

Protein lipoylation is a posttranslational modification that is evolutionarily conserved from bacteria to humans. Lipoic acid modifications are found on five proteins in S. aureus, four of which are components of major metabolic enzymes. In some bacteria, the amidotransferase LipL is critical for the attachment of lipoic acid to these proteins, and yet it is unclear to what extent LipL facilitates the transfer of this cofactor. We find that S. aureus LipL flexibly shuttles lipoic acid among metabolic enzyme subunits, alluding to a dynamic redistribution mechanism within the bacterial cell. This discovery exemplifies a potential means by which bacteria optimize the use of scarce nutrients when resources are limited.
KEYWORDS: Staphylococcus aureus, lipoic acid, lipoyl amidotransferase, metabolism, virulence
ABSTRACT
Staphylococcus aureus competes for myriad essential nutrients during host infection. One of these nutrients is the organosulfur compound lipoic acid, a cofactor required for the activity of several metabolic enzyme complexes. In S. aureus, these include the E2 subunits of three α-ketoacid dehydrogenases and two H proteins, GcvH of the glycine cleavage system and its paralog, GcvH-L. We previously determined that the S. aureus amidotransferase LipL is required for lipoylation of the E2 subunits of pyruvate dehydrogenase (PDH) and branched-chain 2-oxoacid dehydrogenase (BCODH) complexes. The results from this study, coupled with those from Bacillus subtilis, suggested that LipL catalyzes lipoyl transfer from H proteins to E2 subunits. However, to date, the range of LipL targets, the extent of LipL-dependent lipoic acid shuttling between lipoyl domain-containing proteins, and the importance of lipoyl relay in pathogenesis remain unknown. Here, we demonstrate that LipL uses both lipoyl-H proteins as the substrates for lipoyl transfer to all E2 subunits. Moreover, LipL facilitates lipoyl relay between E2 subunits and between H proteins, a property that potentially constitutes an adaptive response to nutrient scarcity in the host, as LipL is required for virulence during infection. Together, these observations support a role for LipL in facilitating flexible lipoyl relay between proteins and highlight the complexity of protein lipoylation in S. aureus.
IMPORTANCE Protein lipoylation is a posttranslational modification that is evolutionarily conserved from bacteria to humans. Lipoic acid modifications are found on five proteins in S. aureus, four of which are components of major metabolic enzymes. In some bacteria, the amidotransferase LipL is critical for the attachment of lipoic acid to these proteins, and yet it is unclear to what extent LipL facilitates the transfer of this cofactor. We find that S. aureus LipL flexibly shuttles lipoic acid among metabolic enzyme subunits, alluding to a dynamic redistribution mechanism within the bacterial cell. This discovery exemplifies a potential means by which bacteria optimize the use of scarce nutrients when resources are limited.
INTRODUCTION
Staphylococcus aureus is the causative agent of a wide array of diseases, including skin and soft tissue infections, infective endocarditis, pneumonia, and severe sepsis. The versatility of S. aureus as a pathogen hinges not only on its release of virulence factors that compromise host immune defenses but also on its capacity to adapt to host nutritional restriction by scavenging essential nutrients (1–8). S. aureus disease is generally controlled by antibiotic treatment, but alleviation of infection is increasingly challenging with the rise of antibiotic resistance (9). The study of bacterial trace nutrient acquisition seeks to exploit its importance for survival and proliferation in host tissues for therapeutic design (10–15). The goal is to develop treatment strategies that divert the metabolic flux of bacteria, such that they remain nonpathogenic or are rendered replication defective (16).
To colonize tissues, S. aureus scavenges myriad essential nutrients from the host, one of which is lipoic acid (3, 17). Lipoic acid is an organosulfur compound that is a cofactor in enzyme complexes involved in central metabolism and is an antioxidant with immunosuppressive properties (18–21). There is mounting evidence that lipoic acid metabolism is required for microbial pathogenesis (22). For example, in malaria-causing Plasmodium species, lipoic acid synthesis and salvage are essential during liver-stage and asexual blood-stage development, respectively (22–27). In Listeria monocytogenes, a lipoic acid auxotroph, salvage of the cofactor contributes to growth in the host cytosol and disease in mice (28–30). In addition, S. aureus differentially uses bacterial and host-derived lipoic acid to support bloodstream infection while also releasing the lipoylated E2 subunit of the pyruvate dehydrogenase complex (E2-PDH) to dampen the activation of proinflammatory macrophages (3, 31). Other pathogens that rely on lipoic acid metabolism for virulence include Burkholderia pseudomallei, Pseudomonas aeruginosa, and Mycobacterium tuberculosis (32–35).
There are four lipoylated enzyme complexes in S. aureus, as follows: the pyruvate dehydrogenase complex (PDH) converts pyruvate into acetyl coenzyme A (acetyl-CoA), the 2-oxoglutarate dehydrogenase complex (OGDH) converts α-ketoglutarate into succinyl-CoA, the branched-chain 2-oxoacid dehydrogenase complex (BCODH) catabolizes deaminated derivatives of branched-chain amino acids for synthesis of branched-chain fatty acids, and the glycine cleavage system (GCS) degrades excess glycine (3, 36, 37). Lipoic acid is covalently attached to a conserved lysine residue on the E2 subunits of α-ketoacid dehydrogenase complexes (E2-PDH, E2-OGDH, and E2-BCODH) and the H protein of GCS (GcvH). Previously, we dissected the mechanisms of lipoic acid acquisition in S. aureus using a genetic approach and found that this bacterium is capable of both de novo synthesis and salvage of lipoic acid (3). Like Bacillus subtilis, S. aureus initiates lipoic acid synthesis by transferring the eight-carbon saturated fatty acid, octanoic acid, from an acyl carrier protein (ACP) to GcvH with the octanoyltransferase LipM (3, 38). The lipoyl synthase LipA then catalyzes the attachment of two sulfhydryl groups to C-6 and C-8 of the pendant octanoyl chain on GcvH (3, 39, 40). This is followed by incorporation of the lipoyl moiety from GcvH onto E2 subunits by the amidotransferase LipL (3, 41, 42). Similar to L. monocytogenes, S. aureus also encodes two lipoic acid salvage enzymes, LplA1 and LplA2 (3, 28, 29, 42). However, only LplA1 is essential for lipoic acid salvage in vitro (3). In contrast, either enzyme is sufficient to promote renal infection by S. aureus in a murine bloodstream infection model. S. aureus also synthesizes a GcvH-like protein, GcvH-L, which is encoded in the same operon as LplA2 (43). The operon is thought to contribute to redox homeostasis, but the exact mechanism remains unclear (43). The ability to produce two lipoic acid salvage enzymes and two H proteins is a unique feature of S. aureus not encountered thus far in other Gram-positive Firmicutes.
In a previous study, we used biochemical assays to demonstrate that LplA1 and LplA2 exhibit differential targeting of lipoyl domain-containing proteins. LplA1 primarily targets H proteins, while LplA2 primarily targets E2 subunits (44). Furthermore, we uncovered genetic evidence that both H proteins facilitate lipoyl transfer to E2 subunits. However, GcvH-L appears to exclusively serve as a node in the lipoic acid salvage pathway (44). Despite elucidation of the lipoic acid biosynthesis and salvage pathways of S. aureus, several outstanding questions remain related to the amidotransferase LipL. The importance of LipL for S. aureus pathogenesis has yet to be determined, and the only direct biochemical evidence of its function to date comes from the following three organisms: octanoyl transfer from GcvH to E2-PDH in B. subtilis, octanoyl transfer from GcvH to the lipoyl domain of E2-BCODH in L. monocytogenes, and lipoyl transfer from the glycine cleavage system H (GCSH) protein to the lipoyl domain of E2-PDH in humans (29, 42, 45). Therefore, we sought to determine the range of lipoyl substrates used by S. aureus LipL to transfer lipoic acid and identify which apoproteins are targeted for lipoylation.
In this study, we performed a biochemical assessment of LipL activity. We found that LipL uses either lipoyl-GcvH or lipoyl-GcvH-L as a donor for transfer of lipoic acid to all three full-length E2 subunits. In addition, LipL promotes lipoyl relay between E2 subunits and between H proteins. In all cases, the reaction is reversible. However, lipoyl transfer to GcvH-L is least favored. The requirement of LipL for optimal S. aureus virulence is demonstrated by the attenuation of a ΔlipL mutant strain during murine infection. Overall, these findings suggest a flexible mechanism for redistributing an essential cofactor that facilitates adaptation to host-imposed lipoic acid limitation.
RESULTS
Identifying the lipL open reading frame of S. aureus.
