

Abstract
Developing genomic insights is challenging in nonmodel species for which resources are often scarce and prohibitively costly. Here, we explore the potential of a recently established approach using Pool‐seq data to generate a de novo genome assembly for mining exons, upon which Pool‐seq data are used to estimate population divergence and diversity. We do this for two pairs of sympatric populations of brown trout (Salmo trutta): one naturally sympatric set of populations and another pair of populations introduced to a common environment. We validate our approach by comparing the results to those from markers previously used to describe the populations (allozymes and individual‐based single nucleotide polymorphisms [SNPs]) and from mapping the Pool‐seq data to a reference genome of the closely related Atlantic salmon (Salmo salar). We find that genomic differentiation (F ST) between the two introduced populations exceeds that of the naturally sympatric populations (F ST = 0.13 and 0.03 between the introduced and the naturally sympatric populations, respectively), in concordance with estimates from the previously used SNPs. The same level of population divergence is found for the two genome assemblies, but estimates of average nucleotide diversity differ ( ≈ 0.002 and ≈ 0.001 when mapping to S. trutta and S. salar, respectively), although the relationships between population values are largely consistent. This discrepancy might be attributed to biases when mapping to a haploid condensed assembly made of highly fragmented read data compared to using a high‐quality reference assembly from a divergent species. We conclude that the Pool‐seq‐only approach can be suitable for detecting and quantifying genome‐wide population differentiation, and for comparing genomic diversity in populations of nonmodel species where reference genomes are lacking.
Keywords: genetic diversity, genome sequencing, population genomics, Salmo trutta, salmonid, single nucleotide polymorphism
We explore the potential of a newly established approach that relies on Pool‐seq data to generate de novo genome assemblies in nonmodel species upon which Pool‐seq data are used to estimate population divergence and diversity. Estimates of population differentiation between sympatric brown trout (Salmo trutta) populations are qualitatively similar to those found from a previously used SNP panel and highly concordant to estimates from mapping our data to the publicly available Atlantic salmon (Salmo salar) reference genome. The Pool‐seq only approach is deemed suitable for detecting population differentiation in nonmodel species for which reference genomes are lacking.

1. INTRODUCTION
Understanding the importance of genetic variation for species' persistence continues to be a major research goal in population genetic and evolutionary studies (Allendorf & Ryman, 2002; Bernatchez, 2016; Soulé & Wilcox, 1980). Quantifying genetic variation within and among populations is important for conservation and management (Fuentes‐Pardo & Ruzzante, 2017; Ovenden, Berry, Welch, Buckworth, & Dichmont, 2015; Volckaert, 2015), and for understanding mechanisms of local adaptation, hybridization, and introgression (Allendorf & Hard, 2009; Waples, Punt, & Cope, 2008).
Developing population genetic insights from enough markers to represent whole genomes quickly and cost‐effectively is challenging in nonmodel species (Schlötterer, Tobler, Kofler, & Nolte, 2014). An alternative to whole‐genome sequencing (WGS) for such species is subsampling of the genome, which provides insights into genome‐level variation at comparably lower cost per individual, thereby enabling assessment across more individuals (Davey et al., 2011; Gagnaire, Pavey, Normandeau, & Bernatchez, 2013; Martinez, Buonaccorsi, Hyde, & Aguilar, 2017; Wang, Shashikant, Jensen, Altman, & Girirajan, 2017). Another alternative, while staying at the genome‐wide scale, is the pooling of individuals for WGS. This enables the sampling of many chromosomes per base pair and thereby accurate estimates of the site frequency spectrum, at a low individual cost (Kofler, Langmüller, Nouhaud, Otte, & Schlötterer, 2016).
Reference genomes provide crucial information needed for organizing, orienting, and annotating WGS reads. When there is none available, as is the case for many nonmodel organisms, the assembly of a closely related species is often used. Relatedness between focal and reference species has implications for the representation of markers found (Recknagel, Jacobs, Herzyk, & Elmer, 2015). As an alternative to using reference genomes of closely related species or for situations when no such reference is available, we here apply a newly developed exon mining via Pool‐seq approach to acquire a draft genome assembly both for the focal species and for single nucleotide polymorphism (SNP) frequency estimation. The approach leverages the power of Pool‐seq to subsample the genome to obtain high‐resolution genomic insights quickly and at a reasonable cost (Neethiraj, Hornett, Hill, & Wheat, 2017). This pipeline has been utilized for elucidating the genomics underlying phenotypic differences between populations of several butterfly species (Keehnen, Hill, Nylin, & Wheat, 2018; Pruisscher, Nylin, Gotthard, & Wheat, 2018; Woronik & Wheat, 2017).
Pool‐seq data generate highly fragmented assemblies, and in order to reduce fragmentation, the method explored here uses transcriptome data from the same species to scaffold contigs that are annotated to nonoverlapping regions of the same protein. The genome assembly is subsequently reduced to only contain scaffolds with identified and unique gene models (including introns and untranslated regions). Pool‐seq data from populations are mapped against the final gene‐models‐only genome assembly which contains both protein‐coding and noncoding sequences, for estimation of population diversity and differentiation (akin to mapping RNA‐seq data against a de novo transcriptome of the same data). This method (Neethiraj et al., 2017) has similarities with that of Therkildsen and Palumbi (2017) who mapped Pool‐seq reads to a reference transcriptome. In our case, we generate a transcriptome from published RNA‐seq data and use it to scaffold and annotate a draft genome assembly from Pool‐seq data.
We explore the Pool‐seq‐only approach of Neethiraj et al. (2017) using the brown trout (Salmo trutta) which belongs to the family Salmonidae that is characterized by large genomes (c. 3 Gbp) with the added complexity of a whole‐genome duplication event that occurred roughly 90 million years ago (MYA) followed by subsequent, and ongoing, rediploidization (Berthelot et al., 2014; Lien et al., 2016; Nugent, Easton, Norman, Ferguson, & Danzmann, 2017). Currently, there are genome assemblies available for Atlantic salmon (Salmo salar; Davidsson et al., 2010; Lien et al., 2016), rainbow trout (Oncorhynchus mykiss; Berthelot et al., 2014), chinook salmon (Oncorhynchus tshawytscha; Christensen, Leong, et al., 2018; Narum, Genova, Micheletti, & Maass, 2018), Arctic charr (Salvelinus alpinus; Christensen, Rondeau, et al., 2018), coho salmon (Oncorhynchus kisutch; GenBank assembly accession: GCA_002021735.1), and grayling (Thymallus thymallus; Sävilammi et al., 2019). The separation of brown trout and its closest relative the Atlantic salmon occurred c. 6–7 MYA (Pustovrh, Snoj, & Bajec, 2014), and nucleotide divergence between the two is below 2% (Leitwein et al., 2017). However, chromosomal rearrangements (Leitwein et al., 2017), number of chromosomes (S. trutta 2n = 80, Phillips & Ráb, 2001; S. salar 2n = 54–58, Brenna‐Hansen et al., 2012), and degrees of residual tetrasomy (Lien et al., 2016) differ significantly between the two species.
Similar to other species of Salmonidae, the brown trout is highly substructured (Laikre, 1999; Lerceteau‐Köhler, Schliewen, Kopun, & Weiss, 2013; Ryman, 1981; Ryman, Allendorf, & Ståhl, 1979; Vøllestad, 2018). Genetically distinct populations maintain separation across limited geographic areas (Palmé, Laikre, & Ryman, 2013; Stelkens, Pompini, & Wedekind, 2012), and the disparity of habitats occupied by brown trout has enabled population differentiation along a variety of phenotypic axes (Hansen, 2002; Hindar, Ryman, & Utter, 1991; Meier, Hansen, Bekkevold, Skaala, & Mensberg, 2011; Meier et al., 2014; Palm & Ryman, 1999). Understanding the role of genetic variation for sustainable management and conservation monitoring is crucial for this socioeconomically important species (Charlier, Laikre, & Ryman, 2012; Hansen, Ruzzante, Nielsen, & Mensberg, 2000; Leitwein, Gagnaire, Desmares, Berrebi, & Guinand, 2018; Petereit et al., 2018).
Our aim is to explore the potential of an exon mining through Pool‐seq approach to characterize the genomic variation and differentiation among brown trout populations. We ask whether this approach is suitable for answering population genomics questions by studying two pairs of sympatric populations for which we can make well informed hypotheses based on previous work (Andersson, Jansson, et al., 2017; Andersson, Johansson, Sundbom, Ryman, & Laikre, 2017; Palm & Ryman, 1999; Palmé et al., 2013). One pair of populations is naturally and cryptically sympatric while the other consists of two experimentally released populations, and we expect differentiation to be greater among the latter. We use Pool‐seq data from one of these populations to generate a de novo brown trout assembly, and then map the Pool‐seq data to this reference to estimate pool‐specific diversity and pairwise differentiation. These results are compared to the differentiation found from previous analyses of the same populations using allozymes and SNPs (Andersson, Jansson, et al., 2017; Andersson, Johansson, et al., 2017; Palm & Ryman, 1999; Palmé et al., 2013). We further contrast the outcome from mapping the Pool‐seq data to our draft assembly to mapping against the reference genome of a related species by repeating the analyses for Pool‐seq data mapped to an available Atlantic salmon genome (Lien et al., 2016).
2. MATERIALS AND METHODS
2.1. Populations studied
Two pairs of sympatric brown trout (Figure 1) populations inhabiting small freshwater lakes in the mountainous regions of the County of Jämtland, central Sweden, were studied. One pair consists of two populations that co‐occur in a natural setting due to an artificial release to this environment whereas the other pair is comprised of naturally sympatric populations. The first pair of populations was collected from the lake system Bävervattnen (Figure 2, Appendix S1, Figure S1) into which fish had been introduced as fry in 1979 for experimental purposes. The released individuals were from two separate populations that had been isolated from each other since the last glaciation (c. 5,000–9,000 years ago) and were ecologically diverged and genetically marked by contrasting homozygosity at the allozyme locus AGP‐2 (Palm & Ryman, 1999). This pair will be referred to as the introduced populations I and II. The second pair of populations comes from the lake system Trollsvattnen (Figure 2, Appendix S1) where the two main lakes are inhabited by a population pair that has previously been described as cryptically sympatric because no phenotypical or ecological difference between them has been possible to detect in spite of extensive screenings (Andersson, Jansson, et al., 2017; Andersson, Johansson, et al., 2017; Palmé et al., 2013). Their existence was initially detected through a consistent heterozygote deficiency at multiple allozyme loci (Jorde & Ryman, 1996), later validated by extensive allozyme monitoring showing consistent population divergence as measured by the fixation index (F ST) over several decades (Palmé et al., 2013) and by a 3 K SNP panel (Andersson, Jansson, et al., 2017). This pair of populations will henceforth be referred to as the natural populations A and B.
