

Primary cells cultured in vitro currently provide the highest available relevance for examining molecular and genetic requirements for the establishment, maintenance, and reactivation of HCMV latency. However, their expense, heterogeneity, and intransigence to both long-term culture and molecular or genetic modification create rigor and reproducibility challenges for HCMV latency studies. There are several cell line models for latency not obstructed by deficiencies inherent in primary cells. However, many researchers view cell line studies of latency to be physiologically irrelevant because of the perception that these models display numerous and significant differences from primary cells. Here, we show that the very first step in a latent HCMV infection, entry of the virus into cells, occurs in cell line models in a manner indistinguishable from that in which it occurs in primary CD34+ hematopoietic progenitor cells. Our data argue that experimental HCMV latency is much more similar than it is different in cell lines and primary cells.
KEYWORDS: cell line models, endocytosis, entry, HCMV, latency, macropinocytosis
ABSTRACT
Human cytomegalovirus (HCMV) enters primary CD34+ hematopoietic progenitor cells by macropinocytosis, where it establishes latency in part because its tegument-transactivating protein, pp71, remains associated with endosomes and is therefore unable to initiate productive, lytic replication. Here we show that multiple HCMV strains also enter cell line models used to study latency by macropinocytosis and endocytosis. In all latency models tested, tegument-delivered pp71 was found to be colocalized with endosomal markers and was not associated with the seven other cytoplasmic localization markers tested. Like the capsid-associated pp150 tegument protein, we initially detected capsid proteins in association with endosomes but later detected them in the nucleus. Inhibitors of macropinocytosis and endocytosis reduced latent viral gene expression and precluded reactivation. Importantly, we utilized electron microscopy to observe entry by macropinocytosis and endocytosis, providing additional visual corroboration of the findings of our functional studies. Our demonstration that HCMV enters cell line models for latency in a manner indistinguishable from that of its entry into primary cells illustrates the utility of these cell lines for probing the mechanisms, host genetics, and small-molecule-mediated inhibition of HCMV entry into the cell types where it establishes latency.
IMPORTANCE Primary cells cultured in vitro currently provide the highest available relevance for examining molecular and genetic requirements for the establishment, maintenance, and reactivation of HCMV latency. However, their expense, heterogeneity, and intransigence to both long-term culture and molecular or genetic modification create rigor and reproducibility challenges for HCMV latency studies. There are several cell line models for latency not obstructed by deficiencies inherent in primary cells. However, many researchers view cell line studies of latency to be physiologically irrelevant because of the perception that these models display numerous and significant differences from primary cells. Here, we show that the very first step in a latent HCMV infection, entry of the virus into cells, occurs in cell line models in a manner indistinguishable from that in which it occurs in primary CD34+ hematopoietic progenitor cells. Our data argue that experimental HCMV latency is much more similar than it is different in cell lines and primary cells.
INTRODUCTION
Human cytomegalovirus (HCMV) productively replicates in many differentiated cell types but also establishes a latent (nonproductive) infection within incompletely differentiated cells of the myeloid lineage (1–7). Productive replication causes disease but can be effectively treated with antivirals until resistant strains emerge (8–11). However, no antiviral intervention targets the latent reservoir that permits lifelong viral persistence within infected hosts. Understanding the molecular parameters of latency is paramount to developing novel antilatency strategies.
HCMV latency in vitro is studied in two broad classes of cells, primary myeloid lineage populations and clonal cell lines of various origins. The primary cells utilized are generally CD34+ hematopoietic progenitor cells derived from umbilical cord blood or bone marrow aspirates (1, 12–16) or CD14+ monocytes isolated from peripheral blood (3, 13, 17–20). Cell lines include naturally transformed clones of the primary cell types used to study HCMV latency (KG-1 and Kasumi-3 CD34+ cells [21–25] and THP-1 monocytes [26–33]), as well as NTera2 (NT2) embryonal carcinoma cells (34–38) and embryonic stem cells (ESCs) (24, 39).
The importance of data acquired from cell line studies of latency has been debated because some researchers view these cells to be less physiologically relevant than primary cells. The recent demonstration that HCMV enters primary CD34+ cells by macropinocytosis prior to establishing latency (40) prompted us to examine whether or not HCMV enters cell line models in a similar fashion as a prospective test for the congruence of results obtained between a primary cell population and various cell line models used to study latency.
Here we show that HCMV enters cell lines by macropinocytosis and endocytosis, processes through which extracellular fluids and solids are routinely internalized by cells into vesicles called endosomes, in a manner indistinguishable from that in which it enters primary CD34+ cells. Our data demonstrate that HCMV virions enter cells with a fluid-phase marker and during entry are surrounded by membrane extrusions, both of which are hallmarks of macropinocytosis. After entry, virion components colocalize with markers of macropinosomes and endosomes. While capsids and the capsid-associated (41) tegument protein pp150 escape from maturing endosomes and migrate to the nucleus, other tegument proteins (pp28, pp65, and pp71) remain associated with the endosome. Inhibitors of macropinocytosis or endocytosis decrease latency-associated gene expression and reduce differentiation-induced reactivation, indicating that entry through these routes results in the establishment of experimental viral latency in vitro. We conclude that HCMV enters these cell line models for latency by macropinocytosis and endocytosis. Our results highlight the similarities between in vitro latent HCMV infections of primary cell populations and cell lines.
RESULTS
Tegument-delivered pp71 is found in cytoplasmic endosomes of cells that support HCMV experimental latency.
Tegument-delivered pp71 reaches the nucleus in 97% of fully differentiated fibroblasts infected with HCMV (Fig. 1A and B). However, when HCMV enters incompletely differentiated cells that support latency, tegument-delivered pp71 remains in the cytoplasm (Fig. 1A). For example, 99% of primary CD34+ cells infected with HCMV retained pp71 in the cytoplasm (Fig. 1B). Similar numbers were achieved in other cell types that support latency (THP-1 cells, 96% cytoplasmic; NT2 cells, 89%; embryonic stem cells, 98%) (Fig. 1B).
FIG 1.

Tegument-delivered pp71 localizes to the cytoplasm of incompletely differentiated cells after HCMV infection. (A) The indicated cells were infected with HCMV AD169 at an MOI of 1 (NHDFs and NT2 cells) or 3 (hESCs and CD34+ THP-1 cells) and then fixed after 2 h (NHDFs and NT2 cells), 5 h (hESCs), or 6 h (CD34+ and THP-1 cells). Indirect immunofluorescence (IF) was performed to visualize tegument-delivered pp71 (red). Nuclei were stained with Hoechst (blue). Representative images of three independent biological replicates are shown. (B) At least 30 cells from each of the three independent biological replicates described above were quantitated for pp71 subcellular localization. The average percentage of cells with pp71 in the cytoplasm (C) or nucleus (N) is presented. Error bars represent standard deviations.
In primary CD34+ cells, tegument-delivered pp71 introduced into cells by infection with the clinical strain TB40/E colocalized with endosomal markers (40). To begin to probe the congruities (or incongruities) between the entry of a clinical strain of HCMV into a primary cell and the entry of a laboratory strain of HCMV into a transformed cell, we identified the subcellular localization of tegument-delivered pp71 in NT2 cells infected with AD169. NT2 cells represent the HCMV latency model preferable for imaging by fluorescence microscopy because they are adherent and flat and have a large cytoplasm while still displaying both the cytoplasmic sequestration of pp71 and the differentiation-dependent activation of IE1 expression that are hallmarks of latency (38). AD169 represents the most commonly used HCMV strain as it grows to the highest titers and the most viral recombinant mutants are available for this strain. This experimental design allowed us to utilize a battery of markers for cytoplasmic organelles and structures that would be impractical to use in primary CD34+ cells because of their small size and high cost. We found that tegument-delivered pp71 failed to colocalize with ribosomes, microtubules, the Golgi apparatus, mitochondria, lysosomes, the plasma membrane, or the endoplasmic reticulum (ER) in NT2 cells infected with HCMV but did colocalize with Rab5-positive endosomes (Fig. 2). The endosomal localization of tegument-delivered pp71 in AD169-infected NT2 cells matched the results in TB40/E-infected primary CD34+ cells (40).
FIG 2.

HCMV tegument protein pp71 fails to colocalize with nonendosomal cytoplasmic markers in NT2 cells. NT2 cells grown on coverslips were incubated with HCMV (AD169; MOI = 1) for 2 h at 37°C. The cells were then fixed and processed for indirect immunofluorescence for pp71 (red) and the indicated cytoplasmic protein (green). Nuclei were stained with Hoechst (blue). The boxed area is shown in magnification (zoom). White arrowheads indicate colocalization. Representative images of three independent biological replicates are shown.
We confirmed the colocalization of pp71 with Rab5 but not with any of the other tested cytoplasmic markers in AD169-infected THP-1 monocytes (Fig. 3) that fully support all known and tested aspects of experimental HCMV latency observed in primary cells (27). In addition to Rab5, the pp71 delivered by AD169 infection also colocalized with the EEA1 and Rab7 endosomal proteins (42, 43) in NT2 cells at both 2 and 8 h postinfection (Fig. 4). In fibroblasts, the pp71 delivered by AD169 colocalized with EEA1, Rab5, and Rab7 at 2 h postinfection, but the colocalization disappeared by 8 h postinfection, when pp71 was found in the nucleus (Fig. 4). Thus, while the initial events of AD169 entry into NT2 cells and fibroblasts are similar, the processes diverge shortly after internalization, mimicking what was observed during comparisons of TB40/E entry into fibroblasts and primary CD34+ cells (40). In every other latency model tested (THP-1 cells, human embryonic stem cells [hESCs], Kasumi-3 cells, and primary CD34+ cells), we also found that AD169-delivered pp71 colocalized with EEA1, Rab5, and Rab7 (Fig. 5). We conclude that, in a manner indistinguishable from that of TB40/E infection of primary CD34+ cells, the tegument-delivered pp71 introduced by AD169 infection localizes to endosomes during in vitro experimental HCMV latent infections in all five cell types tested.
FIG 3.

