

Abstract
The degeneration of injured axons involves a self-destruction pathway whose components and mechanism are not fully understood. Here, we report a new regulator of axonal resilience. The transmembrane protein Raw is cell autonomously required for the degeneration of injured axons, dendrites, and synapses in Drosophila melanogaster. In both male and female raw hypomorphic mutant or knock-down larvae, the degeneration of injured axons, dendrites, and synapses from motoneurons and sensory neurons is strongly inhibited. This protection is insensitive to reduction in the levels of the NAD+ synthesis enzyme Nmnat (nicotinamide mononucleotide adenylyl transferase), but requires the c-Jun N-terminal kinase (JNK) mitogen-activated protein (MAP) kinase and the transcription factors Fos and Jun (AP-1). Although these factors were previously known to function in axonal injury signaling and regeneration, Raw's function can be genetically separated from other axonal injury responses: Raw does not modulate JNK-dependent axonal injury signaling and regenerative responses, but instead restrains a protective pathway that inhibits the degeneration of axons, dendrites, and synapses. Although protection in raw mutants requires JNK, Fos, and Jun, JNK also promotes axonal degeneration. These findings suggest the existence of multiple independent pathways that share modulation by JNK, Fos, and Jun that influence how axons respond to stress and injury.
SIGNIFICANCE STATEMENT Axonal degeneration is a major feature of neuropathies and nerve injuries and occurs via a cell autonomous self-destruction pathway whose mechanism is poorly understood. This study reports the identification of a new regulator of axonal degeneration: the transmembrane protein Raw. Raw regulates a cell autonomous nuclear signaling pathway whose yet unknown downstream effectors protect injured axons, dendrites, and synapses from degenerating. These findings imply that the susceptibility of axons to degeneration is strongly regulated in neurons. Future understanding of the cellular pathway regulated by Raw, which engages the c-Jun N-terminal kinase (JNK) mitogen-activated protein (MAP) kinase and Fos and Jun transcription factors, may suggest new strategies to increase the resiliency of axons in debilitating neuropathies.
Keywords: axon degeneration, axon injury, Drosophila, MAP kinase signaling, motoneuron, Wallerian degeneration
Introduction
Damaged axons that become separated from their cell body undergo a stereotyped fragmentation of cytoskeleton and membrane called Wallerian degeneration. The molecular mechanism of this cell-autonomous destruction process is still poorly understood, however is a subject of great interest for insight into how axonal loss might be curtailed in neuropathies as well as neurodegenerative diseases (Conforti et al., 2014).
Previous studies have implicated a small number of molecules in Wallerian degeneration mechanism. A central player is the NAD+ biosynthetic enzyme Nmnat (nicotinamide mononucleotide adenylyl transferase). Nmnat2 is considered to function as a “survival factor” for axons, because its depletion leads to spontaneous axonal degeneration, whereas increasing Nmnat enzymatic activity in axons inhibits degeneration (Sasaki et al., 2009; Coleman and Freeman, 2010; Gilley and Coleman, 2010). Recent studies have identified the TIR domain containing protein Sarm1 as a key mediator of degeneration downstream of Nmnat2 (Osterloh et al., 2012; Gilley et al., 2015). Sarm1's TIR domains are capable of enzymatically degrading NAD+ and thus may promote degeneration via active destruction of an important metabolite in neurons (Gerdts et al., 2015; Essuman et al., 2017, 2018). Recent studies in Drosophila have also revealed that mutations in Axundead (Axed) can inhibit axonal degeneration even when Sarm1 is activated (Neukomm et al., 2017), however the molecular action of Axed in axonal degeneration is not yet known.
Wallerian degeneration is also strongly influenced by mitogen-activated protein kinase (MAPK) signaling. Studies in different models have suggested multiple points of influence for MAPK signaling in the Wallerian degeneration pathway. These include a role for Jun N-terminal kinases (JNKs) in the execution of axonal degeneration: the presence of JNK inhibitors at the time of injury is sufficient to inhibit axonal degeneration (Miller et al., 2009), and genetic inhibition of all three mammalian JNK kinases (JNK1, JNK2, JNK3) strongly protects axons even when Sarm1 is constitutively activated (Yang et al., 2015). However JNK signaling also regulates the protein stability of Nmnat2 (Walker et al., 2017), implying an upstream regulatory role in the degeneration program. In contrast, in Drosophila neurons JNK signaling regulates a protective pathway that makes injured axons and dendrites more resilient to degeneration (Chen et al., 2012; Xiong and Collins, 2012). In addition, retrograde MAPK signaling in multiple model organisms regulates the ability of injured neurons in the PNS to initiate new axonal growth (Abe and Cavalli, 2008) and, separately, retrograde MAPK signaling regulates axonal degeneration following trophic factor withdrawal (Geden and Deshmukh, 2016; Simon et al., 2016). These observations suggest responses to axonal injury invoke MAPK signaling for multiple functions whose mechanisms are challenging to study separately.
To study axonal injury signaling in a genetic model organism, we have previously established a Drosophila larval nerve crush assay, in which injured axons undergo a highly stereotyped degeneration process (Xiong and Collins, 2012). Using this assay we discovered a new mutation on the second chromosome that strongly inhibits axon degeneration in vivo, and have mapped this mutation via whole-genome sequencing to the Drosophila gene raw. Raw is previously known to regulate multiple developmental processes including embryonic dorsal closure, gonad development, glial development, and dendrite morphogenesis (Bates et al., 2008; Jemc et al., 2012; Lee et al., 2015; Hans et al., 2018; Luong et al., 2018). Here we find an essential function for Raw in axonal degeneration via restraint of the JNK MAP kinase and the transcription factors Fos and Jun. These findings shed light on transcriptional pathways that influence the susceptibility of axons to degeneration.
Materials and Methods
Fly stocks.
The following strains were used in this study: Canton-S (WT), dcp-1prev1 (rawdcp-1) originally from Laundrie et al. (2003), dcp-1k05606 (BL10390), repo-Gal4 (BL7415), UAS-wnd-RNAi (BL35369), and second chromosome deficiencies from the Bloomington fly stock center, raw134.47, raw155.27, UAS-Raw-RA and UAS-Raw-RB from (Jemc et al., 2012), hiwND8 and hiwΔN from (Xiong et al., 2012), m12-Gal4 (P(Gal4)5053A) (Ritzenthaler et al., 2000), BG380-Gal4 (Budnik et al., 1996), ppk-Gal4 (Kuo et al., 2005), D42-Gal4 (Sanyal, 2009), 221-Gal4, UAS-dcr2 (Baumgardt et al., 2007), UAS-JNKDN (Weber et al., 2000), UAS-FosDN (Eresh et al., 1997), UAS-JunDN (Eresh et al., 1997), and puc-lacZE69 (Martín-Blanco et al., 1998). The puc-GFP reporter was obtained from the FlyTrap Project (http://flytrap.med.yale.edu/), line G00462. UAS-nmnat-RNAi (32255), UAS-Rtnl2 RNAi (33320), UAS-raw-RNAi (KK) (101255) and UAS-wnd-RNAi (26910) were from the Vienna RNAi center (Dietzl et al., 2007).
The dcp-1prev1 recombinant lines were generated via standard meiotic recombination in dcp-1prev1 heterozygotes generated from crossing dcp-1prev1 to a w− strain background. Three independent recombinant chromosomes containing the dcp-1prev1 mutation were isozygosed using a second chromosome balancer. As either homozygotes or heterozygotes with the original dcp-1prev1 chromosome, these recombinants showed no axonal degeneration phenotype (Table 1).
Table 1.
Genotypes assessed for axonal and synaptic degeneration after injury, as shown in Figure 1 and Figure 2
| Genotype | Protection from degeneration? |
|---|---|
| dcp-1prev1/dcp-1prev1 | Yes |
| dcp-12/dcp-12 | No |
| dcp-13/dcp-13 | No |
| dcp-1k5606/dcp-1k5606 | No |
| dcp-1prev1/dcp-1k5606 | No |
| dcp-1prev1-recombinant 1/dcp-1prev1 | No |
| dcp-1prev1-recombinant 2/dcp-1prev1 | No |
| dcp-1prev1-recombinant 3/dcp-1prev1 | No |
| dcp-1prev1/raw134.47 | Yes |
| dcp-1prev1/raw155.27 | Yes |
| dcp-1prev1/raw134.47; UAS-Raw-RA/D42-Gal4 | No |
| dcp-1prev1/raw134.47; UAS-Raw-RB/Repo-Gal4 | Yes |
| dcp-1prev1/raw134.47; UAS-Raw-RA/m12-Gal4 | No |
| dcp-1prev1/raw134.47; UAS-Raw-RB/m12-Gal4 | No |
Genotypes in bold were analyzed for polymorphisms by whole-genome sequence analysis.
