

Summary
Metastasis is the leading cause of cancer mortality. Chromatin remodeling provides the foundation for the cellular reprogramming necessary to drive metastasis. However, little is known about the nature of this remodeling and its regulation. Here, we show that metastasis-inducing pathways regulate histone chaperones to reduce canonical histone incorporation into chromatin, triggering deposition of H3.3 variant at the promoters of poor-prognosis genes and metastasis-inducing transcription factors. This specific incorporation of H3.3 into chromatin is both necessary and sufficient for the induction of aggressive traits that allow for metastasis formation. Together, our data clearly show incorporation of histone variant H3.3 into chromatin as a major regulator of cell fate during tumorigenesis, and histone chaperones as valuable therapeutic targets for invasive carcinomas.
Graphical Abstract
In Brief
Gomes et al. reveal metastatic stimuli reduce histone H3.1/H3.2 deposition on chromatin by suppressing the CAF-1 complex in breast cancer cells, leading to increased incorporation of non-canonical histone H3.3, which in turn induces chromatin remodeling and expression of metastatic genes.
Introduction
Cancer is one of the world’s deadliest diseases. While primary tumors can be treated with surgery and adjuvant therapy, metastases are highly resistant to therapy and account for the largest proportion of cancer induced mortality (Lamouille et al., 2014; Valastyan and Weinberg, 2011). Hence, the ability to effectively treat cancer is largely dependent on treating metastases, which are strikingly the least understood aspect of cancer.
In the recent years, multiple genes and signaling pathways have been shown to have the ability to influence cancer progression; however, few signaling events have been established as truly essential to the metastatic process. Among these, ERK signaling has been shown to be of critical importance in cancer progression and metastasis formation. In fact, gain of function mutations in this pathway that result in activated ERK signaling are common, appearing in nearly 30–50% of metastatic human cancers (Hoshino et al., 1999; Samatar and Poulikakos, 2014), and metastasis inducing signals often result in the activation of ERK (Janda et al., 2002; Lamouille et al., 2014). Therefore, it comes as no surprise that a significant effort has been devoted to identifying small molecules targeting the ERK signaling pathway. Although several drugs have shown promising short-term results in the treatment of certain cancers, the emergence of drug resistance upon long-term treatments has severely impacted their use as effective cancer therapies (Roberts and Der, 2007; Samatar and Poulikakos, 2014). Consequently, understanding key events downstream of this signaling pathway is of particular interest for the identification of mechanisms that are fundamental for tumor progression and therefore yield more effective therapeutic targets for invasive carcinomas.
Epigenetic changes are the first line of cellular reprograming that enable complex cell fate decisions. In fact, as a tumor progresses the genome of cancer cells must undergo a series of epigenetic alterations to enable cell fate decisions that allow cancer cells to leave the primary tumor, survive in circulation, resist therapy and colonize distal organs (Bedi et al., 2014; Chaffer et al., 2013; Tam and Weinberg, 2013). Remarkably, very little is known about the nature of these epigenetic alterations, their contribution to tumor progression, and their regulation by oncogenic signaling. Histone variants and histone chaperones have become the latest addition to a growing list of epigenetic regulators. Histone exchange allows chromatin to be highly responsive to changes in the environment, which are particularly dynamic throughout the tumorigenic process (Li, 2002; Morgan et al., 2005; Surani, 2001). Among the histone variants known thus far, histone H3.3 has been gaining interest in the chromatin field due to its unique features (Turinetto and Giachino, 2015; Wen et al., 2014). Unlike its canonical counterparts H3.1/H3.2, H3.3 incorporation into chromatin is cell cycle independent, and it can be deposited at replication sites when canonical H3.1/H3.2 deposition is impaired. H3.3 has also been consistently associated with an active state of chromatin (Burgess and Zhang, 2013; Gurard-Levin et al., 2014; Henikoff and Ahmad, 2005). Highlighting the importance of H3.3 for complex diseases, mutations in H3.3 have been found in pediatric brain cancers and associated with their malignancy (Yuen and Knoepfler, 2013). Despite these observations, the role of H3 variants in tumor progression and their regulation by oncogenic signaling remains largely unexplored.
Results
Metastatic inducers promote a switch in H3 variants incorporated into chromatin
Cancer cells are known to exploit epigenetic regulatory mechanisms for survival, drug-resistance and metastasis (Bedi et al., 2014; Chaffer et al., 2013; Tam and Weinberg, 2013). Considering the growing body of evidence showing that chromatin accessibility by transcriptional machinery is heavily influenced by histone variants (Hu et al., 2013; McKittrick et al., 2004; Venkatesh and Workman, 2015), we hypothesized that histone H3 variants are differentially regulated during tumor progression to promote the transcriptional plasticity necessary to sustain the cellular reprogramming that enables acquisition of metastatic properties. To address this question, we first tested the composition of H3 variants in the chromatin in response to established inducers of epithelial-to-mesenchymal transition (EMT) and metastasis formation (Asiedu et al., 2011; Balkwill, 2006; Borthwick et al., 2012; Korpal and Kang, 2010; López-Novoa and Nieto, 2009) in different carcinoma cells. This analysis demonstrated that classic metastasis inducers, TGFβ/TNFα treatment or expression of an ERK2 mutant that induces EMT (ERK2 D319N mutant (Dimitri et al., 2005)), increased the total levels of chromatin-bound H3.3 (Figure 1A). Surprisingly, we detected a pronounced decrease in chromatin bound H3.1/H3.2 under the same conditions (Figure 1A), which was accompanied with a decrease in the mRNA levels of H3.1 encoding genes (Figure S1A). To further assess the role of the H3 switch in metastasis, we took advantage of a specific clone (LM2) of a breast cancer cell line (MDA-MB-231), which was isolated from a metastatic site and displays higher metastatic capacity in mice compared to its parental cell line (Minn et al., 2005). Strikingly, the LM2 clone shows a similar chromatin-bound histone pattern to what we observed in cells undergoing EMT displaying a reduction in canonical histone H3.1/H3.2 and total H3, and an increase in chromatin-bound H3.3 (Figure S1B), suggesting that the switch in histone H3 variants dictates metastatic potential. The levels of other histones such as H4 and H2A1 were similarly reduced, indicating that despite an increase in H3.3, metastatic signaling induces a decrease in total nucleosome abundance (Figure 1A). To further validate this observation, we quantified the abundance of histones in histone extracts by mass spectrometry and observed a general decline in canonical histones H1, H2A, H3 and H4 (Figure 1B), which was consistent with a decline in total histone levels detected by Coomassie Blue stain (Figure 1A). To explore whether the changes in histone levels accompanying metastatic potential and EMT induction are reflected in chromatin compaction, we performed global mapping of chromatin accessibility using ATAC-seq. Upon ERK2 D319N-mediated EMT induction, 145,607 peaks were identified compared to 109,484 in the control condition, which indicates a 33% increase in chromatin accessibility, a phenomenon detected across the whole genome (Figure 1C and Table S1). In depth analysis of the differentially accessible genes showed multiple EMT-inducing factors including ZEB1, a master regulator of the EMT process (Figure 1D). Together, these data support a model where metastatic signaling regulates nucleosome abundance and composition to establish a chromatin environment primed for alterations necessary to sustain cell fate decisions associated with tumor progression.
Figure 1. EMT induction promotes a global decline in histone levels promoting H3.3-mediated gap filling and increasing chromatin accessibility.
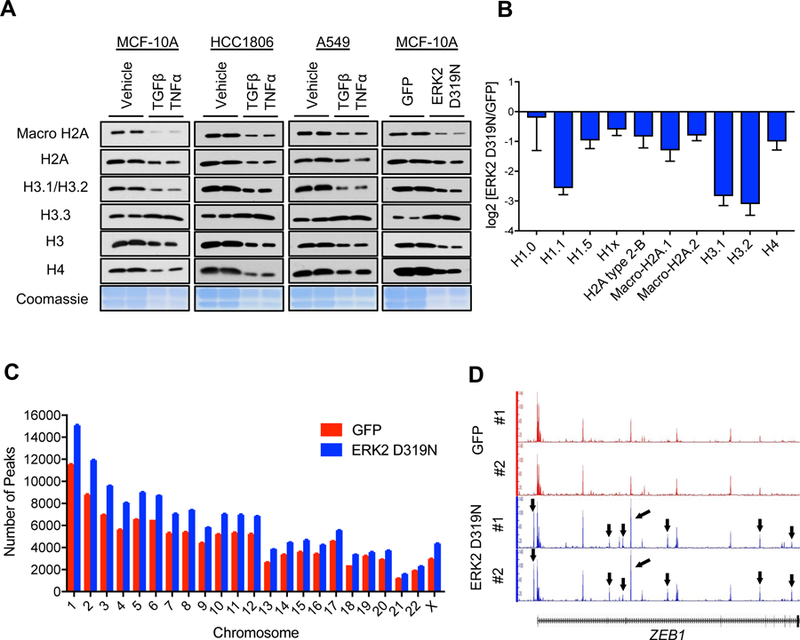
(A) Levels of various histones and Coomassie Blue stain of total histones in histone extracts of cells treated with TGFβ + TNFα (MCF-10A for 5 days, HCC1806 and A549 for 10 days) or MCF-10A expressing ERK2 D319N inducibly for 3 days; representative images (n = 4).
(B) Histone quantification by mass spectrometry in MCF-10A expressing ERK2 D319N inducibly for 3 days (n = 3). All values are expressed as mean ± SEM.
(C and D) Summary of genome-wide nucleosome occupancy showing distribution of detected peaks on chromosomes (All values are expressed as mean ± SEM) (C), and signal tracks for ZEB1 (D) determined by ATAC-seq in MCF-10A expressing ERK2 D319N inducibly for 3 days; arrows indicate the presence of novel peaks.
Histone H3.3 regulates a pro-metastatic transcriptional reprogramming and is essential for acquisition of metastatic properties
The increase in chromatin bound histone H3.3 observed in models of EMT suggested that, similarly to what has been observed in other models of reduced canonical histone abundance (Ray-Gallet et al., 2011; Schneiderman et al., 2012), H3.3 is “gap filling” naked DNA to maintain chromatin integrity, and therefore, alter nucleosome properties and rewire gene expression. To elucidate the role of H3.3 during tumor progression, we performed chromatin immunoprecipitation and genome-wide sequencing (ChIP-seq) in the LM2 cells to globally assess the location of H3 variants. Interestingly, while canonical histone H3.1/H3.2 preferentially localized to gene bodies and introns, H3.3 was enriched at promoter regions (Figures 2A, 2B and S2A), supporting the idea of H3.3 as a transcriptional regulator. Gene set enrichment analysis (GSEA) showed that in highly metastatic cells, H3.3 was deposited mainly at genes belonging to inducible programs essential for tumor progression and metastasis, such as chemotaxis, cell adhesion and movement, morphogenesis and wound healing (Figures 2C, 2D and S2B) (Arwert et al., 2012; Cavallaro and Christofori, 2004; Ribatti, 2017; Roussos et al., 2011; Yamaguchi et al., 2005); conversely, H3.1/H3.2 localizes primarily to genes involved in housekeeping programs such as RNA and DNA metabolism, DNA repair and organelle maintenance (Figures 2C and S2C). Wound healing is a transcriptional program of particular interest, as it is hijacked by tumor cells to induce EMT and allow for the acquisition of important metastatic traits under physiological settings (Arwert et al., 2012; Kalluri and Neilson, 2003; Kalluri and Weinberg, 2009). In fact, we found that in metastatic cells, H3.3 is deposited at the promoters of many EMT-inducing transcription factors and poor-prognosis genes, including ZEB1, SNAI1, SOX9, POU2F1, FOSL2, SOX4 and KLF6 (Figure 2D and Table S2), supporting an essential role of H3.3 for EMT and metastasis formation. These results were also validated by ChIP-PCR with antibodies against the endogenous proteins (Figures S2D and S2E) and in LM2 cells expressing tagged H3.1 or H3.3 (Figures S2F, S2G and S2H). To further assess the role for histone H3.3 enrichment at poor-prognosis and metastasis-inducing genes observed in these metastatic cells, we performed ChIP experiments in chromatin extracts of MCF-10A cells either treated with TGFβ/TNFα or expressing the EMT-inducing ERK2 D319N mutant. In both conditions, we observed an enrichment of H3.3 at the promoters of the EMT-inducing transcription factors ZEB1, SNAI1 and SOX9, which correlated with their transcriptional activation, as evidenced by enrichment of the active form of RNA Pol II (S5 phosphorylation) at their promoters and their increased protein levels (Figures 2E–2H). Remarkably, we observed that in these cells, knockdown of H3.3 blocked the ability of TGFβ/TNFα or ERK2 D319N to induce EMT as evidenced by suppression of the mesenchymal marker fibronectin and re-expression of the epithelial marker E-cadherin (Figures 2I and 2J) (Lamouille et al., 2014; Polyak and Weinberg, 2009). Thus, demonstrating an essential role of histone H3.3 as an effector of metastatic signals to regulate acquisition of aggressive traits.
Figure 2. Histone H3.3 mediates metastatic traits by regulating aggressive factors.

(A-D) Summary of H3.1/H3.2 and H3.3 ChIP-seq analysis in LM2 cells showing genomic distribution of H3.1/H3.2 and H3.3 peaks (A), density heat maps of H3.1/H3.2 and H3.3 peaks across transcriptional starting sites (TSS) (Darker blue indicates higher enrichment) (B), GSEA analysis of genes enriched for H3.1/H3.2 or H3.3 (C), and H3.1/H3.2 and H3.3 signal tracks (D).
(E) Levels of the EMT-inducing transcription factors ZEB1, SNAI1 and SOX9 in MCF-10A treated with TGFβ + TNFα for 5 days or expressing ERK2 D319N for 6 days after transduction; representative images (n = 4).
(F-H) H3.3 and RNA Pol II pS5 enrichment at the ZEB1 (F), SNAI1 (G) and SOX9 (H) promoters in MCF-10A treated with TGFβ + TNFα or expressing inducible ERK2 D319N for 3 days; fold enrichment was determined using IgG as a control for the ChIP (n = 4). All values are expressed as mean ± SEM (**p < 0.01, ***p < 0.001).
(I and J) EMT induction determined by protein levels of the mesenchymal marker fibronectin and the epithelial marker E-cadherin after treatment with TGFβ + TNFα for 5 days (I) or expression of ERK2 D319N for 6 days after transduction (J) in MCF-10A with H3.3 knockdown; histone levels are detected in whole cell lysates to show knockdown efficiency of H3.3; representative images (n = 4).
