

Abstract
Protein–protein interactions (PPIs) are vital to all biological processes. These interactions are often dynamic, sometimes transient, typically occur over large topographically shallow protein surfaces, and can exhibit a broad range of affinities. Considerable progress has been made in determining PPI structures. However, given the above properties, understanding the key determinants of their thermodynamic stability remains a challenge in chemical biology. An improved ability to identify and engineer PPIs would advance understanding of biological mechanisms and mutant phenotypes and also provide a firmer foundation for inhibitor design. In silico prediction of PPI hot-spot amino acids using computational alanine scanning (CAS) offers a rapid approach for predicting key residues that drive protein–protein association. This can be applied to all known PPI structures; however there is a trade-off between throughput and accuracy. Here we describe a comparative analysis of multiple CAS methods, which highlights effective approaches to improve the accuracy of predicting hot-spot residues. Alongside this, we introduce a new method, BUDE Alanine Scanning, which can be applied to single structures from crystallography and to structural ensembles from NMR or molecular dynamics data. The comparative analyses facilitate accurate prediction of hot-spots that we validate experimentally with three diverse targets: NOXA-B/MCL-1 (an α-helix-mediated PPI), SIMS/SUMO, and GKAP/SHANK-PDZ (both β-strand-mediated interactions). Finally, the approach is applied to the accurate prediction of hot-spot residues at a topographically novel Affimer/BCL-xL protein–protein interface.
Protein–protein interactions (PPIs) play a central regulatory role in the majority of cellular signaling processes. Aberrant PPIs lead to disease and represent intervention points for targeted therapeutics, motivating efforts to discover small-molecule or peptide/peptidomimetic ligands.1−7 PPIs generally occur between significantly larger and topographically shallower surfaces than the lock-and-key-like pockets found in conventional drug targets, for example, enzymes and receptors, such as G-protein coupled receptors (GPCRs), making PPIs a challenge for ligand discovery.2,5,8−12 Significant recent successes, for example, ABT-19913 and RG7112,14 have countered the perception that PPIs are undruggable. Nonetheless, it is not clear which PPIs will be therapeutically tractable and relevant to disease,15−17 a problem that may be explored using chemical probes.18 In turn, this prompts the question: which PPIs might feasibly be modulated with small molecules or peptides/peptidomimetics? Though significant progress has been made in structurally characterizing PPIs, in order to understand the molecular basis of recognition it is also necessary to establish the determinants of favorable interaction, that is, which amino-acid residues contribute most to binding. Such information is essential in understanding biological mechanism, rationalizing dynamic conformational transitions, interpreting the functional role of mutation, and identifying starting points for inhibitor design.
Cunningham and Wells first used experimental alanine scanning to map PPIs.19 In this approach, variant proteins or peptides with Xaa-to-Ala mutations are used to evaluate the role of each amino acid side chain in a PPI. Assuming the absence of any changes in the backbone conformation of the protein, the changes indicate the contribution that the side chain makes to the binding free energy. Such studies have established that the thermodynamic driving force for PPIs can be dominated by a few key amino acid residues, termed hot-spots20 or hot regions.21 This experimental approach has been widely applied,22 but it is time-consuming and difficult to implement across entire protein–protein interactomes.23 Moreover, assay-dependent variations in binding affinities make comparative analyses of the literature problematic.24
In silico methods can be implemented to predict hot-spot residues from extant protein structures,24−28 and efforts have been made to classify PPIs in the Protein Data Bank (PDB)29 by secondary or tertiary structure at the interfaces and to predict hot-spot residues en masse.30−32 For computational alanine scanning (CAS), the binding free energies for the native and variant PPIs are calculated and the difference in these two values (ΔΔG) is used to evaluate the participation of the variant amino acid in the interface. Several approaches to CAS have been based on classical molecular dynamics simulations to capture the free energy change on amino acid variation. These methods include: Free Energy Perturbation,33−35 Thermodynamic Integration,34−36 the popular hybrid MM/PBSA method37 that has recently been improved by inclusion of interaction-entropy calculation,38 and by localizing the simulation volume around the residue in question.39 The Flex_ddG method in Rosetta28 uses a combination of sophisticated Monte Carlo sampling, minimization, and specialized force fields to provide one of the most accurate current methods for CAS. We compare our new fast approach, BudeAlaScan, with this method and other rapid methods that use a variety of scoring functions, which fall broadly into the following categories: physicochemical energy functions or statistical functions. Examples currently accessible via web servers include KFC,40 HotPoint,41 DrugScore-PPI,42 and PredHS.43 We chose FoldX44,45 as one of the first methods for rapid computational alanine scanning using a physicochemical force field, Rosetta Flex_ddG28 as the most recent tool exploiting physicochemical energy terms, and BeAtMuSiC46 and mCSM47 as tools employing statistical potentials and machine learning. We have avoided CAS methods that rely on molecular dynamics simulations for conformational sampling as being slow for high throughput calculations.
Typically, fast CAS methods operate on single structures and thus do not account for protein dynamics. In some cases, the methods can be, and have been, performed on multiple structures,25,48−50 for example, from NMR ensembles or MD trajectories. This is an important consideration given that many PPIs exploit intrinsically disordered regions (IDRs)51 and transient or dynamic noncovalent contacts can play important mechanistic roles in recognition.52,53 Here we perform a comparative analysis of common fast tools for CAS enabling informed consensus selection of hot-spot residues. This is benchmarked against an extensive literature data set and further validated through experimental analyses of four diverse PPIs, for which further analyses of NMR ensembles or molecular dynamics trajectories provides insight on dynamics to identify bona fide hot-spot residues.
FoldX is based on empirical potentials built from optimized combinations of various physical energy terms.44,45 The original Robetta method54 is a physical energy function parametrized on a monomeric protein data set (ProTherm).55,56 The Rosetta method used herein is Flex_ddG, based on the ΔΔG monomer method, using both the current general Rosetta force field,28 Ref2015, and the specialized force field Talaris2014.28,57−59 BeAtMuSiC46 is a coarse-grained predictor of changes in binding free energy induced by point mutations and uses a set of statistical potentials derived from known protein structures. The statistical potentials are trained on data from ProTherm56 and validated using the SKEMPI database.60 SKEMPI is a database of 3047 binding free energy changes upon mutation collated from the literature, for PPIs with known structure. In this study, we used all the ΔΔG data for the 748 single mutations to alanine present in the SKEMPI database that were compiled, ratified, and published recently.26 The method mCSM47 describes the protein environment upon mutation using signature vector patterns for each amino acid and was trained and tested on SKEMPI.60 Finally, BudeAlaScan, which we introduce here, is an empirical free-energy approach adapted from a small-molecule-docking algorithm, BUDE,61 using the standard force field (version heavy_by_atom_2016.bhff) provided with the current BUDE release 1.2.9. BudeAlaScan is a command-line application where the user assigns each protein of the complex as the receptor and the ligand. The application varies the ligand residues for alanine and reports the interfacial ΔΔG as ΔGALA – ΔGWT (hence a hot residue will have a positive value). For residues in the wild-type complex that may carry a charge (types DERKH) a rotamer library is used to estimate and account for the configurational entropy loss on forming an interfacial salt bridge. We will make a web-based graphical user interface available for BudeAlaScan in due course. All our selected tools for this comparison could be used to perform calculations on the SKEMPI data set or had been trained on this data set, further supporting our choice of in silico tools.
