

Abstract
The Escherichia coli cAMP receptor protein, CRP, is a homodimeric global transcription activator that employs multiple mechanisms to modulate the expression of hundreds of genes. These mechanisms require different interfacial interactions among CRP, RNA, and DNA of varying sequences. The involvement of such a multiplicity of interfaces requires a tight control to ensure the desired phenotype. CRP-dependent promoters can be grouped into three classes. For decades scientists in the field have been puzzled over the differences in mechanisms between class I and II promoters. Using a new crystal structure, IR spectroscopy, and computational analysis, we defined the energy landscapes of WT and 14 mutated CRPs to determine how a homodimeric protein can distinguish nonpalindromic DNA sequences and facilitate communication between residues located in three different activation regions (AR) in CRP that are ∼30 Å apart. We showed that each mutation imparts differential effects on stability among the subunits and domains in CRP. Consequently, the energetic landscapes of subunits and domains are different, and CRP is asymmetric. Hence, the same mutation can exert different effects on ARs in class I or II promoters. The effect of a mutation is transmitted through a network by long-distance communication not necessarily relying on physical contacts between adjacent residues. The mechanism is simply the sum of the consequences of modulating the synchrony of dynamic motions of residues at a distance, leading to differential effects on ARs in different subunits. The computational analysis is applicable to any system and potentially with predictive capability.
Keywords: thermodynamics, transcription factor, transcription regulation, bacterial transcription, bacterial signal transduction, allosteric regulation, energy landscapes, long-range communication, RNA polymerase, signaling pathway
Introduction
The Escherichia coli cAMP receptor protein (CRP),5 is a homodimeric global transcription activator that controls the transcription of numerous genes involving carbon utilization (1, 2). The CRP-dependent promoters can be grouped into three classes (3–5). In class I promoters, CRP binds upstream of RNA polymerase (RNAP), at a site centered close to position −61.5 on the DNA (3, 6). In class II promoters, CRP is sandwiched between the α-CTD and σ70 subunit of RNAP, at a site centered close to position −41.5 on the DNA (4, 7). In class III promoters, two or more CRP dimers bind at multiple sites through class I or a combination of class I/class II CRP-dependent promoters (5, 6). Fig. 1 shows the models of class I and II promoters with respect to the relative positions of CRP, RNAP, and the two DNA half-sites for CRP binding. Three surface-exposed regions of CRP, designated as activating regions 1–3 (AR1, AR2, and AR3), modulate the contacts with RNAP (4). The contacts between ARs and RNAP are promoter-dependent. Fig. 2A shows the relative locations of these surfaces in CRP and the distances between key residues in these surfaces. The architectural designs of these promoters and the extensive results of mutation studies of class II promoters (4–6, 8–13) enabled the field to conclude that RNAP contacts AR1 of the CRP upstream subunit. Furthermore, through analogous modeling, Ebright and co-workers (13) showed that the interfaces between RNAP and the CRP downstream subunit are AR2 and AR3, as shown in Fig. 1. Hence, the contacts between AR1 surface and RNAP are different and are dependent on the class of CRP-dependent activating promoter. The additional intriguing properties for CRP-dependent promoters are as follows. The directions of transcription are different: lacPI is 5′-ATTAATGTGAGTTAGCTCACTCATTA-3′ (the direction of transcription is ←); and galPI is 3′-AAAGTGTGACATGGAATAAATTAGT-5′ (the direction of transcription is →).
Figure 1.
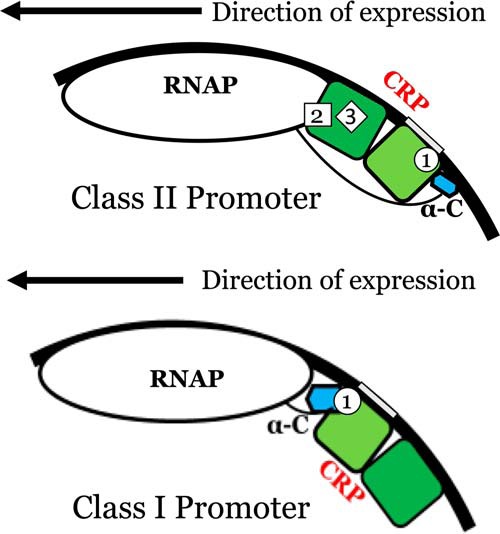
Models of the structural architectures of class II and class I CRP-dependent promoters. The dark and light green subunits of CRP represent subunits A and B, respectively. The blue structure represents α-CTD of RNAP. The black line is the DNA molecule, and the gray box in the DNA represents the –TGTGA– half-site.
Figure 2.

A, structure of subunit A of holo WT CRP showing the spatial locations of ARs, key residues, and the distances between them. B, crystallographic structure of holo WT CRP (PDB code 1I5Z) at pH 7.5 and 1.9 Å resolution. Subunits A and B are shown in green and magenta, respectively. NBD and DBD denote the cAMP- and DNA-binding domain, respectively.
In class II promoters, the interfacial interactions between CRP and the α-subunit of RNAP are via both the upstream and downstream CRP subunits instead of just the downstream CRP subunit as in class I promoters.
In some cases, same mutations are defective for class II but not class I. This observation implies that these mutations exert differential effects on CRP subunits. The question yet to be resolved is the underlying physical principle behind the unique properties displayed in these CRP-dependent class II promoters. How does a homodimeric CRP with apparent symmetrical dimers distinguish the nonpalindromic DNA sequences in class I and II promoters arranged in opposite directions for initiation? How do the same regulatory residues seemingly exert differential effects on the CRP dimer subunits of identical sequence? Currently, there are reports presenting evidence suggesting that the homodimeric CRP might in some way behave as an asymmetric molecule (14, 15). However, there is still no clear understanding of the underlying principles that govern the long-range effects exerted by these ARs that physically occupy different spaces on the CRP surface. Because the structural and molecular genetic studies did not uncover the basic underlying mechanism to address these unresolved issues, we decided to attack them from the viewpoint of the energy landscape. In a more in-depth analysis, we computationally simulated the change in energy landscapes of CRP domains induced by 14 mutants that have been investigated in a series of detailed studies to elucidate the mutual allosteric regulatory mechanism employed by these ARs. In this study, we provide evidence that the CRP subunits are energetically asymmetric and that mutations exert their effects differentially between the two subunits through networks of long-range communication. Each CRP subunit consists of a pre-existing connectivity pattern among residues. The connectivity pattern can be expressed as the stability landscapes of the protein. Thus, the different allosteric behavior between class I and II CRP-dependent promoters is the consequence of differential modulation of the roughness of the energetic landscape.
Results
WT holo CRP at 1.9 Å resolution (PDB code 1I5Z)
Because our solution studies were conducted at pH 7.8 and the structure of CRP is pH-dependent,6 we determined the CRP–cAMP co-complex at pH 7.8 by X-ray crystallography. The crystal was orthorhombic with space-group P212121 as were the previously determined WT–CRP crystals obtained at pH 7.5 (16, 17). A total of 240 one-half-degree–wide frames of 21-min exposure each were collected on a single crystal of WT–CRP. The X-ray diffraction image data were collected with an overall Rmerge of 5.0 and 99.9 completion and with an average redundancy of 4.4. A total of 38,852 unique reflections were measured in the 50–1.9-Å range. Data processing statistics are summarized in Table 1. Fig. 2B shows the structure of CRP and the identities of the pertinent secondary structural elements. Furthermore, Fig. 2A shows the locations of the key residues in ARs and the distances between them. To test for asymmetry between CRP subunits, the cAMP-binding domains (NBD, residues 6–137) were superimposed on one another while allowing the DNA-binding domains (DBD) to float. The Cα RMSD between the A and B subunits of PDB code 1I5Z by superimposing the cAMP-binding domains was 1.96 Å. However, the Cα RMSD between just the NBDs was 0.34 Å. For a similar analysis between just the DBDs (residues 138–206), the Cα RMSD was 0.78 Å. These results indicate that the NBDs are very similar in structure, although there are some differences between the DBDs among the two subunits. The larger RMSD observed while comparing the whole subunits implies that besides the minor secondary/tertiary structural differences there are most likely differences in the relative orientations between domains in each subunit. Hence, CRP is an asymmetric dimer. This difference is amplified further in energy landscapes (see under “Computer-derived energy landscapes of WT and mutant CRP”).