Fly rearing.
For dendrite degeneration and microtubule dynamics assays, Drosophila embryos were collected and aged for 3 d at 25°C in standard CSY media. L3 larvae were immobilized without anesthetic on a microscope slide with agarose pad and tape. For axon injury and puc-LacZ reporter experiments involving RNAi-mediated knock-downs, embryos and larvae were reared on yeast–glucose media at 29°C. For all other assays flies, embryos and larvae were reared on yeast–glucose media at 25°C.
Larval nerve crush assay and immunocytochemistry.
The nerve crush assay was previously described in details in (Hao et al., 2016). Briefly, wandering third instar larvae were anesthetized with CO2 gas, and the segmental nerves were pinched with Dumostar no. 5 forceps. Larvae were then kept on plates containing yeast–glucose food in a 25°C incubator for specified time periods.
Drosophila larvae were dissected in ice-cold PBS and then fixed in 4% paraformaldehyde for 25 min. After fixation, the samples were incubated in blocking buffer (PBS with 0.3% Triton X-100 and 5% normal goat serum) for 30 min at room temperature. Primary antibodies were used at the following concentrations: ms anti-Futsch (22c10, Developmental Studies Hybridoma Bank (DSHB) 1:100, ms anti-lacZ (40–1a, DSHB) 1:100, and guinea pig (gp) anti-Nmnat (gift from Grace Zhai) 1:1000. For secondary antibodies, Cy3-Gt anti-HRP (Jackson Laboratories) were used at 1:1000, A488-Gt anti-mouse or A488-Gt anti-gp (Invitrogen) were used at 1:1000.
Confocal images were collected on an Improvision spinning disk confocal microscope, consisting of a Hamamatsu C9100-50 EMCCD camera, a Yokagawa Nipkow CSU10 scanner, and a Zeiss Axio Observer. All images were taken using the 40× (1.3 numerical aperture, NA) oil objective. Similar settings were used to collect compared genotypes and conditions.
Analysis of whole-genome sequencing data.
Whole-genome sequencing data were obtained for the original dcp-1prev1 line, dcp-1prev1/dcp-1k05606 heterozygotes, and three recombinant lines that contained the dcp-1prev1 mutation but had lost the axonal degeneration phenotype (Table 1). The sequences were aligned to the reference Drosophila melanogaster genome (DM3) using Burrows-Wheerler Alignment tool (BWA) (Li and Durbin, 2009). Reads that map to multiple locations were removed. Samtools was used to call variants (Li et al., 2009). There were 332 variants on chromosome 2 that cosegregated with the axonal degeneration phenotype. (These were present in the dcp-1prev1 line, but absent in dcp-1k05606 parent strain and the three recombinant lines). These variants were then annotated to genes using software SnpEff (Cingolani et al., 2012) and only nonsynonymous mutations were kept as candidates. The six candidate genes were raw, eya, msp-300, CG10874, CG9525, and CG17211. Each candidate was then tested by complementation using deficiency lines that remove each candidate gene (summarized in Table 2).
Table 2.
Complementation tests of mutations identified dcp-1prev1 with deficiency lines
| Candidate gene (chromosomal location) | Mutation locus | Deficiency line(s) | Complementation test: Does dcp-1prev/Df show protection from degeneration? |
|---|---|---|---|
| CG10874 (22D1) | chr2L:2190481(C->A) | Df(2L)BSC688 (22B1-22D6) | No |
| Msp-300 (25C6-25C10) | chr2L:5161154(C->A) | Df(2L)Exel611(25C8-25D5) | No |
| Df(2L)BSC109(25D4-25D8) | |||
| Eya (26E1-26E2) | chr2L:6530148(T->C) | Df(2L)BSC354 (26D7-26E3) | No |
| Raw (29E4-29E6) | chr2L:8712009(G->T) | Df(2L)BSC204(29D5-29F8) | Yes |
| CG9525 (29F5-29F6) | chr2L:8960733(A->C) | Df(2L)BSC204 (29D5-29F8) | No |
| Df(2L)ED678 (29F5-30B12) | |||
| CG17211 (33D2) | chr2L:12127146(C->G) | Df(2L)ED775(33B8-34A3) | No |
Whole-genome sequencing identified predicted loss-of-function mutations in six genes that segregated with the degeneration phenotype in dcp-1prev1. Each was tested by complementation with the listed deficiency lines. Heterozygous Df/dcp-1prev1 animals were injured and tested for the NMJ degeneration phenotype. Only the deficiency line deleted for raw recapitulated the NMJ protective phenotype in homozygous dcp-1prev1 animals, suggesting that the mutation on the gene raw causes the axonal protective phenotype.
Experimental design, imaging, and statistical analysis.
To quantify axon degeneration, we scored the m12-Gal4, UAS-mCD8::GFP labeled axons while blinded to genotype and condition within the segmental nerves according to one of four categories between 0 and 100%: axons that were completely intact showing no signs of degeneration were scored as 0; continuous but with varicosities were assigned a score of 33%; partially fragmented, but partially continuous scored as 67%; and completely fragmented were assigned a degeneration score of 100%. For each specific genotype and condition, >25 axons from at least five animals were quantified.
Degeneration of neuromuscular junction (NMJ) terminals was scored based on the presence to the cytoskeletal marker Futsch and neuronal membrane into one of three categories: complete degeneration (complete loss of Futsch staining and fragmentation of synaptic membrane), partial degeneration (partial loss of Futsch staining and partial fragmentation of synaptic membrane) and no degeneration (both Futsch and synaptic membrane are intact). For each specific genotype and condition, >20 NMJs from at least five animals were quantified. Percentages of NMJs in each category are shown.
For intensity quantification of the puc-lacZ reporter (Martín-Blanco et al., 1998), the mean intensities for lacZ staining in the nuclei of motoneurons located along the dorsal midline in segments 3–7 of the larval nerve cord were measured using the Volocity software (PerkinElmer), and normalized to background signal. Reported values were standardized to the uninjured Canton S condition. Eight or more animals were measured for each genotype. Measurements in ddaE neurons, using a similar puc-GFP reporter (Stone et al., 2014) were performed in L3 larva and quantified via fluorescence intensity (FI) normalized to background signal as follows: nuclear FI − background FI = normalized FI.
For measurement of Nmnat staining intensity in the neuropil, the neuropil area in each image was selected. We used auto-selection function of the Volocity software to select objects that meet the intensity criteria of >1 SDs above the mean within the neuropil region. The mean Nmnat intensity was calculated by total intensity divided by total area in each animal.
Measurement of the regenerative sprouting response was performed in m12-Gal4, UAS-mCD8::GFP labeled axons while blinded to genotype as described in (Hao et al., 2016). Pixels within the most distal 100 μm of the injured proximal stump were selected based on mCD8-GFP intensity criteria of >3 SDs above the mean, and then summed to measure total membrane volume. Figure 6B reports changes in total membrane volume normalized to WT control animals at T = 0 (immediately after injury). For each genotype, at least 11 axons from ≥ 5 independent animals were measured.
Figure 6.

Raw has no effect upon axonal injury signaling and axonal regeneration. A, Representative examples of proximal stump of sprouting m12-Gal4-expressing motoneuron axons 20 h after nerve crush. B, Quantification of new axonal growth based on measured volume of axonal membrane within 100 μm of the injured axon tip. C, Example images showing puc-lacZ reporter expression in motoneuron nuclei that express UAS-raw-RNAi (vdrc 101255), UAS-wnd-RNAi (BL 35369) or both, together with UAS-Dcr2, via the pan-neuronal driver BG380-Gal4. D, Distribution of puc-lacZ expression levels measured in individual motoneuron nuclei coexpressing raw-RNAi; wnd-RNAi (green) compared with raw-RNAi alone (black). E, Quantification of relative puc-lacZ intensities (normalized to uninjured control) in different genotypes. Individual triangles represent mean intensities per larva measured from 30 to 35 dorsal motoneurons in segments 3–7 of the larval nerve cord. Scale bars, 20 μm. Error bars indicate SEM. ****p < 0.0001, ns, one-way ANOVA test (p > 0.05), n ≥ 11 axons in ≥ 5 animals per genotype in B, n ≥ 8 animals in 3 independent trials in E.
For all assays, we report statistical significance as calculated via one-way ANOVA with Tukey's multiple-comparisons test. The adjusted p < 0.05 is considered significant for confidence interval of 95%. ****p < 0.0001, ***0.0001 < p < 0.001, **0.001 < p < 0.01, *0.01 < p < 0.05; “ns” represents “not significant” (p > 0.05). All histograms indicate mean ± SEM.