A prior genetic analysis found that LipL is required for lipoylation of E2-PDH and E2-BCODH in S. aureus (3). In addition, a ΔlipL mutant cannot grow in lipoic acid-deficient or -replete medium. Attempts to complement these phenotypes with lipL were unsuccessful, an outcome we attributed to potential translational coupling of lipL with its upstream gene, pta, which encodes phosphate acetyltransferase (3). The NCBI-annotated sequence of lipL contains a 74-nucleotide intergenic region between the pta and lipL genes (Fig. 1A). Upon inspection, we noticed another viable in-frame start codon for lipL two nucleotides after the stop codon of pta (Fig. 1A). To determine if the longer lipL sequence [lipL(ATG-1)] encodes a functional protein, we complemented a ΔlipL mutant with this lipL sequence and assessed bacterial growth of both the ΔlipL+lipL(ATG-1) and ΔlipL+lipL(ATG-2) mutant strains. We found that the ΔlipL+lipL(ATG-1) mutant strain exhibited wild-type (WT) growth characteristics, whereas the ΔlipL+lipL(ATG-2) mutant strain was unable to replicate (Fig. 1B). Furthermore, lipoylation profiles were fully restored for a ΔlipL+lipL(ATG-1) mutant strain (Fig. 1C, lane 3) grown in lipoic acid-deficient RPMI medium. These data suggest that the S. aureus lipL coding sequence begins two nucleotides downstream of the pta stop codon.
FIG 1.
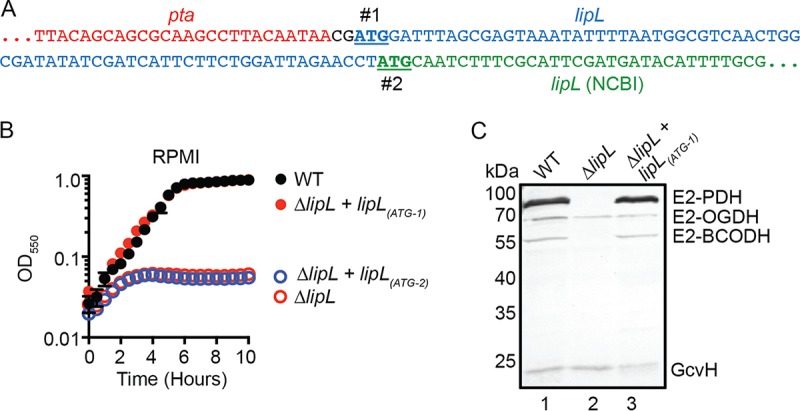
The start codon of lipL is two nucleotides downstream of the pta stop codon in S. aureus. (A) DNA sequences showing two potential in-frame start codons of lipL (#1 and #2) are underlined. The 5′ end of lipL, as published by NCBI, is in green, while additional nucleotides that would form the 5′ end of lipL with the earlier start codon are in blue. (B) Growth of WT, ΔlipL mutant, ΔlipL+lipL(ATG-1) mutant, and ΔlipL+lipL(ATG-2) mutant strains in RPMI medium over 10 h. (C) In vivo lipoylation profiles of WT, ΔlipL mutant, and ΔlipL+lipL(ATG-1) mutant strains after 9 h subculture growth in RPMI medium plus BCFA and NaAc was assessed by immunoblotting SDS-PAGE-resolved whole-cell lysates with rabbit anti-lipoic acid antibody. The presented blot is representative of at least three independent experiments.
LipL cannot use free lipoic acid as a lipoyl substrate.
The lipoic acid salvage enzymes LplA1 and LplA2 use free lipoic acid to modify lipoyl domain-containing proteins, but we have not ruled out the possibility of LipL being able to perform the same reaction (44). To this end, we conducted a lipoylation assay by mixing LipL with each of the five S. aureus apo-E2 and H proteins in the presence of free lipoic acid. In contrast to reactions with LplA1 or LplA2, none of the LipL-containing reactions generated substantial lipoyl-H or lipoyl-E2 subunits (see Fig. S1A in the supplemental material). From this outcome, we concluded that S. aureus LipL, similar to B. subtilis and L. monocytogenes LipL, must use protein-bound lipoic acid as a substrate (29, 41, 42).
LipL uses lipoyl-GcvH and lipoyl-GcvH-L for lipoic acid transfer to E2 subunits.
Only E2-OGDH and GcvH are lipoylated in a ΔlipL mutant, suggesting that LipL is essential for the lipoylation of E2-PDH and E2-BCODH (Fig. 1C, lane 2). Nonetheless, the extent of LipL-mediated lipoic acid transfer remains untested. Previous work with B. subtilis showed that purified LipL catalyzes octanoyl transfer from GcvH to E2-PDH, while another study found that LIPT1, the LipL analog in humans, catalyzes lipoyl transfer from GCSH to a lipoyl domain derived from human PDH (42, 45). We purified recombinant S. aureus lipoyl-GcvH and lipoyl-GcvH-L from E. coli for use in lipoyl transfer assays with purified S. aureus apo-E2-PDH, apo-E2-OGDH, and apo-E2-BCODH. After purification, GcvH was determined to be mostly lipoylated, while purified GcvH-L was partially lipoylated (Fig. S1B). Using lipoyl-GcvH, we observed LipL-dependent lipoyl transfer to E2-PDH, E2-OGDH, and E2-BCODH (Fig. 2A to D and S2A to C). The same was true when lipoyl-GcvH-L was used. However, transfer from lipoyl-GcvH and lipoyl-GcvH-L to E2-PDH appeared to be less efficient than the transfer to E2-OGDH and E2-BCODH (Fig. 2E to H and S2D to F). Taken together, these data indicate that LipL transfers the lipoyl moieties from GcvH or GcvH-L to all three E2 subunits, although lipoyl transfer to E2-PDH is less efficient.
FIG 2.

LipL-dependent modification of E2 subunits with lipoyl-H proteins as lipoyl donors. (A to C and E to G) Lipoyl transfer from lipoic acid (LA)-GcvH (10 μM) (A to C) and LA-GcvH-L (40 μM) (E to G) to E2-PDH (A and E), E2-OGDH (B and F), and E2-BCODH (C and G), with LipL (1 μM; right lanes) and without LipL (left lanes), was assessed by immunoblotting with rabbit anti-lipoic acid antibody. A portion (2.5 μl) of a 50-μl reaction mixture was loaded into each lane. Presented blots are representative of at least three independent experiments. (D and H) Densitometric quantification of lipoyl acceptor band intensities relative to lipoyl-E2-BCODH. The average and standard deviation from three independent blots are shown.
LipL-dependent lipoyl transfer from E2 subunits to H proteins.
The only direct biochemical evidence of amidotransfer from E2 subunits to H proteins comes from a demonstration of octanoyl transfer from the lipoyl domain of E2-BCODH to GcvH in L. monocytogenes (29). Thus, to establish the bidirectionality of lipoyl relay between H proteins and E2 subunits, we conducted a lipoyl transfer assay with each lipoyl-E2 protein and determined whether LipL catalyzes the lipoylation of H proteins using these substrates. Lipoylation of both GcvH and GcvH-L is more pronounced when the lipoyl donor is E2-PDH (Fig. 3A and B and S3A and B, lane 3), followed by E2-OGDH (Fig. 3A and B and S3A and B, lane 5). We detected minimal lipoyl-H proteins when lipoyl-E2-BCODH was used as a substrate (Fig. 3A and B and S3A and B, lane 7). Overall, LipL-dependent lipoylation of H proteins occurs with some, but not all, lipoyl-E2 protein substrates.
FIG 3.

LipL-dependent modification of H proteins with lipoyl-E2 subunits as lipoyl donors. Lipoyl transfer from LA-E2-PDH (10 μM) (lanes 2 and 3), LA-E2-OGDH (10 μM) (lanes 4 and 5), and LA-E2-BCODH (10 μM) (lanes 6 and 7) with LipL (1 μM) (lanes 3, 5, and 7) and without LipL (lanes 2, 4, and 6) to GcvH (20 μM) (A) and GcvH-L (20 μM) (B) was assessed by immunoblotting with rabbit anti-lipoic acid (α-LA) antibody. For positive controls, GcvH (20 μM) and GcvH-L (20 μM) were lipoylated with free lipoic acid (2.4 mM) by LplA1 (1 μM) (lanes 1). A portion (2.5 μl) of a 50-μl reaction mixture was loaded into each lane. Presented blots are representative of at least three independent experiments. Graphs contain densitometric quantification of the relative reduction in lipoylation after the addition of LipL. The average and standard deviation from three independent blots are shown.
LipL catalyzes lipoyl relay between E2 subunits.
The results from our genetic and biochemical analyses are consistent with a model where LipL transfers the lipoyl moiety from H proteins to the lipoyl domains of E2 subunits (3, 41, 42). However, it is unknown whether a lipoyl relay exists between E2 subunits due to a lack of suitable genetic mutants. Thus, we purified all three E2 subunits in apo- and lipoyl-forms and tested if lipoyl transfer between E2 subunits occurs in vitro. With lipoyl-E2-PDH, we observed comparable LipL-dependent modification of E2-OGDH and E2-BCODH (Fig. 4A to C and S4A and B). However, LipL-catalyzed lipoyl transfer from lipoyl-E2-OGDH to E2-PDH was less robust than was the transfer to E2-BCODH (Fig. 4D to F and S4C and D). We also detected more transfer to E2-OGDH than E2-PDH in reactions that used lipoyl-E2-BCODH (Fig. 4G to I and S4E and F). These results provide evidence that LipL relays the lipoyl moiety from one E2 subunit to another, although lipoyl transfer to E2-PDH is less efficient.
