

Abstract
Activation of ferroptosis, a recently described mechanism of regulated cell death, dramatically inhibits growth of ovarian cancer cells. Given the importance of lipid metabolism in ferroptosis and the key role of lipids in ovarian cancer, we examined the contribution to ferroptosis of steroyl CoA desaturase (SCD1), an enzyme that catalyzes the rate-limiting step in monounsaturated fatty acid synthesis, in ovarian cancer cells. SCD1 was highly expressed in ovarian cancer tissue, cell lines, and a genetic model of ovarian cancer stem cells. Inhibition of SCD1 induced lipid oxidation and cell death. Conversely, over-expression of SCD1 or exogenous administration of its C16:1 and C18:1 products, palmitoleic acid or oleate, protected cells from death. Inhibition of SCD1 induced both ferroptosis and apoptosis: inhibition of SCD1 decreased CoQ10, an endogenous membrane antioxidant whose depletion has been linked to ferroptosis, while concomitantly decreasing unsaturated fatty acyl chains in membrane phospholipids and increasing long chain saturated ceramides, changes previously linked to apoptosis. Simultaneous triggering of two death pathways suggests SCD1 inhibition may be an effective component of anti-tumor therapy, since overcoming this dual mechanism of cell death may present a significant barrier to the emergence of drug resistance. Supporting this concept, we observed that inhibition of SCD1 significantly potentiated the anti-tumor effect of ferroptosis inducers in both ovarian cancer cell lines and a mouse orthotopic xenograft model. Our results suggest that the use of combined treatment with SCD1 inhibitors and ferroptosis inducers may provide a new therapeutic strategy for patients with ovarian cancer.
Keywords: cell death, phospholipids, palmitic acid, ceramides, oleic acid, ovarian neoplasms, apoptosis, neoplastic stem cells
INTRODUCTION
Ferroptosis is a recently-described form of cell death that is distinct from other known cell death pathways (1). Interest in ferroptosis has been stimulated by its potential to be activated by cancer treatment (1). Although the precise mechanism of ferroptotic cell death is not completely understood, lipid peroxidation has been identified as a critical mediator of ferroptosis. More specifically, elevated levels of phosphatidylethanolamine (PE) containing oxidized polyunsaturated fatty acyl chains (PUFAs), particularly oxidized arachidonate (C20:4) and adrenate (C22:4), are necessary to trigger ferroptosis (2, 3).
Two key proteins limit the generation of lipid peroxides that induce ferroptosis. These are glutathione peroxidase 4 (GPX4), which reduces esterified oxidized fatty acids to their corresponding alcohols; and SLC7A11, a cystine antiporter that provides the cysteine needed for synthesis of glutathione, a cellular antioxidant and co-factor in the activity of GPX4 (1). Accordingly, small molecule inhibitors of GPX4 or SLC7A11, such as RSL3 or erastin, respectively, induce ferroptosis (1). Other enzymes implicated in ferroptosis also center on lipid pathways: for example, inhibition of acyl-CoA synthetase long chain family member 4 (ACSL4), which activates long chain PUFAs for lipid synthesis, protects cells from ferroptosis (4), as does lysophosphatidylcholine acyltransferase 3 (LPCAT3), which is involved in the generation of PUFA-PEs(5). Conversely, inhibition of squalene synthase, which affects multiple metabolites in the mevalonate pathway including the lipophilic antioxidant CoQ10, sensitizes cells to ferroptosis (6). Given the essential role of lipid peroxidation in ferroptosis, modulation of cellular lipid composition might be expected to perturb ferroptotic cell death.
Sterol CoA desaturase (SCD1) is a lipid modifying enzyme that is upregulated in numerous malignancies, including prostate, liver, and breast cancer (7). SCD1 catalyzes the desaturation of saturated fatty acids, principally stearic acid (18:0) and palmitic acid (16:0) to their Δ9-monounsaturated counterparts, oleic acid (18:1) and palmitoleic acid (16:1) (8). We considered that this enzyme might affect the sensitivity of cells to ferroptosis. In particular, we hypothesized that upregulation of SCD1 is an intrinsic cytoprotective mechanism that protects cancer cells from the cytotoxic action of ferroptosis inducers.
To test this hypothesis, we focused on ovarian cancer and ovarian cancer stem cells, since our earlier work demonstrated that ovarian cancer stem cells are sensitive to the ferroptosis inducer erastin in vitro and in vivo (9). Cancer stem cells are believed to be a small, treatment-refractory subpopulation of tumor cells that seed metastases and give rise to treatment resistance. Thus, our experiments demonstrating sensitivity of cancer stem cells to ferroptosis, as well as results from other groups (10), suggest that there may be a role for ferroptosis inducers in the treatment of ovarian cancer. Further, recent work has shown that lipid desaturation is increased and contributes to the maintenance of stemness in ovarian cancer cells (11). Thus ovarian cancer represents a particularly pertinent model in which to assess the role of SCD1 in the sensitivity to ferroptosis inducers.
Here, we demonstrate that SCD1, a lipid desaturase, alters lipid membrane composition and modulates ferroptosis. Further, inhibition of SCD1 enhances the anti-tumor effect of ferroptosis inducers in ovarian cancer cells.
MATERIALS AND METHODS
Cell culture.
MDAH2774, SW626, SKOV3, TOV-112D cells were purchased from ATCC (on March 6th 2013). Cells were frozen at low passage and used within 2-3 months after thawing. Cells were cultured in DMEM (GIBCO) supplemented with 10% FBS (Gemini Bio-Products). COV362 cells were purchased from Sigma on May 18th 2015 and cultured in DMEM (GIBCO) containing 10% FBS. FT-t and FT-i cells (isolated by transfection of primary fallopian tube stem cells as described in (12)) were cultured in DMEM containing 10% FBS. Human Ovarian Surface Epithelial (HOSE) cells (ScienCell Research Laboratories) were cultured in Ovarian Epithelial Cell Medium (ScienCell Research Laboratories). OVCAR-4 and OVCAR-8 cells were obtained from NCI (distributed by Charles River Labs) on February 25th 2018 and OVCAR5 cells were obtained on May 8, 2019. OVCAR-4, OVCAR-5 and OVCAR-8 cells were cultured in RPMI 1640 + L-Glutamine (GIBCO) supplemented with 10% FBS (Gemini Bio-Products).
Infection and isolation of SCD1-expressing FT-t cells and COV362.
Human SCD1 cDNA was amplified using GE Dharmacon clone (cat# MHS6278-202830110) and introduced into the lentiviral tetracycline (tet) inducible vector pLVX-TetOne-Puro (Takara-Clontech, Mountain View, CA) prior to infection of FT-t cells. For over-expression of SCD1 in COV362 cells SCD1 cDNA was amplified and inserted into the pLVX-TetOn-Puro vector. Lentivirus particles were produced by transient cotransfection of the SCD1 tet-on expression vector and packaging vectors (VSVG, pMDLG, and RSV-REV) into 293T cells. Viral particles containing control empty vector were prepared similarly. Cells were infected and selected for puromycin resistance for two weeks before experiments were performed.
shRNA knockdown of OVCAR4 cells.
Knockdown of SCD1 In OVCAR4 cells was performed using a lentiviral shRNA vector (13) designed to target the sequence GCATTCCAGAATGATGTCTAT in the SCD1 coding region. Stable knockdown cells were isolated by selecting for puromycin resistance.
Quantitative real-time PCR (qRT-PCR).
qRT-PCR was performed as previously described (14) Primers used were: human SCD1 forward: AAACCTGGCTTGCTGATG; human SCD1 reverse: GGGGGCTAATGTTCTTGTCA; human β-Actin forward: TTG CCG ACA GGA TGC AGA AGG A; human β-Actin reverse: AGG TGG ACA GCG AGG CCA GGA T.
Western blot.
