

Antibiotic resistance is a growing threat to global health. To optimize the use of our existing antibiotics and identify new targets for future inhibitors, understanding the fundamental drivers of bacterial growth in the context of the host immune response is paramount. Historically, these genetic drivers have been difficult to manipulate precisely, as they are requisite for pathogen survival. Here, we provide the first application of Mobile-CRISPRi to study conditionally essential virulence genes in mouse models of lung infection through partial gene perturbation. We envision the use of Mobile-CRISPRi in future pathogenesis models and antibiotic target discovery efforts.
KEYWORDS: CRISPRi, conditionally essential genes, type III secretion, Pseudomonas aeruginosa, infection model, virulence lifestyle genes
ABSTRACT
Conditionally essential (CE) genes are required by pathogenic bacteria to establish and maintain infections. CE genes encode virulence factors, such as secretion systems and effector proteins, as well as biosynthetic enzymes that produce metabolites not found in the host environment. Due to their outsized importance in pathogenesis, CE gene products are attractive targets for the next generation of antimicrobials. However, the precise manipulation of CE gene expression in the context of infection is technically challenging, limiting our ability to understand the roles of CE genes in pathogenesis and accordingly design effective inhibitors. We previously developed a suite of CRISPR interference-based gene knockdown tools that are transferred by conjugation and stably integrate into bacterial genomes that we call Mobile-CRISPRi. Here, we show the efficacy of Mobile-CRISPRi in controlling CE gene expression in an animal infection model. We optimize Mobile-CRISPRi in Pseudomonas aeruginosa for use in a murine model of pneumonia by tuning the expression of CRISPRi components to avoid nonspecific toxicity. As a proof of principle, we demonstrate that knock down of a CE gene encoding the type III secretion system (T3SS) activator ExsA blocks effector protein secretion in culture and attenuates virulence in mice. We anticipate that Mobile-CRISPRi will be a valuable tool to probe the function of CE genes across many bacterial species and pathogenesis models.
IMPORTANCE Antibiotic resistance is a growing threat to global health. To optimize the use of our existing antibiotics and identify new targets for future inhibitors, understanding the fundamental drivers of bacterial growth in the context of the host immune response is paramount. Historically, these genetic drivers have been difficult to manipulate precisely, as they are requisite for pathogen survival. Here, we provide the first application of Mobile-CRISPRi to study conditionally essential virulence genes in mouse models of lung infection through partial gene perturbation. We envision the use of Mobile-CRISPRi in future pathogenesis models and antibiotic target discovery efforts.
INTRODUCTION
All pathogenic bacteria require essential and conditionally essential (CE) genes for survival in the host environment (1). Essential genes are typically defined as genes that are indispensable for growth in rich culture media, whereas CE genes are required only in specific conditions, such as maintenance in a host niche (2). Next-generation sequencing of bacterial transposon (Tn)-mutant libraries (e.g., transposon sequencing [Tn-Seq] [3] and insertion sequencing [INSeq] [4]) from infected animals has enabled the comprehensive identification of essential and CE genes in a single experiment, rapidly increasing our knowledge of which genes are required for pathogenesis (5–16). There are two major limitations of using Tn-Seq to study CE genes, both arising from the complete loss of function usually caused by Tn mutagenesis. First, core essential genes are by definition excluded from the analysis of environment-specific essentiality. Second, all-or-nothing mutations preclude our ability to observe the relationship between expression levels of the gene product and fitness in the host environment; this information could be valuable in identifying CE genes for which the organism is highly sensitive to slight perturbations, which would be ideal candidates for inhibitors. Thus, methods that can partially perturb CE gene function in the context of pathogenesis are highly valuable.
Gene repression tools that are currently used to study CE genes during infection have provided numerous insights into gene function but have key technical limitations. Antisense RNAs (17, 18) have variable efficacy, substantial off-target effects (19–21), and cannot be rationally designed (22). Methods to trigger protein degradation (i.e., degrons) [23–26] require each gene of interest to be tagged at its native locus and suffer from toxicity due to interference with protein function and stability (26). Gene depletion from inducible promoters also requires the insertion of the promoter upstream of all genes of interest and is limited by the inability to optimize both the control of noninduced promoter expression (leakiness) and the maximal amount of induced gene product (27).
In contrast, CRISPR interference (CRISPRi)—the use of a catalytically inactive variant of the Cas9 nuclease (dCas9) to repress transcription (28)—is highly efficacious and specific in bacteria (29), is easily programmable by substituting the first 20 nucleotides of the guide RNA (sgRNA) (30), does not require modification of the chromosome at each targeted gene, and maintains the native regulation of targeted genes. We previously developed Mobile-CRISPRi, (31) a technology that enables the transfer and stable integration of CRISPRi systems into diverse bacteria (Fig. 1A). Here, we optimize Mobile-CRISPRi for targeting CE genes in a Pseudomonas aeruginosa PA14 murine pneumonia model of infection.
FIG 1.