Electrophysiology and analysis.
Electrophysiology procedures were performed as described previously (Li et al., 2017). Third instar larvae were dissected within 3 min in HL3 solution containing 0.65 mm calcium at 22°C. Muscle 6 at segment A3 was located by the use of an Olympus BX51WI scope with a 10× water objective and then recorded intracellularly with an electrode made of thick wall glass (1.2 mm × 0.69 mm) pulled by Sutter Instruments Puller P-97. Amplifier GeneClamp 500B and digitizer Digidata 1440A were used. The recording was only used if the resting potential was negative to −60 mV and muscle resistance was >5 mΩ. A Grass Instruments S48 stimulator was used to obtain a large range of stimulation voltage range (1–70 V). To ensure that we could always recruit both axons innervating muscle 6, we first tested a range of stimulation voltage (1–70 V) to find the threshold that triggered the largest response within the testing range. A stimulus slightly larger than this threshold was then used at a frequency of 0.2 Hz and duration of 1 ms for evoked junctional potential (EJP) measurements. Axon Laboratory software was used for acquisition and the Mini Analysis program (Synaptosoft) was used for analysis of and spontaneous mini-EJP (mEJP) frequency and amplitude. Parameters for mini-analysis were set as follows: 0.2 (threshold), 1 (area threshold), 30,000 (period to search a local maximum), 40,000 (period to search a decay time), 40,000 (time before a peak for baseline), 20,000 (period to average a baseline), and 0.6 (fraction of peak to find a decay time and detect complex peak).
Dendrite injuries and microtubule dynamics in dendrites.
Dendrite injuries were performed using a pulsed UV laser (0.001W, 435 nm; Andor Tech) to cut the dorsal comb dendrite of the ddaE class I neuron expressing mCD8-GFP under control of 221-Gal4. Injured larvae were placed in food at 20°C to recover for 18 h. Images were acquired as z-stack projections 18 h postinjury with a 40× 1.4 NA objective on a confocal LSM700 microscope and Zen 2 software (Carl Zeiss) and were processed using the Fiji distribution of ImageJ.
Microtubule dynamics were assessed with EB1-GFP using the UAS-Gal4 expression system with 221-Gal4 driving expression. Microtubule plus end comets were counted in the dorsal comb dendrite of uninjured class I PNS (ddaE) neurons expressing either control (rtnl2-RNAi, VDRC 33320) or raw-RNAi (VDRC 101255).
Drug treatment.
DLK inhibitor GNE-3511 was used as described previously (Feng et al., 2019). L3 larvae were reared on normal CSY fly food at 25°C then transferred for 24 h to drug vehicle (0.5% DMSO) or DLK inhibitor (50 μm GNE-3511 in DMSO to 0.5% final concentration). Puc-GFP imaging and dendrite degeneration assays were then performed as described above.
Results
Mutations in raw, identified from the strain background of dcp-1 mutants, strongly inhibit axonal and synaptic degeneration in Drosophila
Previous studies have suggested that Wallerian degeneration involves mechanisms that are molecularly distinct from apoptosis (Finn et al., 2000; Schoenmann et al., 2010), but to further test this hypothesis, we probed the effects of mutations in the dcp-1 caspase on axonal survival. We observed that loss-of-function mutations in dcp-1, dcp-12, and dcp-13 provided no protection from degeneration (Table 1); however, axonal degeneration was strongly inhibited in animals homozygous for the dcp-1-null allele dcp-1prev1 (Fig. 1). Drosophila motoneuron (Fig. 1A,B) and sensory neuron (Fig. 1C) axons and nerve terminals (Fig. 1C–E) are usually fully degenerated within 20 h after disconnection from their cell bodies by nerve crush injury. However, in dcp-1prev1 mutants, injured axons and synapses remain intact for at least 48 h after injury, which is the limit of what can be assayed in larvae before pupation. This survival of injured axons and synapses in dcp-1prev1 mutants was much longer-lasting than previous observations for manipulations to caspase machinery (Finn et al., 2000; Schoenmann et al., 2010). The unexpected properties of dcp-1prev1 mutants suggested the possibility that this phenotype might be caused by a second site mutation on chromosome 2, rather than by a mutation in dcp-1. The dcp-1prev1 line was originally generated by an imprecise P-element excision from dcp-1k05606 (Laundrie et al., 2003). Because dcp-1k05606 homozygotes and dcp-1k05606 /dcp-1prev1 trans-heterozygotes did not show the degeneration phenotype (Fig. 1D,E, Table 1), the second site mutation responsible for the degeneration phenotype should be represented by a new mutation that in the dcp-1prev1 line, which is absent in the dcp-1k05606 parent line. To find the relevant mutation, we used next-generation sequencing to identify variants between dcp-1prev1, dcp-1k05606 and 3 recombinant lines that cosegregated with the degeneration phenotype. We identified nonsynonymous variants in 6 candidate genes, and then tested each candidate by complementation using available deficiency lines (Table 2). When paired with dcp-1prev1, only the deficiency in the raw gene allowed long lasting survival of damaged axons (Fig. 1D,E). Degeneration was also strongly inhibited when two previously characterized mutations in raw (raw134.47 and raw155.27) (Jemc et al., 2012) were used to make the trans-heterozygotes: raw134.47/dcp-1prev1 and raw155.27/dcp-1prev1 (Fig. 1D,E). To confirm that mutations in raw were the cause of the degeneration phenotype, we tested whether this phenotype could be rescued by introduction of Raw cDNA. We used the Gal4/UAS system to drive expression of UAS-Raw-RA and Raw-RB (two alternatively spliced isoforms of Raw cDNA) in larval motoneurons. Expression of either isoform in neurons was sufficient to confer nearly complete rescue the synaptic degeneration phenotype (Figs. 1D,E, 2). We could therefore conclude that inhibition of degeneration observed in the dcp-1prev1 strain is caused by a mutation in raw, and we named this allele rawdcp-1. This implicates a new function for Raw in promoting Wallerian degeneration.
Figure 1.

A new mutation that strongly inhibits Wallerian degeneration of motoneuron and sensory neuron axons and synapses, identified in dcp-1prev1 animals, maps to raw. A, Single motoneuron axons in third instar larvae are labeled with a membrane-targeted reporter transgene, UAS-mCD8::GFP, driven by m12-Gal4. In control (WT) animals, axons distal to the injury sites are completely fragmented within 20 h after nerve crush injury. However, axons remain completely intact in dcp-1prev1 mutant background. B, Quantification of the degree of axonal degeneration in different genotypes and different time points is shown in black, whereas white bars indicate the percentage of axons that remain intact. C, The axons and nerve terminals of class IV sensory neurons in larval ventral nerve cord were labeled by membrane bound mCD8::GFP with the ppk-Gal4 driver. In WT animals, these terminals were completely fragmented and cleared within 20 h after injury (the remaining terminals are from uninjured segments) but remain intact in dcp-1prev1 animals. D, Representative images of the degeneration phenotype at muscle 4 NMJs 20 h after injury. In WT animals, the cytoskeleton marker Futsch (shown in green) disappeared from the NMJ, whereas the neuronal membrane (labeled by anti-HRP in red) became highly fragmented. In contrast, homozyogous dcp-1prev1 mutations failed to degenerate, however other homozygous mutations in dcp-1: dcp-12 and dcp-13, and trans-heterozygous mutants dcp-1k05606/ dcp-1prev1 showed normal degeneration. NMJs in the dcp-1prev1 animals show no sign of degeneration. Genotypes are described in further detail in Table 1. Trans-heterozygotes of dcp-1prev1 with a deficiency deleted for raw and with previously identified mutations in raw, raw155.27/ dcp-1prev1 and raw134.47/ dcp-1prev1, uncovered the degeneration phenotype. Expression of UAS-Raw in raw134.47/ dcp-1prev1 background using a motoneuron driver D42-Gal4 significantly rescues the protective phenotype, whereas expression of UAS-Raw using a pan-glia driver repo-Gal4 fails to rescue. E, Quantification of NMJ degeneration. Black bars represent the percentage of NMJs that were completely degenerated 20 h after injury, gray bars represent percentage of NMJs that were partially degenerated, and white bars represent the percentage of intact NMJs. F, Schematic of the protein structure of Raw, which has a single transmembrane domain indicated by the black dotted line. The point mutation in raw in the dcp-1prev1 (rawdcp-1) changes amino acid 532, which lies in the extracellular domain, from Alanine to Aspartic acid. Previously described missense mutations raw134.47 and raw155.27 lie with the intracellular domain. Scale bars, 20 μm, error bars represent SEM; ****p < 0.0001, one-way ANOVA test; n ≥ 25 axons or axon terminals from 5 animals per condition.