Figure 1.

The brown trout (Salmo trutta) from Swedish mountain lakes was used as a case study to explore the potential of a recently presented Pool‐seq‐only approach for gaining genomic insights in nonmodel species. Photograph by Anastasia Andersson
Figure 2.
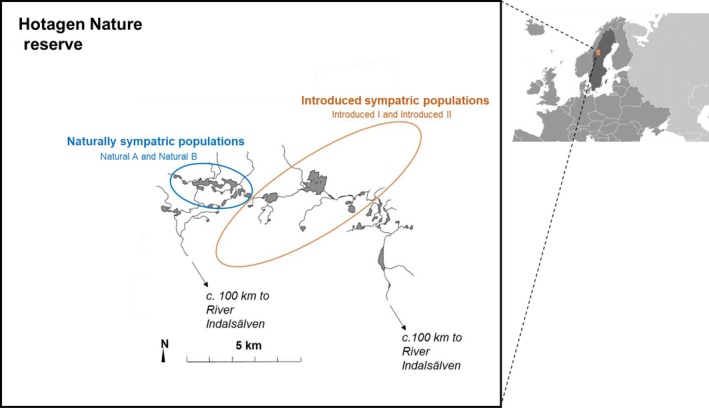
Map of study sites located in Hotagen Nature Reserve, Sweden. Circles indicate sampled lakes inhabited by introduced and naturally sympatric populations, respectively. Both waters are connected to the River Indalsälven which drains into the Baltic Sea c. 400 km from the study site
2.2. Samples
We used n = 50 individuals from each of the two introduced populations (Palm & Ryman, 1999) caught during 1988–1995; frozen tissue had been stored since collection. The fish were assigned to either of the introduced populations based on their allozyme genotype at the marker locus AGP‐2 and their age based on otolith readings. Only individuals representing the parental generation (P) or the F1 generation for which population assignment (no hybrids) was possible using the AGP‐2 genotype were considered. A total of 41 individuals representing the P generation were available, and 59 F1 fish were randomly selected out of c. 700 available fish to provide n = 50 per population.
The natural populations have been monitored for many years with access to thousands of fish. We used n = 50 individuals per natural population collected during 2002–2007 and assigned to their respective population by Andersson, Jansson, et al. (2017) based on a STRUCTURE (Falush, Stephens, & Pritchard, 2003; Pritchard, Stephens, & Donnelly, 2000) analysis from 14 polymorphic allozyme loci and an assignment score above 0.8 (for more details, see Appendix S1).
2.3. DNA extraction, library preparation, and sequencing
DNA was isolated from muscle tissue that had been stored at −80°C since sampling 1–3 decades ago using Qiagen's DNeasy Blood and Tissue Kit according to the manufacturer's protocol (Qiagen) with an additional RNase A treatment. DNA quality was assessed by visual inspection of DNA fragmentation on agarose gels and absorbance at 260/280. DNA with high molecular weight from each of 50 individuals per population was quantified using fluorometry (Qubit; Thermo Scientific) and pooled at equal concentrations to achieve 3 μg pooled genomic DNA in a volume in the range of 65–120 μl. The National Genomics Infrastructure (NGI), Uppsala, Sweden (Science for Life Laboratory), conducted the construction of PCR‐free paired‐end libraries with an average insert size of 350 bp (TruSeq) followed by sequencing using read length 150 bp on an Illumina HiSeq 2000 machine.
2.4. Short‐read data preparation and de novo assembly
Illumina short reads from the four population pools were cleaned for adapters and low‐quality bases using BBDuk implemented in BBTools version 37.31 (http://sourceforge.net/projects/bbmap/). Pool‐seq short‐read data from natural population A were used to generate a draft de novo genome assembly using CLC Genomics version 5.5.1 with default settings: k‐mer size 20, bubble size 50, and minimum contig length 200.
2.5. Transcriptome assembly
We used publicly available RNA‐seq data (Carruthers et al., 2018; https://www.ncbi.nlm.nih.gov/sra/SRX3421649%5Baccn%5D) from whole organism tissue of hatchery strain juveniles of undetermined sex (Table S1) to generate a S. trutta transcriptome. The paired‐end reads, from 8 accessions available at the time of study prior to the official release of the S. trutta transcriptome, were cleaned for adapters in BBMap's implementation version 37.31 (http://sourceforge.net/projects/bbmap/) following default recommendations. Rcorrector version 2 (Song & Florea, 2015) was used at default settings to further filter the data for singleton kmers and ribosomal RNA filtered by BBDuk in the BBTools suite version 37.53 (http://sourceforge.net/projects/bbmap/). The resulting data were used as input for a transcriptome assembly using Trinity version 2.5.1 (Grabherr et al., 2011) with default parameters. The transcriptome assembly was then collapsed into unique protein sequences using the Evigene software (Gilbert, 2013). In order to assess its quality, we compared this unique protein set from S. trutta to the Atlantic salmon protein sequence predictions from the available genome assembly (Lien et al., 2016; accession number GCF_000233375.1). Before analysis, salmon protein sequences were collapsed using CD‐hit into clusters of 90% identity, keeping only the longest member of each cluster for subsequent analysis; this is hereafter referred to as the salar90 protein dataset. This approach is identical to the clustering of UniRef (Suzek, Huang, McGarvey, Mazumder, & Wu, 2007) database to make the UniRef90 dataset, and in this case, it allows us to keep only the longest isoforms and only single members of recent gene duplications. By comparing the S. trutta protein sequences against this salar90 protein dataset, we quantitatively assessed how many genes we assembled compared to expected numbers. We determined whether each gene was assembled at partial or full length by dividing the length of the assembled S. trutta protein by the length of the salmon homolog identified using BLASTP (e‐value cutoff 10e‐5; protein–protein BLAST; version 2.2.28+).
2.6. Scaffolding, annotation, and quality assessment of the Pool‐seq de novo genome assembly
Scaffolding involves joining contigs that belong to nonoverlapping regions of the same protein and thus reduces assembly fragmentation. The Pool‐seq de novo assembly was scaffolded using the S. trutta protein sequences via the MESPA pipeline (Neethiraj et al., 2017). MESPA uses the software SPALN version 2.1.4 (http://www.genome.ist.i.kyoto-u.ac.jp/~aln_user/spaln/; Gotoh, 2008) for protein to genome alignment, and then uses this output to guide further scaffolding per protein based on exons from a single protein that are located on different scaffolds. Gene models for the resulting superscaffolded assembly are thus based on the S. trutta protein dataset. To avoid complications in mapping and variant calling caused by the partially tetraploid characteristics of the S. trutta genome, we used a collapsed version of the full Pool‐seq de novo assembly containing only scaffolds with identified gene models, and of those only retained regions with unique gene models. We used this haploid assembly for subsequent analyses of SNPs within or near our gene model annotations. The completeness of this draft S. trutta assembly was assessed based on gene content from near‐universal single‐copy orthologs using BUSCO version 1.22 (Simão, Waterhouse, Ioannidis, Kriventseva, & Zdobnov, 2015), with the library of ray finned fish proteins (Actinopterygii; Danio rerio) and default settings.
2.7. Read mapping and quality filtering
Paired‐end short reads from each pool filtered for adapters and minimum base quality 20 were mapped to the draft S. trutta assembly and the Atlantic salmon genome (Lien et al., 2016; accession number GCF_000233375.1). We tested three different mapping algorithms: BBMap version 37.31 (http://sourceforge.net/projects/bbmap), bwa mem available in bwa version 0.7.17 (Li & Durbin, 2009), and NextGenMap (NGM) version 0.5.4 (Sedlazeck, Rescheneder, & Von Haeseler, 2013). The mapping success for each algorithm was assessed in Qualimap version 2.2.1 (García‐Alcalde et al., 2012) before and after filtering the bam files at varying levels of mapping quality. Based on these comparisons (Table S2) and visual inspection of bam files in the Integrative Genomics Viewer (IGV; Robinson et al., 2011), bwa mem and filtering for mapping quality 20 were chosen for all subsequent analyses, trading off evenly distributed read coverage across the assembly with mapping accuracy.
Bam files with short‐read data mapped to the two genome assemblies were filtered to keep only properly paired reads. Mpileup files were generated from these bam files using samtools version 1.6 (Li et al., 2009), filtering for mapping quality 20 and base quality 20, as well as invoking the parameter “‐B” to reduce false SNPs from misalignments. The mpileup files obtained from mapping to the S. trutta assembly were inspected for insertions or deletions (indels) using the identify‐genomic‐indel‐regions.pl script in POPOOLATION2 version 1201 (Kofler, Pandey, & Schlötterer, 2011). Indels and 5 bp downstream and upstream every indel were subsequently removed using the filter‐pileup‐by‐gtf.pl tool from POPOOLATION2. Coverage with reads mapped to the S. salar reference genome was highly uneven. Therefore, indels ≥32,766 bp had to be removed from the mpileup file using a custom script before running the scripts implemented in POPOOLATION2 which cannot handle indels of this size and larger. Read depth histograms were assessed for each bam file to define minimum and maximum depth thresholds for subsequent population genetic calculations (Appendix S2).