Western blotting was performed and quantified as previously described (9). Membranes were probed with antibodies to β-actin (Sigma cat# A3854) and SCD1 (Abcam catalog #ab39969). HRP-conjugated goat anti-rabbit was used as a secondary antibody (BioRad cat# 170-5046).
Cell viability and cell death.
2 × 103 − 3 × 103 cells were seeded in 96 well plates and treated with the following reagents: RSL3 (Selleckchem), erastin (Selleckchem), ferrostatin-1 (Selleckchem), MF-438 (Sigma), CAY10566 (Cayman), A939572 (Cayman), fatty acids (oleic acid, palmitoleic acid, stearic acid, or palmitic acid (Sigma)), z-VADFMK (Selleckchem). Cell viability was assessed 24-72 hours post-treatment using calcein-AM (Millipore). FACS analysis of propidium iodide-stained cells was used to directly measure cell death (15).
Synergy.
Synergism was computed using the Chou-Talalay method (16) with CalcuSyn software.
Caspase 3/7 activity.
10, 000 cells were treated with MF-438 in the presence or absence of z-VADFMK (Selleckchem) for 48 hrs or staurosporine (Sigma) for 4 hours. Caspase 3/7 activity was analyzed using Caspase-Glo® 3/7 Assay Systems from Promega.
siRNA knock-down experiments.
All reagents were obtained from GE Dharmacon (Lafayette, CO, USA). 12 ng of ON-TARGETplus Human SCD1 siRNA (L-005061-00-0010), and siNTC (cat#: D-001810-10-05) were used for knockdown experiments. Transfections were performed according to the manufacturer’s recommendations using Dharmafect #1 transfection reagent (cat: T-2001). Knockdown efficiencies were confirmed at the time of harvest by RT-qPCR and/or by western blotting.
Immunohistochemistry.
Formalin-fixed paraffin-embedded slides of de-identified human tissues from HGSOC (9 subjects), and normal ovary (6 subjects) were obtained from the biorepository of UCHC (IRB IE-08-310-1). Tissues were immunostained with antibodies to human SCD1 followed by anti-Rabbit secondary antibody (Biocare Medical MACH2 Rabbit HRP-Polymer; catalog # RHRP520#) , staining with 3,3’-diaminobenzidine (Biocare Medical) and counterstaining with Gill”s Hematoxylin III and Lithium Blue (Poly Scientific) . Competition with specific peptide (Abcam # ab40137) was used as a control. Staining was quantified as described (17) (see Supplemental Materials and Methods).
C11-BODIPY staining.
Cells were plated in 8-chamber slides (BD Falcon) and treated with 1 - 5 μM RSL3 in the presence or absence 2 μM Ferrostatin-1 for 4 hours prior to incubation with 2 μM C11-BODIPY (Thermo Fisher Scientific) for 30 minutes. Slides were washed, fixed with 4% paraformaldehyde (Thermo Fisher Scientific) and mounted using ProLong Gold anti-fade reagent (Invitrogen). Images were acquired using inverted microscopy (Zeiss Axio Vert.A1). For flow cytometry, cells were washed subsequent to staining with C11-bodipy analyzed using a BD LSRII flow cytometer (Becton Dickinson).
Animal Experiments.
Female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (NSG; ~ 6 weeks of age) were obtained from Jackson Laboratory. 100,000 FT-t cells were injected intraperitoneally (i.p) (n=8/group). The next day, mice were injected intraperitoneally with 1 mg/kg A939572 in corn oil (Sigma) in the presence or absence of 20 mg/kg erastin in 2% DMSO (Thermo Fisher Scientific) in corn oil. Vehicle (2% DMSO in corn oil) was used as a control. Treatment was continued at five doses per week for 18 days, when control mice began to develop ascites and become moribund. Mice were sacrificed, tumors were counted, and the combined weight of all tumors within the abdomen of each mouse was measured. Histological evaluation of representative nodules by a board-certified pathologist (PH) confirmed that they were high grade tumors with multiple mitotic figures and areas of necrosis. Group size was based on power calculations and was designed to provide 80% power to detect an effect size of 0.5 gm difference in tumor mass using two-sided t-tests. Two animals in the A9393572 group and the combined treatment group died for unknown reasons before the experiment was completed. No surviving mice were excluded from the analysis. For treatment of established tumors, mice were injected with FT-t cells as described above. Seven days later, two mice were sacrificed and examined for the presence of tumors. Tumors were observed visually and were confirmed by histology. Treatment with vehicle or the combination of erastin and A939572 was then initiated in the remaining mice (n=5/group) and continued as described above until sacrifice at day 18.
Animal Ethics Statement.
Animal studies were conducted in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the AAALAC. The experimental protocol was approved by the Institutional Animal Care and Use Committee at the University of Connecticut Health Center (protocol # 100881).
Statistical analysis of cell culture experiments.
Statistical analyses were performed using Excel or Prism 6 (Graphpad software). All experiments were performed at least three times using a minimum of three replicates per condition in each experiment. Comparison tests were performed between two groups and statistical significance assessed using two-tailed unpaired Student’s t tests. Statistics are reported as the mean ± standard deviation.
Database analyses.
Publicly available Affymetrix GeneChip U133A Plus 2.0 expression profile array of laser-capture microdissected normal fallopian epithelial cells (with and without known BRCA1/2 mutations) and high-grade serous ovarian or fallopian epithelial cells (GEO accession no. GSE109071) were preprocessed as described (18). Preprocessed TCGA Affymetric GeneChip U133A expression profile array data was obtained from the curated OvarianData database (19). Further analysis of these datasets was performed as described in Supplemental Materials and Methods.
Fatty Acid Methyl Ester Analysis (FAME) and Lipidomics.
FAME analysis was performed using GC-MS and untargeted lipidomics analysis was performed using ultra high-performance liquid chromatography - tandem mass spectrometry (UHPLC-MS). Details are provided in Supplemental Materials and Methods.
RESULTS
SCD1 is overexpressed across histological and molecular subtypes of ovarian cancer and in a genetic model of ovarian cancer stem cells
We first assessed the expression of SCD1 in ovarian cancer. There are multiple histological subtypes of ovarian cancer, including adenocarcinoma, endometriod, and high grade serous ovarian cancer (HGSOC). We tested whether SCD1 was broadly upregulated across these ovarian cancer subtypes using cell lines derived from these subtypes. As shown in Figure 1A,B, when compared to normal human ovarian surface epithelial (HOSE) cells, there was an increase in both levels of SCD1 mRNA and protein in all but one ovarian cancer cell line tested. Although the extent of upregulation was variable, there was a general concordance between levels of mRNA and protein, suggesting that the increase in SCD1 is transcriptionally driven. Analysis of a publicly available gene expression database (GEO accession GSE109071) demonstrated that SCD1 was similarly elevated in samples from patients with HGSOC (Figure 1C). Consistent with these results, immunohistochemical analysis revealed that SCD1 protein was increased in tumors from patients with HGSOC when compared to non-malignant ovarian epithelium (Figure 1D, Supplemental Figure 1).
Figure 1. SCD1 is upregulated in ovarian cancer.