HCMV tegument protein pp71 fails to colocalize with nonendosomal cytoplasmic markers in THP-1 cells. THP-1 cells were incubated with HCMV (AD169; MOI = 3) for 4 h at 37°C. The cells were then fixed and processed for indirect immunofluorescence for pp71 (red) and the indicated cytoplasmic protein (green). Nuclei were stained with Hoechst (blue). The boxed area is shown in magnification (zoom). The white arrowhead indicates colocalization. Representative images of three independent biological replicates are shown.
FIG 4.

HCMV tegument protein pp71 colocalizes with endosomal markers in NT2 cells. NT2 cells or normal human dermal fibroblasts (NHDFs) grown on coverslips were incubated with HCMV (AD169; MOI = 1) for 2 h or 8 h, as indicated, at 37°C. The cells were then fixed and processed for indirect immunofluorescence for pp71 (red) and the indicated endosomal marker (green). Nuclei were stained with Hoechst (blue) and are circled by a white dotted line. Boxed areas are shown in magnification (zoom). White arrowheads indicate colocalization. Representative images of three independent biological replicates are shown. hpi, hours postinfection.
FIG 5.

HCMV tegument protein pp71 colocalizes with endosomal markers in cell types where latency is established. The indicated cells were incubated with HCMV (AD169; MOI = 3) for 6 h at 37°C. The cells were then fixed and processed for indirect immunofluorescence for pp71 (red) and the indicated endosomal marker (green). Nuclei were stained with Hoechst (blue). Boxed areas are shown in magnification (zoom). White arrowheads indicate colocalization. Representative images of three independent biological replicates are shown.
HCMV virions are internalized into cells that support latency by macropinocytosis and endocytosis.
The colocalization of tegument-delivered pp71 with endosomal markers during latency most likely results from HCMV virions entering these cells by some form of endocytosis. During the process of vaccinia virus entry by endocytosis, between 8% and 26% of the virion core protein A4 was found to be colocalized with the EEA1, Rab5, and Rab7 markers (44). An incomplete association was observed because endosomes are dynamic structures (42, 43) and the proteins that associate with their cytoplasmic surfaces do so transiently. Therefore, at any single time point, not all internalized virions are expected to colocalize with endosomal markers. Our colocalization studies after AD169 infection of NT2 cells (Fig. 6) showed slightly higher levels of pp71 colocalization with these markers (38% to 46%), as well as with the additional endosomal markers clathrin (45%) and caveolin-1 (41%), than the previous study (44). These results mimic what was previously observed during TB40/E infection of primary CD34+ cells (40) and prompted us to visualize HCMV entry into cells that support latency.
FIG 6.

Quantitation of tegument-delivered pp71 colocalization with cytoplasmic proteins. NT2 cells grown on coverslips were incubated with HCMV (AD169; MOI = 1) for 2 h at 37°C. The cells were then fixed and processed for indirect immunofluorescence for pp71 and the indicated marker (images not shown). Colocalization was determined with Fiji software for at least 10 cells from each of three independent biological replicates. The average percent colocalization is shown. Error bars represent standard deviations.
Electron microscopy (EM) is often used to visualize entry. Most EM studies of viral entry use exceedingly high multiplicities of infection (MOIs) to ensure that all cells are infected with multiple virions to aid in the capture of the short-lived entry events. HCMV latency poses a distinct challenge in this regard because the amount of virus sufficient for productive infection of the vast majority of fibroblasts in culture results in virion entry into <5% of CD34+ cells (1). Despite this technical challenge, we were able to observe HCMV entry into primary CD34+ cells by EM. The clinical strain FIX entered primary CD34+ cells through a process visually resembling endocytosis (Fig. 7A). The laboratory strain AD169 grows to higher titers, allowing for higher MOIs and the capture of more primary CD34+ cell entry events. Images of AD169 entry revealed extracellular virions adjacent to plasma membrane lamellipodium-like ruffles and internalized virions in large, translucent cavities in primary CD34+ cells resembling intermediates in the processes of macropinocytosis and endocytosis (Fig. 7B). To observe even larger numbers of entry events, we employed the commonly utilized THP-1 cell model for HCMV latency, where establishment, maintenance, and reactivation are indistinguishable from those in primary monocytes or primary CD34+ cells (29, 32, 33, 45–50). In THP-1 cells, both FIX (Fig. 8A) and AD169 (Fig. 8B) entered the cells in structures reminiscent of those seen in macropinocytosis or endocytosis. Importantly, tegument-delivered pp71 remained in the cytoplasm under identical experimental conditions (Fig. 8C), indicating that these cells supported an early hallmark of latency (pp71 cytoplasmic sequestration) even at the highest multiplicity of infection utilized for the EM experiments. Fusion events at the plasma membrane were never observed in THP-1 or primary CD34+ cells with either virus strain. We conclude that HCMV laboratory and clinical strain virions enter primary and transformed cells in which latency is established in vitro by processes that visually resemble macropinocytosis and endocytosis.
FIG 7.
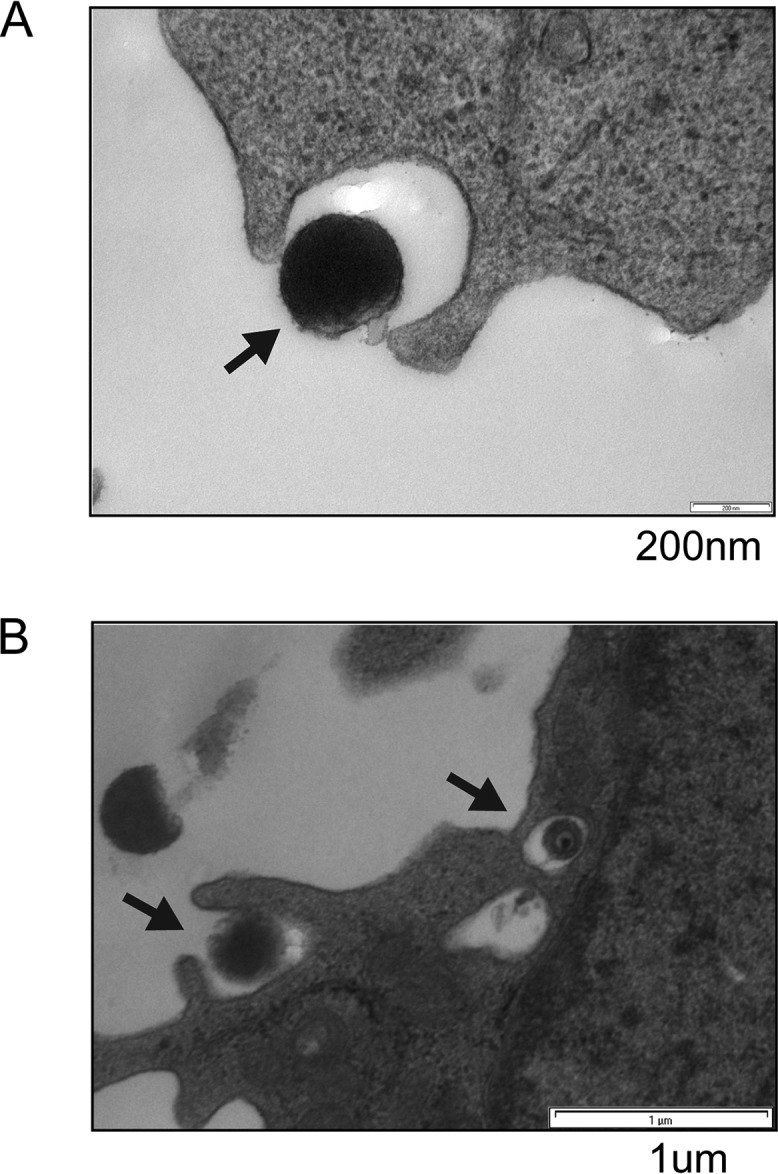
Electron microscopy visualizes HCMV laboratory strain AD169 and clinical strain FIX entering primary CD34+ cells by macropinocytosis and endocytosis. (A) Primary CD34+ cells were infected with HCMV (FIX; MOI = 10) for 2 h at 37°C and then processed for electron microscopy as described in Materials and Methods. The image is representative of two entry events detected during two independent infections. (B) Primary CD34+ cells were infected with HCMV (AD169; MOI = 20) for 2 h at 37°C and then processed for electron microscopy as described in Materials and Methods. The image is representative of nine entry events detected during three independent infections.
FIG 8.