FIG 4.
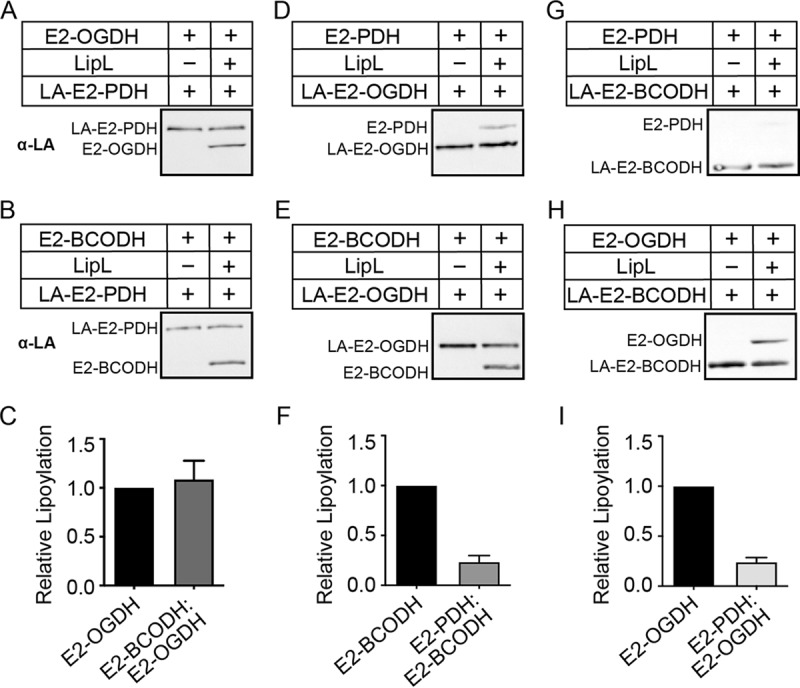
LipL-dependent lipoyl relay between E2 subunits. (A, B, D, E, G, and H) Lipoyl transfer from LA-E2-PDH (10 μM) (A and B), LA-E2-OGDH (10 μM) (D and E), and LA-E2-BCODH (10 μM) (G and H), to E2-OGDH (20 μM) (A and H), E2-BCODH (20 μM) (B and E), and E2-PDH (20 μM) (D and G), with LipL (1 μM) (right lanes) and without LipL (left), was assessed by immunoblotting with rabbit anti-lipoic acid antibody. A portion (2.5 μl) of a 50-μl reaction mixture was loaded into each lane. Presented blots are representative of at least three independent experiments. (C, F, and I) Densitometric quantification of relative lipoyl transfer efficiency compared to lipoyl-E2-OGDH (C and I) and lipoyl-E2-BCODH (F). The average and standard deviation from three independent blots are shown.
LipL catalyzes lipoyl transfer from GcvH-L to GcvH but not vice versa.
Since LipL catalyzes lipoyl relay between E2 subunits, we hypothesized that such a circuit also exists between H proteins. Therefore, we performed a lipoyl transfer assay with either lipoyl-GcvH or lipoyl-GcvH-L to determine if LipL uses one H protein to lipoylate the other. When lipoyl-GcvH-L was used, we observed complete loss of lipoylation on GcvH-L that coincided with the emergence of lipoyl-GcvH (Fig. 5 and S5, lane 8). In contrast, when lipoyl-GcvH was the donor, we neither detected a change in its levels nor observed the formation of lipoyl-GcvH-L (Fig. 5 and S5, lane 6). This finding suggests that the lipoyl moiety is preferentially transferred from GcvH-L to GcvH (3, 44).
FIG 5.
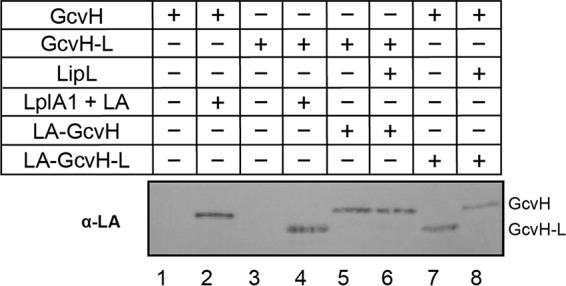
LipL-dependent lipoyl relay between H proteins. Lipoyl transfer from LA-GcvH (10 μM) to GcvH-L (20 μM) (lanes 5 and 6) and from LA-GcvH-L (40 μM) to GcvH (20 μM) (lanes 7 and 8), with LipL (1 μM) (lanes 6 and 8) and without LipL (lanes 5 and 7), was assessed by immunoblotting with rabbit anti-lipoic acid antibody. Apo- (lanes 1 and 3) and LplA1-lipoylated (lanes 2 and 4) GcvH and GcvH-L (20 μM) were included as controls. A portion (2.5 μl) of a 50-μl reaction mixture was loaded into each lane. The presented blot is representative of at least three independent experiments.
In vitro reconstitution of the complete LipL-dependent lipoyl relay network.
Thus far, we have explored LipL function in a one-to-one lipoyl donor-acceptor configuration. To recapitulate the complexity of LipL-dependent lipoyl relay, we conducted a lipoyl transfer assay using one lipoyl protein combined with the other four S. aureus apo-lipoyl domain-containing proteins in a reaction with LipL. When lipoyl-GcvH served as the lipoyl donor, E2-OGDH and E2-BCODH, and, to a lesser degree, E2-PDH but not GcvH-L, were modified (Fig. 6 and S6, lane 2). When lipoyl-GcvH-L was the donor, all of its lipoyl moiety was transferred onto E2-OGDH, E2-BCODH, and GcvH, but we were unable to detect lipoyl-E2-PDH (Fig. 6 and S6, lane 4). The use of lipoyl-E2-PDH resulted in transfer of the cofactor to E2-BCODH and GcvH but not E2-OGDH and GcvH-L (Fig. 6 and S6, lane 6). When lipoyl-E2-OGDH was used, E2-BCODH and GcvH were modified to a minor degree (Fig. 6 and S6, lane 8). Last, GcvH was the only lipoyl recipient from lipoyl-E2-BCODH (Fig. 6 and S6, lane 10). Taken together, LipL-dependent transfer of lipoyl moieties from either GcvH or GcvH-L to E2 subunits approximates what is seen in vivo, appears to be favored over the reverse reaction, and aligns well with outcomes of the one-to-one lipoyl donor-acceptor transfer assays. It is worth noting that reconstitution reactions will achieve equilibrium based on the varied efficiencies of lipoyl transfer; thus, not all transfers may be visible under each condition. In all cases, GcvH-L is a strong donor and weak acceptor of lipoic acid.
FIG 6.
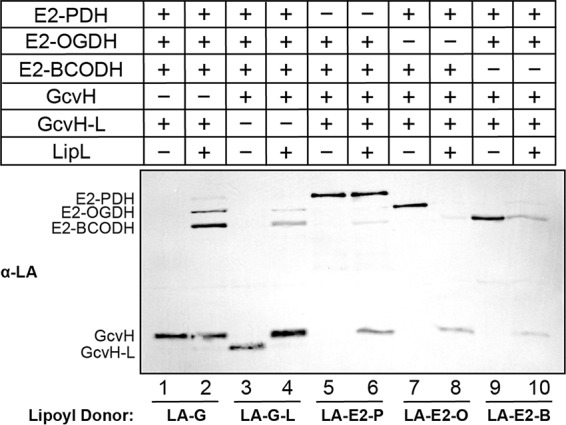
Simultaneous modification of multiple lipoyl domain-containing proteins by LipL. Lipoyl transfer from LA-GcvH (5 μM) (lanes 1 and 2), LA-GcvH-L (20 μM) (lanes 3 and 4), LA-E2-PDH (5 μM) (lanes 5 and 6), LA-E2-OGDH (5 μM) (lanes 7 and 8), and LA-E2-BCODH (5 μM) (lanes 9 and 10) to the other four apo-lipoyl domain-containing proteins (10 μM of each) with LipL (2 μM) (even-numbered lanes) and without LipL (odd-numbered lanes) was assessed by immunoblotting with rabbit anti-lipoic acid antibody. Five microliters of a 100-μl reaction mixture was loaded into each lane. The presented blot is representative of at least three independent experiments.
Lipoyl relay is dynamic in vivo.
