

Abstract
Nuclear depletion of TDP-43, an essential RNA binding protein, may underlie neurodegeneration in amyotrophic lateral sclerosis (ALS). As several functions have been ascribed to this protein, the critical role(s) of TDP-43 in motor neurons that may be compromised in ALS remains unknown. We show here that TDP-43 mediated splicing repression, which serves to protect the transcriptome by preventing aberrant splicing, is central to the physiology of motor neurons. Expression in Drosophila TDP-43 knockout models of a chimeric repressor, comprised of the RNA recognition domain of TDP-43 fused to an unrelated splicing repressor, RAVER1, attenuated motor deficits and extended lifespan. Likewise, AAV9-mediated delivery of this chimeric rescue repressor to mice lacking TDP-43 in motor neurons delayed the onset, slowed the progression of motor symptoms, and markedly extended their lifespan. In treated mice lacking TDP-43 in motor neurons, aberrant splicing was significantly decreased and accompanied by amelioration of axon degeneration and motor neuron loss. This AAV9 strategy allowed long-term expression of the chimeric repressor without any adverse effects. Our findings establish that splicing repression is a major function of TDP-43 in motor neurons and strongly support the idea that loss of TDP-43-mediated splicing fidelity represents a key pathogenic mechanism underlying motor neuron loss in ALS.
Keywords: Amyotrophic lateral sclerosis, cryptic exon, Drosophila, motor neuron, mouse, TDP-43
Introduction
Tar DNA-binding protein 43 (TDP-43, encoded by the gene TARDBP), an essential, highly-conserved RNA binding protein [10, 53, 65], has been implicated in amyotrophic lateral sclerosis, a progressive neurodegenerative disease characterized by the death of upper and lower motor neurons. In nearly all cases of sporadic ALS, TDP-43 in motor neurons depletes from the nucleus and aggregates in the cytoplasm [59]. Missense mutations in TDP-43, which mostly cluster within its C-terminal domain, are linked to familial ALS [26, 55]. Various other genetic mutations associated with familial ALS are also associated with TDP-43 pathology, supporting the notion that TDP-43 mislocalization is central to its pathogenesis [9, 48]. In addition, TDP-43 pathology is evident in cases with frontotemporal dementia [45], inclusion body myositis [51], and Alzheimer’s disease [2]. However, the precise mechanism underlying TDP-43 pathology remains unclear. While initial efforts focused on studies of transgenic wild-type human and ALS-linked mutant human TDP-43 overexpression models [3, 54, 64, 67], subsequent research suggests that loss of TDP-43 function also plays a critical role in motor neuron degeneration [14, 52, 68].
TDP-43 is thought to play important roles in several cellular processes, including cellular stress response pathways [37], mRNA delivery to dendritic or axonal compartments [1, 41], or phase separation of membrane-less organelles [18, 19, 35, 38, 42]. As a member of the heterogeneous ribonuclear protein (hnRNP) family, nuclear TDP-43 is concentrated in transcriptionally-active euchromatin regions [8], and is thought to regulate alternative splicing [7, 46, 47, 60]. TDP-43 interacts with many proteins and RNAs, potentially regulating numerous pathways [15, 17]. Previously, we and others discovered that TDP-43 acts as a guardian of the transcriptome by repressing the splicing of nonconserved, unannotated ‘cryptic’ exons [33, 57], a function that is compromised in cases of ALS and other neurodegenerative diseases with TDP-43 pathology [29, 56]. However, since TDP-43 is involved in multiple intracellular roles [1, 37, 42], it remains unknown whether splicing repression is a major function of TDP-43 in motor neurons. Using multiple model systems and an AAV9-mediated gene delivery approach [16], we establish here that TDP-43-mediated splicing repression is central to the physiology of motor neurons. These findings support the notion that TDP-43 mediated splicing repression is a novel mechanism-based therapeutic target for ALS and potentially other human diseases exhibiting nuclear depletion of TDP-43.
Materials and Methods
Fly stocks and transgenic flies
Flies were maintained at 25°C on a standard cornmeal/agar medium. TBPH null mutant TBPHΔ23 was kindly provided by Dr. Fabian Feiguin. All Gal4 driver lines were obtained from the Bloomington Stock Center at Indiana University (Bloomington, IN). To construct the fly CTR expression vector, a DNA fragment corresponding to aa 1-267 of human TDP-43 and aa450-643 of RAVER1 was obtained from CTR construct [33] using the following primers: forward primer: CCGCTCGAGACCATGGCCTCTGAATATATTCG; reverse primer: CGTCTAGACTACCGTTCGCCGAAGCCACTTG. For the NTDP construct, the following primers were used: forward primer, ATCTCGAGATGGTGAGCAAGGGCGAGGA; reverse primer: ATCTAGACTATCTATTGCTATTGTGCTTAGGTTCGGC. For the CTRF147L/F149L construct, the DNA fragment corresponding to aa 1-267 of human TDP-43 carrying F147L and F149L double mutations in RRM domain and aa450-643 of RAVER1 was synthesized by GeneArt Gene Synthesis (Invitrogen, Waltham, MA). The above DNA fragments were subsequently cloned into pUAST at XhoI and XhaI sites. All transgenic fly strains were generated by BestGene Inc., CA.
Library construction and massively parallel sequencing
RNA was extracted from fly heads using TRIzol. Total RNA for RNA-seq was then processed using the TruSeq Stranded Total RNA Library Prep Kit (Illumina) to construct 100-bp paired end stranded RNA-seq libraries. Sample libraries were sequenced on a HiSeq 2000 to generate approximately 70 million reads per sample which was de-multiplexed and converted into fastq files. Fastq files were aligned to Drosophila genomes using TopHat and annotated using Cufflinks on Galaxy, an open-source, web-based bioinformatics platform. Cryptic exons were initially identified through manual screening of novel exons annotated by Cufflinks that were highly abundant in the TBPH null dataset but not control. TopHat aligned data was then displayed to the UCSC Genome Browser to visualize RNA-seq coverage.
RNA Analysis (Drosophila)
Total RNA from fly heads was purified using RNeasy mini kit (Qiagen) according to manufacturer’s instructions. The cDNA synthesis was performed starting from lug of each RNA sample using RevertAid RT reverse transcription Kit (Thermo Scientific). PCR analysis for cryptic exons has been performed using the following primers: Rugose-F TGTTGCATGTGTGAGCGTGG; Rugose-R GCTGTTGGAGCGTTTGATGT. Pkc53E-F TTCGAGCCATTCACATACGC; PKC53E-R TTCCCACGCAGACACTCACA; Sev-F AGTTCTGCTGCGATCGCGCC; Sev-R CAGCAACAACAACAGCACAG; Pyd-F CTAGATCCTCAGTCCGTTAAC; Pyd-R CTGATCGATATCATTGCCAAGA. CG42450-F TGTGTGTGTTTGTGGGAGTGTG; CG42450-R GTGTCAGGACCTTAAAGGCGGT; Uif-F GCGCGTTGTGGACGTTAAGA; Uif-R GCCGTACTGAAGAAAACCCA; Dyb-Fl CGCGATAAGTGCAGAGAAACAG; Dyb-Rl GCTATGGATGGGAATTCCGCAT; CG8045-F1 GGTGTGTGTGTGTGTGTGTGTGTGTGCAC; CG8045-R ACCTGGTGCTGGGCCATGAT
Video-assisted movement tracking
Tracking arenas were 3.5 cm diameter Petri dish (Corning) filled with transparent silicon elastomer (Dow Corning) with a 3-mm space so that flies could walk freely but not fly. A CCD camera positioned above the arenas was connected to a PC. Three flies were briefly anaesthetized with CO2, placed in the arena and left to recover at 22°C for 1 hour before being tested. Tracking was carried out at 22°C. Recorded videos were converted to fly movie format using the motmot package and loaded into Ctrax software to analyze the positions of the flies throughout the video. Position data for the 3-minute file was exported as a matrix file. Errors in the fracking were fixed using Matlab (Mathworks) as well as FixErrors GUI, which is described in further detail at http://ctrax.sourceforge.net/fixerrors.html. Fixed trajectories were analyzed in Matlab to calculate the mean cumulative distance travelled by the population of flies in the arena. Significance was calculated using the Mann–Whitney U-test with a Bonferroni correction to account for multiple comparisons.
Lifespan analysis
Flies were maintained on standard medium at 25°C. Each group of flies was aged in individual vials containing no more than 20 flies and were transferred to a new vial every 1–2 days. The number of dead flies was recorded for longevity analysis.
Drosophila neuromuscular junction (NMJ) staining
Wandering third instar larval NMJs were dissected out in 1 × PBS and fixed in 4% paraformaldehyde for 40 minutes. After three 5-minute washes, preparations were blocked with 5% normal goat serum in 0.1 M phosphate buffer (pH 7.2) containing 0.3% TritonX-100 for 1 h. Next, a mixture of primary antibodies was added and incubated overnight at 4°C Primary antibodies used are anti-ELAV (DSHB, 1:20), anti-TDP-43 (Proteintech, 10782-2-AP, 1:200), anti-HRP (Jackson ImmunoResearch, 323-001-021, 1:1000), anti-DLG (DSHB, 4F3, 1:50), and anti-Futsch (DSHB, 22C10, 1:20).
Mouse Generation
All mouse procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Johns Hopkins University Animal Care and Use Committee.
We crossbred our previously described conditional Tardbp knockout mice (tardbpF/F, Jax stock 017591) with ChAT-IRES-Cre transgenic mice on a C57BL/6J background (Jax stock 006410) to obtain a cohort of ChAT-IRES-Cre;TardbpF/+ mice. These were subsequently crossed again with tardbpF/F mice to generate the final cohort of ChAT-IRES-Cre;TardbpF/+ (control) and ChAT-IRES-Cre;tardbpF/F (TDP-43 knockout) mice. All mice were housed under a 12L:12D daily cycle and managed by Johns Hopkins University Research Animal Resources (RAR).
