; Mueller G; Wiessler M; Braun K. Inverse‐Electron‐Demand Diels−Alder Reaction as a Highly Efficient Chemoselective Ligation Procedure: Synthesis and Function of a BioShuttle for Temozolomide Transport into Prostate Cancer Cells. J. Pept. Sci. 2009, 15, 235–241. [DOI] [PubMed] [Google Scholar]
- 5.Initial report: [Google Scholar]; (a) Carboni RA; Lindsey RV Reactions of Tetrazines with Unsaturated Compounds. A New Synthesis of Pyridazines. J. Am. Chem. Soc. 1959, 81, 4342–4346. [Google Scholar]; (b) Boger DL; Coleman RS; Panek JS; Yohannes D. Thermal Cycloaddition of Dimethyl 1,2,4,5-Tetrazine-3,6-dicarboxylate with Electron-Rich Olefins: 1,2-Diazine and Pyrrole Introduction. Preparation of Octamethylporphin (OMP). J. Org. Chem. 1984, 49, 4405–4409. [Google Scholar]; (c) Boger DL; Sakya SM Inverse Electron Demand Diels–Alder Reactions of 3,6-Bis(methylthio)-1,2,4,5-tetrazine. 1,2-Diazine Introduction and Direct Implementation of a Divergent 1,2,4,5-Tetrazine → 1,2-Diazine → Benzene (Indoline/Indole) Diels–Alder Strategy. J. Org. Chem. 1988, 53, 1415–1423. [Google Scholar]; (d) Boger DL; Schaum RP; Garbaccio RM Regioselective Inverse Electron Demand Diels−Alder Reactions of N-Acyl 6-Amino-3-(methylthio)-1,2,4,5-tetrazines. J. Org. Chem. 1998, 63, 6329–6337. [DOI] [PubMed] [Google Scholar]
- 6.Initial report: [Google Scholar]; (a) Steigel A; Sauer J. (4+2)-Cycloadditionen 6-Gliedriger Heterocyclen mit Inaminen. Tetrahedron Lett. 1970, 11, 3357–3360. [Google Scholar]; (b) Boger DL; Panek JS Diels–Alder Reaction of Heterocyclic Azadienes. I. Thermal Cycloaddition of 1,2,4-Triazine with Enamines: Simple Preparation of Substituted Pyridines. J. Org. Chem. 1981, 46, 2179–2182. [Google Scholar]; (c) Boger DL; Panek JS; Meier MM Diels–Alder Reaction of Heterocyclic Azadienes. 2.” Catalytic” Diels–Alder Reaction of in situ Generated Enamines with 1,2,4-Triazines: General Pyridine Annulation. J. Org. Chem. 1982, 47, 895–897. [Google Scholar]
- 7.Initial report: [Google Scholar]; (a) Neunhoeffer H; Bachmann M. Cycloadditionen mit Azabenzolen, X. Cycloadditionen mit 1,3,5‐Triazinen. Chem. Ber. 1975, 108, 3877–3882. [Google Scholar]; (b) Boger DL; Schumacher J; Mullican MD; Patel M; Panek JS Thermal Cycloaddition of 1,3,5-Triazine with Enamines: Regiospecific Pyrimidine Annulation. J. Org. Chem. 1982, 47, 2673–2675. [Google Scholar]
- 8.(a) Wilkie GD; Elliott GI; Blagg BSJ; Wolkenberg SE; Soenen DR; Miller MM; Pollack S; Boger DL Intramolecular Diels−Alder and Tandem Intramolecular Diels−Alder/1,3-Dipolar Cycloaddition Reactions of 1,3,4-Oxadiazoles. J. Am. Chem. Soc. 2002, 124, 11292–11294. [DOI] [PubMed] [Google Scholar]; (b) Elliott GI; Fuchs JR; Blagg BSJ; Ishikawa H; Tao H; Yuan Z-Q; Boger DL Intramolecular Diels−Alder/1,3-Dipolar Cycloaddition Cascade of 1,3,4-Oxadiazoles. J. Am. Chem. Soc. 2006, 128, 10589–10595. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sears JE; Boger DL Tandem Intramolecular Diels–Alder/1,3-Dipolar Cycloaddition Cascade of 1,3,4-Oxadiazoles: Initial Scope and Applications. Acc. Chem. Res. 2016, 49, 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Initial report: [Google Scholar]; (a) Neunhoeffer H; Werner G. Reaktion von Pyridazinen mit 1‐Diäthylamino‐propin. Liebigs Ann. Chem. 1973, 437–442. [Google Scholar]; (b) Boger DL; Coleman RS Intramolecular Diels–Alder Reactions of 1,2-Diazines: General Indoline Synthesis. Studies on the Preparation of the Central and Right-hand Segments of CC-1065. J. Org. Chem. 1984, 49, 2240–2245. [Google Scholar]; (c) Kessler SN; Wegner HA Lewis Acid Catalyzed Inverse Electron-Demand Diels−Alder Reaction of 1,2-Diazines. Org. Lett. 2010, 12, 4062–4065. [DOI] [PubMed] [Google Scholar]