Toxicity, efficacy, and specificity of Mobile-CRISPRi in Pseudomonas aeruginosa. (A) Mobile-CRISPRi is comprised of an antibiotic resistance cassette (ABR), sgRNA spacers specific to the gene of interest, a promoter driving dcas9 expression, and dcas9. These components can be substituted before chromosomal integration into a pathogen to generate a knockdown strain. (B) Wild-type PA14 growth was compared to that of Mobile-CRISPRi PA14 strains featuring arabinose-inducible promoters driving dCas9 activity, two different variants of dCas9 (S. pyogenes and S. thermophilus), and the presence or absence of mRFP-targeting sgRNA. To induce the promoter, these strains were incubated with no arabinose, 0.1% arabinose, or 1% arabinose. (C) mRFP was cloned into Mobile-CRISPRi strains with constitutive promoters driving dCas9 expression. The median fluorescence of strains without sgRNA was compared to that of strains with mRFP-targeting sgRNA after 14 h of growth. The 10× knockdown associated with P1 is statistically different from the 14× (**, significant P value) and 17× knockdown (****, significant P value) associated with P2 and P3, respectively. (D) RNA was extracted from mutants featuring P3 with and without mRFP-targeting sgRNA. Gene counts from RNA-seq are plotted for each strain with a dashed line of slope = 1 for reference.
RESULTS
Optimized dCas9 expression eliminates toxicity and allows for graded knockdowns.
dCas9 overexpression often causes nonspecific toxicity in bacteria (32), which would likely complicate the interpretation of our CRISPRi experiments in infection models. Indeed, we found that the full induction of an arabinose-inducible promoter (PBAD) driving the expression of dCas9 variants from Streptococcus pyogenes (dCas9Spy) (28) or Streptococcus thermophilus (dCas9sth) (33) resulted in reduced growth of PA14 in rich culture medium, whereas partial induction showed no apparent toxicity (Fig. 1B). We reasoned that titrating chemical inducers (e.g., arabinose) in a murine infection model could be impractical due to variable tissue penetration (34–38). Instead, we focused on expressing dCas9Spy from a series of weak constitutive promoters from the BioBrick Registry (http://parts.igem.org/Main_Page) to reduce toxicity (see Fig. S1 in the supplemental material) and achieve partial knock down. To assess Mobile-CRISPRi efficacy using the BioBrick promoter strains, we employed a “test” version of Mobile-CRISPRi expressing monomeric red fluorescent protein (mRFP) and an sgRNA targeting the mRFP gene (31). Knockdown levels were quantified for each promoter through comparing the mutants’ fluorescence normalized to growth over time. After 12 hours, we found stable fluorescence ratios between mutants without and with mRFP-targeting sgRNA (see Fig. S2 in the supplemental material). The gradient of knockdown ranged from 10- to 17-fold at the 14-hour timepoint, which roughly corresponded to the BioBrick promoter strength used to express dCas9 (Fig. 1C). We performed RNA sequencing (RNA-seq) on cells expressing dCas9 from the strongest of the three BioBrick promoters in our set and confirmed that CRISPRi retained specificity for RFP (Fig. 1D). We imaged cells expressing dCas9Spy from all three promoters and found no apparent defects in morphology (see Fig. S3 in the supplemental material). We conclude that Mobile-CRISPRi optimized with BioBrick promoters driving dCas9Spy enables a nontoxic gradient of constitutive knockdowns in P. aeruginosa PA14.
Mobile-CRISPRi targeting of CE genes in a murine pneumonia model.
A major goal for developing Mobile-CRISPRi in infection models is to identify CE genes for which a modest perturbation has a substantial impact on pathogenesis. To do so, the system must enable the stable repression of the gene of interest over the course of infection. As a test case, we targeted exsA, which encodes the key activator of type III secretion system (T3SS) genes that are required for pathogenesis in P. aeruginosa. Because the exsA gene is positively autoregulated by the ExsA protein (39), we reasoned that modest knockdown would cause a large reduction in the transcription of T3SS genes, resulting in a loss of effector secretion and impaired virulence. Consistent with this, we found that CRISPRi knockdown of exsA reduced the expression of T3SS genes by more than 100-fold (Fig. 2A), similar to the expression levels observed in a strain with an exsA disruption (exsA::Tn; the Tn insertion position is shown in Fig. S4 in the supplemental material) (40). We found that all three BioBrick promoters driving dCas9Spy expression were equally effective at reducing exsA transcript levels, likely because even modest reductions in ExsA protein levels disrupt positive autoregulation. The esxA::Tn transposon mutation is insertional rather than deletional, which may have led to high levels of the nonfunctional exsA transcript, as measured by quantitative PCR (qPCR) (see Table S1 in the supplemental material). CRISPRi appeared to be slightly more effective at reducing exsA transcript levels than the exsA::Tn allele, possibly because CRISPRi can repress both ExsA-dependent transcription from the exsC promoter and ExsA-independent transcription from the exsA promoter (41). Knock down of exsA also eliminated the detectable production of T3SS pilus (PopB/D) and effector (ExoT/U) proteins (Fig. 2B). Neither the exsA knockdown nor the nontargeting control sgRNA strains showed a growth defect in rich culture medium (Fig. S1).
FIG 2.

T3SS-associated gene transcription, protein secretion profiles, and clearance of PA14 strains in infection model. (A) Reverse transcriptase quantitative PCR (RT-qPCR) analysis for T3SS-related genes across PA14 strains, normalized to WT PA14 RNA levels. (B) Immunoblot analysis of exoenzyme U and exoenzyme S secretion for PA14 strains grown in MinS medium (for type III protein secretion induction).