Electron microscopy visualizes HCMV laboratory strain AD169 and clinical strain FIX entering THP-1 cells by macropinocytosis and endocytosis. (A) THP-1 cells were infected with HCMV (FIX; MOI = 10) for 2 h at 37°C and then processed for electron microscopy as described in Materials and Methods. The image is representative of six entry events detected in a single experiment. (B) THP-1 cells were infected with HCMV (AD169; MOI = 20) for 2 h at 37°C and then processed for electron microscopy as described in Materials and Methods. The image is representative of 57 entry events detected during five independent infections. (C) THP-1 cells were infected with HCMV (AD169; MOI = 20) for 2 h at 37°C and then processed for indirect immunofluorescence for pp71 (red). Nuclei were stained with Hoechst (blue). This nonconfocal image is representative of the images found in two independent biological replicates.
If HCMV virions are indeed internalized by macropinocytosis or endocytosis, then virion components in addition to pp71 should colocalize with endosomal markers. Consistent with virion entry by macropinocytosis and endocytosis, all virion components tested in AD169-infected NT2 cells colocalized with the endosomal markers EEA1 (Fig. 9), Rab5 (Fig. 10), and Rab7 (Fig. 11). The tested proteins included the major capsid protein (UL86), the minor capsid protein (UL85), the capsid-associated (41) tegument protein pp150 (UL32), and other tegument proteins, pp28 (UL99), pp65 (UL83), and pp71 (UL82). This result mimics the colocalization of multiple TB40/E tegument proteins with endosomal markers within infected primary CD34+ cells (40) while expanding the repertoire of virion components analyzed to include two distinct capsid proteins. Similar to pp71 (Fig. 6), the major capsid protein showed the expected level of colocalization with endosomal markers in AD169-infected NT2 cells (Fig. 12).
FIG 9.

HCMV virion proteins colocalize with the EEA1 marker of macropinosomes in NT2 cells. NT2 cells grown on coverslips were incubated with HCMV (AD169; MOI = 1) for 2 h at 37°C. The cells were then fixed and processed for indirect immunofluorescence for the indicated virion protein (red) and EEA1 (green). Nuclei were stained with Hoechst (blue). Boxed areas are shown in magnification (zoom). White arrowheads indicate colocalization. Representative images of three independent biological replicates are shown.
FIG 10.

HCMV virion proteins colocalize with the Rab5 marker of endosomes in NT2 cells. NT2 cells grown on coverslips were incubated with HCMV (AD169; MOI = 1) for 2 h at 37°C. The cells were then fixed and processed for indirect immunofluorescence for the indicated virion protein (red) and Rab5 (green). Nuclei were stained with Hoechst (blue). Boxed areas are shown in magnification (zoom). White arrowheads indicate colocalization. Representative images of three independent biological replicates are shown.
FIG 11.

HCMV virion proteins colocalize with the Rab7 marker of endosomes in NT2 cells. NT2 cells grown on coverslips were incubated with HCMV (AD169; MOI = 1) for 2 h at 37°C. The cells were then fixed and processed for indirect immunofluorescence for the indicated virion protein (red) and Rab7 (green). Nuclei were stained with Hoechst (blue). Boxed areas are shown in magnification (zoom). White arrowheads indicate colocalization. Representative images of three independent biological replicates are shown.
FIG 12.
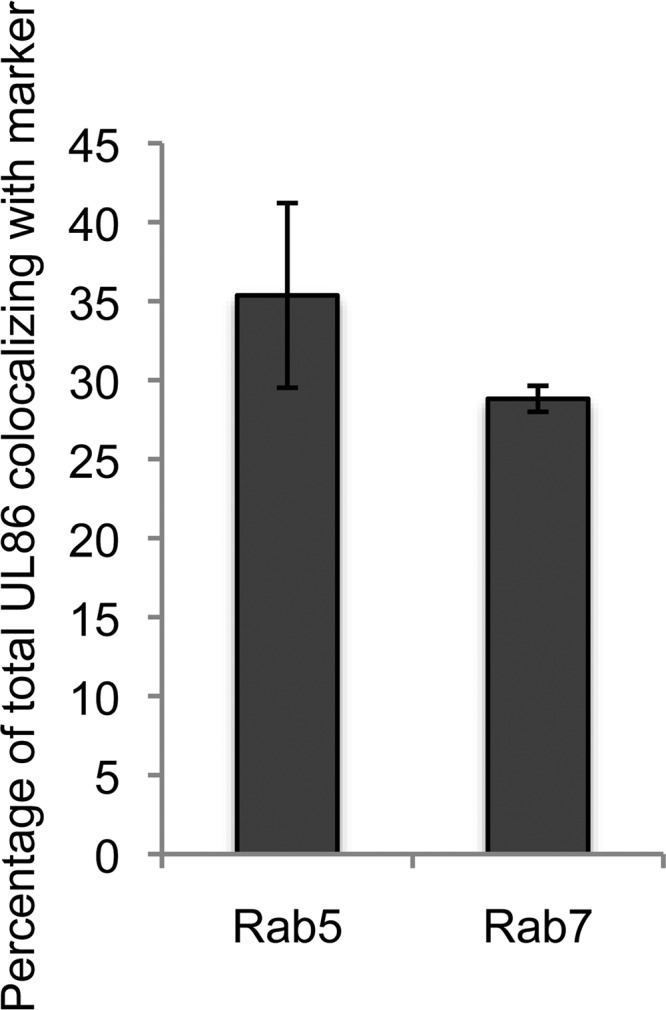
Quantitation of major capsid protein UL86 colocalization with cytoplasmic proteins. NT2 cells grown on coverslips were incubated with HCMV (AD169; MOI = 1) for 2 h at 37°C. The cells were then fixed and processed for indirect immunofluorescence for UL86 and the indicated marker (images not shown). Colocalization was determined with Fiji software for at least 10 cells from each of three independent biological replicates. The average percent colocalization is shown. Error bars represent standard deviations.
Because macropinocytosis is a means for fluid uptake into cells (51, 52), virion components entering by this method should colocalize with a fluid-phase marker. We found that pp71 and pp150, tegument protein markers for entering virions, colocalized with the fluid-phase marker dextran in AD169-infected NT2 cells (Fig. 13), which is indicative of viral entry by macropinocytosis and which reflects previous results in TB40/E-infected primary CD34+ cells (40).
FIG 13.

HCMV tegument proteins coenter NT2 cells with the fluid-phase marker dextran. NT2 cells grown on coverslips were incubated with HCMV (AD169; MOI = 2) for 1 h at 4°C and then shifted to 37°C for 20 min in the presence of 5 μg/ml dextran (molecular weight, 10,000; dextran was conjugated to Texas Red). The cells were then fixed and processed for indirect immunofluorescence for the indicated viral tegument protein (green). Nuclei were stained with Hoechst (blue). Boxed areas are shown in magnification (zoom). White arrowheads indicate colocalization. Representative images of three independent biological replicates are shown.
In order to initiate infection, virions entering cells by macropinocytosis or endocytosis must eventually escape endosomes to permit the release of one or more virion components. For example, herpesviruses must dock their capsids at the nuclear pore complex to allow for the entry of their virion-delivered genomes into the nucleus to initiate viral transcription. For HCMV, the minor capsid protein (UL85) and the capsid-associated pp150 tegument protein enter the nucleus after capsid docking at the nuclear pore, while the major capsid protein (UL86) remains juxtanuclear but cytoplasmic. During HCMV AD169 entry into NT2 cells, we observed UL86 colocalizing with the nuclear pore protein NUP98 at the nuclear rim and UL85 and pp150 within the nucleus (Fig. 14A). pp150 entered the nucleus in the same AD169-infected NT2 cells in which the outer tegument protein pp71 remained in the cytoplasm (Fig. 14B), similar to the results obtained in TB40/E-infected primary CD34+ cells (40). Therefore, sometime during endosomal maturation, the capsid and capsid-associated tegument must dissociate from the rest of the virion, escape the endosome, be released into the cytoplasm, and migrate to the nucleus. We conclude that laboratory and clinical strains of HCMV enter into the primary and transformed cells in which the virus establishes latency by macropinocytosis and endocytosis.
FIG 14.

HCMV capsid and capsid-associated tegument protein pp150 travel to the nucleus in NT2 cells. (A) NT2 cells grown on coverslips were incubated with HCMV (AD169; MOI = 1) for 4 h at 37°C. The cells were then fixed and processed for indirect immunofluorescence for the indicated virion protein (red) and NUP98 (green). Nuclei were stained with Hoechst (blue). Boxed areas are shown in magnification (zoom). White arrowheads indicate colocalization. Representative images of three independent biological replicates are shown. (B) NT2 cells grown on coverslips were incubated with HCMV (AD169; MOI = 1) for 2 h at 37°C. The cells were then fixed and processed for indirect immunofluorescence for pp150 (red) and pp71 (green). Nuclei were stained with Hoechst (blue). Representative images of three independent biological replicates are shown.
Inhibition of macropinocytosis or endocytosis prevents viral transcription during latency and reactivation.
We complemented our visual determination of HCMV internalization into in vitro models of HCMV latency by macropinocytosis and endocytosis with functional experiments to determine whether these types of entry events actually lead to the establishment of latency. The viral long noncoding RNA B2.7 is not incorporated into virions (53) but is de novo expressed (54) when HCMV enters latency. Therefore, we quantitated the accumulation of this latency-associated transcript as an indicator of viral genome delivery to the nucleus and the initiation of infection. We did not detect B2.7 transcripts in mock-infected NT2 cells or when AD169 entry was inhibited by maintaining the cells at 4°C but did detect B2.7 transcripts when NT2 cells treated with the solvent dimethyl sulfoxide (DMSO; as a negative control) were infected with AD169 (Fig. 15A). Ethylisopropyl amiloride (EIPA), an inhibitor of Na+/H+ exchangers that blocks macropinocytosis, significantly reduced B2.7 expression (Fig. 15A) as it did in TB40/E-infected primary CD34+ cells (40) and appeared to prevent the internalization of tegument-delivered pp71 into NT2 cells (Fig. 15B), consistent with a block in viral entry. The magnitude of EIPA inhibition of B2.7 expression in NT2 cells seen here (∼59%) is similar to that observed for B2.7 expression in primary CD34+ cells (53%) (40) and for IE1 during productive infection of HS-578T cells (∼68%) treated with the same concentration of drug (55).
FIG 15.