2.8. Population genomic analyses
Population genomic variation was assessed for each pool using POPOOLATION version 1.2.2 (Kofler, Orozco‐ter Wengel, et al., 2011), including estimates of nucleotide diversity (π; Tajima, 1983) which quantifies the degree of polymorphisms at a locus within a population and Tajima's D (T D), which measures deviations from mutation–drift equilibrium at segregating sites due to selection or demographic events (Tajima, 1989). Since estimates of π and T D are sensitive to sequencing errors (Kofler, Orozco‐ter Wengel, et al., 2011), we subsampled each mpileup file to uniform coverage based on depth histograms (targeting the mode of each pools' coverage distribution and omitting sites with coverage exceeding the mode +½ of the mode; Appendix S2) by running the subsample‐pileup.pl script without replacement (Kofler, Orozco‐ter Wengel, et al., 2011). The script Variance‐sliding.pl implemented in POPOOLATION was used to detect SNPs from subsampled mpileup files and to simultaneously calculate π (including invariant sites) and T D. Calculations were made for nonoverlapping 500‐bp windows across each of the assemblies, using a minor allele count of 2 for a SNP to be called and stringent depth filters for variant and invariant sites to be included (the mode of each pools' depth distribution ±½ of the mode; Appendix S2, Figures S2 and S3). Only windows of >90% coverage with data after applying depth and minor allele frequency filters were included in the analyses. All summary statistics were calculated and statistical tests were performed in R (R Core Team, 2017). A Kruskal–Wallis rank‐sum test for independence of π and T D with respect to populations was performed. If the null‐hypothesis of samples coming from the same distribution was rejected, Wilcoxon rank‐sum tests were performed between all pairs of populations with p‐values adjusted using Bonferroni correction. The scripts used for the population genomic analyses are available in Appendix S3.
The fixation index (F ST; Nei, 1973) was estimated for each population pair in POPOOLATION2 version 1201 (Kofler, Pandey, et al., 2011). First, indel‐filtered mpileup files (not subsampled) were converted to the POPOOLATION2‐specific sync format using the script mpileup2sync.jar. F ST was calculated using the script fst‐sliding.pl with the following parameters, while simultaneously detecting SNPs. Variant and invariant sites with a read depth lower or higher than the thresholds identified from read depth histograms (the mode of the coverage distribution ±½ of the mode; Appendix S2) were excluded from the analyses, and a minor allele count of 3 was used as cutoff for a SNP to be called. F ST was estimated for nonoverlapping windows to avoid increased stochastic error rates associated with small window size (Kofler, Pandey, et al., 2011). A range of window sizes (1 bp–5 kb) and window coverages with data after applying all quality filters (0.5–1.0) were tested before choosing a window size of 500 bp and restricting the analysis to windows of >90% coverage with data after applying depth and minor allele frequency filters (Appendix S2, Table S3). Using the same parameters as described above, F ST was also obtained for noncoding and coding regions, the latter of which to enable a fairer comparison to results obtained from previously published allozyme data. A Kruskal–Wallis rank‐sum test for independence of pairwise F ST values from the full assembly, coding regions, and noncoding regions was performed.
2.9. Comparison to F ST estimates from previously used markers
We compared F ST values from Pool‐seq data mapped to the S. trutta assembly to previous divergence estimates using SNP genotyping of individuals from the same populations (n = 2,832 SNPs genotyped for 18 individuals and n = 2,852 SNPs genotyped for 30 individuals from each of the introduced and natural populations respectively; Andersson, Jansson, et al., 2017; L. Laikre & N. Ryman, unpublished data). All n = 18 individuals from each of the introduced populations were also included in the Pool‐seq samples from those populations. Similarly, the majority of the n = 30 individuals genotyped previously for the natural populations were included in the pools for those populations (overlap of n = 27 and n = 28 for natural populations A and B, respectively). We identified the putative locations of these SNPs in the S. trutta assembly by blasting the sequence surrounding each SNP to the S. trutta assembly and retaining the best hits (BLASTN; version 2.2.28+). For each location, we retrieved the corresponding F ST value estimated in POPOOLATION2 per base pair for Pool‐seq data using BEDTools intersect version 2.25.0 (Quinlan & Hall, 2010). F ST per locus for already available SNP data from individual genotyping had previously been calculated in GENEPOP version 4.0.7 (Rousset, 2008) but was recalculated here using Nei's (1973) F ST = 1−H S/H T since this F ST was used by POPOOLATION2 for the Pool‐seq data according to the manual. We also compared global F ST estimates from allozymes and SNPs to our assembly‐wide and window‐based averages.
3. RESULTS
3.1. Transcriptome assembly
RNA‐seq data from whole organism tissue of hatchery strain juveniles were used (Carruthers et al., 2018) and removal of singleton kmers and rRNA from these raw data reduced the total number of paired‐end reads by c. 6%, resulting in a total of 188 million (M) paired‐end reads (on average 47 M paired‐end reads per accession). The initial transcriptome assembly (254,432 contigs with an N50 of 1,779 bp and an average contig length of 963 bp) was reduced to 82,052 protein‐coding contigs with an N50 of 1,077 bp, average contig length of 600 bp, and total length of 49 Mbp in the final S. trutta protein dataset. Seventy‐eight percent of complete Actinopterygian core genes were recovered in our S. trutta transcriptome assembly. A total of 12,711 (35%) of the proteins in the salar90 protein dataset were represented by nearly full‐length (>90%) sequences in our S. trutta protein dataset.
3.2. De novo assembly of Pool‐seq data
Protein‐based superscaffolding of the S. trutta assembly reduced fragmentation, as reflected in a reduced number of contigs and increased N50 (Table 1). Before scaffolding, we recovered 63,636 S. trutta protein‐coding gene predictions including isoforms at near full length (>90%), and this number increased to 70,376 postscaffolding. A total of 87,191 unique, that is, single‐copy, gene models were identified in the final, collapsed S. trutta assembly. These gene models were located on in total 38,888 scaffolds with an N50 of 17,722 bp, and a total length of 446,412,000 bp (Table 1).
Table 1.
Genome assembly statistics for the de novo assembly of natural pool A from CLC Genomics version 5.5.1 and for the superscaffolded assemblies from MESPA (Neethiraj et al., 2017)
| Metric | Prescaffolding genome assembly | Postscaffolding MESPA genome assemblies | |
|---|---|---|---|
| CLC assembly | Full assembly | Gene‐models‐only assembly | |
| n (contigs) | 1,096,446 | 1,085,382 | 38,888 |
| N50 (bp) | 5,944 | 6,194 | 17,722 |
| Total contigs length (bp) | 1,847,698,765 | 1,848,805,165 | 446,412,000 |
| Percentage (%) of non‐ATGC charactersa | 0.77 | 0.83 | 0.57 |
The number (n) of contigs is specified but refers to the number of scaffolds in the annotated MESPA genome assemblies where contigs have been joined to form scaffolds. The gene‐models‐only assembly is the S. trutta genome assembly used for population genomics in the present study.
Non‐ATGC characters: for example, ambiguous nucleotides or unknown nucleotides (N).
Of the 4,584 Actinopterygian single‐copy orthologs, the initial S. trutta assembly had 58% complete (45% single‐copy and 14% duplicated), 20% fragmented, and 22% missing orthologs. After superscaffolding, 71% of matches were complete (59% single, 12% duplicated), 12% fragmented, and 17% missing.
3.3. Processing of short‐read data for population genomic analyses
On average, 69 giga base pairs (Gbp) of raw data were generated per population pool, corresponding to c. 9 M reads. After cleaning, 30% of reads (on average 2.8 M read pairs) mapped to the S. trutta gene models as proper pairs and c. 50% of reads mapped as pairs against the S. salar reference (on average 5.0 M read pairs; Table 2). Coverage of depth was lower for reads mapped to the S. salar reference (mode of depth distribution: ~50× to S. trutta, and ~45× to S. salar), which is nearly 6 times the size of the S. trutta assembly, and its annotation contains 97,918 gene model predictions. Although reads had been filtered for mapping quality 20, average mapping quality was 11 for reads mapped to S. salar due to the large proportion of reads that did not map (Table 2; mapping quality histograms in Figure S4). The edit distance between the reads and the reference was similar for both reference genomes (2.6%–2.7% and 2.9%, respectively; Table 2).
Table 2.
BAM file statistics from Qualimap version 2.2.1 (García‐Alcalde et al., 2012) for Pool‐seq data from each of the four S. trutta population pools filtered for minimum base quality 20 and mapped to the generated S. trutta assembly and the previously available S. salar assembly, respectively, using bwa mem and mapping quality 20
| Population pool | Introduced I | Introduced II | Natural A | Natural B |
|---|---|---|---|---|
| S. trutta | ||||
| Reference size (bp) | 446,412,000 | 446,412,000 | 446,412,000 | 446,412,000 |
| Number of reads mapped as pairs | 259,292,315 | 254,225,047 | 280,138,464 | 325,530,950 |
| Percentage of reads mapped as pairs | 29.8% | 29.7% | 30.3% | 30.5% |
| Mode of depth of coverage | 47 | 46 | 50 | 57 |
| Mean mapping quality | 55 | 56 | 56 | 55 |
| General error ratea | 2.6% | 2.6% | 2.6% | 2.7% |
| S. salar | ||||
|---|---|---|---|---|
| Reference size (bp) | 2,966,890,203 | 2,966,890,203 | 2,966,890,203 | 2,966,890,203 |
| Number of reads mapped as pairs | 468,965,008 | 457,736,395 | 497,860,918 | 573,473,218 |
| Percentage of reads mapped as pairs | 54.3% | 53.8% | 54.2% | 54.0% |
| Mode of depth of coverage | 40 | 41 | 45 | 50 |
| Mean mapping quality | 11 | 11 | 11 | 11 |
| General error ratea | 2.9% | 2.9% | 2.9% | 2.9% |
The mode of depth of coverage was obtained from BEDTools version 2.25.0 (Quinlan & Hall, 2010).
General error rate is estimated from the ratio of total collected edit distance to the number of mapped bases.
3.4. Population genomic analyses
On average, 53,538 SNPs were called in POPOOLATION from quality‐filtered and subsampled mpileup files for each pool mapped against our constructed S. trutta assembly (Table 3), and 231,548 SNPs on average when mapping to the S. salar reference genome, taking depth (Appendix S2) and minor allele frequency filters into account for each site. Average nucleotide diversity, , ranged between 1.65 and 1.95 with the S. trutta reference (Table 3; Figure S5). Although several confidence intervals overlap, the distributions differ significantly among populations (Kruskal–Wallis H = 145.26, df = 3, p < 2.2e‐16) with the lowest value observed in introduced population II. Pairwise comparisons from Wilcoxon signed‐rank tests showed that all population pairs except that of introduced population I versus natural population A differed, to a degree that results in statistical significance. With the S. salar reference genome, average were considerably lower—between 0.97 and 1.14 (Table 3)—but here too the lowest variability is noted in introduced population II and the population distributions differ (H = 2,916.7, df= 3, p < 2.2e‐16); all pairwise comparisons provided statistically significant differences among all population pairs.
Table 3.