Metastatic signaling alters histone H3 chaperone levels
Because the chromatin-bound levels of histone H3 variants were drastically altered in response to metastatic signaling, we reasoned that histone chaperones that deposit the different variants into chromatin are at the nexus of metastatic signaling and the rewiring of gene expression necessary for the dramatic cell fate alterations that sustain tumor progression and metastasis. CAF-1 deposits the canonical variants H3.1/H3.2 into chromatin in a cell cycle dependent manner while other histone chaperones, HIRA at genic and DAXX at telomeric regions, deposit the H3.3 variant throughout the cell cycle (Figure 3A) (Burgess and Zhang, 2013; Gurard-Levin et al., 2014; Henikoff and Ahmad, 2005). Consistent with the observed changes in H3 variants, we detected a pronounced decrease in CAF-1 and an upregulation of HIRA in TGFβ/TNFα and ERK2 D319N-induced EMT models (Figures 3B and S3A–D). To examine whether histone H3.3 or its chaperones are sufficient to promote tumor progression we overexpressed H3.3, HIRA or DAXX in MCF-10As and measured EMT markers as a surrogate for acquisition of metastatic properties. Surprisingly, none of these were sufficient to induce EMT in this model (Figure S3E), suggesting that the regulation of H3.3-promoting aggressiveness and tumor progression lies elsewhere, and demonstrating that metastatic signaling-induced chromatin remodeling goes far beyond a simple increase in H3.3 abundance.
Figure 3. ERK mediates a switch in histone H3 variants and their chaperones in response to metastatic inducers.
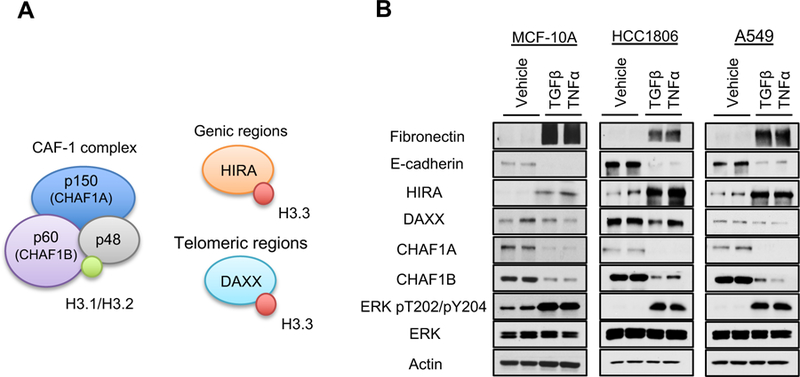
(A) Schematic representation of the histone H3 chaperones: H3.1/H3.2 chaperone CAF-1 (comprised of CHAF1A, CHAF1B and p48) and H3.3 chaperones HIRA and DAXX.
(B) Levels of the histone H3 chaperones in cells treated with TGFβ + TNFα (MCF-10A for 5 days, HCC1806 and A549 for 10 days); representative images (n = 4).
See also Figure S3.
CAF-1 suppression triggers EMT and metastatic-like properties
Since overexpression of H3.3 did not induce EMT, we reasoned that a mechanism of gap filling needs to be triggered to induce H3.3 enrichment at pro-metastatic genetic programs. Consistent with this idea, CAF-1 suppression was observed as early as 24 hours post induction of metastatic signaling (Figure S4A), suggesting that suppression of the CAF-1 complex and the consequent decrease in canonical Histone H3.1/H3.2 might be the trigger for the chromatin remodeling necessary for EMT induction. To test this, we alternately silenced two of the CAF-1 subunits: the 150 kDa subunit (CHAF1A) or the 60 kDa subunit (CHAF1B) in MCF-10A cells. Knockdown of either subunit resulted in suppression of the other, suggesting that CAF-1 components modulate the levels of one another (Figure 4A). More importantly, we observed that knockdown of CAF-1 was sufficient to induce EMT across a variety of cancer cells and breast epithelial cells, as demonstrated by the change from their epithelial morphology to a mesenchymal phenotype evidenced by loss of polarity and a spindle-shaped morphology (Figure 4B). Consistent with the morphological changes resembling EMT, knockdown of CAF-1 was sufficient to promote an increase in the mesenchymal marker fibronectin and a decline in the epithelial marker E-cadherin (Figures 4A, 4C and S4B–S4D) (Lamouille et al., 2014; Polyak and Weinberg, 2009). Not only did the cells become mesenchymal, but suppression of the CAF-1 complex conferred a robust resistance to two commonly used chemotherapeutic agents, paclitaxel and carboplatin (Figures 4D–4F and S4E-S4G). Moreover, suppression of the CAF-1 complex was sufficient to promote stemness, as shown by an upregulation of CD44 and a decline in CD24 (Figures 4G and 4H) (Polyak and Weinberg, 2009), and was sufficient to induce metastatic properties such as increased migration and invasion in cell-based assays (Figure 4I). To exclude off-target effects of the shRNAs, we engineered CHAF1B cDNA resistant to one of the hairpins used to deplete CHAF1B. Reintroduction of CHAF1B blocked the ability of the shRNA to induce EMT and resistance to chemotherapeutic agents, thus showing that this phenotype is specific to depletion of CAF-1 (Figures S4H and S4I). Collectively, our results show that suppression of CAF-1 is a potent inducer of aggressive traits.
Figure 4. Suppression of the CAF-1 complex induces aggressive traits.

(A) EMT induction determined by the protein levels of the mesenchymal marker fibronectin and the epithelial marker E-cadherin in MCF-10A with CHAF1B knockdown for 10 days; representative images (n = 4).
(B) Morphology of MCF-10A with CHAF1A or CHAF1B knockdown for 10 days; representative images (n = 4), scale bar = 200 µm.
(C) EMT induction determined by the protein levels of the mesenchymal marker fibronectin and the epithelial marker E-cadherin in HCC1806 or A549 with CHAF1B knockdown for 10 days; representative images (n = 4).
(D-F) Viability of MCF-10A (D), HCC1806 (E), and A549 (F) with CHAF1B knockdown for 10 days treated with the chemotherapeutic drugs carboplatin and paclitaxel (n = 4).
(G and H) Stemness evaluated by the increase in the CD44 marker and decrease in the CD24 in MCF-10A (G) and A549 (H) both with CHAF1B knockdown for 10 days (n = 4).
(I) Quantification of migration and invasion of MCF-10A with CHAF1B knockdown for 10 days evaluated by transwell assays (left); representative images (right) (n = 4), scale bar = 1 mm.
All values are expressed as mean ± SEM (ns: not significant, *p < 0.05, **p < 0.01, ***p < 0.001).
See also Figure S4.
Metastatic signaling suppresses CAF-1 through regulation of the CHAF1B promoter
Despite the crucial role of CAF-1 as a regulator of cell cycle and chromatin assembly (Kaufman et al., 1995), its regulation remains largely unknown. While EMT and acquisition of metastatic properties has long been associated with cell cycle alterations (Lovisa et al., 2015; Vega et al., 2004), metastatic signaling altered CAF-1 levels before any cell cycle changes were observed as evidenced by lack of differences in the mitotic marker histone H3 S10 phosphorylation (Figure 5A). Likewise, artificially arresting various cell lines in S-phase did not produce suppression of CAF-1 within the same time frame (Figure S5A). Thus, our data support a model where CAF-1 suppression by metastatic signaling is initiated independently of cell cycle. Time-course experiments showed that the regulation of CAF-1 by metastatic signaling lies with CHAF1B, as CHAF1B protein and mRNA levels decline before CHAF1A levels do (Figures 5A and 5B). To determine if this regulation of CHAF1B mRNA is mediated by a transcriptional mechanism, we measured the promoter activity of CHAF1B in our model of ERK2 D319N-induced EMT. The CHAF1B promoter activity was significantly decreased (Figure 5C), demonstrating that ERK2 suppresses CHAF1B transcriptionally. Consistent with the idea that the primary regulation of the CAF-1 complex through ERK signaling lays on CHAF1B, no changes were detected in the activity of the CHAF1A promoter (Figure 5C). While our results cannot discard a potential regulation of CHAF1A levels at a later point, they clearly show that metastatic signaling by ERK2 regulates the transcription of CHAF1B and thereby the CAF-1 complex.
Figure 5. Metastatic signaling controls the CAF-1 complex through an ERK-mediated regulation of the CHAF1B promoter.

(A and B) Time-course analysis of CAF-1 complex and the cell cycle marker H3 pS10 protein levels (A), and CHAF1A and CHAF1B mRNA levels evaluated by qPCR (B) in MCF-10A expressing inducible ERK2 D319N for up to 24 hours; representative images (n = 4).
(C) CHAF1A and CHAF1B promoter activity measured via a luciferase reporter assay in MCF-10A expressing inducible ERK2 D319N for 3 days; luciferase values are normalized to GFP control cells (n = 6).
(D) Schematic of Sp1 and EGR1 binding sites in CHAF1B promoter.
(E) CAF-1 complex, EGR1 and Sp1 p-T739 protein levels in MCF-10A expressing inducible ERK2 D319N for 3 days; representative images (n = 4).
(F) CAF-1 complex, EGR1, Sp1 p-T739 and ERK2 p-T202/Y204 protein levels in cells treated with TGFβ + TNFα (MCF-10A for 5 days, HCC1806 and A549 for 10 days); representative images (n = 4).
(G) CHAF1A and CHAF1B protein levels in MCF-10A with shRNA-mediated Sp1 knockdown for 3 days; representative images (n = 4).
(H) Binding of Sp1 and/or EGR1 to biotinylated DNA fragments of either the CHAF1B promoter containing the overlapping Sp1/EGR1 site or a scrambled control in lysates from MCF-10A expressing inducible ERK2 D319N for 3 days; IgG control for immunoprecipitation of the DNA fragments with streptavidin; representative images (n = 4).
(I) CAF-1 complex protein levels in MCF-10A expressing inducible ERK2 D319N and either Sp1 WT or the Sp1 T453/T739 phosphorylation site mutants for 3 days; representative images (n = 4).
All values are expressed as mean ± SEM (**p < 0.01, ***p < 0.001).
See also Figure S5.
To understand how ERK2 signaling leads to CHAF1B transcriptional suppression, we analyzed the CHAF1B promoter in silico and found three Sp1 binding motifs, one of which overlaps with an EGR1 binding motif (Figure 5D). Intriguingly, both EGR1 and Sp1 are known downstream effectors of ERK signaling. Interestingly, while ERK2-driven metastatic signaling triggers induction of EGR1 levels, it inhibits phosphorylation of Sp1 (Figures 5E, 5F, S5B and S5C), supporting the idea that metastatic signaling creates an ERK2-mediated signaling hub that promotes CAF-1 suppression. Inducing EGR1 levels alone was not sufficient to suppress CHAF1B levels in a consistent manner (Figure S5D). However, inhibition of Sp1 either genetically or chemically led to a significant reduction of CHAF1A/CHAF1B levels mimicking the effects of ERK2 (Figures 5G and S5E) and suggesting that Sp1 is the link between ERK2 and CHAF1B regulation. Interestingly, in monocytes, EGR1 has been shown to antagonize Sp1 in response to growth factors at the promoters of genes with overlapping binding sites (Kubosaki et al., 2009). We considered the possibility that upon metastatic signaling, the combination of EGR1 induction and absence of Sp1 phosphorylation creates a specific environment that leads to repression of CHAF1B transcription and CAF-1 suppression. DNA binding experiments with ERK2 D319N expressing MCF-10A lysates illustrated an interaction between EGR1 and the CHAF1B promoter region within the overlapping EGR1/Sp1 site (Figure 5H). Conversely, an interaction was absent between EGR1 and a control region of the CHAF1B promoter lacking the EGR1/Sp1 site (Figure 5H). We speculated that phosphorylation of Sp1 regulated by ERK2 D319N could act as a switch that dictates whether Sp1 or EGR1 binds to the overlapping site of the CHAF1B promoter, and therefore determines CHAF1B promoter activity. In support of this, we found that when two Sp1 phosphorylation sites were mutated from threonine to alanine (T453A/T739A, AA), Sp1 binding to the CHAF1B promoter region with the overlapping EGR1/Sp1 sites was completely abolished, allowing EGR1 to bind to this region; conversely, phosphomimetic Sp1 mutations (T453E/T739E, EE) enhanced Sp1 binding to the overlapping CHAF1B promoter site, preventing EGR1 binding (Figure S5F). Importantly, CHAF1B and CHAF1A protein levels were restored in ERK2 D319N-induced cells when the Sp1 phosphomimetic mutant was expressed, while the non-phosphorylatable mutant and wild type Sp1 proteins were not able to restore CHAF1B expression (Figure 5I). It is important to note that this mechanism of CAF-1 regulation is independent of a previously described SRC-dependent regulation of CHAF1A (Figure S5G) (Endo et al., 2017). Together, our data show that ERK2 signaling mediates suppression of CAF-1 by silencing CHAF1B transcription through regulation of the dynamics between EGR1 and Sp1 at the CHAF1B promoter, illustrating a complete signaling mechanism that regulates histone variant switch in order to promote cancer progression.
CAF-1 levels dictate metastatic colonization
Having shown that suppression of CAF-1 is sufficient to induce the EMT process and acquisition of aggressive properties, and demonstrated how cancer cells place a significant emphasis on developing intricate mechanisms to regulate cell fate through deposition of histones onto chromatin, it became clear that suppression of CAF-1 is essential for carcinoma cells to acquire and maintain their aggressive properties. Supporting this idea, evaluation of human breast cancer samples showed a pronounced decrease in the levels of CHAF1B in metastases versus their matched primary tumors (Figure 6A and Table S3), indicating a direct relevance to cancer progression in humans. Likewise, in LM2 cells, CAF-1 levels were significantly lower compared to parental cells (Figure 6B). This suggested that CAF-1 levels dictate the ability of these cells to form metastases. Knockdown of CHAF1B in the parental clone led to a significant increase in migration and invasion in vitro (Figure S6A), and robustly increased the ability of these cells to colonize the lungs in mouse xenograft experiments (Figure 6C). Conversely, in the more metastatic LM2 clone, overexpression of the CAF-1 complex significantly decreased their ability to migrate and invade in vitro (Figure S6B) and abrogated their metastatic colonization ability in vivo (Figure 6D). Together these data show that CAF-1 levels dictate the metastatic capability of breast cancer cells, a phenomenon that is not only relevant in cells in culture and xenograft experiments, but also for human disease.
Figure 6. CAF-1 levels regulate metastatic colonization.

(A) CHAF1B levels in a tissue array of primary cancers and matched lymph node metastases from breast cancer patients (left); representative images (right) (n = 50), scale bar = 200 µm.
(B) CAF-1 complex protein levels in MDA-MB-231 parental versus the more metastatic LM2 clone; representative images (n = 4).
(C) Quantification of lung metastatic lesions of parental and CHAF1B knockdown MDA-MB-231 cells (left) (p/s indicates photons/second); representative images (right) (n = 10).
(D) Quantification of lung metastatic lesions of LM2 cells with or without CHAF1B overexpression (left) (p/s indicates photons/second); representative images (right) (n = 7).
All values are expressed as mean ± SEM (ns: not significant, *p < 0.05, **p < 0.01, ***p < 0.001).
