

Dynamic single-molecule imaging reveals NaV1.7 distribution and microtubule-dependent vesicular trafficking in sensory axons.
Abstract
Sodium channel NaV1.7 controls firing of nociceptors, and its role in human pain has been validated by genetic and functional studies. However, little is known about NaV1.7 trafficking or membrane distribution along sensory axons, which can be a meter or more in length. We show here with single-molecule resolution the first live visualization of NaV1.7 channels in dorsal root ganglia neurons, including long-distance microtubule-dependent vesicular transport in Rab6A-containing vesicles. We demonstrate nanoclusters that contain a median of 12.5 channels at the plasma membrane on axon termini. We also demonstrate that inflammatory mediators trigger an increase in the number of NaV1.7-carrying vesicles per axon, a threefold increase in the median number of NaV1.7 channels per vesicle and a ~50% increase in forward velocity. This remarkable enhancement of NaV1.7 vesicular trafficking and surface delivery under conditions that mimic a disease state provides new insights into the contribution of NaV1.7 to inflammatory pain.
INTRODUCTION
Dorsal root ganglia (DRG) house sensory neurons of diverse modalities, including nociceptors, which require precise electrogenic tuning by a distinct set of voltage-gated sodium (NaV) channels. NaV1.7 is arguably the best validated peripheral sodium channel involved in human pain (1, 2). This channel is highly expressed in nociceptors, where it is found at axonal endings in the skin, along the axonal shaft and soma, and in the presynaptic terminals in the dorsal horn of the spinal cord (3, 4). NaV1.7 contributes 60 to 70% of the tetrodotoxin-sensitive (TTX-S) sodium current in small DRG neurons (5–7), is considered a threshold channel (7, 8), and plays a role in transmitter release at the central termini of DRG neurons in the dorsal horn (8, 9). The functional importance of NaV1.7 in nociceptors has sparked intense interest as a therapeutic target in the development of isoform-selective analgesics.
Both channel function and subcellular distribution are important in pain transduction, with well-documented up-regulation of NaV1.7 channels in response to injury or disease. For example, NaV1.7 channels accumulate within blind axonal endings in neuromas in human tissue and in experimental animal models of traumatic nerve injury (10, 11); neuromas are sites of ectopic firing that have been associated with spontaneous pain (12). Increased NaV1.7 expression has also been reported in multiple animal models of inflammation (13–15), and this enhanced expression of NaV1.7 can be detected within a few hours after insult and can persist for days or weeks (16). Moreover, NaV1.7 is required for thermal pain after burn injury (6). Despite the functional importance of NaV1.7 subcellular and surface distribution, mechanisms underlying channel trafficking under normal conditions and after injury or disease are poorly understood.
Because of the length of peripheral sensory axons, which can be more than a meter long, sensory neurons have unique challenges in terms of regulating precise spatial and temporal channel distribution and delivery. The view that NaV biogenesis occurs in the soma and mature sodium channels are then transported to distal axons where they are inserted in the plasma membrane is supported by limited studies using biochemical and immunolabeling techniques and the use of reporter proteins carrying fragments of NaV channels that are predicted to carry trafficking motifs (17, 18). These studies provide limited information, as they do not distinguish between intracellular- and membrane-localized channels and do not support detailed studies of full-length channel behavior in real time.
This study provides the first report of NaV1.7 channel surface distribution, cell surface dynamics, and long-distance axonal transport. We used tagged NaV1.7 constructs suitable for high-resolution, live-neuron imaging combined with a microfluidic culture system. We found that NaV1.7 localizes to small membrane puncta, or nanoclusters, on distal axon termini. Furthermore, we introduced an optical pulse-chase axonal long-distance (OPAL) imaging assay to provide the first visualization of long-distance, microtubule-based transport of NaV1.7 channels and demonstrate that their anterograde movement is via Rab6A-containing vesicles. Given the importance of NaV1.7 in inflammatory pain (1, 19), we capitalized on the temporal and spatial information provided by these assays to assess whether exposure to inflammatory mediators (IM) substantially enhances NaV1.7 vesicular trafficking and surface delivery to distal axons. Our results indicate that IM trigger a remarkable increase in the number of NaV1.7-carrying vesicles per axon, together with a threefold increase in the median number of NaV1.7 channels per vesicle and a ~50% increase in forward velocity.