Endocytosis inhibitors reduce HCMV latency-associated transcription. (A) NT2 cells were pretreated with EIPA (50 μM), dynasore (75 μM), chlorpromazine (25 μM), bafilomycin A1 (1 nM), or DMSO for 30 min at 37°C prior to infection. The cells were chilled at 4°C for 1 h and then mock infected or infected with HCMV (AD169; MOI = 10) for 1 h in the presence of inhibitors before shifting to 37°C (or not) for 1 h to allow entry. Extracellular virions were washed away with heparin, and the cells were incubated for 8 h in the absence of inhibitors. Harvested RNA was analyzed by quantitative reverse transcription-PCR for the HCMV B2.7 transcript. The average from three independent biological replicates relative to the DMSO control is displayed. Error bars represent standard deviations. P values were determined by Student's t test. ns, not significant (P > 0.1); *, P ≤ 0.05; **, P < 0.01; ***, P < 0.001. (B) NT2 cells grown on coverslips were treated with EIPA (50 μM) or DMSO for 30 min and then infected with HCMV (AD169; MOI = 1) for 2 h at 37°C. The cells were then fixed and processed for indirect immunofluorescence for pp71 (red) and EEA1 (green). Nuclei were stained with Hoechst (blue). Boxed areas are shown in magnification (zoom). White arrowheads indicate colocalization. Representative images of three independent biological replicates are shown.
The endocytosis inhibitors chlorpromazine (an inhibitor of clathrin) and dynasore (an inhibitor of dynamin) also inhibited B2.7 expression in AD169-infected NT2 cells (Fig. 15A), but inhibition of endosome acidification with bafilomycin A1 did not. Consistent with the inhibitor data, we found AD169 tegument-delivered pp71 associated with caveolin, clathrin, and dynamin in both NT2 cells (Fig. 16A) and THP-1 cells (Fig. 16B). Furthermore, dynasore, like EIPA (Fig. 15B), appeared to impede the internalization of AD169 tegument-delivered pp71 into NT2 cells (Fig. 16A) and THP-1 cells (Fig. 16B), consistent with a block in viral entry. We conclude that entry by macropinocytosis and endocytosis leads to viral transcription consistent with latency when laboratory and clinical strain virions enter transformed or primary cells.
FIG 16.

Endocytosis inhibitors impair HCMV virion internalization. (A) NT2 cells grown on coverslips were treated with dynasore (75 μM) or DMSO for 30 min and then infected with HCMV (AD169; MOI = 1) for 2 h at 37°C. The cells were then fixed and processed for indirect immunofluorescence for pp71 (red) and the indicated endosomal marker (green). Nuclei were stained with Hoechst (blue). Boxed areas are shown in magnification (zoom). White arrowheads indicate colocalization. Representative images of three independent biological replicates are shown. (B) THP-1 cells were treated with dynasore (75 μM) or DMSO for 30 min and then infected with HCMV (AD169; MOI = 10) for 4 h at 37°C. The cells were then fixed and processed for indirect immunofluorescence for pp71 (red) and the indicated endosomal marker (green). Nuclei were stained with Hoechst (blue). Boxed areas are shown in magnification (zoom). White arrowheads indicate colocalization. Representative images of three independent biological replicates are shown.
Finally, we tested reactivation, the definitive hallmark of latency, in hESCs, where reactivation assays deliver robust, reproducible, and statistically analyzed readouts (39, 48, 49). Chlorpromazine, an inhibitor of clathrin-coated pit formation (56) that is also an antipsychotic drug approved for use in humans (57), inhibited both B2.7 expression (Fig. 17A) and reactivation (Fig. 17B) after AD169 infection of ESCs. We used 1-h treatments with chlorpromazine to inhibit endocytosis either concurrently with HCMV infection or at 6 h after viral infection. We found that chlorpromazine treatment concurrently with HCMV infection reduced subsequent reactivation events by 76%, consistent with an early inhibitory effect on latency prior to the maintenance phase. Treatment at the 6-h time point had a reduced but measurable effect (39%), likely resulting either from toxicity or from nonsynchronous viral entry over a protracted time frame. We conclude that preventing HCMV AD169 entry into ESCs by endocytosis decreases the number of latently infected cells available for reactivation, similar to previous results with TB40/E infection of primary CD34+ cells (40). In summary (Fig. 18), we conclude that HCMV clinical and laboratory strains use endocytosis events, including macropinocytosis, to enter primary and transformed cells that support in vitro latency and release their genome-containing capsids and a capsid-associated tegument protein (but not other tegument proteins) from endosomes for travel to the nucleus and the establishment of a latent infection competent for reactivation.
FIG 17.
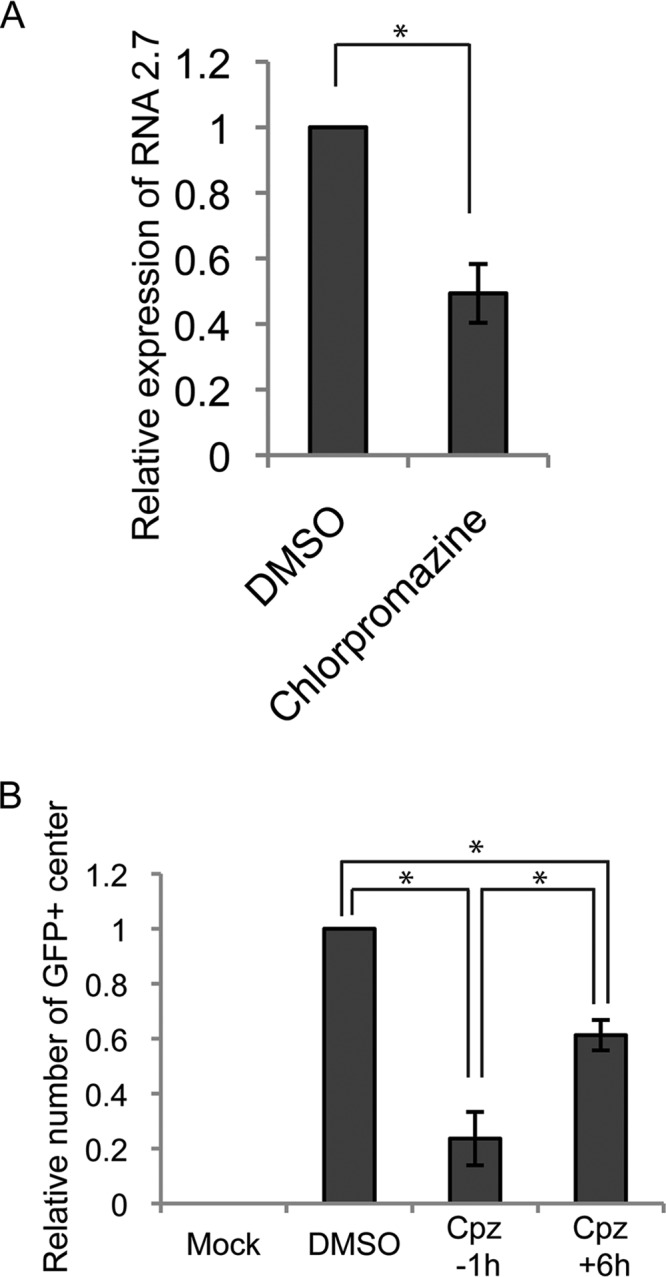
The endocytosis inhibitor chlorpromazine reduces HCMV latency reactivation, likely by inhibiting entry. (A) hESCs were pretreated with chlorpromazine (100 μM) or DMSO for 1 h at 37°C, and then the cells were infected with HCMV (AD169-IE2-GFP; MOI = 3) for 1 h in the presence of the inhibitor. Extracellular virions were washed away with heparin, and the cells were incubated for 9 h in the absence of inhibitors. Harvested RNA was analyzed by quantitative reverse transcription-PCR for the HCMV B2.7 transcript. The average from three independent biological replicates relative to the DMSO control is displayed. Error bars represent standard deviations. *, P ≤ 0.05 by Student's t test. (B) hESCs were incubated with chlorpromazine (Cpz; 100 μM) or DMSO for 1 h at 37°C either 1 h prior to infection (inhibitor pretreatment) of 6 h after infection (inhibitor posttreatment) with HCMV (AD169-IE2- GFP; MOI = 3) for 1 h. Extracellular virions were washed away with heparin, and the cells were incubated for 10 days in the absence of inhibitors. Differentiation was induced with TPA for 3 days, followed by 7 days of incubation. Collected cells were cocultured with fibroblasts for 7 days before the GFP-positive foci were counted. The average from three independent biological replicates relative to the DMSO control is displayed. Error bars represent standard deviations. *, P ≤ 0.05 by Student's t test.
FIG 18.