To compare the predictive capability of the five methods further, we perform detailed experimental analyses from the predictions on three diverse PPI targets: (i) the NOXA-B/MCL-162 interaction, an important current target in oncology;63 (ii) the SIMS/SUMO64 interaction, representative of a number of regulatory PPIs that depend on the SUMOylation post-translational modification;65 (iii) the GKAP/SHANK-PDZ interaction,66 which performs a scaffolding function at synaptic junctions.67 Example i is an α-helix-mediated PPI, while examples ii and iii are both β-strand-mediated interactions. Thus, these provide diversity in secondary structure interfaces and biological functions. Comparison of predicted and experimental data reveals that averaging the ΔΔG values for each residue across the five methods leads to more accurate prediction than any single method alone. Finally, we present further support for the implementation of this approach through prediction of hot residues for a topographically novel Affimer/BCL-xL PPI in which two loops project into a hydrophobic cleft. This allowed selection of a minimal number of residues that were experimentally validated.
Results and Discussion
Scope of the Methods
The CAS methods used here process structure types differently, that is, single X-ray crystal structures, NMR ensembles, MD trajectories. Figure 1 illustrates the capabilities of each in silico prediction tool and how structures are processed (see also Table S1 and Computational Methods in Supporting Information (SI)). BudeAlaScan is the only tool that allows processing of structure ensembles and scanning of multiple mutations to alanine at a time (i.e., hot-residue clusters).
Figure 1.

Schematic overview of approach for experimentally validated predictive alanine scanning using different methods. Different structural starting points can be used (single structure, black; NMR ensemble, blue; MD ensemble, orange) together with different in silico hot-spot residue prediction tools. Users can navigate through this workflow in different ways according to their requirements.
Different in Silico Prediction Tools Function in Different Ways and Provide Varying Results
First, we compared the results of alanine scanning on the SKEMPI data set for each of the tools, focusing on PPIs with natural amino acids and without post-translational modifications. Data for FoldX, mCSM, BeAtMuSiC, and BudeAlaScan were calculated in-house, while data for the Rosetta ddG methods were taken from the literature.28,46 This analysis took 8 min for FoldX, 5 min for BudeAlaScan, and 1–2 h per amino acid variation for Rosetta Flex_ddG on a single core of a linux X86_64 workstation running at 3.5 GHz. The Pearson correlation coefficient between the computed and experimental data in Figure 2 are given in Table 1.
Figure 2.

Comparison of experimental (SKEMPI) and predicted ΔΔG values for different prediction tools: (a–f) correlation plots (solid line is the 1:1 correlation) for predicted versus experimental ΔΔG for each of (a) BudeAlaScan, (b) FoldX, (c) Flex_ddG Talaris, (d) Flex_ddG Ref2015, (e) BeAtMuSiC, (f) mCSM, (g) Average, (h) Fraction Correct overall, and (i) Fraction Correct by residue type. Data for panels c and d are taken from Barlow et al.28 and plotted assuming 1 REU = 1 kcal/mol.
Table 1. Pearson Correlation Coefficient between Experimental and Predicted ΔΔG Values for Mutations to Alanine in the SKEMPI Dataset.
| Flex_ddG |
|||||||
|---|---|---|---|---|---|---|---|
| BUDE | FoldX | Talaris2014 | Ref2015 | BeAtMuSiC | mCSM | Average | |
| Pearson R | 0.50 | 0.28 | 0.51 | 0.49 | 0.47 | 0.66 | 0.60 |
| fraction correct | 0.76 | 0.51 | 0.76 | 0.74 | 0.72 | 0.77 | 0.78 |
The predictive performance of BudeAlaScan is comparable with Rosetta Flex_ddG and BeAtMuSiC. mCSM outperforms the physicochemical based methods, but it should be emphasized that this machine-learning approach is trained on the SKEMPI data it is fitting. Classifying the SKEMPI data as being positive (a hot residue) ΔΔG ≥ 4.184 kJ/mol, neutral (little effect) 4.184 kJ/mol < ΔΔG > −4.184 kJ/mol, or negative (affinity enhancing) ΔΔG ≤ −4.184 kJ/mol allows the determination of the fraction of correct predictions (FC). These results vary across the series (FC 0.51–0.78, see Figure 2h and Tables S2 and S3), although crucially different residues are correctly or incorrectly predicted from tool to tool. Calculating the fraction correct within each of these three categories (positive, neutral, and negative, Supporting Information, see Figure S1) reveals that the accuracy in correctly predicting a hot-residue decreases for all methods (FC ≈ 0.6) with FoldX performing least well. Conversely, the accuracy in predicting neutral residues increases for all methods (FC ≈ 0.8) apart from FoldX. There are only four negative examples in the SKEMPI set where mutation to alanine improves binding, and only FoldX predicts 2 of these, albeit in the context of many false negative predictions. BudeAlaScan uses a set of side-chain rotamers to estimate the configurational entropy loss on forming interactions involving DERKH residues and a fixed backbone. FoldX performs a local side-chain relaxation and Rosetta Flex_ddG allows local backbone and side-chain sampling. When operating on a single structure, none of the methods has sufficient conformational sampling, consideration of dynamics, or explicit solvation to provide the accuracy that might be achieved through more computationally intensive methods.26,28,40−47
PPI’s may possess interfaces where the binding energy is evenly distributed across the surface or concentrated in a few hot residues or regions. Prediction of the most influential residues in the former case is likely to be hampered by the accumulation of systematic errors. Thus, when using a single method, it is necessary to exercise caution in prioritizing one predicted residue over another based on the magnitude of its predicted ΔΔG.
Comparison of Computational and Experimental Alanine Scanning for Three Representative PPIs
While the SKEMPI data set provided an extensive set of data from the literature, as noted previously this is drawn from multiple different studies exploiting different assay methods, motivating our choice to supplement our comparative analysis with high-quality in-house experimental data. For comparison between prediction and experiment, we focused on protein/peptide interactions for several reasons: (1) peptide interacting motifs (PIMS)68 play a crucial role in many PPIs and are known to be effective starting points69 for chemical probe discovery campaigns; (2) a significant proportion of structures in the PDB are protein/peptide interactions with the peptide excised from the full-length protein; (3) ease of preparation/purification and standard N-terminal fluorescent labeling support quantification and a consistent assay format.
Experimental Alanine Scanning
Experimental alanine scanning was performed for structural representatives of three target systems: NOXA-B/MCL-1 (2JM6), SIMS/SUMO (2LAS), and GKAP/SHANK-PDZ (1Q3P). Each of the proteins was expressed in E. coli, and a series of labeled and unlabeled variant peptides was synthesized and purified (see SI). A number of different biophysical assays were implemented for each target (Table 2 and Figure 3). For NOXA-B/MCL-1 (Figure 3a), a competition fluorescence anisotropy (FA) assay (Figure 3b and SI, Figure S2a and Table S4) was used in which titration of variant peptides was used to displace a FITC-labeled NOXA-B sequence from MCL-1 (Kd of the FITC-labeled tracer = 80 nM). ΔΔG values were calculated using the difference in IC50 values. We also carried out a smaller number of direct titrations using FITC-labeled variant peptides with results concordant to those obtained by competition titration (see SI, Figure S2b and Table S5). Circular dichroism (CD) spectroscopy (see SI Figure S4) established that the effects of the amino-acid changes on helicity of the peptides were small compared to effects arising from side-chain contacts with the protein target.