RESULTS
Fluorescently tagged constructs enable live-cell imaging of NaV1.7
To enable dynamic live-cell imaging of NaV1.7 channel surface distribution and mobility within the plasma membrane, we developed a fluorescently tagged construct with an N-terminal fluorescent protein and an extracellular tag. Venus–NaV1.7–BAD (biotin acceptor domain) contains a Venus fluorescent protein fused in-frame with the N-terminus and a BAD incorporated into the extracellular S1-S2 loop of domain IV (Fig. 1A). The biotinylation of the BAD by a biotin ligase (20) enables live-cell labeling for studying the distribution and dynamics of surface channels using streptavidin-conjugated fluorophores. To visualize vesicular trafficking, we made a tagged NaV1.7 construct containing HaloTag, an enzyme that binds covalently with synthetic ligands (Fig. 2A), by creating a 25-transmembrane channel protein, with the HaloTag present on the extracellular face of the membrane (Halo-NaV1.7).
Fig. 1. Venus-NaV1.7-BAD localizes to nanoclusters on distal axons.
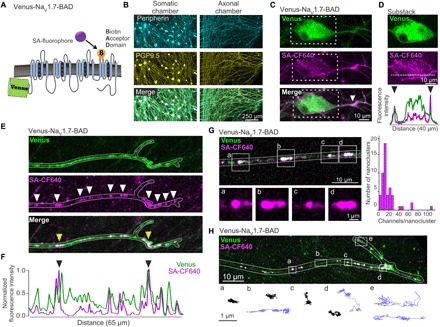
(A) Diagram of the Venus-NaV1.7-BAD construct showing the fusion of Venus to the N-terminus and the insertion of the BAD into the S1-S2 loop of domain IV. (B to H) DRG neurons were cultured for 7 to 10 days in microfluidic chambers (MFCs). (B) Immunolabeling of DRG neurons in MFCs express both peripherin (cyan) and PGP9.5 (yellow) in both the somatic (left) and axonal (right) chambers. (C) Compressed confocal z-stack of a DRG neuron within the MFC somatic chamber expressing Venus-NaV1.7-BAD, showing the distribution of the Venus signal (top), surface labeling with SA-CF640 (middle), and merge (bottom). The surface labeling reveals a region of the proximal axon with increased density of NaV1.7 (white arrow). (D) A substack of the z-slices from (C) chosen from the center of the soma to demonstrate the surface labeling (magenta) surrounding the somatic cytoplasm containing the total NaV1.7 protein (green). Bottom panel shows line scans of the normalized fluorescence intensity of the Venus (green) and SA-CF640 (magenta) signals along the line scan (dashed white line; middle). The black arrowheads indicate the increased fluorescence intensity of the SA-CF640 surface labeling at the plasma membrane. (E) Compressed confocal z-stack of a distal axon from the MFC axonal chamber. Both the Venus signal (top) and surface labeling (middle) demonstrate localization of channels to nanoclusters along the axonal membrane (white arrows). (F) Line profile of the fluorescence intensity of the distal axon in (E), normalized to the maximum fluorescence intensity. Peaks in the line profile corresponding to the clusters of channels along the axon. This example shows two clusters that are noticeably brighter than the others [yellow arrows in (E) and black arrows in (F)]. (G) Estimation of the number of channels per axonal nanocluster. The average fluorescence intensity of single molecules was determined by creating a histogram of individual channel intensities. Fluorescence intensities of nanoclusters were divided by this value to approximate the number of channels in each nanocluster. (H) Single-molecule tracks of individual surface labeled Venus-NaV1.7-BAD channels on a distal axon. A compressed z-stack was used to demonstrate the overall morphology of a distal axon (outlined in white) expressing Venus-NaV1.7-BAD where the overlay between the Venus signal (green) and surface labeling (magenta) appears as white. Tracks are representative molecule behaviors from the regions indicated by the white boxes (boxes a to e). Molecules near the distal end and between nanoclusters show relatively high mobility (blue trajectories), while channels within the nanoclusters display low mobility (black trajectories).