Viral Vector Packaging
AAV9 packaging was performed by Virovek (Hayward CA), and CTR or GFP exp: independently confirmed by HeLa cell transduction and western blot prior to all experiments.
Intracerebroventricular (ICV) Injection
All injections were performed on mouse pups within 8 hours of birth. Pups were cryo-anaesthetized on wet ice for no longer than 2 minutes. A latex barrier between pups and ice prevented skin damage. A sterile, single-use pulled glass needle (Drummond microcaps, 41 mm length, approx. 0.5 mm minimum diameter) was penetrated 3mm into the lateral ventricle of each cryo-anaesthetized mouse pup to slowly deliver 3 μl of AAV9 (1E1013 vg/ml) carrying either our chimeric splicing repressor protein, termed CTR, or GFP into the lateral ventricles over 15 seconds; solutions contained 0.05% trypan blue dye for localization. Pups recovered under a heat lamp with bedding from their home cage in order to restore scent, after which they were returned to their mother cage and monitored after 6 and 12 hours and daily afterwards. A successful injection was identified by dye-induced darkening of the spinal column at 6 hours post-injection. Approximately 80% of pups successfully recovered from injection, with 10% insufficiently recovering from anesthesia at p0-p1 and 10% displaying signs of hydrocephaly at p14-p21. All pups showing signs of distress at any time following recovery were euthanized.
Sample Size
Sample sizes for all mouse cohorts examined in this study are presented in Table S2. All litters were injected prior to genotyping. For motor function and survival analyses, a total of 40 mice from 8 litters were injected for cohort 1. Every pup in each litter was injected at random with an AAV9 vector carrying either our CTR fusion protein or GFP as a control, and all pups in each litter received either CTR or GFP. A total of 8 ChAT-IRES-Cre;TardbpF/+ mice were injected with GFP, 8 ChAT-IRES-Cre;TardbpF/+ mice were injected with CTR, 11 ChAT-IRES-Cre;tardbpF/F were injected with GFP, and 11 ChAT-IRES-Cre;tardbpF/F mice were injected with CTR. The size of cohort 1 was sufficiently powered to measure a mean survival increase of 50% with 25% standard deviation (l-β = 0.994). We then replicated our results by injecting a second cohort of mice from the same breeder pairs, termed ‘cohort 2’. Each litter in cohort 2 received the opposite payload as its cohort 1 counterpart. Four additional cohorts, each from the same breeder pairs, were bred and injected for analysis of spinal cord pathology at 1 month, 3 months, and 5 months (pathology analysis cohorts) and for injection of an AAV9 vector carrying only the N-terminal fragment (NTF) of TDP-43 as another control (NTF cohort).
Hanging Wire Test
Hanging wire tests were performed weekly beginning at p30. Investigators were blindo group of each animal. Each mouse was placed on the center of a metal grid that then was prompt the mouse to hold on before being turned upside down 30 cm over an empty cage. Each mouse was allowed up to three attempts to hold on to the inverted grid for an arbitrary maximum of 60 seconds. Mice were given >2 minutes of rest between attempts, and the best attempt was used for analyses.
Accelerating Rotarod Test
Rotarod tests were performed once every two weeks with an initial acclimatization session beginning at p30. Investigators were blinded to the treatment group of each animal. Mice were placed on the apparatus (Rotamex 5, Columbus Instruments) with a rod diameter of 3 cm, minimum speed of 4 rpm, and accelerating at 20 rpm/min. Latency to fall was recorded. Each mouse was given three attempts with >30 minutes of rest between attempts, and the best attempt was used for analyses.
Survival
Upon showing symptoms of hindlimb paralysis, mice were provided wet chow and Dietgel on the cage floor. End-stage was defined as a failure of a mouse to right itself within 10 seconds when placed on its back on the cage floor and was tested daily after hindlimb paralysis was observed. Investigators were blinded to the treatment group of each animal. All mice were sacrificed and processed for biochemical and pathological analysis upon reaching end-stage.
RNA Analysis (Mouse)
Whole spinal cords of p45 ChAT-IRES-Cre;TardbpF/+ and ChAT-IRES-Cre;tardbpF/F mice were dissected, titurated using a 1 mL syringe with a 20-gauge needle, and placed in TRIzol. Total poly-A-containing messenger RNA was extracted using an RNeasy Mini kit (Qiagen) protocol under RNAse-free conditions and converted to cDNA using the Protoscript II First Strand cDNA Synthesis Kit (NEB). Quantitative PCR for cryptic exon mRNA was performed using the PowerUp SYBR Green protocol (Applied Biosystems) with the following primers: GGCT F: GAGGGGTGTTGGAAGGCTGT; GGCT R: TACCACTCCCCACACTTCGT; SYNJ2BP F: CTCCAACGACAGTGGCATCT; SYNJ2BP R: TCTTCCTGAGGACCTCCGTT; IFT81 F: AAGTGCGAGGACTTCGTGAG; IFT81 R: CAGCGATCTGTCTGCTTTGC; GAPDH F: AGGTCGGTGTGAACGGATTTG; GAPDH R: GGGGTCGTTGATGGCAACA; TBP F: AAGGGAGAATCATGGACCAG; TBP R: CCGTAAGGCATCATTGGACT. Each reaction was performed in duplicate, and each RT-PCR experiment independently repeated three times. Relative transcript levels were calculated in Microsoft Excel according to the formula: (POWER(2, −(Ct mean cryptic exon target)))/(POWER(2, −(AVERAGE(Ct mean TBP, Ct mean GAPDH))).
Histological and Immunohistochemical Analysis
All mice from the pathology analysis cohorts were anaesthetized and perfused with 4% paraformaldehyde. The cervical (C5-C8) and lumbar (L1-L3) enlargements and their corresponding spinal ganglions from each mouse were dissected and post-fixed for 24 hours. Spinal ganglions were embedded in epoxy resin and sagittally sectioned at 1 μm thickness. Spinal cords were embedded in paraffin and sagittally sectioned at 10 μm thickness. Sections were stained with hematoxylin/eosin or Cresyl violet for histological analysis. For immunohistochemical analysis, sections were deparaffmized and incubated in 10 mM citric acid at 95 C for 10 minutes followed by a 30-minute incubation in 0.3% hydrogen peroxide in methanol to quench endogenous peroxidase activity. Normal goat serum (5%) in PBS-T was used to block nonspecific binding, after which primary antibody in blocking buffer was applied to each section overnight at 4 C in a humid chamber. Secondary antibodies were applied at room temperature for 2 hours. A Vectastain Universal Elite ABC kit (Vector Laboratories) was used to amplify signal.
Sections were stained with the following primary antibodies: Human-specific N-terminus TDP-43 (hTDP43, 1:500; AB57105, Abeam), Choline acetyltransferase (ChAT, 1:1000; AB144, Millipore), C-terminus TDP-43 (1:500; 12892-1AP, Proteintech), microtubule-associated protein 2 (Map2, 1:1,000; AB5622, Millipore), phosphorylated neurofilament (Smi31, 1:1,000; BioLegend), synaptophysin (1; 1000; SY38/ab8049, Abeam), ionized calcium-binding adapter molecule 1(IBA1, 1:500; 10904-1AP, Proteintech), glial fibrillary acidic protein (GFAP, 1:500; AB7260, Abeam), phosphorylated tau (Tau422, 1:1000; AB79415, Abeam).
Immunoblot Analysis
Whole spinal cords from mice were flushed and homogenized using a 1 mL syringe with a 20-gauge needle in cold RIPA buffer with protease inhibitor cocktail (Roche). Protein concentration in the supernatants was determined via BCA assay (Pierce), and 10 μg protein was loaded on a 10% Bis-Tris SDS-PAGE gel (Novex) and transferred to a PVDF membrane, which was probed with the following antibodies: N-terminal TDP-43 (1:2000; 10782-2-AP, Proteintech), β-tubulin III (1:20,000; T2200, Sigma).
Adult fly heads were collected using dry ice and homogenized with lysis buffer, and centrifuged at 13,000 rpm at 4°C for 10 minutes. The supernatant were electrophoresed on 10% precast gels (Bio-rad, 456-1033) and electrotransferred onto polyvinylidene difluoride membranes. Immobilized proteins on the membrane were probed with the anti-TBPH (F. Feiguin, International Centre for Genetic Engineering and Biotechnology, Trieste, Italy), anti-TDP-43 (Proteintech, 10782-2-AP) and anti-FLAG (Sigma, F7425 ) and then incubated with HRP-conjugated secondary antibodies.
Statistics
Histological data was analyzed using the unpaired, two-tailed Student’s t-test with Tukey’s multiple comparison where appropriate, and one-way analysis of variance (ANOVA) test using Stata 10 for Mac (Statacorp) and Graphpad Prism for Mac (Graphpad Software). RNA data was analyzed using the Mann-Whitney test. Error bars represent standard deviation unless otherwise mentioned. Mouse hanging wire, rotarod, and weights were analyzed using two-way ANOVA with Tukey’s multiple comparison. Kaplan-Meier survival curves were analyzed using the log-rank test. P values of < 0.05 were considered significant.