Table 2. Kd and IC50 Values for NOXA-B/MCL-1 (2JM6), SIMS/SUMO (2LAS), and GKAP/SHANK-PDZ (1Q3P) Variantsa.
| NOXA | wt | L78A | R79A | R80A | I81A | D83A | V85A |
|---|---|---|---|---|---|---|---|
| Kd (nM)b | 10 ± 1 | e | 19 ± 3 | e | 1300 ± 50 | 4300 ± 200 | e |
| IC50 (μM)c | 1.77 ± 0.03 | 7000 ± 2000 | 8.3 ± 0.3 | 1.9 ± 0.3 | 1220 ± 70 | 13000 ± 8000 | 1200 ± 200 |
| SIM | wt | D2705A | N2706A | E2707A | I2708A | E2709A | V2710A |
|---|---|---|---|---|---|---|---|
| Kd (μM)d | 3.7 ± 0.3 | 4.2 ± 0.1 | 4.0 ± 0.2 | 6.6 ± 0.2 | 2.7 ± 0.1 | 18.9 ± 0.9 | 71 ± 9 |
| IC50 (μM)c | 30 ± 1 | e | e | 45 ± 4 | e | e | 205 ± 7 |
| SIM | I2711A | I2712A | V2713A | W2714A | E2715A | K2716A | K2717A |
|---|---|---|---|---|---|---|---|
| Kd (μM)b | 26 ± 2 | 65 ± 8 | 110 ± 20 | 60 ± 20 | 12.5 ± 0.7 | 10.7 ± 0.8 | 6.4 ± 0.6 |
| IC50 (μM)c | e | 351 ± 12 | e | 684 ± 264 | e | e | e |
| GKAP | wt | E1A | A2G | Q3A | T4A | R5A | L6A | CONH2 |
|---|---|---|---|---|---|---|---|---|
| Kd (μM)d | 2.8 ± 0.1 | 10.2 ± 0.5 | 7.0 ± 0.6 | 1.46 ± 0.08 | 1200 ± 200 | 9.5 ± 0.7 | f | 260 ± 50 |
| IC50 (μM)c | 11 ± 1 | 91 ± 5 | 14.0 ± 0.3 | 10.3 ± 0.9 | f | 14.1 ± 0.9 | f | f |
Amino acid numbering taken from PDB ID.
Fluorescence anisotropy direct titration.
Fluorescence anisotropy competition assay.
Isothermal titration calorimetry.
Not tested.
Did not bind/inhibit.
Figure 3.
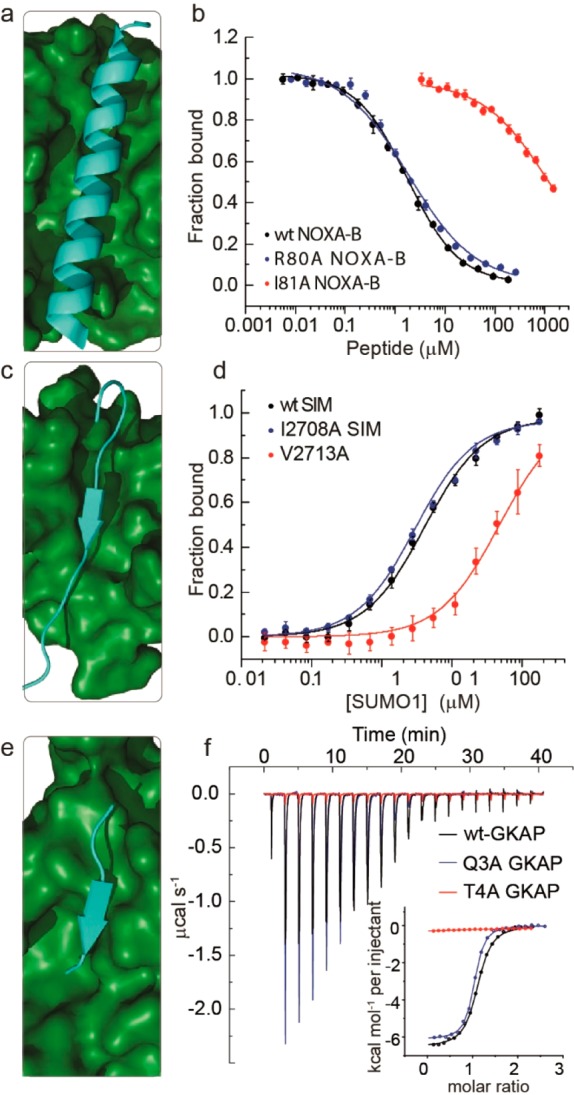
Experimental alanine scanning results for PPIs (amino acid numbering taken from PDB ID): (a) NMR structure for NOXA-B/MCL-1 (PDB ID 2JM6, model 1)), (b) representative competition fluorescence anisotropy inhibition curves for inhibition of the FITC–Ahx–NOXA-B/MCL-1 (50 mM Tris, 150 mM NaCl, 0.01% Triton-X, pH 7.4. using FITC–Ahx–NOXA-B as a tracer at 25 nM concentration and MCL-1 at 200 nM concentration) interaction using variant NOXA-B peptides, (c) NMR structure for SIMS/SUMO (PDB ID 2LAS, model 1), (d) representative fluorescence anisotropy titration curves for interaction of FAM labeled variant SIMS peptides with SUMO (20 mM Tris, 150 mM NaCl, 0.01% Triton-X, pH 7.4, using 50 nM FAM–PEG–SIM tracer), (e) crystal structure for GKAP/SHANK-PDZ (PDB ID 1Q3P), (f) representative isothermal titration calorimetry curves for interaction of acetylated GKAP variant peptides with SHANK-PDZ (20 mM Tris, 150 mM NaCl, pH 7.5, at 25 °C, 150 μM SHANK-PDZ).
For SIMS/SUMO (Figure 3c), a full series of FAM-labeled 13-residue SIMS peptides was prepared with each amino acid sequentially exchanged for alanine. Direct FA titration against SUMO (Figure 3d and SI, Figure S5, Table S6) provided Kd values that were used to determine ΔΔG based on comparison with the wild-type (Kd of the FAM-labeled wild-type tracer = 3.7 μM). As a secondary evaluation, several unlabeled peptides were used to compete against the FAM-labeled wild-type peptide for the SUMO binding site, and again the results were consistent with those obtained from direct titration (see SI, Figure S6 and Table S7). Here, CD spectra (see SI, Figure S7) indicated that all peptides were unstructured in solution except for the Glu2709 → Ala and Glu2715 → Ala; both of these variants promoted an alternative conformation with significant β-strand content. Given that such a conformation might be expected to bind more effectively due to preorganization, we interpret the moderate loss in potency observed for both variants as indicating a minor role for the side chains in the PPI.