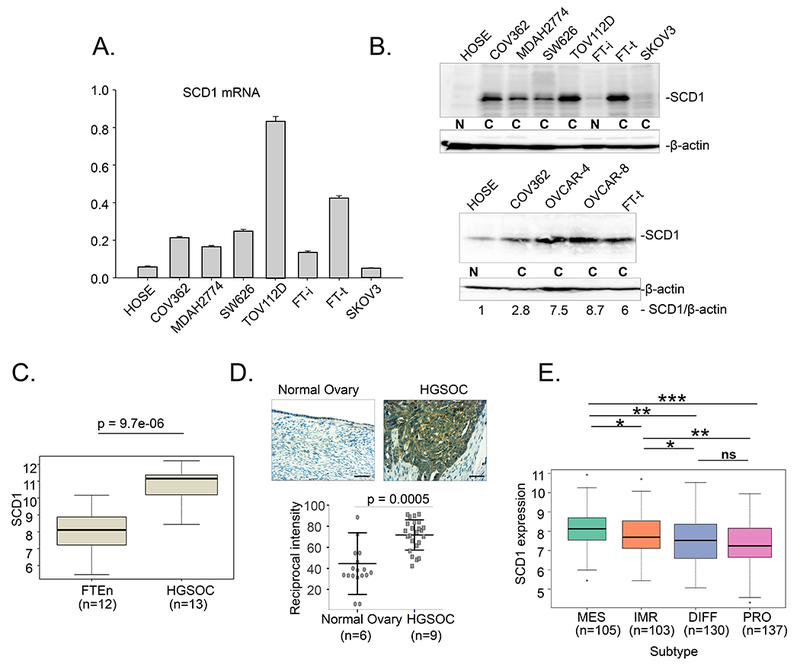
A. SCD1 mRNA in multiple ovarian cancer cell lines and normal ovarian cells assessed using RT-qPCR (FT-i- immortalized fallopian tube cells; FT-t- transformed fallopian tube cells; (cancer stem cells, see Basuli et. al., 2017) B. Western blot analysis of SCD1 in ovarian cells; N, non-malignant cell lines; C, ovarian cancer cell lines. Histological subtypes represented by these cell lines are as follows: adenocarcinoma, SW626, SKOV3; endometriod, MDAH2774, TOV112D; high grade serous ovarian cancer (HGSOC), COV362, OVCAR4, OVCAR8. C. Expression of SCD1 in GEO (GSE 109071). HGSOC compared to normal fallopian tube epithelium (FTEn). D. Immunohistochemical staining of SCD1 in patient tissues (scale bar =100 μm). Upper panel, representative image of normal ovary and HGSOC; lower panel, staining quantification (normal ovary n=6; HGSOC n=9). Two to three different images were captured per patient slide and quantified. Dot plot depicts means and standard deviations of 17 images of normal tissues and 23 images of HGSOC patient tissues. E. Differences in SCD1 expression in prognostic TCGA subtypes (MES, mesenchymal; IMR, immunoreactive; DIFF, differentiated; PRO, proliferative). Boxes represent first and third quartiles, line is median, whiskers indicate data spread, and outliers are represented by dots. Width of boxes is proportional to sample size. *p<0.05; **p<1.0 E-03; ***p=1.2E-E07
We also examined SCD1 in a genetic model of ovarian cancer stem cells created by introduction of h-TERT, SV40 large T and c-myc into normal human fallopian stem cells (12). Cells of the fallopian tube represent at least one of the precursor cells for ovarian cancer (20), and we have previously shown that these FT-t cells form tumors with the characteristic features of HGSOC (12). SCD1 was similarly elevated in these FT-t cancer stem cells when compared to immortalized but not transformed fallopian stem cells (FT-i) (Figure 1A,B).
Ovarian cancer has been divided into prognostic subtypes based on gene expression profile (21). Subtypes identified were proliferative, differentiated, immunoreactive, and mesenchymal (21). To test whether SCD1 was differentially expressed in ovarian cancer subtypes, we queried the TGCA database. As shown in Fig 1E, levels of SCD1 transcripts differed across subtypes, with the highest level observed in the mesenchymal subtype. The mesenchymal phenotype has been broadly associated with stem cell characteristics and with sensitivity to ferroptosis inducers in other cancers (22, 23).
Inhibition of SCD1 induces ferroptotic cell death
The high levels of expression of SCD1 in ovarian cancer cells suggested that SCD1 may play an important role in these cells. To test this hypothesis, we inhibited SCD1 using MF-438 and CAY10566, two chemically distinct pharmacological inhibitors of SCD1 enzymatic activity. As shown in Figure 2A–E, treatment of OVCAR4 HGSOC cells, as well as ovarian cancer stem cells, FT-t, with MF-438 and CAY10566 for 72 hours reduced cell viability and increased cell death. Viability was restored by providing cells with oleic acid, one of the endproducts of SCD1 activity. Consistent with the higher levels of SCD1 in FT-t cells compared to FT-i cells, FT-t cells were more sensitive to SCD1 inhibitors than FT-i cells (Supplemental Figure 2). Cell death was also reduced by treatment with FER-1, an inhibitor of ferroptosis, suggesting that reduction of SCD1 activity induces cell death at least in part by triggering ferroptosis. Consistent with this interpretation, treatment with an SCD1 inhibitor increased lipid oxidation, a characteristic feature of ferroptosis (Figure 2F). To confirm and expand these results beyond a single ovarian cancer cell line, we inhibited SCD1 in COV362 cells using siRNA (Supplemental Figure 3A) and measured effects on cell viability. As shown in Figure 3A and 3B, knockdown of SCD1 reduced cell viability in both COV362 ovarian cancer cells and ovarian cancer stem cells. Viability was restored by providing cells with either oleic acid or FER-1, confirming that SCD1 prevents ferroptotic cell death in both ovarian cancer cells and ovarian cancer stem cells.
Figure 2. Blocking SCD1 activity using small molecule inhibitors causes cell death that can be rescued by oleic acid and the ferroptosis inhibitor Fer-1.
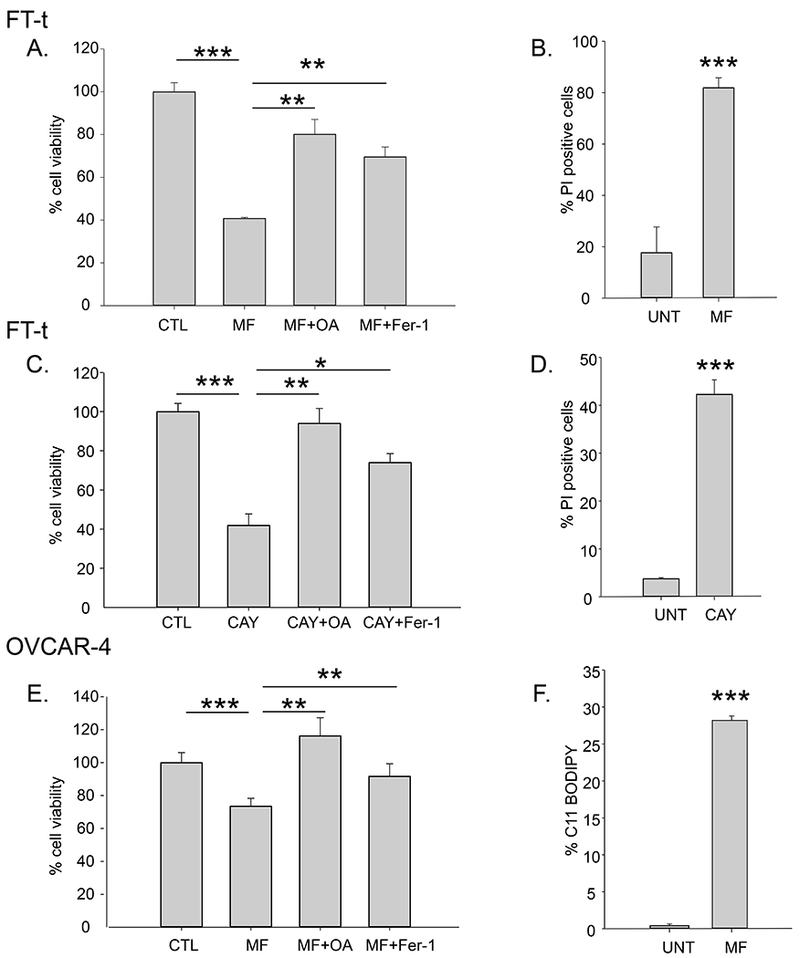
Cells were treated with 1 μM MF-438 (MF) or 5 μM CAY10566 (CAY) in the presence or absence of 5 μM fer-1 or 80 μM oleic acid (OA) for 48 to 72 hours and cell viability assessed by calcein-AM (A,C, E); and cell death was assessed using Propidium iodide( B,D). F. C11-BODIPY staining of OVCAR-4 cells following treatment with 1 μM RSL3 in the presence or absence of 2 μM Fer-1 for 2 hours. Experiments are representative of three independent experiments. Shown are means and standard deviation of eight replicates in one representative experiment. *p<2 E-04; **p<6 E-05; ***p<4E-06
Figure 3. Modulation of SCD1 affects ferroptosis in ovarian cancer cells.
