

Gram-negative bacteria are defined by their asymmetric outer membrane that consists of phospholipids on the inner leaflet and lipopolysaccharide (LPS) in the outer leaflet. LPS is essential in all but a few Gram-negative species; the reason for this differential essentiality is not well understood. One species that can survive without LPS, Acinetobacter baumannii, shows characteristic growth and morphology phenotypes. We show that these phenotypes can be suppressed under conditions of slow growth and describe how LPS loss is connected to the growth defects. In addition to better defining the challenges A. baumannii cells face in the absence of LPS, we provide a new hypothesis that may explain the species-dependent conditional essentiality.
KEYWORDS: Acinetobacter baumannii, lipopolysaccharide loss, suppressors
ABSTRACT
Lipopolysaccharide (LPS) is normally considered to be essential for viability in Gram-negative bacteria but can be removed in Acinetobacter baumannii. Mutant cells lacking this component of the outer membrane show growth and morphological defects. Here, we report that growth rates equivalent to the wild type can be achieved simply by propagation in minimal medium. The loss of LPS requires that cells rely on phospholipids for both leaflets of the outer membrane. We show that growth rate in the absence of LPS is not limited by nutrient availability but by the rate of outer membrane biogenesis. We hypothesize that because cells grow more slowly, outer membrane synthesis ceases to be rate limiting in minimal medium.
IMPORTANCE Gram-negative bacteria are defined by their asymmetric outer membrane that consists of phospholipids on the inner leaflet and lipopolysaccharide (LPS) in the outer leaflet. LPS is essential in all but a few Gram-negative species; the reason for this differential essentiality is not well understood. One species that can survive without LPS, Acinetobacter baumannii, shows characteristic growth and morphology phenotypes. We show that these phenotypes can be suppressed under conditions of slow growth and describe how LPS loss is connected to the growth defects. In addition to better defining the challenges A. baumannii cells face in the absence of LPS, we provide a new hypothesis that may explain the species-dependent conditional essentiality.
INTRODUCTION
The outer membrane of Gram-negative bacteria is asymmetric, displaying lipopolysaccharide on the outer surface and phospholipids on the inner leaflet (1). For many decades, lipopolysaccharide was believed to be essential. Therefore, it was unexpected when, almost 20 years ago, a strain of Neisseria meningitidis completely lacking lipopolysaccharide (LPS) was constructed, demonstrating that an outer membrane could be built with phospholipids in both leaflets (2, 3). Shortly after, clinical strains of the pathogen Acinetobacter baumannii resistant to an LPS-binding antibiotic were isolated with loss-of-function mutations in genes early in the LPS biosynthetic pathway (4). These cells completely lacked LPS, and yet they survived not only under laboratory conditions but also in patients.
Although A. baumannii can survive in the absence of LPS, the cells do show growth and morphological defects under laboratory conditions (5–7, 22). A significant amount of work has been done to understand the synthesis of LPS and subsequent transport to the cell surface (8, 9). The first committed step in the biosynthetic pathway of LPS involves the gene lpxC (which encodes a UDP N-acetylglucosamine deacetylase). Using an lpxC mutant, we sought to understand the basis for its impaired growth and altered morphology. Here, we report that under conditions of propagation in minimal medium, the mutant cells behave almost identically to the wild type. We propose a model in which the loss of LPS makes synthesis of the outer membrane, which now consists of phospholipids in both leaflets, rate limiting for growth.
RESULTS
The growth defect of cells lacking LPS is partially suppressed by mutations in mla and pldA.
As previously reported and confirmed here, mutant cells lacking LPS (ΔlpxC) have a growth defect (5, 7). In rich medium (LB), mutant cells both grow more slowly than the wild type and exhibit clumping and shape abnormalities (Fig. 1, left side of panels A and B). We wondered whether we could isolate suppressors of ΔlpxC that would restore normal growth in rich medium. We passaged several replicates of ΔlpxC mutant cells every 24 hours for 1 week and succeeded in isolating, in every case, strains that grew more rapidly than the ΔlpxC parental strain (see Fig. S1 in the supplemental material). Whole-genome sequencing revealed that each of these more rapidly growing strains had acquired a loss-of-function mutation in pldA or an insertion in the immediate upstream gene (DJ41_RS17650), which we interpreted as having polar effects on pldA (Table 1) (10). In addition, almost all of these strains harbored a loss-of-function mutation in one or another of the genes in the mla pathway. PldA is a phospholipase that in Escherichia coli is known to be responsible for degrading phospholipids in the outer leaflet of the outer membrane, whereas the Mla pathway is responsible for recycling phospholipids back to the inner membrane (11–15).