The loss of exsA function is known to strongly attenuate virulence in a murine pneumonia model (42, 43). To test whether Mobile-CRISPRi can be used to probe the functions of CE genes, such as exsA, in a host environment, we intratracheally instilled C57BL/6 mice with a range of 105 to 107 CFU of wild-type (WT) P. aeruginosa PA14, an isogenic exsA::Tn mutant, or Mobile-CRISPRi strains containing dCas9Spy driven by the P3 BioBrick promoter, and either an exsA-targeting sgRNA or a nontargeting control sgRNA. Although CRISPRi using all three BioBrick promoters resulted in similar levels of exsA knockdown, we chose P3 because we reasoned that it would serve as the most stringent test of potential dCas9Spy toxicity in the context of a mouse infection. We collected the lungs 18 hours after infection and plated lung homogenates to estimate the number of viable bacteria (44) (see Fig. S5A in the supplemental material). Strains with the exsA::Tn allele or Mobile-CRISPRi-targeted exsA were highly attenuated for virulence and yielded similar recovery rates (Fig. 3). This demonstrates that Mobile-CRISPRi is an effective tool to knock down CE genes in PA14 during a mouse infection and implies that Mobile-CRISPRi is as stable during in vivo infection as it is during growth in culture (31). Furthermore, levels of CFU recovery were similar between WT and nontargeting Mobile-CRISPRi, suggesting that the nonspecific toxicity of dCas9 was mitigated by reduced expression. Other general indicators of infection, including hypothermia and leukopenia, were observed for the nontargeting and WT controls (Fig. S5B and S5C). In contrast, both the exsA::Tn and Mobile-CRISPRi-targeted exsA strains produced similar levels of white blood cell counts (equivalent or higher than those seen in the phosphate-buffered saline [PBS] control) and similar body temperatures, altogether indicative of reduced virulence (Fig. S5B and S5C). Consistent with this, WT and nontargeting strains showed severe lung injury not seen in the exsA::Tn and exsA-targeting strains (Fig. S5D). We conclude that Mobile-CRISPRi can probe CE gene phenotypes in infection models.
FIG 3.
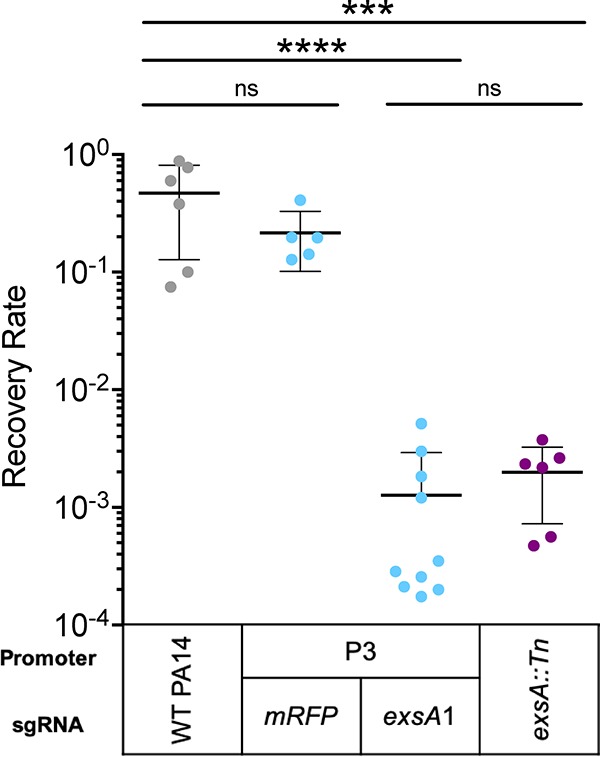
Recovery rates following murine lung infection. Following infection, lung homogenate serial dilutions were plated to estimate the CFU of bacteria recovered from the lung. Recovery rate is output CFU relative to input CFU.
DISCUSSION
A lack of genetic tools that enable facile and precise control over CE and essential gene expression has severely hampered our progress toward understanding bacterial pathogenesis past the point of simply identifying virulence factors. Our work demonstrates that Mobile-CRISPRi is a valuable genetic tool for characterizing CE genes in the context of an animal infection. We establish a synthetic biology approach for generating CRISPRi knockdown gradients and mitigating nonspecific toxicity by using promoters from the BioBrick Registry to control dCas9 expression. Furthermore, we show that Mobile-CRISPRi repression remains stable despite the stringent fitness constraints imposed during growth in a murine infection model—a key prerequisite to large-scale CRISPRi screens for bacterial gene function in pathogenesis. Finally, as a proof of principle, we successfully use Mobile-CRISPRi to modulate the pathogenesis of P. aeruginosa in a mouse pneumonia model by targeting the CE gene exsA. Our studies lay the groundwork for future CRISPRi screens that probe the molecular details of pathogenesis.
Mobile-CRISPRi is an excellent complement to established methods of gene function analysis during pathogenesis. Tn-based techniques (e.g., Tn-Seq [3]/INSeq [4]), transposon site hybridization (45), and signature-tagged mutagenesis (46) have been enormously successful at identifying genes required for growth in mouse models of infection. Mobile-CRISPRi partial knockdowns can be used to further characterize these gene sets by modulating expression to determine the amount of gene product required for virulence. Multiplexed CRISPRi can be used to dissect the genetic pathways by which CE genes operate (29) and will be particularly valuable for characterizing synergies between partially redundant secreted effector proteins. CRISPRi is currently the only method of systematically perturbing essential gene function that can be rationally designed and involves only a single step of strain construction. Thus, a combination of Mobile-CRISPRi partial knockdowns and Tn libraries will enable a comprehensive characterization of all genes required for pathogenesis—including essential genes.