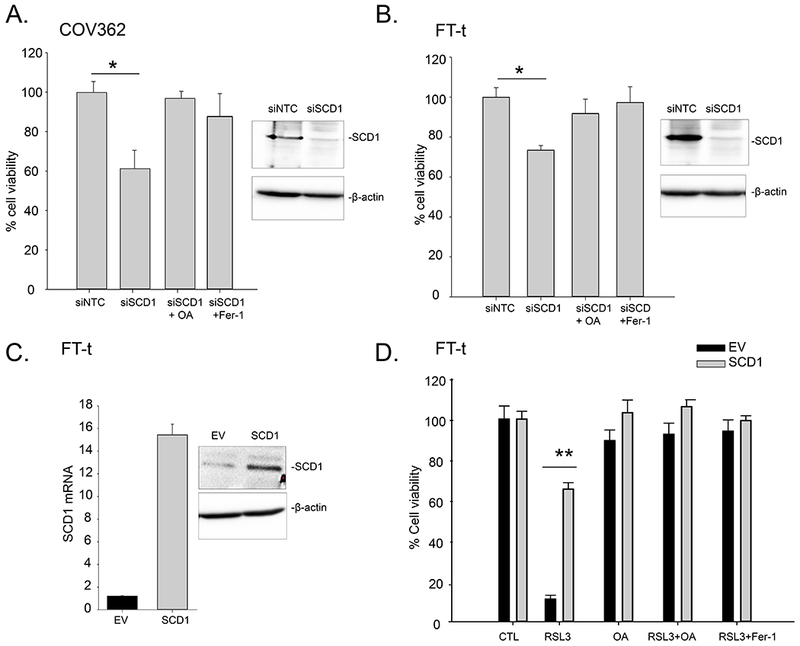
A. COV362 and B. FT-t cells were transfected with siRNA targeted to SCD1 (siSCD1) or non-targeting siRNA (siNTC) for 72 hours in the presence and absence of 5 μM Fer-1 or 80 μM oleic acid (OA). C. Expression of SCD1 in Tet-on constructs analyzed using RT-qPCR and western-blotting (EV, empty vector). D. FT-t cells over-expressing SCD1 or empty vector controls were treated with 5 μM RSL3 in the presence or absence of 10 μM Fer-1 or 80 μM oleic acid (OA) for 24 hours and viability measured. Graphs are representative of four independent experiments. Shown are means and standard deviations of eight replicates in one representative experiment.; *p<2.5E-07; **p<5.3E-11.
Expression of SCD1 protects cells from ferroptosis by increasing the formation of monounsaturated fatty acids
To further probe the influence of SCD1 on ferroptotic cell death, we performed the converse experiment by overexpressing SCD1 in HGSOC and ovarian cancer stem cells. As seen in Figure 3C,D and 4A,B, overexpression of SCD1 led to a dramatic protection from ferroptotic cell death induced by RSL3 or erastin.
Figure 4. Over-expression of SCD1 protects cells from RSL3-mediated lipid peroxidation.
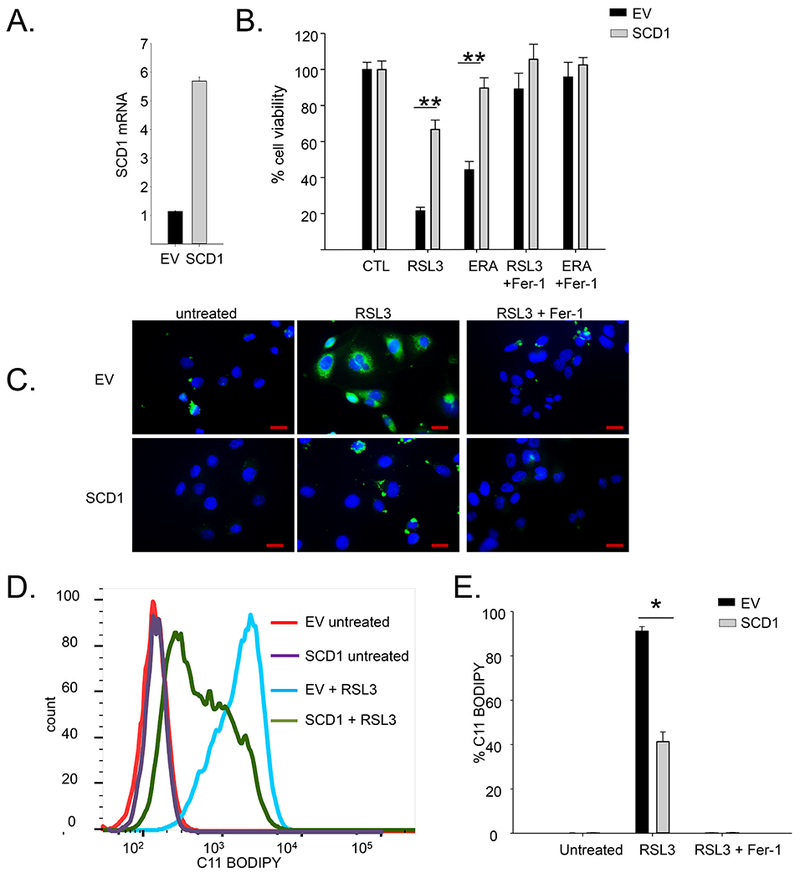
A. SCD1 mRNA in COV362 cells constitutively over-expressing SCD1 (SCD1) or empty vector control (EV) measured by RT-qPCR. B. Viability of COV362 cells over-expressing SCD1 or empty vector controls following treatment with 5 μM of RSL3 or 5μM erastin in the presence or absence of 10 μM Fer-1 for 48 hours. C. C11-BODIPY staining of COV362 SCD1 and COV362 EV cells following treatment with 5 μM RSL3 in the presence or absence of 2 μM Fer-1 for 4 hours. Blue=DAPI; green=C11 BODIPY. Scale bar =20 μm; D. C11-BODIPY staining analyzed by flow cytometry. E. Quantification of C11-BODIPY positive cells . Means and standard deviation of at least three replicas. *p<5.5E-05; **p<3.7E-9
Because the formation of oxidized membrane PUFAs are a hallmark of ferroptosis, we next asked whether SCD1 prevented ferroptotic cell death by preventing the formation of these lipid species. SCD1 over-expressing and control cells were treated with RSL3, and lipid oxidation measured by staining with C11-BODIPY, a dye that detects lipid ROS in cell membranes. This experiment demonstrated a significant decrease in oxidized lipids in cells over-expressing SCD1 (Figure 4C–E).
Consistent with these results, cell death triggered by exposure to the ferroptosis inducer RSL-3 could be rescued by supplementation with palmitoleic or oleic acid, two products of SCD1 activity (Figure 5A,B). Rescue was specific to monounsaturated fatty acids (MUFAs), and could not be recapitulated with palmitic or stearic acid, the corresponding saturated fatty acids (Figure 5C,D). In fact, exposure to these saturated fatty acids was toxic in itself (Figure 5C–F). In contrast to results with MUFAs, the toxic effect of saturated fatty acids could not be inhibited by FER-1, but could successfully be rescued by zVAD-fmk, an inhibitor of apoptosis (Figure 5E,F).
Figure 5. Effects of exogenous lipids, death pathway inhibitors, and SCD1 inhibitors on viability and caspase activity in ovarian cancer cells and ovarian cancer stem cells.