CAF-1 suppression induces an H3.3-dependent pro-metastatic transcriptional reprogramming
The main role of CAF-1 has been reported to be the deposition of newly synthesized histones during S-phase specific chromatin duplication (Gurard-Levin et al., 2014; Kaufman et al., 1995). However, simply arresting cells in G1/S-phase did not induce EMT (Figure S7A), indicating that alterations in cell cycle are unlikely to be the cause of CAF-1-mediated cellular reprogramming that drives tumor progression and metastasis. CAF-1 has also been implicated in regulation of heterochromatin by association with HP1 proteins (Huang et al., 2010; Quivy et al., 2004; Yan et al., 2018), but knockdown of HP1 proteins failed to prevent EMT induced by suppression of the CAF-1 complex (Figure S7B), indicating that heterochromatin regulation also does not mediate the effects of CAF-1 observed. To gain a better understanding of the CAF-1 mediated mechanism of tumor progression and acquisition of aggressive properties, we performed a global analysis of RNA levels. Strikingly, this analysis showed a dramatic transcriptional reprogramming in MCF-10A cells upon CAF-1 suppression (Figures 7A, S7C and Table S4). GSEA analysis showed enrichment of pathways similar to the ones enriched for H3.3 in metastatic cells, including chemotaxis, migration, development, and wound healing (Figures S7D and S7E). Further analysis of the differentially regulated transcripts showed a significant increase in the mRNA of many EMT-inducing factors and poor-prognosis genes (Figures 7B and S7F), demonstrating the powerful role of the CAF-1 complex as a transcriptional regulator downstream of metastatic signaling.
Figure 7. Suppression of the CAF-1 complex triggers an H3.3-dependent global transcriptional reprogramming that underlies the acquisition of aggressive properties.

(A) Heatmap representation of wound-healing-related and poor-prognosis genes detected in RNA-seq analysis in MCF-10A with CHAF1B knockdown for 3 days, up regulated genes are indicated with red and down regulated genes are indicated with blue, (n = 3).
(B) Relative mRNA levels of ZEB1, SNAI1 and SOX9 evaluated by qPCR in MCF-10A with CHAF1B knockdown for 3 days (n = 3).
(C) Levels of H3 histone variants in chromatin extracts and Coomassie Blue stain of total histones in histone extracts of MCF-10A and A549 both with CHAF1B knockdown for 3 days; representative images (n = 4).
(D) H3.3 and RNA Pol II pS5 enrichment at the ZEB1, SNAI1 and SOX9 promoters in MCF-10A with CHAF1B knockdown for 3 days; fold enrichment was determined using IgG as a control for the ChIP (n = 4).
(E) EMT induction determined by the protein levels of the mesenchymal marker fibronectin and the epithelial marker E-cadherin in MCF-10A cells with H3.3 suppression after CHAF1B knockdown for 10 days; representative images; histone levels are detected in whole cell lysate to show knockdown efficiency of H3.3 (n = 4).
(F) mRNA levels of ZEB1 and SOX9 evaluated by qPCR in LM2 cells overexpressing CHAF1B for 3 days (n = 4).
(G) H3.3 and RNA Pol II pS5 enrichment at the ZEB1 and SOX9 promoters in LM2 cells overexpressing CHAF1B for 10 days; fold enrichment was determined using IgG as a control for the chromatin immunoprecipitation (n = 4).
(H) Levels of epithelial markers E-cadherin and zona occludens 1 (ZO1) in LM2 cells overexpressing CHAF1B for 10 days; representative images (n = 4).
All values are expressed as mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001).
The parallels between the transcriptional reprogramming elicited by suppression of the CAF-1 complex and the genetic programs enriched for chromatin-bound H3.3 in metastatic cells led us to reason that reduction of CAF-1 is the connection between the global decline in canonical histone levels induced by metastatic signaling and the transcriptional reprogramming that causes acquisition of aggressive properties during tumor progression, by triggering the deposition of H3.3 onto specific regions of chromatin. It is important to note that H3.3-gap filling requires active replication of DNA (Ray-Gallet et al., 2011) and EMT-induction has long been shown to reduce proliferation (Shin et al., 2019). Our data show that although EMT-inducers slow proliferation, the cells are still actively dividing just at a slower pace (data not shown). Moreover, direct CAF-1 depletion has only mild effects on cell cycle progression in the cell lines used in this study (data not shown) thus offering the conditions necessary for H3.3 to be incorporated onto chromatin. Supporting this idea, CAF-1 suppression mimicked the histone switch observed in response to metastatic signaling: a pronounced decrease in chromatin-bound canonical histone H3.1/H3.2 and an increase in chromatin-bound H3.3 (Figure 7C). ChIP-PCR experiments corroborated this hypothesis, and we observed that upon CAF-1 suppression, H3.3 is enriched at the promoters of the EMT-inducing transcription factors ZEB1, SOX9 and SNAI1 (Figure 7D). H3.3 enrichment also correlated with their transcriptional activation, as evidenced by enrichment of the active form of RNA Pol II and increased mRNA levels of said transcription factors (Figure 7B and 7D). Moreover, knockdown of H3.3 completely prevented CAF-1 suppression-induced EMT, establishing the necessity of H3.3 for CAF-1 induced EMT (Figure 7E). Further supporting this hypothesis, CAF-1 overexpression in LM2 cells, which we have previously shown to abrogate their metastatic capability (Figure 6D), led to a repression of aggressiveness-promoting transcription factors ZEB1 and SOX9 (Figure 7F) which was correlated with a pronounced decrease in H3.3 and activated RNA Pol II bound to the promoter of these transcription factors (Figure 7G) and the re-expression of epithelial markers E-cadherin and zona occludens 1 (ZO1) (Figure 7H). Collectively, our data show that suppression of the CAF-1 complex in response to metastatic signaling triggers incorporation of H3.3 at the promoters of pro-metastatic genes, which is both necessary and sufficient for acquisition of aggressive properties in breast cancer.
HIRA mediates CAF-1 suppression-induced acquisition of aggressive properties and metastatic colonization
Having shown that H3.3 gap filling mediates the transcriptional reprogramming induced by suppression of CAF-1 during tumor progression, we sought to understand how this incorporation was regulated. Analysis of H3.3 histone chaperones in CAF-1 overexpressing LM2 cells, which have a reduced propensity to form metastatic colonies, showed that HIRA levels, but not DAXX, were decreased (Figure 8A). This was in line with our previous observation that upon induction of EMT, either by treatment with TGFβ/TNFα or expression of ERK2 D319N, HIRA levels become upregulated while DAXX levels remained largely unaltered (Figures 3B and S3A–D). Thus, suggesting that HIRA mediates the incorporation of H3.3 induced by the CAF-1 mediated decline in canonical histones. To test this hypothesis, we knocked down HIRA in cells with CAF-1 suppressed and observed that similarly to knockdown of H3.3, HIRA is necessary for the pro-EMT effects of CAF-1 (Figure 8B). This idea was further supported by knockdown of the RPA, a binding partner of HIRA that regulates HIRA-mediated H3.3 incorporation (Zhang et al., 2017), which also blocked the ability of CAF-1 suppression to induce EMT (Figure S8A). Highlighting the role of HIRA as the effector chaperone that mediates H3.3 gap filling triggered by metastatic signaling, HIRA knockdown, but not DAXX knockdown, blocked the ability of TGFβ/TNFα to induce EMT (Figures 8C and S8B). Further strengthening the role of HIRA as the effector of metastatic signaling, we observed that HIRA levels are substantially elevated in LM2 cells when compared to its less metastatic parental clone, which was correlated with high levels of EMT-inducing transcription factors ZEB1, SNAI1 and SOX9 (Figure 8D). Moreover, HIRA knockdown in LM2 cells was sufficient to suppress the expression of these aggressiveness-inducing transcription factors, promote the re-expression of the epithelial markers E-cadherin and ZO1 (Figure 8E), and their transition from a mesenchymal morphology to an epithelial morphology (Figure S8C). This data suggest that suppression of HIRA might constitute a potential effective therapeutic target for metastatic breast carcinomas. In fact, knockdown of HIRA in the LM2 cells blocked their ability to migrate and invade in transwell-based assays (Figure 8F) and their ability to form metastatic colonies when injected in the tail vein of immunocompromised mice (Figure 8G).
Figure 8. HIRA mediates the pro-metastatic effects of CAF-1 and is necessary for EMT induction and metastatic colonization.

(A) Levels of H3.3 chaperones in LM2 cells overexpressing CHAF1B for 10 days; representative images (n = 4).
(B) EMT induction determined by the protein levels of the mesenchymal marker fibronectin and the epithelial marker E-cadherin after 10 days of CHAF1B and HIRA knockdown in MCF-10A cells; representative images (n = 4).
(C) EMT induction determined by protein levels of the mesenchymal marker fibronectin and the epithelial marker E-cadherin after 5 days of treatment with TGFβ + TNFα in MCF-10A cells with HIRA knockdown; representative images (n = 4).
(D) Protein levels of HIRA and the EMT-inducing transcription factors ZEB1, SOX9 and SNAI1 in MDA-MB-231 parental versus the more metastatic LM2 clone; representative images (n = 4).
(E) Levels of epithelial markers E-cadherin and zona occludens 1 (ZO1) and EMT-inducing transcription factors ZEB1, SOX9 and SNAI1 in LM2 cells with HIRA knockdown for 10 days; representative images (n = 4).
(F) Quantification of migration and invasion of LM2 cells with HIRA knockdown for 10 days evaluated by transwell assays (left); representative images (right) (n = 3), scale bar = 1 mm.
(G) Quantification of lung metastatic lesions of LM2 cells with HIRA knockdown for 10 days (left); represented images (right) (n = 9).
All values are expressed as mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001). See also Figure S8.
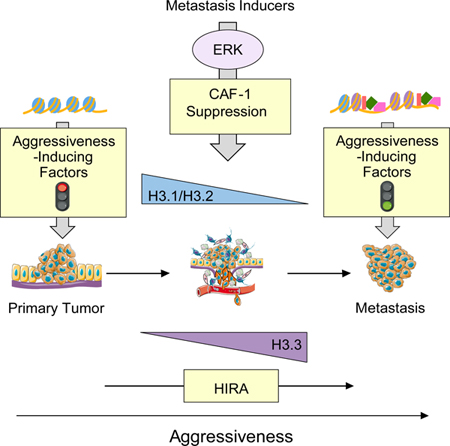
Collectively, these results show that CAF-1 is suppressed during breast cancer progression leading to a drastic reduction of canonical H3.1/H3.2 incorporation into chromatin. This functions as a signal to induce HIRA and creates the “space” for gap-filling with H3.3, leading to a HIRA-dependent H3.3 enrichment at the promoter of EMT and aggressiveness-inducing transcription factors, resulting in tumor progression and metastasis formation. These findings connect metastatic signaling to the emerging field of histone variants and demonstrate how cancer cells rely on the dynamics of histone chaperones to establish a chromatin landscape conducive to plasticity, enabling cancer progression and metastasis while promoting resistance to therapy.
Discussion
In this study we identify CAF-1 complex as an important node of signal integration and describe a cell cycle independent mechanism of regulation for the CAF-1 complex, which is hijacked in pathological conditions to promote breast cancer progression and is distinct from the previously established SRC-mediated regulation of CHAF1A (Endo et al., 2017). While additional studies will be required to pinpoint the precise kinase downstream of ERK that regulates Sp1 phosphorylation by metastatic signaling, our study demonstrates the importance of this phosphorylation event as a hub for transcriptional regulation. It will be interesting to determine if other genes that have been identified to have EGR1/Sp1 overlapping binding sites (Kubosaki et al., 2009) are also regulated by this mode of facilitated inhibition and whether they play a role in tumor progression.
One of the most surprising observations in our study was the reduction of canonical histone proteins during EMT, which was correlated with a general increase in chromatin accessibility. A tightly packaged chromatin structure reduces access to DNA and limits gene expression and genomic instability, while an open chromatin structure promotes gene expression and genomic instability (Dekker et al., 2002; Hendrich and Willard, 1995). Although unanticipated, the loss of canonical histones has been observed in other pathological conditions, such as aging, and shown to be causal (Feser et al., 2010). But what role could a global decline in histones be serving for tumor progression? Histone replacement at promoters and enhancers, for example, provides an opportunity for transient access of factors to the genome, enabling changes in transcriptional activation and repression. Histone replacement also allows effective erasing of histone modifications, rendering chromatin structure highly responsive to changes in the environment (Li, 2002; Morgan et al., 2005; Surani, 2001), a quality essential for the plasticity required for tumor progression and metastasis formation. In addition, reduction of histone levels in yeast have been shown to cause significant genomic instability (Hu et al., 2014), suggesting that a decrease in total histone levels might advance mutations of important oncogenic drivers that further promote tumor progression and metastasis. Intriguingly, the drop of total histone levels brought on by metastatic inducers is countered by a HIRA-mediated increase in chromatin-bound histone H3.3, and its presence is essential for EMT and acquisition of aggressive properties, supporting the idea that the regulation of total histone levels functions as an important transcriptional regulator. Although further work is necessary to establish their value as bona-fide therapeutic targets, this study suggests that targeting histone chaperones, particularly targeting the H3.3 chaperone HIRA, might be a much-needed effective therapy for at least metastatic breast carcinomas.
How broadly applicable are these findings? Cells have developed complex and highly regulated mechanisms to control histone replacement. Previous studies in cultured cells have long suggested that the major function of CAF-1 is to facilitate cell cycle progression by incorporating the canonical H3/H4 into chromatin during S-phase specific chromatin duplication (Quivy et al., 2008; Smith and Stillman, 1989; Ye et al., 2003). Recent studies, however, modify the notion that CAF-1 is merely a DNA replication regulator needed for cell proliferation. Suppression of CAF-1 has been recently shown to be a regulator of somatic cell identity during transcription factor induced cell fate transitions and provides a potential strategy to modulate cellular plasticity (Cheloufi et al., 2015). Along the same line, this study showed that histone replacement and particularly the CAF-1 complex are determinants of cell fate and play a key role in orchestrating cell proliferation and differentiation, a mechanism that goes awry during tumor progression and allows carcinoma cells to acquire metastatic properties.
Interestingly, CAF-1 overexpression has been shown to be pro-leukemia by promoting proliferation and preventing myeloid differentiation (Volk et al., 2018). Moreover, high levels of CAF-1 are associated with melanoma metastasis (Mascolo et al., 2010), predict adverse behavior in prostate cancer (Walden et al., 1991) and knockdown of CHAF1B was shown to reduce cell migration and tumor growth in hepatocellular carcinoma (Peng et al., 2018). These observations are in conflict with the ones presented in this study where CAF-1 suppression in breast and non-small-cell lung cancer cells was found to be pro-tumor progression. While further work will be necessary to determine the role of CAF-1 in different tumors and the pathways that differentially regulate CAF-1 levels in tumors of different origin, it is clear that CAF-1 is at the nexus of cell fate determination in cancer.