Table 1.
Data collection and refinement
| WT | |
|---|---|
| PDB entry | 1I5Z |
| Space group | P212121 |
| Cell dimensions | |
| a (Å) | 43.06 |
| b (Å) | 93.02 |
| c (Å) | 105.5 |
| Resolution range (Å) | 50–1.9 |
| No. of reflections | 38,852 |
| Redundancya | 4.4 (4.2) |
| I/σ(I)a | 11.5 (5.1) |
| Completeness (%)a | 99.9 (99.9) |
| Rmerge (%)a,b | 5.0 (27.0) |
| Refinement | |
| No. of reflections | 38,852 |
| Rcryst (%)a,c | 21.5 (29.3) |
| Rfree (5% of data) (%)a,c | 24.2 (33.2) |
| No. of atoms | |
| Protein, nonhydrogen | 3223 |
| Nonprotein | 334 |
| Average B-factor | 38.5 |
| RMSDs | |
| Bond lengths (Å) | 0.006 |
| Bond angles (°) | 1.2 |
a Data in parentheses are for highest-resolution shell.
bRmerge = Σ(I − 〈I〉)/ΣI, where 〈I〉 is the weighted average of all the symmetry related reflections.
c r = Σ(|Fo| − |Fc|)/Σ|Fo|, where Fo and Fc are observed and calculated structure factors. Rfree is calculated the same way with the test set reflections.
Protein dynamics monitored by FTIR
An elegant NMR study showed that the mechanism of allosteric regulation of protein is intimately linked to entropy (18). Before we invested time for a detailed study on energy landscapes, we decided to probe whether a change in protein dynamics was a manifestation of the effects of Lys-52 and His-159 mutations and whether the K52N mutation acted by reversing the change in protein dynamics induced by K159L. The amide I band, which arises predominantly from the C=O stretching vibrations of peptide bonds of protein backbone (19, 20), is known to be conformation-sensitive and is used extensively in the studies of secondary structural composition and conformational changes of proteins (20). For CRP, the employment of FTIR to monitor H–D exchange has another distinctive advantage. The DBD and NBD of CRP consist of predominantly α-helix and β-sheets, respectively. Hence, it is possible to evaluate the impact of mutation on the change in structural dynamics of each domain, although FTIR does not have the resolving power to identify the subunit in which the domain resides if the subunit structures were asymmetric.
FTIR spectra of H159L, K52N, and H159L/K52N mutants in an H2O/D2O (50:50 v/v) buffer solution were recorded as functions of time from 1 to 60 min (scan starting time) after mixing of the protein in H2O with D2O. Our previous study showed that by reducing the availability of deuterium in bulk solution relative to 100% D2O in H–D exchange experiment, a faster amide proton exchange phase can be singled out for analysis (21). For more detailed secondary structure analysis, a curve-fitting procedure was performed on a selected set of spectra of the H159L mutant recorded at 60 min after the initiation of H–D exchange. All parameters from the curve-fitting analysis are listed in Table 2. Fig. 3A shows the inverted second derivative amide I spectrum of H159L for curve fitting. Fig. 3, B–D, shows the second derivative amide I spectrum of H159L, K52N, and H159L/K52N mutants, respectively, as a function of time. It depicts H–D exchange-induced changes in various secondary structural elements in CRP species, namely apo-CRP and cAMP–CRP. The bands at 1656 and 1649 cm−1 can be assigned to the α-helix of un-exchanged hydrogen and exchanged amide deuterium, respectively (20–22). Similarly, the bands at 1641 and 1634 cm−1 are derived from β-sheet structures. The small band components near 1685, 1677, and 1669 cm−1 can be assigned to β-turn structures. For all three mutants, the cAMP–CRP complexes exhibit the strongest β-sheet band at 1634 cm−1 and the weakest α-helix band near 1653 cm−1 (combination of α-helices with exchanged and un-exchanged amide protons). These results indicate that the binding of cAMP triggered conformational and structural dynamic changes in two opposite directions regarding the two dominant secondary structures of CRP. We compared the time-dependent intensity changes at the 1634 cm−1 (β-sheet) and 1653 cm−1 (α-helix) bands among all four proteins. Fig. 4A shows the plots depicting the relative intensity change at the 1634 cm−1 β-sheet bands of three mutants and WT CRP as functions of time. Intensity of the β-sheet band was calculated as a difference in the second-derivative spectrum between the negative peak at maximum and baseline. The intensity values for WT CRP were adopted from our previous work (20). The values of relative intensities were normalized using the highest intensity value from WT CRP as 100%. The errors for β-sheet and α-helix were 2 and 5%, respectively. The difference was due to greater dynamics of α-helix, which is the predominant secondary structure in the DBD. Hence, it was more difficult to precisely synchronize the start time. For either the H159L or K52N single mutation, the H–D exchange-induced intensity change deviated significantly from that of the WT CRP for all three species. For apo-CRPs, the result indicates that both mutations decrease, in various degrees, the conformational flexibility of the DBD and NBD. For cAMP–CRP complexes, both mutants reduce the conformational flexibility of the NBDs but increase that of the DBDs. The effects of the H159L mutation on the conformational flexibility of the NBD were, as expected, less than that of the K52N mutation, because residue His-159 is located on a surface-loop of DBD distant from the cAMP-binding site. This result provides further evidence indicating inter-domain and/or inter-subunit communication of CRP (23, 24). Although the K52N or H159L single mutation alone disrupts significantly the conformational and structural dynamics of the protein, the K52N/H159L double mutation restored the conformational flexibility (apo-CRP) or compatibility (cAMP–CRP) of DBD close to the level of wide type CRP. Fig. 4B shows the plots depicting the relative intensity changes at the band assigned to α-helix structures of the three mutants, as well as the WT CRP, as function of time. The intensity of the α-helix band was calculated as a difference between the negative peak at maximum (1656–1653 cm−1) and baseline in the second-derivative spectra. Data for WT CRP were adopted from our previous work (20). The values of the relative intensities were normalized using the value of WT apo-CRP in H2O solution as 100%. Because of the predominance of α-helices in the DBD (16, 17), the H–D exchange-induced changes in the relative intensity of the α-helix band reflect changes in the conformational and structural dynamics of the DBD (19). As the amide proton exchange proceeds, the intensity of the α-helix band decreases as it changes from a single component at 1656 (H) cm−1 to two components at 1656 (H) and ∼1649 (D) cm−1, even though the overall integrated area remains relatively constant, as summarized in Table 2.
Table 2.
Frequencies and relative areas of amide I components of H159L CRP after 60 min of H–D exchange in 50:50 H2O/D2O solutions
| Apo |
cAMP |
Assignment | ||
|---|---|---|---|---|
| ν | Area | ν | Area | |
| cm−1 | % | cm−1 | % | |
| 1684.5 | 5.4 | 1684.5 | 6.8 | β-Turn |
| 1677.6 | 7.8 | 1676.6 | 7.4 | β-Turn |
| 1668.7 | 1.3 | 1668.7 | 2.1 | β-Turn |
| 1656.0 | 20.0 | 1656.4 | 19.8 | α-Helix (H)a |
| 1650.1 | 12.2 | 1648.6 | 17.6 | α-Helix (D)b |
| 1641.1 | 17.3 | 0 | β-Sheet (D) | |
| 1633.5 | 36.0 | 1633.5 | 46.8 | β-Sheet |
a Secondary structure with unexchanged amide hydrogen is shown.
b Secondary structure with exchanged amide deuterium is shown.
Figure 3.

A, curve-fitted and inverted second-derivative amide I spectrum of apo-CRP. The inversion of the second-derivative spectrum was done by factoring by −1. The parameters for curve fitting are summarized in Table 2. B, amide I spectra as a function of time of apo- and cAMP-liganded H159L mutant in a 50:50 H2O/D2O. The spectra were recorded at 1, 10, 20, 30, 40, and 60 min (starting times) after the sample mixing. The spectrum of WT apo-CRP in H2O, as shown as a black short broken line, is included for reference. The arrows indicate the direction of change in absorbance as a function of time. C, amide I spectra as a function of time of apo- and cAMP-liganded K52N CRP mutant in a 50:50 H2O/D2O. The spectrum of WT apo-CRP in H2O, as shown as a black short broken line, is included for reference. The arrows indicate the direction of change in absorbance as a function of time. D, amide I spectra as a function of time of apo- and cAMP-liganded K52N/H159L CRP mutant in a 50:50 H2O/D2O. The spectrum of WT apo-CRP in H2O, as shown as black short broken line, is included for reference. The arrows indicate the direction of change in absorbance as a function of time.