FIG 1.
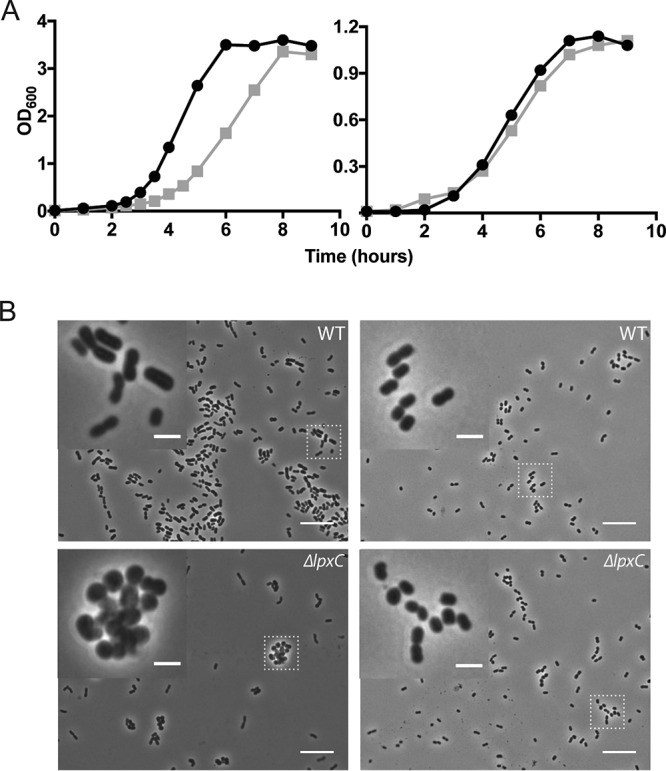
Growth of LPS-deficient cells in rich and minimal medium. (A) Wild-type (circles) and ΔlpxC (squares) cells grown in LB (left) or M9+ (right) medium at 37°C. (B) Phase microscopy with a 100× objective of cells grown under the same conditions. Insets are a 4× enlargement of the regions identified by the boxes. Scale bars are 10 μm for the large images and 2 μm for the insets.
TABLE 1.
Mutations in pldA and mla pathwaya
| Strain genotype | No. of replicates with loss-of-function mutations in: |
||
|---|---|---|---|
| pldA | DJ41_RS17650 | mla pathway | |
| ΔlpxC | 6/15 | 9/15 | 13/15 |
| ΔmlaA ΔlpxC | 7/12 | 5/12 | |
ΔlpxC replicates (15 total) were passaged for 7 days, while the ΔmlaA ΔlpxC replicates (12 total) were passaged for 3 days. Whole-genome sequencing of colonies revealed loss-of-function mutations in the mla pathway (mostly mlaA). Mutations also occurred in either pldA or the adjacent upstream gene (DJ41_RS17650) but never both.
The simplest interpretation of these results is that the observed increase in growth rate was due to the loss of pldA in combination with a mla mutation. To test this idea, we constructed deletions of both pldA and mlaA, a gene in the mla pathway. In the absence of lpxC, only the removal of both pldA and mlaA improved growth; single deletions of either one conferred no growth improvement (see Fig. S2 in the supplemental material). We note that the same suppressor mutations were also reported in a recent publication (7).
Next, we sought to determine how fully the suppressor mutations restored the growth of the ΔlpxC mutant in rich medium. Measurements of generation time showed that the pldA and mlaA mutations only partially restored the growth rate to that of the wild type (38 versus 25 minutes) (black bars in Fig. 2C). We have been unable to find a set of suppressors that will fully restore growth when the cells are growing quickly in rich medium.
FIG 2.