Both Tn mutagenesis and CRISPRi screens have potential pitfalls that should be considered when interpreting single-gene data. When passaged, Tn-mutagenized strains can accumulate second-site suppressors that distort phenotypic analysis. Relatedly, CRISPRi can be inactivated by mutation; the most frequent type spontaneously occurs in the dcas9 gene and is enriched in a population of strains when CRISPRi causes a strong fitness defect (47). To circumvent these fitness defects, the weak P1 version of Mobile-CRISPRi can be used to target “sensitive” genes, for which a modest knock down causes a strong fitness defect (48). Finally, Tn mutagenesis and CRISPRi can both alter the expression of downstream genes in an operon (i.e., polarity), but the CRISPRi effect is much more predictable because the knock down of downstream genes is generally proportional to that of the targeted gene (29).
We previously demonstrated that Mobile-CRISPRi could be used to repress gene expression in a number of bacterial pathogens associated with antibiotic resistance (e.g., the Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species [ESKAPE] pathogens) (31, 49). Our optimized Mobile-CRISPRi system opens the door to systematic analysis of CE genes in these pathogens during infection, enabling drug-gene interaction studies and, in principle, a screen for new inhibitors that synergize with the host immune system.
MATERIALS AND METHODS
Construction of Mobile-CRISPRi plasmids and strains.
Plasmids encoding nuclease-null Streptococcus pyogenes and Streptococcus thermophilus dCas9s were gifted by Lei Qi and Sarah Fortune, respectively. The vectors containing a Tn7-based Mobile-CRISPRi system were constructed as previously described by Peters (31). dCas9 was expressed from the arabinose-inducible PBAD promoter and three constitutive promoters, namely, Anderson BBa_J23117 (P1), Anderson BBa_J23114 (P2), and Anderson BBa_J23115 (P3). The chimeric sgRNA was expressed by a constitutive derivative of the Ptrc promoter with no LacI operator site. In this study, all Pseudomonas aeruginosa UCBPP-PA14 Mobile-CRISPRi strains were constructed by tri-parental mating as previously described (31). Complete lists of plasmids and strains used in the study can be found in Tables S2 and S3, respectively, in the supplemental material. The PA14 exsA::Tn strain was obtained from a transposon insertion library (40).
Toxicity measurements.
For dCas9 toxicity measurements, WT PA14 and the mutants were streaked onto Pseudomonas isolation agar (PIA) plates and incubated for 20 hours at 37°C. On the second day, one colony from each plate was cultured in 2 ml LB and incubated at 37°C with shaking at 350 rpm for 12 hours. Then, cultures were diluted in 100 μl LB medium with no inducer, 0.1% arabinose, or 1% arabinose to yield a mixture with an optical density at 600 nm (OD600nm) of 0.05 in a 96-well plate (catalog no. 351177; Corning, NY). These cultures were grown with a lid for 9 to 10 hours on a plate shaker (OrbiShaker MP, Benchmark Scientific, NJ) at 37°C and 900 rpm. The OD600nm of the plate cultures was measured every hour using a microplate reader (SpectraMax 340PC; Molecular Devices, CA).
RFP knockdown efficiency.
Following triparental mating, two P. aeruginosa colonies were picked from each strain to serve as biological replicates and were incubated overnight in 3 ml of LB with 100 μg/ml gentamicin selective medium at 37°C with shaking. These cultures were diluted to 0.01 OD600nm into fresh LB medium, and 200 μl of this culture was added in triplicate to a clear bottom, black, 96-well plate (Corning Costar). This plate was covered with an optically clear seal, and a needle was used to poke holes in each of the wells. Fluorescence (excitation, 557 nm; emission, 592 nm) and OD600nm were monitored during incubation in a microplate reader (Synergy H1; BioTek Instruments, VT) with continuous, fast, double orbital shaking. Samples were blanked with a well containing LB medium. For each replicate, the fluorescence value was divided by OD600nm values at each time point and plotted in 30-minute intervals.
RNA extraction.
PA14 Mobile-CRISPRi strains, exsA::Tn, and WT were streaked onto Vogel Bonner minimal medium (VBMM) or LB agar plates and incubated overnight at 37°C. One colony from each plate was grown in MinS (T3SS-inducing minimal medium supplemented with nitrotriacetic acid and lacking calcium medium) (50) or LB medium at 37°C for 16 hours with shaking at 250 rpm. Then, the strains were subcultured in 400 μl fresh MinS or LB medium until the OD600nm reached 1.0. Total RNA was extracted from cell pellets using the RNeasy minikit (Qiagen) according to the manufacturer’s instructions with on-column DNase I digestion (Qiagen). The RNA extracts were aliquoted and stored at –80°C.