Results
Splicing Repression is a Major Function of TBPH in Drosophila
We previously showed that TDP-43 mediated splicing repression can be restored using a chimeric protein, consisting of the RNA-recognizing N-terminal domain of TDP-43 fused with an unrelated, structurally distinct but well characterized splicing repressor, RAVER1 [20, 33, 49]. In contrast to replicating cells, it is not known whether splicing repression is a major function of TDP-43 in post mitotic cells such as motor neurons. We took a genetic approach to test whether this unrelated splicing repressor, termed CTR, could complement the loss of TDP-43 function in motor neurons. That either construct encoding the N-terminal domain of TDP-43 or RAVER1 failed to restore splicing repression in cells lacking TDP-43 [33] indicates that this CTR repressor would provide a powerful tool to selectively probe TDP-43 splicing repression in vivo. First, we assessed the ability of CTR to rescue the motor phenotype and lethality occurring in the Drosophila model lacking TBPH, the fruit fly homolog of TDP-43 (TBPHΔ23 [14]). Flies globally lacking TBPH mostly failed to eclose, and the few surviving adults exhibited impaired locomotion and early lethality along with cryptic exon incorporation (evidence of impaired splicing repression) as identified through RNA sequencing (Figures 1–2, Supplemental Figure S1, Supplemental Table S1). The CTR fusion protein or a control protein with just the N-terminal fragment of TDP-43 alone (NTDP) (Figure 2a) was then expressed using the binary GAL4/UAS expression system under control of either a restricted motor neuron specific driver D42-Gal4, or an Hsp70-Ga14 driver to achieve ubiquitous expression [12]. We first confirmed the expression and cell localization of the fusion protein to the nucleus, as expected (Supplemental Figure S2). Whereas NTDP expression under both drivers failed to show any rescue effect, expression of CTR in motor neurons was efficient to mitigate the motility defects (Figure 2b) and extend the life span to almost 30 days, close to one half of a normal life span (Figure 2c). Expression of CTR ubiquitously by Hsp70- Ga14 further extended the life span to over 45 days (Figure 2c).
Figure 1.
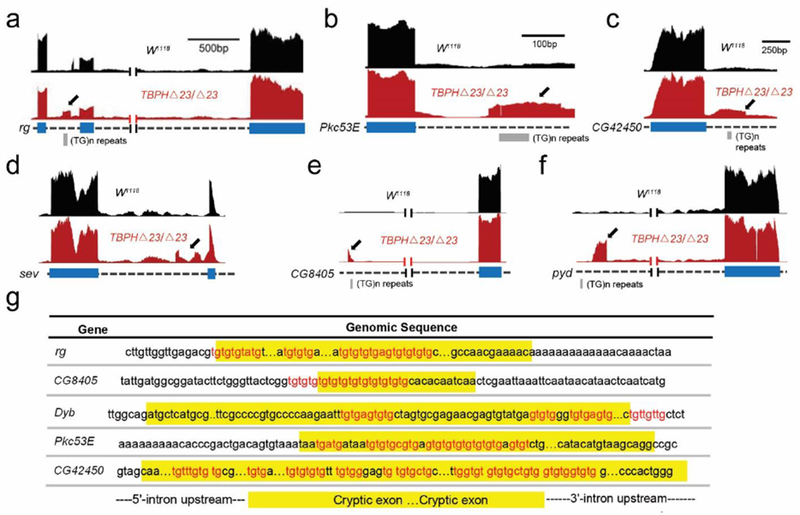
Cryptic exon incorporation in TBPH-deficient flies. Visualization of the cryptic exons (arrows) located in representative genes (red bar). Gene annotation is shown below, labeling exons (thick) and introns (dashed). Cryptic exons may have standard 5’ and 3’ splice sites [rg, (a) and Pkc53E, (b)], or it may present as exon extension [CG42450, (c)], 5’ untranslated region [sev, (d)], polyadenylation site [CG8405, (e)], or inflame insertion [pyd (f)]. (g) As predicted [34], cryptic exons are flanked by UG tandem repeats that exist upstream, downstream, or internally.
Figure 2.
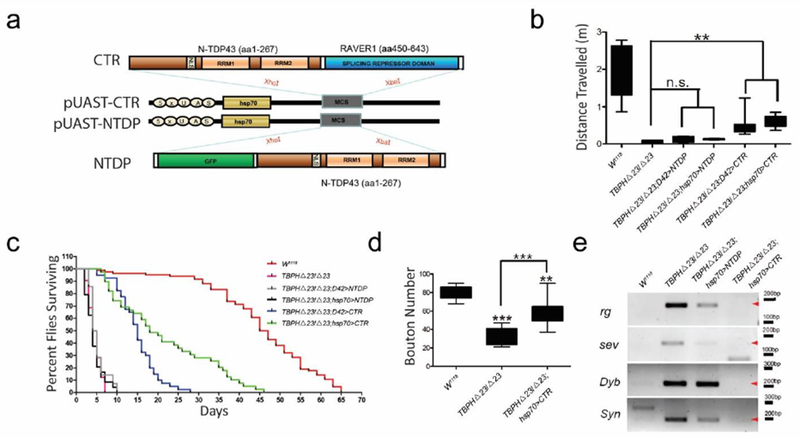
Expression of CTR in TBPH-deficient flies. (a) Diagram of the UAS-CTR and UAS-NTDP chimeric constructs. Expression of CTR fusion protein rather than NTDP could significantly ameliorate locomotive function (b), extend lifespan (c), increased synaptic boutons (d) and (e) repress a variety of cryptic exons including standard cassette (rg), 5’ untranslated region (sev), transcriptional start site (Dyb) and exon extensions (Syn) in a homozygous TBPHΔ23 null background. (** p < 0.01, *** p < 0.001).
Consistent with previous observations that TBPH is required to form and maintain the presynaptic terminal structures [14]. CTR expression resulted in recovery of presynaptic complexity with increased formation of synaptic boutons and axonal terminal branching (Figure 2d, Supplemental Figure S3). Importantly, a subset of splicing abnormalities was restored (Figure 2e, Supplemental Figure S4). Finally, in order to confirm that CTR represses splicing through binding to the mRNAs of TDP-43 targets, we introduced two point mutations, F147L and F149L, in the first RNA recognition motif (RRM1) domain to abolish its nucleotide binding ability [6, 22]. Indeed, expression of CTRF147L/F149L by Hsp70- Ga14 failed to show any rescue effects in TBPH-deficient flies (Supplemental Figure S5). Together, these results strongly support the idea that splicing repression is a major function of TBPH in motor neurons.
An AAV9-Mediated Strategy to Restore TDP-43 Mediated Splicing Repression in Mammalian Motor Neurons
To establish this notion in mammalian motor neurons, we took advantage of our Tardbp conditional knockout mice, in which exon 3 of Tardbp was flanked by loxp sites(Chiang et al., 2010) (TardbpF/F), and crossbred them with a choline acetyltransferase (ChAT) dependent Cre driver line(Rossi et al., 2011) (ChAT-IRES-Cre) to generate a line lacking TDP-43 in >95% of ChAT-positive spinal motor neurons (ChAT-IRES-Cre;tardbpF/F mice, Figure 3a, Supplemental Figure S6). These TDP-43 conditional knockout mice exhibited progressive motor neuron loss accompanied by reduced body weight, tremor, hindlimb weakness, and paralysis with death occurring around 8-10 months of age (Supplemental Figure S7; Supplemental Video S1). This phenotype is consistent with other, previously reported motor neuron Tardbp knockout mouse models [24, 66, 68]. Because TDP-43 is tightly regulated through an autoregulatory mechanism [4, 46]. TDP-43 levels remained normal and no overt phenotype was observed in mice lacking one allele of Tardbp (ChAT-IRES-Cre ;TardbpF/+ heterozygous mice) [10]. As predicted, we found evidence of aberrant splicing and cryptic exon incorporation in spinal cords of ChAT-IRES-Cre;tardbpF/F mice (Figure 5e, red bars).
Figure 3.
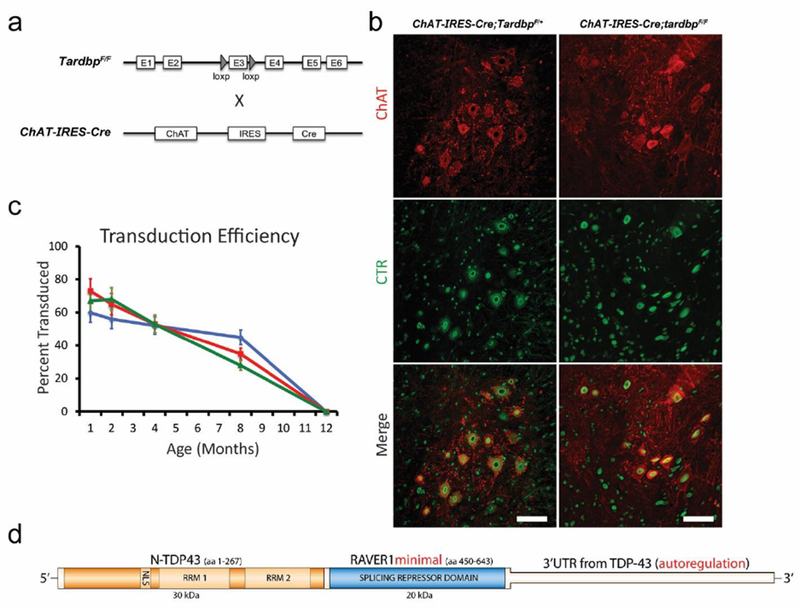
Expression of CTR in Tardbp knockout mice. (a) Schematic of ChAT-IRES-Cre and tardbpF/F alleles in our conditional Tardbp knockout mouse. Cre-mediated excision of exon 3 leads to nonsense-mediated decay of the mRNA transcript. (b) Immunostaining of p30 ChAT (red) and CTR (recognized by human-specific N-terminal TDP-43 antibody, green) in representative lumbar ventral horn sections of ChAT-IRES-Cre;TardbpF/+ and ChAT-IRES-Cre;tardbpF/F mice. At this early age, preceding motor neuron loss, no difference in CTR transduction efficiency between genotypes was observed (mean difference in transduction efficiency = 2±5.5%, p = 0.76, N=5 animals per genotype, 5 cervical and lumbar spinal sections per region per animal) (c) Quantification of nuclear CTR in ChAT-positive neurons in ChAT-IRES-Cre;tardbpF/+ mice in cervical (green), lumbar (red), and dorsal horn neurons (blue). Our protocol allowed for a ~60% efficiency of targeting motor neurons until at least 8 months, with no difference observed between cervical and lumbar regions. (N=3 animals per time point, 5 spinal sections per region per animal, scale bar: 100 μm). (d) Diagram of the CTR chimeric protein construct packaged in AAV9, with the N-terminal fragment of human TDP-43 (orange), the splicing repression domain of RAVER1 (blue), and the 3’ untranslated region (3’UTR) from human TDP-43.