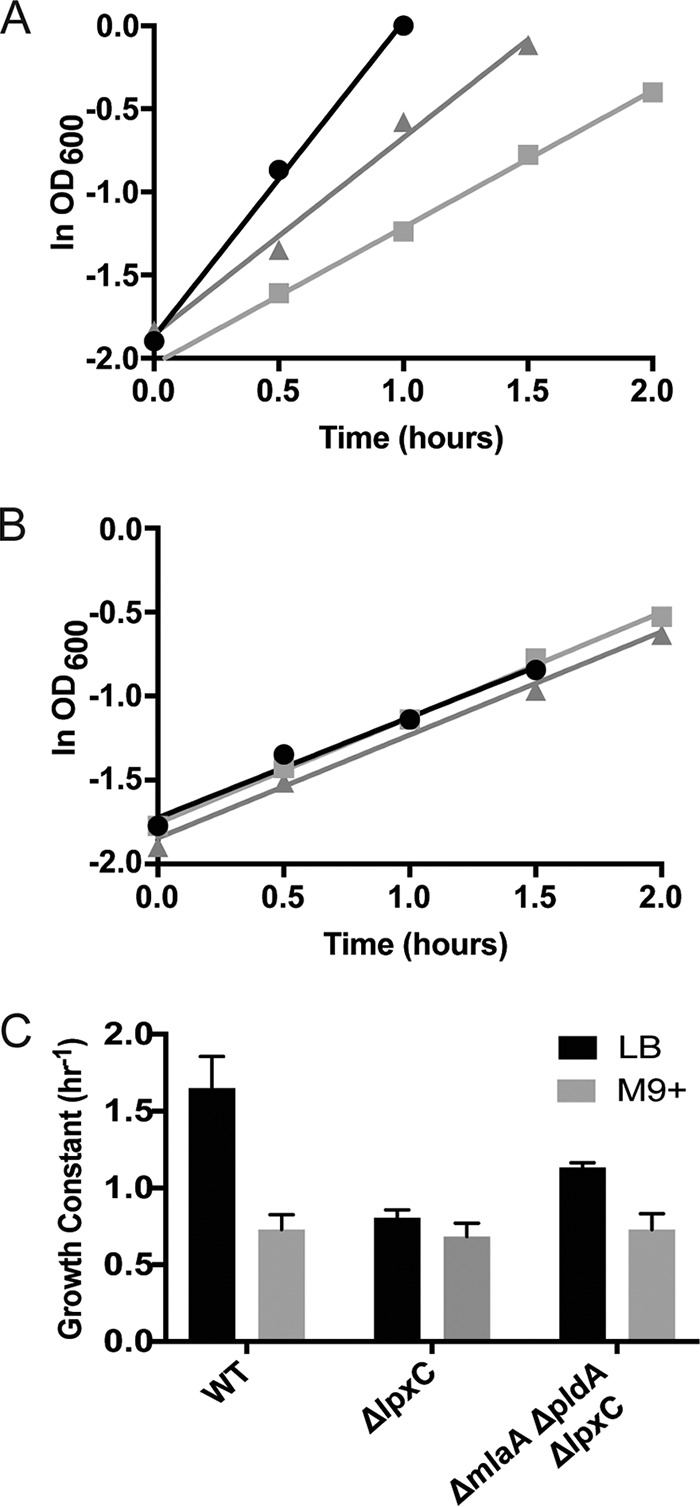
Growth of a ΔlpxC mutant in rich medium is only partially restored by mlaA and pldA suppressor mutations. (A) Semilog plot for early- to mid-log growth of wild-type (WT; circles), ΔlpxC (squares), and ΔmlaA ΔpldA ΔlpxC (triangles) strains grown in LB at 37°C. (B) Same as above for strains grown in M9+ at 37°C. (C) Growth constants calculated for each strain.
Switching to minimal medium restores morphology and growth to wild-type levels.
A striking observation of this investigation was the discovery that growth and morphology were restored to wild-type levels when cells lacking LPS were grown in minimal medium (Fig. 1, right side of panels A and B). Not unexpectedly, the suppressor mutations had no further effect on the growth rate, as the ΔlpxC cells were already growing at wild-type levels (Fig. 2B and C, gray bars).
Importantly, the results of Fig. 2C also showed that the growth rates for the ΔlpxC strain in rich and minimal medium were indistinguishable. Apparently, the absence of LPS sets an upper bound on the rate of growth, a finding that we interpret as indicating that outer membrane synthesis has now become rate limiting for growth.
Lowering temperature in rich medium suppresses the growth defect.