Fig. 2. Anterograde Halo-NaV1.7 vesicles accumulate at the distal axon.
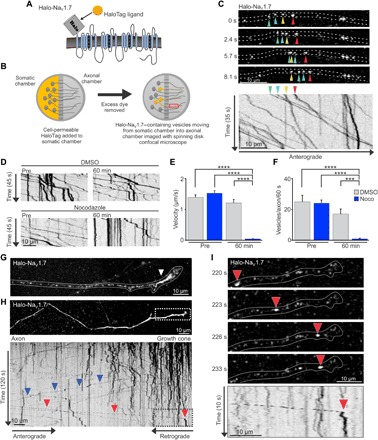
(A) Diagram of the Halo-NaV1.7 construct showing addition of the β4 transmembrane and extracellular location of the HaloTag enzyme. (B) Schematic of the OPAL imaging technique used to visualize anterograde vesicles containing NaV1.7. DRG neurons expressing Halo-NaV1.7 were cultured in MFCs for 7 to 10 days. Immediately prior to imaging, JF549-Halo was added to the somatic chamber for 15 min. After removal of excess dye, axons within the axonal chamber were imaged using spinning disk microscopy to visualize anterograde vesicles traveling from the somatic chamber into the axonal chamber. (C) Representative axon in the axonal chamber labeled as described in (B). Colored arrowheads show locations of the identified vesicles over time. The corresponding kymograph of the Halo-NaV1.7 vesicles was created from a time-lapse image sequence taken at 3.3 frames/s. It shows vesicle position (x axis) as a function of time (y axis). Negative slopes on the kymograph represent movement in the anterograde direction, while vertical lines represent stationary vesicles. (D) Kymographs generated from an axon before and 60 min after either dimethyl sulfoxide (DMSO) or Noco treatment. (E) Average vesicle velocities before and after DMSO or Noco treatment. (F) The number of vesicles with a velocity >0.1 μm/s within a 30-μm segment of axon per 60 s. ****P < 0.0001, ***P < 0.001, two-way ANOVA with Tukey’s multiple comparisons test. (G) Compressed z-stack of an axon terminal in the axonal chamber of the MFC at 1 hour after somatic labeling, demonstrating the accumulation of vesicles near the distal end of the axon (white arrowhead). (H) Image sequence of Halo-NaV1.7 compressed over time to allow visualization of the distal axon. The kymograph of a line scan through this axon shows the behavior of vesicles within this region over time. The vertical lines demonstrate that the vesicles that have accumulated within the distal axon are generally stationary. A small number of channels leave the distal axon as retrograde vesicles (blue arrowheads). Anterograde vesicles move along the axon until reaching the distal axon where they become immobilized (red arrowhead in the black box that corresponds to the region delineated by the white box in the top panel). (I) Time series of a vesicle destined for the distal axon corresponding to the region indicated by the white box in (H), as well as the kymograph region outlined by the back box (H). The red arrowheads follow the position of the vesicle over time as it travels along the axon and then stops at the neck of the distal axon. The vesicle behavior over time is illustrated by the corresponding kymograph with the red arrowhead, denoting where it transitions from mobile to stationary.