Figure 5.
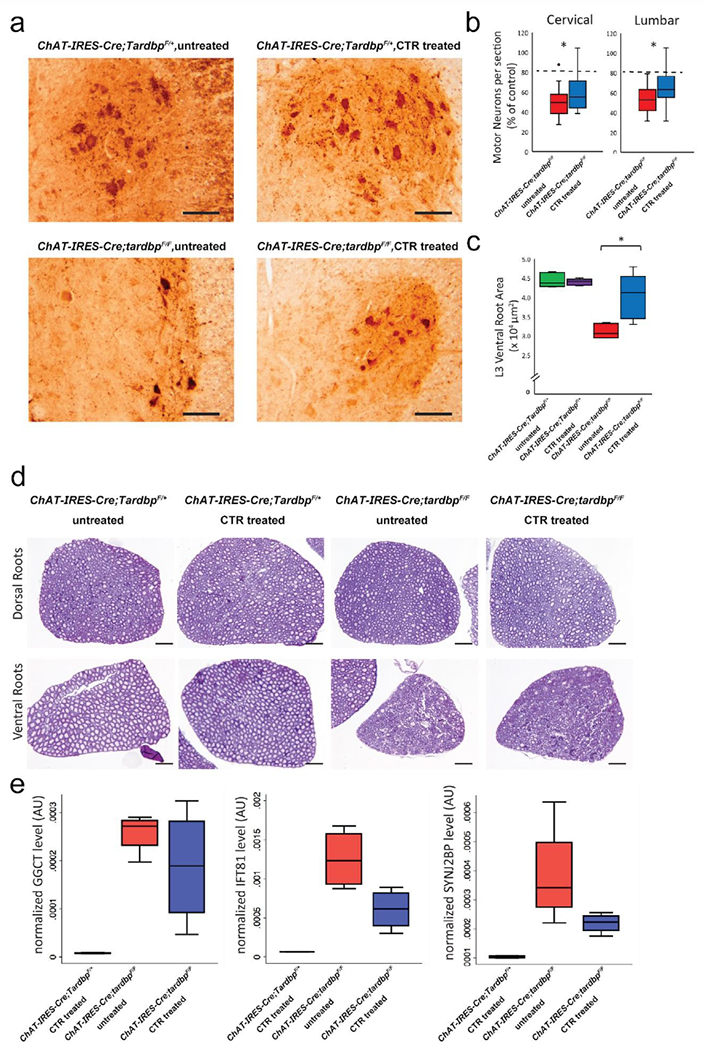
Expression of CTR in Tardbp knockout mice attenuates motor neuron loss and restores splicing repression. (a) Representative ChAT immunostaining of an L3 lumbar ventral horn section at 3 months’ age. (scale bar: 100 μm). (b) Quantification of ChAT-positive motor neurons in CTR-treated (blue) and untreated (red) knockout mice at 3 months’ age. As no difference was observed between CTR-treated and untreated ChAT-IRES-Cre;TardbpF/+ mice, results are shown as a percentage of each group’s respective control. Untreated knockout mice showed a 50% decrease in motor neuron number at p90, whereas the motor neuron abundance of CTR-treated knockout mice is 63% (lumbar) and 61% (cervical) of controls (*p < 0.05). With our observed transduction efficiency of 60 percent, a complete cell-autonomous rescue of motor neuron death by CTR would still only result in a motor neuron rescue of ~80% (dashed line). (c,d) Representative cresyl violet stained dorsal and ventral L3 root sections and quantification of cross-sectional area. Ventral, but not dorsal, root area was diminished in untreated ChAT-IRES-Cre;tardbpF/F mice and restored with CTR treatment (scale bar: 200 μm). (e) Relative levels of cryptic exon mRNA targets predicted to be incorporated in TDP-43-deficient motor neurons in p45 mice, normalized to an average of GAPDH and TBP genes as determined by quantitative RT-PCR. A reduction in cryptic exon incorporation in all three tested targets was observed in CTR-treated ChAT-IRES-Cre;tardbpF/F mice, indicating a partial restoration of splicing repression (* p < 0.05).
To determine whether splicing repression is a major function of TDP-43 in mammalian motor neurons, we elected to use AAV9 to deliver CTR to central neurons of mice lacking TDP-43 in spinal motor neurons. Perinatal unilateral intracerebroventricular injection of AAV9 (3x1010 vg/mouse) carrying our CTR chimeric construct under a ubiquitous chicken beta-actin hybrid (CBhA) promoter selected for its small size and robust long-term expression resulted in CTR protein expression in 50-60% of cervical and lumbar mouse motor neurons that persisted to at least 8 months (Figure 3c,d). CTR expression was strongest in the nucleoplasm, with no evidence of cytoplasmic CTR aggregates (Figure 3b). No difference was observed between cervical and lumbar expression efficiency at any age (Figure 3c) or between ChAT-IRES-Cre;TardbpF/+ and ChAT-IRES-Cre;tardbpF/F mice at p30, an early age preceding neuronal loss (Figure 3b–c). CTR expression had no effect on Cre-mediated Tardbp knockout efficiency (Supplemental Figure S6). While viral transduction was broadly distributed throughout the CNS, no behavioral (Figure 4) or pathological evidence of acute or chronic toxicity from CTR protein expression (data not shown) was observed in ChAT-IRES-Cre;TardbpF/+ mice within the CNS or any major organ system; these treated ChAT-IRES-Cre;TardbpF/+ mice exhibited a normal lifespan (Figure 4a).
Figure 4.
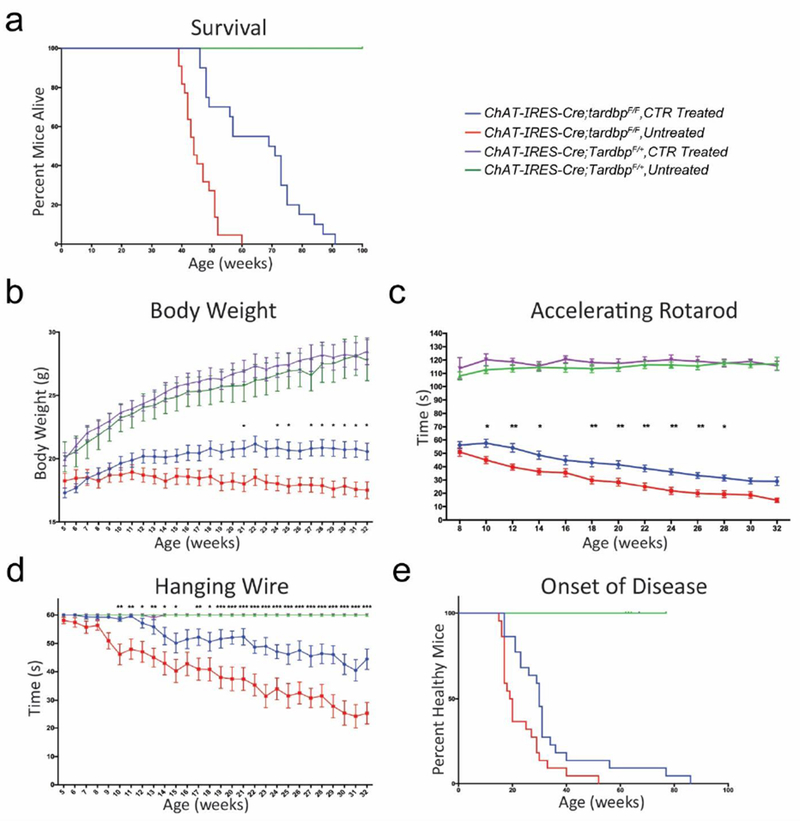
Expression of CTR in Tardbp knockout mice prolong survival and attenuates behavioral deficits. (a) Kaplan-Meier survival curve of ChAT-IRES-Cre;TardbpF/+ and ChAT-IRES-Cre;tardbpF/F mice administered AAV9 containing either CTR (treated) or GFP control (untreated). Data from both cohorts are shown together. Median untreated ChAT-IRES-Cre;tardbpF/F survival was extended from 44 weeks to 73 weeks in treated mice (p < 0.001 for all analyses). (b) CTR treatment mitigated the age-dependent body weight loss of knockout mice while having no effect on control mice. Hanging wire (c) and rotarod performance (d) show a mitigation of motor deficits in CTR-treated knockout mice compared to untreated controls (*p < 0.05, ** p < 0.01, *** p < 0.001. Tukey’s multiple comparison test). Progression of hanging wire deficits was also significantly attenuated in CTR-treated mice (untreated slope=−1.19, CTR slope= −0.68, p < 0.01) (e) Kaplan-Meier survival curve of onset of motor dysfunction, as defined as two consecutive weeks of <60s hanging wire time. Onset was delayed in CTR-treated knockout mice (median onset: 30 versus 19 weeks, p < 0.001).