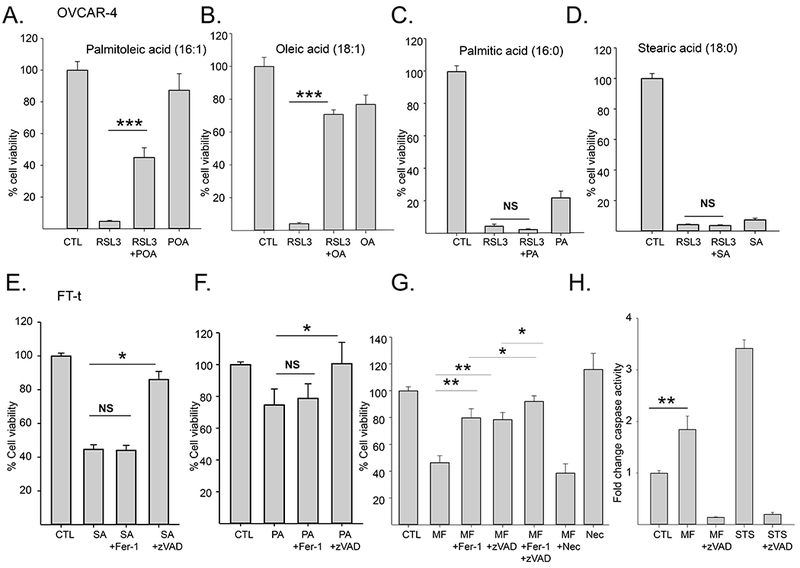
A-D. Viability of OVCAR-4 cells treated with 2μM RSL3 in the presence or absence of 80μM of the indicated lipids for 24 hours; E, F. Viability of FT-t cells treated with 80 μM stearic acid (SA) or palmitic acid (PA) in the presence or absence of 10 μM z-VADfmk or 5 μM Fer-1 for 24hrs; G. Viability of FT-t cells treated with 10 μM MF-438 in the presence or absence of 5 μM Fer-1, 10 μM z-VADfmk, or 10 μM necrostatin for 48 hrs. H. Relative caspase 3/7 activity in FT-t cells treated with 10 μM MF-438 for 48 hr or 1 μM staurosporin for 4 hours in the presence or absence of 10μM z-VADfmk. Data shown is representation of at least three independent experiments. Graphs are means and standard deviation of 8 replicas in one representative experiment. *p<0.024; **p<1 E-04; ***p<7E-14
Work from other groups has shown that blockade of SCD1 induces apoptosis (24, 25). We sought to reconcile these findings with our observations that inhibition of SCD1 induces ferroptosis. We therefore treated cells with the combination of an SCD1 inhibitor and zVAD-fmk, an apoptosis inhibitor, or FER-1, a ferroptosis inhibitor. As shown in Figure 5G, the decrease in cell viability caused by inhibition of SCD1 could be partially rescued by either zVAD-fmk or FER-1 but not by necrostatin, an inhibitor of necrosis (Figure 5G). This observation suggests that inhibition of SCD1 triggers two specific death pathways: apoptosis and ferroptosis. To support these findings, we measured caspase activation, a specific marker of apoptosis, following inhibition of SCD1. Staurosporine, a known inducer of apoptosis, was used as a positive control. As shown in Figure 5H, inhibition of SCD1 increased caspase activity, consistent with induction of apoptosis. Further, combined treatment with zVAD-fmk and FER-1 almost completely blocked cell death (Figure 5G), indicating that ferroptosis and apoptosis are the principal modes of cell death induced by inhibition of SCD1.
Mechanistic links between SCD1, ferroptosis, and apoptosis
We next explored mechanisms by which SCD1 inhibition triggers dual cell death pathways. We focused on lipid metabolism due to the biochemical properties of SCD1 and its known role in lipid remodeling (7). Lipids have been linked to both apoptosis and ferroptosis: ceramides are apoptosis mediators (26, 27), whereas oxidized PUFAs mediate ferroptosis (2). Further, the mevalonate pathway, particularly CoQ10 has been implicated in protection from ferroptosis (6). We therefore probed changes in lipid composition induced by SCD1 blockade using both an analysis of fatty acid composition (FAME analysis) and ultra-performance liquid chromatography/tandem mass spectrometry (UHPLC-MS), a global untargeted lipidomic analysis of major lipid classes.
We first confirmed that perturbation of SCD1 had the anticipated downstream consequences on fatty acid composition. In particular, SCD1 catalyzes the formation of a double bond in the cis-Δ9 position of saturated fatty acyl-CoAs, particularly palmitoyl CoA and stearolyl CoA (C16:0 and C18:0), to produce the monounsaturated fatty acids palmitoyl CoA and oleoyl CoA (C16:1 and C18:1, respectively). Inhibition of this reaction would therefore be expected to decrease the ratio of monounsaturated to saturated fatty acids. To confirm this we performed fatty acid methyl ester (FAME) analysis. SCD1 knockdown indeed caused an overall decrease in the ratio of 16:1/16:0 and 18:1/18:0 monounsaturated/saturated fatty acids (average decrease of 55±7% and 38±17%, respectively).
To clarify and distinguish the roles of SCD1 in apoptosis and ferroptosis, we next investigated effects of SCD1 knockdown on overall lipid composition using an untargeted lipidomic analysis that compared relative levels of over 1700 individual lipid species, including ceramides, phospholipids, free fatty acids, and triglycerides, in cells that had been treated with three chemically distinct inhibitors of SCD1.
We first noted that consistent with our FAME analysis, the ratio of SFA/MUFA increased across all lipid classes with SCD1 inhibition (Figure 6A). Among lipids that increased with SCD1 inhibition (i.e. that were positively associated with SCD1-mediated cell death), the most notable were ceramides containing saturated fatty acids (Figure 6B). An increase in long chain saturated ceramide levels has been previously linked to apoptotic cell death (24), consistent with our observation that SCD1 inhibition induces apoptosis (Figure 5G,H). We also observed a decrease in membrane phospholipids containing unsaturated fatty acids (Figure 6C), a change that has also been linked to apoptosis in cells deprived of SCD1 (28). These results implicate SCD1-mediated effects on ceramides and saturation of phospholipid acyl chains as likely contributors to apoptotic cell death in ovarian cancer cells depleted of SCD1.
Figure 6. Inhibition of SCD1 modulates lipid content of ovarian cancer cells.
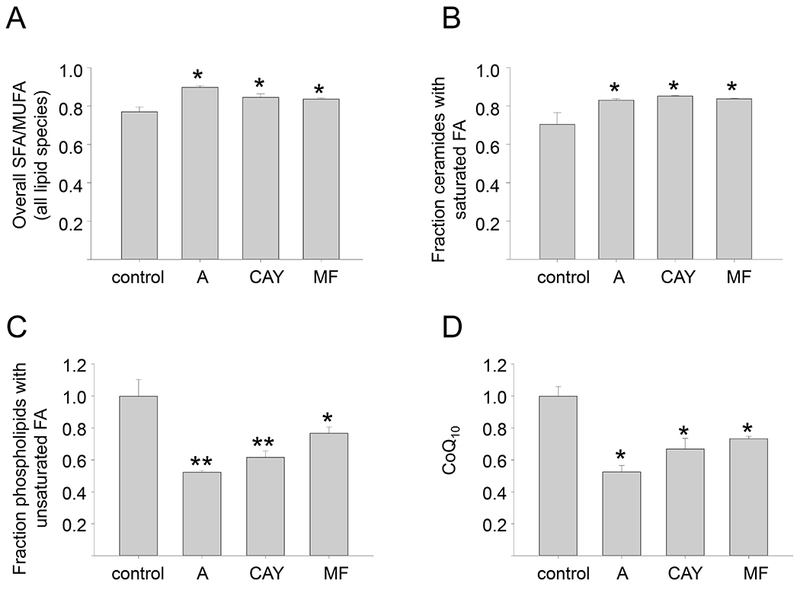
FT-t Cells were treated in triplicate with vehicle (control) or three chemically unrelated, commercially available SCD1 inhibitors: A939572 (A), CAY10566 (CAY), and MF-438 (MF), for 72hours prior to harvest for untargeted lipid analysis by UHPLC-MS. A. Ratio of saturated/monounsaturated fatty acids in all lipid species; B. Fraction of ceramides with saturated fatty acids; C. Fraction of phospholipids with unsaturated fatty acids; D. levels of coenzyme Q10. Means and standard deviations of triplicate determinations are shown. *p<0.04; **p<0.01
Ferroptotic cell death has been linked to oxidation of PUFAs, particularly PE containing arachidonic acid (C20:4) and adrenic acid (C22:4)(2) (29). We therefore asked whether arachidonic or adrenic acid increased following SCD1 inhibition. However, we noted no increase in either free arachidonic acid (adrenic acid was not detected); arachidonoyl- or adrenoyl-esterified PE; or arachidonic and adrenic acid-containing lipids of any class (Supplemental Table 1). In fact the converse appeared true: arachidonic and arachidonic acid-containing lipids either remained unchanged or tended to decrease following SCD1 inhibition (Supplemental Table 1). This may reflect the oxidative destruction of these species during ferroptosis (29).