Both the Venus-NaV1.7-BAD and Halo-NaV1.7 constructs displayed wild-type (WT) currents [produced by the green fluorescent protein (GFP)–2A-NaV1.7 construct] and expressed well in cultured DRG neurons (fig. S1). Current-voltage relationships were obtained by applying a series of 100 ms depolarizing steps from −80 to +50 mV in 5 mV increments (fig. S1A) from a holding potential of −120 mV. Although the maximum current density was 22 to 30% smaller for the tagged channels, the differences did not reach statistical significance: Venus-NaV1.7-BAD, −186 ± 27 mV; Halo-NaV1.7, −163 ± 21 mV; WT, −236 ± 30 mV [F = 2.1, P = 0.14, one-way analysis of variance (ANOVA); Venus, n = 17; Halo, n = 21; WT, n = 15]. Normalized conductance values were fit with the Boltzmann equation to determine the voltage dependence of activation (fig. S1B). Half-activation values were similar between the three constructs: Venus-NaV1.7-BAD, −23 ± 1 mV; Halo-NaV1.7, −23 ± 1 mV; WT, −21 ± 1 mV (F = 1.3, P = 0.3, one-way ANOVA; Venus, n = 16; Halo, n = 20; WT, n = 14). Steady-state fast-inactivation curves were generated by applying 500 ms inactivating potentials from −120 to −10 mV, followed by a 40 ms step to −10 mV. Normalized conductance values were fit with the Boltzmann equation to determine the voltage dependence of steady-state fast-inactivation (fig. S1C). Half-maximal inactivation values were similar between the three constructs: Venus-NaV1.7-BAD, −75 ± 2 mV; Halo-NaV1.7, −74 ± 2 mV; WT, −75 ± 1 mV (F = 0.2, P = 0.8, one-way ANOVA; Venus, n = 9; Halo, n = 10; WT, n = 9). Recovery from fast-inactivation was studied using a twin-pulse protocol, where two identical voltage steps were separated by an incremental time delay (recovery period). Current elicited by the second step (P2) was normalized to the peak current elicited by the first pulse (P1) and plotted versus the length of the recovery period. We generated currents from a holding potential of −90 mV by 20 ms steps to 0 mV (fig. S1D). The rate of recovery from inactivation was not different between the three constructs (F = 0.4, P = 0.7, one-way repeated-measures ANOVA; Venus-NaV1.7-BAD, n = 7; Halo-NaV1.7, n = 10; WT, n = 8). Together, these data demonstrate that the tagged NaV1.7 constructs are functional and suitable for live-cell imaging studies.
NaV1.7 channels localize to nanoclusters on distal axons
We cultured DRG neurons in microfluidic chambers (MFCs), where the somas are plated within one chamber (somatic chamber) and the axons extend through a 450-μm microgroove barrier into the axonal chamber, thus allowing studying of channel trafficking and membrane distribution in distal neuronal compartments. Figure 1B shows day in vitro (DIV) 7 DRG neurons in an MFC that have been immunolabeled with peripherin (top), a marker for small-diameter, unmyelinated sensory neurons, and with PGP9.5 (middle), a pan-neuronal marker. The merged image shows, as expected, that the majority of the neurons in this culture produce unmyelinated axons. To look at the surface distribution of NaV1.7, DRG neurons transfected with Venus-NaV1.7-BAD and the biotin ligase BirA were cultured in MFCs and then labeled with the streptavidin-conjugated fluorophore CF640R (SA-CF640) to visualize the surface distribution of NaV1.7. The Venus signal (green) is expressed strongly throughout the soma and neurites, and the surface labeling (magenta) shows channels localized within the plasma membrane (Fig. 1C). Surface labeling is shown in Fig. 1D, which depicts a substack of z-slices containing the somatic region to show the surface labeling (magenta) encircling the Venus signal (green). This is emphasized by the line scan (Fig. 1D, bottom), showing the normalized fluorescence intensity of the Venus (green) and SA-CF640 (magenta) signals along the line scan (dashed white line). The black arrowheads indicate the increased fluorescence intensity of the SA-CF640 surface labeling at the plasma membrane. Control experiments ruled out bleed-through artifacts, and the intracellular signal represents autofluorescent structures that can be observed in nontransfected DRG neurons (fig. S2E). A concentration of surface-labeled NaV1.7 can be seen on one neurite (Fig. 1C, white arrowhead). This structure is most likely the proximal axonal segment, which has been previously described in cultured DRG neurons (21).
