

Regulation of metabolism and virulence in C. burnetii is not well understood. Here, we show that C. burnetii small RNA 12 (CbsR12) is highly transcribed in the metabolically active large-cell variant compared to the nonreplicative small-cell variant. We show that CbsR12 directly regulates several genes involved in metabolism, along with a type IV effector gene, in trans. In addition, we demonstrate that CbsR12 binds to CsrA-2 in vitro and induces autoaggregation and biofilm formation when transcribed ectopically in Escherichia coli, consistent with other CsrA-sequestering sRNAs. These results implicate CbsR12 in the indirect regulation of a number of genes via CsrA-mediated regulatory activities. The results also support CbsR12 as a crucial regulatory component early on in a mammalian cell infection.
KEYWORDS: AhcY, CarA, CsrA, CvpD, MetK, Coxiella burnetii, noncoding RNA, posttranscriptional regulation, small RNA
ABSTRACT
Coxiella burnetii is an obligate intracellular gammaproteobacterium and zoonotic agent of Q fever. We previously identified 15 small noncoding RNAs (sRNAs) of C. burnetii. One of them, CbsR12 (Coxiella burnetii small RNA 12), is highly transcribed during axenic growth and becomes more prominent during infection of cultured mammalian cells. Secondary structure predictions of CbsR12 revealed four putative CsrA-binding sites in stem loops with consensus AGGA/ANGGA motifs. We subsequently determined that CbsR12 binds to recombinant C. burnetii CsrA-2, but not CsrA-1, proteins in vitro. Moreover, through a combination of in vitro and cell culture assays, we identified several in trans mRNA targets of CbsR12. Of these, we determined that CbsR12 binds and upregulates translation of carA transcripts coding for carbamoyl phosphate synthetase A, an enzyme that catalyzes the first step of pyrimidine biosynthesis. In addition, CbsR12 binds and downregulates translation of metK transcripts coding for S-adenosylmethionine synthetase, a component of the methionine cycle. Furthermore, we found that CbsR12 binds to and downregulates the quantity of cvpD transcripts, coding for a type IVB effector protein, in mammalian cell culture. Finally, we found that CbsR12 is necessary for expansion of Coxiella-containing vacuoles and affects growth rates in a dose-dependent manner in the early phase of infecting THP-1 cells. This is the first characterization of a trans-acting sRNA of C. burnetii and the first example of a bacterial sRNA that regulates both CarA and MetK synthesis. CbsR12 is one of only a few identified trans-acting sRNAs that interacts with CsrA.
IMPORTANCE Regulation of metabolism and virulence in C. burnetii is not well understood. Here, we show that C. burnetii small RNA 12 (CbsR12) is highly transcribed in the metabolically active large-cell variant compared to the nonreplicative small-cell variant. We show that CbsR12 directly regulates several genes involved in metabolism, along with a type IV effector gene, in trans. In addition, we demonstrate that CbsR12 binds to CsrA-2 in vitro and induces autoaggregation and biofilm formation when transcribed ectopically in Escherichia coli, consistent with other CsrA-sequestering sRNAs. These results implicate CbsR12 in the indirect regulation of a number of genes via CsrA-mediated regulatory activities. The results also support CbsR12 as a crucial regulatory component early on in a mammalian cell infection.
INTRODUCTION
Coxiella burnetii is a Gram-negative, obligate intracellular bacterium and etiological agent of Q fever in humans. Q fever most often manifests as an acute, flu-like illness, which in rare cases progress to potentially life-threatening endocarditis (1). C. burnetii undergoes a biphasic life cycle in which it alternates between a metabolically active, replicative large-cell variant (LCV) and a nonreplicative, spore-like small-cell variant (SCV) (2). Upon aerosol transmission of SCVs to a mammalian host, C. burnetii is primarily endocytosed by alveolar macrophages, after which it survives acidification of the host phagolysosome and metamorphoses to LCVs. C. burnetii then utilizes the fusion of its Coxiella-containing vacuole (CCV) with lysosomes and autophagosomes in order to expand the intracellular niche (3, 4). CCV expansion is dependent on C. burnetii protein synthesis, but independent of replication, so expansion of the CCV is facilitated by a repertoire of Dot/Icm effector proteins secreted by a type IV-B secretion system (T4BSS) (5, 6). Many Dot/Icm substrates have been identified in recent years (7) and shown to modulate the host inflammasome (8), influence autophagosomal/lysosomal fusion with the CCV by various mechanisms (9–13), and regulate the host transcriptome after localizing to the nucleus (14, 15). Little is known about regulation of C. burnetii’s T4BSS, although the PmrA response regulator has been shown to enhance synthesis of the T4BSS apparatus, as well as certain Dot/Icm substrates (16).
Bacterial sRNAs are small transcripts (<500 nucleotides [nt]) that usually do not code for functional proteins. Instead, they serve as cis- and/or trans-acting regulators through a variety of mechanisms (reviewed in reference 17). For example, cis-acting sRNAs are often coded antisense to a functional gene target. Upon transcription, the sRNA binds to the mRNA with perfect complementarity, usually culminating in RNase degradation of the target. This effectively limits the free mRNA molecules available for translation (reviewed in reference 18). Alternatively, trans-acting sRNAs are often coded in distant intergenic regions and bind to a variety of mRNAs through a more limited base-pairing mechanism involving a seed region of ca. 7 to 12 nt. Many trans-acting sRNAs have been discovered in bacteria since Escherichia coli MicF was first described in 1984 (19). These regulatory RNAs have been implicated in a variety of processes, including virulence (20), global regulation of transcription (21), iron homeostasis (22), protein degradation (23), and stress response (24, 25).
Typically, trans-acting sRNAs require assistance in “finding” their respective mRNA targets. In most bacteria, this is accomplished by the RNA chaperone Hfq, which binds to both sRNAs and mRNAs and plays the role of a molecular matchmaker (reviewed in reference 26). Hfq is not obligatory, however. For example, Staphylococcus aureus has several sRNAs but does not require Hfq protein for their activities (27). Similarly, C. burnetii does not have a readily apparent hfq gene, although this does not rule out the possibility of an atypical Hfq or some other novel RNA chaperone.
Some sRNAs act by binding to and titrating RNA-binding proteins, effectively sequestering them away from regulatory activities. For instance, C. burnetii codes for two homologs (CsrA-1 and CsrA-2) of the RNA-binding protein CsrA (RsmA), which has been shown to regulate metabolism, biofilm formation, and type 4 secretion in other bacteria (28–30). CsrA is, in turn, regulated by CsrA-binding sRNAs, termed CsrB/C (RsmY/Z). Classical CsrB/C sRNAs consist of a series of stem-loops containing exposed AGGA or ANGGA motifs that bind and sequester CsrA, effectively limiting its mRNA regulatory capabilities (31). Some RsmY/Z sRNAs, however, differ in the number of stem-loop regions containing CsrA-binding sites and can harbor far fewer motifs than the classical CsrB/C counterparts of E. coli (32, 33). The CsrA regulatory cascade has not been studied in C. burnetii, in large part due to the absence of readily discernible RsmY/Z sRNAs, although the CsrA regulon in Legionella pneumophila, a close relative of C. burnetii, has been extensively studied (34, 35).
A previous study by our group revealed 15 novel C. burnetii sRNAs that were differentially transcribed either in LCVs versus SCVs or in host cell infections versus growth in ACCM-2 medium (36, 37). Among these, CbsR12 was found to be markedly upregulated in the intracellular niche compared to ACCM-2. Northern blots also showed that CbsR12 was upregulated in SCVs versus LCVs in ACCM-2 and revealed two distinct sizes of the sRNA, suggesting that either an alternative transcription start site (TSS) or RNase processing of the sRNA was responsible. In a subsequent study, CbsR3 and CbsR13 were found to originate from transcribed loci of a selfish genetic element, termed QMITE1 (38). However, despite the identification and verification of several CbsRs, none has been functionally characterized to date.
In this study, we describe activities of a highly transcribed, infection-specific sRNA of C. burnetii, termed CbsR12. Our analyses show that CbsR12 binds to CsrA-2 but not CsrA-1 in vitro. We also establish that CbsR12 binds and upregulates carA (CBU_1282) and downregulates metK (CBU_2030) transcripts in trans. The bacterial carA gene codes for carbamoyl-phosphate synthetase (small) subunit A (CarA), which forms a heterodimer with carbamoyl-phosphate synthetase (large) subunit B (CarB). The CarAB complex catalyzes the first step in pyrimidine biosynthesis and is involved in arginine biosynthesis in some bacteria (39). The bacterial metK gene codes for S-adenosyl l-methionine (SAM) synthetase, an enzyme responsible for catalyzing production of SAM, the major donor of methyl groups during metabolism in prokaryotic cells. As a methyl donor, SAM affects DNA methylation and thus global transcription (40). It has also been implicated in virulence, being necessary for the production of N-acyl homoserine lactones involved in bacterial quorum sensing (reviewed in reference 41). We also implicate CbsR12 in expansion of the CCV, as its size is directly correlated with levels of the sRNA. Furthermore, we find that CbsR12 binds ahcY (CBU_2031) and cvpD (CBU_1818) transcripts, which are components of the methionine cycle and a T4BSS effector protein, respectively (9). Overall, this study highlights CbsR12 as a crucial component in early stages of a Coxiella infection.
RESULTS
CbsR12 is a principal non-rRNA/tRNA/tmRNA transcript during C. burnetii infection of Vero and THP-1 cells.
CbsR12 was first described as a highly transcribed, infection-specific sRNA that was upregulated in SCVs compared to LCVs when analyzed by Northern blotting (36). The impetus for our study came when we analyzed previous transcriptome sequencing (RNA-Seq) data (SRP041556) (36) by converting raw read data into transcripts per million (tpm), a normalized measure of gene expression (42). These results showed that CbsR12 was the most highly transcribed non-transfer-messenger RNA (non-tmRNA) transcript in both LCVs and SCVs during C. burnetii infection of African green monkey kidney epithelial cells (Vero cells). Additional data from LCVs obtained from a C. burnetii infection of monocytic THP-1 cells corroborates the observation that CbsR12 is a principal transcript during infection (R. Raghavan, unpublished data). Moreover, we were surprised to find that CbsR12 was more abundant in LCVs, not SCVs (Table 1).
TABLE 1.
Top ten expressed genes across various C. burnetii growth conditions, ranked
| Rank | Gene, RNA, or region (tpm)a
|
||||
|---|---|---|---|---|---|
| ACCM-2* LCV (3 dpi) | ACCM-2* SCV (21 dpi) | Vero LCV (3 dpi) | Vero SCV (21 dpi) | THP-1 LCV (3 dpi) | |
| 1 | tmRNA (104,383) | tmRNA (4,430) | tmRNA (153,422) | tmRNA (22,500) | tmRNA (128,928) |
| 2 | CBU_1183 (32,842) | Intergenic (2,632) | CbsR12 (79,870) | CbsR12 (8,521) | RNase P RNA (63,637) |
| 3 | CBU_0307 (13,902) | CBU_1538 (2,133) | RNase P RNA (25,715) | CBU_0089a (8,012) | CbsR12 (33,331) |
| 4 | CBU_1224a (11,023) | Intergenic (1,977) | CBU_0089a (13,882) | CBU_2034 (4,375) | 6S RNA (22,540) |
| 5 | RNase P RNA (10,3923) | tRNA (1,902) | CbsR1 (11,445) | CBU_1170 (3,704) | CBU_0456 (17,603) |
| 6 | CBU_0311 (7,544) | CBU_0183 (1,593) | CBU_0718 (10,370) | CBU_0718 (3,703) | CBU_0474 (16,267) |
| 7 | CBU_0474 (7,120) | Intergenic (1,543) | CBU_1932 (9,734) | CBU_1280a (3,522) | CBU_1183 (13,349) |
| 8 | Intergenic (6,928) | CBU_0157 (1,390) | CBU_1272 (9,011) | RNase P RNA (3,429) | CBU_0307 (12,753) |
| 9 | CBU_0306 (6,666) | Intergenic (1,340) | CBU_0711 (5,530) | Intergenic (3,340) | CBU_0311 (12,146) |
| 10 | CbsR12 (3,522) | Intergenic (1,203) | CBU_1170 (4,633) | CBU_1272 (3,134) | CBU_0473 (11,449) |
Genes as annotated for RSA493 (NCBI accession no. NC_002971.4) are listed, with the corresponding transcripts per million (tpm). *, ACCM-2 is an axenic growth medium (37). CbsR12 genes are indicated in boldface. n = 2 biological replicates per condition.