Figure 2.

Raw functions cell autonomously in neurons to promote Wallerian degeneration. A, Representative images of the degeneration phenotype for single motoneuron axons labeled via the m12-Gal4 driver expressing UAS-mCD8::GFP 20 h after injury. Expression of either of two spliced isoforms of Raw cDNA (Raw-RA or Raw-RB) in m12-Gal4-expressing neurons rescued the protective phenotype in raw134.47/dcp-1prev1 background animals. Moreover, depletion of Raw in motoneurons using raw-RNAi lines in combination with UAS-Dcr2 also strongly delays axonal degeneration. B, Quantification of axonal degeneration of the animals with genotypes shown in A. Scale bars, 20 μm. Error bars indicate SEM. ****p < 0.0001, one-way ANOVA test; n ≥ 25 axons from 5 animals per condition.
Raw functions cell autonomously in injured neurons to promote axonal degeneration
Raw encodes a type I transmembrane domain that lacks identified structural domains (Fig. 1F). The rawdcp-1 allele contains a missense mutation, A532D, which lies in Raw's extracellular domain. Previously characterized mutations in raw are embryonic lethal and cause defects in embryonic dorsal closure, gonad morphogenesis, glial development, and dendrite branching (Bates et al., 2008; Jemc et al., 2012; Lee et al., 2015; Hans et al., 2018; Luong et al., 2018); however, rawdcp-1 mutants are viable and fertile. Because the degeneration phenotype is recessive, we suspected that the rawdcp-1 mutations confer a partial loss in raw function. Consistent with the interpretation that partial loss of raw function in neurons leads to impaired axonal degeneration, knock-down of raw expression in neurons by RNA interference was sufficient to inhibit axonal degeneration (Fig. 2). Two independent UAS-raw-RNAi lines conferred similar inhibition of axonal degeneration when expressed in individual motoneurons via the m12-Gal4 driver (Fig. 2A,B). These observations, together with observation that expression of two different spliced forms of Raw cDNA (Raw-RA and Raw-RB) in defined neurons could restore the ability of injured axons and synapses to degenerate (Figs. 1D,E, 2A,B), indicates that Raw function is required cell autonomously in injured neurons to promote the process of axonal degeneration.
raw mutations preserve functional synapses from injured axons
To test whether raw mutants preserve the function of injured synapses, we conducted electrophysiology studies on NMJs at larval muscle 6 both before and 20 h after injury. We observed no obvious synaptic defect in uninjured raw mutants either by morphology (Fig. 1D) or by recording of EJPs or mEJPs (Fig. 3). In WT animals, NMJs were completely silent for both spontaneous and evoked events at 20 h after injury. In contrast, raw mutants showed robust evoked EJPs and spontaneous mEJPs, similar in amplitude to uninjured NMJs (Fig. 3). Therefore, axons and synapses are functionally intact in raw mutants after injury. In addition, NMJ structure in injured raw mutants showed no difference from uninjured animals (Fig. 1D).
Figure 3.

Injured synapses retain functional neurotransmission properties in raw mutants. A, Example traces for EJP and mEJP potentials recorded from muscle 6 NMJs in control/WT (Canton S), rawdcp-1(dcp-1prev1) homozygous animals and animals expressing UAS-Raw-RA cDNA in motoneurons (via the D42-Gal4 driver) in a rawdcp-1/raw134.47 mutant background. Recordings were done at segment 3 or 4 from naive (uninjured) animals, and in B, in animals 20 h after a ventral nerve crush that injured the nerves innervating these segments. NMJs in rawdcp-1/raw134.47 mutants expressing Raw cDNA in motoneurons showed three phenotypic classes: 6/20recorded NMJs of this genotype retained both EJPs and mEJPs similarly to uninjured NMJs, in left example traces; 12/20 retained mEJPs but lost EJPs, in right traces, and 2/20 lacked both EJPs and mEJPs, similarly to WT injured, indicating partial rescue of the raw phenotype. C, Quantification of EJP amplitude, mEJP amplitude, quantal content (corrected for nonlinear summation), and mEJP frequency (Hz) for recordings from uninjured (black) and 20 h postinjury (red) larvae. D, Percentage of NMJs in the three different phenotypic classes depicted in A. n ≥ 15 NMJs per condition. ****p < 0.0001.
When BG380-Gal4 was used to drive expression of raw cDNA in motoneurons of raw mutants, variable results at 20 h after injury were obtained (Fig. 3B,C). Approximately 30% of synapses had both EJPs and mEJPs (and thus remained similar to the mutant phonotype) and ∼10% of the synapses had neither evoked EJPs nor miniature EJPs (and thus were similar to the WT phenotype). However, the majority of synapses showed the intermediate phenotype of having mEJPs but not EJPs, indicating that expressing the cDNA usually accelerated degeneration compared with the mutant. The inability of UAS-Raw cDNA to completely restore rapid synaptic degeneration may be limited by the timing, level or cell specificity of the Gal4 driver (BG380-Gal4 initiates expression motoneurons in first instar laval stages (Budnik et al., 1996)).
Protection in raw mutants is not sensitive to Nmnat levels
The NAD+ biosynthetic enzyme Nmnat is a central regulator of axonal degeneration, and many previously known manipulations that inhibit axonal degeneration act by altering the stability and turnover of Nmnat protein (Xiong et al., 2012; Babetto et al., 2013; Walker et al., 2017). For example, loss-of-function mutations in the E3 ubiqutin ligase hiw strongly delay axonal degeneration by causing elevated levels of Nmnat enzyme in axons (Xiong et al., 2012; Babetto et al., 2013). We therefore tested whether Nmnat protein levels are altered in raw mutants and whether the raw phenotype is sensitive to changes in nmnat mRNA levels.
We observed no detectable change in the levels of Nmnat protein in raw mutant neurons (Fig. 4A,B). This contrasts with hiw mutants, in which Nmnat protein is dramatically increased in the neuropil (Fig. 4A,B and Xiong et al., 2012). We manipulated Nmnat expression by using the m12-Gal4 driver to express UAS-nmnat-RNAi in subsets of labeled motoneurons, a manipulation that previous studies had demonstrated successfully reduces Nmnat function in Drosophila neurons (Xiong et al., 2012), and which leads to a modest enhancement in the degeneration rate of WT synapses (Fig. 4D). This manipulation did not interfere with the protection from axon degeneration by rawdcp-1 (Fig. 4C,D), even though we were able to replicate the previous finding (Xiong et al., 2012) that it substantially attenuates the protection from degeneration by hiw mutants (Fig. 4D). These data suggest that Raw may regulate degeneration via a mechanism that is not sensitive to Nmnat protein levels, however it may still in theory be sensitive to Nmnat function.
Figure 4.

Protection in raw mutants is not sensitive to levels of Nmnat enzyme. A, Endogenous Nmnat levels in neuropil are increased in hiw mutants (noted with yellow brackets), however are not detectably changed in raw mutants (rawdcp-1). B, Quantification of the relative levels of Nmnat protein in the neuropil regions of WT, hiwΔN and rawdcp-1 animals. C, Example axons from m12-Ga4 expressing neurons, labeled by coexpression of UAS-mCD8::GFP, UAS-Dcr2 and UAS-nmnat-RNAi, shown 20 h after injury. D, Quantification of axonal degeneration in different time points for the noted genotypes. Comparing to WT animals (black line), knock-down of nmnat by RNAi (green line) modestly promotes axonal degeneration. Although knock-down of nmnat in the hiw mutant background significantly rescues the axonal protection phenotype in hiw mutant (compare pink line to blue line), knock-down of nmnat in rawdcp-1 mutant background only modestly changes the axonal protective effect of rawdcp-1 (compare red line to blue line). Scale bar, 20 μm. Error bars indicate SEM. ****p < 0.0001; ns (p = 0.5314), one-way ANOVA test; n = 5 animals per genotype in B, n ≥ 25 axons from 5 animals per condition in D.
The delay to degeneration in raw mutants is not quite as strong as hiw mutants at the NMJ (comparing our data in Fig. 1E to similar assays in Xiong et al., 2012), but it is stronger than the protection caused by overexpressing the DLK/Wnd kinase or by inhibiting JNK (Xiong and Collins, 2012), which are discussed further below. The protection caused by overexpression of DLK/Wnd was also not altered by coexpression of nmnat-RNAi (Xiong et al., 2012), suggesting a mechanism of protection that is distinct from hiw mutants.