We next explored the possibility that downregulation of SCD1 might induce ferroptosis by decreasing synthesis of cytoprotective lipids. CoQ10 is an endogenous antioxidant produced by the mevalonate pathway. Depletion of CoQ10 has previously been linked to ferroptosis triggered by the synthetic ferroptosis inducer FIN56(6). As shown in Figure 6D, after downregulation of SCD1 we observed a striking reduction of the lipophilic antioxidant coenzyme Q10.
Collectively, these results are consistent with a model in which SCD1 depletion triggers apoptosis by increasing synthesis of ceramides enriched in saturated fatty acids and altering the ratio of saturated to unsaturated fatty acids, whereas ferroptosis is triggered by depleting CoQ10.
Blocking SCD1 sensitizes ovarian cancer cells to ferroptosis inducers in vitro and in vivo
The effects of SCD1 on lipid metabolism suggested that inhibition of SCD1 might potentiate the cytotoxic effect of ferroptosis inducers. To test this prediction, we pretreated both ovarian cancer stem cells and ovarian tumor cells with two different SCD1 inhibitors for short periods of time, with the aim of inducing only modest toxicity. We then assessed sensitivity to RSL3 and erastin, two ferroptosis inducers that act at different steps in the ferroptosis cascade (1). Pretreatment of both ovarian cancer stem cells and ovarian tumor cells with the SCD1 inhibitors MF-438, CAY10566, or A939572 dramatically sensitized cells to RSL3 (Figure 7A–C) and erastin (Supplemental Figure 3B). Combined treatment with SCD1 inhibitors and RSL3 resulted in a synergistic cytotoxic effect on FT-t cells as calculated by the Chou- Talalay method (16) (Supplemental Figure 4). The decrease in viability in cells treated with the combination of RSL3 and SCD1 inhibitors was characterized by an increased oxidation of membrane lipids (Figure 7D), consistent with an increase in ferroptotic cell death. Further, blockade of SCD1 using siRNA or shRNA similarly sensitized cells to RSL3 (Supplemental Figure 5), ruling out non-specific effects of SCD1 inhibitors on ferroptosis sensitization. A similar augmentation of erastin cytotoxicity was observed using the SCD1 inhibitor A939572 (Figure 7E). We also tested the sensitivity of OVCAR8 and OVCAR5 cells, two cell lines representative of the mesenchymal subtype of ovarian cancer (30, 31) to the combination of a ferroptosis inducer and SCD1 inhibitor. As shown in Supplemental Figure 6A, both these cells exhibited an enhanced response to a ferroptosis inducer following pharmacological inhibition of SCD1.
Figure 7. Blocking the activity of SCD1 with small molecule inhibitors increases lipid peroxidation, ferroptotic cell death, and the anti-tumor effect of a ferroptosis inducer in vivo.
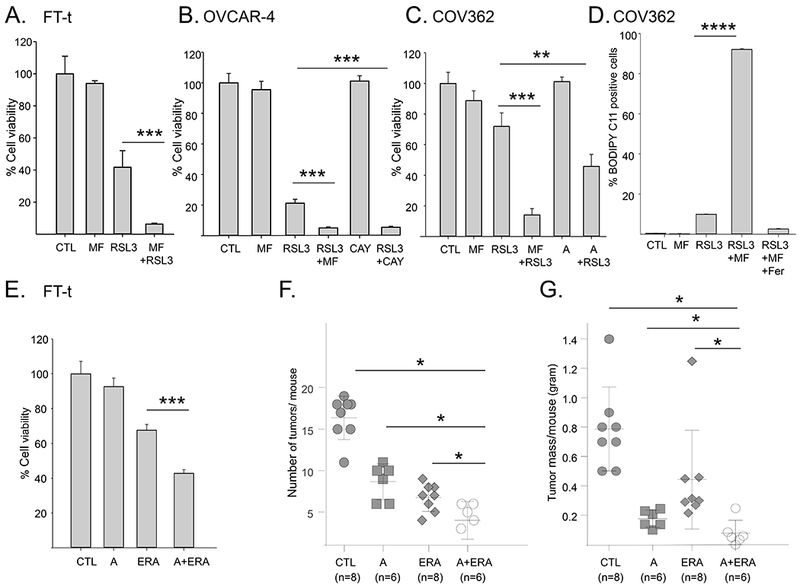
A-C. Viability of FT-t, COV362, or OVCAR-4 cells pretreated with 1μM MF-438 (MF), 1 μM CAY10566 (CAY), or 5 μM A939572 (A) for 24 hours followed by the addition of RSL3 for an additional 24 hours. For each cell type, dose of RSL3 was adjusted to attain modest cytotoxicity in the absence of SCD1 inhibitors (FT-t: 2 μM RSL3;OVCAR-4:1μM RSL3; COV362: 5 μM RSL3). Control cells were treated with SCD1 inhibitor for 48 hours or RSL3 for 24 hours. Data shown is representative of at least three independent experiments each with 8 to 12 replicas D. C11-BODIPY staining of COV362 cells treated with 1 μM MF-438 for 24 hrs followed by 1 μM RSL3 for 4 hrs in the presence or absence of Fer1. E. Viability of FT-t cells treated with A939572 (A) alone, 1μM erastin (ERA) alone, or A939572 followed by erastin. Graphs are representative of at least three independent experiments each with 3 to 8 replicas. F,G. Mice were injected intraperitoneally with FT-t cells and treated for 18 days with either vehicle control, A939572 (A), erastin (E) or the combination of A939572 and erastin. (F) depicts the number of tumors/mouse and (G) depicts total tumor mass/mouse. *p<0.04; **p<0.00027; ***p<2 E-05; ****p<7.2 E-10.
To test whether blockade of SCD1 could potentiate the anti-tumor effects of ferroptosis inducers in vivo, we induced ovarian tumors in mice by injecting ovarian cancer stem cells into the peritoneal cavity, as we have previously described (9). The next day, mice were treated with either vehicle alone, erastin alone, the SCD1 inhibitor A939572 alone, or the combination of erastin and A939572. Mice were sacrificed after 18 days and tumor number and weight assessed. As shown in Figure 7, inhibition of SCD1 exerted substantial anti-tumor effects on its own, and significantly potentiated the antitumor effect of erastin, reducing both tumor number (Figure 7F) and tumor mass (Figure 7G) when compared to erastin treatment alone. We next examined whether this drug combination was also effective in established tumors. Following tumor cell injection, tumors were allowed grow for 7 days, which is approximately a third of the total time required for the development of ascites, morbidity and death in this model. We sacrificed two mice and confirmed that peritoneal tumors were present with visual examination and histological evaluation. We then initiated treatment with the combination of a ferroptosis inducer and SCD1 inhibitor (or vehicle control) in the remaining mice (n=5/group). This treatment remained highly effective even in this more challenging experimental scenario, resulting in a statistically significant 6 fold decrease in tumor number (p<0.028) and 5 fold decrease in total tumor mass (p≤0.026) compared to controls (Supplemental Figure 6B).