Biochemical assessment of LipL function suggests that the amidotransferase engages in flexible relay of protein-bound lipoic acid. We wondered if changing lipoyl protein production in vivo would perturb the lipoyl relay network. S. aureus can grow under oxygen-limiting conditions, where the tricarboxylic acid (TCA) cycle enzyme OGDH is not produced. Therefore, we used oxygen limitation as well as genetic mutants of E2 and H subunits to monitor changes in the efficiency of LipL-dependent lipoyl relay (46). We assessed the lipoylation profiles of the S. aureus WT and ΔlipL, ΔgcvH, ΔpdhC (E2-PDH null), ΔodhB (E2-OGDH null) mutant strains and their complements, grown anaerobically or aerobically, in lipoic acid bypass medium (RPMI medium supplemented with branched-chain fatty acid precursors [BCFAP] and sodium acetate [NaAc]). When grown anaerobically, lipoyl-E2-OGDH was not detected in the WT strain but was detected in a ΔodhB mutant constitutively expressing odhB, confirming the reduction of odhB expression in the WT strain grown in the absence of oxygen (Fig. 7A, lanes 1 and 9). Despite conditional or genetic depletion of E2-OGDH, lipoylation of E2-PDH, E2-BCODH, and GcvH remained unchanged (Fig. 7A, lanes 1 and 8). When grown aerobically, the ΔpdhC and ΔodhB mutant strains also maintained the capacity to lipoylate all other proteins in a LipL-dependent manner (Fig. 7B, lanes 6 and 8).
FIG 7.

Flexibility of lipoyl relay in vivo and requirement of LipL for optimal infection. (A to C) Immunoblot with anti-lipoic acid antibody of whole-cell lysates derived from the indicated strains grown in RPMI medium supplemented with branched-chain fatty acid precursors (10 mM isobutyric acid, 9 mM 2-methylbutyric acid, 9 mM isovaleric acid) and 10 mM sodium acetate, under anaerobic conditions (−O2) at 37°C for 48 h (A), under aerobic conditions (+O2) at 37°C for 9 h after 1:100 subculture from overnight cultures (B), and under aerobic conditions in the presence of 5 μM lipoic acid (+O2 +LA) at 37°C for 9 h after 1:100 subculture from overnight cultures (C). Presented blots are representative of at least three independent experiments. (D) Bacterial burden in kidneys of mice at 96 h postinfection with 1 × 107 CFU WT (n = 8), ΔlipL mutant (n = 8), and ΔlipL+lipL mutant (n = 8) strains. (E) Bacterial burden in skin abscesses of mice at 120 h postinfection with 1 × 107 CFU WT (n = 16), ΔlipL mutant (n = 16), and ΔlipL+lipL mutant (n = 16) strains. Horizontal bars represent median values. P values were determined by Kruskal-Wallis test with Dunn’s posttest. **, P < 0.01; ***, P < 0.001.
Biochemical data suggest that the lipoic acid salvage enzymes LplA1 and LplA2 differentially target all three S. aureus E2 proteins for lipoylation with exogenous lipoic acid (44). Despite their protein targets in vitro, LplA2 does not contribute appreciably to lipoylation, while LplA1 lipoylates only E2-OGDH and GcvH under standard growth conditions (3). However, S. aureus maintains its ability to lipoylate all E2 proteins and does not appear to require GcvH to serve as an intermediate for lipoyl transfer (3, 44). This observation suggests that E2-OGDH must be capable of serving as a lipoyl donor for LipL-dependent modification of E2-PDH and E2-BCODH in vivo. To this end, we generated a ΔlipL ΔgcvH mutant to assess its lipoylation profile when grown in the presence of exogenous lipoic acid. We observed a loss of lipoyl-E2-PDH and lipoyl-E2-BCODH in a ΔlipL ΔgcvH mutant compared to a ΔgcvH mutant (Fig. 7C). Altogether, these findings imply that the flexibility of LipL amidotransferase activity allows for maintenance of protein lipoylation, even when lipoyl domain proteins are conditionally or genetically depleted, and that lipoyl transfer between E2 subunits occurs in vivo, which is supported by recent findings in B. subtilis (47).
LipL is important for S. aureus virulence.
A ΔlipL mutant is defective for growth in broth culture. This defect cannot be rescued by supplementing exogenous lipoic acid, although cell growth can be restored by culturing in bypass medium containing BCFAP and NaAc (3). Given that lipoic acid biosynthesis and salvage together promote tissue-specific infectivity during murine sepsis, we surmised that a ΔlipL mutant would be compromised for colonization in murine infection (3). We infected mice intravenously or intradermally with WT, ΔlipL mutant, and ΔlipL+lipL(ATG-1) complemented strains. At 96 h post-intravenous infection, kidneys of ΔlipL mutant-infected animals had ∼10,000-fold fewer CFU than those infected with WT and ΔlipL+lipL(ATG-1) mutant strains (Fig. 7D). At 120 h post-intradermal infection, ∼10-fold fewer CFU were recovered from abscesses of ΔlipL mutant-infected animals (Fig. 7E). Thus, LipL is required for optimal S. aureus infection.
DISCUSSION
In this study, we investigated the role of the putative S. aureus amidotransferase, LipL, in lipoic acid transfer and virulence. We found that the lipL open reading frame begins two nucleotides downstream of the phosphate acetyltransferase-encoding gene, pta, hinting at a functional relationship between LipL activity and overflow metabolism. Furthermore, we demonstrated that LipL not only shuttles lipoic acid from H proteins to E2 subunits and vice versa, but it also catalyzes inter-E2 and H protein lipoyl transfer. Hence, the ability of LipL to facilitate flexible circulation of endogenous protein-bound lipoic acid represents a potential mechanism used by S. aureus to adjust to changing metabolic demands, especially when the cofactor is limiting. In addition, we present evidence that the efficacy of LipL-dependent lipoyl transfer is dictated by the identities of both the lipoyl donor and acceptor. These features, combined with differential targeting of lipoyl domain-containing proteins by the salvage enzymes LplA1 and LplA2, underscores the complexity of the lipoyl relay network in S. aureus (44).
Among the three E2 subunits, E2-PDH appears to weakly accept lipoic acid via LipL regardless of which lipoyl protein is the donor (Fig. 2D and H, 4F and 4I, and 6). To our knowledge, the only previous attempt to assess lipoyl transfer from GcvH to E2-PDH was performed with the ∼100-amino-acid-residue inner lipoyl domain of human E2-PDH, which resulted in its robust modification (45). However, our ability to purify full-length E2-PDH in good yield and modify the protein with free lipoic acid in the presence of LplA2 leads us to believe that protein stability is not the primary reason for reduced lipoyl transfer efficiency (44). It is possible that the optimal modification of full-length E2-PDH requires it to be in complex with the E1 and E3 subunits of PDH. Similar to E2-PDH, GcvH-L is the weaker lipoyl acceptor of the two H proteins, although it is a viable lipoyl donor for LipL-dependent lipoyl relay (Fig. 2, 5, and 6). Most, if not all, of the lipoyl moiety from GcvH-L is transferred to other lipoyl domain-containing proteins in reactions that use lipoyl-GcvH-L as the donor. This observation could explain why, to date, lipoyl-GcvH-L remains undetectable in lysates from cells grown under standard in vitro culture conditions and supports a model where GcvH-L serves as a transient reservoir of salvaged lipoic acid (3, 44). Moreover, other groups have found that amino acid residues in the vicinity of the lipoyl-lysine dictate the efficiency of protein lipoylation (47, 48). For instance, whether the residue situated three positions to the N-terminal side of the lipoyl-lysine is charged determines the rate of lipoylation and substrate specificity (47, 48). Although this residue is identical in GcvH and GcvH-L, a comparison of their lipoyl domains shows significant amino acid differences that could account for the observed contrast in lipoyl transfer outcomes between both proteins (Fig. S7). Upon being lipoylated, GcvH-L can also be reversibly ADP ribosylated on a residue near the lipoyl-lysine (43). We surmise that this modification, which is not known to occur on GcvH, could regulate the capacity of GcvH-L to be lipoylated.
While we show that LipL catalyzes lipoyl transfer from H proteins to E2 subunits (Fig. 2), the amidotransferase is also capable of catalyzing the reverse reaction from E2 subunits to H proteins (Fig. 3). In this case, H proteins are most readily modified when lipoyl-E2-PDH is the donor, followed by lipoyl-E2-OGDH. In general, based on the intensity of lipoic acid antibody-reactive bands that correspond to the lipoyl-accepting proteins, the reverse reaction is less favorable than the forward reaction. This observation is supported by genetic evidence in which a GcvH-deficient mutant has diminished lipoyl-E2 protein levels in the absence of exogenous lipoic acid, but the ΔpdhC and ΔodhB mutants retain levels of lipoyl-GcvH comparable to those of the WT strain when grown in RPMI medium plus BCFAP and NaAc (3) (Fig. 7B). Nonetheless, reversible protein lipoylation may allow cells to respond to environmental changes by transferring the lipoyl moiety to accommodate metabolic demands, especially in the event of lipoic acid starvation or reduced de novo synthesis. Our demonstration of inter-E2 and H protein lipoyl transfer (Fig. 4 and 5) would likely enable rapid adaptation to metabolic shifts by promoting unhindered lipoyl relay regardless of the substrate. Such a shift could be elicited by carbon source flux, which is known to influence the expression of α-ketoacid dehydrogenases (49–51).