CbsR12 is processed by RNase III in vitro.
Previous Northern blot analyses showed that CbsR12 produced two bands of approximately 170 and 50 nt, regardless of C. burnetii growth conditions or developmental stage (36). We therefore sought to determine whether these RNAs arose from alternative TSSs for the cbsR12 gene or from RNase III processing of the full-length CbsR12 transcript. We first utilized 5′ rapid amplification of cDNA ends (RACE) with total RNA derived from C. burnetii LCVs infecting Vero cells (at 3 days postinfection [dpi]) in order to determine the TSS of CbsR12. This experiment revealed the full-size CbsR12 (∼200 nt), as expected, but also indicated that two potential alternative TSSs existed ∼110 nt upstream of the cbsR12 gene’s Rho-independent terminator (see Fig. S1A in the supplemental material). To determine whether the TSSs were generated by RNase-mediated cleavage, we treated in vitro-transcribed CbsR12 with recombinant C. burnetii RNase III (43) and commercially available E. coli RNase III (New England BioLabs). The results showed that CbsR12 was similarly processed by both kinds of RNase III into two RNA fragments with sizes that closely resembled those in the previously reported Northern blot analysis (Fig. S1B) (36). These results strongly suggest that the two sites are not alternative TSSs but are instead generated by RNase III processing.
CbsR12 binds to C. burnetii recombinant CsrA-2, but not CsrA-1, in vitro.
The predicted secondary structure of CbsR12 also revealed four conserved single-strand sequence motifs among the various stem-loop structures (Fig. S1A). This motif, AGGA/ANGGA, corresponds exactly to the conserved CsrA-binding motif of many bacteria (30, 32). C. burnetii contains two annotated and distinct types of CsrA (termed CsrA-1 [CBU_0024] and CsrA-2 [CBU_1050]) that share 65% primary sequence identity. To test the functionality of these domains, we used an in vitro binding assay and a RNA-protein electrophoretic mobility shift assay (EMSA) to determine whether CbsR12 binds to natively purified recombinant CsrA-1 (rCsrA-1) and/or rCsrA-2. The EMSA results clearly showed that CbsR12 binds to rCsrA-2, but not rCsrA-1, in vitro (Fig. 1). In addition, a CbsR10 negative-control sRNA did not bind either rCsrA. Furthermore, the Kd for rCsrA-2 was determined to be 130 nM, consistent with published values for CsrA-binding sRNAs (44).
FIG 1.

CbsR12 binds to CsrA-2, but not CsrA-1, protein in vitro. EMSAs show RNA-protein interactions between biotin-labeled, in vitro-transcribed CbsR12 (0 or 1 nM) and increasing concentrations of purified, rCsrA-1 (A) or rCsrA-2 (B). CbsR10 (at 0 or 1 nM; right of vertical black line) is included as a negative control, as the sRNA contains only a single discernible CsrA-binding motif.
A cbsR12 mutant shows prolonged lag phase in axenic media.
A cbsR12 mutant of C. burnetii (strain RSA439 with transposon 327 [Tn327], here referred to as MB-cbsR12), as well as an otherwise isogenic transposon insertional control strain (strain RSA439 with Tn1832, here referred to as MB-WT), was previously generated using a Himar1-based transposon system (11). The location of the transposon insertion of strain MB-cbsR12 is shown in Fig. S2A. We also constructed a transposon-directed complement of strain MB-cbsR12 (here referred to as MB-cbsR12-Comp) containing the wild-type cbsR12 gene plus ∼100 bp of 5′ and 3′ flanking sequences to include any potential transcriptional regulator element(s) that could influence cbsR12 expression. PCR was used to confirm the transposon insertions in the MB-cbsR12 and MB-cbsR12-Comp (Fig. S2B). Furthermore, we confirmed that the cbsR12 cassette inserted into an intergenic region between CBU_1788 and CBU_1789 (RSA493 [accession number NC_002971.4]), and we utilized copy number quantitative PCR (qPCR) to confirm the single insertional event in MB-cbsR12-Comp (Fig. S2C).
Next, we conducted growth curve analyses of the MB-WT, MB-cbsR12, and MB-cbsR12-Comp strains grown axenically in ACCM-2 (Fig. 2A; see also Fig. S3A and B) and assayed the production of CbsR12 at incremental time points from the LCV stage (ca. 1 to 6 days) through the SCV stage (≥7 days) by quantitative real-time PCR (qRT-PCR) (Fig. 2B). Growth curve results showed that MB-cbsR12 displayed a prolonged lag phase from 1 to 3 days postinoculation that was not observed in MB-WT or MB-cbsR12-Comp strains (Fig. 2A; see also Fig. S3A and B). After the lag phase, MB-cbsR12 grew at a slightly increased rate relative to the other strains (6 to 9 days dpi) but failed to reach cell numbers seen in the other two strains throughout the assay. The “wild-type” (MB-WT) and complemented (MB-cbsR12-Comp) strains produced essentially indistinguishable growth curves. The qRT-PCR results showed that the Tn insertion in MB-cbsR12 completely abrogated CbsR12 production (Fig. 2B). The results also confirmed that cbsR12’s increased expression in LCVs compared to SCVs as copies of CbsR12 per C. burnetii genome were highest at 3 days postinoculation.
FIG 2.

CbsR12 production and growth effects on C. burnetii grown in ACCM-2. (A) Growth curves for MB-WT, MB-cbsR12, and MB-cbsR12-Comp in ACCM-2 as determined by qPCR. The 0-dpi time point refers to the inoculum. Values represent means ± the standard error of means (SEM) from three technical replicates. The data are representative of three independent experiments with consistent and indistinguishable results. (B) CbsR12 production over time for MB-WT, MB-cbsR12, and MB-cbsR12-Comp grown in ACCM-2, as determined by qRT-PCR. Values represent means ± the SEM of three independent determinations.
CbsR12 impacts the intracellular replication of C. burnetii.
C. burnetii typically infects alveolar macrophages during human infection. We therefore infected a differentiated human monocyte cell line (THP-1) with MB-WT, MB-cbsR12, and MB-cbsR12-Comp strains. Comparative growth curves showed that MB-cbsR12 has a lower growth rate in exponential phase (1 to 3 dpi) compared to the two other strains and never attained the bacterial cell numbers seen in infections with MB-WT or MB-cbsR12-Comp (Fig. 3A; see also Fig. S3C and D). Furthermore, CbsR12 production in THP-1s correlated with replication efficiency of the individual strain. For example, production of CbsR12 in MB-WT and MB-cbsR12-Comp strains increased between 1 and 3 dpi, and CbsR12 levels directly correlated to growth rates of the respective strains between these two time points. However, we observed a dysregulation of CbsR12 in MB-cbsR12-Comp infecting THP-1 cells that was strikingly different from what we observed during axenic growth. Specifically, we observed a maintenance of CbsR12 production throughout the infection of THP-1s (Fig. 3B), whereas in axenic growth there was a progressive drop-off in synthesis after 3 dpi (Fig. 2B). These results suggest that cbsR12 expression differs in this host cell type and that a transcriptional regulatory motif may exist outside the bounds of the complementation insertion, resulting in dysregulation due to the genomic context of cbsR12 in MB-cbsR12-Comp compared to MB-WT in a THP-1 infection.
FIG 3.

CbsR12 production and growth effects on C. burnetii infecting THP-1 cells. (A) Growth curves for MB-WT, MB-cbsR12, and MB-cbsR12-Comp in THP-1 cells as determined by qPCR. The 0-dpi time point refers to the inoculum. Values represent means ± the SEM from three technical replicates. The data are representative of three independent experiments with consistent and indistinguishable results. (B) Cbsr12 production over time for MB-WT, MB-cbsR12, and MB-cbsR12-Comp infecting THP-1 cells, as determined by qRT-PCR. Values represent means ± the SEM from three independent determinations.
CCV size correlates with CbsR12 production in THP-1 infection.
To more closely examine bacterial-host cell interactions, we used immunofluorescence assays (IFAs) of C. burnetii infecting THP-1 cells. C. burnetii colonies and CCV boundaries were visualized using anti-Coxiella (anti-Com1 [45]) and anti-LAMP1 antibodies at both 3 dpi (late LCVs) and 7 dpi (SCVs). Here, we define a C. burnetii colony as multiple C. burnetii organisms inhabiting a LAMP1-decorated intracellular vacuole. LAMP1 is a host cell protein recruited to lysosomes and found on CCVs after lysosome fusion (46). We observed a robust infection at 3 dpi for MB-WT and MB-cbsR12-Comp strains, whereas the MB-cbsR12 strain only produced a few, small CCVs with relatively unclear boundaries (Fig. 4A). In contrast, the MB-cbsR12-Comp strain produced CCVs that were similar in size to those generated by the MB-WT strain, reflecting the trend observed in their respective growth curves (Fig. 3A). Quantitatively, differences in CCV sizes between MB-cbsR12 and the other two strains were significant at 3 dpi (Fig. 4B). However, by 7 dpi the CCVs were similar in size in MB-cbsR12 and MB-WT infections, indicating some compensatory or redundant mechanisms allowing for CCV expansion as the infection proceeded, even though MB-cbsR12 genome counts never reached wild-type levels (Fig. 3A). Interestingly, the MB-cbsR12-Comp strain formed consistently larger CCVs at 3 dpi (significantly greater than MB-WT and MB-cbsR12) and 7 dpi (significantly greater than MB-cbsR12), which meshes well with the sustained expression of cbsR12 evidenced throughout the course of infection (Fig. 3B). Taken as a whole, these results suggest that CbsR12 is important for optimum growth and establishment of CCVs early in the course of infection of THP-1 cells, and CbsR12 can influence CCV expansion throughout a THP-1 infection.
FIG 4.

CbsR12 affects CCV expansion in infected THP-1 cells. (A) Representative IFAs of MB-WT, MB-cbsR12, and MB-cbsR12-Comp infecting THP-1 cells at 3 and 7 dpi. C. burnetii was probed with anti-Com1 antibodies coupled to Alexa Fluor 488 (green), CCV boundaries were labeled with anti-LAMP1 antibodies coupled to rhodamine (red), and host cell nuclei were labeled with DAPI (blue). Scale bars, 20 μm. (B) Sizes of individual CCVs in log10 (pixels) for MB-WT, MB-cbsR12, and MB-cbsR12-Comp. Measurements were taken from 46 individual images of random fields of view spanning three different experiments for each C. burnetii strain. Crossbars represent means ± the SEM (**, P < 0.01, one-way analysis of variance).
CbsR12 binds to carA, metK, and cvpD transcripts in vitro.
Although CbsR12 was identified as a CsrA-binding sRNA, nothing was known about the CsrA regulon in C. burnetii, making it difficult to ascribe intracellular phenotypes to regulation by CsrA. Therefore, we wanted to determine whether CbsR12 could act by regulating mRNAs in trans. To identify potential mRNA targets of CbsR12, we first used three in silico sRNA target discovery algorithms. Each algorithm takes into consideration the extent of sRNA-mRNA hybridization, conservation of the sRNA, and the accessibility of both the sRNA and its target, although TargetRNA2 and IntaRNA (v2) prioritize accessibility, whereas CopraRNA prioritizes comparative interaction predictions among different strains of the indicated bacterium. From these search results, we omitted genes annotated as hypothetical and chose cvpD, metK, carA, purH, rpsA, and dnaA as potential targets based on conserved predictions (Table 2). To get a sense of CbsR12’s ability to bind to these potential mRNA targets, we next performed an RNA-RNA hybridization, followed by EMSAs. The results clearly showed that CbsR12 bound to carA, metK, and cvpD transcripts in vitro but did not interact with rpsA, purH, or dnaA mRNAs (Fig. 5). We further tested CbsR12’s specificity for these transcripts by performing dose-dependent and unlabeled-chase experiments. The results of the EMSA analyses showed that CbsR12 specifically bound carA, metK, and cvpD transcripts in a dose-dependent manner (Fig. S4).