HCMV enters latency by macropinocytosis and endocytosis. The cartoon diagram shows HCMV entry into the undifferentiated cells, where the virus establishes latency. Plasma membrane lamellipodium-like ruffles engulf virions into endosomes. Tegument proteins (pp28, pp65, and pp71) remain associated with the endosome, but capsids and capsid-associated tegument protein pp150 escape the endosomes and travel to the nucleus. Viral genomes enter the nucleus, generate the latency-associated B2.7 transcript, and establish latency.
DISCUSSION
Here we show that clinical and laboratory strain virions entering cell lines used to study HCMV latency are enclosed by membrane ruffles into large cytoplasmic vacuoles that contain extracellular fluid and that associate with the Na+/H+ exchanger EEA1, the endosomal GTPases Rab5 and Rab7, as well as clathrin and dynamin. We also demonstrate that inhibitors of macropinocytosis or endocytosis lower the level of accumulation of an HCMV latency-associated transcript and reduce the level of reactivation. Based on the findings of these complementary visual and functional assays, we conclude that HCMV enters these cell line models of latency via macropinocytosis and other endocytosis pathways.
The data displayed here mimic previous data reported for HCMV strain TB40/E infection of primary CD34+ hematopoietic cells (40). Thus, despite the substantial differences in the cell types employed, as well as differences in the functional glycoprotein content of the infecting virions, the very first step of latency, entry into the cell, appears to be indistinguishable among all virus strains examined and all cell types infected.
The entry of HCMV into myeloid cells is often reported to be dependent upon the presence of a pentamer glycoprotein complex (gH, gL, UL128, UL130, and UL131) within the envelope of the infecting virion (58). However, multiple studies have demonstrated that virions lacking the pentamer are capable of entering and infecting myeloid cells. Pioneering work on in vitro latency models in primary hematopoietic cells used the pentamer-deficient AD169 strain to identify CD34+ cells as a progenitor population experimentally infected with HCMV, as well as CD14+ cells as the lineage-specific subset of hematopoietic cells most susceptible to HCMV (1). AD169 infection of CD34+ and CD14+ cells was confirmed by subsequent work from the same group (3, 59). Furthermore, independent laboratories, including our own, have also verified AD169 infection of primary CD34+ cells (12, 21, 48, 49, 60–62) and AD169 entry into cells that support productive infection by macropinocytosis (55, 63). The FIX strain of HCMV, which has been reported to be functionally null for the pentamer (64), is also commonly used to infect myeloid cells for latency studies (3, 12, 54, 59, 65–71). Therefore, while the pentamer may increase the efficiency of entry and, importantly, may initiate signal transduction cascades that modulate infection, it is clearly not required for entry into myeloid cells.
Primary cells (CD34+ hematopoietic progenitor cells or CD14+ monocytes) are preferred by some researchers for latency studies because latency in these cells is logically thought to most closely imitate latency in vivo, although how accurately they actually do mimic in vivo latency is not known. However, these primary cell types are heterogeneous, are difficult and expensive to obtain, cannot be continuously expanded in culture, infect poorly, are difficult to genetically manipulate or modify, and spontaneously differentiate. Therefore, using primary cells to study HCMV latency substantially reduces the experimental approaches that can be routinely employed, and experiments in primary cells are frequently limited to infections with recombinant viruses or in the presence of small-molecule inhibitors. Indeed, we could find only three studies that have used exogenous RNA interference to modify cellular gene expression in primary cells during HCMV latency (12, 60, 72). Likewise, while the humanized mouse model for HCMV latency (73, 74) is state of the art, it is also imperfect. Infections are introduced in a nonphysiologic manner, the mouse tissue is not infected, and experiments are mainly limited to quantifying viral DNA at various anatomical sites. Consequently, the model has not been widely employed.
Clearly, the use of primary cells is an important component of HCMV latency research, but the field must consider how best to utilize the much more versatile cell line models to accelerate novel discoveries. The demonstrated differences between latency in primary cells and latency in cell lines are few. They include the ability of TB40/E and FIX to reactivate in CD34+/CD38+ cell lines but not in primary CD34+/CD38+ cells, the ability of AD169 to transcribe IE1 in response to histone deacetylase inhibition in primary cells but not in KG-1 or Kasumi-3 cell lines, the ability of AD169 to reactivate from primary cells but not from KG-1 or Kasumi-3 cells, and the ability of FIX to reactivate from primary cells but not from KG-1 cells (21, 24). However, the similarities between latency in primary cells and latency in cell lines are numerous. They include the mode of entry into the cell (this work), the cytoplasmic sequestration of tegument-delivered pp71 (12, 21, 27, 39) (Fig. 1), the activation of latency-associated transcription (22, 24, 39, 48, 49, 75–80), the suppression of productive-phase transcription (21, 22, 24–26, 29, 32, 33, 45, 48–50, 62, 75, 79–83), differentiation-induced reactivation (12, 21, 22, 24, 25, 29, 39, 45, 49, 50, 84–86), and the viral genetic requirements for efficient latency establishment, maintenance, and reactivation (48, 49, 59, 75, 80, 87).
Cell line models are consistently used to study Epstein-Barr virus and Kaposi’s sarcoma-associated herpesvirus latency, and even herpes simplex virus 1 researchers are beginning to embrace the utility of cell lines to study latency (88–92). The use of cell lines to study HCMV latency should not supplant or decrease the use of primary cell models. However, all available evidence indicates that the HCMV latency results obtained in cell line models should not be summarily dismissed.
MATERIALS AND METHODS
Cells and viruses.
Normal human dermal fibroblasts (NHDFs), NTERA-2 (NT2) embryonal carcinoma cells, THP-1 monocytes, and Kasumi-3 cells (all purchased from ATCC), primary CD34+ hematopoietic progenitor cells (HPCs; purchased from Lonza), and WA01 human embryonic stem cells (hESCs; WiCell) were maintained as previously described (21, 28, 39). The HCMV strains used were wild-type strains AD169 and FIX and an AD169 strain in which IE2 was labeled with green fluorescent protein (GFP) (AD169-IE2-GFP) (93). All viral stocks were grown and titers were determined by plaque assay on NHDFs as previously described (94). Multiplicities of infection were based on NHDF-derived titers, were selected for the ease of signal detection with negligible background or toxicity, and were determined empirically.
Electron microscopy.
Experiments were performed at the University of Wisconsin—Madison School of Medicine and Public Health Electron Microscopy Facility. THP-1 monocytes (5 × 106) or CD34+ HPCs (1.5 × 106) were collected by centrifugation and infected at 37°C and 5% CO2 for 2 h with AD169 (MOI = 20) or FIX (MOI = 10) in a minimal volume. The cells were then collected by centrifugation, washed twice with phosphate-buffered saline (PBS), and fixed in 2% (wt/vol) paraformaldehyde–2.5% (wt/vol) glutaraldehyde in 0.1 M phosphate buffer (PB) for 1 h at 37°C. Fixed cells were washed five times with PB and then postfixed with 1% osmium tetroxide and 1% potassium ferrocyanide in PB for 1 h at 37°C. Postfixed cells were washed five times with PB, dehydrated in an ethanol gradient, and transitioned with propylene oxide. Dehydrated cells were infiltrated in a 1:1 propylene oxide and Durcupan ACM epoxy resin for 1 h, followed by 100% Durcupan ACM for 40 min, and polymerized in fresh Durcupan ACM for 24 h at 60°C. Embedded cells were sectioned into 90-nm sections using a Leica EM UC6 ultramicrotome, poststained in lead acetate-uranyl citrate, visualized on a Phillips CM120 electron microscope at 80 kV, and imaged using a Soft Imaging Solutions Megaview III digital camera.
Indirect immunofluorescence assay.
Adherent cells (NHDFs, NT2 cells, ESCs) were grown on glass coverslips, infected with HCMV at an MOI of 1 for 2 h, fixed with 1% paraformaldehyde, and stained as previously described (28). Suspension cells (THP-1 and Kasumi-3 cells and primary CD34+ HPCs) were infected in microcentrifuge tubes in a 100-μl volume at the MOIs indicated above for 1 h, plated for the times indicated above, and then washed with Dulbecco’s phosphate buffered saline (DPBS; Gibco), left for 1 h at room temperature to attach to coverslips, and fixed and stained as previously described (27). The images were obtained with a Nikon confocal laser scanning microscope using Prairie View software and processed with ImageJ software. Colocalization was quantitated with Fiji software.
Inhibitors and antibodies.