Raw regulates axonal degeneration via Fos and Jun transcription factors
Raw has been previously demonstrated to regulate dorsal closure, gonad morphogenesis and glial development by quenching a JNK signaling cascade and the downstream activity of the basal activator protein 1 (AP-1) transcription factor (Bates et al., 2008; Jemc et al., 2012; Luong et al., 2018). Because these downstream components are predicted to have unrestrained activity in raw mutants, we tested their role in the axonal protection phenotype of raw mutants. Because JNK (also known as Bsk in Drosophila) and the components of AP-1, Fos (kayak) and Jun (Jra), are essential for embryonic development in Drosophila, we used the Gal4/UAS system to restrict the expression of dominant negative forms of JNK, Fos, and Jun to small subset of larval motoneurons assayed for axonal degeneration.
Consistent with Raw's previously characterized function in restraining Fos and Jun during dorsal closure (Bates et al., 2008), we observed that the protection from axonal degeneration in raw mutants requires cell autonomous function of Fos and Jun. Expression of dominant negative transgenes FosDN or JunDN alone had no effect upon axonal degeneration in a WT background (Fig. 5), however expression of either dominant negative transgene significantly rescued the protective phenotype of rawdcp-1 mutants (Fig. 5).
Figure 5.

Protection of injured axons from degeneration in raw mutants requires the transcription factors Fos and Jun and the JNK MAP kinase. A, Single motoneuron axons are labeled by membrane bound mCD8::GFP driven by the m12-Gal4 driver. As shown in Figure 1, axonal degeneration is strongly delayed in rawdcp-1 animals. Expression of dominant negative forms of Jun and Fos (JunDN and FosDN) partially rescue the protective effect in rawdcp-1 animals. B, Quantification of axon degeneration scores in indicated genotypes. Scale bar, 20 μm. Error bars indicate SEM. ****p < 0.0001, *0.01 < p < 0.05 (p = 0.0445). ns (p > 0.05), one-way ANOVA test; n ≥ 25 axons from 5 animals per condition.
The role of JNK in the raw protective phenotype was more complicated to address since, as reported previously, both genetic and pharmacological methods to inhibit JNK in mammalian as well as Drosophila neurons can inhibit axonal degeneration (Miller et al., 2009; Xiong et al., 2010; Yang et al., 2015). We note that the protection from degeneration caused by JNKDN expression is not as strong as the raw mutant phenotype (Fig. 5). Strikingly, whereas JNKDN inhibited degeneration on its own, it caused a modest increase in degeneration in the raw mutants and the appearance of varicosities within axons, which typically occurs as a precursor to complete degeneration (Griffin et al., 1995) (Fig. 5A,B). This suggests that JNK might play dual roles in axonal degeneration: it promotes axonal degeneration in WT animals, but an unrestrained version of JNK activity in raw mutants promotes a protective phenotype which delays axonal degeneration. In line with this idea, dichotomous roles for JNK kinase in both axonal degeneration and maintenance of axonal integrity have been previously described in other contexts (for review, see Coffey, 2014).
Protection in raw mutants does not require the Wnd/DLK injury signaling kinase
Previous work has demonstrated that JNK and Fos are required cell-autonomously in the same injured Drosophila neuron to promote new axonal growth after axonal injury (Xiong et al., 2010; Stone et al., 2014). In this case, JNK signaling becomes activated via the mixed linage kinase Wnd/DLK, which becomes activated in response to axonal injury (for review, see Asghari Adib et al., 2018). In addition, it has been previously shown that activation of Wnd and downstream signaling by axonal injury promotes a protective response which inhibits axonal degeneration - this response inhibits degeneration in axons that have sustained a previous injury (Xiong and Collins, 2012). We therefore hypothesized that the protection in raw mutants could involve activation of Wnd signaling. However, we found that RNAi knock-down of wnd, which is sufficient to inhibit downstream signaling and an axonal sprouting response (Fig. 6A,B and Xiong et al., 2010), had no effect on the protective phenotype of raw mutants (Fig. 5). The proximal stumps of raw mutants showed no identifiable differences in the sprouting response to injury (Fig. 6A,B). Although this response was inhibited by knock-down of wnd, within the same axons, the distal stump in raw mutants remained protected from degeneration and unchanged by knock-down of wnd (Fig. 5A,B).
An additional probe of the relationship between Raw and Wnd-regulated axonal injury signaling was enabled by use of the puc-lacZ reporter for JNK signaling (Martín-Blanco et al., 1998; Xiong et al., 2010). Knock-down of Raw expression in neurons (using a pan neuronal BG380-Gal4 driver to drive expression of UAS-raw-RNAi) led to a >3-fold increase in expression from the puc-lacZ reporter. This adds further evidence for the interpretation that JNK signaling becomes activated in raw mutants. Axonal injury leads to a similar increase in puc-lacZ expression (Xiong et al., 2010) and (Fig. 6C–E, at 24 h after injury), and caused no additional increase in raw-RNAi expressing neurons (Fig. 6C–E). The induction of puc-lacZ following injury is abolished in wnd-RNAi-expressing neurons (Xiong et al., 2010) and (Fig. 6C–E). By contrast, coexpression of wnd-RNAi with raw-RNAi lead to an increased variability in puc-lacZ expression between neurons (Fig. 6D), but did not alter the mean intensity (Fig. 6E). Neurons coexpressing wnd-RNAi with raw-RNAi remained resilient to degeneration (data not shown), similarly to raw mutants expressing wnd-RNAi in Figure 5A,B). We interpret these results to mean that Raw restrains JNK signaling downstream or in parallel to regulation by Wnd (see Fig. 8).
Figure 8.
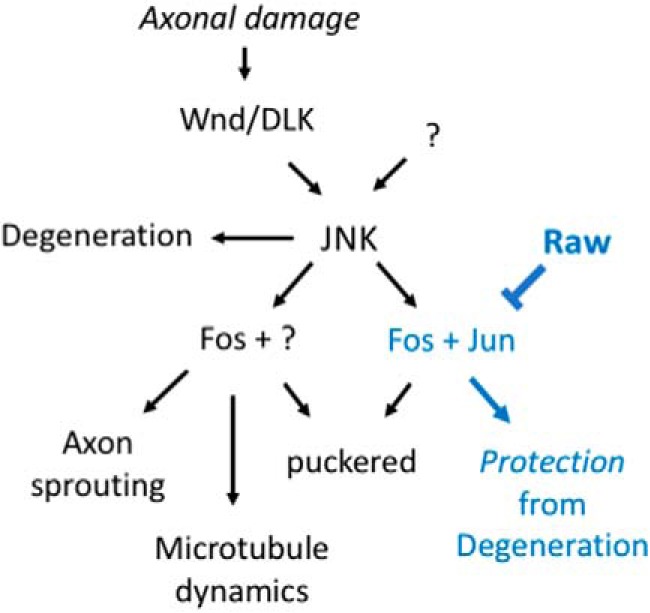
Working model: Raw regulates the ability of injured axons and dendrites to degenerate by restraining a JNK-dependent transcriptional program. Signaling downstream of the Wallenda/DLK kinase after axonal injury and in other conditions of stress (Asghari Adib et al., 2018) regulates multiple JNK-dependent responses. These include promotion of axonal degeneration, which may occur locally in injured axons independently of the nucleus (Miller et al., 2009; Yang et al., 2015), and downstream events that involve transcription factors including Fos and Jun: regenerative axon sprouting (Xiong et al., 2010; Stone et al., 2014), global upregulation of microtubule dynamics (Stone et al., 2010; Chen et al., 2012), and protection of neurites from degeneration (Chen et al., 2012, 2016; Xiong and Collins, 2012). Our data suggest that Raw regulates protective signaling without influencing sprouting or increasing microtubule dynamics. Because the nuclear localization of Jun(Jra) is altered (increased) in raw mutants (Jemc et al., 2012), we propose that this specificity might occur via the regulation of different transcription factor complexes. Raw may therefore provide a new inroad to functionally separate pathways that share dependence upon the JNK MAP kinase.
We also tested whether overexpression of Raw could impair axonal injury signaling. We observed no effect for either the Raw-RA or Raw-RB isoforms upon puc-lacZ expression (data not shown). However, we note that we have been unable to observe any overexpression phenotype for Raw, including any effect upon axonal degeneration.
raw mutations inhibit dendrite degeneration
Previous work has linked protection of neurites from degeneration to a cytoskeletal response that can be detected as a global increase in the number of growing microtubules (Stone et al., 2010; Chen et al., 2012, 2016). Expression of deleterious proteins and axonal damage results in a neuronal response that inhibits degeneration and increases microtubule dynamics throughout the cell (Chen et al., 2012). Because neuroprotection and increased microtubule dynamics in these scenarios are mediated by JNK signaling and inhibited by dominant negative Fos(Chen et al., 2016), and because raw mutants have unrestrained JNK signaling, we investigated whether Raw influences microtubule dynamics and dendrite degeneration.