STAR★Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. John Blenis: job2064@med.cornell.edu
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
MDA-MB-231 parental and LM2 (4175) clones (female) described previously (Minn et al., 2005) were obtained from Dr. Massague’s lab, and HEK293T cells were obtained from GenHunter. All other cell lines were obtained from the American Type Culture Collection (ATCC). MCF-10A human breast epithelial cells (female) were cultured in DMEM:F12 media (Corning) supplemented with 5% horse serum (Gibco), 20 ng/mL EGF (Peprotech), 100 ng/mL cholera toxin (Sigma-Aldrich), 10 µg/mL insulin (Sigma-Aldrich), 0.5 mg/mL hydrocortisone (Sigma-Aldrich) and penicillin-streptomycin (Gibco) as previously described (Debnath et al., 2003). NMuMG normal murine mammary gland cells (female) were cultured in high glucose DMEM (Gibco) supplemented with 10% FBS (Sigma-Aldrich), 10 µg/mL insulin (Sigma-Aldrich) and penicillin-streptomycin (Gibco). The human breast cancer cell lines HCC1806, HCC38 and HCC1937 (all female) were cultured in RPMI 1640 medium (Corning) supplemented with 10% FBS (Sigma-Aldrich) and penicillin-streptomycin (Gibco). SKBR3 human breast cancer (female) and HCT116 human colon cancer (male) cell lines were cultured in modified McCoy’s 5A medium (Gibco) supplemented with 10% FBS (Sigma-Aldrich) and penicillin-streptomycin (Gibco). HEK293T (female), A549 human lung cancer (male), and MDA-MB-231 human breast cancer parental and LM2 cell lines were cultured in high glucose DMEM (Gibco) supplemented with 10% FBS (Sigma-Aldrich) and penicillin-streptomycin (Gibco). All cell lines were maintained at 37 °C and 5% CO 2. All cell lines were routinely tested for mycoplasma using the MycoAlert mycoplasma detection kit (Lonza) and were at all times mycoplasma negative. Cell lines obtained from ATCC had been authenticated by ATCC using morphology, karyotyping, and PCR based approaches.
Mice
Female nu/nu athymic mice were purchased from Envigo at the age of 4–6 weeks. Female mice were utilized since xenograft experiments were performed with breast cancer cell lines. The mice were randomly assigned to experimental groups and xenograft experiments were started 7–10 days after the mice were received. Mice were group housed (5 maximum) in conventional cages with unrestricted food and water access in a human xenograft designated area following animal biosafety level-2 procedures. The room was maintained at 21–23 °C and a 12 hours light-dark cycle. PicoLab Rodent Diet 5053 (Labdiet, Purina) containing 20% protein and 5% fat was used. Animal husbandry was carried out by Weill Cornell Medicine Belfer Research Building Vivarium technical staff. The mice were maintained and treated in compliance to Weill Cornell Medicine Institutional Animal Care and Use Committee protocols.
METHOD DETAILS
Plasmids Generation, Cloning and Mutagenesis
Human EGR1, DAXX and H3F3A open reading frame clones were obtained from the human ORFeome collection (PlasmID, Harvard Medical School) and human H-RAS G12V and GFP were obtained from Addgene (Addgene plasmid 22252 and 15301, respectively) in Gateway compatible pDONR223 or pDONR221 vectors. Human CHAF1B and HIRA cDNA (PlasmID, Harvard Medical School) were subcloned into the pDONR223 vector (kind gift from Dr. David Sinclair (Yoon et al., 2014)) through BP reaction (Life Technologies).Gateway compatible DONR vector containing rat ERK2 D319N was constructed by subcloning HA-tagged ERK2 from pBABE-HA-ERK2 (Shin et al., 2010) into the pENTR1A vector (Addgene plasmid 17398). Likewise, gateway compatible human Sp1 vector was also constructed by subcloning HA-tagged Sp1 from pCMV3-HA-Sp1 (Sino Biological) into the pENTR1A vector. Sp1 point mutants (T453A/T739A or T453E/T739E, Table S5) and CHAF1B shRNA-resistant mutant (T192A/T195C/C198T/A201G/C204A/C2017T, Table S5) were generated by site-directed mutagenesis in pENTR1A using the Quikchange II XL site directed mutagenesis kit (Agilent Technologies). These DONR vectors were then recombined into the appropriate Gateway destination vectors (pCW57.1 – Addgene plasmid 41393; pInducer20 -Addgene plasmid 44012; pLenti Blast – Addgene plasmid 17451) using LR clonase II (Life Technologies). A blasticidin resistant pLKO.1 shCHAF1B vector was made through ligating the hairpin sequence into pLKO.1-blast (Addgene plasmid 26655). The hairpin structure was generated by annealing of oligonucleotide sequences obtained from the genetic perturbation platform (GPP portal, Broad Institute) corresponding to the shCHAF1B TRCN0000074279 (target sequence: CGTCATACCAAAGCCGTCAAT). All vectors were sequence verified prior to use.
Cell Culture Treatments
To induce EMT with TGFβ and TNFα, cells were treated with 5 ng/mL of recombinant human TGF-β1 (PeproTech) and 5 ng/mL of recombinant human TNFα (PeproTech) for the indicated time period up to 10 days. During the course of these treatments the cells were split every other day. To artificially arrest cells in S-phase MCF-10A, HCC1806 and A549 cells were treated with 1 mM thymidine for 24 hours or 7 days. Cells were seeded and treated the next day; during the 7 day treatment they were split every other day. To inhibit Sp1, MCF-10A cells were plated and treated the following day with the vehicle (0.1% DMSO) or 5 µM mithramycin A (Sp1 inhibitor –Enzo Life Sciences) for 24 hours. To inhibit SRC during EMT induction, MCF-10A cells were plated and pretreated the following day with the vehicle (0.1% DMSO) or 25 nM KX2–391 (SRC inhibitor – Selleckchem) for 1 hour before adding TGFβ and TNFα for an additional 24 hours.
Stable Overexpression Cell Lines
For constitutive expression, pBabe empty and ERK2 D319N retrovirus were produced by co-transfection of HEK293T cells with plasmids encoding gagpol (Addgene plasmid 14887) and vsvg (Addgene plasmid 8454) using X-tremeGENE HP (Roche) in accordance with the manufacturer’s protocol. For constitutive expression pLenti Blast GFP and pLenti Blast HIRA; pLv105-empty (GeneCopoeia, EX-NEG-Lv105) and pLv105-CHAF1B (GeneCopoeia, EX-T0093-Lv105); pLvRFP-puro, pLvHIST1H3B-HA-puro, pLvGFP-blast and pLvH3F3A-V5-blast (Custom made vectors from Vector Builder) lentivirus were produced by co-transfection of HEK293T cells with plasmids encoding pRSV-Rev Addgene plasmid 12253), pMDLg/pRRE (Addgene plasmid 12251) and pMD2.G (Addgene plasmid 12259) using X-tremeGENE HP (Roche) in accordance with the manufacturer’s protocol. For inducible expression Inducer20 GFP, ERK2 D319N and H-RAS G12V, or pCW 57.1 GFP, CHAF1B, CHAF1B shRNA resistant mutant, EGR1, DAXX, HIRA, H3F3A, wild type Sp1, Sp1 AA and Sp1 EE lentivirus were produced by co-transfection of HEK293T cells with plasmids encoding psPAX2 (Addgene plasmid 12260), and pMD2.G (Addgene plasmid 12259) using X-tremeGENE HP (Roche) in accordance with the manufacturer’s protocol. During all virus production, media was changed 24 hours post-transfection and the virus was harvested after 48 hours and filtered. Cells were infected in the presence of 8 µg/mL polybrene (Sigma-Aldrich), and the selection of resistant colonies was initiated 24 hours later using the appropriate antibiotic marker (2 µg/mL puromycin (Sigma-Aldrich) or 300 µg/mL G418 (Sigma-Aldrich)). Cells were maintained for the time points indicated. All cells carrying an inducible transgene were maintained un-induced in culture. To induce the expression of the transgenes the cells were treated daily with 0.5 µg/mL doxycycline for the time points indicated. To test if Sp1 mediates ERK2’s effects on CHAF1B, MCF-10A cells expressing pInducer20 GFP or ERK2 D319N were infected with pCW 57.1 GFP, Sp1 wild type, Sp1 AA or Sp1 EE lentivirus as described above and selected 24 hours after the second infection using 300 µg/mL G418 and 2 µg/mL puromycin (both from Sigma-Aldrich). After selection the expression of both transgenes was induced with 1 µg/mL doxycycline for 24 hours and the cells were harvested for the appropriate assays. For the HIRA overexpression experiments in the context of CHAF1B overexpression LM2 cells were co-infected with pLv105-CHAF1B and pLenti Blast HIRA, or pLV105-empty and pLenti Blast GFP. Selection was achieved 24 hours later using 2 µg/mL puromycin and 5 µg/mL blasticidin (Sigma-Aldrich).
Constitutive Gene Silencing
pLKO.1-puro shNT (shGFP: TRCN0000072181), shCHAF1A #1 (TRCN0000074273), shCHAF1A #2 (TRCN0000234600), shCHAF1B #1 (TRCN0000074279), shCHAF1B #2 (TRCN0000074278), shDAXX #1 (TRCN0000279732), shDAXX #2 (TRCN0000279733), shSp1 #1 (TRCN0000285151), shSp1 #2 (TRCN0000274208) were obtained from Sigma, and shHIRA #1 (TRCN0000020514) and shHIRA #2 (TRCN0000020515) were obtained from OpenBiosystems. The lentiviruses were produced and cells were infected as described above for lentiviral vectors and selected with 2 µg/mL puromycin for 24 hours. The stable knockdown cells were maintained for the indicated time periods. To test the specificity of the shCHAF1B #1 construct MCF-10A cells induced with 0.5 µg/mL of doxycycline for 24 hours to express the RNAi-resistant CHAF1B mutant were infected with pLKO.1-Blast shCHAF1B #1 or pLKO.1-Blast shScramble (Addgene plasmid 26701). 24 hours after infection, the cells expressing both the shRNA and the transgene were selected with 5 µg/mL of blasticidin (Thermo Fisher) and 2 µg/mL puromycin (Sigma-Aldrich), and maintained for an additional 10 days prior to assessing EMT markers. For double knockdown of CHAF1B and HIRA, MCF-10A cells were infected with pLKO.1-Blast shCHAF1B #1, shNT #2 (pLKO.1-Blast shScramble – Addgene plasmid 26701) and pLKO.1-Puro shHIRA#1/shHIRA#2 (or the corresponding control plasmids). 24 hours after infection, the cells expressing both shRNA were selected with 5 µg/mL of blasticidin (Thermo Fisher) and 2 µg/mL puromycin (Sigma-Aldrich), and maintained for 10 days.
Inducible H3.3 Gene Silencing
To knockdown H3.3, lentiviruses targeting H3F3A, H3F3B, and Luciferase (control) were produced using doxycycline-inducible mirE-based shRNA constructs (Dow et al., 2012) obtained from the RNAi Core facility at Memorial Sloan Kettering Cancer Center (New York, NY). Antisense sequences: H3F3A #1: TTATATTAAAAATTCACACACA; H3F3A #2: TTATTATAAAGTTGAACCGCTG; H3F3B #1: TTAGTTAAGATGATGCTGGTGT; H3F3B #2: TTAAGACTGAGTTCTACACCTG. These lentiviruses were produced by co-transfection of HEK293T cells with plasmids encoding shPASCHA (a kind gift from Dr. Lukas Dow, WCM), psPAX2 (Addgene plasmid 12260), and pMD2.G (Addgene plasmid 12259) using X-tremeGENE HP (Roche) in accordance with the manufacturer’s protocol. Media was changed 24 hours post-transfection, and the virus was harvested and filtered after 48 hours. To effectively knockdown H3.3, MCF-10A cells were infected with shLuciferase, or shH3F3A and shH3F3B at a 1:1 ratio in the presence of 8 µg/mL polybrene (Sigma-Aldrich). Selection of resistant colonies was initiated 24 hours later using 2 µg/mL puromycin (Sigma-Aldrich). To test whether induction of EMT by TGFβ and TNFα is dependent on H3.3, the cells expressing shH3.3 (shH3F3A+shH3F3B) or shLuciferase for 1 day were subsequently treated with TGFβ and TNFα as described above for 5 additional days. The expression of shH3.3 and shLuciferase was maintained by replenishing doxycycline in the media every day. To test whether ERK2 D319N is dependent on H3.3 to induce EMT, the cells expressing shH3.3 or shLuciferase were subsequently infected with pInducer20 GFP or ERK2 D319N and after selection with 300 µg/mL G418 and 2 µg/mL puromycin (both from Sigma-Aldrich) were induced with 1 µg/mL of doxycycline (Sigma-Aldrich) for 10 days. To test the dependence of suppression of the CAF-1 complex on H3.3 to induce EMT, the cells expressing shH3.3 or shLuciferase for 1 day were subsequently infected with pLKO.1-Blast shCHAF1B or shScramble (Addgene plasmid 26701) and selected with 5 µg/mL of blasticidin (Thermo Fisher) and 2 µg/mL of puromycin (Sigma-Aldrich) to specifically select for cells carrying both mirE- and pLKO.1-driven shRNAs. The expression of shH3.3 and shLuciferase was maintained by replenishing doxycycline in the media every day, and the cells were maintained for 10 days after infection with pLKO.1-Blast.
Immunoblots For Total Cell Lysates
Proteins were isolated directly from intact cells via acid extraction using a 10% TCA solution (10% trichloroacetic acid, 25 mM NH4OAc, 1 mM EDTA, 10 mM Tris⋅HCl pH 8.0). Precipitated proteins were harvested and solubilized in a 0.1 M Tris⋅HCl pH 11 solution containing 3% SDS and boiled for 10–15 minutes. Protein content was determined with the DC Protein Assay kit II (BioRad), and 20 µg total protein from each sample were run on SDS-PAGE under reducing conditions. The separated proteins were electrophoretically transferred to a nitrocellulose membrane (GE Healthcare), which was blocked in TBS-based Odyssey Blocking buffer (LI-COR). Proteins of interest were probed with specific antibodies: CHAF1B (HPA021679, Sigma-Aldrich), CHAF1A (ab126625, Abcam), EGR1 (ab133695, Abcam), Sp1 pS739 (ab195733, Abcam), total Sp1 (39058, Active Motif), HA (sc-7392, Santa Cruz), V5 (V8012, Sigma-Aldrich), SRC pY416 (6943S, Cell Signaling), SRC (2109S, Cell Signaling), HP1α (ab109028, Abcam), HP1β (ab10478, Abcam), HP1γ (ab10480, Abcam), RPA1 (ab79398, Abcam), DAXX (4533, Cell Signaling), HIRA (ab129169, Abcam), ERK1/2 (9102, Cell Signaling), ERK1/2 pT202/pY204 (M9692, Sigma-Aldrich), E-cadherin (610181, BD Biosciences), ZO1 (5406S , Cell Signaling), p48 (ab79416, Abcam), fibronectin (610077, BD Biosciences), RAS (3339, Cell Signaling), actin (sc1615, Santa Cruz), vinculin (V9264, Sigma-Aldrich), H3 pS10 (12201S, Cell Signaling), histone H3 (61475, Active Motif), histone H3.1/H3.2 (ABE154, Millipore), histone H3.3 (09–838, Millipore). Membranes were incubated with the primary antibodies overnight at 4°C, and then with the appropriate horseradish peroxidase–conjugated (HRP) anti-rabbit, anti-mouse (both from GE Healthcare) or anti-goat (Millipore) immunoglobulin for 2 hours at room temperature. The signals were developed using Amersham ECL detection system (GE Healthcare).