The flexibility of lipoyl relay in S. aureus was further highlighted by our reconstitution of the complete LipL-dependent lipoyl relay network in vitro, where each lipoyl protein served as a donor for all remaining proteins (Fig. 6). In this scenario, GcvH was a primary recipient of lipoic acid, even in the presence of apo-E2 proteins (Fig. 6, lanes 6, 8, and 10). This finding suggests that reversible lipoyl relay from E2 subunits to GcvH is favored over relay between E2 subunits. The apparent centrality of GcvH in reversible lipoyl transfer, as well as its position as a major node in de novo synthesis and salvage of lipoic acid, is consistent with the suggested role of GcvH as the primary reservoir of lipoic acid within the cell (36).
Our studies using anaerobic growth conditions to experimentally reduce the expression of lipoyl-E2-OGDH demonstrated that WT S. aureus still maintains an otherwise-normal lipoylation profile (Fig. 7A, lane 1). Thus, the reduced expression of one major E2 protein, via a change in environmental conditions, does not compromise the ability of S. aureus to lipoylate critical E2 proteins needed in anaerobiosis, such as E2-PDH. This observation is consistent with evidence from a prior study in Escherichia coli that reported an important role of PDH in fermentative metabolism of glucose and the downregulation of genes encoding tricarboxylic acid cycle enzymes in S. aureus during anoxia (46, 52). Likewise, the deletion of pdhC and odhB does not disrupt overall protein lipoylation, which likely occurs as a result of lipoyl transfer from GcvH (Fig. 7A and B, lanes 6 and 8). However, WT levels of E2 subunit lipoylation are also observed in a ΔgcvH mutant in the presence of exogenous lipoic acid, suggesting that inter-E2 transfer during salvage can compensate to maintain adequate protein lipoylation (44). This observation is supported by the loss of E2-PDH and E2-BCODH lipoylation in a ΔlipL ΔgcvH mutant (Fig. 7C). Thus, both conditional and genetic manipulation of lipoyl relay donors and recipients highlight the compensating effects of LipL plasticity that permit the maintenance of metabolic enzyme lipoylation.
The importance of LipL to S. aureus metabolism is further demonstrated in a disease environment, where loss of the amidotransferase caused a marked reduction in bacterial burden during bloodstream infection, and, to a lesser degree, during skin and soft tissue infection (Fig. 7D and E). We hypothesize that the modest defect of lipL mutant colonization in skin may be due to the ability of S. aureus to incorporate host-derived fatty acids into its membrane, which could compensate for the loss of branched-chain fatty acid synthesis by BCODH in a lipL mutant (10, 53, 54). Indeed, sebaceous glands in the skin produce unique lipid species not found in other tissues that could potentially serve as a source of fatty acids for S. aureus (55). Further investigation into this question is under way. Considering the lack of homology between S. aureus LipL and human LIPT1 (Fig. S8) and that LipL is only found in Gram-positive bacteria, the amidotransferase makes an attractive target for novel approaches to combat S. aureus disease (45). Therefore, our studies to better understand bacterial metabolism both shed light on conserved aspects of metabolic pathways across organisms and uncover previously unappreciated roles for bacterial metabolic components during pathogenesis that can be exploited for clinical benefit.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All bacterial strains used in this paper are listed in Table S1 in the supplemental material. E. coli strains were routinely grown in Miller’s lysogeny broth (LB; BD), with antibiotics added as necessary. S. aureus strains were grown in either tryptic soy broth (TSB; BD) or in RPMI medium (Corning) supplemented with 1% Casamino Acids (VWR). All strains were grown overnight with shaking, unless stated otherwise. To probe in vivo lipoylation, S. aureus strains were subcultured 1:100 from overnight cultures into 5 ml fresh RPMI medium containing branched-chain fatty acid precursors (BCFAP) (10 mM isobutyric acid [Sigma-Aldrich], 9 mM 2-methylbutyric acid [Alfa Aesar], 9 mM isovaleric acid [Sigma-Aldrich]) and 10 mM sodium acetate (NaAc; Sigma-Aldrich) in 15-ml conical tubes and grown with shaking (220 rpm) at 37°C for 9 h. To determine protein lipoylation in cells grown under anaerobic conditions, 5 ml RPMI medium plus BCFAP and NaAc were added to 15-ml conical tubes and reduced in a BBL GasPak jar (BD) at 37°C overnight before inoculation with a single S. aureus colony. The inoculated tubes were incubated in the same anaerobic jar with fresh palladium catalyst (BD), an anaerobe container system sachet (BD), and resazurin anaerobic indicator strip (Oxoid) at 37°C for 48 h. When needed, cultures were supplemented with the following agents for selection: 100 μg ml−1 ampicillin (GoldBio), 10 μg ml−1 chloramphenicol (Amresco), 150 μg ml−1 kanamycin (Amresco), and 0.1 mM cadmium chloride (Alfa Aesar).
Growth curve.
WT and ΔlipL, ΔlipL+lipL(ATG-1), and ΔlipL+lipL(ATG-2) mutant strains were grown in triplicate in 200 μl of fresh RPMI medium plus BCFAP and NaAc in a 96-well, round-bottom plate (Celltreat) overnight at 37°C with shaking. The bacteria were washed three times and resuspended in the same volume of fresh RPMI medium before being subcultured 1:100 into 198 μl of the same medium in a 96-well, flat-bottom plate (Celltreat) at 37°C with shaking in a Synergy H1 hybrid multimode microplate reader (Biotek). The optical density at 550 nm (OD550) was measured every 15 min over a 10-h period.
Generation of ΔlipL+lipL(ATG-1) mutant strain.
The ΔlipL+lipL(ATG-1) mutant strain was generated using the pJC1111 plasmid (56). Using the primers listed in Table S2, the lipL gene was amplified from WT S. aureus genomic DNA, while the constitutive PHELP promoter was amplified from pIMAY plasmid (57). The resulting amplicons were subjected to splicing by overlap extension (SOE) PCR to obtain PHELP-lipL(ATG-1), which was cloned into pJC1111 at the PstI and SalI restriction sites (58). The recombinant construct was propagated in E. coli DH5α and transformed into S. aureus pathogenicity island 1 (SaPI-1) integrase-expressing S. aureus (RN9011) to allow single-copy integration at the SaPI-1 site in the chromosome (56, 58). Bacteriophage Φ11 was used to package the integrated complementation plasmid from RN9011 and was subsequently transduced into the ΔlipL mutant strain. ΔlipL+lipL(ATG-1) transductants were selected for based on cadmium chloride resistance and confirmed by PCR.
Generation of ΔpdhC+pdhC and ΔodhB+odhB mutant strains.
The ΔpdhC+pdhC and ΔodhB+odhB mutant strains were generated using the pOS1 plasmid (59). Using the primers listed in Table S2, the pdhC and odhB genes were amplified from WT S. aureus genomic DNA, while the nucleotide sequence corresponding to the constitutive S. aureus sarA promoter fused to the sod ribosome binding site (PsarA-sodRBS) was amplified from pOS1-PsarA-sodRBS-sgfp (60). The resulting amplicons were fused by SOE PCR, and the products were cloned into pOS1 at the EcoRI and SalI restriction sites (58). The recombinant constructs were propagated in E. coli DH5α and transformed into S. aureus RN4220 (61) for isolation and subsequent transformation into the ΔpdhC and ΔodhB mutant strains.
Generation of a ΔlipL ΔgcvH strain.
A marked ΔgcvH::kan mutation was transduced into a ΔlipL mutant strain, as described previously, to generate a ΔlipL ΔgcvH mutant strain (3).
Whole-cell lysate preparation.
S. aureus cultures were measured for OD550 and centrifuged at 4,200 rpm for 15 min to pellet the bacteria. The supernatants were removed, and the bacterial cell pellets were resuspended in 250 μl of phosphate-buffered saline. The resuspended bacteria were then added to screw-cap microcentrifuge lysing tubes (Fisher Scientific) containing 250 μl of 0.1-mm glass cell disruption beads (Scientific Industries, Inc.). Cells were lysed using a Fast Prep-24 5G homogenizer (MP Biomedicals) at a speed of 5.0 m/s for 20 s with subsequent incubation on ice for 5 min and then disrupted a second time at a speed of 4.5 m/s for 20 s. Thirty microliters of 6× sodium dodecyl sulfate (SDS) sample buffer (0.375 M Tris [pH 6.8], 12% SDS, 60% glycerol, 0.6 M dithiothreitol [DTT], 0.06% bromophenol blue) was added to the 90 μl of cell lysates, and the mixture was boiled for 10 min. Samples were stored at –20°C for no longer than a few days.
Purification of 6×His-LipL.