Figure 4.

Relative intensity changes of the β-sheet band (∼1634 cm−1) (A) and the α-helix band (1656–1653 cm−1) (B) of three mutants and WT CRP in a 50:50 H2O/D2O solution as functions of time. The values of relative intensities were normalized. The samples and symbols are as follows: WT (1); D159L (2); K52N/D159L (3); and K52N (4). Data points in red and black are for holo and apo-CRP, respectively. The error for the short time points (1–5 min) is ±2% for the β-sheet band but is ±5% for the α-helix band.
Compared with apo-CRP, a faster amide proton exchange at the α-helix structure was observed for the cAMP–CRP complexes of the three mutants, indicating a flexible DBD domain like that of WT CRP. After a 40-min exchange, the α-helix band intensities of the cAMP–CRP complexes of the three mutants, as well as the WT CRP, were finally stabilized showing a rank order of decreasing flexibility, K52N, H159L/K52N, WT, H159L, within a narrow range of distribution of 71–76% relative intensity in respect to the common reference (100% exchange of WT CRP). This result is consistent with the observations that these mutations resulted in no defect on specific DNA binding (23, 24). It implies that the conformational flexibility of DBD may play a key role in determining the DNA-binding ability of the cAMP–CRP complex.
In summary, the FTIR data indicate minor structural changes in CRP induced by these mutations. However, the dynamic motions of the α-helices and β-sheets were differentially perturbed by single mutations. The double mutations of K52N/H159L restored the dynamic changes induced by single mutations to resemble that of WT CRP. Nevertheless, besides providing information on changes in protein dynamics, these results do not elucidate the underlying molecular mechanism(s) on the regulatory roles of mutations at the energetics level.
Computer-derived energy landscapes of WT and mutant CRP
Stability profiles of residues
To acquire an in-depth understanding from the viewpoint of the energy landscape, we probed for information on the change of energy landscapes of CRP induced by these various mutations. We employed the algorithm COREX/BEST, which has proven successful in capturing the essence of allostery for multiple systems (25–28).
The algorithm simulated the stability constants of individual residues, lnKi, f, of CRP based on the structures of holo CRP (PDB code 1I5Z). Typical results of COREX/BEST simulations for WT subunits are shown in Fig. 5, subunits A and B. The vertical line at residue 138 marks the beginning of the DNA-binding domain. The energetic landscapes of WT NBD are very similar in both subunits, as shown by the black lines in Fig. 5, subunits A and B, although the loops encompassing residues 50–60 and 72–80 in one subunit (for convenience it will be referred to as A) were less stable, i.e. had lower values for lnKi, f. The more obvious differences in the energetic landscapes were in the DBDs, particularly in the region encompassing residues 140–160, i.e. the D-helix and part of the loop region between the D- and E-helices. This seems to be a key region involved in positioning the DNA-binding F-helix. This difference in the energy landscape of the DBDs among the two subunits of CRP is evidence of asymmetry of the CRP subunits.
Figure 5.

Residue stability of holo CRP residues for the A and B subunits, respectively. Thick black line and thick red or blue lines are residues for WT and K52N mutant, respectively. Numbers in boxes show the residues.
We examined the effects of mutations according to the reported functional perturbations as listed in Table 3, which summarized the residues target for mutation, nature of mutation, location of these residues in the various activating regions (ARs), and the biological effects induced by these mutations.
Table 3.
List of mutations, location, and biological effects
| Mutation residue | AR location | Biological effects | Refs. |
|---|---|---|---|
| H19L | 2 | Defective for class II, not class I | 33 |
| H21L | 2 | Defective for class II, not class I | 33 |
| K52N | 3 | Positive charge for maximum expression of class II | 8 |
| H58A | 3 | Negative charge for optimal expression of class II, not class I | 8 |
| H58K | 3 | ||
| E96L | 2 | Restore the repressive effect of H159L on class II but not class I | 29 |
| K101D | 2 | Defective for class II, not class I | 33 |
| T158A | 1 | Defective for class II and class I | 33 |
| H159L | 1 | Defective for class I and restored by K52N or E96L | 10 |
| K52N/H159L | 1, 3 | Positive charge for maximum expression of class II | 8 |
| K52N/H159L/K101D | 1, 2, 3 | Defective for class II, not class I | 33 |
| H58A/K52N/H159L/K101D | 1, 2, 3 | Negative charge for optimal expression of class II, not class I | 8 |
| H58K/K52N/H159L/K101D | 1, 2, 3 | ||
| E181V | Induced non-WT binding specificity | 29 |
The K52N mutation induced significant changes in the magnitudes of lnKi, f of the subunits, as shown by the red and blue lines in Fig. 5, subunits A and B, respectively. Compared with WT CRP, the K52N mutation lowered the general stability of subunit A but significantly elevated that of the other subunit (arbitrarily assigned as B). Nevertheless, the general profiles of the energetic landscapes for NBDs remained the same as shown by the boxes identifying these residues. The more significant change in profile was observed again in the DBDs around the similar region as in the WT, namely encompassing residues 140–160. Hence, K52N exerted a differential effect on the stability of these two subunits and exacerbated the asymmetry in the stability of these subunits. This general pattern of mutation effects was repeated for other mutants. Results of all these simulations of various mutants are shown in Figs. S1–S5. We expressed the data for a more global view by calculating the average lnKi, f (〈lnKi, f〉) of each domain.
Effects on stability profiles of domains
Fig. 6 summarizes the results of 〈lnKi, f〉x, where x is NBD or DBD, and the numerical values of 〈lnKi, f〉x are listed in Table 4. The common trends from these results are as follows: in subunit A the mutants did not induce significant changes in the stability of the NBD, i.e. ∼0.6 lnK unit, nor in the DBD with an ∼2 lnK unit increase only. The mutation effects on subunit B exhibited a more complex pattern and more significant change in magnitude, e.g. 3–4 lnK units in response to mutation. Hence, mutations exacerbated the asymmetry of stability of not only subunits but also the domains. Regardless of the location of the residues or nature of the side chain of substitution, all 14 mutations affected the stability of subunit B more significantly, particularly the DBD of subunit B.
Figure 6.
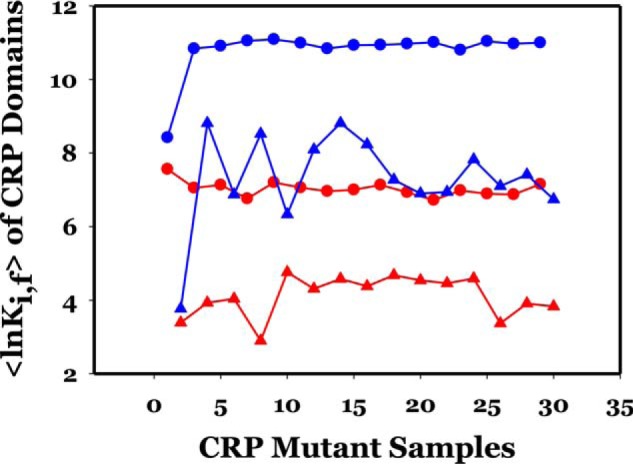
〈lnKi, f〉 of CRP subunits and domains as a function of mutations. Odd- and even-numbered samples represent those of subunits A and B, respectively. The color codes are red and blue for subunits A and B, respectively; ○ and ▵ for NBD and DBD, respectively. The order of mutant presented is as follows: 1–2 (WT); 3–4 (H19L); 5–6 (H21L); 7–8 (K52N); 9–10 (H58A); 11–12 (H58K); 13–14 (E96A); 15–16 (K101D); 17–18 (T158A); 19–20 (H159L); 21–22 (K52N-H159L); 23–24 (52–159-101); 25–26 (T58K + 23/24); 27–28 (T58A + 23/24); 29–30 (E181V).
Table 4.