NaV1.7 localizes to distinct puncta on the distal axon
Action potentials are generated in peripheral sensory neurons at distal axon terminals and propagate toward the central nervous system (CNS) along the axon. This contrasts with CNS neurons where action potentials are generally initiated at the axon initial segment (AIS), a region with a high density of NaV channels on the proximal axon, and are propagated in the anterograde direction (22). Here, we investigated whether NaV1.7 is concentrated near the axon distal terminal that might be responsible for action potential initiation. We observed the Venus signal throughout the axon and terminal region, with a bead-like pattern of fluorescence signal (Fig. 1E, top). The regions with higher fluorescence intensity often corresponded to surface-labeled channels (Fig. 1E, middle), representing the accumulation of channels into distinct puncta (white arrowheads). Figure 1F shows a line profile representing the normalized fluorescence intensity along the axon with peaks demonstrating the location of the NaV1.7 channel puncta. We refer to these puncta as nanoclusters, defining them as small membrane regions containing multiple NaV1.7 channels (further clarified in the next section). These nanoclusters differ in fluorescence intensity, suggesting that different numbers of channels localize to the different nanoclusters. On the axon shown, there are two nanoclusters that are brighter than the rest, denoted by the black arrowheads on the line scan and the yellow arrowheads on the picture (Fig. 1, E and F). The fluorescence intensity of the Venus signal and that of the SA-CF640 surface labeling mirror each other, although the Venus signal is often brighter, which suggests the presence of an additional intracellular pool of channels.
It is noteworthy that the organization of surface channels in nanoclusters is not an artifact of the use of the multivalent streptavidin reagent, which could have cross-linked several channels together, because similar nanoclusters are also observed in DRG neurons expressing Halo-NaV1.7 after surface labeling with the cell impermeable ligand, JF549i (fig. S3B). In contrast to the multivalent streptavidin reagent, there is no known mechanism by which the monovalent Halo ligands would cause channel cross-linking. Further, nanoclusters are not observed in human embryonic kidney (HEK) 293 cells expressing the Venus-NaV1.7-BAD construct even though these cells produce a robust NaV1.7 current (fig. S3C). This is similar to the observation that nanoclusters of NaV1.6-BAD-GFP in the soma of hippocampal neurons are also not artifacts of the use of the streptavidin reagents (23).
Number of channels per nanocluster
To further examine the concentration of NaV1.7 at the distal axons, we investigated the number of channels present within the nanoclusters. We first measured the fluorescence intensity of isolated particles demonstrating random diffusion characteristic of single channels within the plasma membrane and created a histogram of fluorescence intensities for individual particles (fig. S4). This was used to determine the average fluorescence intensity representing a single channel, and we then divided the total fluorescence intensity of each nanocluster by that of a single channel to approximate the number of channels per cluster. Of 44 nanoclusters measured (seven axons from two independent cultures), sizes ranged from 3 to 108 channels per nanocluster with a median of 12.5 channels per nanocluster and only a few carrying >30 channels per nanocluster (Fig. 1G).