6×His-LipL was purified using Ni2+ affinity chromatography. The lysY/Iq E. coli strain containing pET15b-lipL was grown in 10 ml LB with 100 μg ml−1 ampicillin at 37°C with shaking overnight. The following day, bacteria were subcultured 1:100 into 500 ml fresh LB with 100 μg ml−1 ampicillin and 2 mM MgSO4 and allowed to grow for 3 h at 37°C shaking. Protein synthesis was induced by the addition of 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG), followed by incubation for another 4 h at 37°C with shaking. Bacteria were pelleted by centrifugation at 8,801 × g for 10 min at 4°C and stored overnight at −80°C. To purify the recombinant proteins, bacterial pellets were thawed in a 37°C water bath and resuspended in lysis buffer (25 mM imidazole, 50 mM Tris-HCl, 300 mM NaCl [pH 8]) supplemented with 1 mM DTT and 1 mM phenylmethylsulfonyl fluoride (PMSF). Bacteria were lysed at a constant rate of 0.8 s per pulse and an output of 340 W in 20-s intervals for 15 min on ice using a Branson S-450A large-tip sonicator. Debris from lysed bacteria was pelleted by centrifugation for 30 min at 16,000 × g, followed by filtration of the clarified lysate using a 0.45-μm syringe filter. The lysate was then incubated with 1 ml equilibrated nickel-nitrilotriacetic acid (nickel-NTA) resin slurry (Qiagen) on a rotisserie for 1 h at 4°C. Nickel-NTA-bound 6×His-LipL was washed with lysis buffer and eluted using the same buffer containing 500 mM imidazole. Samples were dialyzed with 10-kDa molecular weight cutoff (MWCO) regenerated cellulose dialysis tubing (Spectrum) using the following scheme: the sample-filled tubing was first placed in lysis buffer containing 100 mM imidazole for 3 h, in the same buffer containing 25 mM imidazole overnight, and then another 3 h in lysis buffer only. The concentration of purified 6×His-LipL was measured using a bicinchoninic acid (BCA) kit (Thermo Fisher Scientific) and stored at −80°C. 6×His-LipL was confirmed to be purified to apparent homogeneity by resolving ∼2 μg of protein by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), followed by staining of the gel with GelCode blue stain reagent (Thermo Fisher Scientific).
Purification of 6×His-LplA1 and 6×His-LplA2.
6×His-LplA1 and 6×His-LplA2 were purified as previously described (44).
Purification of 6×His-apo-E2 subunits and 6×His-apo-H proteins.
6×His-apo-E2 subunits and 6×His-apo-H proteins were purified from the lysY/Iq ΔlipA::kan E. coli strain using Ni2+ affinity chromatography. lysY/Iq ΔlipA::kan E. coli strains containing pET15b-pdhC, pET15b-odhB, pET15b-bmfBB, pET15b-gcvH, and pET15b-gcvH-L were grown overnight in 20 ml LB with 100 μg ml−1 ampicillin at 37°C with shaking. The following day, strains were subcultured 1:100 into 1 liter LB with 100 μg ml−1 ampicillin and grown for 3 h. Protein expression was induced with 0.4 mM IPTG, followed by incubation for another 3 h at 37°C with shaking. The subsequent steps were the same as described above for the purification of 6×His-LipL.
Purification of 6×His-lipoyl-E2 subunits and 6×His-lipoyl-H proteins.
6×His-lipoyl-E2 subunits and 6×His-lipoyl-H proteins were purified from lysY/Iq E. coli strains containing the same constructs used for purification of 6×His-apo-E2 subunits and 6×His-apo-H proteins, using Ni2+ affinity chromatography. Strains were grown in 10 ml LB with 100 μg ml−1 ampicillin at 37°C with shaking overnight. The following day, bacteria were subcultured 1:100 into 500 ml fresh LB with 100 μg ml−1 ampicillin and 2 mM MgSO4 and allowed to grow for 3 h at 37°C shaking. Lipoyl protein expression was induced by addition of 0.4 mM IPTG and 100 μM lipoic acid (Sigma-Aldrich) and grown for another 3 h at 30°C with shaking. Further protein synthesis was inhibited by addition of 150 μg ml−1 kanamycin and cells were incubated for an additional hour. The subsequent steps were the same as described above for the purification of 6×His-LipL.
Assay of LipL function with free lipoic acid as the substrate.
The lipoic acid ligation assay was set up as previously described with 1 μM each of 6×His-LipL, 6×His-LplA1, and 6×His-LplA2 (44).
Assay of LipL function with one-to-one lipoyl donor-acceptor configuration.
Lipoylation assays were set up as previously described, with some modifications (44). All assays were conducted in 50-μl reaction volumes in reaction buffer (50 mM Tris-HCl, 300 mM NaCl [pH 8]) containing 1 mM DTT, 1 mM MgCl2, 20 μM lipoyl acceptor (6×His-apo-E2-PDH, 6×His-apo-E2-OGDH, 6×His-apo-E2-BCODH, 6×His-apo-GcvH, or 6×His-apo-GcvH-L), and 10 μM lipoyl donor (6×His-lipoyl-E2-PDH, 6×His-lipoyl-E2-OGDH, 6×His-lipoyl-E2-BCODH, or 6×His-lipoyl-GcvH). Forty micromolar 6×His-GcvH-L was used as a lipoyl donor to account for incomplete in vivo lipoylation of the recombinant protein. Reaction mixtures were incubated with or without 1 μM 6×His-LipL for 2 h at 37°C with shaking. After incubation, 5 μl of the reaction mixtures was resolved on 4 to 15% Mini-Protean TGX stain-free gels (Bio-Rad), followed by staining with the GelCode blue stain reagent (Thermo Fisher Scientific), and another 5 μl on 12% SDS-PAGE gels at 120 V for immunoblotting to determine protein lipoylation (see below).
Assay of LipL function with one-to-four lipoyl donor-acceptor configuration.
Lipoylation assays were conducted in 100-μl reaction volumes under the same buffer conditions as described above, with 10 μM each lipoyl acceptor (6×His-apo-E2-PDH, 6×His-apo-E2-OGDH, 6×His-apo-E2-BCODH, 6×His-apo-GcvH, or 6×His-apo-GcvH-L) and 5 μM lipoyl donor (6×His-lipoyl-E2-PDH, 6×His-lipoyl-E2-OGDH, 6×His-lipoyl-E2-BCODH, or 6×His-lipoyl-GcvH). A concentration of 20 μM 6×His-GcvH-L was used as lipoyl donor to account for incomplete in vivo lipoylation of the recombinant protein. Reaction mixtures were incubated with or without 2 μM 6×His-LipL for 2 h at 37°C with shaking. After incubation, 10 μl of the reaction mixtures was resolved on 4 to 15% Mini-Protean TGX stain-free gels (Bio-Rad), followed by staining with GelCode blue stain reagent (Thermo Fisher Scientific) and another 10 μl on 12% SDS-PAGE gels at 120 V for immunoblotting to determine protein lipoylation (see below).
Determination of protein lipoylation and densitometry.
Proteins from optical density (OD)-normalized whole-cell lysates or in vitro lipoylation assays were separated by SDS-PAGE in 12% polyacrylamide gels at 120 V. Separated proteins were transferred from gels to 0.2-μm Immobilon polyvinylidene difluoride (PVDF) membranes (Millipore Sigma) at 100 V for 30 min in a Criterion blotter (Bio-Rad). After transfer, membranes were incubated for 1 h with TBST (0.1% Tween 20 [Amresco] in Tris-buffered saline) supplemented with 5% bovine serum albumin (BSA; GoldBio). Rabbit polyclonal anti-lipoic acid antibody (Calbiochem) was added to the membranes at a 1:7,500 dilution, followed by incubation for 1 h and three subsequent 15-minute washes in ∼20 ml of TBST. Alkaline phosphatase (AP)-conjugated goat anti-rabbit IgG (H+L) (Invitrogen) was then added at a 1:5,000 dilution in 5% BSA in TBST for 45 min, followed by three 15-minute washes in ∼20 ml of TBST and another 5-min wash in AP buffer (100 mM Tris [pH 9.5], 100 mM NaCl, 5 mM MgCl2). Membranes were developed with 5-bromo-4-chloro-3-indoylphosphate–nitroblue tetrazolium (BCIP-NBT) color development substrate (GoldBio). Densitometry analysis was performed using the ImageJ software on three independent experiments to determine the lipoylation of each band relative to the control band set at a value of 1.
Murine infection models.
Murine skin and soft tissue and systemic infections were performed as previously described with WT, ΔlipL mutant, and ΔlipL+lipL(ATG-1) mutant strains (3, 62).
Ethics statement.
All experiments were performed following the ethical standards of the Institutional Biosafety Committee and the Institutional Animal Care and Use Committee (IACUC) at Loyola University Chicago Health Sciences Division. The institution is approved by the Public Health Service (PHS; A3117-01 through 28 February 2022), is fully accredited by the AAALAC International (000180, certification dated 17 November 2016), and is registered/licensed by the USDA (33-R-0024 through 24 August 2020). All animal experiments were performed in animal biosafety level 2 (ABSL2) facilities with IACUC-approved protocols (IACUC no. 2017028) under the guidance of the Office of Laboratory Animal Welfare following USDA and PHS policy and guidelines on the humane care and use of laboratory animals.
Quantification and statistical analyses.