Summary of values of lnK of subunits and domains induced by mutations
| CRP no. | Mutation | 〈lnKi, f〉 |
|||||
|---|---|---|---|---|---|---|---|
| S.U. | Subunit | NBD | DBD | ΔNBDa | ΔDBDb | ||
| 1 | WT | A | 6.11 | 7.56 | 3.39 | ||
| 2 | WT | B | 6.86 | 8.42 | 3.77 | ||
| 3 | H19L | A | 5.98 | 7.05 | 3.93 | −0.51 | 0.54 |
| 4 | H19L | B | 10.16 | 10.84 | 8.81 | 3.28 | 5.04 |
| 5 | H21L | A | 6.07 | 7.13 | 4.04 | −0.43 | 0.65 |
| 6 | H21L | B | 9.55 | 10.91 | 6.87 | 3.35 | 3.38 |
| 7 | K52N | A | 5.46 | 6.76 | 2.9 | −0.8 | −0.49 |
| 8 | K52N | B | 9.98 | 11.05 | 8.52 | 3.49 | 5.13 |
| 9 | H58A | A | 6.36 | 7.2 | 4.76 | −0.36 | 1.37 |
| 10 | H58A | B | 9.49 | 11.09 | 6.33 | 2.67 | 2.56 |
| 11 | H58K | A | 6.12 | 7.06 | 4.31 | −0.5 | 0.92 |
| 12 | H58K | B | 10.02 | 10.99 | 8.09 | 2.57 | 4.32 |
| 13 | E96A | A | 6.14 | 6.96 | 4.58 | −0.6 | 1.19 |
| 14 | E96A | B | 10.16 | 10.84 | 8.81 | 2.42 | 5.04 |
| 15 | K101D | A | 6.1 | 7 | 4.38 | −0.56 | 0.99 |
| 16 | K101D | B | 10.02 | 10.93 | 8.23 | 2.51 | 4.46 |
| 17 | T158A | A | 6.29 | 7.13 | 4.68 | −0.43 | 1.29 |
| 18 | T158A | B | 9.71 | 10.94 | 7.27 | 2.52 | 3.9 |
| 19 | H159L | A | 6.11 | 6.93 | 4.54 | −0.63 | 1.15 |
| 20 | H159L | B | 9.61 | 10.97 | 6.9 | 2.55 | 3.13 |
| 21 | K52N/H159L | A | 5.95 | 6.72 | 4.46 | −0.84 | 1.07 |
| 22 | K52N/H159L | B | 9.65 | 11.01 | 6.94 | 2.59 | 3.17 |
| 23 | 52-159-101 | A | 5.68 | 6.89 | 3.37 | −0.67 | -0.02 |
| 24 | 52-159-101 | B | 9.72 | 11.04 | 7.1 | 2.62 | 3.33 |
| 25 | T58K + #25 | A | 5.85 | 6.87 | 3.91 | −0.69 | 0.52 |
| 26 | T58K + #25 | B | 9.78 | 10.97 | 7.41 | 2.55 | 3.64 |
| 27 | T58A + #25 | A | 6 | 7.15 | 3.83 | −0.41 | 0.44 |
| 28 | T58A + #25 | B | 9.57 | 11 | 6.74 | 2.58 | 2.97 |
| 29 | E181V | A | 6.13 | 6.98 | 4.59 | −0.58 | 1.2 |
| 30 | E181V | B | 9.8 | 10.8 | 7.82 | 2.38 | 4.05 |
aΔNBD = NBDmu − NBDwt.
bΔDBD = DBDmu − DBDwt.
In view of the current models of organizations of class I and II CRP-dependent promoters shown in Fig. 1, it is imperative to investigate the effects of these 14 mutations on residues encompassed in these ARs. Because AR1 resides in DBD and AR2, 3 in NBD, are these changes in stabilities reflected by the changes of residues residing in their respective domains?
Effects of mutations on energy landscapes of activating regions
AR1 consists of residues Asn-65, Gln-66, Leu-148, Lys-152, Ala-156, Thr-158, and Met-163. Fig. 7, subunits A and B, shows the effects of the 14 mutations on the values of lnKi, f of the residues in AR1 of subunits A and B, respectively. The corresponding information for WT CRP was also shown as a reference in sample 1. It is obvious that the patterns were different between the two subunits, i.e. the same mutation exerted a differential effect on the equivalent residue as a function of the subunit in which it resides. The responses were not only dependent on the locations of these residues in the subunit but were also dependent on the specific mutation. Stability of some residues was affected in a synchronized manner, e.g. residues 65 and 66 and residues 158 and 163. In subunit B, all mutations increased the stability of Asn-65 and Gln-66 significantly by ∼2–4 kcal/mol. The response of Leu-148, Lys-152, Ala-156, Thr-158, and Met-163 was quite synchronized; however, K52N, E58A, and the K52N–E58A–K101E–H159L mutations have no effect, but the others enhanced the stability of this group of residues.
Figure 7.

lnK of AR1 residues in subunits A and B as a function of mutations. The identities of mutations are the same as in Fig. 6. The color codes for residues are as follows: red circle, 62; black circle, 66; green circle, 148; blue circle, 152; pink circle, 156; light green circle, 158; and gray circle, 163.
AR2 consists of residues His-21, Glu-37, and Ser-98. Fig. 8 shows the results of the effects of mutations on the residues in AR2. In subunit A, all three residues responded to all mutations by lowering their stability by ∼0.5 kcal/mol or less, i.e. quite insignificantly. In subunit B, His-21 and Ser-98 were induced by all mutations to a higher stability by ∼1 and ∼3 kcal/mol, respectively. Glu-37 was not affected by any of these mutations.
Figure 8.
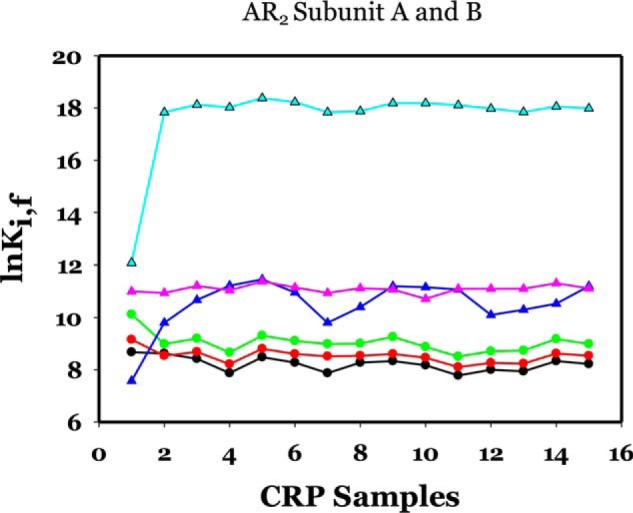
lnK of AR2 residues in subunits A and B as a function of mutations. The identities of mutations are the same as in Fig. 6. The color codes for residues are as follows: subunit A: black circle, 21; red circle, 37; green circle, 98; subunit B: blue triangle, 21; pink triangle, 37; cyan triangle, 98.
AR3 consists of residues Ser-27, Thr-28, Gln-32, Leu-34, Lys-52, Glu-54, and Glu-55. Fig. 9, subunits A and B, show these results. The responses of Ser-27, Thr-28, Gln-32, and Leu-34 were quantitatively synchronized in subunit A but less so in subunit B. In subunit A, the responses of Lys-52, Glu-54, and Glu-55 were synchronized with minor fluctuation, although the increases in stability of those residues in subunit B were more substantial.
Figure 9.

lnK of AR3 residues in subunits A and B as a function of mutations. The identities of mutations are the same as in Fig. 6. The color codes for residues are as follows: subunit A: black circle, 19, 21, 32, and 34; black triangle, 52, 54, and 55; subunit B: red circle, 19; green circle, 21; blue circle, 32; *, 34; black triangle, 52, 54, and 55.
Table 5 summarizes the results on the perturbations of stability of residues in ARs. These results show, together with Fig. 6, that mutations impose great differential effects on residues within the ARs and predominantly affect the stability of domains in subunit B. Because all these mutations were designed to uncover their roles in modulating the expression of class II CRP-dependent promoters, perturbations of subunit B were expected and are consistent with the model shown in Fig. 1. Furthermore, these mutations were shown not to affect class I promoters except H159L. Hence, it was not surprising that the stability of subunit A residues was not significantly altered.
Table 5.