Chromatin Extraction for DNA-bound Histones
Chromatin extracts from cell-number normalized samples were prepared with the ChromaFlash Chromatin Extraction Kit according to manufacturer’s instructions (Epigentek). The correspondent to 100 ng DNA of the chromatin extracts were run on SDS-PAGE under reducing conditions and immunoblotted (as described above) with the following antibodies: histone H3.1/H3.2 (ABE154, Millipore), histone H3.3 (09–838, Millipore), histone H3 (61475, Active Motif), histone H4 (ab10158, Abcam), histone H2A (ab18255, Abcam) and histone macro H2A.1 (ab37264, Abcam). Blots for total histone H3 were performed in the same membrane as histone H3.1/H3.2 blots after stripping of the membranes.
Acid Extraction of Histone Proteins
Cells were harvested, normalized for cell number and washed twice with ice-cold PBS. The cell pellets were resuspended in Triton Extraction Buffer (TEB: PBS containing 0.5% Triton × 100 (v/v), 2 mM phenylmethylsulfonyl fluoride (PMSF), 0.02% (w/v) NaN3) at a cell density of 107 cells per mL and lysed on a rotator at 4°C for 10 min. Nuclei were spun down at 6,500 × g for 10 min at 4°C, and washe d once with half the volume of TEB. Acid extraction of the histones was achieved in 0.2 N HCl at a density of 4×107 nuclei per mL overnight at 4°C. Samples were centri fuged at 6,500 × g for 10 min at 4°C to pellet debris. The supernatant, which contains the histone protein, was collected and neutralized with 2 M NaOH at 1/10 of the volume of the supernatant. 5 µl of each sample was resolved on a 15% polyacrylamide gel and visualized by Coomassie Blue staining using SimplyBlue SafeStain (Invitrogen) according to manufacturer’s protocol.
Mass Spectrometry Analysis
For Mass Spectrometry (MS) analysis, the acid extracted histones (as described above) were precipitated with trichloroacetic acid (TCA, 20% w/v), rinsed three times with acetone, and dried at room temperature. The pellets were re-suspended in 100 µL resuspension buffer (8 M urea, 50 mM ammonium bicarbonate, and 5 mM DTT) and subjected to reduction and alkylation. 15 mM iodoacetamide was added to each sample for 30 min in the dark at room temperature, followed by addition of 5 mM DTT to quench the reaction. Samples were diluted to a final concentration of 2 M urea and digested with LysC at room temperature overnight, and then diluted further at 1 M urea and digested with Trypsin at 37°C overnight (for each e nzyme a ratio of 1:125 enzyme:protein was used). Samples were labeled using reductive demethylation (Boersema et al., 2008). Labeling was done while the peptides were bound to the solid phase C18 resin in self-packed STAGE Tip micro-columns (Rappsilber et al., 2003). Stage tips were washed with: 1) methanol, 2) acetonitrile (ACN) 70% v/v and formic acid (FA) 1% v/v, and finally 3) FA 1% v/v. Samples were acidified by adding 100% FA to a final concentration of 2% FA before loading. After sample loading, stage tips were washed with 1% FA and phosphate/citrate buffer (0.23 M sodium phosphate and 86.4 mM citric acid [pH 5.5]). At this point, the “light” solution (0.4% CH2O and 60 mM NaBH3CN), or “heavy” solution (0.4% CD2O and 60 mM NaBD3CN) was added twice on each stage tip to label the peptides. A final wash with 1% FA was performed prior to elution with 70% ACN and 1% FA. Samples were dried under vacuum, resuspended in 5% FA, and mixed together in equal amounts for analysis using an Orbitrap Fusion Mass Spectrometer. Peptides were introduced into the mass spectrometer by nano-electrospray as they eluted off a self-packed 40 cm, 75 µm (ID) reverse-phase column packed with 1.8 µm, 120 Å pore size, SEPAX C18 resin. Peptides were separated with a gradient of 5–13-23% buffer B (99.9% ACN, 0.1% FA) with a flow rate of 350 nl/min for 85 min. For each scan cycle, one high mass resolution full MS scan was acquired in the Orbitrap mass analyzer at a resolution of 120K, AGC value of 500000, in a m/z scan range of 375–1400, max acquisition time of 100 ms and up to 20 parent ions were chosen based on their intensity for collision induced dissociation (normalized collision energy=35%) and MS/MS fragment ion scans at low mass resolution in the linear ion trap. Dynamic exclusion was enabled to exclude ions that had already been selected for MS/MS in the previous 40 s. Ions with a charge of +1 and those whose charge state could not be assigned were also excluded. All scans were collected in centroid mode. Five biological replicates for each condition were processed and analyzed.
MS2 spectra were searched using SEQUEST (version 28 revision 13) (Eng et al., 1994) against a composite database containing all Swiss-Prot reviewed human protein sequences (20,193 target sequences, downloaded from www.uniprot.org March 18, 2016) and their reversed complement, using the following parameters: a precursor mass tolerance of ±25 ppm; 1.0 Da product ion mass tolerance; tryptic digestion; up to two missed cleavages; static modifications of carbamidomethylation on cysteine (+57.0214) and dimethylation on n-termini and lysines (+28.0313); dynamic modifications of methionine oxidation (+15.9949) and heavy dimethylation on n-termini and lysines (+6.03766). Peptide spectral matches (PSMs) were filtered to 1% FDR using the target-decoy strategy (Elias and Gygi, 2007) combined with linear discriminant analysis (LDA) (Huttlin et al., 2010) using several different parameters including Xcorr, ∆Cn’, precursor mass error, observed ion charge state, and predicted solution charge state. Linear discriminant models were calculated for each LC-MS/MS run using peptide matches to forward and reversed protein sequences as positive and negative training data. PSMs within each run were sorted in descending order by discriminant score and filtered to a 1% FDR as revealed by the number of decoy sequences remaining in the data set. The data were further filtered to control protein level FDRs. Peptides were combined and assembled into proteins. Protein scores were derived from the product of all LDA peptide probabilities, sorted by rank, and filtered to 1% FDR as described for peptides. We required all peptides to have a sum of heavy and light signal-to-noise (SN) ≧ 10 and we search for unique peptides. Protein ratios for histone proteins were calculated as the log2 ratio of the total SN of all experimental sample peptide values over that for GFP control sample peptides. For those histone proteins that have control value equal to zero, we imputed a value of 5 for ratio calculations. Subsequent visualization and statistical analysis was done with Perseus and R program (Tyanova et al., 2016). The raw data can be accessed at ftp://massive.ucsd.edu/MSV000084205/.
Chemotherapeutic Drug Assays
MCF-10A cells expressing inducible H-RAS G12V were treated with 0.5 µg/mL doxycycline for 5 days before introducing lentiviral constructs for shCHAF1B. The cells were selected with 2 µg/mL puromycin for 24 hours and maintained for additional 5 days in the presence of 0.5 µg/mL doxycycline. All other cells with silenced CHAF1B were established as described above and were maintained for 10 days after infection. Cells were seeded into 96-well plates in technical triplicates and treated the next day with either vehicle control, DMSO (0.1%), or carboplatin (0–200 µM) or paclitaxel (0–7.5 nM) at various concentrations. The media containing the treatments was replaced every day for 4 days. At the end of the treatments the cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) diluted in PBS for 30 minutes. After removing the fixative solution the plates were washed with PBS and stained with 0.1% crystal violet solution for 15 minutes. The staining solution was removed and the plates washed 3 times under running water, to remove the excess stain, and allowed to dry at room temperature. To quantify the biomass, crystal violet staining was eluted with 100% methanol and the absorbance at 590 nm was measured using an Envision plate Reader (Perkin Elmer).
Gene Expression Analysis
RNA was isolated using the PureLink RNA isolation kit (Life Technologies) and contaminant DNA was digested with DNAse I (Amplification grade, Sigma-Aldrich). cDNA was synthesized using the iSCRIPT cDNA synthesis kit (BioRad) and analyzed by quantitative PCR (qPCR) using SYBR green master mix (Life Technologies) on a QuantStudio6 Real-Time PCR system (Life Technologies). Target gene expression was normalized to actin and TATA-binding protein (TBP) expression. Primer sequences are listed in Table S6.
Global Gene Expression Analysis by RNA-seq
RNA was isolated as described above. RNA quality was assessed by BioAnalyzer, and only RNAs with RIN > 9 were used. RNA-sequencing libraries were prepared using the Illumina TruSeq RNA Sample Preparation v2 Guide (Illumina Part # 15026495). Polyadenylated RNA was enriched from 1 µg total RNA. Purified mRNAs were fragmented and a first strand DNA synthesized as per the protocol. RNA was degraded and a second DNA strand was synthesized, followed by end repair, adenylation of 3’ ends and adapter ligation. Optimal number of PCR amplification cycles was determined using Kapa Library Quantification Kit (Roche, KK4824). Amplified libraries were quantified and pooled for sequencing in one lane of HiSeq2500 System (Illumina), at SR50 base pair read length. Sequencing was performed at the Epigenomics Core of Weill Cornell Medicine. RNA-seq data was aligned to the human reference genome (GRCh38/hg38) using STAR with standard input parameters. Aligned reads were filtered for reads that mapped uniquely. Transcript counts were produced using HTseq against the Ensembl reference transcriptome. Transcript counts were normalized for library size and differential expression was determined using DESeq2 (Love et al., 2014). All downstream analyses and visualization on RNA-seq data were performed on variance-stabilizing transformed data obtained from DESeq2. Gene Set Enrichment Analyses was performed using the GSEA algorithm as described (Mootha et al., 2003; Subramanian et al., 2005). Enrichment of genes was assessed using the GSEA algorithm against a gene list ranked by signal-to-noise (difference of means scaled by the standard deviation). Gene sets based on annotations from the Gene Ontology database (http://www.geneontology.org) and oncogenic signatures available within the Molecular Signatures Database (MSigDB) were used. GOseq (Young et al., 2010) was used for functional enrichment analysis of differentially expressed genes which passed the threshold of FDR <0.05 and log2 fold change > 1. Only gene sets with 20 genes or more, and with 300 genes or less were evaluated. The accession number for the raw sequencing data reported in this paper is GEO: GSE119030.
CHAF1B and CHAF1A Promoter Assays
CHAF1B and CHAF1A promoter activity was evaluated using luciferase-based CHAF1B promoter construct (SwitchGear plasmid S718588) and CHAF1A promoter construct (SwitchGear plasmid S719853). MCF-10A cells expressing GFP or ERK2 D319N for 1 day were transfected with the CHAF1B and CHAF1A promoter constructs or an empty vector to establish a blank (SwitchGear plasmid S790005) using X-tremeGENE HP (Roche) in accordance with the manufacturer’s protocol. The day after the transfection the media was replaced and 0.5 µg/mL doxycycline was maintained in the media for the expression of GFP or ERK2 D319N. Luciferase activity was measured at day 3 of GFP or ERK2 D319N expression using a LighSwitch Dual Assay kit (Active Motif) according to manufacturer’s instructions and using Cypridine TK control vector (Active Motif plasmid 32036) as reference on an Envision plate reader (PerkinElmer). The data are presented normalized to the signal of the empty vector and to cypridine to normalize for variation between transfection replicates.
DNA-Binding Assay
ERK2 D319N-mediated differential binding of EGR1 and Sp1 to the CHAF1B promoter was evaluated in vitro utilizing a biotinylated DNA fragment of the CHAF1B promoter containing the overlapping Sp1 and EGR1 binding sites (Biotin-5’-TGGCGTCTATGAGTGGGCGGGGCTTC-CCTGGGGTAC) and using a biotinylated DNA fragment of the CHAF1B promoter without Sp1 and/or EGR1 consensus binding sequences (Biotin-5’-AAAAATCCTAGAGCAGTTGGCTCC-TCAGATAACCCT) as a control. Whole cell lysates from MCF-10A cells expressing ERK2 D319N or GFP for 3 days were prepared using a low stringency IP buffer (0.05% NP-40, 50 mM NaCl, 0.5 mM EDTA, 50 mM Tris HCl, pH 7.4) supplemented with EDTA-free protease inhibitor and phosphatase inhibitor cocktails (Roche) and 100 U of microccocal nuclease (Thermo Fisher) per mL of lysate. 10 µg of each double-stranded DNA fragment was incubated with 1 mg of whole cell lysate in binding buffer (0.5 % Triton X-100, 150 mM NaCl, 5 µM ZnCl2, 10 mM Tris⋅HCl, pH 7) supplemented with EDTA-free protease inhibitor and phosphatase inhibitor cocktails (Roche) overnight at 4°C. Following binding, DNA fragments were immunoprecipitated using 25 µL of streptavidin-agarose beads (Sigma-Aldrich) for 3 hours at 4 °C using 1 m g of the same cell lysate without incubation with the biotinylated DNA fragment as IgG control. Immunoprecipitated material was washed ten times with binding buffer (5 minutes incubation per wash), after which it was eluted in SDS-PAGE buffer supplemented with 50 mM DTT and boiled for 10 minutes. Input proteins, proteins bound to the DNA fragments, and IgG control were run on SDS-PAGE and immunoblotted (as described above) with the following primary antibodies, anti-Sp1 (39058, Active Motif), anti-EGR1 (ab133695, Abcam) and anti-HA (sc-7392, Santa Cruz). To evaluate the effect of Sp1 phosphorylation on Sp1 binding to the CHAF1B promoter, the same protocol was followed with whole cell lysates from MCF-10A cells expressing ERK2 D319N and the WT Sp1, Sp1 AA mutant, Sp1 EE mutant or GFP as a control for 24 hours.