Two cohorts of ChAT-IRES-Cre;tardbpF/F mice (Supplemental Table S2) injected with our CTR fusion protein gained greater weight as compared to their untreated breeder-matched knockout controls (Figure 4b, Supplemental Figure S8). Treated ChAT-IRES-Cre;tardbpF/F mice also performed better on the hanging wire and accelerating rotarod tests, with a delayed onset and slower progression of motor deficits (Figure 4c–e, Supplemental Video S1). Consequently, treated ChAT-IRES-Cre;tardbpF/F mice showed a robust extension of their lifespan, with a median survival increase of 29 weeks (66%) (Figure 4a). Notably, a cohort of ChAT-IRES-Cre;tardbpF/F mice treated with just the N-terminal fragment of TDP-43 showed no such motor improvements or weight gain, suggesting that splicing repression, and not some other function of the N-terminal fragment of TDP-43, underlies the rescue effect (Supplemental Figure S9).
Pathological analysis of 1, 3, and 5-month old cohorts of ChAT-IRES-Cre;TardbpF/+ and ChAT-IRES-Cre;tardbpF/F mice (Supplemental Table S2) revealed that while knockout mice lose 50% of ChAT-positive spinal motor neurons between 1 and 3 months’ age, CTR treatment significantly mitigated this loss (Figure 5a, b). Importantly, with our observed transduction efficiency of ~60%, this motor neuron rescue in CTR-treated ChAT-IRES-Cre;tardbpF/F mice represents about 45% of the predicted maximum cell-autonomous rescue effect (Figure 5b, dotted line). The observed rescue effect of CTR on spinal motor neurons was most robust at the 3-month time point (Supplemental Figure S10). While the size of L3 dorsal roots remained unchanged, L3 ventral spinal root cross-sectional area was markedly preserved in treated ChAT-IRES-Cre;tardbpF/F mice (Figure 5c–d), corroborating the behavioral benefits offered by the CTR splicing repressor.
While the downstream effects of TDP-43 mediated splicing repression occur on a transcriptome-wide level, we selected three transcripts with cryptic exon incorporation identified through previous RNA-seq analysis [25] to serve as a biochemical correlate of TDP-43 mediated splicing repression. As predicted, treatment with our CTR protein significantly re-repressed these aberrant cryptic exon splicing events as determined by quantitative RT-PCR (Figure 5e). Expression of the N-terminal fragment of TDP-43 alone did not result in any such re-repression, suggesting that CTR expression does not interfere with normal TDP-43 function (Supplemental Figure S9). Taken together, our results support the notion that splicing repression is a major function of TDP-43 in motor neurons and suggest that loss of this splicing repression could underlie neurodegeneration in diseases such as ALS.
Discussion
We validate here for the first time that TDP-43 mediated splicing repression is a major function of TDP-43 in motor neurons. Our findings strongly support the idea that compromised transcriptomic integrity following the loss of TDP-43 mediated splicing repression represents a key pathogenic mechanism underlying motor neuron degeneration.
A crucial mechanistic question regarding TDP-43 pathology is whether nuclear loss of TDP-43 - i.e., loss of splicing repression - independent of its cytoplasmic aggregates drives neuronal loss in neurodegenerative diseases exhibiting TDP-43 pathology. While gain- and loss-of-function studies have been performed in efforts to clarify mechanism of TDP-43 pathology over the past decade [30, 59], its precise pathophysiology in human disease remain elusive. Several lines of evidence from human studies strongly support the idea that loss of TDP-43 splicing repression drives neuron loss. We initially disclosed that TDP-43 is a splicing repressor of nonconserved cryptic exons [33], a novel finding that have been replicated by several groups [28, 31, 39, 58]. An emerging view is that TDP-43 is a founding member of growing family RNA binding proteins (including another ALS-linked gene encoding Matrin3) [13, 32, 36, 61] which serve as the “guardians of the transcriptome”, ensuring the generation of a normal proteome for cell function. Importantly, this repressor function of TDP-43 is compromised in ALS, FTLD-TDP and AD exhibiting TDP-43 pathology [33, 56]. Interestingly, TDP-43 nuclear depletion in brain neurons occurs at pre-symptomatic stage in a C90RF72 linked ALS-FTD case [62], supporting the notion that loss of TDP-43 repression represents an early event that drives neuron loss. Consistent with this idea, we found that TDP-43-associated cryptic exons are also incorporated in brains of AD cases with TDP-43 pathology, notably those exhibiting nuclear clearance of TDP-43 without cytoplasmic inclusions [56]. That von Economo neurons and fork cells within the frontoinsular and anterior cingulate cortices lacking nuclear TDP-43 inclusion observed in behavioral variant FTD cases showed atrophy comparable to inclusion-bearing neurons [43] further strengthen this notion. ALS-linked TDP-43 mutants from fibroblast-derived human motor neurons fail to repress TDP-43 cryptic exons, including that of Stcithmin2 as a critical target for motor axon regeneration, supporting the view that loss of TDP-43 repression independent of TDP-43 cytoplasmic aggregates drives neuron loss [39]. Together, these data from studies of human disease strongly support the notion that nuclear depletion of TDP-43, i.e., compromised splicing repression from neurons represents an early event (as opposed to cytoplasmic inclusions marking a late- or end-stage cell) that contributes to the pathogenesis of human disease exhibiting TDP-43 pathology.
As no compensatory mechanism exists for TDP-43 repression when such function is loss in motor neurons, complementing TDP-43 splicing repression via gene therapy approach, such as the FDA-approved AAV platform for human disease [23, 40], would be a rational mechanism-based therapeutic strategy for ALS. Alternatively, ASO designed to block specific TDP-43 cryptic exon targets, such as Stathmin2 [28, 39], may prove to be a valuable therapeutic approach. As multiple TDP-43 cryptic exons are affected when TDP-43 repression is compromised [33], it will be important in the future to evaluate whether targeting multiple TDP-43 targets will be critical, particularly within the context of motor neuron degeneration. Given that multiple cell types are impacted in ALS-FTD [43, 45], and existence of cell type specific TDP-43 cryptic exons [25], a more general strategy designed to repress multiple cryptic exon targets from multiple cell types may provide greater clinical outcomes.
Our finding that the lack of acute or chronic toxicity in any major organ system and normal lifespan observed in CTR-treated control mice suggests that any off-target splicing effects of CTR in TDP-43 knockout mice are likely outweighed by the therapeutic effects of CTR treatment. It will be also important in the future to assess the benefit of this therapeutic approach in glial cells exhibiting loss of nuclear TDP-43 mediated splicing repression, as evidenced by glial cytoplasmic TDP-43 inclusions in cell of presumed oligodendroglial lineage [44] and help set the stage for future gene therapy trials for ALS.
We previously demonstrated that cryptic exons are highly variable between different cell types and organisms, suggesting that TDP-43 loss may impair cell-type specific pathways in unique ways and complicating efforts at developing treatments targeting any particular downstream pathway [25]. We previously showed that Tbc1d1, a gene involved in fat metabolism and down-regulated in the absence of TDP-43 and a target of TDP-43 [10], is highly incorporated in mouse stem cells, but low in myocytes and neurons [25]. However, the splicing repression function of TDP-43 is highly conserved across species [33], making our proposed mechanism-based therapeutic approach, which rescues lethality in both Drosophila and murine models, a more attractive upstream strategy. In this study, we assessed cryptic exon incorporation in selected TDP-43 targets as a reflection of a cell’s loss of TDP-43 splicing repression; in ALS patients and our animal models, both normal and cryptic splicing abnormalities mediated by nuclear loss of TDP-43 may contribute to neuronal death. Notably, loss of TDP-43 repression impacts both cryptic exons as well as conserved exons in a cell-type specific manner. We have observed exon 17b-containing Sort1 in mouse Tardbp knockout hippocampal tissue, but not in spinal cord motor neurons (Supplemental Figure S11a). Importantly, CTR expression markedly attenuated the aberrant exon 17b-containing Sort1 variant (Supplemental Figure S11a). Our results do not exclude other important functions of TDP-43, as this protein has been suggested to be involved in nuclear and nonnuclear cellular processes besides splicing repression. For example, TBPH interacts with futsch mRNA and this interaction modulates the intracellular transport/localization and translation of this mRNA in fly motor neurons [50]. As expected, CTR expression could not ameliorate the significant reduction of Futsch protein in TBPH-null flies (Supplemental Figure S11b). Similarly, our results do not exclude the potential toxic effects of cytoplasmic TDP-43 aggregation [11, 63]. Nuclear TDP-43 depletion could be an early pathogenic event, possibly preceding its cytoplasmic aggregation [56, 62], and while promising therapeutic options for reducing TDP-43 aggregation mediated toxicity are being developed [5, 21, 27, 38], further effort is needed to clarify the different contributions of toxic gain of function and nuclear loss of function of TDP-43 to the pathogenesis of neurodegenerative disease. Assessing the importance of splicing repression as well as the ability of CTR to avoid cytoplasmic sequestration and perform this role in other models of TDP-43 dysfunction will improve our understanding of the complex interplay between cytoplasmic accumulation and nuclear loss of TDP-43.
Supplementary Material
Figure S10. Quantification of ChAT-positive motor neurons in CTR-treated (blue) and untreated (red) knockout mice at 1 months’ age (a) and 5 months’ age (b). As no difference was observed between motor neuron numbers of CTR-treated and untreated ChAT-IRES-Cre;TardbpF/+ mice at any time point, results are shown as a percentage of each group’s respective control. While no changes were observed at the 1-month time point, untreated knockout mice showed a decrease in motor neuron number at 5 months’ age to 47% of that of controls. The motor neuron abundance of CTR-treated knockout mice is 54% (lumbar) and 56% (cervical) of controls (*p < 0.05).
Figure S6. Immunofluorescent stain of ChAT (red) and TDP-43 (blue) in p30 ChAT-IRES-Cre;TardbpF/+ mice, CTR treated and untreated ChAT-IRES-Cre;tardbpF/F mice. In all three groups, Cre-dependent Tardbp knockout rate was ~95% in both cervical and lumbar sections and not significantly different between groups (mean difference in knockout rate = 1±1.2%, p = 0.88, N=5 mice per genotype, 5 cervical/lumbar sections per animal).