Finally, for GKAP/SHANK-PDZ (Figure 3e), isothermal titration calorimetry (Figure 3f and SI, Figure S8 and Table S8) was used to obtain Kd values (WT Kd = 800 nM) from the titration of variant peptides (including the C-terminal amide and Ala2 → Gly variants) against SHANK-PDZ, while a competition assay using a FITC-labeled GKAP peptide was elaborated to give IC50 values (see SI, Figure S9 and Table S9). The two methods yielded similar values, while CD spectra (see SI, Figure S10) indicated that the 6-residue peptides were unstructured in solution as expected. Average ΔΔG values for each of the six predictive methods and the experimental data were then compared (Figures 4 and 5).
Figure 4.

Comparison of experimental (from this work) and predicted ΔΔG values for different prediction tools: (a–f) correlation plots (dotted line is the 1:1 correlation) for predicted versus experimental ΔΔG for each of (a) BudeAlaScan, (b) FoldX, (c) Flex_ddG Talaris, (d) Robetta, (e) BeAtMuSiC, (f) mCSM, (g) Average, (h) Fraction Correct overall, and (i) Fraction Correct by residue type. There are 6 fewer points for panel d due to Robetta automatically defining the interface. Data for panels c and d are plotted assuming 1 REU = 1 kcal/mol.
Figure 5.

Comparison of predictive and experimental ΔΔG values (bars represent experimental data for each target): (a) NOXA-B/MCL-1 (PDB model 1); (b) SIMS/SUMO (PDB model 1); (c) GKAP/SHANK-PDZ (chains A and C). Two data points for FoldX are missing from the plot since they are less than −5.0 kJ/mol (R79A and W2714A).
Computational Alanine Scanning
Computational alanine scans were carried out for the same three target systems: NOXA-B/MCL-1 (2JM6), SIMS/SUMO (2LAS), and GKAP/SHANK-PDZ (1Q3P). We chose the simplest approach, namely, a single structure, in the first round of CAS; hence only the first structure of each NMR ensemble for 2JM6 and 2LAS was used along with the crystal structure 1Q3P. Each structure was subjected to each of six representative computational alanine-scanning methods. The GKAP sequence had an alanine (Ala2), and Gly was used to evaluate the role of this side chain. Crucially, crystallographic analyses suggested a key role for the C-terminal carboxylate, not just the side chains. Since BudeAlaScan is built on BUDE, it is simple to submit the modified backbone to BUDE itself to dissect out the roles of the side chain and carboxylate independently yielding a ΔΔG of 19.6 kJ/mol for replacing the carboxylate by the corresponding amide compared with the experimental value of 11.4 kJ/mol (Table 2). This feature is unique to BudeAlaScan among the in silico methods and, therefore, applicable to multiple other unconventional changes to peptide structure (vide infra).
Based on comparison of the in silico and experimental data, in each case, most of the computational methods correctly identified the majority of experimentally defined hot residues (Figures 4 and 5), but they differed in the residues predicted incorrectly. For example, mCSM predicted Thr4 in GKAP as a hot-residue particularly well and with good accuracy, but wrongly predicted Arg5 as a hot-residue in GKAP. On the other hand, BudeAlaScan over-predicted the contribution of Arg79 in NOXA-B, but under-predicted Asp83. Encouragingly, by taking the average ΔΔG of all the methods for each residue, all but two hot-spot residues were correctly predicted giving a fraction correct of 0.92 (Table 3). One residue (Ile2711) is predicted rather well, but the experimental value is close to the selection criterion of 4.184 kJ/mol and the predicted and experimental values straddle this cutoff. The other (Val2713) is solvent exposed and hydrophobic, favors beta structure, and is poorly predicted by all methods; hence it may perform a structural role in supporting the extended β-strand conformation required for binding, rather than contributing to the PPI per se.
Table 3. Pearson Correlation Coefficient between Experimental and Predicted ΔΔG Values for Mutations to Alanine in This Work.
| BUDE | FoldX | Flex_ddG Talaris2014 | Robetta | BeAtMuSiC | mCSM | Average | |
|---|---|---|---|---|---|---|---|
| Pearson R | 0.58 | 0.52 | 0.56 | 0.71 | 0.71 | 0.36 | 0.76 |
| fraction correct | 0.80 | 0.68 | 0.72 | 0.88 | 0.84 | 0.68 | 0.92 |
The Use of Multiple Structures Improves Predictive Alanine Scanning and Includes the Role of Dynamic Conformational Variation
A single protein structure, for example, that determined by X-ray crystallography, represents a single point on the global potential-energy landscape, but it is well established that proteins in their native state occupy a family of conformations clustered around such a local minimum. In other words, proteins are dynamic and show thermally induced fluctuations in their native folded states. This dynamic behavior makes accurate predictions of the free energy difference between two variants difficult to achieve with a single pair of structures. A number of the tools we evaluate could be used for this purpose, however, here as a proof-of-concept we used BudeAlaScan to probe these fluctuations. The BUDE force field goes some way to mitigating the effects of dynamics by using a soft-core potential to ameliorate geometrical inaccuracies in a structure. BudeAlaScan also employs multiple rotamers for side chains that can carry charge (DERKH) to account for entropic loss on freezing such side chains in interfacial salt bridges. However, we anticipated that greater accuracy would be achieved using an ensemble of conformations that include backbone flexibility. There are two ways to do this: by using (a) structures from solution phase NMR ensembles or (b) snapshots from molecular dynamics (MD) simulations. Although the two methods typically report on different time scales, the purpose here is not to compare the data from NMR with MD, rather to highlight that either can be useful in establishing where potential hot-residues are persistent. Crucially, such an approach begins to acknowledge the dynamic and conformationally varied nature of PPIs. The calculation of interaction energies with BUDE is very fast; hence BudeAlaScan rapidly processes ensembles of conformations. For our analysis, each structure was subjected to a 1 μs MD simulation, and the BudeAlaScan calculation was carried out on each of 100 snapshots (every 10 ns of a 1 μs trajectory). In addition, BudeAlaScan was performed on each of the submitted models in the NMR ensembles for the two PPIs with NMR structures. In general, the predictions improved through the use of this approach for all targets (Figure 6).
Figure 6.

Comparison of BudeAlaScan predicted and experimental ΔΔG values (gray bars). Green, average and standard deviation of 100 structures from 1 μs MD simulations; blue average and standard deviation of 20 (2JM6) and 10 (2LAS) NMR structures; (a) NOXA-B/MCL-1; (b) SIMS/SUMO; (c) GKAP/SHANK-PDZ.
These data also allow the average strength and persistence of interactions to be assessed as a standard deviation: residues that are tightly packed into the interface with little conformational freedom tended to show low standard deviations. This behavior is exemplified by Leu78, Ile81, and Val85 for the NOXA-B/MCL-1 interaction (Figure 6a). In contrast, the surface-exposed residues Arg79 and Asp83 for the NOXA-B/MCL-1 interaction that transiently form salt bridges exhibit higher standard deviations. SIMS/SUMO shows similar behavior (Figure 6b). The smaller interface in GKAP/SHANK (Figure 6c) is much more mobile and shows large standard deviations throughout the sequence. In the absence of a corresponding NMR structure, we are unable to say if this is a consequence of excessive mobility in the MD simulation, although the poorer match with experimental alanine scanning results suggests this is the case. The match between prediction and experiment is generally somewhat improved by using ensembles of structures. Hence, the Pearson correlation coefficient for NOXA-B/MCL-1 and SIMS/SUMO between the BudeAlaScan data and experiment is 0.58 for single structures (Figure 5a,b) and 0.66 and 0.62 for the MD and NMR ensembles, respectively (Figure 6a,b).