First, we investigated dendrite degeneration after laser axotomy in ddaE sensory neurons expressing UAS-raw-RNAi. Injured dendrites were maintained for at least 18 h after dendrotomy, showing strong resistance to degeneration (Fig. 7A). Therefore, Raw function influences the resilience of dendrites as well as axons. Dendrite degeneration in raw-RNAi-expressing neurons was unaffected in animals raised in the presence of the DLK inhibitor GNE-3511 (Fig. 7B), which caused a modest reduction in expression of the puc-GFP reporter in raw-RNAi expressing ddaE neurons (Fig. 7C). In contrast this DLK inhibitor strongly inhibits puc-GFP expression and Wnd-dependent responses in patronin knock-down neurons (Feng et al., 2019). These observations suggest that Raw regulates dendrite protection similarly to axon protection, independently or downstream of Wnd signaling (Fig. 8).
Figure 7.

Raw is required for dendrite degeneration. Individual dendrite branches of ddaE sensory neurons, labeled by expression of UAS-mCD8-GFP with the 221-Gal4 driver, were surgically transected using a pulsed dye laser in flies coexpressing expressing Dcr2 and either control RNAi (rtnl2 RNAi, VDRC 33320) or raw RNAi (VDRC 101255). A, Example images of ddaE neurons 0 and 18 h postdendrotomy are shown. Degeneration is characterized by membrane blebbing and loss of continuity in the primary comb (red line). Scale bar, 20 μm. B, Quantification of dendrites that remain intact 18 h postdendrotomy is shown in the graph. Reduction of raw expression via RNAi protects dendrites comparably to overexpression of the protective enzyme Nmnat, as in Chen et al. (2016). Rearing animals in the presence of GNE-3511, which inhibits DLK (Patel et al., 2015) and Wnd-mediated phenotypes in ddaE neurons (Feng et al., 2019), caused no significant change to the raw-RNAi phenotype compared with animals reared in the presence of the DMSO vehicle (B), but did cause a modest reduction in expression of the puc-GFP reporter (C). D, In contrast to a previously described mechanism for dendrite protection (Chen et al., 2012), raw mutants do not show increased microtubule dynamics. The number of growing microtubule plus ends labeled by EB1::GFP were measured in UAS-Dicer2;221Gal4, UAS-EB1::GFP larvae expressing UAS- rtnl2 (control) RNAi or UAS-raw-RNAi. Statistical significance was calculated using one-way ANOVA. ***p < 0.001, **0.001 < p < 0.01, ns represents p > 0.05 (p = 0.15). n ≥ 14 ddaE neurons per condition.
Because the microtubule response requires JNK signaling and correlates with the protective phenotype (Chen et al., 2012, 2016), we hypothesized that raw mutants would also share an increase in microtubule dynamics. However, knock-down of raw led to a reduction rather than increase the number of growing microtubules measured by EB1-GFP (Fig. 7D). This suggests that the protective pathway restrained by Raw may not depend upon increased microtubule dynamics (Fig. 8).
Discussion
Since the discovery of the gain-of-function mutation WldS, which delays Wallerian degeneration for weeks in mammals, many studies have been focused on identifying loss-of-function mutants to better understand the molecular mechanisms by which injured axons self-destruct(Conforti et al., 2014; Neukomm and Freeman, 2014; Gerdts et al., 2016; Ding and Hammarlund, 2019). In this study, we identified Raw as a new regulator of axonal degeneration in Drosophila. We have demonstrated that partial reductions in raw function lead to misregulated signaling that enables long survival of injured axons. This signaling requires transcription factors Fos and Jun, consistent with previous findings that Raw functions as a negative regulator of the AP-1 transcription factor (Bates et al., 2008; Jemc et al., 2012). The involvement of transcription factors implies that the susceptibility of axons to degeneration is regulated in neurons by transcriptional programs and signaling pathways. This idea alone is not new: the lag phase between injury and onset of degeneration varies between different types of neurons, whether they are myelinated, and whether they have sustained previous injuries (Conforti et al., 2014). Recent work has identified another transcription factor, Pebbled, which influences degeneration of glutamatergic but not cholinergic neurons (Farley et al., 2018). Raw represents a new inroad to studying these pathways and the mechanisms that influence axon resiliency. Candidate effectors include channels that affect neuronal excitability and calcium signaling(George et al., 1995; Moldovan et al., 2009; Mishra et al., 2013; Vargas et al., 2015), which are classical targets of immediate early genes including Fos and Jun, and can strongly affect bioenergetics and metabolism in axons (Flavell and Greenberg, 2008). Interestingly, the raw mutant phenotype is not sensitive to levels of Nmnat protein, so the protective pathway restrained by Raw may potentially act in parallel or downstream of the protective actions of the Nmnat enzyme. The lack of upregulated microtubule dynamics in raw knock-down neurons is consistent with this type of protective pathway acting independently from Nmnat. Increased Nmnat levels are sufficient to upregulate microtubule dynamics (Chen et al., 2016); if Nmnat activity were increased by Raw reduction, then an increase in EB1 comet number would be expected.
Previous studies in Drosophila have noted a correlation between protection of neurites and a JNK- mediated increase in microtubule dynamics (Chen et al., 2012, 2016; Hao and Collins, 2017). Because this feature is not shared in raw mutants, the protective pathway restrained by Raw may not require microtubule dynamics per se. Recent work has suggested that Raw may regulate adhesion in Drosophila dendrites (Lee et al., 2015). Because JNK signaling regulates cell adhesion during developmental axon remodeling (Bornstein et al., 2015), the relationship of this function to neurite degeneration is an interesting question for future studies.
Raw and its ortholog Olrn-1 in C. elegans are both neuronally expressed proteins (Bauer Huang et al., 2007; Lee et al., 2015) and Drosophila Raw is capable of localizing to axons, dendrites and cell bodies (Lee et al., 2015). The rawdcp-1 point mutation lies in the large extracellular domain of the Raw protein. The evidence that this mutation results in partial but not complete disruption of Raw's function is twofold. First, the rawdcp-1 mutations are not lethal (in contrast to other characterized mutations in raw) and, second, partial knock-down of Raw expression by RNAi yields a similar delay in degeneration (Fig. 2). Previous studies suggest that Raw regulates cell–cell interactions in several developmental events, including dorsal closure, gonad morphogenesis, and dendrite patterning (Bates et al., 2008; Jemc et al., 2012; Lee et al., 2015; Hans et al., 2018; Luong et al., 2018). Recent work has implicated functions for Raw in glial development (Hans et al., 2018; Luong et al., 2018). It is therefore tempting to hypothesize that Raw participates in glia-neuron interactions, which can influence the onset of axonal degeneration as well as the clearance of axonal debris (Beirowski, 2013; Corty and Freeman, 2013). Although roles for JNK and AP-1 signaling have been noted for glial cells undergoing phagocytosis of axonal debris (Macdonald et al., 2013; Lu et al., 2017; Hilu-Dadia et al., 2018), our rescue assays indicate that Raw functions cell autonomously in neurons to promote degeneration. We have not detected a role for glial Raw in axonal degeneration.
Although Raw does not have an obvious homolog in mammals, Raw's downstream targets, JNK and the AP-1 complex of Fos and Jun transcription factors, are highly conserved in mechanism and function and are strongly implicated in axonal injury responses in both vertebrate and invertebrate neurons. Confoundingly, JNK signaling has been implicated in multiple dichotomous responses to axonal injury: in promoting axonal degeneration (Miller et al., 2009; Yang et al., 2015), protective signaling (Chen et al., 2012; Xiong and Collins, 2012), and in axonal injury signaling and regeneration (Abe and Cavalli, 2008; Xiong et al., 2010; Lee et al., 2015). We found that raw mutants uncover a distinct role for JNK signaling in protecting axons and dendrites from degeneration that can be separated from other JNK-mediated phenotypes (Fig. 8).
Distinct responses that share requirements for JNK presumably work via additional factors that are unique in context and possibly in cellular localization in different scenarios. Different scaffolding molecules suggest one possible mechanism for context specificity (Whitmarsh, 2006; Dhanasekaran et al., 2007); it is also possible that different subunits or isoforms of transcription factor complexes contribute to context specificity and different downstream responses to JNK activation. Prior work has noted that the localization of Jun (Jra) is altered in raw mutants (Jemc et al., 2012). We propose that Raw may regulate one aspect of JNK signaling by modulating a specific transcription factor subunit (Fig. 8). Regardless of the mechanism, the specificity of the raw phenotype presents a new inroad to future study and understanding of how unique JNK signaling pathways can be regulated simultaneously.