ATAC-Seq
MCF-10A cells carrying the inducible GFP or ERK2 D319N transgenes were treated daily with 0.5 µg/mL doxycycline for 3 days. Healthy adherent cells were trypsinized, normalized according to cell numbers and frozen in growth media containing serum and 5% DMSO. Cryopreserved cells were sent to Active Motif to perform the ATAC-seq assay. The cells were then thawed in a 37°C water b ath, pelleted, washed with cold PBS, and tagmented as previously described (Buenrostro et al., 2013), with some modifications based on (Corces et al., 2017). Briefly, cell pellets were resuspended in lysis buffer, pelleted, and tagmented using the enzyme and buffer provided in the Nextera Library Prep Kit (Illumina). Tagmented DNA was then purified using the MinElute PCR purification kit (Qiagen), amplified with 10 cycles of PCR, and purified using Agencourt AMPure SPRI beads (Beckman Coulter). Resulting material was quantified using the KAPA Library Quantification Kit for Illumina platforms (KAPA Biosystems), and sequenced with PE42 sequencing on the NextSeq 500 sequencer (Illumina).
Reads were aligned to the human genome (hg38) using the BWA algorithm (Li and Durbin, 2009) (mem mode; default settings). Duplicate reads were removed, only reads mapping as matched pairs and only uniquely mapped reads (mapping quality >= 1) were used for further analysis. Alignments were extended in silico at their 3’-ends to a length of 200 bp and assigned to 32-nt bins along the genome. The resulting histograms (genomic “signal maps”) were stored in bigWig files. Peaks were identified using the MACS 2.1.0 algorithm (Zhang et al., 2008) at a cutoff of p value 1e-7, without control file, and with the –nomodel option. Peaks that were on the ENCODE blacklist of known false ChIP-Seq peaks were removed. Signal maps and peak locations were used as input data to Active Motifs proprietary analysis program, which creates Excel tables containing detailed information on sample comparison, peak metrics, peak locations and gene annotations. Signal tracks were visualized using Integrated Genome Browser (IGB) (Freese et al., 2016). The accession number for the raw sequencing data reported in this paper is GEO: GSE132046.
ChIP-seq in MDA-MB-231 LM2 cells
Cells were fixed with 1% formaldehyde for 15 min and quenched with 0.125 M glycine. Cell pellets were sent to Active Motif for further processing for ChIP-seq analysis and ChIP-qPCR. Chromatin was isolated by the addition of lysis buffer, followed by disruption with a Dounce homogenizer. Lysates were sonicated and the DNA sheared to an average length of 300–500 bp. Genomic DNA (Input) was prepared by treating aliquots of chromatin with RNase, proteinase K and heat for de-crosslinking, followed by ethanol precipitation. Pellets were resuspended and the resulting DNA was quantified on a NanoDrop spectrophotometer. Extrapolation to the original chromatin volume allowed quantitation of the total chromatin yield. An aliquot of chromatin (30 µg) was precleared with protein A (for H3.3) or G (for H3.1/3.2) agarose beads (Invitrogen). Genomic DNA regions of interest were isolated using 4 µg of antibody against H3.1/3.2 (61629, Active Motif, Lot#: 31814001) or 6 µg of antibody against H3.3 (17–10245, Millipore, Lot#: 2914462). Complexes were washed, eluted from the beads with SDS buffer, and subjected to RNase and proteinase K treatment. Crosslinks were reversed by incubation overnight at 65°C, and ChIP DNA was p urified by phenol-chloroform extraction and ethanol precipitation. Illumina sequencing libraries were prepared from the ChIP and Input DNAs by the standard consecutive enzymatic steps of end-polishing, dA-addition, and adaptor ligation. After a final PCR amplification step, the resulting DNA libraries were quantified and sequenced on Illumina’s NextSeq 500 (75 nt reads, single end). Reads were aligned to the human genome (hg38) using the BWA algorithm (Li and Durbin, 2009) (default settings). Duplicate reads were removed and only uniquely mapped reads (mapping quality >= 25) were used for further analysis. Alignments were extended in silico at their 3’-ends to a length of 200 bp, which is the average genomic fragment length in the size-selected library, and assigned to 32-nt bins along the genome. The resulting histograms (genomic “signal maps”) were stored in bigWig files. H3 enriched regions were identified using the SICER algorithm (Li and Durbin, 2009) at a cutoff of FDR 1×10^(−10) and a max gap parameter of 600 bp (for some analyses 0 bp). Alternatively, to identify promoter peak regions, the MACS algorithm (Zhang et al., 2008) (v1.4.2) with a cutoff of p value = 1×10^(−7) was used, followed by intersecting peak regions with promoter regions (−1000 bp to 0 relative to TSS). Peaks that were on the ENCODE blacklist of known false ChIP-Seq peaks were removed. Signal maps and peak locations were used as input data to Active Motif’s proprietary analysis program, which creates Excel tables containing detailed information on sample comparison, peak metrics, peak locations and gene annotations. Signal tracks were visualized using Integrated Genome Browser (IGB) (Freese et al., 2016). A list of genes that are enriched for either H3.1/H3.2 or H3.3 (fold difference of at least 2, with maximum tag numbers above 75) were analyzed for gene set enrichment using GSEA software (Mootha et al., 2003; Subramanian et al., 2005). “Classic” enrichment statistics and “ratio of classes” metrics were used for analysis. Number of permutations were set to 1000 as suggested, and “gene set” was used as permutation type (1533865063713 was used as the seed for permutations). Gene Ontology (GO)-Biological Processes (BP) gene set database was utilized. The accession number for the raw sequencing data reported in this paper is GEO: GSE120313.
ChIP-seq validation in MDA-MB-231 LM2 cells
qPCR reactions were carried on ChIP DNA (prepared as described above) in triplicate on specific genomic regions using SYBR Green Supermix (Bio-Rad). The resulting signals were normalized for primer efficiency by carrying out qPCR for each primer pair using Input DNA. The primer sets are listed in Table S7. To further validate ChIP-seq results chromatin immunoprecipitation in LM2 cells expressing a HA-tagged version of H3.1, an V5-tagged version of H3.3, or the control constructs was performed using a commercially available kit (ChIP-IT Express Enzymatic Shearing Kit, Active Motif) according to the manufacturer’s instructions. Chromatin was sheared enzymatically for 10 minutes. Immunoprecipitation was performed using anti-V5 Magnetic Beads (MBL International) or anti-HA Magnetic Beads (Thermo Scientific Pierce). DNA was purified with a Chromatin IP DNA purification kit (Active Motif) and qPCR was performed with SYBR Green master mix on a Step One Real-Time PCR system (all Life Technologies) using the primer sets listed in Table S7.
ChIP-PCR in MCF-10A and MDA-MB-231 LM2 cells
Chromatin immunoprecipitation was performed using a commercially available kit (ChIP-IT Express, Active Motif) according to the manufacturer’s instructions. For MCF-10A cells chromatin was sheared using a Bioruptor sonicator (Diagenode) until DNA was an average size of 600 base pairs, while for LM2 cells chromatin was sheared enzymatically for 10 minutes (ChIP-IT Express Enzymatic Shearing Kit, Active Motif). For both cells Dynabeads (1:1 protein A to protein G; Life Technologies) were used instead of the magnetic beads from the kit. Immunoprecipitation was performed using anti-H3.3 (17–10245, Millipore), anti-RNA pol II pS5 (ab5131, Abcam) or anti-Rabbit IgG (sc-2027, Santa Cruz) antibodies. DNA was purified with a Chromatin IP DNA purification kit (Active Motif) and qPCR was performed with SYBR Green master mix on a Step One Real-Time PCR system (all Life Technologies) using the primer sets for ZEB1, SNAI1 and SOX9 listed in Table S7.
Analysis of CD24 and CD44
Cells were dissociated using Cell Stripper (Corning), collected on ice and pelleted by centrifugation. After removing the Cell Stripper and washing the cell pellet with ice cold PBS, the cells were stained on ice for 30 minutes in 100 µL DMEM/F12 (without phenol red) with an APC mouse anti-human CD44 (559942, BD Biosciences) and FITC mouse anti-human CD24 (555427, BD Biosciences) or an APC mouse IgG2b (555745, BD Biosciences) and FITC mouse IgG2a (553456, BD Biosciences) as isotype controls. After labeling, each sample was washed twice with ice cold PBS and resolved on BD Accuri B6 flow cytometer (BD Biosciences). Data analysis to determine the medium fluorescence intensity (MFI) of CD24 and CD44 positive cells was performed using the FlowJo software package.
Transwell Migration and Invasion Assays
MCF-10A cells were trypsinized and collected as previously described (Shin et al., 2010). Resuspension media was aspirated and cells were resuspended in assay media (DMEM/F12 (Corning), 0.5% Horse Serum (Gibco), 500 ng/mL hydrocortisone (Sigma-Aldrich), 100 ng/mL cholera toxin (Sigma-Aldrich). For migration assays, Boyden chamber inserts (BD Biosciences, 8 µm pore size) were pre-coated with 25 µg/µl rat tail collagen 1 (Corning). Assay media supplemented with 5 ng/mL EGF (Peprotech) was added to the bottom chamber of the cell culture inserts. Cells (5 × 104 cells/ 250 µL assay media) were then added to the top chamber of cell culture inserts in a 24-well companion plate. After 6 hours of incubation, the cells that had migrated to the lower surface of the membrane were fixed with ethanol and stained with 0.2% crystal violet in 2% ethanol. For cell invasion assays, BD BioCoat invasion chambers coated with growth factor reduced Matrigel were used. Invasion chambers were prepared according to manufacturer’s specifications and assays were performed as described for migration assays except that 20 ng/mL of EGF (Peprotech) was added to MCF-10A assay media to serve as the chemo-attractant and cells were allowed to invade for 24 hours.
For MDA-MB-231 cells, transwell migration and invasion assays were performed as described above with minor changes. For MDA-MB-231 cells, high-glucose DMEM (Gibco) supplemented with 250 µg/mL BSA (Sigma-Aldrich) was used as the assay media, and high glucose DMEM media supplemented with 10% FBS (Sigma-Aldrich) was used as the chemoattractant for both migration and invasion assays.
Images of crystal violet stained cells were captured using a Nikon DS-Fi2 camera, and quantification were carried out in an automated way using Fiji/ImageJ (Rueden et al., 2017; Schindelin et al., 2012). Briefly, binary images of the area covered by crystal violet-positive cells was generated using thresholding and settings that were appropriate for control samples, and these settings were used throughout the analysis. The percentage area covered by crystal violet-positive cells was quantified for each condition, using a minimum of three technical replicates.
Immunohistochemistry
Immunohistochemistry (IHC) analysis of primary and matched metastatic breast carcinoma tissue array (BR10010e from US Biomax) was performed by US Biomax. Briefly, slides were stained on a Ventana Discovery Ultra automated system, using the manufacturer’s protocol with proprietary reagents. Heat-induced antigen retrieval (HIER) was performed with CC1, pH 8.5 (Ventana) for 56 minutes at 95 °C. Peroxidase inhibitor was applied. The primary antibody (CHAF1B-HPA021679, Sigma-Aldrich at 1:50) was incubated at 37 °C for 28 minutes. The secondary an tibody used was Discovery anti-Rabbit HQ and anti-HQ HRP (Ventana), incubated at 37 °C for 8 minutes each. Tyramide signal amplification was applied using Discovery Amp HQ kit (Ventana), for 8 minutes. DAB chromagen was applied using Discovery ChromoMap DAB Kit (Ventana). Slides were then counterstained with Hematoxylin II (Ventana). Images were quantified using Visiopharm, an automated quantification software. Nuclei were scored as Negative, 1+, 2+, and 3+ using the algorithm for ER/PR scoring. For each sample an H-score (Sum of percentages of 1+ 2+ 3+ nuclei weighted by a factor of 1, 2, and 3 respectively) was calculated.
Lung Colonization in Mice
Luciferase-expressing MDA-MB-231 parental cells (less metastatic) and LM2 cells (more metastatic) (Minn et al., 2005) were utilized for these experiments. The parental cells were infected with shCHAF1 #1/#2 to silence the CAF-1 complex; LM2 cells were infected with pLv105-CHAF1B for overexpression of CAF-1 complex or with shHIRA #1/#2 to silence HIRA. For these experiments empty pLKO.1 (Er et al., 2018) rather than shGFP-pLKO.1 construct was used as a control to avoid interference with the luciferase construct present in the MDA-MB-231 parental cells (Minn et al., 2005). Cells were maintained in culture for 7 days after lentiviral transduction. Lung colonization was evaluated as described before (Minn et al., 2005; Oskarsson et al., 2011). Briefly, female nu/nu athymic mice were injected with 100,000 cells in 100 µL PBS through tail vein injections. Metastases were monitored using IVIS Spectrum CT Pre-Clinical In Vivo Imaging System (Perkin-Elmer). 7–10 mice were used in each experimental group. After 6 weeks, luminescence was measured and quantified using the Living Image Software (Perkin-Elmer) to determine lung colonization.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analyses were performed using Microsoft Excel and GraphPad Prism7. A two-tailed paired Student’s t test was used to determine significance when two conditions were compared; for experiments with more than two conditions a one-way ANOVA was used to determine significance. In both types of statistical analysis values of p < 0.05 were considered significant. Data are represented as the mean ± SEM (standard error of the mean) of at least three independent experiments performed. Number of replicates and animals are reported in the figure legends. For all experiments similar variances between groups were observed. Normal distribution of samples was not determined. In the GSEA analyses for the ChIP-seq and RNA-seq experiments FDR corrected p values are used to determine significance.
DATA AND SOFTWARE AVAILABILITY
The accession number for the raw sequencing data for the RNA-seq is GEO: GSE119030, the ATAC-seq is GEO: GSE132046, and the ChIP-seq is GEO: GSE120313. The raw data for the mass spectrometry analysis can be accessed at ftp://massive.ucsd.edu/MSV000084205/.