Use of Multiple Tools to Predict Hot-Residues at a Topographically Distinct PPI
Having experimentally validated the approach with three model systems, we applied the workflow to a novel protein–protein interface with a different topography. For this, we selected a recently described PPI between an Affimer and BCL-xL,70 which, like MCL-1, is an antiapoptotic member of the BCL-2 family of apoptotic regulators (Figure 7a).71 In this structure, two nine-residue loops from the Affimer interact with the BH3 helix binding cleft on BCL-xL.
Figure 7.

Application of predictive workflow to a topographically distinct protein–protein interaction. (a) Affimer/BCL-xL cocrystal structure (PDB ID 6HJL), highlighting residues Trp41, Trp44, and Trp76 (orange) identified for experimental characterization (Affimer in cyan, BCL-xL in green). (b) Predicted ΔΔG values calculated using each predictive tool together with average values for alanine variants of residues within the two 9-residue variable loops of the Affimer scaffold; gray boxes show ΔΔG limits estimated from the experimental W to A changes; residues 41 and 44 ≥ 15 kJ/mol, residue 76 ≤ 1 kJ/mol, (3 points for Foldx, D43A, E46A, and W76A, are missing from the plot as their values are less than −5 kJ/mol). (c) Competition fluorescence anisotropy inhibition curves for inhibition of the FITC–BID/BCL-xL interaction (20 mM Tris, 150 mM NaCl, 0.01% Triton X-100, pH 7.4, using FITC–BID as a tracer at 25 nM and BCL-xL at 100 nM concentration) using variant Affimer sequences.
Predictions for each loop were performed using each of the tools, and the average ΔΔG was determined (Figure 7b). We then selected three residues to test experimentally: two predicted to be hot-residues (Trp41 and Trp44), and one predicted as not being a hot-residue (Trp76) according to the average values of ΔΔG across the six methods. The parent and variant Affimer sequences were cloned, expressed, and purified and then tested in a competitive FA assay, that is, by displacement of FITC–BID fluorescently labeled peptide from BCL-xL. These experiments fully confirmed the predictions (Figure 7c and SI, Table S9). Crucially, at least one of the individual prediction tools would not have predicted these residues accurately, further validating the use of comparative analyses.
Conclusions
We outline effective approaches for rapid predictive in silico examination of PPI interfaces. This employs multiple computational tools, allowing comparative analyses to predict hot-residues accurately. In the broader context of providing improved and accessible methods for in silico Ala scanning, we introduce BudeAlaScan. This is a versatile tool for the rapid analysis of PPIs using X-ray structures, NMR ensembles, or snapshots from MD trajectories as input files. Exploiting six in silico prediction tools illustrates that use of multiple methods leads to improved accuracy in the prediction of hot-residues. We demonstrate this further by comparing the predictions with experimental studies for three different PPIs. We note that the predicted ΔΔG values for single-alanine variants vary between the in silico methods and also change through structural ensembles analyzed with any single method. Thus, in terms of informing on the role of individual amino acids in PPIs and developing starting points for ligand design, we advocate using a combination of methods to predict hot-residues and hot-spots. Additional use of NMR ensembles or snapshots from MD trajectories allows an assessment of the persistence of noncovalent interactions. Taken together this allows prioritization of potential hot-residues to reduce the number of predictions that require experimental validation and provides greater confidence in those that should be selected for mimicry in ligand based design.
Methods
Computational Methods and MD
SKEMPI Data Set and Predictions
The set of 748 single mutations to alanine from the SKEMPI database with associated binding data is that described by Barlow et al.28 and provided in that paper’s Supporting Information (jp7b11367_si_002.xlsx).
FoldX
The program FoldX was downloaded from http://FoldXsuite.crg.eu and used in AlaScan mode with default parameters.
Rosetta
Flex_ddG
Data were taken from the Supporting Information of previously published work28 for benchmarking against SKEMPI. Mutations specific to this work were calculated using the flex_ddG method in Rosetta Commons release and the flex_ddG.xml script provided in the SI of Barlow et al.28 The Talaris 2014 force field was used, and each result is the average of 50 repeats.
Robetta
Mutations specific to this work were also calculated with the Robetta server, http://robetta.bakerlab.org or via the Rosetta Commons release.
BudeAlaScan
BudeAlaScan is a command-line python application for computational alanine scanning. It employs ISAMBARD5 for structure manipulation and a customized version of the Bristol University Docking Engine (BUDE)6 for energy calculations. The program was run in scan mode with default parameters. The application is available via the BAlaS server: http://coiledcoils.chm.bris.ac.uk/balas
BeAtMuSiC
Data used in the BeAtMuSiC publication were taken from the MCSM Web site, http://biosig.unimelb.edu.au/mcsm/data, for benchmarking against SKEMPI. Mutations specific to this work were calculated using the BeAtMuSiC server: http://babylone.ulb.ac.be/beatmusic.
mCSM
The mCSM data were calculated in-house by querying the server http://biosig.unimelb.edu.au/mcsm/protein_protein with a python script.
Molecular Dynamics Simulations
All simulations were performed using the GROMACS 5.1.4 suite and the following general protocols. Structures from the protein database (SIMS/SUMO 2LAS; GKAP/SHANK-PDZ 1Q3P; NOXA-B/MCL-1 2JM6) were processed with the GROMACS tool chain. The utility pdb2gmx was used to add hydrogen atoms consistent with pH 7 and virtual-site hydrogens for mobile groups and parametrize the protein with the amber99SB-ildn force field. The protein was placed in an orthorhombic box 2 nm larger than the protein in each dimension and filled with TIP3P water containing 0.15 M sodium chloride ions to give a charge-neutral system overall. After 10000 steps of steepest descent minimization, molecular dynamics was initiated with random velocities while restraining the protein backbone to its original position with a force constant of 1000 kJ/nm for 0.2 ns. Simulations were developed for a further 1 μs without backbone position restraints under periodic boundary conditions. The Particle Mesh Ewald’s method was used for long-range electrostatic interactions, while short-range Coulombic and van der Waals energies were truncated at 1.2 nm. The temperature was maintained at 310 K using the v-rescale method and the pressure at 1 bar with the Berendsen barostat. The use of virtual-site hydrogens allowed a 5 fs time step for the leapfrog integrator. Bond constraints were implemented with the LINCS method, and SETTLE was used for waters. Trajectories were processed and analyzed with the GROMACS tools and visualized with VMD 1.9.3.
Expression and Purification of Proteins
Proteins were expressed using standard protocols and characterized as described in the Supporting Information.
Peptide Synthesis and Purification
Peptides were prepared using microwave assisted solid-phase FMoc based synthesis using a CEM Liberty Blue peptide synthesizer and purified by reverse phase HPLC. Full details and characterization are available in the Supporting Information.