As an independent test of the idea that outer membrane biogenesis is not rate limiting when cells are growing slowly, we investigated the effect of lowering temperature in rich medium. When quantified using optical density, the growth rate of the ΔlpxC cells was significantly lower than that of wild type (Fig. 3A). Because ΔlpxC cells aggregate substantially more when grown in in LB at 25°C than 37°C, optical density is a poor representation of cell number (Fig. 3B). Calculating growth rates from total ATP levels, which is independent of cell aggregation, showed that cells were doubling at a rate similar to wild type. Moreover, the suppressors reduced the amount of clumping, and the growth rate of this strain was as rapid as the wild type when measured by either optical density or ATP levels (Fig. 3). These observations reinforced the view that when growing slowly, membrane biogenesis is no longer rate limiting for growth.
FIG 3.
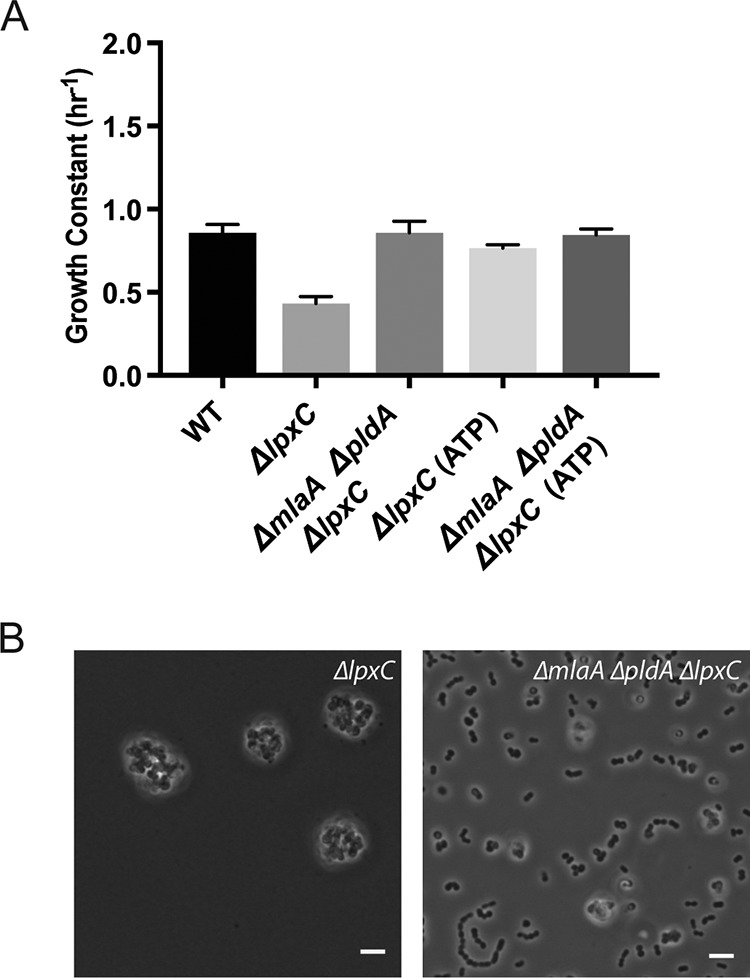
Growth at low temperatures is sufficient for suppression. (A) Growth constants calculated for each strain grown in LB at 25°C from measurements of either OD600 or total ATP levels. (B) Phase microscopy with a 60× objective of ΔlpxC and ΔmlaA ΔpldA ΔlpxC strains grown in LB at 25°C. Scale bars are 10 μm.
DISCUSSION
It is known that LPS is not essential in A. baumannii, although cells lacking LPS grow slowly and display significant morphological aberrations under normal growth conditions (LB; 37°C). Prior studies on LPS-deficient cells were principally carried out using rich medium. A major finding of our present investigation is that under conditions of slow growth in minimal medium, LPS-deficient cells (that is, otherwise wild-type cells simply lacking lpxC) grow just as rapidly as the wild type and without morphological defects. Importantly, no suppressor mutations were required to achieve equivalent growth rates to the wild type in minimal medium.