Reverse transcriptase quantitative PCR.
cDNA was synthesized using random hexamer primers and a RevertAid first-strand cDNA synthesis kit (Thermo Scientific, Waltham, MA). To check the amplification efficiency of the primers, a 1:50 dilution of WT PA14 cDNA was mixed with PowerUp SYBR green master mix (Thermo Scientific) and detected by the MX3000P qPCR System (Stratagene, La Jolla, CA). Primers for T3SS-related genes (see Table S3 in the supplemental material) had amplification efficiencies between 90% and 110%. A PA14 housekeeping gene, nadB, was used as internal control for normalization of total RNA levels (51). The relative efficiency of each primer pair was tested and compared with that of nadB, and the threshold cycle (2−ΔΔCT) data analysis was used (52). All reactions were performed in triplicates and repeated at least twice using independent cultures, with average values of biological replicates and error bars representing standard deviation of ΔΔCT.
cDNA library preparation and RNA-seq.
The RNA concentration for each sample was determined with a NanoDrop spectrophotometer (Thermo Fisher Scientific). A total of 10 ng of RNA of each sample was fragmented for 6 min, cDNA libraries were prepared using a NEBNext Ultra RNA library prep kit for Illumina (New England BioLabs [NEB] number E7770S). Libraries were sequenced in collaboration with the Chan Zuckerberg Biohub in San Francisco on an Illumina MiSeq instrument in 150-bp paired-end runs. Approximately 1,000,000 reads were collected for each of the two samples, with ∼94% alignment to PA14 WT by Bowtie2 (53), and transcripts were counted with HTSeq (54). Only genes with a nonnormalized read count greater than 1 in both samples were included in analysis, with a coverage of 1,286 genes (∼20% genome).
Type III secretion profile of Pseudomonas aeruginosa by immunoblotting.
To knock down the exsA gene, two specific sgRNAs, exsA1 and exsA2, were designed. PA14 Mobile-CRISPRi mutants, exsA::Tn, and WT were streaked onto VBMM agar plates and incubated at 37°C overnight. One colony from each plate was grown at 37°C for 16 hours in a shaking incubator at 250 rpm in MinS medium (50). Bacteria were removed by centrifugation at 6,000 × g for 15 min. Then, the supernatant was collected and the secreted proteins were precipitated by the addition of ammonium sulfate. The protein pellets were dissolved in sample buffer. After boiling, samples were loaded onto ExpressPlus 4% to 20% PAGE gels (Genscript, Piscataway, NJ) and run under denaturing conditions. PAGE gels were transferred to a polyvinylidene difluoride (PVDF) membrane and immunoblotted with polyclonal rabbit antiserum against ExoU, ExoT/ExoS, PopB, and PopD proteins, as previously described (50).
Murine infection model.
Pathogen-free male C57BL/6J mice, 8 weeks of age, were purchased from Jackson Laboratories. Animal experiments were conducted in accordance with the approval of the Institutional Animal Care and Use Committee (IACUC) at UCSF. A total of 29 mice were randomly assigned in the following 5 groups: G1, WT PA14, 6 mice; G2, P3 with mRFP-targeting sgRNA, 5 mice; G3, P3 with exsA1 targeting sgRNA, 10 mice; G4, exsA::Tn, 6 mice; and G5, saline control, 2 mice. Mice were anesthetized with isofluorane prior to intratracheal instillation with bacteria at a range of 1 × 105 to 1 × 107 CFU/animal in a volume of 50 μl, per an established protocol (44). Animal weights and rectal temperatures were measured prior to euthanasia. The lungs were collected in 1 ml of sterile PBS and processed with a handheld homogenizer (Polytron PT1200E; Kinematica). A total of 50 μl of lung homogenate with appropriate dilutions were spread onto PIA plates with and without gentamicin to count output CFU. The bacterial recovery rate was calculated as the ratio of output CFU to input CFU. For whole-blood analysis, blood was collected by cardiac puncture into acid citrate dextrose (Sigma-Aldrich), and white blood cells (WBCs) were measured by a hematology analyzer (Genesis; Oxford Science).
Microscopy.
An overnight culture was diluted 1:100 and grown to mid-log phase in LB medium. This culture was diluted to an OD600nm of 0.1 before being added to an agar pad composed of 1% agarose and LB medium. No. 1 coverslips were used, and the slide was sealed with Valap. Thereafter, 250 μl of LB medium was added to the agar pad to prevent desiccation. Sample slides were mounted in the stage top of a Nikon Ti microscope warmed to 37°C. All images were collected on a Nikon Ti-E inverted microscope equipped with a Plan Apo VC 100×/1.4. Images were acquired with an Andor Zyla 4.2 sCMOS camera controlled with MicroManager. Multiple stage positions were collected using an ASI XYZ stage. Brightness and contrast were adjusted (identically for compared image sets) using Fiji software.
Statistical analysis.
GraphPad Prism (v. 7.0) was used for the statistical analysis of all the data. For log-transformed bacterial recovery rate, temperature, WBC counts, and weight changes, all groups were analyzed by ordinary one-way analysis of variance (ANOVA) followed by Tukey’s multiple-comparison test. For the transcription data, two-way ANOVA followed by Tukey’s multiple-comparison test was implemented. To compare changes in fluorescence at 14 hours, data points associated with mRFP-targeting sgRNAs were normalized to median fluorescence of the respective strain without sgRNAs. One-way ANOVA followed by Tukey’s multiple-comparison test was used to assess the significance of the knockdown levels.
Data availability.
All RNA-seq data have been deposited in the National Center for Biotechnology Information's GEO database (55) and are accessible through accession number GSE134771.