Isothermal Titration Calorimetry (ITC)
ITC experiments were carried out using Microcal ITC200i instrument (Malvern) at 25 °C in 20 mM Tris, 150 mM NaCl, pH 7.5, buffer. SHANK-PDZ was dialyzed against the buffer prior to experiment; lyophilized peptides were dissolved in the same buffer. SHANK-PDZ (150 μM) was present in the cell and titrated with 1.4–2 mM peptide solutions loaded into the syringe using 2 μL injections with 120 s spacing between the injections for 20 injections. Heats of peptide dilution were subtracted from each measurement raw data. Data was analyzed using Microcal Origin 8 and fitted to a one binding site model.
Fluorescence Anisotropy
Fluorescence anisotropy assays were performed in 384-well plates (Greiner Bio-one). Each experiment was run in triplicate, and the fluorescence anisotropy was measured using a PerkinElmer EnVisionTM 2103 MultiLabel plate reader with excitation at 480 nm (30 nm bandwidth), polarized dichroic mirror at 505 nm, and emission at 535 nm (40 nm bandwidth, S and P polarized) for FAM and FITC labeled peptides. The excitation and emission wavelengths for BODIPY labeled BAK peptide were set to 531 and 595 nm, respectively. The excitation and emission wavelengths for FITC labeled BID peptide were set to 490 and 535 nm, respectively.
Direct Binding Assays
Fluorescence anisotropy direct titration assays were performed with protein concentration diluted over 16–24 points using 1/2 dilutions. Twenty microliters of buffer was first added to each well. Twenty microliters of a solution of protein was added to the first column. The solution was well mixed, and 20 μL was taken out and added to the next column and so on. This operation consists of serial dilution of the protein across the plate. Finally, 20 μL of tracer was added to the wells. For control wells, the tracer peptide was replaced with an identical volume of assay buffer, and plates were read after 1 h.
Competition Binding Assays
FA competition assays were performed in 384 well plates with the concentration of variant peptide competitor typically from 10 to 1500 μM, diluted over 16–24 points in 1/2 regime with fixed protein and tracer concentrations. For control wells, the tracer peptide was replaced with an identical volume of assay buffer. The total volume in each well was 60 μL. Plates were read after 1 h (and 16 h for BCL-xL assays) of incubation at RT.
Additional details on buffer composition, reagent concentration for individual PPIs, and data analyses are given in the Supporting Information.
Acknowledgments
We thank P. Ramsahye for help with protein expression.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.9b00560.
Raw computational predictions and data for SKEMPI database (XLSX)
Raw computational predictions and data for experimental targets (XLSX)
Protein expression and purification (including characterization), additional details of computational methods (including performance analysis), peptide synthesis and purification protocols (including characterization), additional details on biophysical methods and data, and circular dichroism details and data (PDF)
Author Contributions
∇ A.A.I. and G.J.B. contributed equally to this work. A.J.W., R.B.S., A.N., T.A.E., and D.N.W. conceived and designed the research program, A.A.I., G.J.B., Z.H., S.D., F.H., K.A.H., K.H., and K.S. designed studies and performed research. The manuscript was written by A.J.W. with contributions from all authors. Z.H., R.B.S., and F. H. prepared figures.
This work was supported by the EPSRC (EP/N013573/1, EP/KO39292/1), The ERC (340764), and The Wellcome Trust (097827/Z/11/A, WT094232MA, 094232/Z/10/Z). A.S.N. holds an EPSRC Fellowship (EP/N025652/1). D.N.W. holds a Royal Society Wolfson Research Merit Award (WM140008).
The authors declare no competing financial interest.
Supplementary Material
References
- Azzarito V.; Long K.; Murphy N. S.; Wilson A. J. (2013) Inhibition of α-helix-mediated protein-protein interactions using designed molecules. Nat. Chem. 5, 161–173. 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]
- Arkin M. R.; Tang Y.; Wells J. A. (2014) Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing toward the Reality. Chem. Biol. 21, 1102–1114. 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milroy L.-G.; Grossmann T. N.; Hennig S.; Brunsveld L.; Ottmann C. (2014) Modulators of Protein–Protein Interactions. Chem. Rev. 114, 4695–4748. 10.1021/cr400698c. [DOI] [PubMed] [Google Scholar]
- Pelay-Gimeno M.; Glas A.; Koch O.; Grossmann T. N. (2015) Structure-Based Design of Inhibitors of Protein–Protein Interactions: Mimicking Peptide Binding Epitopes. Angew. Chem., Int. Ed. 54, 8896–8927. 10.1002/anie.201412070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott D. E.; Bayly A. R.; Abell C.; Skidmore J. (2016) Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discovery 15, 533–550. 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- Grison C. M.; Burslem G. M.; Miles J. A.; Pilsl L. K. A.; Yeo D. J.; Imani Z.; Warriner S. L.; Webb M. E.; Wilson A. J. (2017) Double quick, double click reversible peptide “stapling”. Chem. Sci. 8, 5166–5171. 10.1039/C7SC01342F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher J. M.; Horner K. A.; Bartlett G. J.; Rhys G. G.; Wilson A. J.; Woolfson D. N. (2018) De novo coiled-coil peptides as scaffolds for disrupting protein–protein interactions. Chem. Sci. 9, 7656–7665. 10.1039/C8SC02643B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavassoli A. (2011) Targeting the protein-protein interactions of the HIV lifecycle. Chem. Soc. Rev. 40, 1337–1346. 10.1039/C0CS00092B. [DOI] [PubMed] [Google Scholar]
- Nordgren I. K.; Tavassoli A. (2011) Targeting tumour angiogenesis with small molecule inhibitors of hypoxia inducible factor. Chem. Soc. Rev. 40, 4307–4317. 10.1039/c1cs15032d. [DOI] [PubMed] [Google Scholar]