Average nucleotide diversity () and Tajima's D (T D) with 95% confidence intervals in brackets estimated from Pool‐seq data mapped to the S. trutta and S. salar assemblies, respectively
| Reference | Pool | n (windows) | n (SNPs) | (10–3) | T D |
|---|---|---|---|---|---|
| S. trutta | Introduced I | 16,346 | 52,473 | 1.86 (1.81–1.92) | −0.065 (−0.081 to −0.048) |
| Introduced II | 15,564 | 45,783 | 1.65 (1.60–1.70) | −0.180 (−0.200 to −0.163) | |
| Natural A | 17,250 | 55,707 | 1.80 (1.74–1.84) | −0.160 (−0.174 to −0.141) | |
| Natural B | 16,212 | 60,188 | 1.95 (1.90–2.01) | −0.202 (−0.220 to −0.184) | |
| S. salar | Introduced I | 125,934 | 204,956 | 1.07 (1.06–1.08) | 0.103 (0.098–0.108) |
| Introduced II | 127,524 | 200,006 | 0.97 (0.96–0.98) | −0.038 (−0.043 to −0.033) | |
| Natural A | 155,326 | 289,247 | 1.14 (1.13–1.15) | −0.004 (−0.009 to 0.001) | |
| Natural B | 120,600 | 231,984 | 1.09 (1.08–1.10) | −0.030 (−0.036 to −0.024) |
Average values were obtained from estimates of nonoverlapping, 500‐bp windows with >90% coverage after subsampling and quality and depth filtering. n (windows): total number of windows; n (SNPs): total number of SNPs.
Tajima's D values (T D) were all below 0 ranging from −0.202 to −0.065 when mapping to S. trutta with the largest value observed in introduced population I (Table 3, Figure S6); several confidence intervals overlap, but we do find differences between distributions (H = 228.3, df = 3, p < 2.2e‐16). Pairwise comparisons indicate statistically significant differences between the populations except for between introduced population II and each of the natural populations. When mapping to S. salar, T D increases substantially, but the relationship with the largest value observed for introduced I remains (Table 3). Here too, the distributions of T D among the three populations with lower T D overlap, but with significant differences among all four distributions (H = 2,818.6, df = 3, p < 2.2e16) and statistically significant differences in T D distributions between both population pairs.
We used POPOOLATION2 for F ST estimation, and an average of c. 1,500,000 SNPs were called assembly‐wide and per population comparison from quality‐filtered sync files for pools mapped to the S. trutta assembly (Table 4), taking depth (Appendix S2) and minor allele frequency filters into account per site. We compared all possible population pairs (six in total) and found average F ST ≈ 0.10 for 5 of these comparisons, while one population pair—the two natural populations—showed F ST = 0.03 (Figure 3, Table 4). The distribution of F ST for the two main pairs of interest—the introduced versus the naturally sympatric populations—illustrates the considerably higher divergence for the introduced as compared to the natural pair (Figure 4a,b). Average F ST was higher across coding (c. 11,000 SNPs) than noncoding (c. 1,000,000 SNPs) regions (introduced pair: W = 260 × 106; p ≪ .001; natural pair: W = 263 × 106; p ≪ .001), although the mean values differed only slightly for each pair (Table 4).
Table 4.
Average pairwise F ST between introduced and natural populations, respectively
| Marker | n | Pairwise F ST between introduced populations | Pairwise F ST between natural populations |
|---|---|---|---|
| Allozymes | 14 | 0.41a | 0.30b |
| SNP genotyping | n (introduced) = 2,832 | 0.34a | 0.03b |
| n (natural) = 2,852 | |||
| Pool‐seq mapped against S. trutta | |||
| Assembly‐wide | n (introduced) = 1,466,801 | 0.1280 (0.1274–0.1285) | 0.0276 (0.0275–0.0278) |
| n (natural) = 1,525,838 | |||
| Coding regions | n (introduced) = 10,667 | 0.1491 (0.1436–0.1547) | 0.0317 (0.0304–0.0330) |
| n (natural) = 10,637 | |||
| Noncoding regions | n (introduced) = 1,083,064 | 0.1270 (0.1264–0.1277) | 0.0274 (0.0273–0.0276) |
| n (natural) = 1,126,527 | |||
| Pool‐seq mapped against S. salar | |||
| Assembly‐wide | n (introduced) = 2,206,076 | 0.1548 (0.1544–0.1552) | 0.0320 (0.0319–0.0321) |
| n (natural) = 2,510,827 | |||
| Coding regions | n (introduced) = 24,936 | 0.1499 (0.1466–0.1533) | 0.0326 (0.0318–0.0334) |
| n (natural) = 25,676 | |||
| Noncoding regions | n (introduced) = 1,967,836 | 0.1544 (0.1540–0.1548) | 0.0318 (0.0317–0.0319) |
| n (natural) = 2,250,772 |
Estimates from previous studies using allozymes and SNP genotyping are given, as well as estimates from Pool‐seq data mapped to each of the S. trutta assembly and S. salar genome using nonoverlapping, 500‐bp windows with >90% coverage after subsampling, quality and depth filtering. The number (n) of allozyme markers, previously used SNPs, and Pool‐seq SNPs used in each pairwise comparison is specified. 95% confidence intervals for Pool‐seq estimates are given in brackets.
L. Laikre and N. Ryman (unpublished data) using Weir and Cockerham's (1984) F ST.
Figure 3.
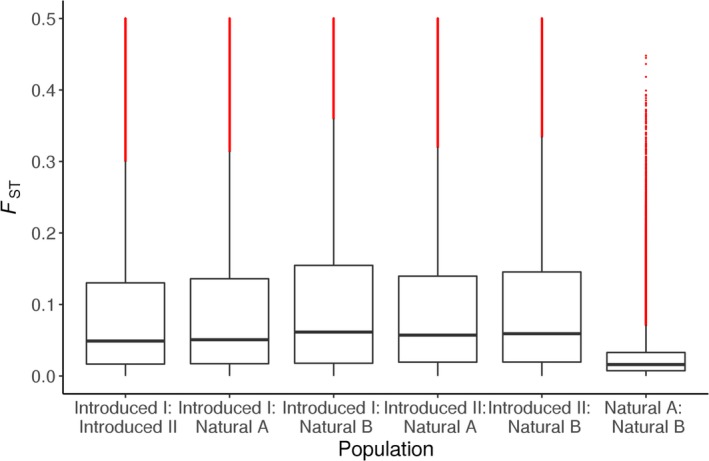
Boxplot of F ST values within 500‐bp windows along the S. trutta assembly for all pairwise comparisons between population pools. The horizontal line at the center of the box is median F ST, and the top and bottom of the box show 25th and 75th percentiles, respectively. Vertical black lines show the boundaries of the interquartile range and red markings show outliers
Figure 4.
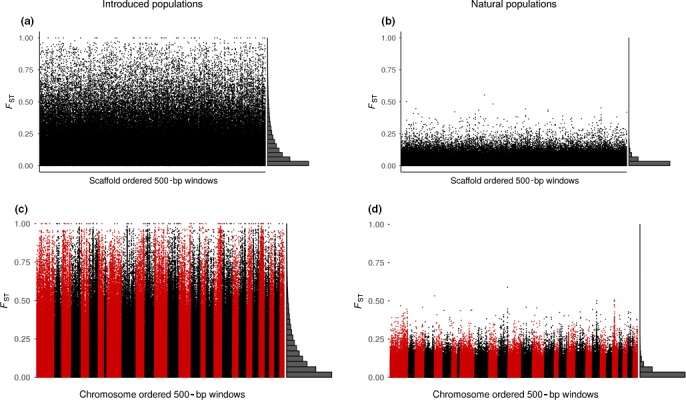
Pairwise F ST values within 500‐bp windows along the (a, b) S. trutta and (c, d) S. salar assemblies for (a, c) introduced and (b, d) naturally sympatric populations. Histograms in the margins represent frequency distributions of F ST values
F ST estimates when mapping the Pool‐seq data to the S. salar reference were similar to those observed using the S. trutta assembly (using on average 2,400,000 SNPS; Table 4), and the distribution of F ST remained higher for the introduced than the natural pair (Figure 3c,d). For both references, F ST for the naturally sympatric population pair was close to 0.03, and for the introduced pair, F ST was around 0.13 and 0.15 when mapping to the S. trutta and S. salar assembly, respectively. With the sort of “pseudochromosomal” context that F ST windows were placed in from the transitory structures formed when using the S. salar reference, outlier regions appear more evident than when using the S. trutta assembly (cf. Figure 4a,b,c,d).
3.5. Comparison to F ST estimates from previously used markers
Estimates of F ST from the previously used allozymes exceeded Pool‐seq averages calculated across assemblies, coding regions, and noncoding regions for both population pairs (Table 4). Average F ST calculated across the previously used SNPs exceeded Pool‐seq F ST for the introduced pair, while for the natural pair, the two estimates were concordant (Table 4). For our comparison of previously used SNPs with Pool‐seq SNPs, 1,974 and 1,985 of the 2,832 and 2,852 SNPs previously genotyped in the introduced and natural population pair, respectively (Andersson, Jansson, et al., 2017), were successfully located in our S. trutta assembly. Of these, F ST estimates were available from the Pool‐seq data for 1,415 and 1,378 individual SNPs per population pair for the introduced and natural populations, respectively. F ST values obtained from previous individual SNP genotyping and from Pool‐seq data were correlated for both population pairs (introduced: linear regression coefficient b = 0.81, r 2 = 0.76; t = 68; p < .001; natural: linear regression coefficient b = 0.66, r 2 = 0.21; t = 19; p < .001; Figure 5).
Figure 5.