Summary of changes induced in lnKi, f of residues by mutations in ARi in subunits A and B
Code for font and magnitude of ΔlnKi (lnKi, mu − lnKi, wt): black and boldface for ΔlnKi <2 lnK units and >6 lnK units, respectively, where i is residue number.
| Mutation residue | AR1 residues in A subunit affected | AR1 residues in B subunit affected | AR2 residues in A subunit affected | AR2 residues in B subunit affected | AR3 residues in A subunit affected | AR3 residues in B subunit affected |
|---|---|---|---|---|---|---|
| H19L | 65, 148–152 | 65, 66, 148–163 | 98 | 21, 98 | 27, 28, 52–55 | |
| H21L | 65, 148–152 | 65, 66 | 98 | 21, 98 | 27, 28, 52–55 | |
| K52N | 148–152, 156 | 65, 66, 148–163 | 98 | 21, 98 | 52, 54, 55 | 52–55 |
| H58A | 148–152 | 65, 66 | 98 | 21, 98 | 52–55 | |
| H58K | 148–163 | 65, 66, 148–163 | 98 | 21, 98 | ||
| E96L | 148–152 | 65, 66, 156, 148–163 | 98 | 21, 98 | ||
| K101D | 65, 66, 156, 148–163 | 98 | 21, 98 | |||
| T158A | 148–156 | 65, 66, 156, 148–163 | 98 | 21, 98 | ||
| H159L | 65, 66, 148–163 | 98 | 21, 98 | |||
| K52N/H159L | 65, 66, 148–163 | 98 | 21, 98 | 52–55 | ||
| K52N/H159L/K101D | 158, 163 | 65, 66, 156, 148–163 | 98 | 21, 98 | 52–55 | |
| H58A/K52N/H159L/K101D | 65, 66, 156, 148–163 | 98 | 21, 98 | 52–55 | ||
| H58K/K52N/H159L/K101D | 148, 152 | 65, 66, 156 | 98 | 21, 98 | 52–55 | |
| E181V | 148, 152, 158, 163 | 65, 66, 148–163 | 98 | 21, 98 |
In summary, the simulation results identify the differential change in the energy landscape of ARs induced by these mutations as the mechanism to induce the observed biological changes. Furthermore, the differential effects are consequences of long-range signal transmission.
Connectivity network among residues
One of the persistent issues in allostery is whether signal transmission is a consequence of a stochastic process without a specific pathway or whether the signal transmission pathway network is engineered into the protein. If a pre-engineered pathway exists, what does mutation do to that pathway? Fig. 5 and Figs. S1–S5 show the consistency of the energy landscapes with specific residues identified. Furthermore, we constructed a pattern of connectivity in the form of a heat map of each residue with respect to all other residues of the WT and mutant CRPs.
Residue-specific connectivity (RSC)
Fig. 10 is a presentation of WT CRP A and B subunits and the corresponding maps for the K52N mutant. If two residues were folding and unfolding in total synchrony in the same time scale, then the value of RSC would be positive and assumes a value of 1, whereas those out of synchrony would be negative. It is obvious that many residues in NBD are in some degree of synchrony in their folding–unfolding dynamic motions. For example, residues in 63–69, 85–103, and 116–123 were in synchronized dynamic motions in a positive manner, as indicated by positive (red) RSC values with the rest of the subunit including those in DBD. There are also residues with negative synchronization shown in negative (blue) RSC values. Asynchronization means that stabilization of residue j is coupled to destabilization of residue k. This is the intrinsic networking in CRP and would be the underlying principle of long-range communications. The K52N mutation did not change the connectivity pattern of subunit A, although it did modulate the magnitudes of synchronization of some of these residues. For example, the value of RSC between residues 50–60 and 130–206 was negative, i.e. asynchronization between these two regions. However, in comparing the magnitude of RSC between those regions, the K52N rendered it less negative than that of WT; hence, the observation indicated that the folding–unfolding process of residues between these two regions was not as out of phase as that in WT. The pattern of RSC for subunit B was different from that of subunit A. There were not that many residues in subunit B that underwent synchronized folding–unfolding motions. However, the pattern observed in WT subunit B was not altered by K52N mutation. Again, in DBD the magnitudes were altered but the general pattern was retained. Figs. S6–S13 summarize the connectivity maps of mutants. It is evident that all the maps show essentially identical connectivity pattern among residues in NBD. The detectable differences were in DBD, which was to be expected due to the observed changes in energetic landscapes induced by the mutations. However, all the observable changes were not in the basic pattern but in changes of the magnitude of the RSC. Mutations affected only the extent of synchronization of their folding–unfolding.
Figure 10.

A and B are connectivity map (RSC) of WT and K52N subunits A and B, respectively. The false color scale indicates positive and negative connectivity between residues. Positive connectivity indicates the folding–unfolding reactions of these residues are synchronizes at the same time scale, whereas negative connectivity indicates asynchronization between these residues.
Discussion
The structural and regulatory architecture of CRP-dependent class II promoters is quite elaborate. Much effort has been devoted to elucidating these issues, and many intriguing observations have been reported. The results of this study uncovered the underlying regulatory mechanism for class II CRP-dependent promoters.
The asymmetric nature of the CRP subunits and the promoter construction, i.e. location and orientation of –GTGTA–, plays key roles in defining the biological consequences in changes of function. The initial work showing the difference in orientation of CRP subunits in its interaction with the α-subunit of RNAP was the result of elegant studies by Busby and Ebright and their respective co-workers (30–35). The Ebright lab engineered a heterodimer harboring an E181V mutation that specifically orients one subunit to bind to the half-site of a specific sequence (34, 35). However, the individual subunits of dimeric WT–CRP are intrinsically asymmetric in their energetic landscapes. Furthermore, the persistent pattern of mutation effects on subunits A versus B is another strong indication of an intrinsic distinction between the two subunits. We show7 that subunit A is the likely binding target of the first cAMP molecule. This conclusion was derived from analysis of apo-CRP structures and solution biophysical measurements. Hence, we hypothesize that the CRP–cAMP1 complex is equivalent to cAMP–subunit A. Furthermore, we propose that the CRP–cAMP1 complex binds to the consensus DNA half-site –TGTGA–. If this hypothesis were correct, it will then serve as the basis of the underlying molecular mechanism in the resulting discussion of the function and regulatory properties of the CRP-dependent class II promoter. Based on crystal structures of the class I and II promoter complexes and according to the nomenclature of our data, the interface between CRP and RNAP in class I is AR1 of the downstream subunit B (29). In class II promotor there are three interfaces: AR2 and AR3 of the downstream subunit A and AR1 of the downstream subunit B (13).
Based on the newly uncovered regulatory mechanism, we can examine in more detail the changes in biological functions in view of changes in CRP energy landscapes. The vital differences between class I and II promoters are the inter-macromolecular interfaces in CRP. In class I promoters, the interface is AR1 of subunit B (29). In class II promoters, the interface is AR1 of subunit A in conjunction with AR2 and AR3 of subunit B. Hence, energetic perturbation of AR1 in subunit A will lead to change in expression in class I promoters, whereas perturbation of AR1 in subunit B does not matter. Perturbation of AR1 in subunit A in addition to AR2 and AR3 in subunit B will modulate the full expression in class II promoters. A lesser degree of defective expression can be observed with mutations in AR1 of subunit A if both AR2 and AR3 retain their WT sequences because CRP would still be able to interact with RNAP with functional AR2 and AR3. Mutations affecting subunit A or B, in regions other than AR1, could exert either positive or negative effects on class II but not class I promoters. Hence, the present level of understanding is limited to changes, be they positive or negative. For mutations that affect only class II not class I expression listed in Table 3, except for H159L, they all induced significant changes in subunit B but not subunit A.
Let's review the reported biological perturbations (Table 3) and correlate that with the results in Table 5 in which the code for font and magnitude of ΔlnKi (lnKi, mu − lnKi, WT) are as follows: black type indicates ΔlnKi = <2 lnK units and ∼3–4 lnK units, respectively; and boldface type indicates ∼6 lnK units, where i is residue number. A cursory view of Table 5 shows that none of the mutants affects the energy landscape of subunit A by more than 2 lnK units, i.e. ∼1 kcal/mol. Nevertheless, all AR1 and AR2 in subunit B are affected by >3 or as large as 6 lnK units. Eight of the 14 mutants affected AR3 in subunit B to the same extent. All the mutants that affected the ARs in subunit B exhibit phenotypes that disturb class II gene expressions. Thus, there is a correlation between the biological phenomena and patterns of the energy landscapes for single and complex mutants. The K52N and the H58A/H58K mutants exhibit dominant effects in some of multiple mutation complexes.