Of particular significance, LPS-deficient cells were found to grow just as rapidly in minimal medium as rich medium. In other words, something other than nutrient availability restricts growth in LPS-mutant cells. A simple interpretation of this finding is that membrane synthesis becomes rate limiting for growth under conditions in which LPS synthesis is blocked. That is, the lpxC mutation impairs the cell’s ability to produce an outer membrane, and it must now presumably rely on phospholipid for both the inner and outer leaflets. To build this membrane, cells must synthesize sufficient phospholipids, transport them to the outer membrane, and then flip them into the outer leaflet. While restrictions at any of these steps would limit the rate at which the membrane could be constructed, only phospholipid flipping is a process unique to cells lacking LPS. There is no reason to presume cells would have an enzymatic mechanism for introducing phospholipids into the outer leaflet because it normally contains only LPS (16). Therefore, we propose that inserting phospholipids into the outer leaflet is challenging because there is an energetic barrier for phospholipid flipping from the inner to the outer leaflet. Since cells can only grow as quickly as they can produce a membrane bilayer, growth of LPS-mutant cells would be limited by their ability to incorporate phospholipids into the leaflet (17). Unlike phospholipid biosynthesis, phospholipid flipping into the outer leaflet would not have specific nutrient requirements, which explains why switching from minimal to rich medium has little or no effect on growth rate.
Our hypothesis also helps to explain how the mla and pldA suppressors we and others have found only partially restore growth in LB and have no effect in minimal medium. Simply put, they do not fix the barrier to phospholipid flipping. Rather, by eliminating the pathways responsible for removing phospholipids from the outer leaflet, cells can somewhat increase the rate at which they build the outer leaflet of the membrane, thus modestly improving their growth rate.
Until recently, it was widely believed that LPS is an indispensable feature of Gram-negative bacteria. However, the discovery that A. baumannii can survive in the absence of this component of the outer leaflet raises the possibility that under the right conditions, LPS is dispensable in other more distantly related Gram-negative bacteria. Perhaps under artificially imposed conditions of slow growth, other well studied Gram-negative organisms will also be found to maintain viability in the absence of LPS.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All strains and plasmids used in this study are described in Table S1 in the supplemental material. The growth media used were LB Miller (BD) and M9 formulated for Acinetobacter spp. (6.78 g/liter Na2HPO4, 3 g/liter KH2PO4, 0.5 g/liter NaCl, 1 g/liter NH4Cl, 0.4% sodium succinate, 2 mM MgSO4, and 0.1 mM CaCl2). M9+ was a formulation of M9 with 2.5% LB added. When preparing cells from glycerol stocks, all LPS-deficient strains were streaked onto LB plates containing 1.2% agarose instead of agar to compensate for a plating defect. All other strains were streaked onto LB agar (1.2%), and all plates were incubated at 37°C. When appropriate, medium was supplemented with antibiotics at the following concentrations: kanamycin (40 μg/ml), apramycin (150 μg/ml), and carbenicillin (100 μg/ml).
Growth curves.
To increase consistency, all strains were freshly streaked from glycerol stocks the day before starting the growth curves and were never refrigerated. Also, it was noticed that good aeration was essential for the growth of the LPS-deficient mutants. Starter cultures (2 ml) of LB were inoculated from the plates and grown overnight (O/N; <16 h) to saturation at 37°C. Cells were then diluted 1:10 into fresh medium (2 ml), namely, LB or M9+ depending on the experiment, and grown for 90 minutes at 37°C. These cultures were diluted again into 20 ml of the appropriate medium in 125-ml flasks, normalizing to an optical density at 600 nm (OD600) of 0.01 (LB) or 0.03 (M9+). The cultures were grown at the indicated temperature with vigorous shaking (220 rpm), and OD600 measurements were taken every 30 minutes to 1 h. Each experiment was repeated at least 3 times, and a representative curve is shown. All growth curves were generated using GraphPad Prism.
Due to the number of replicates, growth was monitored during the passaging experiment using a plate reader. Briefly, 150 μl of each of the newly passaged replicates (1:100 dilution of O/N culture) was transferred to a 96-well plate. The plate was transferred to a Tecan Sunrise plate reader and incubated with shaking at 25°C or 30°C, depending on the replicate set. OD600 measurements were taken every 10 minutes for 24 hours. We believe the exacerbated growth defect observed for the mutants is a result of the poor aeration in the plate. Data from the plate reader were processed using Microsoft Excel, and all growth curves were generated using GraphPad Prism.