Supplementary Material
ACKNOWLEDGMENTS
We thank S. Fortune (Harvard University) for the dCas9sth plasmid, A. Hauser (Northwestern University) for the polyclonal rabbit antiserum for exsA-related proteins, and C. Gross (UCSF) and J. Engel (UCSF) for productive discussions and some strains. The sequencing team at the Chan Zuckerberg Biohub (N. Neff, B. Yu, R. Sit, and M. Tan) assisted with RNA-seq experiments. C. Mahendra and A. Borges in the lab of J. Bondy-Demony (UCSF) provided technical assistance during the fluorescence plate reader assay. Microscopy data were acquired at the Nikon Imaging Center at UCSF.
This work was supported by NIH 1K22AI137122 (to J.M.P.), NIAID R01 AI125445 (to M.R.L.), NSF-GRFP 1650113 (to N.K.P.), the National Natural Science Foundation of China 81400005/H0104 and Special Support Fund of Shenzhen for Introduced High-Level Medical Team and Translational medicine of Biochip in clinical laboratory SZSM201412005 (to J.Q.), and 5R01EB024014, Chan-Zuckerberg Biohub, CF Foundation Research Development Program, and Gilead Sciences Research Scholars Program in Cystic Fibrosis (to O.S.R.).
We declare no competing interests.
Author contributions were as follows: conceptualization, J.M.P. and O.S.R.; methodology and investigation, J.Q., N.K.P., M.A.Y., S.C., N.H., A.L., E.C., and M.R.S.; supervision, E.C., M.R.L., and O.S.R.; draft writing and revision, J.Q., N.K.P., J.M.P., and O.S.R.; funding acquisition, O.S.R.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00304-19.
REFERENCES
- 1.Wassenaar TM, Gaastra W. 2001. Bacterial virulence: can we draw the line? FEMS Microbiol Lett 201:1–7. doi: 10.1111/j.1574-6968.2001.tb10724.x. [DOI] [PubMed] [Google Scholar]
- 2.Tufariello JM, Chapman JR, Kerantzas CA, Wong K-W, Vilchèze C, Jones CM, Cole LE, Tinaztepe E, Thompson V, Fenyö D, Niederweis M, Ueberheide B, Philips JA, Jacobs WR. 2016. Separable roles for Mycobacterium tuberculosis ESX-3 effectors in iron acquisition and virulence. Proc Natl Acad Sci U S A 113:E348–57. doi: 10.1073/pnas.1523321113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Opijnen TK, Bodi L, Camilli A. 2009. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods 6:767–772. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodman AL, Wu M, Gordon JI. 2011. Identifying microbial fitness determinants by insertion sequencing using genome-wide transposon mutant libraries. Nat Protoc 6:1969–1980. doi: 10.1038/nprot.2011.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burrack LS, Higgins DE. 2007. Genomic approaches to understanding bacterial virulence. Curr Opin Microbiol 10:4–9. doi: 10.1016/j.mib.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Bachman MA, Breen P, Deornellas V, Mu Q, Zhao L, Wu W, Cavalcoli JD, Mobley HLT. 2015. Genome-wide identification of Klebsiella pneumoniae fitness genes during lung infection. mBio 6:e00775-15. doi: 10.1128/mBio.00775-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fu YM, Waldor K, Mekalanos JJ. 2013. Tn-Seq analysis of Vibrio cholerae intestinal colonization reveals a role for T6SS-mediated antibacterial activity in the host. Cell Host Microbe 14:652–663. doi: 10.1016/j.chom.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gawronski JD, Wong SMS, Giannoukos GD, Ward V, Akerley BJ. 2009. Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proc Natl Acad Sci U S A 106:16422–16427. doi: 10.1073/pnas.0906627106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamp HD, Patimalla-Dipali B, Lazinski DW, Wallace-Gadsden F, Camilli A. 2013. Gene fitness landscapes of Vibrio cholerae at important stages of its life cycle. PLoS Pathog 9:e1003800. doi: 10.1371/journal.ppat.1003800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skurnik D, Roux D, Aschard H, Cattoir V, Yoder-Himes D, Lory S, Pier GB. 2013. A comprehensive analysis of in vitro and in vivo genetic fitness of Pseudomonas aeruginosa using high-throughput sequencing of transposon libraries. PLoS Pathog 9:e1003582. doi: 10.1371/journal.ppat.1003582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turner KH, Wessel AK, Palmer GC, Murray JL, Whiteley M. 2015. Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc Natl Acad Sci U S A 112:4110–4115. doi: 10.1073/pnas.1419677112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verhagen LM, de Jonge MI, Burghout P, Schraa K, Spagnuolo L, Mennens S, Eleveld MJ, van der Gaast-de Jongh CE, Zomer A, Hermans PWM, Bootsma HJ. 2014. Genome-wide identification of genes essential for the survival of Streptococcus pneumoniae in human saliva. PLoS One 9:e89541. doi: 10.1371/journal.pone.0089541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang N, Ozer EA, Mandel MJ, Hauser AR. 2014. Genome-wide identification of Acinetobacter baumannii genes necessary for persistence in the lung. mBio 5:e01163. doi: 10.1128/mBio.01163-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gutierrez MG, Yoder-Himes DR, Warawa JM. 2015. Comprehensive identification of virulence factors required for respiratory melioidosis using Tn-seq mutagenesis. Front Cell Infect Microbiol 5:78. doi: 10.3389/fcimb.2015.