TABLE 2.
CbsR12 target prediction using various algorithms
| Rank | Target gene (P)a
|
||
|---|---|---|---|
| TargetRNA2 | IntaRNA | CopraRNA | |
| 1 | CBU_1041 (0.001) | cvpD (0.000017) | cvpD (0.00001) |
| 2 | prlC (0.001) | CBU_0537 (0.00018) | CBU_0537 (0.000039) |
| 3 | suhB (0.001) | CBU_1161 (0.00018) | CBU_0922 (0.00053) |
| 4 | mutS (0.002) | CBU_0922 (0.00091) | CBU_0103 (0.00074) |
| 5 | bioD (0.003) | CBU_2028 (0.0012) | CBU_2028 (0.00078) |
| 6 | trmD (0.003) | CBU_0103 (0.0013) | rpsA (0.0013) |
| 7 | dnaA (0.005) | rpsA (0.0022) | metK (0.0014) |
| 8 | yciL (0.007) | metK (0.0023) | purH (0.0019) |
| 9 | metK (0.008) | purH (0.003) | CBU_1741 (0.0025) |
| 10 | CBU_0558 (0.01) | carA (0.015) | carA (0.0067) |
Target genes were identified through the indicated algorithms, as ranked. The calculated P values for each target are indicated. Genes involved in subsequent experiments are indicated in boldface type.
FIG 5.
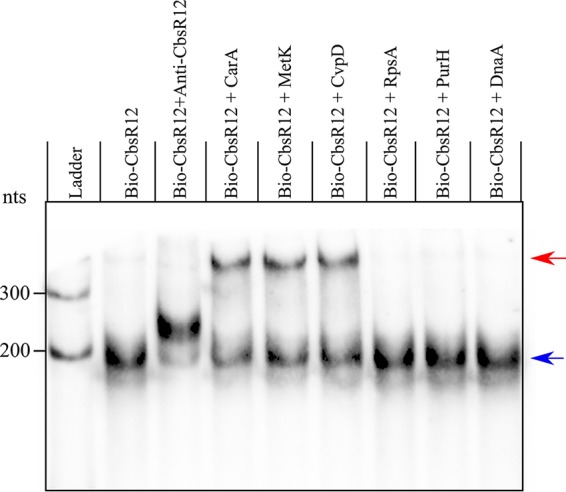
CbsR12 targets carA, metK, and cvpD transcripts in vitro. RNA-RNA EMSA results show hybridization reactions with 10 nM biotin-labeled CbsR12 and 5 nM in vitro-transcribed segments of carA, metK, cvpD, purH, rpsA, or dnaA. Anti-CbsR12 represents a 10 nM positive control consisting of a transcript equal in size, but antisense, to the CbsR12 transcript. Arrows indicate unbound bio-CbsR12 (blue) and bio-CbsR12 bound to RNA targets (red).
CbsR12 binds to metK, carA, cvpD, and ahcY transcripts in C. burnetii cells.
To determine the CbsR12 targetome within C. burnetii cells, we used a Crosslink-Seq technique previously used to detect intracellular mRNA targets of E. coli sRNAs (47). For this procedure, we used C. burnetii LCVs grown in ACCM-2 to produce sufficient volumes to capture CbsR12 target RNAs for cDNA library preparation and RNA-Seq analysis. Hybridized RNAs in lysates from both MB-WT and MB-cbsR12 strains were chemically cross-linked, captured by anti-CbsR12 probes, and analyzed by RNA-Seq in order to identify RNAs enriched in MB-WT compared to MB-cbsR12. The Crosslink-Seq results confirmed that CbsR12 targeted carA, metK, and cvpD transcripts in C. burnetii cells (Fig. 6), as demonstrated in vitro (Fig. 5). We also discovered an additional mRNA target, ahcY, coding for adenosylhomocysteinase, another component of the methionine cycle. Interestingly, ahcY was also predicted as an mRNA target by IntaRNA, although the P value was not significant (P = 0.13). In addition, ahcY is in an operon with and downstream of metK. To address whether ahcY is actually a target of CbsR12 or whether it is a result of CbsR12’s binding to a polycistronic mRNA, we used the Artemis genome browser to observe Crosslink-Seq reads aligned to the C. burnetii RSA439 genome. This analysis showed distinct segments of these genes to which the captured reads mapped, suggesting that they are separate binding events (Fig. S5B). We also confirmed the other identified targets by the same method (Fig. S5A and C).
FIG 6.

Volcano CbsR12 Crosslink-Seq plot highlights mRNAs that occur at different levels between Crosslink-Seq experiments with strains MB-WT and MB-cbsR12. CbsR12 targets several C. burnetii transcripts, including those of metK, carA, and cvpD. Labeled transcripts are indicated by green dots and are significantly enriched in MB-WT versus MB-cbsR12, identifying them as targets of CbsR12. Black dots represent transcripts not significantly different between the strains tested. Orange dots represent transcripts having a log2 fold change of >1 but a false discovery rate (FDR) of >0.05. There were no transcripts indicated by red dots, which would represent mRNAs with an FDR of <0.05 but a log2 fold change in levels of <1. Data shown are representative of two biological replicates each of MB-WT and MB-cbsR12 Crosslink-Seq experiments. The potential CbsR12-binding site in the coding region of the ahcY transcript is indicated in the inset.
Next, we wanted to determine whether CbsR12-mediated regulation of predicted in trans mRNA targets would occur independently of CsrA. To this end, we searched for potential CsrA-binding (AGGA/ANGGA) motifs within the 100 bases up- and downstream of the start codons of carA, metK, cvpD, and ahcY. This search showed that carA, metK, and cvpD contained single potential CsrA-binding sites, whereas cvpD had none. Moreover, the motifs of carA and metK did not occur in predicted ribosome-binding sites (RBS), suggesting that CsrA is unlikely to regulate the corresponding transcripts (35) (Table 3). Thus, CbsR12 regulation of carA, metK, and cvpD transcripts likely occurs as a direct result of in trans binding by CbsR12 and independently of CsrA. In contrast, the ahcY sequence has two potential regulatory ANGGA CsrA-binding sites, so we cannot exclude the possibility of indirect regulatory effects caused by CbsR12-mediated sequestering of CsrA. As such, we did not further explore CbsR12-mediated regulation of ahcY.
TABLE 3.
Predicted CsrA motifs in CbsR12 targets
| CbsR12 target gene | Locus taga | Base rangeb | No. of CsrA motifsc | Position(s) of motifd |
|---|---|---|---|---|
| carA | CBU_1282 | 1234872–1235073e | 1 | +71 to +75 |
| metK | CBU_2030 | 1936983–1937183 | 1 | –12 to –8 |
| cvpD | CBU_1818 | 1748916–1749116 | 0 | NA |
| ahcY | CBU_2031 | 1938166–1938366 | 2 | +1 to +5, +13 to +17 |
Locus tags are as annotated for RSA493 (NCBI accession no. NC_002971.4).
Base range indicates 100 nt up- and downstream of the annotated start codon for RSA493 (NCBI accession no. NC_002971.4).
The consensus CsrA-binding motifs are AGGA or ANGGA.
That is, the position(s) of the CsrA-binding motifs relative to the first nucleotide of the start codon (+1). NA, not applicable.
Gene located on complementary strand.
CbsR12 negatively affects the quantity of cvpD transcripts and regulates the synthesis of CarA and MetK.
Next, we set out to determine whether CbsR12 regulates carA, metK, and cvpD transcripts in C. burnetii. First, we performed 5′ RACE on the three transcripts in total RNA extracted from LCVs infecting mammalian cells (3 dpi). 5′ RACE results for the MB-WT cvpD gene indicated three apparent TSSs, including a TSS for the full-length transcript, several questionable “TSSs” within the CbsR12-binding site, and an alternative TSS for a short transcript downstream of the CbsR12-binding site and with its own predicted promoter element (Fig. S6A). Interestingly, putative RBSs and start codons exist downstream of TSSs for both the full-length and the short transcripts. Moreover, the two start codons are in-frame with each other, and the existence of putative RBSs supports the possibility that translation occurs from both elements. The questionable “TSSs” within the CbsR12-binding region likely result from CbsR12-mediated RNase III degradation of cvpD mRNA, because 5′ RACE results for MB-cbsR12, a strain that lacks CbsR12, did not produce TSSs in this region. We predict that CbsR12 downregulates production of full-length CvpD since the CbsR12-binding site occurs in the coding region. However, CbsR12 would predictably have no effect on production of the putative truncated CvpD, since the CbsR12-binding site occurs upstream of the alternative TSS (Fig. S6A and B).
To determine whether CbsR12 binds to and causes degradation of full-length cvpD transcripts, we performed qRT-PCR on MB-WT, MB-cbsR12, and MB-cbsR12-Comp LCVs obtained from infected THP-1 cells. These results clearly showed that the absence of CbsR12 in strain MB-cbsR12 led to a significant increase in full-length cvpD transcripts in LCVs (3 dpi) (Fig. S6C). At 7 dpi, MB-WT and MB-cbsR12 levels were not significantly different, presumably due to reduced CbsR12 production in MB-WT SCVs (see Fig. 3B). However, cvpD expression was significantly lower in MB-cbsR12-Comp, most likely due to the maintained production of CbsR12 in the strain’s SCVs (see Fig. 3B). Whether or not two different forms of CvpD are produced from cvpD is unknown, although it appears that CbsR12 may negatively regulate the full-length cvpD transcript.
In order to determine whether CbsR12 binds to and regulates carA and metK in a cellular environment, we devised a reporter assay in E. coli that measures the effects of CbsR12 production on translation of carA-luc or metK-luc fusion constructs. 5′ RACE results for both MB-WT and MB-cbsR12 strains revealed that carA has two potential TSSs, a finding that is consistent with transcription of E. coli carA (39). Based on the position of the TSSs, CbsR12 could only regulate the full-length carA transcript and not the shorter mRNA, whose transcription starts immediately upstream of the RBS and downstream of the CbsR12-binding site (Fig. 7A). From these results, we hypothesized that CbsR12 binds to the 5′ untranslated region (UTR) of carA and upregulates translation by relieving the secondary structure that occludes the predicted RBS (Fig. 7A and B). The results of the E. coli reporter assay confirmed our hypothesis, because translation of luciferase enzyme from a carA-5′ UTR-luc fusion was significantly upregulated in the presence of CbsR12 relative to a strain lacking the sRNA (Fig. 7C).
FIG 7.

CbsR12 targets and upregulates translation of a carA-luciferase fusion construct. (A) Secondary structure of the carA 5′ UTR as predicted by Mfold. Red asterisks indicate TSSs for the shorter transcripts as determined by 5′ RACE. (Nucleotide 1 was determined to be the TSS for the full-length transcript by 5′ RACE.) Colored lines represent the start codon (green), the predicted RBS (red), and the determined CbsR12-binding sites (blue). (B) Representation of CbsR12 binding to the carA transcript as determined by IntaRNA, with the respective base numbers indicated. The top strand in the model represents the carA sequence, while the bottom strand represents the complementary CbsR12 sequence. (C) carA-luc reporter assay indicating relative luminescence units produced by pBESTluc constructs with (i) no luciferase production (frameshifted luciferase), (ii) pBESTluc vector (pBEST), (iii) pBESTluc with the carA 5′ UTR upstream of luc but lacking cbsR12 (pBEST+carA 5′ UTR), and (iv) carA 5′ UTR upstream of luc plus the cbsR12 gene driven by a Ptac promoter (pBEST+carA 5′ UTR+cbsR12). Values represent means ± the SEM from three independent determinations (*, P < 0.05 [Student t test]).