Dynasore, EIPA, chlorpromazine, and bafilomycin A1 were purchased from Sigma and dissolved in DMSO. The antibodies used were either previously described (95–97) (pp28 [CMV157 antibody], pp65 [8F6 antibody], pp71 [IE233 antibody], pp150 [CMV127 antibody]), gifts from Bill Britt (UL85, UL86 [28-4 antibody]), or purchased from Abcam (sodium potassium ATPase [catalog number EP1845Y], LAMP1 [catalog number Ab24170]), Cell Signaling Technologies (calnexin [catalog number C5C9], caveolin-1 [catalog number D46G3], clathrin [catalog number D3C6], cytochrome c oxidase subunit IV (Cox IV) [catalog number 3E11], EEA1 [catalog number C45B10], GM130 [catalog number D6B1], NUP98 [catalog number C39A3], Rab5 [catalog number C8B1], Rab7 [catalog number D95F2], S6 [catalog number 5G10], tubulin [catalog number D2N5G]), or Thermo Fisher (dynamin 2 antibody). Secondary antibodies were from Molecular Probes (Alexa Fluor 488 and Alexa Fluor 594).
Successful entry assay.
Cells were pretreated with inhibitors for 30 min at 37°C and then chilled at 4°C for 30 min. Prechilled virus was added at an MOI of 10 for 1 h at 4°C, and then the temperature was shifted to 37°C for 1 h to permit entry. Extracellular virus was removed with two heparin (150 μg/ml) washes, and fresh medium with inhibitor was added. Infected cells were incubated for an additional 8 h before dissociation with TrypLE recombinant enzyme (Gibco) and RNA isolation with a total RNA minikit (IBI). Equal amounts of RNA were treated with DNase I (Progema) according to the manufacturer’s protocols. cDNA was synthesized using SuperScript III first-strand synthesis Supermix for quantitative PCR (Invitrogen) or Maxima H minus cDNA synthesis master mix (Thermo Scientific). Quantitative PCR was performed using an ABI7900HT real-time PCR system (Applied Biosystems) with iTaq SYBR green Supermix (catalog number 172–5124; Bio-Rad) and primer sets B2.7 (forward primer, AGA TGA AAT TAT CCC GTG TCC G; reverse primer, GTG AAA TCT GGC TTG GTT GTG) or GAPDH (glyceraldehyde-3-phosphate dehydrogenase; forward primer, GAG CCA AAA GGG TCA TC; reverse primer, GTG GTC ATG AGT CCT TC) as previously described (54, 98).
Latency reactivation assay.
Latency reactivation assays were performed as described previously (39). Briefly, hESCs were pretreated with inhibitor for 1 h, infected with AD169-IE2-GFP at an MOI of 3 in the presence of the inhibitor for 1 h, and then washed twice with heparin. Alternatively, cells infected in the absence of the inhibitor were treated with inhibitor at 6 h postinfection for 1 h. After 10 days, latently infected cells were treated with 100 ng/ml tetradecanoyl phorbol acetate (TPA) to induce differentiation. After 7 days, the infected cells were harvested and cocultured with fibroblasts for 7 days before counting the GFP-positive foci.
ACKNOWLEDGMENTS
We thank Randall Massey for help with the electron microscopy, Shelby Lyon for help with image quantification, Halena VanDeusen for help with the illustrations, Bill Britt (University of Alabama at Birmingham) for generously providing antibodies, and the members of our laboratory for helpful discussions, especially Emily Albright for help with the manuscript.
J.-H.L. and R.F.K. designed and interpreted all experiments, except the electron microscopy experiments, and wrote the paper. J.-H.L. performed all the experiments, except for the electron microscopy experiments. R.F.K. and J.R.P. designed and interpreted the results of electron microscopy experiments. J.R.P. performed the electron microscopy experiments.
This work was supported by grants from the NIH (grants AI130089 and AI139180) to R.F.K.
REFERENCES
- 1.Goodrum FD, Jordan CT, High K, Shenk T. 2002. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc Natl Acad Sci U S A 99:16255–16260. doi: 10.1073/pnas.252630899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mendelson M, Monard S, Sissons P, Sinclair J. 1996. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J Gen Virol 77:3099–3102. doi: 10.1099/0022-1317-77-12-3099. [DOI] [PubMed] [Google Scholar]
- 3.Hargett D, Shenk TE. 2010. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc Natl Acad Sci U S A 107:20039–20044. doi: 10.1073/pnas.1014509107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kondo K, Kaneshima H, Mocarski ES. 1994. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc Natl Acad Sci U S A 91:11879–11883. doi: 10.1073/pnas.91.25.11879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor-Wiedeman J, Sissons JG, Borysiewicz LK, Sinclair JH. 1991. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J Gen Virol 72:2059–2064. doi: 10.1099/0022-1317-72-9-2059. [DOI] [PubMed] [Google Scholar]
- 6.Hahn G, Jores R, Mocarski ES. 1998. Cytomegalovirus remains latent in a common precursor of dendritic, and myeloid cells. Proc Natl Acad Sci U S A 95:3937–3942. doi: 10.1073/pnas.95.7.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhuravskaya T, Maciejewski JP, Netski DM, Bruening E, Mackintosh FR, St Jeor S. 1997. Spread of human cytomegalovirus (HCMV) after infection of human hematopoietic progenitor cells: model of HCMV latency. Blood 90:2482–2491. [PubMed] [Google Scholar]
- 8.Mercorelli B, Sinigalia E, Loregian A, Palu G. 2008. Human cytomegalovirus DNA replication: antiviral targets and drugs. Rev Med Virol 18:177–210. doi: 10.1002/rmv.558. [DOI] [PubMed] [Google Scholar]
- 9.Andrei G, De Clercq E, Snoeck R. 2009. Drug targets in cytomegalovirus infection. Infect Disord Drug Targets 9:201–222. doi: 10.2174/187152609787847758. [DOI] [PubMed] [Google Scholar]
- 10.Ahmed A. 2011. Antiviral treatment of cytomegalovirus infection. Infect Disord Drug Targets 11:475–503. doi: 10.2174/187152611797636640. [DOI] [PubMed] [Google Scholar]
- 11.Biron KK. 2006. Antiviral drugs for cytomegalovirus diseases. Antiviral Res 71:154–163. doi: 10.1016/j.antiviral.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 12.Saffert RT, Penkert RR, Kalejta RF. 2010. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J Virol 84:5594–5604. doi: 10.1128/JVI.00348-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crawford LB, Kim JH, Collins-McMillen D, Lee BJ, Landais I, Held C, Nelson JA, Yurochko AD, Caposio P. 2018. Human cytomegalovirus encodes a novel FLT3 receptor ligand necessary for hematopoietic cell differentiation and viral reactivation. mBio 9:e00682-18. doi: 10.1128/mBio.00682-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JH, Collins-McMillen D, Buehler JC, Goodrum FD, Yurochko AD. 2017. Human cytomegalovirus requires epidermal growth factor receptor signaling to enter and initiate the early steps in the establishment of latency in CD34(+) human progenitor cells. J Virol 91:e01206-16. doi: 10.1128/JVI.01206-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coronel R, Takayama S, Juwono T, Hertel L. 2015. Dynamics of human cytomegalovirus infection in CD34+ hematopoietic cells and derived Langerhans-type dendritic cells. J Virol 89:5615–5632. doi: 10.1128/JVI.00305-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodrum F, Jordan CT, Terhune SS, High K, Shenk T. 2004. Differential outcomes of human cytomegalovirus infection in primitive hematopoietic cell subpopulations. Blood 104:687–695. doi: 10.1182/blood-2003-12-4344. [DOI] [PubMed] [Google Scholar]
- 17.Kim JH, Collins-McMillen D, Caposio P, Yurochko AD. 2016. Viral binding-induced signaling drives a unique and extended intracellular trafficking pattern during infection of primary monocytes. Proc Natl Acad Sci U S A 113:8819–8824. doi: 10.1073/pnas.1604317113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collins-McMillen D, Stevenson EV, Kim JH, Lee BJ, Cieply SJ, Nogalski MT, Chan GC, Frost RW III, Spohn CR, Yurochko AD. 2017. Human cytomegalovirus utilizes a nontraditional signal transducer and activator of transcription 1 activation cascade via signaling through epidermal growth factor receptor and integrins to efficiently promote the motility, differentiation, and polarization of infected monocytes. J Virol 91:e00622-17. doi: 10.1128/JVI.00622-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soderberg-Naucler C, Streblow DN, Fish KN, Allan-Yorke J, Smith PP, Nelson JA. 2001. Reactivation of latent human cytomegalovirus in CD14(+) monocytes is differentiation dependent. J Virol 75:7543–7554. doi: 10.1128/JVI.75.16.7543-7554.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith MS, Bentz GL, Alexander JS, Yurochko AD. 2004. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J Virol 78:4444–4453. doi: 10.1128/jvi.78.9.4444-4453.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Albright ER, Kalejta RF. 2013. Myeloblastic cell lines mimic some but not all aspects of human cytomegalovirus experimental latency defined in primary CD34+ cell populations. J Virol 87:9802–9812. doi: 10.1128/JVI.01436-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forte E, Swaminathan S, Schroeder MW, Kim JY, Terhune SS, Hummel M. 2018. Tumor necrosis factor alpha induces reactivation of human cytomegalovirus independently of myeloid cell differentiation following posttranscriptional establishment of latency. mBio 9:e01560-18. doi: 10.1128/mBio.01560-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan C, Zhu D, Wang Y, Li L, Li D, Liu F, Zhang CY, Zen K. 2016. Human cytomegalovirus miR-UL148D facilitates latent viral infection by targeting host cell immediate early response gene 5. PLoS Pathog 12:e1006007. doi: 10.1371/journal.ppat.1006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O'Connor CM, Murphy EA. 2012. A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation, resulting in infectious progeny. J Virol 86:9854–9865. doi: 10.1128/JVI.01278-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poole E, Lau JC, Sinclair J. 2015. Latent infection of myeloid progenitors by human cytomegalovirus protects cells from FAS-mediated apoptosis through the cellular IL-10/PEA-15 pathway. J Gen Virol 96:2355–2359. doi: 10.1099/vir.0.000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yee LF, Lin PL, Stinski MF. 2007. Ectopic expression of HCMV IE72 and IE86 proteins is sufficient to induce early gene expression but not production of infectious virus in undifferentiated promonocytic THP-1 cells. Virology 363:174–188. doi: 10.1016/j.virol.