Supplementary Material
Table S1. ATAC-seq peak distributions along chromosomes (Related to Figure 1)
Table S2. ChIP-seq analysis summary (Related to Figure 2)
Table S3. Breast cancer tissue array information and scoring (Related to Figure 6)
Table S4. RNA-seq analysis summary (Related to Figure 7)
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CHAF1B | Sigma-Aldrich | HPA021679 |
| CHAF1A | Abcam | ab126625 |
| EGR1 | Abcam | ab133695 |
| Sp1 pS739 | Abcam | ab195733 |
| Sp1 | Active Motif | 39058 |
| HA | Santa Cruz | sc-7392 |
| V5 | Sigma-Aldrich | V8012 |
| SRC pY416 | Cell Signaling | 6943S |
| SRC | Cell Signaling | 2109S |
| HP1α | Abcam | ab109028 |
| HP1β | Abcam | ab10478 |
| HP1γ | Abcam | ab10480 |
| RPA1 | Abcam | ab79398 |
| DAXX | Cell Signaling | 4533S |
| HIRA | Abcam | ab129169 |
| ERK1/2 | Cell Signaling | 9102L |
| ERK1/2 pT202/pY204 | Sigma-Aldrich | M9692 |
| E-cadherin | BD Biosciences | 610181 |
| ZO1 | Cell Signaling | 5406S |
| p48 | Abcam | ab79416 |
| Fibronectin | BD Biosciences | 610077 |
| RAS | Cell Signaling | 3339S |
| Actin | Santa Cruz | sc-1615 |
| Vinculin | Sigma-Aldrich | V9264 |
| Histone H3 pS10 | Cell Signaling | 12201S |
| Histone H3 | Active Motif | 61475 |
| Histone H3.1/H3.2 | Millipore | ABE154 |
| Histone H3.3 | Millipore | 09–838 |
| Histone H4 | Abcam | ab10158 |
| Histone H2A | Abcam | ab18255 |
| Histone macro H2A.1 | Abcam | ab37264 |
| anti-Rabit HRP secondary | GE Healthcare | NA934 |
| anti-Mouse HRP secondary | GE Healthcare | NA931 |
| anti-Goat HRP secondary | Millipore | AP180P |
| Histone H3.1/H3.2 (for ChIP-seq) | Active Motif | 61629 |
| Histone H3.3 (for ChIP-seq) | Millipore | 17–10245 |
| RNA Pol II pS5 | Abcam | ab5131 |
| Rabbit IgG | Santa Cruz | sc-2027 |
| CD44 | BD Biosciences | 559942 |
| CD24 | BD Biosciences | 555427 |
| APC mouse IgG2b | BD Biosciences | 555745 |
| FITC mouse IgG2a | BD Biosciences | 553456 |
| Discovery anti-Rabbit HQ | Ventana | 760–4815 |
| Anti-V5 Magnetic beads | MBL International | M215–11 |
| Anti-HA Magnetic beads | Pierce Thermo Scientific | 88836 |
| Biological Samples | ||
| Breast cancer and matched metastatic carcinoma tissue array | Biomax | BR10010e |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Human EGF | Peprotech | AF-100–15 |
| Cholera toxin | Sigma-Aldrich | C8052 |
| Insulin | Sigma-Aldrich | I9278 |
| Hydrocortizone | Sigma-Aldrich | H4001 |
| Recombinant Human TGFβ | Peprotech | 100–21 |
| Recombinant Human TNFα | Peprotech | 300–01A |
| Tymidine | Hydrocortizone | Sigma-Aldrich |
| Mithramycin A | Enzo Life Sciences | BML-GR305–0001 |
| KX2–391 | Selleckchem | S2700 |
| Polybrene | Sigma-Aldrich | H9268 |
| Puromycin | Sigma-Aldrich | P7255 |
| G418 Disulfate | Caisson Labs | G030 |
| Blasticidin S HCl | Thermo Fisher | A11139–03 |
| Doxycycline | Millipore | 324385 |
| Carboplatin | Sigma-Aldrich | C2538 |
| Paclitaxel | Sigma-Aldrich | T7191 |
| 16% Paraformaldehyde | Electron Microscopy Sciences | 15710 |
| Crystal Violet | Sigma-Aldrich | C3886 |
| Trypsin | Promega | V511C |
| Lysyl Endopeptidase | Wako | 129–02541 |
| Formalin-D2 | CDN isotopes | D-5933 |
| Formaldehyde solution | Sigma-Aldrich | 252549 |
| Sodium cyanoborohydride | Sigma-Aldrich | 296945 |
| Sodium cyanoborodeuteride | Cambridge Isotope Laboratories Inc | DLM-7364–1 |
| Micrococcal nuclease | Thermo Fisher | 88216 |
| Complete EDTA-free protease inhibitor cocktail | Roche | 5056489001 |
| PhosSTOP phosphatase inhibitor cocktail | Roche | 04906837001 |
| Rat Tail Collagen I | Corning | 354236 |
| BSA | P212121 | CI-00535 |
| Hematoxylin II | Ventana (Roche) | 790–2208 |
| Critical Commercial Assays | ||
| SimplyBlue SafeStain | Invitrogen | LC6065 |
| MycoAlert mycoplasma detection kit | Lonza | LT07–318 |
| X-tremeGENE HP DNA Transfection Reagent | Roche | 06366546001 |
| DC protein Assay kit II | Biorad | 5000112 |
| TBS-based Odyssey blocking buffer | Licor | 927–50150 |
| Amersham ECL detection kit | GE Healthcare | RPN2134 |
| ChromaFlash Chromatin Extraction kit | Epigentek | P-2001–100 |
| PureLink RNA mini kit | Life Technologies | 12183018A |
| QuikChange II XL site directed mutagenesis kit | Agilent Technologies | 200521 |
| iScript cDNA synthesis kit | Bio-Rad | 170–8891BUN |
| SYBR green master mix | Life Technologies | 4312704 |
| TruSeq RNA Sample Prep Kit v2 | Illumina | 15026495 |
| Kapa Library Quantification Kit | Roche | KK4824 |
| LightSwitch Dual Assay kit | Active Motif | 32035 |
| Nextera Library Prep Kit | Illumina | FC-141–1007 |
| MinElute PCR purification kit | Qiagen | 28006 |
| SYBR Green Supermix | Biorad | 1725120 |
| ChIP-IT Express Kit | Active Motif | 53008 |
| ChIP-IT Express Enzymatic Shearing Kit | Active Motif | 53035 |
| Chromatin IP DNA purification kit | Active Motif | 58002 |
| Discovery Amp HQ kit | Ventana (Roche) | 760–052 |
| Discovery ChromoMap DAB kit | Ventana (Roche) | 760–159 |
| LR Clonase II Enzyme Mix | Life Technologies | 11791–100 |
| BP Clonase II Enzyme Mix | Life Technologies | 11789–100 |
| Amplification grade DNase I kit | Sigma-Aldrich | AMPD1–1KT |
| Deposited Data | ||
| ATAC-seq raw data | This paper | GEO: GSE132046 |
| ChIP-seq raw data | This paper | GEO: GSE120313 |
| RNA-seq raw data | This paper | GEO: GSE119030 |
| Mass Spectrometry raw data | This paper | ftp://massive.ucsd.edu/MSV000084205/ |
| Experimental Models: Cell Lines | ||
| MDA-MB-231 parental | (Minn et al., 2005) | parental |
| MDA-MB-231 LM2 | (Minn et al., 2005) | 4175 |
| HEK293T | GenHunter | Q401 |
| MCF-10A | ATCC | CRL-10317 |
| NMuMG | ATCC | CRL-1636 |
| HCC1806 | ATCC | CRL-2335 |
| HCC38 | ATCC | CRL-2314 |
| HCC1937 | ATCC | CRL-2336 |
| SKBR3 | ATCC | HTB-30 |
| HCC116 | ATCC | CCL-247 |
| A549 | ATCC | CCL-185 |
| Experimental Models: Organisms/Strains | ||
| Female nu/nu athymic mice (Hsd:Athymic Nude-Foxn1nu) | Envigo | 069 |
| Oligonucleotides | ||
| Primers Used for Mutagenesis | This paper | See Table S5 |
| Primers Used for Gene Expression qPCR Analysis | This paper | See Table S6 |
| Primers Used for ChIP-qPCR Analysis | This paper | See Table S7 |
| biotinylated DNA fragment of the CHAF1B promoter containing the overlapping Sp1 and EGR1 binding sites (Biotin-5’- TGGCGTCTATGAGTGGGCGGGGCTTC-CCTGGGGTAC) | This paper | N/A |
| biotinylated DNA fragment of the CHAF1B promoter without Sp1 and/or EGR1 consensus binding sequences (Biotin-5’- AAAAATCCTAGAGCAGTTGGCTCC-TCAGATAACCCT) | This paper | N/A |
| Recombinant DNA | ||
| pENTR1A | Addgene | 17398 |
| pCW57.1 | Addgene | 41393 |
| pInducer20 | Addgene | 44012 |
| pLenti Blast | Addgene | 17451 |
| pLKO.1-blast | Addgene | 26655 |
| pLv105-empty | GeneCopoeia | EX-NEG-Lv105 |
| pLvRFP-puro | Vector Builder | This paper |
| pLvGFP-blast | Vector Builder | This paper |
| gagpol | Addgene | 14887 |
| Vsvg | Addgene | 8454 |
| pRSV-Rev | Addgene | 12253 |
| pMDLg/pRRE | Addgene | 12251 |
| pMD2.G | Addgene | 12259 |
| psPax2 | Addgene | 12260 |
| shPASCHA construct | kind gift from Dr. Lukas Dow, WCM | (Dow et al., 2012) |
| pDONR223 | kind gift from Dr. David Sinclair, HMS | (Yoon et al., 2014) |
| pENTRY-GFP | Addgene | 15301 |
| pLenti Blast GFP | This paper | N/A |
| pInducer20 GFP | This paper | N/A |
| pCW57.1 GFP | This paper | N/A |
| pOTB7 CHAF1B | PlasmID HMS | HsCD00330561 |
| pLv105-CHAF1B | GeneCopoeia | EX-T0093-Lv105 |
| pDONR223-CHAF1B | This paper | N/A |
| pCW57.1-CHAF1B | This paper | N/A |
| pENTR1A-CHAF1B T192A/T95C/C198T/A201G/C204A/C207T | This paper | N/A |
| pCW57.1-CHAF1B T192A/T95C/C198T/A201G/C204A/C207T | This paper | N/A |
| pENTR223-EGR1 | PlasmID HMS | HsCD00376208 |
| pCW57.1-EGR1 | This paper | N/A |
| pENTR223-DAXX | PlasmID HMS | HsCD00365258 |
| pCW57.1-DAXX | This paper | N/A |
| pCMV Sport6 HIRA closed | PlasmID HMS | HsCD00337796 |
| pDONR223-HIRA | This paper | N/A |
| pLenti Blast HIRA | This paper | N/A |
| pCW57.1-HIRA | This paper | N/A |
| pDONR221-H3F3A | PlasmID HMS | HsCD00044667 |
| pCW57.1-H3F3A | This paper | N/A |
| pLvH3F3A-V5-blast | Vector Builder | This paper |
| pLvH3F3B-HA-puro | Vector Builder | This paper |
| pENTR1A-H-RAS G12V | Addgene | 22252 |
| pInducer20-H-RAS G12V | This paper | N/A |
| pBABE empty | (Shin et al., 2010) | N/A |
| pBABE-HA-ERK2-D319N | (Shin et al., 2010) | N/A |
| pENTR1A-HA-ERK2-D319N | This paper | N/A |
| pInducer20-HA-ERK2-D319N | This paper | N/A |
| pCMV3-HA-Sp1 | Sino Biologicals | HG12024-NY |
| pENTR1A-HA-Sp1 | This paper | N/A |
| pENTR1A-HA-Sp1 T453A/T739A | This paper | N/A |
| pENTR1A-HA-Sp1 T453E/T739E | This paper | N/A |
| pCW57.1-HA-Sp1 | This paper | N/A |
| pCW57.1-HA-Sp1 T453A/T739A | This paper | N/A |
| pCW57.1-HA-Sp1 T453E/T739E | This paper | N/A |
| empty promoter construct | SwitchGear | S790005 |
| CHAF1B promoter construct | SwitchGear | S718588 |
| CHAF1A promoter construct | SwitchGear | S719853 |
| Cypridine TK control vector | Active Motif | 32036 |
| pLKO.1-Blast shScramble | Addgene | 26701 |
| pLKO.1-puro empty | Kind gift from Dr. Joan Massagué | (Er et al., 2018) |
| pLKO.1-puro shGFP | Sigma-Aldrich | TRCN0000072181 |
| pLKO.1-puro shCHAF1A #1 | Sigma-Aldrich | TRCN0000074273 |
| pLKO.1-puro shCHAF1A #2 | Sigma-Aldrich | TRCN0000234600 |
| pLKO.1-puro shCHAF1B #1 | Sigma-Aldrich | TRCN0000074279 |
| pLKO.1-Blast shCHAF1B #1 | This paper | N/A |
| pLKO.1-puro shCHAF1B #2 | Sigma-Aldrich | TRCN0000074278 |
| pLKO.1-puro shDAXX #1 | Sigma-Aldrich | TRCN0000279732 |
| pLKO.1-puro shDAXX #2 | Sigma-Aldrich | TRCN0000279733 |
| pLKO.1-puro shSp1 #1 | Sigma-Aldrich | TRCN0000285151 |
| pLKO.1-puro shSp1 #2 | Sigma-Aldrich | TRCN0000274208 |
| pLKO.1-puro shHIRA #1 | Open Biosytems | TRCN0000020514 |
| pLKO.1-puro shHIRA #2 | Open Biosytems | TRCN0000020515 |
| dox-inducible mirE-based shH3F3A #1 | RNAi Core facility at MSKCC (Dow et al., 2012) | This paper |
| dox-inducible mirE-based shH3F3A #2 | RNAi Core facility at MSKCC (Dow et al., 2012) | This paper |
| dox-inducible mirE-based shH3F3B #1 | RNAi Core facility at MSKCC (Dow et al., 2012) | This paper |
| dox-inducible mirE-based shH3F3B #2 | RNAi Core facility at MSKCC (Dow et al., 2012) | This paper |
| dox-inducible mirE-based shH3F3A #5 | RNAi Core facility at MSKCC (Dow et al., 2012) | This paper |
| dox-inducible mirE-based shLuciferase | RNAi Core facility at MSKCC (Dow et al., 2012) | This paper |
| Software and Algorithms | ||
| DESeq2 | (Love et al., 2014) | N/A |
| GSEA | (Mootha et al., 2003; Subramanian et al., 2005) | N/A |
| GOseq | (Young et al., 2010) | N/A |
| BWA | (Li and Durbin, 2009) | N/A |
| MACS 2.1.0 | (Zhang et al., 2008) | N/A |
| IGB | (Freese et al., 2016) | N/A |
| SICER | (Li and Durbin, 2009) | N/A |
| SEQUEST v28 revision 13 | (Eng et al., 1994) | N/A |
| Perseus | (Tyanova et al., 2016) | N/A |
| R | The R Foundation | N/A |
| FlowJo v10 | FlowJo LLC | N/A |
| Fiji/Image J | (Rueden et al., 2017; Schindelin et al., 2012) | N/A |
| Visiopharm Image Analysis Apps | Visiopharm | N/A |
| Living Image Software | Perkin-Elmer | N/A |
| Excel 2013 | Microsoft | N/A |
| Prism8 | GraphPad | N/A |
| QuantStudio Real-Time PCR Software v1.3 | Thermo Fisher | N/A |
| Other | ||
| Streptavidin-agarose beads | Sigma-Aldrich | S1638 |
| Nitrocellulose Membrane | GE Healthcare | 10600001/2 |
| Agencourt AMPure SPRI beads | Beckman Coulter | A63882 |
| Protein A magnetic beads (Dynabeads) | Invitrogen | 10002D |
| Protein G magnetic beads (Dynabeads) | Invitrogen | 10004D |
| Cellstripper Non-enzymatic dissociation solution | Corning | 25–056-CI |
| Growth Factor Reduced BioCoat invasion chambers | Corning | 354483 |
| Boyden Chamber inserts | Corning | 353097 |
| Fetal Bovine Serum | Sigma-Aldrich | 12303C |
| Horse Serum | Life Technologies | 16050–122 |
| PicoLab Rodent Diet | Labdiet Purina | 5053 |
Significance.