In contrast, CbsR12 was predicted to downregulate MetK translation by binding to the coding region of the transcript, immediately downstream of its start codon (Fig. 8A). As is often the case with this type of sRNA-mediated regulation, RNase III would likely be recruited and the metK transcript cleaved, resulting in downregulation of the encoded protein product. Unexpectedly, 5′ RACE analyses of metK mRNA also identified apparent alternative “TSSs” within the CbsR12-binding region, suggesting that the truncated mRNAs resulted from CbsR12-mediated RNase III processing (Fig. 8A and B). Indeed, 5′ RACE analysis of RNA from strain MB-cbsR12 infecting THP-1 cells did not detect the “TSSs,” suggesting they are a product of RNase III processing. The results of the reporter assays in E. coli confirmed our hypothesis, since the presence of CbsR12 significantly downregulated translation of luciferase from the metK-luc fusion construct compared to a strain lacking the sRNA (Fig. 8C).
FIG 8.

CbsR12 targets and downregulates translation of a metK-luciferase fusion construct. (A) Secondary structure of the metK 5′ UTR and initial coding sequence as predicted by Mfold. Asterisks indicate apparent alternative “TSSs” determined by 5′ RACE. (Nucleotide 1 was determined to be the TSS for the full-length transcript by 5′ RACE.) Colored lines represent the start codon (green), a predicted RBS (red), and the determined CbsR12-binding site (blue). (B) Representation of CbsR12 binding to the metK transcript, as determined by IntaRNA, with the base numbers indicated. The top strand in the model represents the metK sequence, while the bottom strand represents the complementary CbsR12 sequence. (C) metK-luc reporter assay indicating relative luminescence units produced by pBESTluc constructs with (i) no luciferase production (frameshifted luciferase), (ii) pBESTluc vector (pBEST), (iii) pBESTluc with the CbsR12 binding site cloned in frame into luc but lacking the cbsR12 gene (pBEST+metK), and (iv) pBESTluc with the CbsR12 binding site cloned in frame into luc plus the cbsR12 gene driven by a Ptac promoter (pBEST+metK+cbsR12). Values represent means ± the SEM from three independent determinations (*, P < 0.05 [Student t test]).
Although we determined that CbsR12 targets carA and metK transcripts in vitro and in E. coli, we were curious whether the absence of CbsR12 would also result in differential amounts of CarA and MetK proteins in C. burnetii. To this end, we performed Western blots with polyclonal antibody generated against recombinant C. burnetii CarA and MetK. As predicted, when proteins from whole-cell lysates of MB-WT, MB-cbsR12, and MB-cbsR12-Comp strains were compared, we found that CarA was synthesized in MB-WT and MB-cbsR12-Comp strains at comparable levels but was undetectable in protein profiles of strain MB-cbsR12 (Fig. 9A; see also Fig. S7A). In sharp contrast, MetK was highly synthesized in strain MB-cbsR12 but was produced at relatively lower and comparable levels in the MB-WT and MB-cbsR12-Comp strains (Fig. 9B; see also Fig. S7B).
FIG 9.
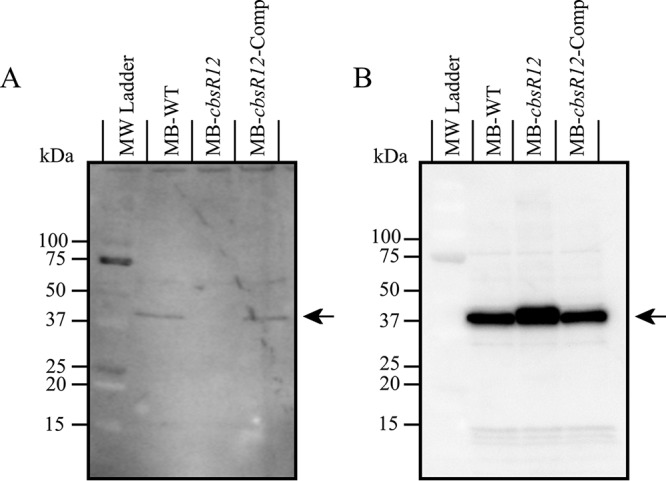
CarA and MetK proteins are differentially synthesized in MB-WT, MB-cbsR12, and MB-cbsR12-Comp strains. (A) Proteins (30 μg total) from MB-WT, MB-cbsR12, and MB-cbsR12-Comp LCVs (mid-log phase; 96 h for MB-WT and MB-cbsR12-Comp and 144 h for MB-cbsR12) grown in ACCM-2 were resolved on a 10 to 20% acrylamide gradient SDS-PAGE gel, blotted, probed with rabbit anti-CarA antibodies, and detected with chemiluminescence. The arrow indicates CarA. (B) Proteins (60 μg total) from MB-WT, MB-cbsR12, and MB-cbsR12-Comp LCVs (mid-log phase; 96 h for MB-WT and MB-cbsR12-Comp and 144 h for MB-cbsR12) grown in ACCM-2 were resolved on 10 to 20% acrylamide gradient SDS-PAGE gels, blotted, probed with rabbit anti-MetK antibodies, and detected using chemiluminescence. The arrow indicates MetK.
DISCUSSION
In this report, we show that CbsR12, a multifunctional sRNA that binds to mRNAs and to the regulatory protein CsrA-2, is important for proper Coxiella replication and CCV expansion during infection of human macrophage-like THP-1 cells. Induction of cbsR12 expression in mammalian cell culture versus in vitro conditions (Table 1) led us to label CbsR12 as “infection specific” and, as a result, we hypothesized that it played an important regulatory role in infection of host cells. The cbsR12 sequence is conserved among all C. burnetii strains sequenced to date, underscoring the potential for an important regulatory role, but from an evolutionary viewpoint. Interestingly, though, the cbsR12 gene is missing or degenerate in Coxiella-like endosymbionts (48), suggesting that the sRNA is important for a mammalian infection but is dispensable in endosymbionts that reside in arthropods.
Interestingly, CbsR12 binds rCsrA-2, but not rCsrA-1, in a dose-dependent manner, in vitro. Why CsrA-1 does not bind to consensus motifs present in CbsR12 is unclear, especially since both CsrA-1 and CsrA-2 maintain the critical L4 and R44 RNA-binding residues, although CsrA-1 has these residues at L4 and R46 (49). There are several examples of pathogens harboring multiple copies of CsrA (50, 51). For example, RsmF of P. aeruginosa is a homolog of RsmA (CsrA) and functions by binding a subset of mRNAs that RsmA also binds (51). However, an rsmF mutant did not display a phenotype during infection (51). Similarly, In C. burnetii, a transposon-mediated csrA-1 mutant was shown to have no intracellular phenotype (12). It is also conceivable that CsrA-1 diverged during C. burnetii’s adaptation to an intracellular lifestyle and is no longer functional. It is also possible that CsrA-1 binds to a noncanonical motif not present in CbsR12, although such CsrA homologs have not been described, to our knowledge. Regardless, it is necessary to examine the CsrA-1 and CsrA-2 regulons in order to determine their respective roles during infection.
In L. pneumophila, a close relative of C. burnetii, successful infection depends on a LetAS-RsmYZ-CsrA regulatory cascade. LetAS is a two-component system (TCS) that regulates the production of two sRNAs, RsmY and RsmZ, which in turn act as RNA “sponges” that soak up CsrA and modulate its activity (35). We determined that CbsR12 possesses only four CsrA-binding sites, similar to the RsmY/Z sRNAs of L. pneumophila. Interestingly, L. pneumophila RsmY/Z was implicated in the formation of cell aggregates and biofilms when the sRNAs were ectopically overproduced in E. coli, mimicking the effects of E. coli’s own CsrA-binding sRNAs (33). Likewise, when we overproduced CbsR12 in E. coli reporter assays (Fig. 7 and 8), we observed a similar autoaggregative phenotype (see Fig. S8A in the supplemental material). Moreover, CbsR12 induced biofilm production in E. coli, reflecting a CsrA-depleting phenotype (Fig. S8B). Together, these results suggest that CbsR12 is a CsrA-sequestering, RsmY/Z-like sRNA, although further research is necessary to determine the exact influence of CbsR12 on the regulatory roles of CsrA-2.
C. burnetii has several potential transcription factors that are known to upregulate bacterial expression, including integration host factor (IHF) (52), response regulator PmrA (53), and transcription factor DksA (54). However, only PmrA has been well studied, to date (16). It is interesting that the cbsR12 gene contains a close approximation to a PmrA-binding site (consensus sequence [with less-conserved nucleotides in lowercase]: cTTAA-N2-TT-N2-cTTAA) (55) immediately upstream of its predicted −10 promoter element (cbsR12 sequence: gTTTA-N2-TT-N1-gTTAA). However, the presence of this sequence does not explain the prolonged CbsR12 production observed during a THP-1 infection by MB-cbsR12-Comp since the putative PmrA-binding sequence is present in the cbsR12 cassette that was inserted. We predict that expression of cbsR12 is regulated by an unidentified TCS in a fashion similar to the L. pneumophila LetAS TCS regulation of RsmYZ sRNAs (35). In fact, the LetAS TCS is not unlike the GacAS TCS involved in RsmYZ-CsrA cascades of other bacteria (56). C. burnetii codes for four different GacA response regulators that could bind upstream elements of cbsR12 and regulate its expression (57). This upstream regulator may, in turn, help to explain the dysregulation of expression seen in MB-cbsR12-Comp during infection of THP-1 cells (Fig. 3B) that is not apparent during axenic growth (Fig. 2B). Alternatively, expression of cbsR12 could be upregulated by PmrA, and some other regulator may be involved in its downregulation, in conjunction with RNase III-mediated decay (see Fig. S1). Together, these would aid in suppression of CbsR12 as the LCV-to-SCV transition occurs, effectively freeing sequestered CsrA-2 to regulate the fate of target transcripts.
It is worth noting that we have identified a second sRNA, Coxiella burnetii small RNA 1 (CbsR1), that possesses five putative CsrA-binding sites with an ANGGA motif (Fig. S9B) (36). Similar to Cbsr12, CbsR1 is also produced at high levels in LCVs infecting Vero cells (Table 1). Furthermore, cbsR1 harbors a putative LetA-binding site similar to that of L. pneumophila RsmY (Fig. S9A) (33). Together, these observations suggest that CbsR1 and CbsR12 may represent orthologs of RsmYZ, although further exploration of CbsR1 and its cooperativity with CbsR12 is required. If CbsR1 does indeed serve as a CsrA-binding sRNA, its potential, redundant regulatory role may help to explain why MB-cbsR12 CCVs expanded to wild-type sizes as the infection progressed (Fig. 4B).
We also found that CbsR12 binds carA transcripts (Fig. 5 and 7) and upregulates the production of C. burnetii CarA (Fig. 9A). Pyrimidine metabolism in C. burnetii presumably requires CarAB to catalyze the conversion of l-glutamine into carbamoyl phosphate and glutamate, since it is unable to shunt this process through the arginine dihydrolase pathway; C. burnetii apparently lacks the necessary enzymes (58). CbsR12-mediated upregulation of CarA in LCVs would result in increased production of pyrimidines that the pathogen requires for robust intracellular growth.
In E. coli, carA expression is tightly controlled by a series of transcriptional regulators and the two distinct promoters that are regulated by feedback from arginine and pyrimidines (39). 5′ RACE analysis showed two distinct TSSs for C. burnetii carA mRNA, with the full-length transcript containing two CbsR12-binding sites and a shorter putative transcript lacking the site (Fig. 7A). We do not believe that the shorter, alternative TSS is due to RNase III-mediated degradation resulting from CbsR12 binding because this alternative TSS remained in 5′ RACE analysis of the MB-cbsR12 strain. We do not know conditions under which the shorter transcript is produced, but it may involve feedback from arginine/pyrimidine in accordance with carA regulation in E. coli.