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moule MG, Spink N, Willcocks S, Lim J, Guerra-Assunção JA, Cia F, Champion OL, Senior NJ, Atkins HS, Clark T, Bancroft GJ, Cuccui J, Wren BW. 2016. Characterization of new virulence factors involved in the intracellular growth and survival of Burkholderia pseudomallei. Infect Immun 84:701–710. doi: 10.1128/IAI.01102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perry BJ, Akter MS, Yost CK. 2016. The use of transposon insertion sequencing to interrogate the core functional genome of the legume symbiont Rhizobium leguminosarum. Front Microbiol 7:1873. doi: 10.3389/fmicb.2016.01873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forsyth RA, Haselbeck RJ, Ohlsen KL, Yamamoto RT, Xu H, Trawick JD, Wall D, Wang L, Brown-Driver V, Froelich JM, C KG, King P, McCarthy M, Malone C, Misiner B, Robbins D, Tan Z, Zhu Zy Z, Carr G, Mosca DA, Zamudio C, Foulkes JG, Zyskind JW. 2002. A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol Microbiol 43:1387–1400. doi: 10.1046/j.1365-2958.2002.02832.x. [DOI] [PubMed] [Google Scholar]
- 18.Ji Y, Zhang B, Van SF, Horn, Warren P, Woodnutt G, Burnham MK, Rosenberg M. 2001. Identification of critical staphylococcal genes using conditional phenotypes generated by antisense RNA. Science 293:2266–2269. doi: 10.1126/science.1063566. [DOI] [PubMed] [Google Scholar]
- 19.Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS. 2003. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol 21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- 20.Doench JG, Petersen CP, Sharp PA. 2003. siRNAs can function as miRNAs. Genes Dev 17:438–442. doi: 10.1101/gad.1064703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sigoillot FD, Lyman S, Huckins JF, Adamson B, Chung E, Quattrochi B, King RW. 2012. A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nat Methods 9:363–366. doi: 10.1038/nmeth.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho SH, Haning K, Contreras LM. 2015. Strain engineering via regulatory noncoding RNAs: not a one-blueprint-fits-all. Curr Opin Chem Eng 10:25–34. doi: 10.1016/j.coche.2015.07.008. [DOI] [Google Scholar]
- 23.de Lorenzo V, Eltis L, Kessler B, Timmis KN. 1993. Analysis of Pseudomonas gene products using lacIq/Ptrp-lac plasmids and transposons that confer conditional phenotypes. Gene 123:17–24. doi: 10.1016/0378-1119(93)90533-9. [DOI] [PubMed] [Google Scholar]
- 24.Castang S, McManus HR, Turner KH, Dove SL. 2008. H-NS family members function coordinately in an opportunistic pathogen. Proc Natl Acad Sci U S A 105:18947–18952. doi: 10.1073/pnas.0808215105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei J-R, Krishnamoorthy V, Murphy K, Kim J-H, Schnappinger D, Alber T, Sassetti CM, Rhee KY, Rubin EJ. 2011. Depletion of antibiotic targets has widely varying effects on growth. Proc Natl Acad Sci U S A 108:4176–4181. doi: 10.1073/pnas.1018301108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cameron DE, Collins JJ. 2014. Tunable protein degradation in bacteria. Nat Biotechnol 32:1276–1128. doi: 10.1038/nbt.3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meisner J, Goldberg JB. 2016. The Escherichia coli rhaSR-PrhaBAD inducible promoter system allows tightly controlled gene expression over a wide range in Pseudomonas aeruginosa. Appl Environ Microbiol 82:6715–6727. doi: 10.1128/AEM.02041-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peters JM, Colavin A, Shi H, Czarny TL, Larson MH, Wong S, Hawkins JS, Lu CHS, Koo B-M, Marta E, Shiver AL, Whitehead EH, Weissman JS, Brown ED, Qi LS, Huang KC, Gross CA. 2016. A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell 165:1493–1506. doi: 10.1016/j.cell.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peters JM, Koo B-M, Patino R, Heussler GE, Hearne CC, Qu J, Inclan YF, Hawkins JS, Lu CHS, Silvis MR, Harden MM, Osadnik H, Peters JE, Engel JN, Dutton RJ, Grossman AD, Gross CA, Rosenberg OS. 2019. Enabling genetic analysis of diverse bacteria with Mobile-CRISPRi. Nat Microbiol 4:244–250. doi: 10.1038/s41564-018-0327-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang S, Voigt CA. 2018. Engineered dCas9 with reduced toxicity in bacteria: implications for genetic circuit design. Nucleic Acids Res 46:11115–11125. doi: 10.1093/nar/gky884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rock JM, Hopkins FF, Chavez A, Diallo M, Chase MR, Gerrick ER, Pritchard JR, Church GM, Rubin EJ, Sassetti CM, Schnappinger D, Fortune SM. 2017. Programmable transcriptional repression in mycobacteria using an orthogonal CRISPR interference platform. Nat Microbiol 2:16274. doi: 10.1038/nmicrobiol.2016.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khlebnikov A, Risa O, Skaug T, Carrier TA, Keasling JD. 2000. Regulatable arabinose-inducible gene expression system with consistent control in all cells of a culture. J Bacteriol 182:7029–7034. doi: 10.1128/jb.182.24.7029-7034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jang S, Jang S, Jung GY. 2018. Toward tunable dynamic repression using CRISPRi. Biotechnol J 13:e1800152. doi: 10.1002/biot.201800152. [DOI] [PubMed] [Google Scholar]