All experiments were repeated at least three independent times. Statistical analyses of data from animal studies were performed using GraphPad Prism 7 with the statistical test specified in each figure legend.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the laboratory for helpful discussions and for critically reviewing the manuscript.
This study was supported by grants NIH R01 AI120994 to F.A. and AHA 19POST34380259 to W.P.T.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00446-19.
REFERENCES
- 1.Thammavongsa V, Kim HK, Missiakas D, Schneewind O. 2015. Staphylococcal manipulation of host immune responses. Nat Rev Microbiol 13:529–543. doi: 10.1038/nrmicro3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seilie ES, Bubeck Wardenburg J. 2017. Staphylococcus aureus pore-forming toxins: the interface of pathogen and host complexity. Semin Cell Dev Biol 72:101–116. doi: 10.1016/j.semcdb.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zorzoli A, Grayczyk JP, Alonzo F. 2016. Staphylococcus aureus tissue infection during sepsis is supported by differential use of bacterial or host-derived lipoic acid. PLoS Pathog 12:e1005933. doi: 10.1371/journal.ppat.1005933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kehl-Fie TE, Chitayat S, Hood MI, Damo S, Restrepo N, Garcia C, Munro KA, Chazin WJ, Skaar EP. 2011. Nutrient metal sequestration by calprotectin inhibits bacterial superoxide defense, enhancing neutrophil killing of Staphylococcus aureus. Cell Host Microbe 10:158–164. doi: 10.1016/j.chom.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dale SE, Doherty-Kirby A, Lajoie G, Heinrichs DE. 2004. Role of siderophore biosynthesis in virulence of Staphylococcus aureus: identification and characterization of genes involved in production of a siderophore. Infect Immun 72:29–37. doi: 10.1128/IAI.72.1.29-37.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coulter SN, Schwan WR, Ng EY, Langhorne MH, Ritchie HD, Westbrock-Wadman S, Hufnagle WO, Folger KR, Bayer AS, Stover CK. 1998. Staphylococcus aureus genetic loci impacting growth and survival in multiple infection environments. Mol Microbiol 30:393–404. doi: 10.1046/j.1365-2958.1998.01075.x. [DOI] [PubMed] [Google Scholar]
- 7.Connolly J, Boldock E, Prince LR, Renshaw SA, Whyte MK, Foster SJ. 2017. Identification of Staphylococcus aureus factors required for pathogenicity and growth in human blood. Infect Immun 85:17. doi: 10.1128/IAI.00337-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beasley FC, Marolda CL, Cheung J, Buac S, Heinrichs DE. 2011. Staphylococcus aureus transporters Hts, Sir, and Sst capture iron liberated from human transferrin by staphyloferrin A, staphyloferrin B, and catecholamine stress hormones, respectively, and contribute to virulence. Infect Immun 79:2345–2355. doi: 10.1128/IAI.00117-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kourtis AP, Hatfield K, Baggs J, Mu Y, See I, Epson E, Nadle J, Kainer MA, Dumyati G, Petit S, Ray SM, Emerging Infections Program MRSA author group, Ham D, Capers C, Ewing H, Coffin N, McDonald LC, Jernigan J, Cardo D. 2019. Vital signs: epidemiology and recent trends in methicillin-resistant and in methicillin-susceptible Staphylococcus aureus bloodstream infections—United States. MMWR Morb Mortal Wkly Rep 68:214–219. doi: 10.15585/mmwr.mm6809e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delekta PC, Shook JC, Lydic TA, Mulks MH, Hammer ND. 2018. Staphylococcus aureus utilizes host-derived lipoprotein particles as sources of fatty acids. J Bacteriol 200:e00728-17. doi: 10.1128/JB.00728-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Juttukonda LJ, Berends ETM, Zackular JP, Moore JL, Stier MT, Zhang Y, Schmitz JE, Beavers WN, Wijers CD, Gilston BA, Kehl-Fie TE, Atkinson J, Washington MK, Peebles RS, Chazin WJ, Torres VJ, Caprioli RM, Skaar EP. 2017. Dietary manganese promotes staphylococcal infection of the heart. Cell Host Microbe 22:531–542.e8. doi: 10.1016/j.chom.2017.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelliher JL, Radin JN, Grim KP, Párraga Solórzano PK, Degnan PH, Kehl-Fie TE. 2018. Acquisition of the phosphate transporter NptA enhances Staphylococcus aureus pathogenesis by improving phosphate uptake in divergent environments. Infect Immun 86:e00631-17. doi: 10.1128/IAI.00631-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grim KP, San Francisco B, Radin JN, Brazel EB, Kelliher JL, Párraga Solórzano PK, Kim PC, McDevitt CA, Kehl-Fie TE. 2017. The metallophore staphylopine enables Staphylococcus aureus to compete with the host for zinc and overcome nutritional immunity. mBio 8:e01281-17. doi: 10.1128/mBio.01281-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Radin JN, Zhu J, Brazel EB, McDevitt CA, Kehl-Fie TE. 2019. Synergy between nutritional immunity and independent host defenses contributes to the importance of the MntABC manganese transporter during Staphylococcus aureus Infection. Infect Immun 87:e00642-18. doi: 10.1128/IAI.00642-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelliher JL, Radin JN, Kehl-Fie TE. 2018. PhoPR contributes to Staphylococcus aureus growth during phosphate starvation and pathogenesis in an environment-specific manner. Infect Immun 86:e00371-18. doi: 10.1128/IAI.00371-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruiz J, Castro I, Calabuig E, Salavert M. 2017. Non-antibiotic treatment for infectious diseases. Rev Esp Quimioter 30(Suppl 1):66. [PubMed] [Google Scholar]
- 17.Cassat JE, Skaar EP. 2012. Metal ion acquisition in Staphylococcus aureus: overcoming nutritional immunity. Semin Immunopathol 34:215–235. doi: 10.1007/s00281-011-0294-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Packer L, Witt EH, Tritschler HJ. 1995. Alpha-lipoic acid as a biological antioxidant. Free Radic Biol Med 19:227–250. doi: 10.1016/0891-5849(95)00017-R. [DOI] [PubMed] [Google Scholar]
- 19.Reed LJ, Hackert ML. 1990. Structure-function relationships in dihydrolipoamide acyltransferases. J Biol Chem 265:8971. [PubMed] [Google Scholar]
- 20.O’Neill HC, Rancourt RC, White CW. 2008. Lipoic acid suppression of neutrophil respiratory burst: effect of NADPH. Antioxid Redox Signal 10:277–285. doi: 10.1089/ars.2007.1890. [DOI] [PubMed] [Google Scholar]
- 21.Moini H, Packer L, Saris NL. 2002. Antioxidant and prooxidant activities of α-lipoic acid and dihydrolipoic acid. Toxicol Appl Pharmacol 182:84–90. doi: 10.1006/taap.2002.9437. [DOI] [PubMed] [Google Scholar]
- 22.Spalding MD, Prigge ST. 2010. Lipoic acid metabolism in microbial pathogens. Microbiol Mol Biol Rev 74:200–228. doi: 10.1128/MMBR.00008-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Afanador GA, Guerra AJ, Swift RP, Rodriguez RE, Bartee D, Matthews KA, Schön A, Freire E, Freel Meyers CL, Prigge ST. 2017. A novel lipoate attachment enzyme is shared by Plasmodium and Chlamydia species. Mol Microbiol 106:439–451. doi: 10.1111/mmi.13776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jhun H, Walters MS, Prigge ST. 2018. Using lipoamidase as a novel probe to interrogate the importance of lipoylation in Plasmodium falciparum. mBio 9:e01872-18. doi: 10.1128/mBio.01872-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Afanador GA, Matthews KA, Bartee D, Gisselberg JE, Walters MS, Freel Meyers CL, Prigge ST. 2014. Redox-dependent lipoylation of mitochondrial proteins in Plasmodium falciparum. Mol Microbiol 94:156–171. doi: 10.1111/mmi.12753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Falkard B, Kumar TRS, Hecht L-S, Matthews KA, Henrich PP, Gulati S, Lewis RE, Manary MJ, Winzeler EA, Sinnis P, Prigge ST, Heussler V, Deschermeier C, Fidock D. 2013. A key role for lipoic acid synthesis during Plasmodium liver stage development. Cell Microbiol 15:1585–1604. doi: 10.1111/cmi.12137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Storm J, Muller S. 2012. Lipoic acid metabolism of Plasmodium–a suitable drug target. Curr Pharm Des 18:3480–3489. doi: 10.2174/138161212801327266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keeney KM, Stuckey JA, O’Riordan MXD. 2007. LplA1‐dependent utilization of host lipoyl peptides enables Listeria cytosolic growth and virulence. Mol Microbiol 66:758–770. doi: 10.1111/j.1365-2958.2007.05956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christensen QH, Hagar JA, O’Riordan MXD, Cronan JE. 2011. A complex lipoate utilization pathway in Listeria monocytogenes. J Biol Chem 286:31447. doi: 10.1074/jbc.M111.273607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Riordan M, Moors MA, Portnoy DA. 2003. Listeria intracellular growth and virulence require host-derived lipoic acid. Science 302:462–464. doi: 10.1126/science.1088170. [DOI] [PubMed] [Google Scholar]