We also determined that CbsR12 binds to metK transcripts and downregulates production of C. burnetii MetK protein (Fig. 5, 8, and 9B). MetK is a key component of the methionine cycle, which converts methionine to SAM via MetK, SAM to S-adenosylhomocysteine (SAH) via various methylases, SAH to homocysteine via AhcY, and homocysteine to methionine via MetH/MetE. Cells produce homocysteine as an input molecule through a series of reactions involving activated homoserines (reviewed in reference 59). C. burnetii is a semiauxotroph for methionine, since it can potentially grow without methionine in axenic media, albeit at a lower growth rate (60). Interestingly, C. burnetii lacks several components of the methionine synthesis pathway, most notably the ability to produce activated homoserines. Most bacteria activate homoserine through addition of an O-succinyl group catalyzed by MetA or an O-acetyl group catalyzed by MetX (reviewed in reference 59). C. burnetii apparently lacks genes coding for these enzymes. An ABC methionine transporter has been hypothesized (60) but not verified in Coxiella. If this is indeed a functional transporter, CbsR12’s negative regulation of metK transcripts makes sense in the context of the sRNA’s high level in LCVs, because any amount of scavenged methionine would be critical to growth. Shifting the equilibrium from SAM synthesis to methionine retention would be necessary as C. burnetii rapidly produces proteins to expand its intracellular niche.
SAM is a major methyl donor, is necessary for regulation of numerous enzymes, and has been implicated as a major contributor to virulence (61, 62). Some bacteria lack metK and instead transport SAM directly (63). There are many uncharacterized transporters encoded in the C. burnetii genome, so it is conceivable that a SAM transporter is present (57). This would allow for SAM scavenging even when MetK production is downregulated by CbsR12. Furthermore, if SAM transport occurs, C. burnetii could synthesize methionine without having to scavenge it, since the amino acid can be synthesized from SAM without activated homoserine via the methionine cycle.
We also determined that CbsR12 actively targets ahcY transcripts (Fig. 6). AhcY is a component of the methionine cycle and catalyzes conversion of SAH into homocysteine and adenosine. Based on the location of the CbsR12-binding site in the coding region of the ahcY transcript (Fig. 6, inset), we predict that CbsR12 negatively regulates AhcY translation. The underlying reason for this negative regulation is unknown, although it could help to suppress adenosine and/or homocysteine accumulation in LCVs.
cvpD mRNA was also identified as a CbsR12 target through Crosslink-Seq (Fig. 6), and this was confirmed by RNA-RNA hybridization/EMSA and qRT-PCR analyses (Fig. 5; see also Fig. S6C). In this study, we found that CbsR12 was necessary for CCV expansion in early stages of a THP-1 infection. The mechanism for this is unclear, although it may involve regulation of cvpD, which is required for C. burnetii’s intracellular replication and CCV expansion in infected THP-1 and HeLa cells (9). CbsR12 is predicted to target the coding region of the cvpD transcript and would negatively regulate translation. However, in the context of cbsR12’s expression pattern, this is an unclear association, since one would expect upregulation of CvpD synthesis at a time when CbsR12 is highly produced in LCVs. However, 5′ RACE analysis of cvpD transcripts in MB-WT and MB-cbsR12 provides a potential explanation, since an alternative cvpD promoter downstream of the CbsR12-binding site occurs that also possesses a putative RBS and start codon (see Fig. S6A). From these results, we hypothesize that there are two gene isoforms of cvpD that are transcribed and differentially expressed depending on the C. burnetii morphotype. Due to high expression of cbsR12 in LCVs, the longer cvpD transcript isoform would be downregulated by RNase III. As expression of cbsR12 decreases as the infection proceeds, the longer transcript isoform would accumulate. qRT-PCR data support this explanation, since a lack of CbsR12 in MB-cbsR12 significantly increased the quantity of long cvpD isoform transcripts (see Fig. S6C). This hypothesis could be confirmed if the two putative CvpD products could be identified and distinguished.
The 2007 to 2010 Dutch outbreak involving C. burnetii yielded several newly annotated genomes specific to that epidemic (64). Curiously, 7 of 13 strains analyzed contained a frameshift deletion in cvpD, leading to premature stop codons (64). Among these, strains 18430 (NZ_CP014557.1), 14160-001 (NZ_CP014551.1), 701CbB1 (NZ_CP014553.1), and 2574 (NZ_CP014555.1) had single-base deletions that only affected the long cvpD isoform. These strains were isolated from aborted placentas of ruminants and cattle in the Netherlands and France (64). Additional related strains include Heizberg (NZ_CP014561.1), Henzerling (NZ_CP014559.1), and RSA 331 (NC_010117.1). These strains, which were isolated from patients with acute Q fever in northern Italy and Greece in the mid-1900s, harbored 4-bp frameshift deletions near the middle of the cvpD coding region, affecting both long and short cvpD isoforms and introducing premature stop codons (64, 65). Apparently, CvpD was dispensable for virulence in the latter strains, whereas the Dutch outbreak strains harbored intact cvpD genes, or cvpD genes with a 1-bp frameshift deletion only affecting the longer gene isoform. Thus, in these Dutch isolate strains it appears that CbsR12 regulation of cvpD is dispensable. Granted, there are many genotypic differences between RSA439 and the Dutch isolates (64), and some compensatory mechanism(s) may exist for the absence of cvpD. Alternatively, cvpD may be necessary during infection of human cell lines and dispensable in host-animal infections. Regardless, the role and regulation of the CvpD effector requires further investigation.
cbsR12’s high level of expression during infection likely facilitates and regulates the many functions we have described. CbsR12 not bound to CsrA-2 presumably acts in trans to facilitate efficient replication through translational upregulation of CarA and downregulation of MetK and perhaps potentiates expansion of the CCV by means of cvpD transcript regulation. Furthermore, regulation is most likely independent of CsrA because these genes lack multiple CsrA-binding sites necessary for regulation (Table 3). It is worth noting that the CsrA-binding sites of CbsR12 do not overlap the metK and cvpD binding sites. Hence, it is feasible that CbsR12 may still regulate metK and cvpD in trans while bound to CsrA-2; in fact, a chaperone-like function such as this has recently been ascribed to CsrA (66).
CbsR12 is one of only a few identified trans-acting sRNAs that also binds CsrA (reviewed in reference 67). We hypothesize that CbsR12’s role in regulating C. burnetii replication and CCV expansion is due to a combination of in trans mRNA (metK, carA, and cpvD) targeting and regulation of CsrA-2 function. Our lab is currently working to elucidate the CsrA-1/CsrA-2 regulons, along with regulation of the putative CbsR12-CsrA-2 cascade of C. burnetii and the nature of CsrA-CbsR1 binding to clarify the interplay between CbsR12’s roles as a trans-acting and CsrA-sequestering sRNA.
MATERIALS AND METHODS
Bacterial strains, cell lines, and growth conditions.
The strains, primers, and plasmids used in this study are listed in Fig. S10 in the supplemental material. E. coli was grown in lysogeny broth (LB) supplemented with ampicillin (100 μg/ml) or kanamycin (50 μg/ml), as needed. When necessary, overnight cultures were expanded to 100 ml of LB, grown for 2 h, and then supplemented with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for induction. C. burnetii Nine Mile phase II (strain RSA439, clone 4), MB-WT, MB-cbsR12, and MB-cbsR12-Comp were grown in ACCM-2 medium (37) supplemented with ampicillin (5 μg/ml) or kanamycin (350 μg/ml) at 5% CO2, 2.5% O2, and 92.5% N2 and at 37°C and 100% humidity with continuous shaking at 75 rpm (37). SCVs collected from Vero cells were used for all C. burnetii infections and growth curve experiments. Briefly, C. burnetii was used to infect Vero cell monolayers for 7 days at 5% CO2 and 37°C; the cultures were then removed to room temperature, and the flask lids were tightened and covered for two additional weeks (68). After this, SCVs were harvested with digitonin, as previously described (69).
African green monkey kidney (Vero) epithelia (CCL-81; American Type Culture Collection [ATCC]) and human monocytic leukemia (THP-1) cells (TIB-202; ATCC) cell lines were maintained in RPMI medium (Gibco) supplemented with 10% fetal bovine serum (FBS; RMBIO) in a humidified atmosphere with 5% CO2 at 37°C. THP-1 cells were differentiated to macrophages by supplementing the growth medium with 200 nM phorbol myristate acetate (PMA; Sigma) overnight.
Plasmid construction.
pBESTluc was used as a backbone for all reporter assay constructs and was included in the luciferase assay system kit (Promega). pBEST+metK was created by inserting nucleotides corresponding to the first 10 codons of C. burnetii metK immediately downstream of the luc start codon using a Q5 site-directed mutagenesis kit, as instructed (New England Biolabs). cbsR12 was cloned into pBESTluc using primers containing XhoI and AfeI restriction sites on the forward and reverse primers, respectively. The forward primer also encoded a Ptac promoter and lac operator. The PCR product was cloned into pBEST+metK using unique XhoI and AfeI restriction sites in an irrelevant intergenic region. A frameshifted luc construct was created as a by-product of the metK Q5 mutagenesis of pBESTluc and contained a 1-bp frameshift deletion in the 5′ end of luc. pBEST+carA 5′ UTR was created using primers specific to the 5′ UTR of carA with HindIII and BamHI restriction sites on the forward and reverse primers, respectively. The forward primer also encoded the Ptac promoter. Nucleotides corresponding to the lac operator were inserted using a Q5 site-directed mutagenesis kit to create the final pBEST+carA 5′ UTR construct. The cbsR12 gene was inserted into this construct in the same fashion as for pBEST+metK+cbsR12.
Recombinant CarA and MetK were generated by PCR amplification of carA and metK using forward and reverse primers containing BamHI and HindIII restriction sites, respectively. The resulting amplicons were cloned into compatible restriction sites of pQE30 (Qiagen) by standard protocol.
Axenic growth of C. burnetii.
For growth curves and Crosslink-Seq experiments, 3.33 × 104 genomic equivalents (GE)/ml of MB-WT, MB-cbsR12, or MB-cbsR12-Comp were inoculated into 300 ml of ACCM-2 in a 1-liter flask with either chloramphenicol (5 μg/ml), kanamycin (350 μg/ml) or both. The GE/ml values were initially determined from frozen cell stocks by qPCR, as previously described (68), although different dotA primers were used (Fig. S10). Cell viability was determined using a BacLight bacterial viability kit, as instructed (Thermo Scientific).
C. burnetii infection of differentiated THP-1 cells.
THP-1 cells were seeded onto four-well chambered glass slides (Lab-Tek) or T-75 flasks. After 2 days of growth, 200 nM PMA was added, along with fresh medium, and the cells were allowed to differentiate overnight. The PMA-supplemented medium was removed, and fresh medium was restored; differentiated THP-1 cells were then allowed to recover for 4 h prior to C. burnetii infection at a multiplicity of infection (MOI) of 10. Initial infections were rocked for 2 h at room temperature before the cells were returned to 5% CO2 and 37°C. At 1 dpi, the supernatant was removed, extracellular C. burnetii was washed away with warmed 1× phosphate-buffered saline (PBS), and fresh medium was added.
Total RNA and genomic DNA extraction and purification.
C. burnetii samples grown in ACCM-2 were centrifuged at 15,000 × g at 4°C for 15 min; the pellets were then resuspended in 1 ml of TRI Reagent (Ambion). The suspension was incubated for 1 h at room temperature, frozen for 2 h at –80°C, thawed for 30 min at room temperature, and then pipetted vigorously until homogenized. Next, 100 μl of BCP (Acros Organics) was added, and the solution vortexed for 30 s, incubated for 5 min at room temperature, and centrifuged at 12,000 × g at 4°C for 10 min. The aqueous phase was then collected, and 300 μl of 100% ethanol added. The mixture was immediately vortexed for 10 s, and a RiboPure RNA purification kit (Ambion) was used to collect, concentrate, and wash the resulting RNA. RNA was collected in nuclease-free H2O and treated with DNase I for 1 h at 37°C. After RNA precipitation in 100% ethanol, the purified RNA was run on a NanoDrop spectrophotometer (Thermo Scientific) to determine the concentration and purity.
In order to purify total RNA from C. burnetii grown in THP-1 cell lines, growth medium was first removed and replaced with 1 ml of TRI Reagent. Flasks containing TRI Reagent were rocked for 1 h at room temperature, and the cells were mechanically scraped and collected into a 15-ml conical tube. The mixture was frozen overnight at –80°C and thawed to room temperature for 30 min, and the RNA purification procedure was continued as described above.