The following CRP samples are discussed in more detail to illustrate the general conclusions. The H19L and H21L mutations in AR2 lead to defective expression of genes regulated by class II but not class I promoters. These two mutations lead to only an ∼1 lnK unit increase in the stability for residues 52, 148, and 152 of subunit A but an increase of 3–4 lnK units, i.e. ∼2 kcal/mol increase in the stability of residues 148–163 in subunit B (Fig. 8, samples 2 and 3 for H19L and H21L, respectively, and Table 5). In addition, the stability of residues 52–55 in AR3 of subunit B is significantly increased. Thus, the effects of mutating these two residues are on subunit B, not subunit A. So, the biological effects are on class II promoters only.
The T158A and H159L mutations in AR1 lead to defective gene expression regulated by class I and II CRP-dependent promoters. This biological consequence is due to the increase of 3–4 lnK units, i.e. ∼2 kcal/mol, in the stability of AR1 residues 148–163 and AR2 residues 21 and 98 in subunit B, whereas the stability of residues 148–156 in subunit A only increased by a minimum of ∼1 lnK unit, i.e. ∼0.5 kcal/mol (Fig. 7, samples 17–18 and 19–20 for T158A and H159L, respectively, and Table 5). These results are expected to only affect class II promoters and not class I because there is minimal effect on AR1 of subunit A, which interacts with RNAP in class I promoters.
The H159L mutation does not lead to changes in the energy landscape in subunit A AR1 but it is detrimental to the expression of class I and only partially to class II. There may be the requirement of a specific stereochemical interaction between α-CTD RNAP and CRP subunit A in class I promoters. The lack of reactivation of the H159L mutation in class I promoters is most likely because the K52N mutant destabilizes the DBD in which His-159 resides, whereas the same mutant stabilizes the same domain in subunit B. The same rationale could apply to the observation for the E96L mutation, i.e. differential effects on the same domain between subunit A and subunit B.
Another novel observation is that CRP apparently consists of a pre-existing connectivity pattern among residues residing in the inter-macromolecular interfaces identified previously as activating regions. Hence, substitutions of side chains in these ARs can affect the magnitudes and nature of connectivity which lead to changes in the transcription processes governed by the class II CRP-dependent promoters. In addition, this study uncovered some key groups residues which seem to be “connected” to all residues in CRP, e.g. 41–46, 63–69, 71–81, 83–103, and 116–123 in subunit A. However, there is still a lot to uncover in terms of the various roles of these residues in modulating the functions of CRP.
The FTIR data indicate that at least the K52N and H159L mutations affect the dynamics of CRP. The double mutant of K52N/H159L restores the dynamic behavior to that of the WT. Change in protein dynamics as a function of mutation is consistent with our earlier study showing a linear correlation between protein dynamics and magnitudes of allostery in cAMP binding (36).
Conclusions
We elucidated an underlying mechanism for allostery. Apparently, an allosteric protein consists of a pre-existing connectivity pattern among residues. The connectivity pattern can be expressed as the stability landscape of the protein. Allosteric behavior is the consequence of modulation of the roughness of the energetic landscape. In a homopolymeric protein, differential modulation of subunits may occur due to a difference in the synchronization regime between subunits that consist of the same pattern for communication. As such, computational simulations can capture the essence of biological observations reported in the literature.
Experimental procedures
cAMP was purchased from Sigma. Deuterium oxide (99.96 atom % D) was the product of Cambridge Isotope Laboratories (Andover, MA).
The stock solution of cAMP (18.4 mm) was prepared by dissolving cyclic nucleotides in 50 mm Tris, 0.1 m KCl, and 1 mm EDTA, pH 7.8, and pH was adjusted with KOH solution. The concentrations of cAMP were determined spectrophotometrically using the extinction coefficients of 14,650 m−1 cm−1 at 258 nm (The Merck Index).
Crystal structure determination of WT CRP
Crystallization
Large, single complex crystals were grown at room temperature using the hanging-drop vapor-diffusion method. Initial crystallization screening was performed with Hampton Research screen kit I, and 8 of 50 conditions grew tiny crystals after 2 days. The optimal conditions that were used to grow the crystals for data collection are as follows. Protein and cAMP, with a molar ratio of 1:20, were mixed in 50 mm Tris, pH 7.8, 100 mm KCl, 1 mm EDTA, with 5 mm DTT (growing buffer); the final protein concentration is 2 mg/ml. 10-μl aliquots of the mixed solution were sealed in vapor diffusion wells and allowed to equilibrate with the growing buffer reservoir solution via the hanging-drop method. Crystals suitable for data collection generally grew within 2 weeks. The crystals formed elongated rectangular rods (∼0.4 × 0.3 × 0.2 mm). Prior to rapid cryo-cooling, crystals were step-soaked with increasing concentrations of glycerol up to a final concentration of 30% (v/v) glycerol and 10 mm cAMP.
Refinement
Structure refinement of the WT CRP structure started with rigid-body refinement of the 2.5 Å resolution WT–CRP structure, 3GAP (17, 18), using CNS (37). This was followed by a separate rigid-body refinement of the four individual domains of the dimer, where each of the two cAMP molecules in the jellyroll site was included in their respective CNB domain rigid-body group. Initial examination of the electron density maps revealed a third cAMP molecule bound to the DBD of subunit A, which is in the “closed” DNA-binding position. This DBD forms a crystal contact that holds it in the closed conformation. Model building proceeded in XtalView (19, 38), followed by rounds of positional and individual B-factor refinement in CNS. Loop 52–55 and the region around β-sheet 9 (148–158) had very poor electron density and were rebuilt in several rounds using omit maps. The C-terminal residues were rebuilt from Gly-200 when it was discovered that the original model had incorporated an excessively sharp turn at Gly-200 causing the following amino acid chain to be mis-registered by one. Waters were added using omit maps and a locally written peak search and analysis program, as well as using the CNS water-pick utility, and then individually examined. Anisotropic scattering and absorption were modeled using a local scaling algorithm (39, 40). This program is available from the author, M. A. W., as part of the PMB suite of utilities. Data were collected on a single crystal cryo-cooled to 93 K on a Mac Science DIP2030H image plate detector system mounted on a Mac Science MX06HF X-ray generator equipped with Bruker Göebel optics.
The coordinates and experimental structure factors of the WT CRP structure were deposited in the RCSB Protein Data Bank with accession code 1I5Z.
Mutagenesis of CRP mutants
K52N, H159L, and H159L/K52N mutations were introduced into the CRP gene by the Promega altered site in vitro mutagenesis system. The DNA sequence 5′-GGG TCT GTG GCA GTG CTG ATC AAC GAC GAA GAG GGT AAA G-3′ was used as the mutagenic oligonucleotide for constructing the K52N mutant; 5′-CAA CCAGAC CCT ATG ACT CTG CCG GAC GGT ATG CAA ATC-3′ was used for constructing the H159L mutant. The underlined triplet sequence was codons for the mutagenic amino acids. The double mutant H159L/K52N was constructed by using K52N as a template and H159L as a primer. The desired mutants were directly screened by DNA sequencing. The mutant CRP alleles were cloned into the expression vector pPLc28. The recombinant plasmids were then used for transformation of E. coli strain CA8445/pRK248cIts. The cells carrying CRP mutants were grown in Terrific Broth medium at 32 °C and induced to express at 42 °C. The CRP mutants were isolated and purified, as described previously (22, 23), and stored in −20 °C freezer until use. The homogeneity of purified CRP mutants was >99% as judged by SDS-PAGE.
Sample preparations
CRP solutions were dialyzed against 50 mm Tris buffer containing 0.1 m KCl and 1 mm EDTA, pH 7.8, overnight to remove the stabilizing agent glycerol and other additives. The resultant was concentrated with a Centricon 10 microconcentrator (Amicon) at 5000 rpm using an SS-34 rotor. The concentration of stock CRP solution was determined spectrophotometrically using the extinction coefficient of 40,800 m−1 cm−1 at 278 nm for the CRP dimer (41). The cAMP-liganded CRP samples were prepared by adding aliquots of a stock solution of cAMP to CRP solutions in the molar ratio of 5:1 prior to H–D exchange or FTIR measurement. This ratio was used to ensure cyclic nucleotide saturation according to their binding constant. The H–D exchange was carried out by mixing aliquots of stock solution of CRPs, after adjusting buffer concentration, with D2O at percentage ratio of 50:50 prior to FTIR measurement (20).