Measuring total ATP levels.
The same cultures that were used to measure OD600 over a period of time were also tested for total ATP levels using the BacTiter-Glo microbial cell viability assay. At each time period, 100 μl of cell culture was added, in duplicate, to a 96-well plate. Reagents were added according to the manufacturer’s instructions. Samples were incubated together for 5 min, and luminescence was measured.
Suppressor screen.
We started with 16 replicates of each of 2 strains, namely, the ΔlpxC and ΔmlaA ΔlpxC strains, half of which were grown at 25°C and half at 30°C. To initiate the experiment, each strain was streaked onto LB agarose plates and grown at 37°C. Single colonies (16 each) were inoculated into 2 ml of LB and were grown O/N at 37°C. Glycerol stocks of these starting populations were saved and sequenced as day 0. The setup for the two temperatures differed slightly, but the 16 replicates at each temperature originated from the same 16 starting populations. For the 25°C populations, 5-ml cultures of LB were inoculated with 50 μl of the O/N started cultures and allowed to grow in a roller drum at 25°C. For the 30°C populations, 150 μl of the starter cultures was added to 15 ml LB in 50-ml Falcon tubes and grown at 30°C (220 rpm). The populations were then passaged into fresh LB (1:100 dilution) every 24 hours. Initially some of the 25°C replicates were clear after 24 hours and were not passaged until there was visible growth (in 24-hour increments). Growth was monitored via a plate reader at days 3 and 7, and glycerol stocks were saved at the same time. After 7 days, all replicates were showing growth profiles that matched the wild type, and thus, the passaging was ended so as not to accumulate additional mutations.
Next-generation sequencing.
DNA was isolated from single colonies of each replicate using the PureLink Pro 96 genomic DNA kit according to the manufacturer’s instructions. Samples were barcoded and prepped for sequencing using a Nextera DNA sample preparation kit following Illumina’s protocols. Sequencing was done at the Bauer Core Facility (Harvard University) using an Illumina HiSeq 2500 instrument. Reads were aligned and mapped to the A. baumannii ATCC 19606 published genome using Breseq software in consensus mode (18). Variants were also identified using Breseq, and aligned reads were visualized using the Integrated Genomics Viewer (Broad Institute) (19). Full sequencing results are available in Table S2 in the supplemental material.
Mutant construction.
All oligonucleotide primers sequences are given in Table S3 in the supplemental material.
(i) Marked deletions. Mutants were constructed by allelic exchange through double-crossover homologous recombination, as reported previously, with some minor changes (20). Briefly, the linear constructs containing either a Kanr (amplified from pIM1440) or Aprr (amplified from pSET152) resistance marker were constructed as follows. Upstream and downstream regions (∼500 bp) of the target gene were amplified with primers designed to create overlap with the resistance marker on the 3′ end of the upstream region and 5′ end of the downstream region. The flanking regions and resistance cassette were subsequently assembled into a linear cassette using Gibson assembly master mix (New England BioLabs [NEB]). The linear cassette was then amplified by PCR to obtain sufficient quantities for transformation. To construct the deletion strains, 20 ml of the recipient strain was grown in LB at 37°C to an OD600 of approximately 0.8. The cells were pelleted and washed 2 times with 2 ml chilled water. A final wash with chilled 10% (vol/vol) glycerol was performed, and the cells were resuspended in 150 μl 10% glycerol. The cell suspension was mixed with ∼8 μg of the linear DNA cassette and transferred to a chilled electroporation cuvette (2-mm gap). It was then pulsed with an Eppendorf Eporator (2.5 kV). A total of 1 ml of LB was quickly added, and the cells were transferred to a culture tube and incubated at 37°C (220 rpm) for 90 min. The entire transformation was plated onto LB plates with kanamycin or apramycin and incubated at 37°C for 24 hours. All isolated colonies were tested by colony PCR for insertion of the resistance cassette. In cases of strains with multiple mutations, the removal of LPS (lpxC) was always done last.