Figure S7. Depletion of TDP-43 in lower motor neurons leads to age dependent motor deficits and early lethality in mice. (a) Kaplan-Meier survival curve of ChAT-IRES-Cre;tardbpF/+ (N=22, 10♀) and ChAT-IRES-Cre;tardbpF/F (N=14, 7♀) mice. Median untreated ChAT-IRES-Cre;tardbpF/F survival was 44 weeks in knockout mice. (b) Hanging wire, (c) body weight, and (d) rotarod performance are all impaired in ChAT-IRES-Cre;tardbpF/F mice. (p < 0.001 for all analyses).
Figure S8. Body weights for CTR treated and untreated ChAT-IRES-Cre;TardbpF/+ and ChAT-IRES-Cre;tardbpF/F mice, separated by sex (* p < 0.05).
Figure S9. N-terminal fragment expression alone confers no therapeutic benefit in Tardbp knockout mice. (a) Hanging wire performance and (b) body weights of ChAT-IRES-Cre;tardbpF/F mice administered AAV9 containing the N-terminal fragment of TDP-43 alone (NTF, blue) showed no improvement over untreated littermates (red). (c) NTF treated ChAT-IRES-Cre;tardbpF/F mice also showed no restoration of splicing repression as evidenced by cryptic exon incorporation into 3 targets tested
Movie S1. Representative video of p90 untreated ChAT-IRES-Cre;tardbpF/F mice (left cage) and a CTR-treated ChAT-IRES-Cre;tardbpF/F mouse (right cage). At the end of the video, a representative ChAT-IRES-Cre;TardbpF/+ mouse is shown as well. The improvement in motor function in CTR-treated Tardbp knockout mice is evident by the increased locomotion and hindlimb mobility.
Figure S11. (a) RT-PCR amplification of abnormal Sort1 splicing variant including exon 17b in mouse spinal cord and hippocampus. Like Tbc1d1, aberrant splicing of Sort1 was not observed in mouse motor neurons, but appeared in hippocampal neurons due to the depletion of TDP-43 which could be restored with the treatment of CTR. Lane 1: ChaT-IRES-Cre;TardbpF/+; lane 2: ChaT-IRES-Cre;TardbpF/F; lane 3: CTR-treated ChaT-IRES-Cre;TardbpF/F mice; lane 4: CamK2a-Cre;tardbpF/+ ; lane 5: CamK2a-Cre;tardbpF/F; lane 6: CTR-treated CamK2a-Cre;tardbpF/F mice. (b) Confocal images showing reduced Futsch (green) expression in the distal boutons (HRP, red) of TBPHΔ23/Δ23 flies (ii) compared with wild type (i). This defect could not be reversed by the expression of CTR fusion protein (iii). Scale bar: 10 μm.
Table S1. List of cryptic exons identified in TBPH-deficient Drosophila.
Table S2. Final sample sizes for all mouse cohorts examined in this study, separated by genotype and injection type.
Figure S1. Sequencing validation of cryptic exon RT-PCR. (a) Diagram of RT-PCR detection strategy. Primers were designed to amplify only the cryptic exon splice junction. (b) Gel purification and sequencing of these RT-PCR bands confirms that these DNA products precisely correspond to the predicted cryptic exon splice junctions. (c) To visualize this, sequencing data was aligned to the Drosophila genome using UCSC BLAT; thick bands in the sequence alignment indicate DNA sequences that are present in the RT-PCR product, thin bands represent sequences that have been spliced out. Sequence alignment demonstrates clear overlap with cryptic exon and completely matches the predicted the splice junctions.
Figure S2. Expression of CTR in TBPH null flies. (a) Western blot confirmation of CTR expression. Both TBPHΔ23 homozygous null and the Hsp-70 CTR line lack the endogenous 58 KD TBPH protein as shown by the N-terminal TDP-43 antibody in wild type flies. Instead, the Hsp-70 CTR line expresses the shorter CTR fusion protein. Hsp-70 CTR expression level remains stable in aged flies as it is shown here at D30 after eclosion. (b) Immunofluorescent staining of CTR (red) and neuron-specific marker ELAV (nuclear, green) in third instar larval ventral nerve cord.
Figure S3. Morphological defects at neuromuscular synapsis in TBPH null flies. (a) Confocal images of motor neuron presynaptic terminals at abdominal segment III in wild type third instar larvae stained with anti-HRP (red) and anti-DLG (green) antibodies, reveals the branching pattern and the presence of synaptic boutons. (b) Similar staining and anatomical position show reduced axonal branching and number of synaptic boutons in TBPHΔ23 homozygous larvae. (c) Recovery of presynaptic complexity with increased formation of synaptic boutons and axonal terminal branching in Hsp-70<CTR larvae.
Figure S4. Certain cryptic exon transcripts are not reversed in CTR rescue. Designing primers across cryptic exon sequences is difficult due to repetitive sequences. Therefore, high levels of background are inevitable. Nevertheless, relatively clean primers were identified for ten of the thirteen genes (Complete list of Drosophila cryptic exons is provided in Table S1), and their RT-PCR products can be clearly identified (red arrowheads). Four cryptic exon transcripts (rg, sev, Dyb, and Syn) were reversed in CTR rescue. The other six transcripts shown here were not completely reversed.
Figure S5. Expression of CTRF147L/F149L in TBPH-deficient flies has no rescue effect. (a) Diagram of the CTRF147L/F149L and UAS-NTDP chimeric constructs. Flagged CTRF147L/F149L strain did not express endogenous TBPH (b), nor did it extend lifespan (c), ameliorate locomotive function (d) or repress cryptic exon incorporations (e) in a homozygous TBPHΔ23 null background.
Acknowledgments
We thank L. Martin, C. Sumner, and T. Lloyd for thoughtful comments and V. Nehus and H. Wu for technical assistance.
Funding
This work was supported by the NIH Grant R01 NS095969 (PCW), McKnight Memory and Cognitive Disorders Award (PCW and LC), Robert Packard Center for ALS Research (LC and PCW) and the Amyotrophic Lateral Sclerosis Association (PCW). JPL is a recipient of a Johns Hopkins Kavli Neuroscience Discovery Institutte fellowship award.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Competing Interests
Authors declare no competing interests.
References
- 1.Alami NH, Smith RB, Carrasco MA, Williams LA, Winbom CS, Han SSW, Kiskinis E, Winbom B, Freibaum BD, Kanagaraj A et al. (2014) Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81: 536–543 Doi 10.1016/j.neuron.2013.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, Graff-Radford NR, Hutton ML, Dickson DW (2007) TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 61: 435–445 Doi 10.1002/ana.21154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ash PE, Zhang YJ, Roberts CM, Saldi T, Hutter H, Buratti E, Petrucelli L, Link CD (2010) Neurotoxic effects of TDP-43 overexpression in C. elegans. Human molecular genetics 19: 3206–3218 Doi 10.1093/hmg/ddq230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ayala YM, De Conti L, Avendano-Vazquez SE, Dhir A, Romano M, D’Ambrogio A, Tollervey J, Ule J, Baralle M, Buratti E et al. (2011) TDP-43 regulates its mRNA levels through a negative feedback loop. The EMBO journal 30: 277–288 Doi 10.1038/emboj.2010.310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, Messing J, Kim HJ, Soriano A, Auburger G et al. (2017) Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544: 367–371 Doi 10.1038/nature22038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buratti E, Baralle FE (2001) Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. The Journal of biological chemistry 276: 36337–36343 Doi 10.1074/jbc.M104236200 [DOI] [PubMed] [Google Scholar]
- 7.Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE (2001) Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. The EMBO journal 20: 1774–1784 Doi 10.1093/emboj/20.7.1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casafont I, Bengoechea R, Tapia O, Berciano MT, Lafarga M (2009) TDP-43 localizes in mRNA transcription and processing sites in mammalian neurons. J Struct Biol 167: 235–241 Doi 10.1016/j.jsb.2009.06.006 [DOI] [PubMed] [Google Scholar]
- 9.Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, Lee CW, Jansen-West K, Kurti A, Murray ME et al. (2015) Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 348: 1151–1154 Doi 10.1126/science.aaa9344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiang PM, Ling J, Jeong YH, Price DL, Aja SM, Wong PC (2010) Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proceedings of the National Academy of Sciences of the United States of America 107: 16320–16324 Doi 10.1073/pnas.1002176107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chou CC, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, Sayegh M, Donlin-Asp PG, Chen YH, Duong DM et al. (2018) TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nature neuroscience 21: 228–239 Doi 10.1038/s41593-017-0047-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duffy JB (2002) GAL4 system in Drosophila: a fly geneticist’s Swiss army knife. Genesis 34: 1–15 Doi 10.1002/gene.10150 [DOI] [PubMed] [Google Scholar]
- 13.Ehrmann I, Crichton JH, Gazzara MR, James K, Liu Y, Grellscheid SN, Curk T, de Rooij D, Steyn JS, Cockell S et al. (2019) An ancient germ cell-specific RNA-binding protein protects the germline from cryptic splice site poisoning. Elife 8: Doi 10.7554/eLife.39304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feiguin F, Godena VK, Romano G, D’Ambrogio A, Klima R, Baralle FE (2009) Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS letters 583: 1586–1592 Doi 10.1016/j.febslet.2009.04.019 [DOI] [PubMed] [Google Scholar]