FTIR spectroscopy
FTIR spectra were measured with a Bomem MB-Series FTIR spectrometer (Quebec, Canada) equipped with a dTGS detector and purged constantly with dry air generated by a Balston (Haverhill, MA) air dryer. Protein samples (3.0 mg/ml) were loaded in a product no. 20500 heatable liquid IR cell (Graseby) with CaF2 windows and a 25-μm spacer. For each spectrum, a 128-scan interferogram was collected in single beam mode with a 4-cm−1 resolution. The scan accumulation time was 6½ min. Reference spectra were recorded under identical scan conditions with only the corresponding buffer in the cell. For the H–D exchange experiment, the time points refer to the scan-starting times. Protein spectra were obtained according to previously established criteria and double-subtraction procedure (20). The residual water vapor signals, if present, in the spectrum of protein were removed by subtracting the spectrum of gaseous water.
Second-derivative spectra were obtained with a seven-point Savitsky-Golay derivative function. Second-derivative spectra (the amide I region only) were baseline corrected and area normalized as described previously (35). Final spectra were treated with a 2× interpolate function and plotted with a SigmaPlot 5 software (Jandel Scientific). Band areas were determined by curve-fitting analysis of the inverted second-derivative spectrum as described previously (20). These spectra were highly reproducible with error within the size of the data point.
Computation analysis of CRP structures
Briefly, the connectivity of different structural elements in CRP was studied by computation to provide a rationale for the functional behavior of the wildtype and mutant CRP. The computation was accomplished by using the COREX/BEST algorithm where conformational ensembles were generated with a modified version of COREX/BEST that employs a Monte Carlo sampling strategy. High-stability constants signify residues that are folded in most highly-probable states under native conditions, whereas lower-stability constants signify residues that are unfolded in many of those states. The templates for mutant CRPs were generated by substituting the appropriate residue side chains in PDB code 1I5Z. The basic approach followed that employed for the study of various proteins in our laboratory (25–28). The COREX/BEST algorithm generates many folded and unfolded states with a window of 10 residues. The simulation process is repeated by sliding the window one residue at a time until the end of the sequence. The weight of each state is defined by Ki = exp(ΔGi/RT). Hence, it is determined from the calculated Gibbs energy, ΔGi, and the probability of each state (Pi) is determined by Equation 1,
| (Eq. 1) |
The residue stability constant, ki, f, is the ratio of the summed probability of all states in the ensemble in which a particular residue i is in a folded conformation (ΣPi, f) to the summed probability of all states in which that residue is in an unfolded conformation (ΣPi, nf), as shown in Equation 2,
| (Eq. 2) |
The important feature of the residue stability constants is the fact that they provide a measure of the local stability around each residue.
Residue-specific connectivity
In describing the coupling between two residues j and k, we introduce the parameter RSC. In the context of the Monte Carlo sampling method, we use the correlation function to define the RSC as shown in Equation 3,
| (Eq. 3) |
where Sj and Sk denote the folding state of residues j and k (if a residue is folded in a particular state, S = 1; if the residue is unfolded, S = (S − 1), and 〈Sj〉 and 〈Sk〉 denote the average folding state of residues j and k over the ensemble. A positive value of the RSC indicates that a stabilization of residue j or k results in a stabilization of residue k or j (i.e. they display positive cooperativity), whereas a negative value indicates a stabilization of residue j or k leads to a destabilization of residue k or j (i.e. they display negative cooperativity). A value of 0 means there is no correlation, and the residues are not energetically coupled.
Author contributions
W. E., A. D., M. A. W., and J. L. data curation; W. E., A. D., M. A. W., J. L., and J. C. L. formal analysis; W. E., M. A. W., and J. L. validation; W. E., A. D., M. A. W., J. L., and J. C. L. writing-original draft W. E., A. D., M. A. W., J. L., and J. C. L. writing-review and editing; J. C. L. supervision; J. C. L. funding acquisition; J. C. L. project administration.
Supplementary Material
Acknowledgments
We thank Drs. Heather Lander, Lavanya Rajagopalan, and Thomas Smith for a critical review.
This work was supported by National Institutes of Health Grant GM-77551 and by The Robert A. Welch Foundation Grant H-0013. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This paper is dedicated to Serge N. Timasheff (1926–2019) for his enormous contributions to biophysical chemistry of biomacromolecules, molecular recognition, and the field of pharmaceutical formulation.
This article contains Figs. S1–S13.
The atomic coordinates and structure factors (code 1I5Z) have been deposited in the Protein Data Bank (http://wwpdb.org/).
W. Evangelista, A. Dong, M. A. White, J. Li, and J. C. Lee, manuscript in preparation.
W. Evangelista, A. Dong, M. A. White, J. Li, and J. C. Lee, unpublished data.
- CRP
- cyclic AMP receptor protein
- AR
- activation region
- PDB
- Protein Data Bank
- RNAP
- RNA polymerase
- RSC
- residue-specific connectivity
- DBD
- DNA-binding domain
- NBD
- nucleotide-binding domain
- RMSD
- root mean square deviation
- α-CTD
- C-terminal domain of α subunit of RNAP.
References
- 1. Kolb A., Busby S., Buc H., Garges S., and Adhya S. (1993) Transcription regulation by cAMP and its receptor protein. Annu. Rev. Biochem. 62, 749–795 10.1146/annurev.bi.62.070193.003533 [DOI] [PubMed] [Google Scholar]
- 2. Harman J. G. (2001) Allosteric regulation of the cAMP receptor protein. Biochim. Biophys. Acta 1547, 1–17 10.1016/S0167-4838(01)00187-X [DOI] [PubMed] [Google Scholar]
- 3. Ebright R. H. (1993) Transcription activation at class I CAP-dependent promoters. Mol. Microbiol. 8, 797–802 10.1111/j.1365-2958.1993.tb01626.x [DOI] [PubMed] [Google Scholar]
- 4. Busby S., and Ebright R. H. (1997) Transcription activation at class II CAP-dependent promoters. Mol. Microbiol. 23, 853–859 10.1046/j.1365-2958.1997.2771641.x [DOI] [PubMed] [Google Scholar]
- 5. Busby S., and Ebright R. H. (1999) Transcription activation by catabolite activator protein. J. Mol. Biol. 293, 199–213 10.1006/jmbi.1999.3161 [DOI] [PubMed] [Google Scholar]
- 6. Lawson C. L., Swigon D., Murakami K. S., Darst S. A., Berman H. M., and Ebright R. H. (2004) Catabolite activator protein: DNA binding and transcription activation. Curr. Opin. Struct. Biol. 14, 10–20 10.1016/j.sbi.2004.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Busby S., and Ebright R. H. (1994) Promoter structure, promoter recognition, and transcription activation in prokaryotes. Cell 79, 743–746 10.1016/0092-8674(94)90063-9, [DOI] [PubMed] [Google Scholar]
- 8. Rhodius V. A., and Busby S. J. (2000) Transcription activation by Escherichia coli cyclic AMP receptor protein: determinants within activation region 3. J. Mol. Biol. 299, 295–310 10.1006/jmbi.2000.3736 [DOI] [PubMed] [Google Scholar]
- 9. Rhodius V. A., West D. M., Webster C. L., Busby S. J., and Savery N. J. (1997) Transcription activation at class II CRP-dependent promoter: the role of different activating regions. Nucleic Acids Res. 25, 326–332 10.1093/nar/25.2.326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bell A., Gaston K., Williams R., Chapman K., Kolb A., Buc H., Minchin S., Williams J., and Busby S. (1990) Mutations that alter the ability of the Escherichia coli cyclic AMP receptor protein to activate transcription. Nucleic Acids Res. 18, 7243–7250 10.1093/nar/18.24.7243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Williams R., Bell A., Sims G., and Busby S. (1991) The role of two surface exposed loops in transcription activation by the Escherichia coli CRP and FNR proteins. Nucleic Acids Res. 19, 6705–6712 10.1093/nar/19.24.6705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rhodius V. A., and Busby S. J. (1998) Positive activation of gene expression. Curr. Opin. Microbiol. 1, 152–159 10.1016/S1369-5274(98)80005-2 [DOI] [PubMed] [Google Scholar]