- Burslem G. M.; Kyle H. F.; Nelson A.; Edwards T. A.; Wilson A. J. (2017) Hypoxia inducible factor (HIF) as a model for studying inhibition of protein–protein interactions. Chem. Sci. 8, 4188–4202. 10.1039/C7SC00388A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pricer R.; Gestwicki J. E.; Mapp A. K. (2017) From Fuzzy to Function: The New Frontier of Protein–Protein Interactions. Acc. Chem. Res. 50, 584–589. 10.1021/acs.accounts.6b00565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. D.; Dugan A.; Gestwicki J. E.; Mapp A. K. (2012) Fine-Tuning Multiprotein Complexes Using Small Molecules. ACS Chem. Biol. 7, 1311–1320. 10.1021/cb300255p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souers A. J.; Leverson J. D.; Boghaert E. R.; Ackler S. L.; Catron N. D.; Chen J.; Dayton B. D.; Ding H.; Enschede S. H.; Fairbrother W. J.; Huang D. C. S.; Hymowitz S. G.; Jin S.; Khaw S. L.; Kovar P. J.; Lam L. T.; Lee J.; Maecker H. L.; Marsh K. C.; Mason K. D.; Mitten M. J.; Nimmer P. M.; Oleksijew A.; Park C. H.; Park C.-M.; Phillips D. C.; Roberts A. W.; Sampath D.; Seymour J. F.; Smith M. L.; Sullivan G. M.; Tahir S. K.; Tse C.; Wendt M. D.; Xiao Y.; Xue J. C.; Zhang H.; Humerickhouse R. A.; Rosenberg S. H.; Elmore S. W. (2013) ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208. 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- Ray-Coquard I.; Blay J.-Y.; Italiano A.; Le Cesne A.; Penel N.; Zhi J.; Heil F.; Rueger R.; Graves B.; Ding M.; Geho D.; Middleton S. A.; Vassilev L. T.; Nichols G. L.; Bui B. N. (2012) Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 13, 1133–1140. 10.1016/S1470-2045(12)70474-6. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R. (2002) The druggable genome. Nat. Rev. Drug Discovery 1, 727–730. 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Dandapani S.; Marcaurelle L. A. (2010) Grand Challenge Commentary: Accessing new chemical space for ’undruggable’ targets. Nat. Chem. Biol. 6, 861–863. 10.1038/nchembio.479. [DOI] [PubMed] [Google Scholar]
- Waring M. J.; Arrowsmith J.; Leach A. R.; Leeson P. D.; Mandrell S.; Owen R. M.; Pairaudeau G.; Pennie W. D.; Pickett S. D.; Wang J.; Wallace O.; Weir A. (2015) An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discovery 14, 475–486. 10.1038/nrd4609. [DOI] [PubMed] [Google Scholar]
- Blagg J.; Workman P. (2017) Choose and Use Your Chemical Probe Wisely to Explore Cancer Biology. Cancer Cell 32, 9–25. 10.1016/j.ccell.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham B.; Wells J. (1989) High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science 244, 1081–1085. 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- Clackson T.; Wells J. (1995) A hot spot of binding energy in a hormone-receptor interface. Science 267, 383–386. 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- London N.; Raveh B.; Schueler-Furman O. (2013) Druggable protein–protein interactions – from hot spots to hot segments. Curr. Opin. Chem. Biol. 17, 952–959. 10.1016/j.cbpa.2013.10.011. [DOI] [PubMed] [Google Scholar]
- DeLano W. L. (2002) Unraveling hot spots in binding interfaces: progress and challenges. Curr. Opin. Struct. Biol. 12, 14–20. 10.1016/S0959-440X(02)00283-X. [DOI] [PubMed] [Google Scholar]
- Rogers J. M.; Passioura T.; Suga H. (2018) Nonproteinogenic deep mutational scanning of linear and cyclic peptides. Proc. Natl. Acad. Sci. U. S. A. 115, 10959–10964. 10.1073/pnas.1809901115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangone A.; Bonvin A. M. J. J. (2015) Contacts-based prediction of binding affinity in protein–protein complexes. eLife 4, e07454. 10.7554/eLife.07454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massova I.; Kollman P. A. (1999) Computational Alanine Scanning To Probe Protein–Protein Interactions: A Novel Approach To Evaluate Binding Free Energies. J. Am. Chem. Soc. 121, 8133–8143. 10.1021/ja990935j. [DOI] [Google Scholar]
- Moreira I. S.; Fernandes P. A.; Ramos M. J. (2007) Computational alanine scanning mutagenesis—An improved methodological approach. J. Comput. Chem. 28, 644–654. 10.1002/jcc.20566. [DOI] [PubMed] [Google Scholar]
- Brender J. R.; Zhang Y. (2015) Predicting the Effect of Mutations on Protein-Protein Binding Interactions through Structure-Based Interface Profiles. PLoS Comput. Biol. 11, e1004494. 10.1371/journal.pcbi.1004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow K. A.; Ó Conchúir S.; Thompson S.; Suresh P.; Lucas J. E.; Heinonen M.; Kortemme T. (2018) Flex ddG: Rosetta Ensemble-Based Estimation of Changes in Protein–Protein Binding Affinity upon Mutation. J. Phys. Chem. B 122, 5389–5299. 10.1021/acs.jpcb.7b11367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins A. M.; Arora P. S. (2014) Anatomy of β-Strands at Protein–Protein Interfaces. ACS Chem. Biol. 9, 1747–1754. 10.1021/cb500241y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergey C. M.; Watkins A. M.; Arora P. S. (2013) HippDB: a database of readily targeted helical protein–protein interactions. Bioinformatics 29, 2806–2807. 10.1093/bioinformatics/btt483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock B. N.; Jochim A. L.; Arora P. S. (2011) Assessing Helical Protein Interfaces for Inhibitor Design. J. Am. Chem. Soc. 133, 14220–14223. 10.1021/ja206074j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukharta L.; Gutiérrez-de-Terán H.; Åqvist J. (2014) Computational Prediction of Alanine Scanning and Ligand Binding Energetics in G-Protein Coupled Receptors. PLoS Comput. Biol. 10, e1003585. 10.1371/journal.pcbi.1003585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ C. D.; Mark A. E.; van Gunsteren W. F. (2010) Basic ingredients of free energy calculations: A review. J. Comput. Chem. 31, 1569–1582. 10.1002/jcc.21450. [DOI] [PubMed] [Google Scholar]
- Chipot C. (2014) Frontiers in free-energy calculations of biological systems. WIREs Comput. Mol. Sci. 4, 71–89. 10.1002/wcms.1157. [DOI] [Google Scholar]
- Khavrutskii I. V.; Wallqvist A. (2011) Improved Binding Free Energy Predictions from Single-Reference Thermodynamic Integration Augmented with Hamiltonian Replica Exchange. J. Chem. Theory Comput. 7, 3001–3011. 10.1021/ct2003786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massova I.; Kollman P. A. (2000) Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discovery Des. 18, 113–135. 10.1023/A:1008763014207. [DOI] [Google Scholar]
- Duan L.; Liu X.; Zhang J. Z. H. (2016) Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein–Ligand Binding Free Energy. J. Am. Chem. Soc. 138, 5722–5728. 10.1021/jacs.6b02682. [DOI] [PubMed] [Google Scholar]
- Dourado D. F. A. R.; Flores S. C. (2014) A multiscale approach to predicting affinity changes in protein–protein interfaces. Proteins: Struct., Funct., Genet. 82, 2681–2690. 10.1002/prot.24634. [DOI] [PubMed] [Google Scholar]