Chromatin remodeling is the first line of cellular reprograming that enables complex cell fate decisions. Despite this fact, the epigenetic changes that underlie metastasis formation remain largely unknown. Here we show that incorporation of histone H3.3 into chromatin is essential for tumor progression and metastatic colonization by eliciting an aggressive transcriptional reprogramming. We identified histone chaperones as the point of regulation of histone H3 replacement with H3.3 by metastatic signaling, thus pointing to histone chaperones as much needed therapeutic targets for invasive carcinomas.
Highlights.
Metastasis inducers lead to a decline in CAF-1 suppressing canonical H3 incorporation
EMT and metastatic colonization occur as a function of CAF-1 levels
Histone H3.3 variant is essential for tumor progression and aggressive phenotypes
HIRA-mediated H3.3 gap filling induces a pro-metastatic transcriptional reprogramming
Acknowledgments
We are grateful to the Blenis Lab members for input on this project, which was supported by NIH grants R01 CA46595 and R03 CA212562 to J.B. A.P.G. is supported by a Susan G. Komen Postdoctoral Fellowship and a Pathway to Independence Award from NCI (K99CA218686–01). T.S. is supported by the NIH F31 pre-doctoral fellowship 1F31CA220750–01.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
J.M. is a scientific advisor and owns company stock in Scholar Rock. A.M.M. receives research funding from Janssen and has consulted for Epizyme and Constellation. L.C.C. owns equity in, receives compensation from, and serves on the Board of Directors and Scientific Advisory Board of Agios Pharmaceuticals and Petra Pharma Corporation. No potential conflicts of interest were disclosed by the other authors.
References
- Arwert EN, Hoste E, and Watt FM (2012). Epithelial stem cells, wound healing and cancer. Nat Rev Cancer 12, 170–180. [DOI] [PubMed] [Google Scholar]
- Asiedu MK, Ingle JN, Behrens MD, Radisky DC, and Knutson KL (2011). TGFbeta/TNF(alpha)-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res 71, 4707–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F (2006). TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev 25, 409–416. [DOI] [PubMed] [Google Scholar]
- Bedi U, Mishra VK, Wasilewski D, Scheel C, and Johnsen SA (2014). Epigenetic plasticity: a central regulator of epithelial-to-mesenchymal transition in cancer. Oncotarget 5, 2016–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersema PJ, Aye TT, van Veen TA, Heck AJ, and Mohammed S (2008). Triplex protein quantification based on stable isotope labeling by peptide dimethylation applied to cell and tissue lysates. Proteomics 8, 4624–4632. [DOI] [PubMed] [Google Scholar]
- Borthwick LA, Gardner A, De Soyza A, Mann DA, and Fisher AJ (2012). Transforming Growth Factor-β1 (TGF-β1) Driven Epithelial to Mesenchymal Transition (EMT) is Accentuated by Tumour Necrosis Factor α (TNFα) via Crosstalk Between the SMAD and NF-κB Pathways. Cancer Microenviron 5, 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RJ, and Zhang Z (2013). Histone chaperones in nucleosome assembly and human disease. Nat Struct Mol Biol 20, 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallaro U, and Christofori G (2004). Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer 4, 118–132. [DOI] [PubMed] [Google Scholar]
- Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, D’Alessio AC, Young RA, and Weinberg RA (2013). Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 154, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloufi S, Elling U, Hopfgartner B, Jung YL, Murn J, Ninova M, Hubmann M, Badeaux AI, Euong Ang C, Tenen D, et al. (2015). The histone chaperone CAF-1 safeguards somatic cell identity. Nature 528, 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, and Brugge JS (2003). Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256–268. [DOI] [PubMed] [Google Scholar]
- Dekker J, Rippe K, Dekker M, and Kleckner N (2002). Capturing chromosome conformation. Science 295, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Dimitri CA, Dowdle W, MacKeigan JP, Blenis J, and Murphy LO (2005). Spatially separate docking sites on ERK2 regulate distinct signaling events in vivo. Curr Biol 15, 1319–1324. [DOI] [PubMed] [Google Scholar]
- Dow LE, Premsrirut PK, Zuber J, Fellmann C, McJunkin K, Miething C, Park Y, Dickins RA, Hannon GJ, and Lowe SW (2012). A pipeline for the generation of shRNA transgenic mice. Nat Protoc 7, 374–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Endo A, Ly T, Pippa R, Bensaddek D, Nicolas A, and Lamond AI (2017). The Chromatin Assembly Factor Complex 1 (CAF1) and 5-Azacytidine (5-AzaC) Affect Cell Motility in Src-transformed Human Epithelial Cells. J Biol Chem 292, 172–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Er EE, Valiente M, Ganesh K, Zou Y, Agrawal S, Hu J, Griscom B, Rosenblum M, Boire A, Brogi E, et al. (2018). Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat Cell Biol 20, 966–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, and Tyler JK (2010). Elevated histone expression promotes life span extension. Mol Cell 39, 724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freese NH, Norris DC, and Loraine AE (2016). Integrated genome browser: visual analytics platform for genomics. Bioinformatics 32, 2089–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurard-Levin ZA, Quivy JP, and Almouzni G (2014). Histone chaperones: assisting histone traffic and nucleosome dynamics. Annu Rev Biochem 83, 487–517. [DOI] [PubMed] [Google Scholar]
- Hendrich BD, and Willard HF (1995). Epigenetic regulation of gene expression: the effect of altered chromatin structure from yeast to mammals. Hum Mol Genet 4 Spec No, 1765–1777. [DOI] [PubMed] [Google Scholar]
- Henikoff S, and Ahmad K (2005). Assembly of variant histones into chromatin. Annu Rev Cell Dev Biol 21, 133–153. [DOI] [PubMed] [Google Scholar]
- Hoshino R, Chatani Y, Yamori T, Tsuruo T, Oka H, Yoshida O, Shimada Y, Ari-i S, Wada H, Fujimoto J, et al. (1999). Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene 18, 813–822. [DOI] [PubMed] [Google Scholar]
- Hu G, Cui K, Northrup D, Liu C, Wang C, Tang Q, Ge K, Levens D, Crane-Robinson C, and Zhao K (2013). H2A.Z facilitates access of active and repressive complexes to chromatin in embryonic stem cell self-renewal and differentiation. Cell Stem Cell 12, 180–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Chen K, Xia Z, Chavez M, Pal S, Seol JH, Chen CC, Li W, and Tyler JK (2014). Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev 28, 396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Yu Z, Zhang S, Liang X, Chen J, Li C, Ma J, and Jiao R (2010). Drosophila CAF-1 regulates HP1-mediated epigenetic silencing and pericentric heterochromatin stability. J Cell Sci 123, 2853–2861. [DOI] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, Beug H, and Grunert S (2002). Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol 156, 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, and Neilson EG (2003). Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest 112, 1776–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, and Weinberg RA (2009). The basics of epithelial-mesenchymal transition. J Clin Invest 119, 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman PD, Kobayashi R, Kessler N, and Stillman B (1995). The p150 and p60 subunits of chromatin assembly factor I: a molecular link between newly synthesized histones and DNA replication. Cell 81, 1105–1114. [DOI] [PubMed] [Google Scholar]
- Korpal M, and Kang Y (2010). Targeting the transforming growth factor-beta signalling pathway in metastatic cancer. Eur J Cancer 46, 1232–1240. [DOI] [PubMed] [Google Scholar]
- Kubosaki A, Tomaru Y, Tagami M, Arner E, Miura H, Suzuki T, Suzuki M, Suzuki H, and Hayashizaki Y (2009). Genome-wide investigation of in vivo EGR-1 binding sites in monocytic differentiation. Genome Biol 10, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S, Xu J, and Derynck R (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 15, 178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E (2002). Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet 3, 662–673. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, Wu CC, Hagos Y, Burckhardt BC, Pentcheva-Hoang T, et al. (2015). Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med 21, 998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Novoa JM, and Nieto MA (2009). Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med 1, 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascolo M, Vecchione ML, Ilardi G, Scalvenzi M, Molea G, Di Benedetto M, Nugnes L, Siano M, De Rosa G, and Staibano S (2010). Overexpression of Chromatin Assembly Factor-1/p60 helps to predict the prognosis of melanoma patients. BMC Cancer 10, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKittrick E, Gafken PR, Ahmad K, and Henikoff S (2004). Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc Natl Acad Sci U S A 101, 1525–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, and Massagué J (2005). Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Morgan HD, Santos F, Green K, Dean W, and Reik W (2005). Epigenetic reprogramming in mammals. Hum Mol Genet 14 Spec No 1, R47–58. [DOI] [PubMed] [Google Scholar]
- Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, Downey RJ, Manova-Todorova K, Brogi E, and Massagué J (2011). Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med 17, 867–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Fu H, Yin J, and Zhao Q (2018). CHAF1B knockdown blocks migration in a hepatocellular carcinoma model. Oncol Rep 40, 405–413. [DOI] [PubMed] [Google Scholar]
- Polyak K, and Weinberg RA (2009). Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 9, 265–273. [DOI] [PubMed] [Google Scholar]
- Quivy JP, Gérard A, Cook AJ, Roche D, and Almouzni G (2008). The HP1-p150/CAF-1 interaction is required for pericentric heterochromatin replication and S-phase progression in mouse cells. Nat Struct Mol Biol 15, 972–979. [DOI] [PubMed] [Google Scholar]
- Quivy JP, Roche D, Kirschner D, Tagami H, Nakatani Y, and Almouzni G (2004). A CAF-1 dependent pool of HP1 during heterochromatin duplication. EMBO J 23, 3516–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, and Mann M (2003). Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75, 663–670. [DOI] [PubMed] [Google Scholar]
- Ray-Gallet D, Woolfe A, Vassias I, Pellentz C, Lacoste N, Puri A, Schultz DC, Pchelintsev NA, Adams PD, Jansen LE, et al. (2011). Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol Cell 44, 928–941. [DOI] [PubMed] [Google Scholar]
- Ribatti D (2017). Epithelial-mesenchymal transition in morphogenesis, cancer progression and angiogenesis. Exp Cell Res 353, 1–5. [DOI] [PubMed] [Google Scholar]
- Roberts PJ, and Der CJ (2007). Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26, 3291–3310. [DOI] [PubMed] [Google Scholar]
- Roussos ET, Condeelis JS, and Patsialou A (2011). Chemotaxis in cancer. Nat Rev Cancer 11, 573–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, and Eliceiri KW (2017). ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samatar AA, and Poulikakos PI (2014). Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov 13, 928–942. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneiderman JI, Orsi GA, Hughes KT, Loppin B, and Ahmad K (2012). Nucleosome-depleted chromatin gaps recruit assembly factors for the H3.3 histone variant. Proc Natl Acad Sci U S A 109, 19721–19726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S, Buel GR, Nagiec MJ, Han MJ, Roux PP, Blenis J, and Yoon SO (2019). ERK2 regulates epithelial-to-mesenchymal plasticity through DOCK10-dependent Rac1/FoxO1 activation. Proc Natl Acad Sci U S A 116, 2967–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S, Dimitri CA, Yoon SO, Dowdle W, and Blenis J (2010). ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol Cell 38, 114–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S, and Stillman B (1989). Purification and characterization of CAF-I, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell 58, 15–25. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surani MA (2001). Reprogramming of genome function through epigenetic inheritance. Nature 414, 122–128. [DOI] [PubMed] [Google Scholar]
- Tam WL, and Weinberg RA (2013). The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med 19, 1438–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turinetto V, and Giachino C (2015). Histone variants as emerging regulators of embryonic stem cell identity. Epigenetics 10, 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
- Valastyan S, and Weinberg RA (2011). Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega S, Morales AV, Ocaña OH, Valdés F, Fabregat I, and Nieto MA (2004). Snail blocks the cell cycle and confers resistance to cell death. Genes Dev 18, 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S, and Workman JL (2015). Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol 16, 178–189. [DOI] [PubMed] [Google Scholar]
- Volk A, Liang K, Suraneni P, Li X, Zhao J, Bulic M, Marshall S, Pulakanti K, Malinge S, Taub J, et al. (2018). A CHAF1B-Dependent Molecular Switch in Hematopoiesis and Leukemia Pathogenesis. Cancer Cell 34, 707–723.e707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walden TA, Knieps LJ, and Baxter A (1991). Contingent provision of social referential information by parents of children with and without developmental delays. Am J Ment Retard 96, 177–187. [PubMed] [Google Scholar]
- Wen D, Banaszynski LA, Liu Y, Geng F, Noh KM, Xiang J, Elemento O, Rosenwaks Z, Allis CD, and Rafii S (2014). Histone variant H3.3 is an essential maternal factor for oocyte reprogramming. Proc Natl Acad Sci U S A 111, 7325–7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Wyckoff J, and Condeelis J (2005). Cell migration in tumors. Curr Opin Cell Biol 17, 559–564. [DOI] [PubMed] [Google Scholar]
- Yan H, Xiang X, Chen Q, Pan X, Cheng H, and Wang F (2018). HP1 cooperates with CAF-1 to compact heterochromatic transgene repeats in mammalian cells. Sci Rep 8, 14141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Franco AA, Santos H, Nelson DM, Kaufman PD, and Adams PD (2003). Defective S phase chromatin assembly causes DNA damage, activation of the S phase checkpoint, and S phase arrest. Mol Cell 11, 341–351. [DOI] [PubMed] [Google Scholar]
- Yoon JC, Ling AJ, Isik M, Lee DY, Steinbaugh MJ, Sack LM, Boduch AN, Blackwell TK, Sinclair DA, and Elledge SJ (2014). GLTSCR2/PICT1 links mitochondrial stress and Myc signaling. Proc Natl Acad Sci U S A 111, 3781–3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MD, Wakefield MJ, Smyth GK, and Oshlack A (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 11, R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen BT, and Knoepfler PS (2013). Histone H3.3 mutations: a variant path to cancer. Cancer Cell 24, 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Gan H, Wang Z, Lee JH, Zhou H, Ordog T, Wold MS, Ljungman M, and Zhang Z (2017). RPA Interacts with HIRA and Regulates H3.3 Deposition at Gene Regulatory Elements in Mammalian Cells. Mol Cell 65, 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. ATAC-seq peak distributions along chromosomes (Related to Figure 1)
Table S2. ChIP-seq analysis summary (Related to Figure 2)
Table S3. Breast cancer tissue array information and scoring (Related to Figure 6)
Table S4. RNA-seq analysis summary (Related to Figure 7)
Data Availability Statement
The accession number for the raw sequencing data for the RNA-seq is GEO: GSE119030, the ATAC-seq is GEO: GSE132046, and the ChIP-seq is GEO: GSE120313. The raw data for the mass spectrometry analysis can be accessed at ftp://massive.ucsd.edu/MSV000084205/.