- 13. Feng Y., Zhang Y., and Ebright R. H. (2016) Structural basis of transcription activation. Science 352, 1330–1333 10.1126/science.aaf4417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gallagher D. T., Smith N., Kim S. K., Robinson H., and Reddy P. T. (2009) Profound asymmetry in the structure of the cAMP-free cAMP receptor protein (CRP) from Mycobacterium tuberculosis. J. Biol. Chem. 284, 8228–8232 10.1074/jbc.C800215200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lanfranco M. F., Gárate F., Engdahl A. J., and Maillard R. A. (20157) Asymmetric configurations in a reengineered homodimer reveal multiple subunit communication pathways in protein allostery. J. Biol. Chem. 292, 6086–6093 10.1074/jbc.M117.776047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weber I. T., and Steitz T. A. (1987) Structure of a complex of catabolite gene activator protein and cyclic AMP refined at 2.5 Å resolution. J. Mol. Biol. 198, 311–326 10.1016/0022-2836(87)90315-9 [DOI] [PubMed] [Google Scholar]
- 17. Passner J. M., Schultz S. C., and Steitz T. A. (2000) Modelling the cAMP-induced allosteric transition using the crystal structure of CAP-cAMP at 2.1 Å resolution. J. Mol. Biol. 304, 847–859 10.1006/jmbi.2000.4231 [DOI] [PubMed] [Google Scholar]
- 18. Tzeng S.-R., and Kalodimos C. G. (2012) Protein activity regulation by conformational entropy. Nature 488, 236–240 10.1038/nature11271 [DOI] [PubMed] [Google Scholar]
- 19. Li J., Cheng X., and Lee J. C. (2002) Structure and dynamics of the modular halves of Escherichia coli cyclic AMP receptor protein. Biochemistry 41, 14771–14778 10.1021/bi026383q [DOI] [PubMed] [Google Scholar]
- 20. Dong A., Malecki J. M., Lee L., Carpenter J. F., and Lee J. C. (2002) Ligand-induced conformational and structural dynamics changes in Escherichia coli cyclic AMP receptor protein. Biochemistry 41, 6660–6667 10.1021/bi020036z [DOI] [PubMed] [Google Scholar]
- 21. Susi H., and Byler D. M. (1986) Resolution-enhanced Fourier transform infrared spectroscopy of enzymes. Methods Enzymol. 130, 290–311 10.1016/0076-6879(86)30015-6 [DOI] [PubMed] [Google Scholar]
- 22. Dong A., and Caughey W. S. (1994) Infrared methods for study of hemoglobin reactions and structures. Methods Enzymol. 232, 139–175 10.1016/0076-6879(94)32047-0 [DOI] [PubMed] [Google Scholar]
- 23. Cheng X., and Lee J. C. (1994) Absolute requirement of cyclic nucleotide in the activation of the G141Q mutant cAMP receptor protein from Escherichia coli. J. Biol. Chem. 269, 30781–30784 [PubMed] [Google Scholar]
- 24. Lin S.-H., Kovac L., Chin A. J., Chin C. C. Q., and Lee J. C. (2002) Ability of E. coli cyclic AMP receptor protein to differentiate cyclic nucleotides: effects of single site mutations. Biochemistry 41, 2946–2955 10.1021/bi0119215 [DOI] [PubMed] [Google Scholar]
- 25. Pan H., Lee J. C., and Hilser V. J. (2000) Binding sites in Escherichia coli dihydrofolate reductase communicate by modulating the conformational ensemble. Proc. Natl. Acad. Sci. U. S. A. 97, 12020–12025 10.1073/pnas.220240297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yeh L.-C. C., Falcon W. E., Garces A., Lee J. C., and Lee J. C. (2012) A host–guest relationship in bone morphogenetic protein receptor-II defines specificity in ligand–receptor recognition. Biochemistry 51, 6968–6980 10.1021/bi3003023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Evangelista W., Yeh L.-C. C., Gmyrek A., Lee J. C., and Lee J. C. (2015) Long-range communication network in the type 1B bone morphogenetic protein receptor. Biochemistry 54, 7079–7088 10.1021/acs.biochem.5b00955 [DOI] [PubMed] [Google Scholar]
- 28. Maillard R. A., Liu T., Beasley D. W., Barrett A. D., Hilser V. J., and Lee J. C. (2014) Thermodynamic mechanism for the evasion of antibody neutralization in flaviviruses. J. Am. Chem. Soc. 136, 10315–10324 10.1021/ja503318x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu B., Hong C., Huang R. K., Yu Z., and Steitz T. A. (2017) Structural basis of bacterial transcription activation. Science 358, 947–951 10.1126/science.aao1923 [DOI] [PubMed] [Google Scholar]
- 30. Williams R. M., Rhodius V. A., Bell A. I., Kolb A., and Busby S. J. (1996) Orientation of functional activating regions in the Escherichia coli CRP protein during transcription activation at class II promoters. Nucleic Acids Res. 24, 1112–1118 10.1093/nar/24.6.1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou Y., Zhang X., and Ebright R. H. (1993) Identification of the activating region of catabolite gene activator protein (CAP): isolation and characterization of mutants of CAP specifically defective in transcription activation. Proc. Natl. Acad. Sci. U.S.A. 90, 6081–6085 10.1073/pnas.90.13.6081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Niu W., Zhou Y., Dong Q., Ebright Y. W., and Ebright R. H. (1994) Characterization of the activating region of Escherichia coli catabolite gene activator protein (CAP): I. Saturation and alanine-scanning mutagenesis. J. Mol. Biol. 243, 595–602 10.1016/0022-2836(94)90034-5 [DOI] [PubMed] [Google Scholar]
- 33. Niu W., Kim Y., Tau G., Heyduk T., and Ebright R. H. (1996) Transcription activation at class II CAP-dependent promotors: two interactions between CAP and RNA polymerase. Cell 87, 1123–1134 10.1016/S0092-8674(00)81806-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou Y., Busby S., and Ebright R. H. (1993) Identification of the functional subunit of a dimeric transcription factor protein by use of oriented heterodimer. Cell 73, 375–379 10.1016/0092-8674(93)90236-J [DOI] [PubMed] [Google Scholar]
- 35. Zhou Y., Pendergrast P. S., Bell A., Williams R., Busby S., and Ebright R. H. (1994) The functional subunit of a dimeric transcription activator protein depends on promoter architecture. EMBO J. 13, 4549–4557 10.1002/j.1460-2075.1994.tb06776.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gekko K., Obu N., Li J., and Lee J. C. (2004) A linear correlation between the energetics of allosteric communication and protein flexibility in the Escherichia coli cyclic AMP receptor protein revealed by mutation-induced changes in compressibility and amide hydrogen-deuterium exchange. Biochemistry 43, 3844–3852 10.1021/bi036271e [DOI] [PubMed] [Google Scholar]
- 37. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J.-S., Kuszewski J., Nilges N., Pannu N. S., Read R. J., Rice L. M., Simonson T., and Warren G. L. (1998) Crystallography and NMR system (CNS): a new software system for macromolecular structure determination. Acta Crystallogr. Sect. D Biol. Crystallogr. 54, 905–921 10.1107/S0907444998003254 [DOI] [PubMed] [Google Scholar]
- 38. McRee D. E. (1992) A visual protein crystallographic software system for X11/Xview. J. Mol. Graph. 10, 4446 [Google Scholar]
- 39. Matthews B. W., and Czerwinski E. W. (1975) Local scaling: a method to reduce systematic errors in isomorphous replacement and anomalous scattering measurements. Acta Cryst. A31, 480–487 10.1107/S0567739475001040 [DOI] [Google Scholar]
- 40. Hynes T. R., and Fox R. O. (1991) The crystal structure of staphylococcal nuclease refined at 1.7 A resolution proteins. 10, 92–105 [DOI] [PubMed] [Google Scholar]
- 41. Heyduk T., and Lee J. C. (1989) Escherichia coli cAMP receptor protein: evidence for three protein conformational states with different promoter binding affinities. Biochemistry 28, 6914–6924 10.1021/bi00443a021 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.