- 31.Grayczyk JP, Harvey CJ, Laczkovich I, Alonzo F. 2017. A lipoylated metabolic protein released by Staphylococcus aureus suppresses macrophage activation. Cell Host Microbe 22:678–687.e9. doi: 10.1016/j.chom.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bryk R, Arango N, Maksymiuk C, Balakrishnan A, Wu Y, Wong CH, Masquelin T, Hipskind P, Lima CD, Nathan C. 2013. Lipoamide channel-binding sulfonamides selectively inhibit mycobacterial lipoamide dehydrogenase. Biochemistry 52:9375–9384. doi: 10.1021/bi401077f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bryk R, Lima CD, Erdjument-Bromage H, Tempst P, Nathan C. 2002. Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science 295:1073–1077. doi: 10.1126/science.1067798. [DOI] [PubMed] [Google Scholar]
- 34.Dacheux D, Epaulard O, de Groot A, Guery B, Leberre R, Attree I, Polack B, Toussaint B. 2002. Activation of the Pseudomonas aeruginosa type III secretion system requires an intact pyruvate dehydrogenase aceAB operon. Infect Immun 70:3973–3977. doi: 10.1128/IAI.70.7.3973-3977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pilatz S, Breitbach K, Hein N, Fehlhaber B, Schulze J, Brenneke B, Eberl L, Steinmetz I. 2006. Identification of Burkholderia pseudomallei genes required for the intracellular life cycle and in vivo virulence. Infect Immun 74:3576–3586. doi: 10.1128/IAI.01262-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cronan JE. 2016. Assembly of lipoic acid on its cognate enzymes: an extraordinary and essential biosynthetic pathway. Microbiol Mol Biol Rev 80:429–450. doi: 10.1128/MMBR.00073-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perham RN. 2000. Swinging arms and swinging domains in multifunctional enzymes: catalytic machines for multistep reactions. Annu Rev Biochem 69:961–1004. doi: 10.1146/annurev.biochem.69.1.961. [DOI] [PubMed] [Google Scholar]
- 38.Christensen QH, Cronan JE. 2010. Lipoic acid synthesis: a new family of octanoyltransferases generally annotated as lipoate protein ligases. Biochemistry 49:10024–10036. doi: 10.1021/bi101215f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin N, Lombardía E, Altabe SG, de Mendoza D, Mansilla MC. 2009. A lipA (yutB) mutant, encoding lipoic acid synthase, provides insight into the interplay between branched-chain and unsaturated fatty acid biosynthesis in Bacillus subtilis. J Bacteriol 191:7447–7455. doi: 10.1128/JB.01160-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCarthy EL, Booker SJ. 2017. Destruction and reformation of an iron-sulfur cluster during catalysis by lipoyl synthase. Science 358:373–377. doi: 10.1126/science.aan4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin N, Christensen QH, Mansilla MC, Cronan JE, de Mendoza D. 2011. A novel two-gene requirement for the octanoyltransfer reaction of Bacillus subtilis lipoic acid biosynthesis. Mol Microbiol 80:335–349. doi: 10.1111/j.1365-2958.2011.07597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Christensen QH, Martin N, Mansilla MC, de Mendoza D, Cronan JE. 2011. A novel amidotransferase required for lipoic acid cofactor assembly in Bacillus subtilis. Mol Microbiol 80:350–363. doi: 10.1111/j.1365-2958.2011.07598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rack J, Morra R, Barkauskaite E, Kraehenbuehl R, Ariza A, Qu Y, Ortmayer M, Leidecker O, Cameron DR, Matic I, Peleg AY, Leys D, Traven A, Ahel I. 2015. Identification of a class of protein ADP-ribosylating sirtuins in microbial pathogens. Mol Cell 59:309–320. doi: 10.1016/j.molcel.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laczkovich I, Teoh WP, Flury S, Grayczyk JP, Zorzoli A, Alonzo F. 2018. Increased flexibility in the use of exogenous lipoic acid by Staphylococcus aureus. Mol Microbiol 109:150–168. doi: 10.1111/mmi.13970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao X, Zhu L, Song X, Hu Z, Cronan JE. 2018. Protein moonlighting elucidates the essential human pathway catalyzing lipoic acid assembly on its cognate enzymes. Proc Natl Acad Sci U S A 115:E7063–E7072. doi: 10.1073/pnas.1805862115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park MK, Myers RAM, Marzella L. 1992. Oxygen tensions and infections: modulation of microbial growth, activity of antimicrobial agents, and immunologic responses. Clin Infect Dis 14:720–740. doi: 10.1093/clinids/14.3.720. [DOI] [PubMed] [Google Scholar]
- 47.Rasetto NB, Lavatelli A, Martin N, Mansilla MC. 2019. Unravelling the lipoyl-relay of exogenous lipoate utilization in Bacillus subtilis. Mol Microbiol 112:302–316. doi: 10.1111/mmi.14271. [DOI] [PubMed] [Google Scholar]
- 48.Fujiwara K, Okamura-Ikeda K, Motokawa Y. 1996. Lipoylation of acyltransferase components of α-ketoacid dehydrogenase complexes. J Biol Chem 271:12932–12936. doi: 10.1074/jbc.271.22.12932. [DOI] [PubMed] [Google Scholar]
- 49.Ward DE, van Der Weijden CC, van Der Merwe MJ, Westerhoff HV, Claiborne A, Snoep JL. 2000. Branched-chain alpha-keto acid catabolism via the gene products of the bkd operon in Enterococcus faecalis: a new, secreted metabolite serving as a temporary redox sink. J Bacteriol 182:3239–3246. doi: 10.1128/JB.182.11.3239-3246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Resnekov O, Melin L, Carlsson P, Mannerlöv M, von Gabain A, Hederstedt L. 1992. Organization and regulation of the Bacillus subtilis odhAB operon, which encodes two of the subenzymes of the 2-oxoglutarate dehydrogenase complex. Mol Gen Genet 234:285–296. doi: 10.1007/BF00283849. [DOI] [PubMed] [Google Scholar]
- 51.Blencke HM, Homuth G, Ludwig H, Mäder U, Hecker M, Stülke J. 2003. Transcriptional profiling of gene expression in response to glucose in Bacillus subtilis: regulation of the central metabolic pathways. Metab Eng 5:133–149. doi: 10.1016/S1096-7176(03)00009-0. [DOI] [PubMed] [Google Scholar]
- 52.Fuchs S, Pané-Farré J, Kohler C, Hecker M, Engelmann S. 2007. Anaerobic gene expression in Staphylococcus aureus. J Bacteriol 189:4275–4289. doi: 10.1128/JB.00081-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parsons JB, Frank MW, Jackson P, Subramanian C, Rock CO. 2014. Incorporation of extracellular fatty acids by a fatty acid kinase-dependent pathway in Staphylococcus aureus. Mol Microbiol 92:234–245. doi: 10.1111/mmi.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Parsons JB, Broussard TC, Bose JL, Rosch JW, Jackson P, Subramanian C, Rock CO. 2014. Identification of a two-component fatty acid kinase responsible for host fatty acid incorporation by Staphylococcus aureus. Proc Natl Acad Sci U S A 111:10532–10537. doi: 10.1073/pnas.1408797111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pappas A. 2009. Epidermal surface lipids. Dermatoendocrinol 1:72–76. doi: 10.4161/derm.1.2.7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen J, Yoong P, Ram G, Torres VJ, Novick RP. 2014. Single-copy vectors for integration at the SaPI1 attachment site for Staphylococcus aureus. Plasmid 76:1–7. doi: 10.1016/j.plasmid.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Monk IR, Shah IM, Xu M, Tan M, Foster TJ. 2012. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio 3:e00277-11. doi: 10.1128/mBio.00277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sambrook J, Fritsch EF, Maniatis T. 2012. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 59.Schneewind O, Model P, Fischetti VA. 1992. Sorting of protein A to the staphylococcal cell wall. Cell 70:267–281. doi: 10.1016/0092-8674(92)90101-h. [DOI] [PubMed] [Google Scholar]
- 60.DuMont AL, Yoong P, Surewaard BGJ, Benson MA, Nijland R, van Strijp JA, Torres VJ. 2013. Staphylococcus aureus elaborates leukocidin AB to mediate escape from within human neutrophils. Infect Immun 81:1830–1841. doi: 10.1128/IAI.00095-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Novick RP. 1991. Genetic systems in staphylococci. Methods Enzymol 204:587–636. [DOI] [PubMed] [Google Scholar]
- 62.Cosgriff CJ, White CR, Teoh WP, Grayczyk JP, Alonzo F. 2019. Control of Staphylococcus aureus quorum sensing by a membrane-embedded peptidase. Infect Immun 87:e00019-19. doi: 10.1128/IAI.00019-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.