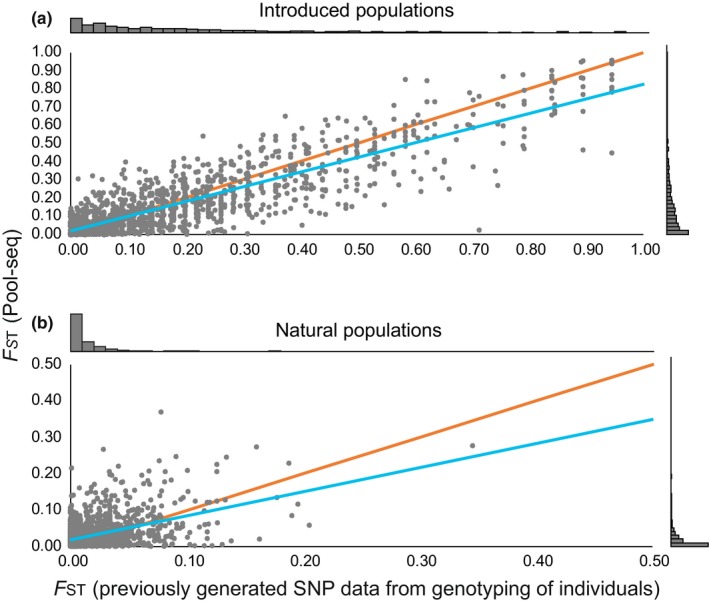
F ST values from Pool‐seq analyses (y‐axes) compared to those from previous individual SNP genotyping of the same SNP loci (x‐axes; data from Andersson, Jansson, et al., 2017 and L. Laikre & N. Ryman, unpublished data). Nei's F ST computed as F ST = 1−H S/H T was used for the previous SNP data, while for Pool‐seq, Nei's F ST was computed using POPOOLATION v. 1.2.2. (a) Pairwise F ST for 1,415 SNP loci for the introduced population (linear regression coefficient b = 0.81, r 2= 0.76, t = 68, p < 0.001). (b) Pairwise F ST for 1,378 SNP loci for the naturally sympatric populations (linear regression coefficient b = 0.66, r 2 = 0.21, t = 19, p < 0.001). The blue lines are linear regression trend lines, and the orange ones represent expected values with r2 = 1. Histograms in the margins represent frequency distributions of F ST values
4. DISCUSSION
4.1. Pool‐seq draft assembly and S. trutta transcriptome assembly
The S. trutta genome was assembled by superscaffolding a highly fragmented de novo assembly of Pool‐seq short‐read data, and simultaneously annotated using publicly available transcriptome data from the same species, using a newly established method for nonmodel organisms with limited genomic resources (Neethiraj et al., 2017). This assembly was used as a reference to map the Pool‐seq data and estimate population genomic metrics, both within and nearby protein‐coding regions. We used publicly available RNA‐seq data recently published in Carruthers et al. (2018) to assemble a draft S. trutta transcriptome. The validation of completeness of our S. trutta transcriptome, wherein 78% of single‐copy Actinopterygian orthologs were found, is comparable to recovery ranges of Carruthers et al. (2018), implying that the S. trutta transcriptome is suitable to annotate and identify coding regions in our final S. trutta assembly. We found nearly 13,000 S. salar proteins represented by nearly full‐length transcripts having >90% coverage in the final S. trutta transcriptome. This corresponds to c. 35% of all S. salar proteins. In their brown trout transcriptome, Carruthers et al. (2018) achieved c. 40% recovery of transcripts at near to full length from the same NCBI protein database for Atlantic salmon. This affirms good coverage and quality of our transcriptome assembly.
To improve mapping and subsequent variant calling, we used a haploid subset of the full S. trutta assembly, comprised only of contigs with gene model predictions. This reduced fragmentation compared to the full assembly is reflected in increased N50 (Table 1) and the representation of over 70 percent complete Actinopterygian core genes which is furthermore comparable to reports for the S. salar reference (Carruthers et al., 2018).
The study species used to explore the present Pool‐seq approach is a salmonid and thus has a genome duplication background. Although the duplication event is old—c. 90 million years—full rediploidization has not occurred (Lien et al., 2016), but we consider the MESPA approach to be careful in this respect since the method implies that only one copy of each contig with a gene model prediction of the original genome assembly remains in the final assembly and related contigs are grouped together (Neethiraj et al., 2017). Also, we were careful to apply stringent quality filters to the short‐read data mapped to the S. trutta draft assembly to avoid potential problems. Such problems could, for example, arise in duplicated regions that are only present as one copy in the S. trutta draft assembly. In such a region, short reads from both duplicates would map, including reads from the paralog region, which can be reduced with stringent mapping quality and read depth filters.
4.2. Population genomics analyses for pools mapped to the S. trutta assembly
We found no strong indications of demographic perturbations or selection in the sampled populations, which agrees with our general knowledge of these populations. Average nucleotide diversity estimated from data mapped to the S. trutta assembly was about 0.16%–0.20% across the four populations (Table 3, Figure S5). This estimate is lower than previous reports for brown trout ( ≈ 0.5%; Leitwein et al., 2016). Estimates of Tajima's D fell slightly below zero for all populations. Positive T D indicates a greater degree of heterozygosity given the number of segregating sites, as expected after population contraction, while negative values of T D may indicate population expansion after a recent bottleneck, though slightly negative values of T D are expected for natural populations (Gillespie, 2004). We did find differences in both nucleotide diversity and T D among populations, but differences are relatively small. Most pronounced is the lower level of nucleotide diversity of introduced population II which originates from a small mountain lake (0.14 km2) at the uppermost part of a water system, and where introduced fish originated from few parent fish caught in the wild and taken to a hatchery for production of fish for release.
Average F ST between the two natural populations was the lowest of all pairwise comparisons, while the highest F ST was found between the two introduced populations (Table 4, Figure 4). Genomic comparisons of sympatric populations where gene flow may occur are often distinguished by low diversity with condensed regions of divergent outliers (Jacobs et al., 2018), which may be indicated for the naturally sympatric pair (Figure 4), although we did not perform an outlier analysis in the present study. The comparably high divergence between the introduced populations was expected based on the source populations' geographic separation. Introduced population I stems from a hatchery population characterized by large piscivorous individuals with potential for long‐distance migration, whereas the source of the introduced population II is characterized by small, lake‐resident fish. One generation after introduction, descendants of introduced I migrated further downstream than descendants of introduced II (Palm & Ryman, 1999), indicating maintenance of the source populations' local adaptations. This, along with slightly higher values of T D, which was positive for introduced I when mapping pools to the S. salar reference, could again be indicative of the disparate genetic background and history of the introduced populations. A deeper understanding of the demographic and selective forces that have shaped the genomic variation of these populations requires further investigation.
4.3. Comparison to F ST from SNP genotyping
One of the main benefits of Pool‐seq is the generation of vast amounts of SNPs per genome for entire populations, with the prospect of improving power of population detection and delineation (Anand et al., 2016). However, average differentiation based on the previously identified SNPs was inflated compared to average estimates based on all Pool‐seq SNPs in the introduced population pair, but not in the natural population pair (Table 4). Such a discrepancy has been found in other studies, for example, in pooled versus individually genotyped SNPs using RAD‐seq (Gaughran et al., 2018) and has multiple possible explanations. Importantly, the previously used SNPs have been selected based on high degree of polymorphisms, while our Pool‐seq includes loci with low minor allele frequencies. Secondly, individually genotyped SNPs are associated with ascertainment biases (Albrechtsen, Nielsen, & Nielsen, 2010; Lachance & Tishkoff, 2013) and the previously used SNPs were developed based on polymorphisms in brown trout of Danish breeding programs and Norwegian rivers (Andersson, Jansson, et al., 2017).
In contrast to assembly‐wide average comparisons of F ST, the correlation of F ST values from previously genotyped SNPs in individuals and the same SNPs in our present Pool‐seq data identified using BLAST showed high consistency between the two methods for the introduced pair, but a weaker correlation for the natural pair where variance in F ST is low. For both pairs, we observe on average somewhat higher F ST values with the individual genotyped SNPs (x‐axes; Figure 5) than from Pool‐seq F ST values (y‐axes; Figure 5). Previous work has used allele frequencies directly to compare Pool‐seq approaches to conventional individual genotyping (Dorant et al., 2019; Hivert, Leblois, Petit, Gautier, & Vitalis, 2018; Zhu, Bergland, González, & Petrov, 2012), and when we compare frequency of the most common allele in each of the SNP loci from the previous individually based genotyping versus those from the pools, we find very strong correlation in all four populations (Figure 6a–d); coefficients of determination (r 2) are over 0.7 in all four populations and linear regression coefficients are well over 0.8 and close to 0.9 for each of the naturally sympatric populations. We suggest that observed differences (Figure 6) are largely due to the small sample sizes of the previous individually genotyped dataset of only n = 18 individuals for each of the introduced populations and n = 30 for each of the natural, sympatric pair. Although the same individuals are predominantly included in the pools, the additional individuals in the pools are likely to affect allele frequency estimates. The consistent observation of on average higher frequencies of the most common allele in the individually based dataset when allele frequencies approach 1, but lower allele frequencies than for the pools when allele frequencies are close to 0.5 is consistent with expectations from smaller sample sizes. Small sample sizes are expected to result in larger allele frequency estimates when the frequency of the most common allele is close to 1, and a contrasting discrepancy at lower allele frequencies. Our observations are in line with this expectation with the smaller sample sizes from the previously genotyped SNPs showing smaller values (Figure 6, x‐axes) of frequency of the most common allele than the allele frequency estimates from Pool‐seq (Figure 6, y‐axes) when close to 0.5 but the opposite when the frequency of the most common allele is close to 1 (compare blue regression lines to orange expected lines in Figure 6). Dorant et al. (2019) observe somewhat better correlations of allele frequencies from Pool‐seq versus conventional genotyping by sequencing than we do. However, they have larger sample sizes (n = 48) in both pools and individual sequencing and use the same individuals in both.
Figure 6.