- 10.(a) Anderson ED; Boger DL Inverse Electron Demand Diels–Alder Reactions of 1,2,3-Triazines: Pronounced Substituent Effects on Reactivity and Cycloaddition Scope. J. Am. Chem. Soc. 2011, 133, 12285–12292. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Glinkerman CM; Boger DL Catalysis of Heterocyclic Azadiene Cycloaddition Reactions by Solvent Hydrogen Bonding: Concise Total Synthesis of Methoxatin. J. Am. Chem. Soc. 2016, 138, 12408–12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J; Shukla V; Boger DL Inverse Electron Demand Diels–Alder Reactions of Heterocyclic Azadienes, 1-Aza-1,3-Butadienes, Cyclopropenone Ketals and Related Systems. A Retrospective. J. Org. Chem. 2019, 84, 9397–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Thomas JR; Quelch GE; Schaefer HF The Unknown Unsubstituted Tetrazines: 1,2,3,4-Tetrazine and 1,2,3,5-Tetrazine. J. Org. Chem. 1991, 56, 539–543. [Google Scholar]; (b) Fabian J; Lewars E. Azabenzenes (Azines) – The Nitrogen Derivatives of Benzene with One to Six N Atoms: Stability, Homodesmotic Stabilization Energy, Electron Distribution, and Magnetic Ring Current; a Computational Study. Can. J. Chem. 2004, 82, 50–69. [Google Scholar]
- 13.William MK; Michael ES Low-Smoke Gas Generating Low Order Pressure Pulse Compositions. US Patent 7220328B1.
- 14.Baydar AE; Boyd GV; Lindley PF; Walton AR Approaches to 1,2,3,5-Tetrazines. Synthesis of 1,2,3,5-Tetrazinones and the Formation of Zwitterionic 1,2,4-Triazolin-3-ones and 1-Arylimino-1,2,4-triazolium Salts. J. Chem. Soc. Perkin Trans. 1 1985, 415–418. [Google Scholar]
- 15.(a) Stevens MFG; Hickman JA; Langdon SP; Chubb D; Vickers L; Stone R; Baig G; Goddard C; Gibson NW; Slack JA; Newton C; Lunt E; Fizames C; Lavelle F. Antitumor Activity and Pharmacokinetics in Mice of 8-Carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (CCRG 81045; M & B 39831), a Novel Drug with Potential as an Alternative to Dacarbazine. Cancer Res. 1987, 47, 5846–5852. [PubMed] [Google Scholar]; (b) Stevens MFG; Hickman JA; Stone R; Gibson NW; Baig GU; Lunt E; Newton CG Antitumor Imidazotetrazines. 1. Synthesis and Chemistry of 8-Carbamoyl-3-(2-chloroethyl)imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one, a Novel Broad-Spectrum Antitumor Agent. J. Med. Chem. 1984, 27, 196–201. [DOI] [PubMed] [Google Scholar]
- 16.(a) Ohsawa A; Heihachiro A; Hidefumi O; Hiroshi I. Synthesis, Oxidation, and Reduction of Monocyclic 1,2,3-Triazines. J. Chem. Soc. Chem. Commun. 1980, 24, 1182–1183. [Google Scholar]; (b) Okatani T; Koyama J; Tagahara K. Modified Synthesis of Monocyclic 1,2,3-Triazine and Cycloaddition Reaction with Enamine: The Application to the Synthesis of Alkaloids, Tortuosamine, N-Formyltortuosamine and N-Acetyltortuosamine. Heterocycles 1989, 29, 1809–1814. [Google Scholar]; (c) Sugimura H; Takeuchi R; Ichikawa S; Nagayama E; Sasaki I. Synthesis of 1,2,3-Triazines Using the Base-Mediated Cyclization of (Z)-2,4-Diazido-2-alkenoates. Org. Lett 2018, 20, 3434–3437. [DOI] [PubMed] [Google Scholar]