Studies using the model organism D. melanogaster have identified several new genes important for axon degeneration, illustrating the power of genetics and significantly influencing our understanding of this degeneration process. In this study, a new regulator of degeneration was discovered not through systematic screening, but fortuitously as a new mutation that arose in the strain background of dcp-1 mutant flies. This discovery illustrates the power of next-generation sequencing approaches combined with functional recombination mapping in a model organism. It also serves as a reminder that extra precautions are needed when studying mutations in balanced lines. Because an entire chromosome is maintained in a heterozygous state, second site mutations that yield interesting phenotypes as homozygotes can arise unnoticed. In this case, a new regulator of axonal degeneration was revealed. Because the basic molecular mechanisms of Wallerian degeneration are highly conserved from invertebrates to mammals (for review, see Neukomm and Freeman, 2014), future understanding of the regulation of degeneration by Raw will shed light on mechanisms for maintaining the resiliency of axons.
Footnotes
This work was supported by the National Institutes of Health (RO1 NS069844 to C.A.C., and RO1 GM085115 to M.M.R.) Stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) and the Vienna Drosophila Resource Center (VDRC) were used in this study. We also thank the TRiP (Transgenic RNAi Project) at Harvard Medical School (NIH/NIGMS R01-GM084947) for providing transgenic RNAi fly stocks used in this study. The 22c10 and 40–1a monoclonal antibodies were obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242. We thank Grace Zhai for sharing Nmnat antibodies and we thank Eric Robertson and Pushpanjali Soppina for technical support in maintaining reagents. We give special thanks to Jennifer Jemc Mierisch for sharing Drosophila stocks and helpful discussions.
The authors declare no competing financial interests.
References
- Abe N, Cavalli V (2008) Nerve injury signaling. Curr Opin Neurobiol 18:276–283. 10.1016/j.conb.2008.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asghari Adib E, Smithson LJ, Collins CA (2018) An axonal stress response pathway: degenerative and regenerative signaling by DLK. Curr Opin Neurobiol 53:110–119. 10.1016/j.conb.2018.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babetto E, Beirowski B, Russler EV, Milbrandt J, DiAntonio A (2013) The phr1 ubiquitin ligase promotes injury-induced axon self-destruction. Cell Rep 3:1422–1429. 10.1016/j.celrep.2013.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates KL, Higley M, Letsou A (2008) Raw mediates antagonism of AP-1 activity in Drosophila. Genetics 178:1989–2002. 10.1534/genetics.107.086298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer Huang SL, Saheki Y, VanHoven MK, Torayama I, Ishihara T, Katsura I, van der Linden A, Sengupta P, Bargmann CI (2007) Left-right olfactory asymmetry results from antagonistic functions of voltage-activated calcium channels and the raw repeat protein OLRN-1 in C. elegans. Neural Dev 2:24. 10.1186/1749-8104-2-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgardt M, Miguel-Aliaga I, Karlsson D, Ekman H, Thor S (2007) Specification of neuronal identities by feedforward combinatorial coding. PLoS Biol 5:e37. 10.1371/journal.pbio.0050037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B. (2013) Concepts for regulation of axon integrity by enwrapping glia. Front Cell Neurosci 7:256. 10.3389/fncel.2013.00256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein B, Zahavi EE, Gelley S, Zoosman M, Yaniv SP, Fuchs O, Porat Z, Perlson E, Schuldiner O (2015) Developmental axon pruning requires destabilization of cell adhesion by JNK signaling. Neuron 88:926–940. 10.1016/j.neuron.2015.10.023 [DOI] [PubMed] [Google Scholar]
- Budnik V, Koh YH, Guan B, Hartmann B, Hough C, Woods D, Gorczyca M (1996) Regulation of synapse structure and function by the Drosophila tumor suppressor gene dlg. Neuron 17:627–640. 10.1016/S0896-6273(00)80196-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Stone MC, Tao J, Rolls MM (2012) Axon injury and stress trigger a microtubule-based neuroprotective pathway. Proc Natl Acad Sci U S A 109:11842–11847. 10.1073/pnas.1121180109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Nye DM, Stone MC, Weiner AT, Gheres KW, Xiong X, Collins CA, Rolls MM (2016) Mitochondria and caspases tune Nmnat-mediated stabilization to promote axon regeneration. PLoS Genet 12:e1006503. 10.1371/journal.pgen.1006503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:80–92. 10.4161/fly.19695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey ET. (2014) Nuclear and cytosolic JNK signalling in neurons. Nat Rev Neurosci 15:285–299. 10.1038/nrn3729 [DOI] [PubMed] [Google Scholar]
- Coleman MP, Freeman MR (2010) Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci 33:245–267. 10.1146/annurev-neuro-060909-153248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Gilley J, Coleman MP (2014) Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat Rev Neurosci 15:394–409. 10.1038/nrn3680 [DOI] [PubMed] [Google Scholar]
- Corty MM, Freeman MR (2013) Cell biology in neuroscience: architects in neural circuit design: glia control neuron numbers and connectivity. J Cell Biol 203:395–405. 10.1083/jcb.201306099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanasekaran DN, Kashef K, Lee CM, Xu H, Reddy EP (2007) Scaffold proteins of MAP-kinase modules. Oncogene 26:3185–3202. 10.1038/sj.onc.1210411 [DOI] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, Couto A, Marra V, Keleman K, Dickson BJ (2007) A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448:151–156. 10.1038/nature05954 [DOI] [PubMed] [Google Scholar]
- Ding C, Hammarlund M (2019) Mechanisms of injury-induced axon degeneration. Curr Opin Neurobiol 57:171–178. 10.1016/j.conb.2019.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eresh S, Riese J, Jackson DB, Bohmann D, Bienz M (1997) A CREB-binding site as a target for decapentaplegic signalling during Drosophila endoderm induction. EMBO J 16:2014–2022. 10.1093/emboj/16.8.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J (2017) The SARM1 Toll/Interleukin-1 receptor domain possesses intrinsic NAD(+) cleavage activity that promotes pathological axonal degeneration. Neuron 93:1334–1343.e5. 10.1016/j.neuron.2017.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essuman K, Summers DW, Sasaki Y, Mao X, Yim AKY, DiAntonio A, Milbrandt J (2018) TIR domain proteins are an ancient family of NAD(+)-consuming enzymes. Curr Biol 28:421–430.e4. 10.1016/j.cub.2017.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley JE, Burdett TC, Barria R, Neukomm LJ, Kenna KP, Landers JE, Freeman MR (2018) Transcription factor Pebbled/RREB1 regulates injury-induced axon degeneration. Proc Natl Acad Sci U S A 115:1358–1363. 10.1073/pnas.1715837115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng C, Thyagarajan P, Shorey M, Seebold DY, Weiner AT, Albertson RM, Rao KS, Sagasti A, Goetschius DJ, Rolls MM (2019) Patronin-mediated minus end growth is required for dendritic microtubule polarity. J Cell Biol 218:2309–2328. 10.1083/jcb.201810155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn JT, Weil M, Archer F, Siman R, Srinivasan A, Raff MC (2000) Evidence that wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. J Neurosci 20:1333–1341. 10.1523/JNEUROSCI.20-04-01333.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell SW, Greenberg ME (2008) Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu Rev Neurosci 31:563–590. 10.1146/annurev.neuro.31.060407.125631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geden MJ, Deshmukh M (2016) Axon degeneration: context defines distinct pathways. Curr Opin Neurobiol 39:108–115. 10.1016/j.conb.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George EB, Glass JD, Griffin JW (1995) Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J Neurosci 15:6445–6452. 10.1523/JNEUROSCI.15-10-06445.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J (2015) SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science 348:453–457. 10.1126/science.1258366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Summers DW, Milbrandt J, DiAntonio A (2016) Axon self-destruction: new links among SARM1, MAPKs, and NAD+ metabolism. Neuron 89:449–460. 10.1016/j.neuron.2015.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Coleman MP (2010) Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol 8:e1000300. 10.1371/journal.pbio.1000300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Orsomando G, Nascimento-Ferreira I, Coleman MP (2015) Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep 10:1974–1981. 10.1016/j.celrep.2015.02.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JW, George EB, Hsieh ST, Glass JD (1995) Axonal degeneration and disorders of the axonal cytoskeleton. In: The axon: structure, function and pathophysiology (Waxman SG, Jeffery DK, Stys PK, eds), pp 375–390. New York: OUP. [Google Scholar]