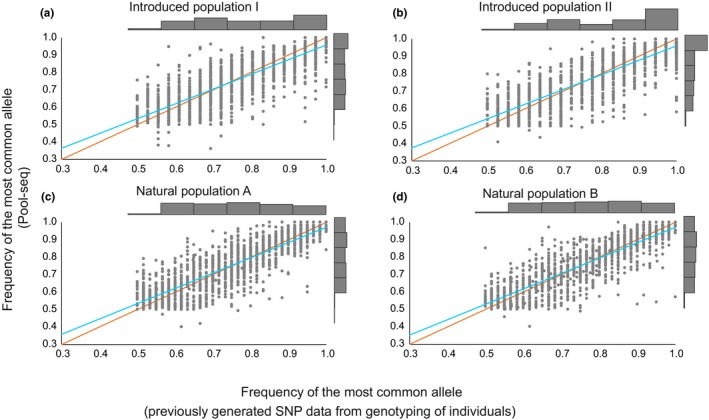
Comparison of frequency of most common alleles of individual SNP loci estimated using Pool‐seq data (y‐axes) versus previously genotyped individuals (x‐axes). (a) Frequency of the most common allele at each of 1,415 individual SNP loci for the introduced population I (linear regression coefficient b = 0.85, r 2 = 0.71, t = 60, p < 0.001), and (b) introduced population II (linear regression coefficient b = 0.84, r 2 = 0.76, t = 66, p < 0.001). (c) Frequency of the most common allele for each of the 1,378 individual SNP loci for the naturally sympatric populations A (linear regression coefficient b = 0.88, r 2 = 0.73, t = 60, p < 0.001) and (d) B (linear coefficient b = 0.88, r 2 = 0.74, t = 63, p < 0.001). Blue lines are linear regression lines, and orange lines represent expected values. The number of individuals was n = 50 for each of the pools and n = 18 for previous data from individual genotyping of the SNP loci for each of the introduced populations, and n = 30 for each of the naturally sympatric populations. Historgrams in the margins represent distributions of allele frequencies
Average assembly‐wide F ST from Pool‐seq data for both pairs of populations were much smaller than the estimates obtained from allozymes (Table 4). Our present data indicate that the previously used allozymes and SNPs do not reflect genome‐wide estimates of average divergence for the introduced populations. Further, our data support expectations and previous findings using SNP markers for the present naturally sympatric populations, namely that relatively few loci appear to be involved in this cryptic substructuring (Andersson, Jansson, et al., 2017). The genomic characteristics of partly reproductively isolated populations are expected to be primarily determined by drift and migration (Jacobs et al., 2018), probably reflected by the previously used SNPs and the assembly‐wide average F ST from Pool‐seq data for the natural pair, and by diversifying selection in limited regions as represented by the allozymes (Andersson, Jansson, et al., 2017). The contention that only a few regions are under selection is further supported by the assembly‐wide patterns of divergence estimated from Pool‐seq data for the natural populations, which showed few sparsely placed peaks of limited differentiation (Figure 4b,d). The loci and genes involved in differentiation of both population pairs remain to be explored, and the present study has provided a tool with which a multitude of SNPs can be detected across the genome.
4.4. Comparison of S. trutta and S. salar references
To validate results obtained based on the S. trutta assembly, we mapped the Pool‐seq data to the Atlantic salmon reference genome (Davidsson et al., 2010; Lien et al., 2016). The relatedness between a resequenced species and the species for which a reference genome is available for mapping has implications for the representation of the resequenced genome (Recknagel et al., 2015). Nucleotide divergence between the Atlantic salmon and brown trout is below 2% (Leitwein et al., 2017). However, since the two Salmo species differ in chromosomal number and structure, as well as degree of tetrasomy, limited mapping success may be expected, for example, due to structural differences (Brenna‐Hansen et al., 2012; Lien et al., 2016). Thus, greater divergence between reads and reference is expected when mapping to the S. salar genome than to the S. trutta assembly, especially since the S. trutta assembly was made from one of the populations analyzed here – the naturally sympatric population A. Indeed, we find differences in mapping success when comparing the two reference genomes (Table 2). In spite of the larger sequence divergence, a higher percentage of reads mapped to the S. salar genome than to the S. trutta assembly. This may be explained by the level of completeness of the two genome assemblies. While the S. salar genome is a highly contiguous assembly at chromosome level (Lien et al., 2016), the S. trutta assembly is reduced to scaffolds harboring gene models that occurred in only one copy, representing c. 20% of the expected size of the S. trutta genome. Reads from genome regions not present in the S. trutta assembly may not map at all or map to the wrong region in the assembly, the latter of which will inflate coverage when mapping to the S. trutta reference.
There are also implications of relatedness between resequencing data and reference assembly for estimates of population genomic variation. Average nucleotide diversity (π) estimates were lower when using the S. salar reference than when mapping pools to the S. trutta assembly (Table 3). This might be attributed to the fact that we are expected to map reads to highly conserved regions between the two species (S. trutta vs. S. salar) and the conserved regions are likely to be less variable. The regions with most sequence diversity, on the other hand, are likely to be those in the process of divergence between the species. Reads mapped to the S. trutta draft assembly are expected to align with higher probability to those regions and may thus be better at reflecting nucleotide diversity. Similarly, the T D values were larger when mapping to the S. salar reference compared to our S. trutta assembly. However, the relative estimates of both π and T D among pools are largely consistent for both references (Table 3).
Divergence estimates were highly concordant when comparing pools mapped to the two references (introduced populations assembly‐wide F ST = 0.13 and 0.15 for S. trutta and S. salar references, respectively, and natural populations assembly‐wide F ST = 0.03 in both cases; Table 4). We were also able to place the F ST results into a “pseudochromosomal” context when using the S. salar genome, which revealed clustered outlier SNPs in the Manhattan plots that were not as apparent when using the S. trutta assembly (Figure 4). We have not pursued outlier analyses here, but plan to return to this issue in forthcoming work. Finally, it is important to note that the genomes we studied here are large and complex and further population genomic studies using the Pool‐seq‐only approach applied here are warranted.
5. CONCLUSIONS
We explored a recently presented Pool‐seq‐only approach to generate a draft genome assembly for a nonmodel species. We then mapped Pool‐seq data to this assembly to estimate population genomic parameters. We used the brown trout (S. trutta) and two pairs of populations from which we had previous population genetic information from allozymes and individually genotyped SNPs. We also mapped our pools to a high‐quality reference genome from Atlantic salmon (S. salar), a closely related species, to compare to our Pool‐seq‐only results. We find high consistency in genome‐wide F ST values between the two population pairs using the Pool‐seq‐only approach versus the S. salar reference. We find less consistency when comparing the genome‐wide Pool‐seq F ST values to those obtained from 14 allozymes and those from c. 3,000 SNPs. In contrast, a high correlation in F ST values and allele frequencies is observed when comparing the exact same SNPs in the pools versus those from previous individual genotyping. Estimates of nucleotide diversity and Tajima's D are higher when mapping to the Pool‐seq assembly versus when mapping to the S. salar reference. However, the relationships between values are largely consistent. We conclude that the Pool‐seq approach explored here is a cost‐effective way to gain basic population genomic insights in nonmodel species where a reference genome is lacking. The method is particularly suitable for exploring population divergence but might also be used to compare relative levels of genome‐wide diversity among populations.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
The biological research initiating this study was led by L.L., S.K., A.A., and N.R. They provided the questions and materials and designed the study guided by L.A., C.W., and C.J.R. S.K. analyzed the data with M.P.C.M., J.H., and V.E.K., supervised by C.W., V.E.K., and L.A. Graphs and statistical analyses were produced by S.K., assisted by A.A. S.K. also led the writing, with all authors contributing to and approving the submitted manuscript. The research was funded by grants to L.L.
Supporting information
ACKNOWLEDGMENTS
We thank Ramprasad Neethiraj for bioinformatic assistance, Atal Saha for valuable suggestions, Mari Edman for proofreading, and three anonymous reviewers for valuable suggestions. Collaboration with Jijnjevaerie local Sami community, the County Administrative Board of Jämtland, and National Genomics Infrastructure (NGI) Uppsala (Science for Life Laboratory) is acknowledged. This research was supported by the Swedish Research Council Formas (L.L.), the Swedish Agency for Marine and Water Management (L.L.), the SciLifeLab Bioinformatics Long‐term Support (L.L.), and the Carl Trygger Foundation (L.L.).
Kurland S, Wheat CW, de la Paz Celorio Mancera M, et al. Exploring a Pool‐seq‐only approach for gaining population genomic insights in nonmodel species. Ecol Evol. 2019;9:11448–11463. 10.1002/ece3.5646
Contributor Information
Sara Kurland, Email: sara.kurland@zoologi.su.se.
Linda Laikre, Email: linda.laikre@popgen.su.se.
DATA AVAILABILITY STATEMENT
Data for this study are available in the Dryad repository: https://doi.org/10.5061/dryad.q1h4k0n. Population genomic scripts are available in Appendix S3.