DISCUSSION
Ovarian cancer is often diagnosed late, and few good therapeutic options exist (32). The discovery of ferroptosis, a mechanism of cell death to which ovarian cancer cells are susceptible (9), opens the door to new treatments for this disease. In this manuscript we report that SCD1 is over-expressed in ovarian cancer, that genetic or pharmacologic blockade of SCD1 induces ferroptosis as well as apoptosis, and that blockade of SCD1 enhances sensitivity to ferroptosis inducers in both a genetic model of ovarian cancer stem cells and in ovarian cancer cell lines. Sensitization to ferroptosis inducers was observed both in vitro and in vivo.
Lipids participate in complex ways in multiple death pathways, acting both as initiators and facilitators of apoptosis, and impacting necroptosis and ferroptosis (27). Lipid oxidation appears to be particularly central to the process of ferroptosis. We observed that interference with lipid homeostasis through blockade of SCD1 induced multiple changes in cellular lipid content. In addition to lipid oxidation (Figures 4, 7) we observed an increase in apoptosis-promoting ceramides and a decrease in the lipid antioxidant coenzyme Q10 (Figure 6). These complex changes conspire to induce two death pathways, ferroptosis and apoptosis, in ovarian cancer cells challenged with SCD1 inhibitors (Figure 5). Simultaneous triggering of two death pathways suggests SCD1 inhibition may be a particularly effective component of anti-tumor therapy, since overcoming this dual mechanism of cell death may present a significant barrier to the emergence of drug resistance.
SCD1 may be regulated through one or more of the complex pathways that link tumor suppressors to ferroptosis(33). For example, p53 has been shown to repress SCD1 (34). Based on our findings, repression of SCD1 might contribute to the tumor-suppressive activity of p53 by disabling the anti-ferroptotic function of SCD1. This would be consistent with work by others, which has shown that in lung cancer, osteosarcoma and breast cancer, p53 cells to ferroptosis by repressing SLC7A11, a cystine/glutamate antiporter required for synthesis of GSH and the activity of GPX4 (35). However the role of p53 is complex, and in colorectal cancer, p53 antagonizes rather than fosters ferroptosis by blocking DPP4 activity (36). Further, recent work has shown that effects of p53 may also depend on its ability to induce p21(37). BAP1, another potent tumor suppressor, has also been linked to ferroptosis (38, 39), although a connection between BAP1 and SCD1 has not yet been established. Dissecting the multiple links between tumor suppressors, SCD1 and ferroptosis promises to be a fruitful avenue for further investigation.
We propose a model for the mechanism by which SCD1 blockade potentiates effects of ferroptosis inducers. We hypothesize that inhibiting SCD1 alters membrane phospholipid composition, decreasing the content of monounsaturated fatty acyl chains at the expense of increased polyunsaturated fatty acyl chains, as well as decreasing membrane-localized antioxidants (Figure 6). This shift in membrane composition provides more substrate for membrane lipid oxidation, thus sensitizing cells to ferroptosis inducers, which trigger cell death by increasing lipid oxidation (Figure 4,7). Induction of apoptosis through modulation of ceramide content further contributes to the anti-tumor effect of combined SCD1 blockade and ferroptosis induction (Figure 6).
In contrast to our finding that inhibition of SCD1 causes cell death in ovarian cancer cells, others have reported that SKOV3 cells are relatively insensitive to cell death induced by SCD1 blockade (24). There are two potential explanations for this discrepancy. First, although the SKOV3 cell line is used to model high grade serous ovarian cancer (HGSOC), the validity of this cell line as a model of HGSOC has been questioned on the basis of genomic profiling (40). Thus it is unclear that results obtained with SKOV3 cells (which may be representative of a different histologic subtype of ovarian cancer(40)) should be expected to be concordant with results reported here, which are based on OVCAR 4 and COV362 cell lines, which exhibit genetic as well as histologic features that approximate HGSOC (40, 41). Second, we noted that the SKOV3 cell line expresses the lowest level of SCD1 of all the ovarian cancer cell lines we tested (Figure 1), suggesting that SKOV3 cells may be less dependent on this pathway than other ovarian cancer cells, and consequently less sensitive to its inhibition.
There are several promising features of the use of SCD1 inhibitors as an adjunct to ferroptosis inducers in ovarian cancer therapy. First, SCD1 inhibitors can inhibit MUFA formation despite the presence of other SCD isoforms: although SCD5 is also expressed in humans, it appears unable to compensate for loss of SCD1 (42).
Second, it may be possible to manage systemic toxicities engendered by SCD1 blockade. SCD1 knockout mice are viable, suggesting that normal tissues can tolerate long-term loss of SCD1 function (43). However, knockout mice do exhibit toxicities, including fur loss, skin abnormalities and weight reduction, effects that closely mimic effects in mice treated long-term with candidate SCD1 inhibitors (44). These side effects have presented an obstacle to the development of SCD1 inhibitors as obesity drugs, shifting attention to their use in oncology (45). Use of SCD1 inhibitors in a more temporally and spatially restricted manner in cancer treatment may be feasible, both because these side effects, which appear linked to sustained SCD1 inhibition, may be less evident with shorter term cancer treatments, and also because in ovarian cancer patients, delivery of drugs to the peritoneal cavity, close to the site of the tumor, is a clinically viable option that may reduce drug exposure of peripheral organs and therefore undesired side effects (46). Finally, the use of SCD1 inhibitors in combination with a ferroptosis inducer may allow use of lower doses of both agents, with consequent reduction in systemic toxicity. For example in our experiments, we observed augmentation of RSL3 cytotoxicity with concentrations of A 939572, CAY10566 or MF-438 that under the same conditions exhibited minimal effects as single agents (Figure 7).
Third, anti-cancer drugs that induce ferrroptosis are currently available. These include sorafinib, a multikinase inhibitor (47), as well as drugs such as cisplatin, which was recently shown to trigger ferroptosis (48). Thus although there is an intensive effort to develop new drugs to trigger ferroptosis, implementation of an anti-cancer approach that includes a ferroptosis inducer need not await new drug discovery.
Fourth, combined use of a ferroptosis inducer and SCD1 blockade is effective in inhibiting proliferation of cancer stem cells (Figure 7). Using a genetic model of an ovarian cancer stem cell, we observed that inhibition of SCD1 was highly effective in inducing lipid oxidation and cell death in vitro, and in inhibiting tumor growth in vivo (Figure 7). Since cancer stem cells are hypothesized to contribute to drug resistance and disease recurrence, and the emergence of drug resistance is a major contributor to the poor outcome of patients with ovarian cancer, eradicating these cells may be of particular clinical utility. Supporting this concept, inhibitors of GPX4 (i.e. ferroptosis inducers) were found to be particularly effective in targeting drug-resistant “persister” cells in tumors (22) as well as tumor cells with mesenchymal characteristics (23). We observed that two ovarian cancer cell lines representative of the mesenchymal subtype were sensitive to the combined effect of an SCD1 inhibitor and ferroptosis inducer (Supplemental Figure 6A), suggesting that a therapeutic approach involving inhibition of SCD1 in combination with induction of ferroptosis may be of benefit even in this treatment-refractory subtype of ovarian cancer.
Overall, our results demonstrate that ovarian tumors express high levels of SCD1, and that blockade of SCD1 modulates lipid metabolism and sensitizes ovarian cancer cells to ferroptosis inducers in vitro and in vivo. Treatment of ovarian cancer with the combination of SCD1 inhibitors and ferroptosis inducers merits further investigation.
Supplementary Material
Significance:
The combination of SCD1 inhibitors and ferroptosis inducers may provide a new therapeutic strategy for the treatment of ovarian cancer patients.