Mobility of NaV1.7 channels within the plasma membrane
To determine whether surface NaV1.7 channels are stationary or mobile, we used single-particle tracking to observe the behavior of individual channels in the plasma membrane. While total internal reflection fluorescence (TIRF) microscopy is, in principle, well suited for single-particle tracking, DRG somas and axons do not reliably stay within the TIRF field, especially in older cultures. In contrast to hippocampal neurons where the basal membrane will often adhere tightly to the culture substrate, DRG neurons maintain a rounded somatic morphology, and their axons cross over each other, creating a three-dimensional (3D) network well above the TIRF field. Thus, we used a spinning disk confocal microscope for single-particle tracking by capturing 100-ms exposures and frame averaging of 4 to yield a frame rate of 2.5 Hz. Although this is considered slow for precise single-particle behavior, it allowed us to explore the overall mobility of NaV1.7 channels within the axonal plasma membrane. Figure 1H shows a distal axon expressing Venus-NaV1.7-BAD and surface channels labeled with SA-CF640. Movement of single NaV1.7 molecules was tracked automatically using uTrack (24) from image sequences. We observed channels with both high and low mobility. Representative tracks are shown from the regions outlined by white boxes (Fig. 1H, boxes a to e). To describe these two populations, we grouped trajectories as either low diffusivity (<0.004 μm2/s) or high diffusivity (>0.004 μm2/s) using ad hoc thresholds. Channels showing low mobility (black traces) were often associated with membrane regions where nanoclusters (for example, in box a) were observed in the initial z-stack. These channels showed diffusion coefficients of 0.0012 ± 0.001 μm2/s (mean ± SD; 169 trajectories), which is similar to previous reports of NaV channels in the AIS of hippocampal neurons that are presumably tethered to the cytoskeleton through AnkyrinG binding (25, 26). Other channels showed >10-fold higher mobility (blue traces) and were seen moving throughout the axonal shaft (box b) and within the axon terminal (box e). These channels had diffusion coefficients of 0.0176 ± 0.0170 μm2/s (mean ± SD; 83 trajectories), which is similar to NaV1.6 channel diffusivity outside of somatic nanoclusters in hippocampal neurons (23).
Visualization of anterograde NaV1.7 axonal trafficking
The Venus-NaV1.7-BAD construct allows visualization of the surface channels; however, it cannot be used to observe anterograde vesicular trafficking of NaV1.7 channels. Although bright, Venus has low photostability and bleaches quickly during time-lapse imaging. During these experiments, retrograde, but not anterograde, vesicles can be observed traveling along the photobleached neurites (fig. S5). One possible explanation for our inability to observe anterograde transport of channels might be due to a small number of channels per vesicle relative to the large pool of intracellular channels. If this is true, then it implies that retrograde vesicles contain more channels per vesicle (are brighter) than anterograde vesicles.
To substantially enhance the signal-to-noise ratio for visualizing anterograde vesicular trafficking of NaV1.7, we developed OPAL imaging, an optical pulse-chase method of labeling NaV1.7 vesicles. First, we used Halo-NaV1.7, which binds covalently with synthetic ligands (Fig. 2A). We used cell-permeable JaneliaFluor549- or JaneliaFluor646-HaloTag ligands (JF549-Halo or JF646-Halo, respectively) to label the Halo-NaV1.7, as these synthetic fluorophores are bright and photostable, enabling detection with single-molecule resolution. For the optical pulse-chase experiment, we cultured DRG neurons expressing Halo-NaV1.7 in MFCs and then labeled channels with cell-permeable JF549-Halo (27) only in the somatic chamber. By imaging in the axonal chamber with the spinning disk confocal microscope, we could selectively visualize vesicles that had been labeled within the somatic chamber and then transported into the axonal chamber (Fig. 2B). Thus, this method by design labels anterograde transport events. The use of bright, photostable synthetic fluors combined with low background provided an excellent signal-to-noise ratio for clear visualization of vesicles. Figure 2C shows an example axon visualizing anterograde Halo-NaV1.7 vesicles using OPAL imaging, where the different-colored arrows denote the positions of individual vesicles at each time point. These data are shown in movie S1. The corresponding kymograph below identifies vesicle positions (x axis) over time (y axis), with the arrows denoting the tracks associated with the vesicles marked in the frames above. This high-resolution imaging technique allows visualization of individual Halo-NaV1.7 vesicles with single-molecule resolution. Even with these ideal imaging conditions, some of the vesicles are still quite dim, requiring relatively slow imaging rates of around 3 Hz. This is consistent with a low number of channels per vesicle and explains the difficulties of visualizing these vesicles with fluorescent protein tags.