- Hans VR, Wendt TI, Patel AM, Patel MM, Perez L, Talbot DE, Jemc JC (2018) Raw regulates glial population of the eye imaginal disc. Genesis 56:e23254. 10.1002/dvg.23254 [DOI] [PubMed] [Google Scholar]
- Hao Y, Collins C (2017) Intrinsic mechanisms for axon regeneration: insights from injured axons in Drosophila. Curr Opin Genet Dev 44:84–91. 10.1016/j.gde.2017.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Frey E, Yoon C, Wong H, Nestorovski D, Holzman LB, Giger RJ, DiAntonio A, Collins C (2016) An evolutionarily conserved mechanism for cAMP elicited axonal regeneration involves direct activation of the dual leucine zipper kinase DLK. eLife 5:e14048. 10.7554/eLife.14048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilu-Dadia R, Hakim-Mishnaevski K, Levy-Adam F, Kurant E (2018) Draper-mediated JNK signaling is required for glial phagocytosis of apoptotic neurons during Drosophila metamorphosis. Glia 66:1520–1532. 10.1002/glia.23322 [DOI] [PubMed] [Google Scholar]
- Jemc JC, Milutinovich AB, Weyers JJ, Takeda Y, Van Doren M (2012) raw functions through JNK signaling and cadherin-based adhesion to regulate Drosophila gonad morphogenesis. Dev Biol 367:114–125. 10.1016/j.ydbio.2012.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CT, Jan LY, Jan YN (2005) Dendrite-specific remodeling of Drosophila sensory neurons requires matrix metalloproteases, ubiquitin-proteasome, and ecdysone signaling. Proc Natl Acad Sci U S A 102:15230–15235. 10.1073/pnas.0507393102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laundrie B, Peterson JS, Baum JS, Chang JC, Fileppo D, Thompson SR, McCall K (2003) Germline cell death is inhibited by P-element insertions disrupting the dcp-1/pita nested gene pair in Drosophila. Genetics 165:1881–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Peng Y, Lin WY, Parrish JZ (2015) Coordinate control of terminal dendrite patterning and dynamics by the membrane protein raw. Development 142:162–173. 10.1242/dev.113423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R (2009) Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25:1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup (2009) The sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang YV, Asghari Adib E, Stanchev DT, Xiong X, Klinedinst S, Soppina P, Jahn TR, Hume RI, Rasse TM, Collins CA (2017) Restraint of presynaptic protein levels by Wnd/DLK signaling mediates synaptic defects associated with the kinesin-3 motor Unc-104. eLife 6:e24271. 10.7554/eLife.24271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TY, MacDonald JM, Neukomm LJ, Sheehan AE, Bradshaw R, Logan MA, Freeman MR (2017) Axon degeneration induces glial responses through draper-TRAF4-JNK signalling. Nat Commun 8:14355. 10.1038/ncomms14355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luong D, Perez L, Jemc JC (2018) Identification of raw as a regulator of glial development. PLoS One 13:e0198161. 10.1371/journal.pone.0198161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald JM, Doherty J, Hackett R, Freeman MR (2013) The c-jun kinase signaling cascade promotes glial engulfment activity through activation of draper and phagocytic function. Cell Death Differ 20:1140–1148. 10.1038/cdd.2013.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM, Martinez-Arias A (1998) puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev 12:557–570. 10.1101/gad.12.4.557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, DiAntonio A (2009) A dual leucine kinase-dependent axon self-destruction program promotes wallerian degeneration. Nat Neurosci 12:387–389. 10.1038/nn.2290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra B, Carson R, Hume RI, Collins CA (2013) Sodium and potassium currents influence wallerian degeneration of injured Drosophila axons. J Neurosci 33:18728–18739. 10.1523/JNEUROSCI.1007-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan M, Alvarez S, Krarup C (2009) Motor axon excitability during wallerian degeneration. Brain 132:511–523. 10.1093/brain/awn332 [DOI] [PubMed] [Google Scholar]
- Neukomm LJ, Freeman MR (2014) Diverse cellular and molecular modes of axon degeneration. Trends Cell Biol 24:515–523. 10.1016/j.tcb.2014.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neukomm LJ, Burdett TC, Seeds AM, Hampel S, Coutinho-Budd JC, Farley JE, Wong J, Karadeniz YB, Osterloh JM, Sheehan AE, Freeman MR (2017) Axon death pathways converge on axundead to promote functional and structural axon disassembly. Neuron 95:78–91.e5. 10.1016/j.neuron.2017.06.031 [DOI] [PubMed] [Google Scholar]
- Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, MacDonald JM, Ziegenfuss JS, Milde S, Hou YJ, Nathan C, Ding A, Brown RH Jr, Conforti L, Coleman M, Tessier-Lavigne M (2012) dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337:481–484. 10.1126/science.1223899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Cohen F, Dean BJ, De La Torre K, Deshmukh G, Estrada AA, Ghosh AS, Gibbons P, Gustafson A, Huestis MP, Le Pichon CE, Lin H, Liu W, Liu X, Liu Y, Ly CQ, Lyssikatos JP, Ma C, Scearce-Levie K, Shin YG (2015) Discovery of dual leucine zipper kinase (DLK, MAP3K12) inhibitors with activity in neurodegeneration models. J Med Chem 58:401–418. 10.1021/jm5013984 [DOI] [PubMed] [Google Scholar]
- Ritzenthaler S, Suzuki E, Chiba A (2000) Postsynaptic filopodia in muscle cells interact with innervating motoneuron axons. Nat Neurosci 3:1012–1017. 10.1038/79833 [DOI] [PubMed] [Google Scholar]
- Sanyal S. (2009) Genomic mapping and expression patterns of C380, OK6 and D42 enhancer trap lines in the larval nervous system of Drosophila. Gene Expr Patterns 9:371–380. 10.1016/j.gep.2009.01.002 [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Vohra BP, Baloh RH, Milbrandt J (2009) Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J Neurosci 29:6526–6534. 10.1523/JNEUROSCI.1429-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenmann Z, Assa-Kunik E, Tiomny S, Minis A, Haklai-Topper L, Arama E, Yaron A (2010) Axonal degeneration is regulated by the apoptotic machinery or a NAD+-sensitive pathway in insects and mammals. J Neurosci 30:6375–6386. 10.1523/JNEUROSCI.0922-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DJ, Pitts J, Hertz NT, Yang J, Yamagishi Y, Olsen O, Tešić Mark M, Molina H, Tessier-Lavigne M (2016) Axon degeneration gated by retrograde activation of somatic pro-apoptotic signaling. Cell 164:1031–1045. 10.1016/j.cell.2016.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MC, Nguyen MM, Tao J, Allender DL, Rolls MM (2010) Global up-regulation of microtubule dynamics and polarity reversal during regeneration of an axon from a dendrite. Mol Biol Cell 21:767–777. 10.1091/mbc.e09-11-0967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MC, Albertson RM, Chen L, Rolls MM (2014) Dendrite injury triggers DLK-independent regeneration. Cell Rep 6:247–253. 10.1016/j.celrep.2013.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas ME, Yamagishi Y, Tessier-Lavigne M, Sagasti A (2015) Live imaging of calcium dynamics during axon degeneration reveals two functionally distinct phases of calcium influx. J Neurosci 35:15026–15038. 10.1523/JNEUROSCI.2484-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LJ, Summers DW, Sasaki Y, Brace EJ, Milbrandt J, DiAntonio A (2017) MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2. eLife 6:e22540. 10.7554/eLife.22540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber U, Paricio N, Mlodzik M (2000) Jun mediates frizzled-induced R3/R4 cell fate distinction and planar polarity determination in the Drosophila eye. Development 127:3619–3629. [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ. (2006) The JIP family of MAPK scaffold proteins. Biochem Soc Trans 34:828–832. 10.1042/BST0340828 [DOI] [PubMed] [Google Scholar]
- Xiong X, Collins CA (2012) A conditioning lesion protects axons from degeneration via the Wallenda/DLK MAP kinase signaling cascade. J Neurosci 32:610–615. 10.1523/JNEUROSCI.3586-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Wang X, Ewanek R, Bhat P, Diantonio A, Collins CA (2010) Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J Cell Biol 191:211–223. 10.1083/jcb.201006039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Hao Y, Sun K, Li J, Li X, Mishra B, Soppina P, Wu C, Hume RI, Collins CA (2012) The highwire ubiquitin ligase promotes axonal degeneration by tuning levels of nmnat protein. PLoS Biol 10:e1001440. 10.1371/journal.pbio.1001440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Wu Z, Renier N, Simon DJ, Uryu K, Park DS, Greer PA, Tournier C, Davis RJ, Tessier-Lavigne M (2015) Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell 160:161–176. 10.1016/j.cell.2014.11.053 [DOI] [PMC free article] [PubMed] [Google Scholar]