Genomic DNA was purified from TRI Reagent mixtures according to manufacturer protocols (Ambion). The resulting DNA was purified using a nucleotide removal kit as instructed (Qiagen).
Quantitative PCR and quantitative real-time PCR.
Quantitative PCR (qPCR) and quantitative real-time PCR (qRT-PCR) experiments were performed as previously described (69) using 300 nM concentrations of primers specific to cbsR12 and a volume of iQ SYBR green Supermix (Bio-Rad). The resulting reactions were cycled on a MyiQ single-color real-time PCR detection system (v1.0 software; Bio-Rad). The CbsR12 cDNA copy number was normalized against the dotA copy number derived from C. burnetii genomic DNA to obtain copy numbers/GE values. Growth curve GE/ml and GE/flask values were obtained from genomic DNA purified from the same cells from which total RNA was purified.
For ACCM-2 growth curves, 30-ml aliquots of a 300-ml culture were removed at each time point. gDNA was extracted and resuspended in 30 μl of nuclease-free H2O. Then, 1 μl of gDNA was used in subsequent qPCR reactions producing GE/ml values. For THP-1 growth curves, eight separate T-75 flasks for each C. burnetii strain tested were inoculated simultaneously. At the specified time point, one flask was taken, and 30 μl of gDNA was again extracted. Finally, 1 μl of gDNA was used in subsequent qPCR reactions, and the GE/flask values were calculated. Each growth curve and qPCR reaction was performed in triplicate.
RNase III assay.
RNase III assays were performed as previously described (43) using 200 nM CbsR12 substrate and the C. burnetii intervening sequence RNA as a positive control (43). The resulting reaction products were electrophoresed on 7% denaturing polyacrylamide gels and stained with 2 μg/ml acridine orange to visualize bands.
Identification of transcription start sites.
5′ RACE analysis of carA, metK, cvpD, and cbsR12 transcripts was performed on MB-WT and MB-cbsR12 RNA extracted from infected Vero cells (cbsR12, carA, and metK) or THP-1 cells (cvpD) at 3 dpi using a 5′ RACE system kit (Invitrogen) according to the manufacturer’s protocols and with gene-specific primers (see Fig. S10). The resulting PCR products were cloned into pCR2.1-TOPO as instructed (Invitrogen) and then sequenced with M13 universal primers by Sanger automated sequencing. In general, three biological replicate total RNA samples were obtained from MB-WT and MB-cbsR12 infections of Vero and THP-1 cells. From these pools, 5′ RACE was performed as described above, and 4 clones from each replicate were sequenced, producing 12 total clones analyzed for each total RNA pool for each gene analyzed.
In silico and bioinformatics analyses.
RNA target predictions were carried out using TargetRNA2 (70), IntaRNA (71), and CopraRNA (72) algorithms with default settings. RNA was folded using Mfold (73) and visualized with Visualization Applet for RNA (74). Analyses of RNA-Seq data were carried out as previously described (38). Briefly, raw fastq files were concatenated, quality filtered with the FASTX toolkit (http://hannonlab.cshl.edu/fastx_toolkit/), and then clipped, aligned, and filtered with Nesoni version 0.128 tools (http://www.vicbioinformatics.com/software.nesoni.shtml). Transcripts per million (tpm) were calculated using custom Perl and Python scripts that can be accessed through GitHub (https://github.com/shawachter/TPM_Scripts). Crosslink-Seq enrichment was accomplished by processing .bam files using featureCounts (75), followed by use of the DESeq2 package in R version 3.4.4 to obtain differentially expressed genes (76). The Artemis genome browser was used to visualize generated alignment files (http://www.sanger.ac.uk/science/tools/artemis) (77).
All IFA images were processed and analyzed using Fiji (78) and Cell Profiler (79), respectively. Figures were created using R version 3.4.4, Inkscape (https://inkscape.org/release/inkscape-0.92.4/), and GIMP (https://www.gimp.org/downloads/).
RNA-RNA hybridization and EMSA.
Regions of target genes were first selected for PCR amplification. The following regions were chosen based on inclusion of predicted 5′ UTRs and CbsR12-binding sites (the +1 nucleotide designation is the first nucleotide of the annotated start codon): carA (−143 to −1), metK (−26 to +110), cvpD (−41 to +101), purH (−100 to +44), dnaA (−97 to +61), and rpsA (−65 to +77). PCR products (1 μg) of desired templates were transcribed in vitro overnight at 37°C with 2.5 mM ribonucleotide solution mix (New England Biolabs) and, when needed, 0.5 mM Bio-16-UTP (Invitrogen) using a MAXIscript T7 transcription kit (Invitrogen). The resulting reaction mixtures were incubated for 1 h at 37°C with 1 μl of Turbo DNase (Invitrogen), heated for 4 min at 85°C, and then immediately plunged in ice and electrophoresed on a 7% polyacrylamide gel for 75 min at 100 V. The gels were stained with a 2-μg/ml acridine orange solution, and visualized bands were excised and eluted overnight into probe elution buffer (0.5 M ammonium acetate, 1 mM EDTA, 0.2% sodium dodecyl sulfate [SDS]) at 37°C. The resulting solution was precipitated with ethanol overnight at –20°C, washed with 70% ethanol, and resuspended in nuclease-free H2O. The RNA concentrations were determined using a NanoDrop spectrophotometer. Next, 10 nM biotin-labeled CbsR12 and, unless otherwise noted, 5 nM target RNA were combined and heated for 5 min at 85°C. A high-salt TMN buffer (100 mM NaCl, 50 mM MgCl2, 100 mM Tris-Cl, 0.05% Tween 20) was then added, and the reaction mixtures were immediately plunged on ice for 30 s, followed by incubation for 30 min at 37°C. A nondenaturing loading dye (0.25% bromophenol blue) was added, and the resulting RNA mixtures were resolved on a 7% polyacrylamide gel for 1 h 20 min at 100 V. RNA was transferred to a BrightStar-Plus positively charged nylon membrane (Ambion) using an electroblot transfer system (Bio-Rad) and cross-linked with short-wave UV light in a GS gene linker UV chamber (Bio-Rad). A North2South chemiluminescence hybridization and detection kit (Thermo Scientific) was used to detect resulting bands. The blot was imaged on a LAS-3000 imaging system (Fujifilm).
RNA-protein EMSA.
CbsR12-CsrA1/2 EMSAs were performed as previously described for CsrA-binding RNAs (44). Biotin-labeled CbsR12 was synthesized in vitro as described above for RNA-RNA EMSAs. C. burnetii csrA-1 and csrA-2 genes were cloned into pQE30 and expressed, and the resulting proteins were natively purified as previously described (43). Then, 1 nM biotin-labeled CbsR12 diluted in TE buffer (10 mM Tris-HCl, 1 mM EDTA) was heated at 75°C for 3 min, and the samples were equilibrated to room temperature for 10 min. Purified CsrA1/2 diluted in CsrA-binding buffer (1 μl in 10 mM Tris-HCl, 10 mM MgCl2, 100 mM KCl, 10 mM dithiothreitol, 10% glycerol, and 10 U of RNasin [Promega]) was added, and reaction mixtures were incubated for 30 min at 37°C. The samples were immediately resolved on 10% nondenaturing polyacrylamide gels for 3 h. Membrane transfer and imaging were performed as in the RNA-RNA EMSAs described above. The Kd for CsrA-2 was determined as previously described (44).
Reporter assay.
A luciferase assay system kit (Promega) was used. All pBESTluc constructs were transformed into E. coli TOP10F′. The resulting E. coli strains were grown overnight at 30°C in 10 ml of LB containing ampicillin (100 μg/ml) and 1% glucose in order to mitigate the autoaggregative effects of CbsR12. An aliquot (4.5 ml) of the overnight culture was inoculated into 40.5 ml of LB with 100 μg/ml ampicillin and grown for 1.5 h at 30°C. IPTG was added (to 1 mM), and culture aliquots (100 μl) were removed at the 0-, 1-, and 2-h time points. Next, 80 μl of LB and 20 μl of CCLR lysis solution (1× CCLR, 25 mg of BSA, 12.5 mg of lysozyme, 7.5 ml of water) were added to the aliquots, and the samples were gently inverted until the solution clarified. Then, 50 μl of the resulting lysate was aliquoted to a 96-well plate, 100 μl of luciferase assay substrate was added, and the luminescence was immediately read with a SpectraMax M5 plate reader (Molecular Devices).
Crosslink-Seq analysis.
RNA-RNA cross-linking was performed essentially as previously described (47, 80), except that TRI Reagent was utilized for total RNA extraction as described above and that 10-nmol portions of two distinct biotinylated in vitro-transcribed anti-CbsR12 RNAs were used as probes. The resulting captured RNA was sent to the Yale Center for Genomic Analysis for RNA-Seq analysis.
Immunofluorescence assay.
IFAs on infected THP-1 cells were performed as previously described with modifications (11). Briefly, four-well chambered glass slides were coated for 30 min with a 0.2% solution of Sigmacote (Sigma). THP-1 cells were inoculated into chambered slides, followed by incubation overnight or until 60% confluence was reached and then differentiated with 200 nM PMA. Confluent cells were then infected with MB-WT, MB-cbsR12, or MB-cbsR12-Comp strains at an MOI of 10. At 1 dpi, infections were stopped by washing cells three times for 5 min in prewarmed 1× PBS, and then fresh medium was added. At 3 or 7 dpi, the growth medium was removed, and the cells were fixed with ice-cold 100% methanol for 5 min at room temperature. The cells were washed three times for 5 min each time with ice-cold 1× PBS, blocked for 1 h at room temperature with a 2% BSA solution in 1× PBS, and then incubated with anti-Com1 (1:1,000) and anti-LAMP1 (1:50; H4A3 was deposited into the Developmental Studies Hybridoma Bank by J. T. August and J. E. K. Hildreth) antibodies for 2 h. Cells were washed and incubated with Alexa Fluor 488 (1:200; Thermo Scientific) and rhodamine-conjugated goat anti-mouse antibodies (1:200; Thermo Scientific) along with 300 nM DAPI (4′,6′-diamidino-2-phenylindole; Thermo Scientific) for 1 h. The cells were then washed three times for 5 min each time in ice-cold 1× PBS and immediately imaged. Images were processed using Fiji (78). Cell Profiler was used to measure the CCV areas, as previously described (81). Measurements were taken from 46 individual images of random fields of view spanning three different experiments for each C. burnetii strain.
Protein synthesis, purification, and antibody production.
Recombinant Coxiella RNase III was synthesized from a previously generated pQE30 construct and purified as described previously (43). C. burnetii carA and metK genes were cloned in frame into pQE30 (Qiagen), and the resulting N-terminal His6-tagged proteins were synthesized and purified as previously described for C. burnetii RNA helicase (82). Purified recombinant CarA and MetK proteins were submitted to General Bioscience, Inc., for rabbit polyclonal antibody production.
Western blot analyses.
Western blot analyses were performed as previously described (36) with minor modifications. MB-WT, MB-cbsR12, and MB-cbsR12-Comp strains were grown to mid-log phase (4 days for MB-WT and MB-cbsR12-Comp, 6 days for MB-cbsR12) in ACCM-2. Proteins at 30 μg (CarA blot) or 60 μg (MetK blot) were resolved on 10 to 20% acrylamide gradient Tris-glycine SDS-PAGE gels. Duplicate gels were run in parallel for Coomassie brilliant blue staining (0.1% [wt/vol] in 50% methanol, 7% [vol/vol] acetic acid) in order to present a loading control. Blots were incubated with primary antibody solution (1× PBS [pH 7.4], 0.3% [vol/vol] Tween 20, 1:500 CarA/1:5,000 MetK primary antibody) for 2 h with rocking at room temperature. Blots were washed five times for 5 min in 1× PBS and then incubated for 1 h in secondary antibody solution (1× PBS plus 1:2,000 goat anti-rabbit–HRP). Blots were again washed five times for 5 min in 1× PBS and immediately developed using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) according to the manufacturer’s protocol. Imaging was performed on a ChemiDoc XRS+ system (Bio-Rad).