- 15.Fiesel FC, Voigt A, Weber SS, Van den Haute C, Waldenmaier A, Gorner K, Walter M, Anderson ML, Kern JV, Rasse TM et al. (2010) Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. The EMBO journal 29: 209–221 Doi 10.1038/emboj.2009.324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foust KD, Wang X, McGovern VL, Braun L, Bevan AK, Haidet AM, Le TT, Morales PR, Rich MM, Burghes AH et al. (2010) Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol 28: 271–274 Doi 10.1038/nbt.1610 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Freibaum BD, Chitta RK, High AA, Taylor JP (2010) Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res 9: 1104–1120 Doi 10.1021/pr901076y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gasset-Rosa F, Lu S, Yu H, Chen C, Melamed Z, Guo L, Shorter J, Da Cruz S, Cleveland DW (2019) Cytoplasmic TDP-43 De-mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron 102: 339–357 e337 Doi 10.1016/j.neuron.2019.02.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gopal PP, Nirschl JJ, Klinman E, Holzbaur EL (2017) Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons. Proceedings of the National Academy of Sciences of the United States of America 114: E2466–E2475 Doi 10.1073/pnas.1614462114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gromak N, Rideau A, Southby J, Scadden AD, Gooding C, Huttelmaier S, Singer RH, Smith CW (2003) The PTB interacting protein raver1 regulates alpha-tropomyosin alternative splicing. The EMBO journal 22: 6356–6364 Doi 10.1093/emboj/cdg609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo L, Kim HJ, Wang H, Monaghan J, Freyermuth F, Sung JC, O’Donovan K, Fare CM, Diaz Z, Singh N et al. (2018) Nuclear-Import Receptors Reverse Aberrant Phase Transitions of RNA-Binding Proteins with Prion-like Domains. Cell 173: 677–692 e620 Doi 10.1016/j.cell.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang YC, Lin KF, He RY, Tu PH, Koubek J, Hsu YC, Huang JJ (2013) Inhibition of TDP-43 aggregation by nucleic acid binding. PLoS One 8: e64002 Doi 10.1371/journal.pone.0064002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hudry E, Vandenberghe LH (2019) Therapeutic AAV Gene Transfer to the Nervous System: A Clinical Reality. Neuron 102: 263 Doi 10.1016/j.neuron.2019.03.020 [DOI] [PubMed] [Google Scholar]
- 24.Iguchi Y, Katsuno M, Niwa J, Takagi S, Ishigaki S, Ikenaka K, Kawai K, Watanabe H, Yamanaka K, Takahashi R et al. (2013) Loss of TDP-43 causes age-dependent progressive motor neuron degeneration. Brain 136: 1371–1382 Doi 10.1093/brain/awt029 [DOI] [PubMed] [Google Scholar]
- 25.Jeong YH, Ling JP, Lin SZ, Donde AN, Braunstein KE, Majounie E, Traynor BJ, LaClair KD, Lloyd TE, Wong PC (2017) Tdp-43 cryptic exons are highly variable between cell types. Mol Neurodegener 12: 13 Doi 10.1186/s13024-016-0144-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F et al. (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40: 572–574 Doi 10.1038/ng.132 [DOI] [PubMed] [Google Scholar]
- 27.Kim HJ, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ, Lee VM, Finkbeiner S, Gitler AD, Bonini NM (2014) Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet 46: 152–160 Doi 10.1038/ng.2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R et al. (2019) ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nature neuroscience 22: 167–179 Doi 10.1038/s41593-018-0300-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LaClair KD, Donde A, Ling JP, Jeong YH, Chhabra R, Martin LJ, Wong PC (2016) Depletion of TDP-43 decreases fibril and plaque beta-amyloid and exacerbates neurodegeneration in an Alzheimer’s mouse model. Acta neuropathologica 132: SQSTM1/p62859-62873 Doi 10.1007/s00401-016-1637-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee EB, Lee VM, Trojanowski JQ (2011) Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci 13: 38–50 Doi 10.1038/nrn3121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z, Vuong JK, Zhang M, Stork C, Zheng S (2017) Inhibition of nonsense-mediated RNA decay by ER stress. RNA 23: 378–394 Doi 10.1261/rna.058040.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ling JP, Chhabra R, Merran JD, Schaughency PM, Wheelan SJ, Corden JL, Wong PC (2016) PTBP1 and PTBP2 Repress Nonconserved Cryptic Exons. Cell Rep 17: 104–113 Doi 10.1016/j.celrep.2016.08.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ling JP, Pletnikova O, Troncoso JC, Wong PC (2015) TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349: 650–655 Doi 10.1126/science.aab0983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lukavsky PJ, Daujotyte D, Tollervey JR, Ule J, Stuani C, Buratti E, Baralle FE, Damberger FF, Allain FH (2013) Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nature structural & molecular biology 20: 1443–1449 Doi 10.1038/nsmb.2698 [DOI] [PubMed] [Google Scholar]
- 35.Mann JR, Gleixner AM, Mauna JC, Gomes E, DeChellis-Marks MR, Needham PG, Copley KE, Hurtle B, Portz B, Pyles NJ et al. (2019) RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron 102: 321–338 e328 Doi 10.1016/j.neuron.2019.01.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McClory SP, Lynch KW, Ling JP (2018) HnRNP L represses cryptic exons. RNA 24: 761–768 Doi 10.1261/rna.065508.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, Rouleau GA, Vande Velde C (2011) TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Human molecular genetics 20: 1400–1410 Doi 10.1093/hmg/ddr021 [DOI] [PubMed] [Google Scholar]
- 38.McGurk L, Gomes E, Guo L, Mojsilovic-Petrovic J, Tran V, Kalb RG, Shorter J, Bonini NM (2018) Poly(ADP-Ribose) Prevents Pathological Phase Separation of TDP-43 by Promoting Liquid Demixing and Stress Granule Localization. Molecular cell 71: 703–717 e709 Doi 10.1016/j.molcel.2018.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melamed Z, Lopez-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW et al. (2019) Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nature neuroscience 22: 180–190 Doi 10.1038/s41593-018-0293-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, Lowes L, Alfano L, Berry K, Church K et al. (2017) Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med 377: 1713–1722 Doi 10.1056/NEJMoa1706198 [DOI] [PubMed] [Google Scholar]
- 41.Mitra J, Guerrero EN, Hegde PM, Liachko NF, Wang H, Vasquez V, Gao J, Pandey A, Taylor JP, Kraemer BC et al. (2019) Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proceedings of the National Academy of Sciences of the United States of America: Doi 10.1073/pnas.1818415116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163: 123–133 Doi 10.1016/j.cell.2015.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nana AL, Sidhu M, Gaus SE, Hwang JL, Li L, Park Y, Kim EJ, Pasquini L, Allen IE, Rankin KP et al. (2019) Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta neuropathologica 137: 27–46 Doi 10.1007/s00401-018-1942-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neumann M, Kwong LK, Truax AC, Vanmassenhove B, Kretzschmar HA, Van Deerlin VM, Clark CM, Grossman M, Miller BL, Trojanowski JQ et al. (2007) TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J Neuropathol Exp Neurol 66: 177–183 Doi 10.1097/01.jnen.0000248554.45456.58 [DOI] [PubMed] [Google Scholar]
- 45.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: 130–133 Doi 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- 46.Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C et al. (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nature neuroscience 14: 459–468 Doi 10.1038/nn.2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prudencio M, Belzil VV, Batra R, Ross CA, Gendron TF, Pregent LJ, Murray ME, Overstreet KK, Piazza-Johnston AE, Desaro P et al. (2015) Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nature neuroscience 18: 1175–1182 Doi 10.1038/nn.4065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L et al. (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72: 257–268 Doi 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rideau AP, Gooding C, Simpson PJ, Monie TP, Lorenz M, Huttelmaier S, Singer RH, Matthews S, Curry S, Smith CW (2006) A peptide motif in Raver1 mediates splicing repression by interaction with the PTB RRM2 domain. Nature structural & molecular biology 13: 839–848 Doi 10.1038/nsmb1137 [DOI] [PubMed] [Google Scholar]
- 50.Romano M, Feiguin F, Buratti E (2016) TBPH/TDP-43 modulates translation of Drosophila futsch mRNA through an UG-rich sequence within its 5’UTR. Brain Res 1647: 50–56 Doi 10.1016/j.brainres.2016.02.022 [DOI] [PubMed] [Google Scholar]
- 51.Salajegheh M, Pinkus JL, Taylor JP, Amato AA, Nazareno R, Baloh RH, Greenberg SA (2009) Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis. Muscle Nerve 40: 19–31 Doi 10.1002/mus.21386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmid B, Hruscha A, Hogl S, Banzhaf-Strathmann J, Strecker K, van der Zee J, Teucke M, Eimer S, Hegermann J, Kittelmann M et al. (2013) Loss of ALS-associated TDP-43 in zebrafish causes muscle degeneration, vascular dysfunction, and reduced motor neuron axon outgrowth. Proceedings of the National Academy of Sciences of the United States of America 110: 4986–4991 Doi 10.1073/pnas.1218311110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sephton CF, Good SK, Atkin S, Dewey CM, Mayer P 3rd, Herz J, Yu G (2010) TDP-43 is a developmentally regulated protein essential for early embryonic development. The Journal of biological chemistry 285: 6826–6834 Doi 10.1074/jbc.M109.061846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shan X, Chiang PM, Price DL, Wong PC (2010) Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 107: 16325–16330 Doi 10.1073/pnas.1003459107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E et al. (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319: 1668–1672 Doi 10.1126/science.1154584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun M, Bell W, LaClair KD, Ling JP, Han H, Kageyama Y, Pletnikova O, Troncoso JC, Wong PC, Chen LL (2017) Cryptic exon incorporation occurs in Alzheimer’s brain lacking TDP-43 inclusion but exhibiting nuclear clearance of TDP-43. Acta neuropathologica 133: 923–931 Doi 10.1007/s00401-017-1701-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tan Q, Krishna Yalamanchili H, Park J, De Maio A, Lu H-C, Wan Y-W, White JJ, Bondar VV, Sayegh LS, Liu X et al. (2016) Extensive cryptic splicing upon loss of RBM17 and TDP43 in neurodegeneration models. Human molecular genetics: Doi 10.1093/hmg/ddw337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tan Q, Yalamanchili HK, Park J, De Maio A, Lu HC, Wan YW, White JJ, Bondar VV, Sayegh LS, Liu X et al. (2016) Extensive cryptic splicing upon loss of RBM17 and TDP43 in neurodegeneration models. Human molecular genetics 25: 5083–5093 Doi 10.1093/hmg/ddw337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taylor JP, Brown RH Jr., Cleveland DW (2016) Decoding ALS: from genes to mechanism. Nature 539: 197–206 Doi 10.1038/nature20413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Konig J, Hortobagyi T, Nishimura AL, Zupunski V et al. (2011) Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nature neuroscience 14: 452–458 Doi 10.1038/nn.2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uemura Y, Oshima T, Yamamoto M, Reyes CJ, Costa Cruz PH, Shibuya T, Kawahara Y (2017) Matrin3 binds directly to intronic pyrimidine-rich sequences and controls alternative splicing. Genes Cells 22: 785–798 Doi 10.1111/gtc.12512 [DOI] [PubMed] [Google Scholar]