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 27.Saffert RT, Kalejta RF. 2007. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J Virol 81:9109–9120. doi: 10.1128/JVI.00827-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol 80:3863–3871. doi: 10.1128/JVI.80.8.3863-3871.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagenknecht N, Reuter N, Scherer M, Reichel A, Muller R, Stamminger T. 2015. Contribution of the major ND10 proteins PML, hDaxx and Sp100 to the regulation of human cytomegalovirus latency and lytic replication in the monocytic cell line THP-1. Viruses 7:2884–2907. doi: 10.3390/v7062751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murayama T, Ohara Y, Obuchi M, Khabar KS, Higashi H, Mukaida N, Matsushima K. 1997. Human cytomegalovirus induces interleukin-8 production by a human monocytic cell line, THP-1, through acting concurrently on AP-1- and NF-kappaB-binding sites of the interleukin-8 gene. J Virol 71:5692–5695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geist LJ, Monick MM, Stinski MF, Hunninghake GW. 1994. The immediate early genes of human cytomegalovirus upregulate tumor necrosis factor-alpha gene expression. J Clin Invest 93:474–478. doi: 10.1172/JCI116995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gan X, Wang H, Yu Y, Yi W, Zhu S, Li E, Liang Y. 2017. Epigenetically repressing human cytomegalovirus lytic infection and reactivation from latency in THP-1 model by targeting H3K9 and H3K27 histone demethylases. PLoS One 12:e0175390. doi: 10.1371/journal.pone.0175390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ioudinkova E, Arcangeletti MC, Rynditch A, De Conto F, Motta F, Covan S, Pinardi F, Razin SV, Chezzi C. 2006. Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 384:120–128. doi: 10.1016/j.gene.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 34.Andrews PW. 1984. Retinoic acid induces neuronal differentiation of a cloned human embryonal carcinoma cell line in vitro. Dev Biol 103:285–293. doi: 10.1016/0012-1606(84)90316-6. [DOI] [PubMed] [Google Scholar]
- 35.Odeberg J, Wolmer N, Falci S, Westgren M, Seiger A, Soderberg-Naucler C. 2006. Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J Virol 80:8929–8939. doi: 10.1128/JVI.00676-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Belzile JP, Stark TJ, Yeo GW, Spector DH. 2014. Human cytomegalovirus infection of human embryonic stem cell-derived primitive neural stem cells is restricted at several steps but leads to the persistence of viral DNA. J Virol 88:4021–4039. doi: 10.1128/JVI.03492-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo MH, Schwartz PH, Fortunato EA. 2008. Neonatal neural progenitor cells and their neuronal and glial cell derivatives are fully permissive for human cytomegalovirus infection. J Virol 82:9994–10007. doi: 10.1128/JVI.00943-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Penkert RR, Kalejta RF. 2010. Nuclear localization of tegument-delivered pp71 in human cytomegalovirus-infected cells is facilitated by one or more factors present in terminally differentiated fibroblasts. J Virol 84:9853–9863. doi: 10.1128/JVI.00500-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Penkert RR, Kalejta RF. 2013. Human embryonic stem cell lines model experimental human cytomegalovirus latency. mBio 4:e00298-13. doi: 10.1128/mBio.00298-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee JH, Kalejta RF. 2019. Human cytomegalovirus enters the primary CD34+ hematopoietic progenitor cells where it establishes latency by macropinocytosis. J Virol 93:e00452-19. doi: 10.1128/JVI.00452-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu X, Jih J, Jiang J, Zhou ZH. 2017. Atomic structure of the human cytomegalovirus capsid with its securing tegument layer of pp150. Science 356:eaam6892. doi: 10.1126/science.aam6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huotari J, Helenius A. 2011. Endosome maturation. EMBO J 30:3481–3500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Egami Y, Taguchi T, Maekawa M, Arai H, Araki N. 2014. Small GTPases and phosphoinositides in the regulatory mechanisms of macropinosome formation and maturation. Front Physiol 5:374. doi: 10.3389/fphys.2014.00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rizopoulos Z, Balistreri G, Kilcher S, Martin CK, Syedbasha M, Helenius A, Mercer J. 2015. Vaccinia virus infection requires maturation of macropinosomes. Traffic 16:814–831. doi: 10.1111/tra.12290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arcangeletti MC, Vasile Simone R, Rodighiero I, De Conto F, Medici MC, Maccari C, Chezzi C, Calderaro A. 2016. Human cytomegalovirus reactivation from latency: validation of a “switch” model in vitro. Virol J 13:179. doi: 10.1186/s12985-016-0634-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paijo J, Doring M, Spanier J, Grabski E, Nooruzzaman M, Schmidt T, Witte G, Messerle M, Hornung V, Kaever V, Kalinke U. 2016. cGAS senses human cytomegalovirus and induces type I interferon responses in human monocyte-derived cells. PLoS Pathog 12:e1005546. doi: 10.1371/journal.ppat.1005546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu SE, Miller WE. 2015. The human cytomegalovirus lytic cycle is induced by 1,25-dihydroxyvitamin D3 in peripheral blood monocytes and in the THP-1 monocytic cell line. Virology 483:83–95. doi: 10.1016/j.virol.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee SH, Caviness K, Albright ER, Lee JH, Gelbmann CB, Rak M, Goodrum F, Kalejta RF. 2016. Long and short isoforms of the human cytomegalovirus UL138 protein silence IE transcription and promote latency. J Virol 90:9483–9494. doi: 10.1128/JVI.01547-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee SH, Albright ER, Lee JH, Jacobs D, Kalejta RF. 2015. Cellular defense against latent colonization foiled by human cytomegalovirus UL138 protein. Sci Adv 1:e1501164. doi: 10.1126/sciadv.1501164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poole EL, Kew VG, Lau JCH, Murray MJ, Stamminger T, Sinclair JH, Reeves MB. 2018. A virally encoded DeSUMOylase activity is required for cytomegalovirus reactivation from latency. Cell Rep 24:594–606. doi: 10.1016/j.celrep.2018.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mercer J, Helenius A. 2012. Gulping rather than sipping: macropinocytosis as a way of virus entry. Curr Opin Microbiol 15:490–499. doi: 10.1016/j.mib.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 52.Buckley CM, King JS. 2017. Drinking problems: mechanisms of macropinosome formation and maturation. FEBS J 284:3778–3790. doi: 10.1111/febs.14115. [DOI] [PubMed] [Google Scholar]
- 53.Bresnahan WA, Shenk TE. 2000. UL82 virion protein activates expression of immediate early viral genes in human cytomegalovirus-infected cells. Proc Natl Acad Sci U S A 97:14506–14511. doi: 10.1073/pnas.97.26.14506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rossetto CC, Tarrant-Elorza M, Pari GS. 2013. Cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14 (+) monocytes and CD34 (+) cells. PLoS Pathog 9:e1003366. doi: 10.1371/journal.ppat.1003366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Q, Fischer E, Cohen JI. 2016. Cell surface THY-1 contributes to human cytomegalovirus entry via a macropinocytosis-like process. J Virol 90:9766–9781. doi: 10.1128/JVI.01092-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vercauteren D, Vandenbroucke RE, Jones AT, Rejman J, Demeester J, De Smedt SC, Sanders NN, Braeckmans K. 2010. The use of inhibitors to study endocytic pathways of gene carriers: optimization and pitfalls. Mol Ther 18:561–569. doi: 10.1038/mt.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sawa A, Snyder SH. 2002. Schizophrenia: diverse approaches to a complex disease. Science 296:692–695. doi: 10.1126/science.1070532. [DOI] [PubMed] [Google Scholar]
- 58.Collins-McMillen D, Buehler J, Peppenelli M, Goodrum F. 2018. Molecular determinants and the regulation of human cytomegalovirus latency and reactivation. Viruses 10:E444. doi: 10.3390/v10080444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goodrum F, Reeves M, Sinclair J, High K, Shenk T. 2007. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110:937–945. doi: 10.1182/blood-2007-01-070078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rauwel B, Jang SM, Cassano M, Kapopoulou A, Barde I, Trono D. 2015. Release of human cytomegalovirus from latency by a KAP1/TRIM28 phosphorylation switch. Elife 4:e06068. doi: 10.7554/eLife.06068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Albright ER, Kalejta RF. 2016. Canonical and variant forms of histone H3 are deposited onto the human cytomegalovirus genome during lytic and latent infections. J Virol 90:10309–10320. doi: 10.1128/JVI.01220-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qin Q, Penkert RR, Kalejta RF. 2013. Heterologous viral promoters incorporated into the human cytomegalovirus genome are silenced during experimental latency. J Virol 87:9886–9894. doi: 10.1128/JVI.01726-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hetzenecker S, Helenius A, Krzyzaniak MA. 2016. HCMV induces macropinocytosis for host cell entry in fibroblasts. Traffic 17:351–368. doi: 10.1111/tra.12355. [DOI] [PubMed] [Google Scholar]