- 17.(a) Sauer J; Heldmann DK; Hetzenegger J; Krauthan J; Sichert H; Schuster J. 1,2,4,5‐Tetrazine: Synthesis and Reactivity in [4+2] Cycloadditions. Eur. J. Org. Chem. 1998, 12, 2885–2896. [Google Scholar]; (b) Karver MR; Weissleder R; Hilderbrand SA Synthesis and Evaluation of a Series of 1,2,4,5-Tetrazines for Bioorthogonal Conjugation. Bioconjugate Chem. 2011, 22, 2263–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Selvaraj R; Fox JM An Efficient and Mild Oxidant for the Synthesis of s-Tetrazines. Tetrahedron Lett. 2014, 55, 4795–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Butler RN; Cunningham D; McArdle P; O’Halloran GA 1,2,3,5-Tetrazines and 1,2,3-Triazaspiro[4.4]nonanes: Remarkable Products from 1,3-Dipolar Cycloadditions of N-sulphinylamines with Substituted Triazolium Imides. J. Chem. Soc. Chem. Commun. 1988, 3, 232–234. [Google Scholar]; (b) Butler RN; Evans AM; McNeela EM; Cunningham D; McArdIe P. 8π-Six-Atom Rings: 1,3,4,5-Oxa-and-Thia-triazines and 1,2,3,5-Tetrazines from an Extended Tandem Reaction: Reactions of 1,2,3-Triazolium-1-imides with (E)-Cinnamaldehyde, Methyl Cyanodithioformate, and Aryl-N-sulphinylamines: New Tetrahydro-oxazolo[4,5-d]-1,2,3-Triazoliumides and Triazaspiro-[4.4]nonanes. Azolium 1,3-Dipoles. Part 4. J. Chem. Soc. Perkin Trans. 1 1990, 2527–2536. [Google Scholar]
- 19.The structures of 2 (CCDC 1941636), 8 (CCDC 1941634), and 10 (CCDC 1941635) were established and confirmed in single-crystal X-ray structure determinations conducted on crystals grown from CH2Cl2 (2, yellow needles) or CHCl3/hexanes (8, orange block; 10, yellow block) and have been deposited with the Cambridge Crystallographic Data Center.
- 20.Yang X; Xi C; Jiang Y. Regioselective Nitration of N,N-Dialkylanilines Using Cerium (IV) Ammonium Nitrate in Acetonitrile. Tetrahedron Lett. 2005, 46, 8781–8783. [Google Scholar]
- 21.Differential scanning calorimetric measurements (Supporting Information page S29) at 5 °C min−1 indicate that 2 does not melt prior to decomposition, has an onset decomposition temperature of 138 °C, and does so with a moderate thermal potential (0.177 kcal/g, 744 J/g). Intermediate 8 melts at 233 °C then displays an onset decomposition temperature at 242 °C, decomposes over a broader temperature range (242–400 °C, peak at 305 °C), and releases 0.657 kcal/g (2756 J/g) of energy over the full temperature range of decomposition. Safety precautions should be exercised especially when 8 is prepared on large scale and conducting chemistry with 8 within 100 °C of the onset temperature should be avoided.
- 22.Allen FH; Kennard O; Watson DG; Brammer L; Orpen AG; Taylor R. Tables of Bond Lengths Determined by X-ray and Neutron Diffraction. Part 1. Bond Lengths in Organic Compounds. J. Chem. Soc. Perkin Trans. 2 1987, 12, S1–S19. [Google Scholar]
- 23.Plugge M; Alain-Rizzo V; Audebert P; Brouwer AM Excited State Dynamics of 3,6-Diaryl-1,2,4,5-tetrazines. Experimental and Theoretical Studies. J. Photochem. Photobiol. A 2012, 234, 12–20. [Google Scholar]
- 24.Fischer H; Müller T; Umminger I; Neugebauer FA; Chandra H; Symons MC Radical Cations and Anions of 1,2,4,5-Tetrazines: An Electron Spin Resonance and Cyclic Voltammetric Study. J. Chem. Soc. Perkin Trans. 2 1988, 3413–3421. [Google Scholar]
- 25.Yang YF; Yu P; Houk KN Computational Exploration of Concerted and Zwitterionic Mechanisms of Diels–Alder Reactions Between 1,2,3-Triazines and Enamines and Acceleration by Hydrogen-Bonding Solvents. J. Am. Chem. Soc. 2017, 139, 18213–18221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y-F; Liang Y; Liu F; Houk KN Diels–Alder Reactivities of Benzene, Pyridine, and Di-, Tri-, and Tetrazines: The Roles of Geometrical Distortions and Orbital Interactions. J. Am. Chem. Soc. 2016, 138, 1660–1667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.