- Darnell S. J.; LeGault L.; Mitchell J. C. (2008) KFC Server: interactive forecasting of protein interaction hot spots. Nucleic Acids Res. 36, W265–W269. 10.1093/nar/gkn346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuncbag N.; Keskin O.; Gursoy A. (2010) HotPoint: hot spot prediction server for protein interfaces. Nucleic Acids Res. 38, W402–W406. 10.1093/nar/gkq323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger D. M.; Gohlke H. (2010) DrugScorePPI webserver: fast and accurate in silico alanine scanning for scoring protein–protein interactions. Nucleic Acids Res. 38, W480–W486. 10.1093/nar/gkq471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L.; Zhang Q. C.; Chen Z.; Meng Y.; Guan J.; Zhou S. (2014) PredHS: a web server for predicting protein–protein interaction hot spots by using structural neighborhood properties. Nucleic Acids Res. 42, W290–W295. 10.1093/nar/gku437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymkowitz J.; Borg J.; Stricher F.; Nys R.; Rousseau F.; Serrano L. (2005) The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–W388. 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerois R.; Nielsen J. E.; Serrano L. (2002) Predicting Changes in the Stability of Proteins and Protein Complexes: A Study of More Than 1000 Mutations. J. Mol. Biol. 320, 369–387. 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- Dehouck Y.; Kwasigroch J. M.; Rooman M.; Gilis D. (2013) BeAtMuSiC: prediction of changes in protein–protein binding affinity on mutations. Nucleic Acids Res. 41, W333–W339. 10.1093/nar/gkt450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires D. E. V.; Ascher D. B.; Blundell T. L. (2014) mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30, 335–342. 10.1093/bioinformatics/btt691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedix A.; Becker C. M.; de Groot B. L.; Caflisch A.; Böckmann R. A. (2009) Predicting free energy changes using structural ensembles. Nat. Methods 6, 3. 10.1038/nmeth0109-3. [DOI] [PubMed] [Google Scholar]
- Fogha J.; Marekha B.; De Giorgi M.; Voisin-Chiret A. S.; Rault S.; Bureau R.; Sopková-de Oliveira Santos J. (2017) Towards understanding MCL-1 promiscuous and specific binding modes. J. Chem. Inf. Model. 57, 2885–2895. 10.1021/acs.jcim.7b00396. [DOI] [PubMed] [Google Scholar]
- Bradshaw R. T.; Patel B. H.; Tate E. W.; Leatherbarrow R. J.; Gould I. R. (2011) Comparing experimental and computational alanine scanning techniques for probing a prototypical protein–protein interaction. Protein Eng., Des. Sel. 24, 197–207. 10.1093/protein/gzq047. [DOI] [PubMed] [Google Scholar]
- Wright P. E.; Dyson H. J. (2015) Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 16, 18–29. 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J. M.; Wong C. T.; Clarke J. (2014) Coupled Folding and Binding of the Disordered Protein PUMA Does Not Require Particular Residual Structure. J. Am. Chem. Soc. 136, 5197–5200. 10.1021/ja4125065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicky B. I. M.; Shammas S. L.; Clarke J. (2017) Affinity of IDPs to their targets is modulated by ion-specific changes in kinetics and residual structure. Proc. Natl. Acad. Sci. U. S. A. 114, 9882–9887. 10.1073/pnas.1705105114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. E.; Chivian D.; Baker D. (2004) Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32, W526–W531. 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M. D. S.; Bava K. A.; Gromiha M. M.; Prabakaran P.; Kitajima K.; Uedaira H.; Sarai A. (2006) ProTherm and ProNIT: thermodynamic databases for proteins and protein–nucleic acid interactions. Nucleic Acids Res. 34, D204–D206. 10.1093/nar/gkj103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bava K. A.; Gromiha M. M.; Uedaira H.; Kitajima K.; Sarai A. (2004) ProTherm, version 4.0: thermodynamic database for proteins and mutants. Nucleic Acids Res. 32, D120–D121. 10.1093/nar/gkh082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortemme T.; Baker D. (2002) A simple physical model for binding energy hot spots in protein–protein complexes. Proc. Natl. Acad. Sci. U. S. A. 99, 14116–14121. 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortemme T.; Kim D. E.; Baker D. (2004) Computational alanine scanning of protein-protein interfaces. Sci. Signaling 2004, pl2. 10.1126/stke.2192004pl2. [DOI] [PubMed] [Google Scholar]
- O’Meara M. J.; Leaver-Fay A.; Tyka M. D.; Stein A.; Houlihan K.; DiMaio F.; Bradley P.; Kortemme T.; Baker D.; Snoeyink J.; Kuhlman B. (2015) Combined Covalent-Electrostatic Model of Hydrogen Bonding Improves Structure Prediction with Rosetta. J. Chem. Theory Comput. 11, 609–622. 10.1021/ct500864r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moal I. H.; Fernández-Recio J. (2012) SKEMPI: a Structural Kinetic and Energetic database of Mutant Protein Interactions and its use in empirical models. Bioinformatics 28, 2600–2607. 10.1093/bioinformatics/bts489. [DOI] [PubMed] [Google Scholar]
- McIntosh-Smith S.; Price J.; Sessions R. B.; Ibarra A. A. (2015) High performance in silico virtual drug screening on many-core processors. Int. J. High. Perform. C 29, 119–134. 10.1177/1094342014528252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar P. E.; Lee E. F.; van Delft M. F.; Day C. L.; Smith B. J.; Huang D. C. S.; Fairlie W. D.; Hinds M. G.; Colman P. M. (2007) Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc. Natl. Acad. Sci. U. S. A. 104, 6217–6222. 10.1073/pnas.0701297104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nhu D.; Lessene G.; Huang D. C. S.; Burns C. J. (2016) Small molecules targeting Mcl-1: the search for a silver bullet in cancer therapy. MedChemComm 7, 778–787. 10.1039/C5MD00582E. [DOI] [Google Scholar]
- Namanja A. T.; Li Y.-J.; Su Y.; Wong S.; Lu J.; Colson L. T.; Wu C.; Li S. S. C.; Chen Y. (2012) Insights into High Affinity Small Ubiquitin-like Modifier (SUMO) Recognition by SUMO-interacting Motifs (SIMs) Revealed by a Combination of NMR and Peptide Array Analysis. J. Biol. Chem. 287, 3231–3240. 10.1074/jbc.M111.293118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gareau J. R.; Lima C. D. (2010) The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 11, 861. 10.1038/nrm3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im Y. J.; Lee J. H.; Park S. H.; Park S. J.; Rho S.-H.; Kang G. B.; Kim E.; Eom S. H. (2003) Crystal Structure of the Shank PDZ-Ligand Complex Reveals a Class I PDZ Interaction and a Novel PDZ-PDZ Dimerization. J. Biol. Chem. 278, 48099–48104. 10.1074/jbc.M306919200. [DOI] [PubMed] [Google Scholar]
- Monteiro P.; Feng G. (2017) SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 18, 147. 10.1038/nrn.2016.183. [DOI] [PubMed] [Google Scholar]
- Tompa P.; Davey N. E.; Gibson T. J.; Babu M. M. (2014) A Million Peptide Motifs for the Molecular Biologist. Mol. Cell 55, 161–169. 10.1016/j.molcel.2014.05.032. [DOI] [PubMed] [Google Scholar]
- Nim S.; Jeon J.; Corbi-Verge C.; Seo M.-H.; Ivarsson Y.; Moffat J.; Tarasova N.; Kim P. M. (2016) Pooled screening for antiproliferative inhibitors of protein-protein interactions. Nat. Chem. Biol. 12, 275–281. 10.1038/nchembio.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles J. A., Hobor F., Taylor J., Tiede C., Rowell P. R., Trinh C. H., Jackson B., Nadat F., Kyle H. F., Wicky B. I. M., Clarke J., Tomlinson D. C., Wilson A. J., and Edwards T. A. (2019) Selective Affimers Recognize BCL-2 Family Proteins Through Non-Canonical Structural Motifs, bioRxiv 651364. https://www.biorxiv.org/content/10.1101/651364v1. [DOI] [PMC free article] [PubMed]
- Czabotar P. E.; Lessene G.; Strasser A.; Adams J. M. (2014) Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 15, 49–63. 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.