ACKNOWLEDGEMENTS
We thank Erica Lemler for her contribution to the early phases of this study and Dr. Evan Jellison for expert help in flow cytometry analysis.
Financial support: Supported by NCI grants R01 CA188025 (S.V.T), R01 CA171101 (F.M.T), F32CA214030 (A.K.) and NCI Cancer Center Support Grant P30 CA012197 (PMSR at WFUHS).
Footnotes
Conflict of interest: The authors declare no conflict of interest
Note: While this article was under review, Magtanong et al. demonstrated that exogenous monounsaturated fatty acids promote a ferroptosis resistant state (Cell Chemical Biology 2019;26:1-13).
REFERENCES
- 1.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171:273–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nature chemical biology. 2017;13:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E4966–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nature chemical biology. 2017;13:91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS chemical biology. 2015;10:1604–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nature chemical biology. 2016;12:497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Igal RA. Stearoyl CoA desaturase-1: New insights into a central regulator of cancer metabolism. Biochimica et biophysica acta. 2016;1861:1865–80. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, Klein MG, Zou H, Lane W, Snell G, Levin I, et al. Crystal structure of human stearoyl-coenzyme A desaturase in complex with substrate. Nature structural & molecular biology. 2015;22:581–5. [DOI] [PubMed] [Google Scholar]
- 9.Basuli D, Tesfay L, Deng Z, Paul B, Yamamoto Y, Ning G, et al. Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene. 2017;36:4089–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mai TT, Hamaï A, Hienzsch A, Cañeque T, Müller S, Wicinski J, et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem. 2017;advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J, Condello S, Thomes-Pepin J, Ma X, Xia Y, Hurley TD, et al. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell stem cell. 2017;20:303–14 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamamoto Y, Ning G, Howitt BE, Mehra K, Wu L, Wang X, et al. In vitro and in vivo correlates of physiological and neoplastic human Fallopian tube stem cells. The Journal of pathology. 2016;238:519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng Z, Manz DH, Torti SV, Torti FM. Iron-responsive element-binding protein 2 plays an essential role in regulating prostate cancer cell growth. Oncotarget. 2017;8:82231–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blanchette-Farra N, Kita D, Konstorum A, Tesfay L, Lemler D, Hegde P, et al. Contribution of three-dimensional architecture and tumor-associated fibroblasts to hepcidin regulation in breast cancer. Oncogene. 2018;37:4013–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crowley LC, Scott AP, Marfell BJ, Boughaba JA, Chojnowski G, Waterhouse NJ. Measuring Cell Death by Propidium Iodide Uptake and Flow Cytometry. Cold Spring Harbor protocols. 2016;2016. [DOI] [PubMed] [Google Scholar]
- 16.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Advances in enzyme regulation. 1984;22:27–55. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen DH, Zhou T, Shu J, Mao J. Quantifying chromogen intensity in immunohistochemistry via reciprocal intensity. CancerInCytes. 2013;2. [Google Scholar]
- 18.Konstorum A, Lynch ML, Torti SV, Torti FM, Laubenbacher RC. A Systems Biology Approach to Understanding the Pathophysiology of High-Grade Serous Ovarian Cancer: Focus on Iron and Fatty Acid Metabolism. Omics : a journal of integrative biology. 2018;22:502–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ganzfried BF, Riester M, Haibe-Kains B, Risch T, Tyekucheva S, Jazic I, et al. curatedOvarianData: clinically annotated data for the ovarian cancer transcriptome. Database : the journal of biological databases and curation. 2013;2013:bat013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim J, Park EY, Kim O, Schilder JM, Coffey DM, Cho CH, et al. Cell Origins of High-Grade Serous Ovarian Cancer. Cancers. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verhaak RG, Tamayo P, Yang JY, Hubbard D, Zhang H, Creighton CJ, et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. The Journal of clinical investigation. 2013;123:517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Ren J, Yang L, Li Y, Fu J, Li Y, et al. Stearoyl-CoA desaturase-1 mediated cell apoptosis in colorectal cancer by promoting ceramide synthesis. Scientific reports. 2016;6:19665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mason P, Liang B, Li L, Fremgen T, Murphy E, Quinn A, et al. SCD1 inhibition causes cancer cell death by depleting mono-unsaturated fatty acids. PloS one. 2012;7:e33823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Obeid LM, Linardic CM, Karolak LA, Hannun YA. Programmed cell death induced by ceramide. Science. 1993;259:1769–71. [DOI] [PubMed] [Google Scholar]
- 27.Magtanong L, Ko PJ, Dixon SJ. Emerging roles for lipids in non-apoptotic cell death. Cell death and differentiation. 2016;23:1099–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ariyama H, Kono N, Matsuda S, Inoue T, Arai H. Decrease in membrane phospholipid unsaturation induces unfolded protein response. The Journal of biological chemistry. 2010;285:22027–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doll S, Conrad M. Iron and ferroptosis: A still ill-defined liaison. IUBMB life. 2017;69:423–34. [DOI] [PubMed] [Google Scholar]
- 30.Fang D, Chen H, Zhu JY, Wang W, Teng Y, Ding HF, et al. Epithelial-mesenchymal transition of ovarian cancer cells is sustained by Rac1 through simultaneous activation of MEK1/2 and Src signaling pathways. Oncogene. 2017;36:1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haley J, Tomar S, Pulliam N, Xiong S, Perkins SM, Karpf AR, et al. Functional characterization of a panel of high-grade serous ovarian cancer cell lines as representative experimental models of the disease. Oncotarget. 2016;7:32810–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lengyel E Ovarian cancer development and metastasis. The American journal of pathology. 2010;177:1053–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gnanapradeepan K, Basu S, Barnoud T, Budina-Kolomets A, Kung CP, Murphy ME. The p53 Tumor Suppressor in the Control of Metabolism and Ferroptosis. Frontiers in endocrinology. 2018;9:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mirza A, Wu Q, Wang L, McClanahan T, Bishop WR, Gheyas F, et al. Global transcriptional program of p53 target genes during the process of apoptosis and cell cycle progression. Oncogene. 2003;22:3645–54. [DOI] [PubMed] [Google Scholar]
- 35.Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell reports. 2017;20:1692–704. [DOI] [PubMed] [Google Scholar]
- 37.Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, et al. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell reports. 2018;22:569–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nature cell biology. 2018;20:1181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Affar EB, Carbone M. BAP1 regulates different mechanisms of cell death. Cell death & disease. 2018;9:1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nature communications. 2013;4:2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitra AK, Davis DA, Tomar S, Roy L, Gurler H, Xie J, et al. In vivo tumor growth of high-grade serous ovarian cancer cell lines. Gynecologic oncology. 2015;138:372–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flowers MT, Ntambi JM. Role of stearoyl-coenzyme A desaturase in regulating lipid metabolism. Current opinion in lipidology. 2008;19:248–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ntambi JM, Miyazaki M, Stoehr JP, Lan H, Kendziorski CM, Yandell BS, et al. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:11482–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Z, Dales NA, Winther MD. Opportunities and challenges in developing stearoyl-coenzyme A desaturase-1 inhibitors as novel therapeutics for human disease. J Med Chem. 2014;57:5039–56. [DOI] [PubMed] [Google Scholar]
- 45.Theodoropoulos PC, Gonzales SS, Winterton SE, Rodriguez-Navas C, McKnight JS, Morlock LK, et al. Discovery of tumor-specific irreversible inhibitors of stearoyl CoA desaturase. Nature chemical biology. 2016;12:218–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tewari D, Java JJ, Salani R, Armstrong DK, Markman M, Herzog T, et al. Long-term survival advantage and prognostic factors associated with intraperitoneal chemotherapy treatment in advanced ovarian cancer: a gynecologic oncology group study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33:1460–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends in cell biology. 2016;26:165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, et al. Ferroptosis: A Novel Anti-tumor Action for Cisplatin. Cancer research and treatment : official journal of Korean Cancer Association. 2018;50:445–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.