E. coli biofilm induction assay.
E. coli biofilm induction assays were performed as previously described (33). Plates (96 well) were inoculated with an overnight culture of either pBEST or pBEST+carA 5′ UTR+cbsR12 E. coli strains (see Fig. S10). Cultures were allowed to grow for 3 h until induction with 1 mM IPTG, and then the cultures were allowed to grow an additional 21 h before subsequent staining with crystal violet. The average optical density at 570 nm readings of 10 wells were obtained by spectrophotometry.
Generation of a CbsR12-complemented strain.
MB-cbsR12 was complemented as previously described, with modifications (83). Briefly, wild-type cbsR12, along with 100 bp of flanking sequences, was PCR amplified using primers containing EcoRI and BamHI restriction sites. The amplicon was cloned into compatible restriction sites of pMini-Tn7-KAN by standard protocol (84). The resulting plasmid was transformed into electrocompetent E. coli PIR1 cells for propagation. The pMini-Tn7-CbsR12-KAN plasmid (20 μg), along with 10 μg of a second plasmid containing the transposase, pMini-TnS2-ABCD, was transformed into MB-cbsR12 in a single electroporation reaction (25 kV, 500 Ω, 25 μF). Electroporated cells were allowed to recover for 5 days in ACCM-2 supplemented with 1% FBS, and then dilutions were plated onto ACCM-2 agar containing kanamycin (375 μg/ml). Isolated colonies were picked and recultured on ACCM-2 agar plates for several rounds. Colony PCR was used to screen for the MB-cbsR12-Comp strain, and the location of the cbsR12 cassette was determined by PCR and Sanger automated sequencing. qPCR of MB-cbsR12-Comp genomic DNA utilizing primers specific to cbsR12 was used to ensure that a single transposon insertion event occurred.
Data availability.
The sequencing reads from the Crosslink-Seq experiments are available at the NCBI Sequencing Read Archive (accession number PRJNA522455).
Supplementary Material
ACKNOWLEDGMENTS
We thank Paul Beare for his generous donation of E. coli PIR1 cells, pMini-TnS2-ABCD plasmid, and the pMini-Tn7-KAN plasmid. We thank Jenny Wachter for her contribution of the TPM calculator and Linda D. Hicks for excellent technical assistance.
R.R. was supported by National Institutes of Health (NIH) grants AI123464, AI126385, and AI133023. M.F.M. was supported by NIH grants AI128575, AI123293, and AI119798. S.W. was supported by a research grant from the Montana Academy of Sciences.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00524-19.
REFERENCES
- 1.Angelakis E, Raoult D. 2010. Q fever. Vet Microbiol 140:297–309. doi: 10.1016/j.vetmic.2009.07.016. [DOI] [PubMed] [Google Scholar]
- 2.McCaul TF, Williams JC. 1981. Developmental cycle of Coxiella burnetii: structure and morphogenesis of vegetative and sporogenic differentiations. J Bacteriol 147:1063–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Romano PS, Gutierrez MG, Berón W, Rabinovitch M, Colombo MI. 2007. The autophagic pathway is actively modulated by phase II Coxiella burnetii to efficiently replicate in the host cell. Cell Microbiol 9:891–909. doi: 10.1111/j.1462-5822.2006.00838.x. [DOI] [PubMed] [Google Scholar]
- 4.Gutierrez MG, Vazquez CL, Munafo DB, Zoppino FCM, Beron W, Rabinovitch M, Colombo MI. 2005. Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cell Microbiol 7:981–993. doi: 10.1111/j.1462-5822.2005.00527.x. [DOI] [PubMed] [Google Scholar]
- 5.Howe D, Melnicakova J, Barak I, Heinzen RA. 2003. Maturation of the Coxiella burnetii parasitophorous vacuole requires bacterial protein synthesis but not replication. Cell Microbiol 5:469–480. doi: 10.1046/j.1462-5822.2003.00293.x. [DOI] [PubMed] [Google Scholar]
- 6.Zamboni DS, McGrath S, Rabinovitch M, Roy CR. 2003. Coxiella burnetii express type IV secretion system proteins that function similarly to components of the Legionella pneumophila Dot/Icm system. Mol Microbiol 49:965–976. doi: 10.1046/j.1365-2958.2003.03626.x. [DOI] [PubMed] [Google Scholar]
- 7.Chen C, Banga S, Mertens K, Weber MM, Gorbaslieva I, Tan Y, Luo ZQ, Samuel JE. 2010. Large-scale identification and translocation of type IV secretion substrates by Coxiella burnetii. Proc Natl Acad Sci U S A 107:21755–21760. doi: 10.1073/pnas.1010485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cunha LD, Ribeiro JM, Fernandes TD, Massis LM, Khoo CA, Moffatt JH, Newton HJ, Roy CR, Zamboni DS. 2015. Inhibition of inflammasome activation by Coxiella burnetii type IV secretion system effector IcaA. Nat Commun 6:10205. doi: 10.1038/ncomms10205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larson CL, Beare PA, Voth DE, Howe D, Cockrell DC, Bastidas RJ, Valdivia RH, Heinzen RA. 2015. Coxiella burnetii effector proteins that localize to the parasitophorous vacuole membrane promote intracellular replication. Infect Immun 83:661–670. doi: 10.1128/IAI.02763-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez E, Allombert J, Cantet F, Lakhani A, Yandrapalli N, Neyret A, Norville IH, Favard C, Muriaux D, Bonazzi M. 2016. Coxiella burnetii effector CvpB modulates phosphoinositide metabolism for optimal vacuole development. Proc Natl Acad Sci U S A 113:E3260–E3269. doi: 10.1073/pnas.1522811113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinez E, Cantet F, Fava L, Norville I, Bonazzi M. 2014. Identification of OmpA, a Coxiella burnetii protein involved in host cell invasion, by multi-phenotypic high-content screening. PLoS Pathog 10:e1004013. doi: 10.1371/journal.ppat.1004013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newton HJ, Kohler LJ, McDonough JA, Temoche-Diaz M, Crabill E, Hartland EL, Roy CR. 2014. A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog 10:e1004286. doi: 10.1371/journal.ppat.1004286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weber MM, Chen C, Rowin K, Mertens K, Galvan G, Zhi H, Dealing CM, Roman VA, Banga S, Tan Y, Luo ZQ, Samuel JE. 2013. Identification of Coxiella burnetii type IV secretion substrates required for intracellular replication and Coxiella-containing vacuole formation. J Bacteriol 195:3914–3924. doi: 10.1128/JB.00071-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weber MM, Faris R, McLachlan J, Tellez A, Wright WU, Galvan G, Luo ZQ, Samuel JE. 2016. Modulation of the host transcriptome by Coxiella burnetii nuclear effector Cbu1314. Microbes Infect 18:336–345. doi: 10.1016/j.micinf.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Mahapatra S, Gallaher B, Smith SC, Graham JG, Voth DE, Shaw EI. 2016. Coxiella burnetii employs the Dot/Icm type IV secretion system to modulate host NF-κB/RelA activation. Front Cell Infect Microbiol 6:188. doi: 10.3389/fcimb.2016.00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beare PA, Sandoz KM, Larson CL, Howe D, Kronmiller B, Heinzen RA. 2014. Essential role for the response regulator PmrA in Coxiella burnetii type 4B secretion and colonization of mammalian host cells. J Bacteriol 196:1925–1940. doi: 10.1128/JB.01532-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carrier MC, Lalaouna D, Masse E. 2018. Broadening the definition of bacterial small RNAs: characteristics and mechanisms of action. Annu Rev Microbiol 72:141–161. doi: 10.1146/annurev-micro-090817-062607. [DOI] [PubMed] [Google Scholar]
- 18.Thomason MK, Storz G. 2010. Bacterial antisense RNAs: how many are there, and what are they doing? Annu Rev Genet 44:167–188. doi: 10.1146/annurev-genet-102209-163523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizuno T, Chou MY, Inouye M. 1984. A unique mechanism regulating gene expression: translational inhibition by a complementary RNA transcript (micRNA). Proc Natl Acad Sci U S A 81:1966–1970. doi: 10.1073/pnas.81.7.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Novick RP, Ross HF, Projan SJ, Kornblum J, Kreiswirth B, Moghazeh S. 1993. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J 12:3967–3975. doi: 10.1002/j.1460-2075.1993.tb06074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wassarman KM, Storz G. 2000. 6S RNA regulates E. coli RNA polymerase activity. Cell 101:613–623. doi: 10.1016/s0092-8674(00)80873-9. [DOI] [PubMed] [Google Scholar]
- 22.Massé E, Gottesman S. 2002. A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc Natl Acad Sci U S A 99:4620–4625. doi: 10.1073/pnas.032066599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keiler KC, Waller PRH, Sauer RT. 1996. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science 271:990–993. doi: 10.1126/science.271.5251.990. [DOI] [PubMed] [Google Scholar]
- 24.Sledjeski DD, Gupta A, Gottesman S. 1996. The small RNA, DsrA, is essential for the low temperature expression of RpoS during exponential growth in Escherichia coli. EMBO J 15:3993–4000. doi: 10.1002/j.1460-2075.1996.tb00773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Majdalani N, Chen S, Murrow J, St John K, Gottesman S. 2001. Regulation of RpoS by a novel small RNA: the characterization of RprA. Mol Microbiol 39:1382–1394. doi: 10.1111/j.1365-2958.2001.02329.x. [DOI] [PubMed] [Google Scholar]
- 26.Updegrove TB, Zhang A, Storz G. 2016. Hfq: the flexible RNA matchmaker. Curr Opin Microbiol 30:133–138. doi: 10.1016/j.mib.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bohn C, Rigoulay C, Bouloc P. 2007. No detectable effect of RNA-binding protein Hfq absence in Staphylococcus aureus. BMC Microbiol 7:10. doi: 10.1186/1471-2180-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Romeo T, Gong M, Liu MY, Brun-Zinkernagel AM. 1993. Identification and molecular characterization of csrA, a pleiotropic gene from Escherichia coli that affects glycogen biosynthesis, gluconeogenesis, cell size, and surface properties. J Bacteriol 175:4744–4755. doi: 10.1128/jb.175.15.4744-4755.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jackson DW, Suzuki K, Oakford L, Simecka JW, Hart ME, Romeo T. 2002. Biofilm formation and dispersal under the influence of the global regulator CsrA of Escherichia coli. J Bacteriol 184:290–301. doi: 10.1128/jb.184.1.290-301.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rasis M, Segal G. 2009. The LetA-RsmYZ-CsrA regulatory cascade, together with RpoS and PmrA, posttranscriptionally regulates stationary phase activation of Legionella pneumophila Icm/Dot effectors. Mol Microbiol 72:995–1010. doi: 10.1111/j.1365-2958.2009.06705.x. [DOI] [PubMed] [Google Scholar]
- 31.Liu MY, Gui G, Wei B, Preston JF III, Oakford L, Yuksel U, Giedroc DP, Romeo T. 1997. The RNA molecule CsrB binds to the global regulatory protein CsrA and antagonizes its activity in Escherichia coli. J Biol Chem 272:17502–17510. doi: 10.1074/jbc.272.28.17502. [DOI] [PubMed] [Google Scholar]
- 32.Heeb S, Blumer C, Haas D. 2002. Regulatory RNA as mediator in GacA/RsmA-dependent global control of exoproduct formation in Pseudomonas fluorescens CHA0. J Bacteriol 184:1046–1056. doi: 10.1128/jb.184.4.1046-1056.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sahr T, Bruggemann H, Jules M, Lomma M, Albert-Weissenberger C, Cazalet C, Buchrieser C. 2009. Two small ncRNAs jointly govern virulence and transmission in Legionella pneumophila. Mol Microbiol 72:741–762. doi: 10.1111/j.1365-2958.2009.06677.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Molofsky AB, Swanson MS. 2003. Legionella pneumophila CsrA is a pivotal repressor of transmission traits and activator of replication. Mol Microbiol 50:445–461. doi: 10.1046/j.1365-2958.2003.03706.x. [DOI] [PubMed] [Google Scholar]