REFERENCES
- Albrechtsen, A. , Nielsen, F. C. , & Nielsen, R. (2010). Ascertainment biases in SNP chips affect measures of population divergence. Molecular Biology and Evolution, 27, 2534–2547. 10.1093/molbev/msq148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allendorf, F. W. , & Hard, J. J. (2009). Human‐induced evolution caused by unnatural selection through harvest of wild animals. Proceedings of the National Academy of Sciences of the United States of America, 106, 9987–9994. 10.1073/pnas.0901069106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allendorf, F. W. , & Ryman, N. (2002). The role of genetics in population viability analysis In Beissinger S. R. & McCullough D. R. (Eds.), Population viability analysis (pp. 50–85). Chicago, IL: University of Chicago Press. [Google Scholar]
- Anand, S. , Mangano, E. , Barizzone, N. , Bordoni, R. , Sorosina, M. , Clarelli, F. , & De Bellis, G. (2016). Next generation sequencing of pooled samples: Guideline for variants' filtering. Scientific Reports, 6, 33735 10.1038/srep33735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson, A. , Jansson, E. , Wennerström, L. , Chiriboga, F. , Arnyasi, M. , Kent, M. P. , & Laikre, L. (2017). Complex genetic diversity patterns of cryptic, sympatric brown trout (Salmo trutta) populations in tiny mountain lakes. Conservation Genetics, 18, 1213–1227. 10.1007/s10592-017-0972-4 [DOI] [Google Scholar]
- Andersson, A. , Johansson, F. , Sundbom, M. , Ryman, N. , & Laikre, L. (2017). Lack of trophic polymorphism despite substantial genetic differentiation in sympatric brown trout (Salmo trutta) populations. Ecology of Freshwater Fish, 26, 643–652. 10.1111/eff.12308 [DOI] [Google Scholar]
- Bernatchez, L. (2016). On the maintenance of genetic variation and adaptation to environmental change: Considerations from population genomics in fishes. Journal of Fish Biology, 89, 2519–2556. 10.1111/jfb.13145 [DOI] [PubMed] [Google Scholar]
- Berthelot, C. , Brunet, F. , Chalopin, D. , Juanchich, A. , Bernard, M. , Noël, B. , & Guiguen, Y. (2014). The rainbow trout genome provides novel insights into evolution after whole‐genome duplication in vertebrates. Nature Communications, 5, 3657 10.1038/ncomms4657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenna‐Hansen, S. , Li, J. , Kent, M. P. , Boulding, E. G. , Dominik, S. , Davidson, W. S. , & Lien, S. (2012). Chromosomal differences between European and North American Atlantic salmon discovered by linkage mapping and supported by fluorescence in situ hybridization analysis. BMC Genomics, 13, 432 10.1186/1471-2164-13-432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carruthers, M. , Yurchenko, A. A. , Augley, J. J. , Adams, C. E. , Herzyk, P. , & Elmer, K. R. (2018). De novo transcriptome assembly, annotation and comparison of four ecological and evolutionary model salmonid fish species. BMC Genomics, 19(32), 1–17. 10.1186/s12864-017-4379-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlier, J. , Laikre, L. , & Ryman, N. (2012). Genetic monitoring reveals temporal stability over 30 years in a small, lake‐resident brown trout population. Heredity, 109, 246–253. 10.1038/hdy.2012.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen, K. A. , Leong, J. S. , Sakhrani, D. , Biagi, C. A. , Minkley, D. R. , Withler, R. E. , … Devlin, R. H. (2018). Chinook salmon (Oncorhynchus tshawytscha) genome and transcriptome. PLoS ONE, 13(4), e0195461 10.1371/journal.pone.0195461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen, K. A. , Rondeau, E. B. , Minkley, D. R. , Leong, J. S. , Nugent, C. M. , Danzmann, R. G. , … Koop, B. F. (2018). The Arctic charr (Salvelinus alpinus) genome and transcriptome assembly. PLoS ONE, 13(9), e0204076 10.1371/journal.pone.0204076 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Davey, J. W. , Hohenlohe, P. A. , Etter, P. D. , Boone, J. Q. , Catchen, J. M. , & Blaxter, M. L. (2011). Genome‐wide genetic marker discovery and genotyping using next‐generation sequencing. Nature Reviews Genetics, 12, 499–510. 10.1038/nrg3012 [DOI] [PubMed] [Google Scholar]
- Davidsson, W. S. , Koop, B. F. , Jones, S. J. M. , Iturra, P. , Vidal, R. , Maass, A. , … Omholt, S. W. (2010). Sequencing the genome of the Atlantic salmon (Salmo salar). Genome Biology, 11, 403 10.1186/gb-2010-11-9-403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorant, Y. , Benestan, L. , Rougemont, Q. , Normandeau, E. , Boyle, B. , Rochette, R. , & Bernatchez, L. (2019). Comparing Pool‐seq, Rapture, and GBS genotyping for inferring weak population structure: The American lobster (Homarus americanus) as a case study. Ecology and Evolution, 9, 6606–6623. 10.1002/ece3.5240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush, D. , Stephens, M. , & Pritchard, J. K. (2003). Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics, 164, 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes‐Pardo, A. P. , & Ruzzante, D. E. (2017). Whole‐genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Molecular Ecology, 26, 5369–5406. 10.1111/mec.14264 [DOI] [PubMed] [Google Scholar]
- Gagnaire, P.‐A. , Pavey, S. A. , Normandeau, E. , & Bernatchez, L. (2013). The genetic architecture of reproductive isolation during speciation‐with‐gene‐flow in Lake Whitefish species pairs assessed by RAD sequencing. Evolution, 67, 2483–2497. 10.1111/evo.12075 [DOI] [PubMed] [Google Scholar]
- García‐Alcalde, F. , Okonechnikov, K. , Carbonell, J. , Cruz, L. M. , Götz, S. , Tarazona, S. , … Conesa, A. (2012). Qualimap: Evaluating next‐generation sequencing alignment data. Bioinformatics, 28, 2678–2679. 10.1093/bioinformatics/bts503 [DOI] [PubMed] [Google Scholar]
- Gaughran, S. J. , Quinzin, M. C. , Miller, J. M. , Garrick, R. C. , Edwards, D. L. , Russello, M. A. , … Caccone, A. (2018). Theory, practice, and conservation in the age of genomics: The Galápagos giant tortoise as a case study. Evolutionary Applications, 11, 1084–1093. 10.1111/eva.12551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, D. (2013). EvidentialGene: tr2aacds, mRNA transcript assembly software. Retrieved from http://arthropods.eugenes.org/EvidentialGene/
- Gillespie, J. H. (2004). Population genetics: A concise guide. Baltimore, MD and London, UK: The Johns Hopkins University Press. [Google Scholar]
- Gotoh, O. (2008). Direct mapping and alignment of protein sequences onto genomic sequence. Bioinformatics, 24, 2438–2444. 10.1093/bioinformatics/btn460 [DOI] [PubMed] [Google Scholar]
- Grabherr, M. G. , Haas, B. J. , Yassour, M. , Levin, J. Z. , Thompson, D. A. , Amit, I. , … Regev, A. (2011). Trinity: Reconstructing a full‐length transcriptome without a genome from RNA‐Seq data. Nature Biotechnology, 29(7), 644 10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, M. M. (2002). Estimating the long‐term effects of stocking domesticated trout into wild brown trout (Salmo trutta) populations: An approach using microsatellite DNA analysis of historical and contemporary samples. Molecular Ecology, 11, 1003–1015. 10.1046/j.1365-294X.2002.01495.x [DOI] [PubMed] [Google Scholar]
- Hansen, M. M. , Ruzzante, D. E. , Nielsen, E. E. , & Mensberg, K. L. D. (2000). Microsatellite and mitochondrial DNA polymorphism reveals life‐history dependent interbreeding between hatchery and wild brown trout (Salmo trutta L.). Molecular Ecology, 9, 583–594. 10.1046/j.1365-294x.2000.00898.x [DOI] [PubMed] [Google Scholar]
- Hindar, K. , Ryman, N. , & Utter, F. (1991). Genetic effects of cultured fish on natural fish populations. Canadian Journal of Fisheries and Aquatic Sciences, 48, 945–957. 10.1139/f91-111 [DOI] [Google Scholar]
- Hivert, V. , Leblois, R. , Petit, E. J. , Gautier, M. , & Vitalis, R. (2018). Measuring genetic differentiation from Pool‐seq data. Genetics, 210, 315–330. 10.1534/genetics.118.300900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs, A. , Carruthers, M. , Yurchenko, A. , Gordeeva, N. , Alekseyev, S. , Hooker, O. , … Elmer, K. R. (2018). Convergence in form and function overcomes non‐parallel evolutionary histories in Arctic charr. bioRxiv, 265272 10.1101/265272 [DOI] [Google Scholar]
- Jorde, P. E. , & Ryman, N. (1996). Demographic genetics of brown trout (Salmo trutta) and estimation of effective population size from temporal change of allele frequencies. Genetics, 143, 1369–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keehnen, N. L. , Hill, J. , Nylin, S. , & Wheat, C. W. (2018). Microevolutionary selection dynamics acting on immune genes of the green veined white butterfly, Pieris napi . Molecular Ecology, 27, 2807–2822. 10.1111/mec.14722 [DOI] [PubMed] [Google Scholar]
- Kofler, R. , Langmüller, A. M. , Nouhaud, P. , Otte, A. K. , & Schlötterer, C. (2016). Suitability of different mapping algorithms for genome‐wide polymorphism scans with Pool‐Seq data. G3: Genes, Genomes, Genetics, 6, 3507–3515. 10.1534/g3.116.034488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler, R. , Orozco‐ter Wengel, P. , de Maio, N. , Pandey, R. V. , Nolte, V. , Futschik, A. , & Schlötterer, C. (2011). PoPoolation: A toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS ONE, 6, e15925 10.1371/journal.pone.0015925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler, R. , Pandey, R. V. , & Schlötterer, C. (2011). PoPoolation2: Identifying differentiation between populations using sequencing of pooled DNA samples (Pool‐Seq). Bioinformatics, 27, 3435–3436. 10.1093/bioinformatics/btr589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachance, J. , & Tishkoff, S. A. (2013). SNP ascertainment bias in population genetic analyses: Why it is important, and how to correct it. BioEssays, 35, 780–786. 10.1002/bies.201300014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laikre, L. (Ed.) (1999). Conservation genetic management of brown trout (Salmo trutta) in Europe, TROUTCONCERT EU FAIR CT97‐3882, ISBN 87‐987732‐0‐8. Retrieved from https://www.researchgate.net/publication/305657027_Conservation_Genetic_Management_of_Brown_Trout_Salmo_trutta_in_Europe [Google Scholar]
- Leitwein, M. , Gagnaire, P. A. , Desmarais, E. , Guendouz, S. , Rohmer, M. , Berrebi, P. , & Guinand, B. (2016). Genome‐wide nucleotide diversity of hatchery‐reared Atlantic and Mediterranean strains of brown trout Salmo trutta compared to wild Mediterranean populations. Journal of Fish Biology, 89, 2717–2734. 10.1111/jfb.13131 [DOI] [PubMed] [Google Scholar]
- Leitwein, M. , Gagnaire, P.‐A. , Desmares, E. , Berrebi, P. , & Guinand, B. (2018). Genomic consequences of a recent three‐way admixture in supplemented wild brown trout populations revealed by local ancestry tracts. Molecular Ecology, 27, 3466–3483. 10.1111/mec.14816 [DOI] [PubMed] [Google Scholar]