(ii) Markerless deletion. The pldA markerless deletion was introduced by biparental conjugation following a known protocol with modifications (21). Briefly, the upstream and downstream regions of pldA (∼1 kb) were amplified with overlap to the pEX18ApGW plasmid as well as overlap to one another. The plasmid was also amplified by PCR, with primers originating at the HindIII and KpnI restriction sites. The final plasmid (pEX18ApGW-pldA) was constructed by assembling the three fragments using Gibson Assembly and then transformed into E. coli strain pRK2013 via electroporation. The transformants were plated onto LB agar with kanamycin and carbenicillin (50 μg/ml) and grown at 30°C for 24 hours. The recipient Acinetobacter baumannii mlaA::Kanr strain was streaked at the same time. The two plates were gently scraped, and cells were resuspended in LB to an OD600 of ∼1.0. Equal amounts (100 μl) of the two suspensions were added to 600 μl LB and subsequently collected by centrifugation (7,000 × g; 2 min). Cells were washed 2 times by gentle resuspension in 600 μl LB. After the final spin, the pellet was resuspended in 50 μl LB, spotted onto the center of a dried LB plate, and incubated O/N at 30°C. To select against E. coli, the entire spot was resuspended in 1 ml LB and then 100 μl was plated on a large plate with Simmons citrate agar (BD) containing carbenicillin. After 2 days of incubation at 37°C, the plate contained Acinetobacter colonies that had integrated the plasmid, which could be confirmed by colony PCR. Several colonies were inoculated into LB and grown O/N at 37°C to cure the plasmid. Tenfold dilutions of the cultures were then plated onto LB plates with 10% (vol/vol) sucrose. Surviving colonies were expected to have flipped out the plasmid. Several colonies were checked by PCR to distinguish the mutants from those that had resolved to wild type.
Growth constants.
To calculate growth constants, the growth curve data from the OD600 range ∼0.15 to 0.5 were linearized on a semilog plot. The range varied slightly from sample to sample depending on the OD600 at the time tested and the quality of the linear regression. Only data with R2 of >0.99 were used. The slope of the linear regression line was calculated, which determined the growth constant. Doubling times can be obtained from the growth constant by calculating ln (2) divided by the growth constant. All shown growth constants are the averages of at least 3 separate experiments.
Microscopy.
All images were of overnight cultures grown to saturation in the indicated medium at either 37°C or 25°C. The cells were immobilized on 2% agarose pads made with phosphate-buffered saline. Cells were imaged using an Olympus BX-61 upright microscopy with a 100× objective. Images using a 60× objective were obtained on a Nikon Eclipse Ti inverted microscope. Images were processed using Fiji, and all compared image sets were adjusted identically.
Supplementary Material
ACKNOWLEDGMENTS
We thank The Bauer Core Facility at Harvard University for performing sequencing work.
This work was supported by National Institutes of Health awards GM066174 to D.K. and GM018568 to R.L.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00420-19.
REFERENCES
- 1.Kamio Y, Nikaido H. 1976. Outer membrane of Salmonella typhimurium: accessibility of phospholipid head groups to phospholipase c and cyanogen bromide activated dextran in the external medium. Biochemistry 15:2561–2570. doi: 10.1021/bi00657a012. [DOI] [PubMed] [Google Scholar]
- 2.Steeghs L, den Hartog R, den Boer A, Zomer B, Roholl P, van der Ley P. 1998. Meningitis bacterium is viable without endotoxin. Nature 392:449–450. doi: 10.1038/33046. [DOI] [PubMed] [Google Scholar]
- 3.Peng D, Hong W, Choudhury BP, Carlson RW, Gu XX. 2005. Moraxella catarrhalis bacterium without endotoxin, a potential vaccine candidate. Infect Immun 73:7569–7577. doi: 10.1128/IAI.73.11.7569-7577.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moffatt JH, Harper M, Harrison P, Hale JD, Vinogradov E, Seemann T, Henry R, Crane B, St Michael F, Cox AD, Adler B, Nation RL, Li J, Boyce JD. 2010. Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrob Agents Chemother 54:4971–4977. doi: 10.1128/AAC.00834-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beceiro A, Moreno A, Fernandez N, Vallejo JA, Aranda J, Adler B, Harper M, Boyce JD, Bou G. 2014. Biological cost of different mechanisms of colistin resistance and their impact on virulence in Acinetobacter baumannii. Antimicrob Agents Chemother 58:518–526. doi: 10.1128/AAC.01597-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bojkovic J, Richie DL, Six DA, Rath CM, Sawyer WS, Hu Q, Dean CR. 2015. Characterization of an Acinetobacter baumannii lptD deletion strain: permeability defects and response to inhibition of lipopolysaccharide and fatty acid biosynthesis. J Bacteriol 198:731–741. doi: 10.1128/JB.00639-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Powers MJ, Trent MS. 2018. Phospholipid retention in the absence of asymmetry strengthens the outer membrane permeability barrier to last-resort antibiotics. Proc Natl Acad Sci U S A 115:E8518–E8527. doi: 10.1073/pnas.1806714115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raetz CR, Whitfield C. 2002. Lipopolysaccharide endotoxins. Annu Rev Biochem 71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okuda S, Sherman DJ, Silhavy TJ, Ruiz N, Kahne D. 2016. Lipopolysaccharide transport and assembly at the outer membrane: the PEZ model. Nat Rev Microbiol 14:337–345. doi: 10.1038/nrmicro.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thi Khanh Nhu N, Riordan DW, Do Hoang Nhu T, Thanh DP, Thwaites G, Huong Lan NP, Wren BW, Baker S, Stabler RA. 2016. The induction and identification of novel colistin resistance mutations in Acinetobacter baumannii and their implications. Sci Rep 6:28291. doi: 10.1038/srep28291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abellón-Ruiz J, Kaptan SS, Baslé A, Claudi B, Bumann D, Kleinekathöfer U, van den Berg B. 2017. Structural basis for maintenance of bacterial outer membrane lipid asymmetry. Nat Microbiol 2:1616–1623. doi: 10.1038/s41564-017-0046-x. [DOI] [PubMed] [Google Scholar]
- 12.Dekker N. 2000. Outer-membrane phospholipase A: known structure, unknown biological function. Mol Microbiol 35:711–717. doi: 10.1046/j.1365-2958.2000.01775.x. [DOI] [PubMed] [Google Scholar]
- 13.Malinverni JC, Silhavy TJ. 2009. An ABC transport system that maintains lipid asymmetry in the gram-negative outer membrane. Proc Natl Acad Sci U S A 106:8009–8014. doi: 10.1073/pnas.0903229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thong S, Ercan B, Torta F, Fong ZY, Wong HY, Wenk MR, Chng SS. 2016. Defining key roles for auxiliary proteins in an ABC transporter that maintains bacterial outer membrane lipid asymmetry. Elife 5:e19042. doi: 10.7554/eLife.19042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.May KL, Silhavy TJ. 2018. The Escherichia coli phospholipase PldA regulates outer membrane homeostasis via lipid signaling. mBio 9:e00379-18. doi: 10.1128/mBio.00379-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sutterlin HA, Shi H, May KL, Miguel A, Khare S, Huang KC, Silhavy TJ. 2016. Disruption of lipid homeostasis in the Gram-negative cell envelope activates a novel cell death pathway. Proc Natl Acad Sci U S A 113:E1565–E1574. doi: 10.1073/pnas.1601375113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao Z, Davis RM, Kishony R, Kahne D, Ruiz N. 2012. Regulation of cell size in response to nutrient availability by fatty acid biosynthesis in Escherichia coli. Proc Natl Acad Sci U S A 109:E2561–E2568. doi: 10.1073/pnas.1209742109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deatherage DE, Barrick JE. 2014. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol 1151:165–188. doi: 10.1007/978-1-4939-0554-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aranda J, Poza M, Pardo BG, Rumbo S, Rumbo C, Parreira JR, Rodriguez-Velo P, Bou G. 2010. A rapid and simple method for constructing stable mutants of Acinetobacter baumannii. BMC Microbiol 10:279. doi: 10.1186/1471-2180-10-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carruthers MD, Nicholson PA, Tracy EN, Munson RS Jr. 2013. Acinetobacter baumannii utilizes a type VI secretion system for bacterial competition. PLoS One 8:e59388. doi: 10.1371/journal.pone.0059388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boll JM, Crofts AA, Peters K, Cattoir V, Vollmer W, Davies BW, Trent MS. 2016. A penicillin-binding protein inhibits selection of colistin-resistant, lipooligosaccharide-deficient Acinetobacter baumannii. Proc Natl Acad Sci U S A 113:E6228–E6237. doi: 10.1073/pnas.1611594113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.