- 64.Murrell I, Tomasec P, Wilkie GS, Dargan DJ, Davison AJ, Stanton RJ. 2013. Impact of sequence variation in the UL128 locus on production of human cytomegalovirus in fibroblast and epithelial cells. J Virol 87:10489–10500. doi: 10.1128/JVI.01546-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petrucelli A, Rak M, Grainger L, Goodrum F. 2009. Characterization of a novel Golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J Virol 83:5615–5629. doi: 10.1128/JVI.01989-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Umashankar M, Petrucelli A, Cicchini L, Caposio P, Kreklywich CN, Rak M, Bughio F, Goldman DC, Hamlin KL, Nelson JA, Fleming WH, Streblow DN, Goodrum F. 2011. A novel human cytomegalovirus locus modulates cell type-specific outcomes of infection. PLoS Pathog 7:e1002444. doi: 10.1371/journal.ppat.1002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Keyes LR, Hargett D, Soland M, Bego MG, Rossetto CC, Almeida-Porada G, St Jeor S. 2012. HCMV protein LUNA is required for viral reactivation from latently infected primary CD14(+) cells. PLoS One 7:e52827. doi: 10.1371/journal.pone.0052827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reeves MB, Breidenstein A, Compton T. 2012. Human cytomegalovirus activation of ERK and myeloid cell leukemia-1 protein correlates with survival of latently infected cells. Proc Natl Acad Sci U S A 109:588–593. doi: 10.1073/pnas.1114966108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tarrant-Elorza M, Rossetto CC, Pari GS. 2014. Maintenance and replication of the human cytomegalovirus genome during latency. Cell Host Microbe 16:43–54. doi: 10.1016/j.chom.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 70.Umashankar M, Rak M, Bughio F, Zagallo P, Caviness K, Goodrum FD. 2014. Antagonistic determinants controlling replicative and latent states of human cytomegalovirus infection. J Virol 88:5987–6002. doi: 10.1128/JVI.03506-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhu D, Pan C, Sheng J, Liang H, Bian Z, Liu Y, Trang P, Wu J, Liu F, Zhang CY, Zen K. 2018. Human cytomegalovirus reprogrammes haematopoietic progenitor cells into immunosuppressive monocytes to achieve latency. Nat Microbiol 3:503–513. doi: 10.1038/s41564-018-0131-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kew V, Wills M, Reeves M. 2017. HCMV activation of ERK-MAPK drives a multi-factorial response promoting the survival of infected myeloid progenitors. J Mol Biochem 6:13–25. [PMC free article] [PubMed] [Google Scholar]
- 73.Smith MS, Goldman DC, Bailey AS, Pfaffle DL, Kreklywich CN, Spencer DB, Othieno FA, Streblow DN, Garcia JV, Fleming WH, Nelson JA. 2010. Granulocyte-colony stimulating factor reactivates human cytomegalovirus in a latently infected humanized mouse model. Cell Host Microbe 8:284–291. doi: 10.1016/j.chom.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Crawford LB, Streblow DN, Hakki M, Nelson JA, Caposio P. 2015. Humanized mouse models of human cytomegalovirus infection. Curr Opin Virol 13:86–92. doi: 10.1016/j.coviro.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Krishna BA, Poole EL, Jackson SE, Smit MJ, Wills MR, Sinclair JH. 2017. Latency-associated expression of human cytomegalovirus US28 attenuates cell signaling pathways to maintain latent infection. mBio 8:e01754-17. doi: 10.1128/mBio.01754-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu SE, Miller WE. 2016. The HCMV US28 vGPCR induces potent Galphaq/PLC-beta signaling in monocytes leading to increased adhesion to endothelial cells. Virology 497:233–243. doi: 10.1016/j.virol.2016.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Meshesha MK, Bentwich Z, Solomon SA, Avni YS. 2016. In vivo expression of human cytomegalovirus (HCMV) microRNAs during latency. Gene 575:101–107. doi: 10.1016/j.gene.2015.08.040. [DOI] [PubMed] [Google Scholar]
- 78.Beisser PS, Laurent L, Virelizier JL, Michelson S. 2001. Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected THP-1 monocytes. J Virol 75:5949–5957. doi: 10.1128/JVI.75.13.5949-5957.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Krishna BA, Miller WE, O’Connor CM. 2018. US28: HCMV’s Swiss army knife. Viruses 10:E445. doi: 10.3390/v10080445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Humby MS, O'Connor CM. 2015. Human cytomegalovirus US28 is important for latent infection of hematopoietic progenitor cells. J Virol 90:2959–2970. doi: 10.1128/JVI.02507-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Abraham CG, Kulesza CA. 2013. Polycomb repressive complex 2 silences human cytomegalovirus transcription in quiescent infection models. J Virol 87:13193–13205. doi: 10.1128/JVI.02420-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roche KL, Nukui M, Krishna BA, O'Connor CM, Murphy EA. 2018. Selective 4-thiouracil labeling of RNA transcripts within latently infected cells after infection with human cytomegalovirus expressing functional uracil phosphoribosyltransferase. J Virol 92:e00880-18. doi: 10.1128/JVI.00880-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qin Q, Lee SH, Liang R, Kalejta RF. 2014. Insertion of myeloid-active elements into the human cytomegalovirus major immediate early promoter is not sufficient to drive its activation upon infection of undifferentiated myeloid cells. Virology 448:125–132. doi: 10.1016/j.virol.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Reeves MB, Sinclair JH. 2010. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J Gen Virol 91:599–604. doi: 10.1099/vir.0.015602-0. [DOI] [PubMed] [Google Scholar]
- 85.Reeves MB, MacAry PA, Lehner PJ, Sissons JG, Sinclair JH. 2005. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc Natl Acad Sci U S A 102:4140–4145. doi: 10.1073/pnas.0408994102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kew VG, Yuan J, Meier J, Reeves MB. 2014. Mitogen and stress activated kinases act co-operatively with CREB during the induction of human cytomegalovirus immediate-early gene expression from latency. PLoS Pathog 10:e1004195. doi: 10.1371/journal.ppat.1004195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weekes MP, Tan SY, Poole E, Talbot S, Antrobus R, Smith DL, Montag C, Gygi SP, Sinclair JH, Lehner PJ. 2013. Latency-associated degradation of the MRP1 drug transporter during latent human cytomegalovirus infection. Science 340:199–202. doi: 10.1126/science.1235047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Edwards TG, Bloom DC. 2019. Lund human mesencephalic (LUHMES) neuronal cell line supports herpes simplex virus 1 latency in vitro. J Virol 93:e02210-18. doi: 10.1128/JVI.02210-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Thellman NM, Botting C, Madaj Z, Triezenberg SJ. 2017. An immortalized human dorsal root ganglion cell line provides a novel context to study herpes simplex virus 1 latency and reactivation. J Virol 91:e00080-17. doi: 10.1128/JVI.00080-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pourchet A, Modrek AS, Placantonakis DG, Mohr I, Wilson AC. 2017. Modeling HSV-1 latency in human embryonic stem cell-derived neurons. Pathogens 6:E24. doi: 10.3390/pathogens6020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.D’Aiuto L, Prasad KM, Upton CH, Viggiano L, Milosevic J, Raimondi G, McClain L, Chowdari K, Tischfield J, Sheldon M, Moore JC, Yolken RH, Kinchington PR, Nimgaonkar VL. 2015. Persistent infection by HSV-1 is associated with changes in functional architecture of iPSC-derived neurons and brain activation patterns underlying working memory performance. Schizophr Bull 41:123–132. doi: 10.1093/schbul/sbu032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zimmer B, Ewaleifoh O, Harschnitz O, Lee YS, Peneau C, McAlpine JL, Liu B, Tchieu J, Steinbeck JA, Lafaille F, Volpi S, Notarangelo LD, Casanova JL, Zhang SY, Smith GA, Studer L. 2018. Human iPSC-derived trigeminal neurons lack constitutive TLR3-dependent immunity that protects cortical neurons from HSV-1 infection. Proc Natl Acad Sci U S A 115:E8775–E8782. doi: 10.1073/pnas.1809853115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ibig-Rehm Y, Götte M, Gabriel D, Woodhall D, Shea A, Brown NE, Compton T, Feire AL. 2011. High-content screening to distinguish between attachment and post-attachment steps of human cytomegalovirus entry into fibroblasts and epithelial cells. Antiviral Res 89:246–256. doi: 10.1016/j.antiviral.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 94.Yu D, Smith GA, Enquist LW, Shenk T. 2002. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J Virol 76:2316–2328. doi: 10.1128/jvi.76.5.2316-2328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kalejta RF, Bechtel JT, Shenk T. 2003. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol Cell Biol 23:1885–1895. doi: 10.1128/mcb.23.6.1885-1895.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nowak B, Sullivan C, Sarnow P, Thomas R, Bricout F, Nicolas JC, Fleckenstein B, Levine AJ. 1984. Characterization of monoclonal antibodies and polyclonal immune sera directed against human cytomegalovirus virion proteins. Virology 132:325–338. doi: 10.1016/0042-6822(84)90039-4. [DOI] [PubMed] [Google Scholar]
- 97.Zhu H, Shen Y, Shenk T. 1995. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J Virol 69:7960–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Juckem LK, Boehme KW, Feire AL, Compton T. 2008. Differential initiation of innate immune responses induced by human cytomegalovirus entry into fibroblast cells. J Immunol 180:4965–4977. doi: 10.4049/jimmunol.180.7.4965. [DOI] [PubMed] [Google Scholar]