- 62.Vatsavayai SC, Yoon SJ, Gardner RC, Gendron TF, Vargas JN, Trujillo A, Pribadi M, Phillips JJ, Gaus SE, Hixson JD et al. (2016) Timing and significance of pathological features in C9orf72 expansion-associated frontotemporal dementia. Brain 139: 3202–3216 Doi 10.1093/brain/aww250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.White MA, Kim E, Duffy A, Adalbert R, Phillips BU, Peters OM, Stephenson J, Yang S, Massenzio F, Lin Z et al. (2018) TDP-43 gains function due to perturbed autoregulation in a Tardbp knock-in mouse model of ALS-FTD. Nature neuroscience 21: 552–563 Doi 10.1038/s41593-018-0113-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, Smits V, Ceuterick-de Groote C, Van Broeckhoven C, Kumar-Singh S (2010) TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proceedings of the National Academy of Sciences of the United States of America 107: 3858–3863 Doi 10.1073/pnas.0912417107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu LS, Cheng WC, Hou SC, Yan YT, Jiang ST, Shen CK (2010) TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis 48: 56–62 Doi 10.1002/dvg.20584 [DOI] [PubMed] [Google Scholar]
- 66.Wu LS, Cheng WC, Shen CK (2012) Targeted depletion of TDP-43 expression in the spinal cord motor neurons leads to the development of amyotrophic lateral sclerosis-like phenotypes in mice. The Journal of biological chemistry 287: 27335–27344 Doi 10.1074/jbc.M112.359000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu YF, Gendron TF, Zhang YJ, Lin WL, D’Alton S, Sheng H, Casey MC, Tong J, Knight J, Yu X et al. (2010) Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. The Journal of neuroscience : the official journal of the Society for Neuroscience 30: 10851–10859 Doi 10.1523/JNEUROSCI.1630-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang C, Wang H, Qiao T, Yang B, Aliaga L, Qiu L, Tan W, Salameh J, McKenna-Yasek DM, Smith T et al. (2014) Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the United States of America 111: E1121–1129 Doi 10.1073/pnas.1322641111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S10. Quantification of ChAT-positive motor neurons in CTR-treated (blue) and untreated (red) knockout mice at 1 months’ age (a) and 5 months’ age (b). As no difference was observed between motor neuron numbers of CTR-treated and untreated ChAT-IRES-Cre;TardbpF/+ mice at any time point, results are shown as a percentage of each group’s respective control. While no changes were observed at the 1-month time point, untreated knockout mice showed a decrease in motor neuron number at 5 months’ age to 47% of that of controls. The motor neuron abundance of CTR-treated knockout mice is 54% (lumbar) and 56% (cervical) of controls (*p < 0.05).
Figure S6. Immunofluorescent stain of ChAT (red) and TDP-43 (blue) in p30 ChAT-IRES-Cre;TardbpF/+ mice, CTR treated and untreated ChAT-IRES-Cre;tardbpF/F mice. In all three groups, Cre-dependent Tardbp knockout rate was ~95% in both cervical and lumbar sections and not significantly different between groups (mean difference in knockout rate = 1±1.2%, p = 0.88, N=5 mice per genotype, 5 cervical/lumbar sections per animal).
Figure S7. Depletion of TDP-43 in lower motor neurons leads to age dependent motor deficits and early lethality in mice. (a) Kaplan-Meier survival curve of ChAT-IRES-Cre;tardbpF/+ (N=22, 10♀) and ChAT-IRES-Cre;tardbpF/F (N=14, 7♀) mice. Median untreated ChAT-IRES-Cre;tardbpF/F survival was 44 weeks in knockout mice. (b) Hanging wire, (c) body weight, and (d) rotarod performance are all impaired in ChAT-IRES-Cre;tardbpF/F mice. (p < 0.001 for all analyses).
Figure S8. Body weights for CTR treated and untreated ChAT-IRES-Cre;TardbpF/+ and ChAT-IRES-Cre;tardbpF/F mice, separated by sex (* p < 0.05).
Figure S9. N-terminal fragment expression alone confers no therapeutic benefit in Tardbp knockout mice. (a) Hanging wire performance and (b) body weights of ChAT-IRES-Cre;tardbpF/F mice administered AAV9 containing the N-terminal fragment of TDP-43 alone (NTF, blue) showed no improvement over untreated littermates (red). (c) NTF treated ChAT-IRES-Cre;tardbpF/F mice also showed no restoration of splicing repression as evidenced by cryptic exon incorporation into 3 targets tested
Movie S1. Representative video of p90 untreated ChAT-IRES-Cre;tardbpF/F mice (left cage) and a CTR-treated ChAT-IRES-Cre;tardbpF/F mouse (right cage). At the end of the video, a representative ChAT-IRES-Cre;TardbpF/+ mouse is shown as well. The improvement in motor function in CTR-treated Tardbp knockout mice is evident by the increased locomotion and hindlimb mobility.
Figure S11. (a) RT-PCR amplification of abnormal Sort1 splicing variant including exon 17b in mouse spinal cord and hippocampus. Like Tbc1d1, aberrant splicing of Sort1 was not observed in mouse motor neurons, but appeared in hippocampal neurons due to the depletion of TDP-43 which could be restored with the treatment of CTR. Lane 1: ChaT-IRES-Cre;TardbpF/+; lane 2: ChaT-IRES-Cre;TardbpF/F; lane 3: CTR-treated ChaT-IRES-Cre;TardbpF/F mice; lane 4: CamK2a-Cre;tardbpF/+ ; lane 5: CamK2a-Cre;tardbpF/F; lane 6: CTR-treated CamK2a-Cre;tardbpF/F mice. (b) Confocal images showing reduced Futsch (green) expression in the distal boutons (HRP, red) of TBPHΔ23/Δ23 flies (ii) compared with wild type (i). This defect could not be reversed by the expression of CTR fusion protein (iii). Scale bar: 10 μm.
Table S1. List of cryptic exons identified in TBPH-deficient Drosophila.
Table S2. Final sample sizes for all mouse cohorts examined in this study, separated by genotype and injection type.
Figure S1. Sequencing validation of cryptic exon RT-PCR. (a) Diagram of RT-PCR detection strategy. Primers were designed to amplify only the cryptic exon splice junction. (b) Gel purification and sequencing of these RT-PCR bands confirms that these DNA products precisely correspond to the predicted cryptic exon splice junctions. (c) To visualize this, sequencing data was aligned to the Drosophila genome using UCSC BLAT; thick bands in the sequence alignment indicate DNA sequences that are present in the RT-PCR product, thin bands represent sequences that have been spliced out. Sequence alignment demonstrates clear overlap with cryptic exon and completely matches the predicted the splice junctions.
Figure S2. Expression of CTR in TBPH null flies. (a) Western blot confirmation of CTR expression. Both TBPHΔ23 homozygous null and the Hsp-70 CTR line lack the endogenous 58 KD TBPH protein as shown by the N-terminal TDP-43 antibody in wild type flies. Instead, the Hsp-70 CTR line expresses the shorter CTR fusion protein. Hsp-70 CTR expression level remains stable in aged flies as it is shown here at D30 after eclosion. (b) Immunofluorescent staining of CTR (red) and neuron-specific marker ELAV (nuclear, green) in third instar larval ventral nerve cord.
Figure S3. Morphological defects at neuromuscular synapsis in TBPH null flies. (a) Confocal images of motor neuron presynaptic terminals at abdominal segment III in wild type third instar larvae stained with anti-HRP (red) and anti-DLG (green) antibodies, reveals the branching pattern and the presence of synaptic boutons. (b) Similar staining and anatomical position show reduced axonal branching and number of synaptic boutons in TBPHΔ23 homozygous larvae. (c) Recovery of presynaptic complexity with increased formation of synaptic boutons and axonal terminal branching in Hsp-70<CTR larvae.
Figure S4. Certain cryptic exon transcripts are not reversed in CTR rescue. Designing primers across cryptic exon sequences is difficult due to repetitive sequences. Therefore, high levels of background are inevitable. Nevertheless, relatively clean primers were identified for ten of the thirteen genes (Complete list of Drosophila cryptic exons is provided in Table S1), and their RT-PCR products can be clearly identified (red arrowheads). Four cryptic exon transcripts (rg, sev, Dyb, and Syn) were reversed in CTR rescue. The other six transcripts shown here were not completely reversed.
Figure S5. Expression of CTRF147L/F149L in TBPH-deficient flies has no rescue effect. (a) Diagram of the CTRF147L/F149L and UAS-NTDP chimeric constructs. Flagged CTRF147L/F149L strain did not express endogenous TBPH (b), nor did it extend lifespan (c), ameliorate locomotive function (d) or repress cryptic exon incorporations (e) in a homozygous TBPHΔ23 null background.