- 35.Nevo O, Zusman T, Rasis M, Lifshitz Z, Segal G. 2014. Identification of Legionella pneumophila effectors regulated by the LetAS-RsmYZ-CsrA regulatory cascade, many of which modulate vesicular trafficking. J Bacteriol 196:681–692. doi: 10.1128/JB.01175-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warrier I, Hicks LD, Battisti JM, Raghavan R, Minnick MF. 2014. Identification of novel small RNAs and characterization of the 6S RNA of Coxiella burnetii. PLoS One 9:e100147. doi: 10.1371/journal.pone.0100147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Omsland A, Beare PA, Hill J, Cockrell DC, Howe D, Hansen B, Samuel JE, Heinzen RA. 2011. Isolation from animal tissue and genetic transformation of Coxiella burnetii are facilitated by an improved axenic growth medium. Appl Environ Microbiol 77:3720–3725. doi: 10.1128/AEM.02826-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wachter S, Raghavan R, Wachter J, Minnick MF. 2018. Identification of novel MITEs (miniature inverted-repeat transposable elements) in Coxiella burnetii: implications for protein and small RNA evolution. BMC Genomics 19:247. doi: 10.1186/s12864-018-4608-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piette J, Nyunoya H, Lusty CJ, Cunin R, Weyens G, Crabeel M, Charlier D, Glansdorff N, Piérard A. 1984. DNA sequence of the carA gene and the control region of carAB: tandem promoters, respectively controlled by arginine and the pyrimidines, regulate the synthesis of carbamoyl-phosphate synthetase in Escherichia coli K-12. Proc Natl Acad Sci U S A 81:4134–4138. doi: 10.1073/pnas.81.13.4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Urig S, Gowher H, Hermann A, Beck C, Fatemi M, Humeny A, Jeltsch A. 2002. The Escherichia coli Dam DNA methyltransferase modifies DNA in a highly processive reaction. J Mol Biol 319:1085–1096. doi: 10.1016/S0022-2836(02)00371-6. [DOI] [PubMed] [Google Scholar]
- 41.Churchill ME, Chen L. 2011. Structural basis of acyl-homoserine lactone-dependent signaling. Chem Rev 111:68–85. doi: 10.1021/cr1000817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN. 2010. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 26:493–500. doi: 10.1093/bioinformatics/btp692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Warrier I, Walter MC, Frangoulidis D, Raghavan R, Hicks LD, Minnick MF. 2016. The intervening sequence of Coxiella burnetii: characterization and evolution. Front Cell Infect Microbiol 6:83. doi: 10.3389/fcimb.2016.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yakhnin AV, Yakhnin H, Babitzke P. 2012. Gel mobility shift assays to detect protein-RNA interactions. Methods Mol Biol 905:201–211. doi: 10.1007/978-1-61779-949-5_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Battisti JM, Hicks LD, Minnick MF. 2011. A unique Coxiella burnetii lipoprotein involved in metal binding (LimB). Microbiology 157:966–976. doi: 10.1099/mic.0.046649-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heinzen RA, Scidmore MA, Rockey DD, Hackstadt T. 1996. Differential interaction with endocytic and exocytic pathways distinguish [sic] parasitophorous vacuoles of Coxiella burnetii and Chlamydia trachomatis. Infect Immun 64:796–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kacharia FR, Millar JA, Raghavan R. 2017. Emergence of new sRNAs in enteric bacteria is associated with low expression and rapid evolution. J Mol Evol 84:204–213. doi: 10.1007/s00239-017-9793-9. [DOI] [PubMed] [Google Scholar]
- 48.Smith TA, Driscoll T, Gillespie JJ, Raghavan R. 2015. A Coxiella-like endosymbiont is a potential vitamin source for the Lone Star tick. Genome Biol Evol 7:831–838. doi: 10.1093/gbe/evv016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schubert M, Lapouge K, Duss O, Oberstrass FC, Jelesarov I, Haas D, Allain FH. 2007. Molecular basis of messenger RNA recognition by the specific bacterial repressing clamp RsmA/CsrA. Nat Struct Mol Biol 14:807–813. doi: 10.1038/nsmb1285. [DOI] [PubMed] [Google Scholar]
- 50.Abbott ZD, Yakhnin H, Babitzke P, Swanson MS. 2015. csrR, a paralog and direct target of CsrA, promotes Legionella pneumophila resilience in water. mBio 6:e00595. doi: 10.1128/mBio.00595-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marden JN, Diaz MR, Walton WG, Gode CJ, Betts L, Urbanowski ML, Redinbo MR, Yahr TL, Wolfgang MC. 2013. An unusual CsrA family member operates in series with RsmA to amplify posttranscriptional responses in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 110:15055–15060. doi: 10.1073/pnas.1307217110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Friedman DI. 1988. Integration host factor: a protein for all reasons. Cell 55:545–554. doi: 10.1016/0092-8674(88)90213-9. [DOI] [PubMed] [Google Scholar]
- 53.Lou YC, Weng TH, Li YC, Kao YF, Lin WF, Peng HL, Chou SH, Hsiao CD, Chen C. 2015. Structure and dynamics of polymyxin-resistance-associated response regulator PmrA in complex with promoter DNA. Nat Commun 6:8838. doi: 10.1038/ncomms9838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Parshin A, Shiver AL, Lee J, Ozerova M, Schneidman-Duhovny D, Gross CA, Borukhov S. 2015. DksA regulates RNA polymerase in Escherichia coli through a network of interactions in the secondary channel that includes sequence insertion 1. Proc Natl Acad Sci U S A 112:E6862–E6871. doi: 10.1073/pnas.1521365112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zusman T, Aloni G, Halperin E, Kotzer H, Degtyar E, Feldman M, Segal G. 2007. The response regulator PmrA is a major regulator of the icm/dot type IV secretion system in Legionella pneumophila and Coxiella burnetii. Mol Microbiol 63:1508–1523. doi: 10.1111/j.1365-2958.2007.05604.x. [DOI] [PubMed] [Google Scholar]
- 56.Kay E, Humair B, Denervaud V, Riedel K, Spahr S, Eberl L, Valverde C, Haas D. 2006. Two GacA-dependent small RNAs modulate the quorum-sensing response in Pseudomonas aeruginosa. J Bacteriol 188:6026–6033. doi: 10.1128/JB.00409-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seshadri R, Paulsen IT, Eisen JA, Read TD, Nelson KE, Nelson WC, Ward NL, Tettelin H, Davidsen TM, Beanan MJ, Deboy RT, Daugherty SC, Brinkac LM, Madupu R, Dodson RJ, Khouri HM, Lee KH, Carty HA, Scanlan D, Heinzen RA, Thompson HA, Samuel JE, Fraser CM, Heidelberg JF. 2003. Complete genome sequence of the Q-fever pathogen Coxiella burnetii. Proc Natl Acad Sci U S A 100:5455–5460. doi: 10.1073/pnas.0931379100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schimke RT, Berlin CM, Sweeney EW, Carroll WR. 1966. The generation of energy by the arginine dihydrolase pathway in Mycoplasma hominis 07. J Biol Chem 241:2228–2236. [PubMed] [Google Scholar]
- 59.Ferla MP, Patrick WM. 2014. Bacterial methionine biosynthesis. Microbiology 160:1571–1584. doi: 10.1099/mic.0.077826-0. [DOI] [PubMed] [Google Scholar]
- 60.Sandoz KM, Beare PA, Cockrell DC, Heinzen RA. 2016. Complementation of arginine auxotrophy for genetic transformation of Coxiella burnetii by use of a defined axenic medium. Appl Environ Microbiol 82:3042–3051. doi: 10.1128/AEM.00261-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sánchez-Romero MA, Cota I, Casadesús J. 2015. DNA methylation in bacteria: from the methyl group to the methylome. Curr Opin Microbiol 25:9–16. doi: 10.1016/j.mib.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 62.Garcia-Del Portillo F, Pucciarelli MG, Casadesus J. 1999. DNA adenine methylase mutants of Salmonella typhimurium show defects in protein secretion, cell invasion, and M cell cytotoxicity. Proc Natl Acad Sci U S A 96:11578–11583. doi: 10.1073/pnas.96.20.11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tucker AM, Winkler HH, Driskell LO, Wood DO. 2003. S-Adenosylmethionine transport in Rickettsia prowazekii. J Bacteriol 185:3031–3035. doi: 10.1128/jb.185.10.3031-3035.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuley R, Kuijt E, Smits MA, Roest HIJ, Smith HE, Bossers A. 2017. Genome plasticity and polymorphisms in critical genes correlate with increased virulence of Dutch outbreak-related Coxiella burnetii strains. Front Microbiol 8:1526. doi: 10.3389/fmicb.2017.01526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Glazunova O, Roux V, Freylikman O, Sekeyova Z, Fournous G, Tyczka J, Tokarevich N, Kovacava E, Marrie TJ, Raoult D. 2005. Coxiella burnetii genotyping. Emerg Infect Dis 11:1211–1217. doi: 10.3201/eid1108.041354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Muller P, Gimpel M, Wildenhain T, Brantl S. 2019. A new role for CsrA: promotion of complex formation between an sRNA and its mRNA target in Bacillus subtilis. RNA Biol 16:972–987. doi: 10.1080/15476286.2019.1605811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Romeo T, Babitzke P. 2018. Global regulation by CsrA and its RNA antagonists. Microbiol Spectr 6:RWR-0009-2017. doi: 10.1128/microbiolspec.RWR-0009-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Coleman SA, Fischer ER, Howe D, Mead DJ, Heinzen RA. 2004. Temporal analysis of Coxiella burnetii morphological differentiation. J Bacteriol 186:7344–7352. doi: 10.1128/JB.186.21.7344-7352.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Raghavan R, Hicks LD, Minnick MF. 2008. Toxic introns and parasitic intein in Coxiella burnetii: legacies of a promiscuous past. J Bacteriol 190:5934–5943. doi: 10.1128/JB.00602-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kery MB, Feldman M, Livny J, Tjaden B. 2014. TargetRNA2: identifying targets of small regulatory RNAs in bacteria. Nucleic Acids Res 42:W124–W129. doi: 10.1093/nar/gku317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mann M, Wright PR, Backofen R. 2017. IntaRNA 2.0: enhanced and customizable prediction of RNA-RNA interactions. Nucleic Acids Res 45:W435–W439. doi: 10.1093/nar/gkx279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wright PR, Georg J, Mann M, Sorescu DA, Richter AS, Lott S, Kleinkauf R, Hess WR, Backofen R. 2014. CopraRNA and IntaRNA: predicting small RNA targets, networks and interaction domains. Nucleic Acids Res 42:W119–W123. doi: 10.1093/nar/gku359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Darty K, Denise A, Ponty Y. 2009. VARNA: interactive drawing and editing of the RNA secondary structure. Bioinformatics 25:1974–1975. doi: 10.1093/bioinformatics/btp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 76.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 78.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW, Carpenter AE. 2011. Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics 27:1179–1180. doi: 10.1093/bioinformatics/btr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lustig Y, Wachtel C, Safro M, Liu L, Michaeli S. 2010. ‘RNA walk’ a novel approach to study RNA-RNA interactions between a small RNA and its target. Nucleic Acids Res 38:e5. doi: 10.1093/nar/gkp872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martinez E, Cantet F, Bonazzi M. 2015. Generation and multi-phenotypic high-content screening of Coxiella burnetiii transposon mutants. J Vis Exp 99:e52851. doi: 10.3791/52851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hicks LD, Warrier I, Raghavan R, Minnick MF. 2011. Ribozyme stability, exon skipping, and a potential role for RNA helicase in group I intron splicing by Coxiella burnetii. J Bacteriol 193:5292–5299. doi: 10.1128/JB.05472-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Beare PA, Larson CL, Gilk SD, Heinzen RA. 2012. Two systems for targeted gene deletion in Coxiella burnetii. Appl Environ Microbiol 78:4580–4589. doi: 10.1128/AEM.00881-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Choi KH, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods 2:443–448. doi: 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing reads from the Crosslink-Seq experiments are available at the NCBI Sequencing Read Archive (accession number PRJNA522455).