- Leitwein, M. , Guinand, B. , Pouzadoux, J. , Desmarais, E. , Berrebi, P. , & Gagnaire, P.‐A. (2017). A dense brown trout (Salmo trutta) linkage map reveals recent chromosomal rearrangements in the Salmo genus and the impact of selection on linked neutral Diversity. G3: Genes, Genomes, Genetics, 7, 1365–1376. 10.1534/g3.116.038497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerceteau‐Köhler, E. , Schliewen, U. , Kopun, T. , & Weiss, S. (2013). Genetic variation in brown trout Salmo trutta across the Danube, Rhine, and Elbe headwaters: A failure of the phylogeographic paradigm? BMC Evolutionary Biology, 13, 176 10.1186/1471-2148-13-176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler Transform. Bioinformatics, 25, 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , & Durbin, R. (2009). The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien, S. , Koop, B. F. , Sandve, S. R. , Miller, J. R. , Kent, M. P. , Nome, T. , & Davidson, W. S. (2016). The Atlantic salmon genome provides insights into rediploidization. Nature, 533, 200–205. 10.1038/nature17164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, E. , Buonaccorsi, V. , Hyde, J. R. , & Aguilar, A. (2017). Population genomics reveals high gene flow in grass rockfish (Sebastes rastrelliger). Marine Genomics, 33, 57–63. 10.1016/j.margen.2017.01.004 [DOI] [PubMed] [Google Scholar]
- Meier, K. , Hansen, M. M. , Bekkevold, D. , Skaala, Ø. , & Mensberg, K.‐L.‐D. (2011). An assessment of the spatial scale of local adaptation in brown trout (Salmo trutta L.): Footprints of selection at microsatellite DNA loci. Heredity, 106, 488–499. 10.1038/hdy.2010.164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier, K. , Hansen, M. M. , Normandeau, E. , Mensberg, K.‐L.‐D. , Frydenberg, J. , Larsen, P. F. , … Bernatchez, L. (2014). Local adaptation at the transcriptome level in brown trout: Evidence from early life history temperature genomic reaction norms. PLoS ONE, 9, e85171 10.1371/journal.pone.0085171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narum, S. R. , Genova, A. D. , Micheletti, S. J. , & Maass, A. (2018). Genomic variation underlying complex life‐history traits revealed by genome sequencing in Chinook salmon. Proceedings of the Royal Society B: Biological Sciences, 285, 20180935 10.1098/rspb.2018.0935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neethiraj, R. , Hornett, E. A. , Hill, J. A. , & Wheat, C. W. (2017). Investigating the genomic basis of discrete phenotypes using a Pool‐Seq‐only approach: New insights into the genetics underlying colour variation in diverse taxa. Molecular Ecology, 26, 4990–5002. 10.1111/mec.14205 [DOI] [PubMed] [Google Scholar]
- Nei, M. (1973). Analysis of gene diversity in subdivided populations. Proceedings of the National Academy of Sciences of the United States of America, 70, 3321–3323. 10.1073/pnas.70.12.3321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent, C. M. , Easton, A. A. , Norman, J. D. , Ferguson, M. M. , & Danzmann, R. G. (2017). A SNP based linkage map of the Arctic charr (Salvelinus alpinus) genome provides Insights into the diploidization process after whole genome duplication. G3: Genes, Genomes, Genetics, 7, 543–556. 10.1534/g3.116.038026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovenden, J. R. , Berry, O. , Welch, D. J. , Buckworth, R. C. , & Dichmont, C. M. (2015). Ocean's eleven: A critical evaluation of the role of population, evolutionary and molecular genetics in the management of wild fisheries. Fish and Fisheries, 16, 125–159. 10.1111/faf.12052 [DOI] [Google Scholar]
- Palm, S. , & Ryman, N. (1999). Genetic basis of phenotypic differences between transplanted stocks of brown trout. Ecology of Freshwater Fish, 8(3), 169–180. 10.1111/j.1600-0633.1999.tb00068.x [DOI] [Google Scholar]
- Palmé, A. , Laikre, L. , & Ryman, N. (2013). Monitoring reveals two genetically distinct brown trout populations remaining in stable sympatry over 20 years in tiny mountain lakes. Conservation Genetics, 14, 795–808. 10.1007/s10592-013-0475-x [DOI] [Google Scholar]
- Petereit, C. , Bekkevold, D. , Nickel, S. , Dierking, J. , Hantke, H. , Hahn, A. , & Puebla, O. (2018). Population genetic structure after 125 years of stocking in sea trout (Salmo trutta L.). Conservation Genetics, 19, 1–14. 10.1007/s10592-018-1083-6 [DOI] [Google Scholar]
- Phillips, R. , & Ráb, P. (2001). Chromosome evolution in the Salmonidae (Pisces): An update. Biological Reviews, 76, 1–25. 10.1111/j.1469-185X.2000.tb00057.x [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K. , Stephens, M. , & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruisscher, P. , Nylin, S. , Gotthard, K. , & Wheat, C. W. (2018). Genetic variation underlying local adaptation of diapause induction along a cline in a butterfly. Molecular Ecology, 27, 3613–3626. 10.1111/mec.14829 [DOI] [PubMed] [Google Scholar]
- Pustovrh, G. , Snoj, A. , & Bajec, S. S. (2014). Molecular phylogeny of Salmo of the western Balkans, based upon multiple nuclear loci. Genetics Selection Evolution, 46, 7 10.1186/1297-9686-46-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan, A. R. , & Hall, I. M. (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2017). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; Retrieved from https://www.r-project.org/ [Google Scholar]
- Recknagel, H. , Jacobs, A. , Herzyk, P. , & Elmer, K. R. (2015). Double‐digest RAD sequencing using Ion Proton semiconductor platform (ddRADseq‐ion) with nonmodel organisms. Molecular Ecology Resources, 15, 1316–1329. 10.1111/1755-0998.12406 [DOI] [PubMed] [Google Scholar]
- Robinson, J. T. , Thorvaldsdóttir, H. , Winckler, W. , Guttman, M. , Lander, E. S. , Getz, G. , & Mesirov, J. P. (2011). Integrative genomics viewer. Nature Biotechnology, 29, 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset, F. (2008). GENEPOP'007: A complete re‐implementation of the GENEPOP software for Windows and Linux. Molecular Ecology Resources, 8, 103–106. 10.1111/j.1471-8286.2007.01931.x [DOI] [PubMed] [Google Scholar]
- Ryman, N. (1981). Conservation of genetic resources: Experiences from the brown trout (“Salmo trutta”) In Ryman N. (Ed.), Fish gene pools: Preservation of genetic resources in relation to wild fish stocks (vol. 34, pp. 61–74). Stockholm, Sweden: Ecological Bulletins; The Editorial Service / FRN. Retrieved from http://www.jstor.org/stable/43908647 [Google Scholar]
- Ryman, N. , Allendorf, F. W. , & Ståhl, G. (1979). Reproductive isolation with little genetic divergence in sympatric populations of Brown trout (Salmo trutta). Genetics, 92, 247–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sävilammi, T. , Primmer, C. R. , Varadharajan, S. , Guyomard, R. , Guiguen, Y. , Sandve, S. R. , … Lien, S. (2019). The chromosome‐level genome assembly of European grayling reveals aspects of a unique genome evolution process within salmonids. G3: Genes, Genomes, Genetics, 9, 1283–1294. 10.1534/g3.118.200919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer, C. , Tobler, R. , Kofler, R. , & Nolte, V. (2014). Sequencing pools of individuals‐mining genome‐wide polymorphism data without big funding. Nature Reviews Genetics, 15, 749–763. 10.1038/nrg3803 [DOI] [PubMed] [Google Scholar]
- Sedlazeck, F. J. , Rescheneder, P. , & Von Haeseler, A. (2013). NextGenMap: Fast and accurate read mapping in highly polymorphic genomes. Bioinformatics, 29(21), 2790–2791. 10.1093/bioinformatics/btt468 [DOI] [PubMed] [Google Scholar]
- Simão, F. A. , Waterhouse, R. M. , Ioannidis, P. , Kriventseva, E. V. , & Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. 10.1093/bioinformatics/btv351 [DOI] [PubMed] [Google Scholar]
- Song, L. , & Florea, L. (2015). Rcorrector: Efficient and accurate error correction for Illumina RNA‐seq reads. GigaScience, 4, 1–8. 10.1186/s13742-015-0089-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulé, M. E. , & Wilcox, B. A. (1980). Conservation biology: An evolutionary‐ecological perspective. Sunderland, MA: Sinauer Associates Inc. [Google Scholar]
- Stelkens, R. B. , Pompini, M. , & Wedekind, C. (2012). Testing for local adaptation in brown trout using reciprocal transplants. BMC Evolutionary Biology, 12, 247 10.1186/1471-2148-12-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzek, B. E. , Huang, H. , McGarvey, P. , Mazumder, R. , & Wu, C. H. (2007). UniRef: Comprehensive and non‐redundant UniProt reference clusters. Bioinformatics, 23, 1282–1288. 10.1093/bioinformatics/btm098 [DOI] [PubMed] [Google Scholar]
- Tajima, F. (1983). Evolutionary relationship of DNA sequences in finite populations. Genetics, 105, 437–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics, 123, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therkildsen, N. O. , & Palumbi, S. R. (2017). Particular low‐coverage genomewide sequencing hundreds of individually barcoded samples for population and evolutionary genomics in nonmodel species. Molecular Ecology Resources, 17, 194–208. 10.1111/1755-0998.12593 [DOI] [PubMed] [Google Scholar]
- Volckaert, F. A. M. (2015). Applying the hidden treasures of population biodiversity to decision making and management. Ocean and Coastal Management, 118, 47–50. 10.1016/j.ocecoaman.2014.12.018 [DOI] [Google Scholar]
- Vøllestad, L. A. (2018). Understanding brown trout population genetic structure: A Northern‐European perspective In Lobón‐Cerviá J. & Sanz N. (Eds.), Brown trout: Biology, ecology and management (pp. 127–144). West Sussex, UK: John Wiley & Sons, Ltd; 10.1002/9781119268352.ch5 [DOI] [Google Scholar]
- Wang, Q. , Shashikant, C. S. , Jensen, M. , Altman, N. S. , & Girirajan, S. (2017). Novel metrics to measure coverage in whole exome sequencing datasets reveal local and global non‐uniformity. Scientific Reports, 7(1), 1–11. 10.1038/s41598-017-01005-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples, R. , Punt, A. E. , & Cope, J. M. (2008). Integrating genetic data into management of marine resources: How can we do it better ? Fish and Fisheries, 455, 424–435. 10.1111/j.1467-2979.2008.00303.x [DOI] [Google Scholar]
- Weir, B. S. , & Cockerham, C. C. (1984). Estimating F‐statistics for the analysis of population structure. Evolution, 38(6), 1358–1370. 10.1111/j.1558-5646.1984.tb05657.x [DOI] [PubMed] [Google Scholar]
- Woronik, A. , & Wheat, C. W. (2017). Advances in finding Alba: The locus affecting life history and color polymorphism in a Colias butterfly. Journal of Evolutionary Biology, 30, 26–39. 10.1111/jeb.12967 [DOI] [PubMed] [Google Scholar]
- Zhu, Y. , Bergland, A. O. , González, J. , & Petrov, D. A. (2012). Empirical validation of pooled whole genome population re‐sequencing in Drosophila melanogaster . PLoS ONE, 7(7), e41901 10.1371/journal.pone.0041901 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data for this study are available in the Dryad repository: https://doi.org/10.5061/dryad.q1h4k0n. Population genomic scripts are available in Appendix S3.