- 36.Loessner H, Leschner S, Endmann A, Westphal K, Wolf K, Kochruebe K, Miloud T, Altenbuchner J, Weiss S. 2009. Drug-inducible remote control of gene expression by probiotic Escherichia coli Nissle 1917 in intestine, tumor and gall bladder of mice. Microbes Infect 11:1097–1105. doi: 10.1016/j.micinf.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 37.Loessner H, Endmann A, Leschner S, Westphal K, Rohde M, Miloud T, Hämmerling G, Neuhaus K, Weiss S. 2007. Remote control of tumour-targeted Salmonella enterica serovar Typhimurium by the use of l-arabinose as inducer of bacterial gene expression in vivo. Cell Microbiol 9:1529–1537. doi: 10.1111/j.1462-5822.2007.00890.x. [DOI] [PubMed] [Google Scholar]
- 38.Seri K, Sanai K, Matsuo N, Kawakubo K, Xue C, Inoue S. 1996. l-arabinose selectively inhibits intestinal sucrase in an uncompetitive manner and suppresses glycemic response after sucrose ingestion in animals. Metab Clin Exp 45:1368–1374. doi: 10.1016/S0026-0495(96)90117-1. [DOI] [PubMed] [Google Scholar]
- 39.Hovey AK, Frank DW. 1995. Analyses of the DNA-binding and transcriptional activation properties of ExsA, the transcriptional activator of the Pseudomonas aeruginosa exoenzyme S regulon. J Bacteriol 177:4427–4436. doi: 10.1128/jb.177.15.4427-4436.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, Wu G, Villanueva J, Wei T, Ausubel FM. 2006. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci U S A 103:2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marsden AE, Intile PJ, Schulmeyer KH, Simmons-Patterson ER, Urbanowski ML, Wolfgang MC, Yahr TL. 2016. Vfr directly activates exsA transcription to regulate expression of the Pseudomonas aeruginosa type III secretion system. J Bacteriol 198:1442–1450. doi: 10.1128/JB.00049-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Apodaca G, Bomsel M, Lindstedt R, Engel J, Frank D, Mostov KE, Wiener-Kronish J. 1995. Characterization of Pseudomonas aeruginosa-induced MDCK cell injury: glycosylation-defective host cells are resistant to bacterial killing. Infect Immun 63:1541–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kudoh I, Wiener-Kronish JP, Hashimoto S, Pittet JF, Frank D. 1994. Exoproduct secretions of Pseudomonas aeruginosa strains influence severity of alveolar epithelial injury. Am J Physiol 267:L551–L556. doi: 10.1152/ajplung.1994.267.5.L551. [DOI] [PubMed] [Google Scholar]
- 44.Ortiz-Muñoz G, Looney MR. 2015. Non-invasive intratracheal instillation in mice. Bio Protoc 5:e1504. doi: 10.21769/BioProtoc.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murry JP, Sassetti CM, Lane JM, Xie Z, Rubin EJ. 2008. Transposon site hybridization in Mycobacterium tuberculosis. Methods Mol Biol 416:45–59. doi: 10.1007/978-1-59745-321-9_4. [DOI] [PubMed] [Google Scholar]
- 46.Hensel M, Shea JE, Gleeson C, Jones MD, Dalton E, Holden DW. 1995. Simultaneous identification of bacterial virulence genes by negative selection. Science 269:400–403. doi: 10.1126/science.7618105. [DOI] [PubMed] [Google Scholar]
- 47.Zhao H, Sun Y, Peters JM, Gross CA, Garner EC, Helmann JD. 2016. Depletion of undecaprenyl pyrophosphate phosphatases disrupts cell envelope biogenesis in Bacillus subtilis. J Bacteriol 198:2925–2935. doi: 10.1128/JB.00507-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rousset F, Cui L, Siouve E, Becavin C, Depardieu F, Bikard D. 2018. Genome-wide CRISPR-dCas9 screens in E. coli identify essential genes and phage host factors. PLoS Genet 14:e1007749. doi: 10.1371/journal.pgen.1007749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rice LB. 2008. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J Infect Dis 197:1079–1108. doi: 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- 50.El Solh AA, Akinnusi ME, Wiener-Kronish JP, Lynch SV, Pineda LA, Szarpa K. 2008. Persistent infection with Pseudomonas aeruginosa in ventilator-associated pneumonia. Am J Respir Crit Care Med 178:513–519. doi: 10.1164/rccm.200802-239OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lequette Y, Lee JH, Ledgham F, Lazdunski A, Greenberg EP. 2006. A distinct QscR regulon in the Pseudomonas aeruginosa quorum-sensing circuit. J Bacteriol 188:3365–3370. doi: 10.1128/JB.188.9.3365-3370.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 53.Langmead B, Salzberg S. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Edgar R, Domrachev M, Lash AE. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RNA-seq data have been deposited in the National Center for Biotechnology Information's GEO database (55) and are accessible through accession number GSE134771.