Using OPAL imaging, we can observe the behavior of anterograde vesicles along the axon and as they reach the axon terminal. While some vesicles pause or stop along the axon shaft (vertical lines on the kymograph; Fig. 2C), the majority continue to move along the axon to the distal end. To determine whether this transport is microtubule dependent, we treated the axons with nocodazole (Noco), a microtubule-destabilizing agent, and imaged the same axons before and 1 hour after treatment (Fig. 2D). The average velocity of vesicles within the Noco-treated axons was significantly reduced (pre-Noco: 1.53 ± 0.09 μm/s, n = 143 vesicles, nine axons; post-Noco: 0.01 ± 0.01 μm/s, n = 69 vesicles, nine axons; P ≤ 0.0001, two-way ANOVA with Tukey’s multiple comparisons test), while dimethyl sulfoxide (DMSO)–treated axons displayed stable velocity (pre-DMSO: 1.39 ± 0.09 μm/s, n = 111 vesicles, seven axons; post-DMSO: 1.21 ± 0.11 μm/s, n = 90 vesicles, seven axons) (Fig. 2E). The number of forward-moving vesicles (with an anterograde velocity >0.1 μm/s) per axon was also significantly inhibited by Noco (pre-Noco: 23.80 ± 2.07 moving vesicles per axon per 60 s, n = 9 axons; post-Noco: 0.74 ± 0.39 moving vesicles per axon per 60 s, n = 9 axons; P ≤ 0.0001, two-way ANOVA with Tukey’s multiple comparisons test), while DMSO-treated axons continued to have forward trafficking (pre-DMSO: 24.71 ± 4.16 moving vesicles per axon per 60 s, n = 7 axons; post-DMSO: 16.82 ± 3.27 moving vesicles per axon per 60 s, n = 7 axons) (Fig. 2F).
Next, we sought to determine the destination of these anterograde vesicles. Within an hour after labeling, there is a buildup of vesicles at the distal end of the axons, suggesting that many vesicles travel the length of the axon and stop when they reach the distal axon end (Fig. 2G). Figure 2H shows an image series that has been averaged over time. The shape of the axon is visible, with the signal intensity increasing toward the axonal end due to the accumulation of vesicles in this region. The corresponding kymograph of the vesicular movement along the axon is shown below. The majority of the vesicles near the distal axon are generally stationary (vertical lines). A few vesicles can be seen moving in the retrograde direction (blue arrowheads). The signal could only have come from Halo-NaV1.7 that was in the somatic chamber at the time of labeling, suggestive of vesicle recycling, perhaps without delivering their cargo channels. We can also visualize some anterograde vesicles that quickly move down the axon and then stop directly at or near the neck of the distal axon (red arrowheads). An example of this behavior is shown in Fig. 2I, where the vesicle position is shown over time, indicated by the red arrowheads. The time sequence corresponds to the region denoted by the white outline box in Fig. 2H, and the kymograph is enlarged from the black outline box, also from Fig. 2H. Together, these data suggest that the anterograde NaV1.7 vesicles visualized using OPAL imaging preferentially travel to and accumulate at the distal end of the axon.
Anterograde NaV1.7 vesicles are Rab6A positive
To demonstrate that NaV1.7 trafficking is via conventional vesicular trafficking and to assess potential selective sorting, we sought to identify Rab protein cotrafficking with NaV1.7. Rab proteins are small cytoplasmic guanosine triphosphatases (GTPases) that are markers for vesicular trafficking (28). On the basis of RNA sequencing data from mouse DRG neurons (29, 30), we chose eight of the abundantly expressed Rabs for an initial screen for cotrafficking with Halo-NaV1.7. We cotransfected neurons with Halo-NaV1.7 and one of the EGFP (enhanced green fluorescent protein)–Rab constructs (Rab1A, Rab2, Rab3A, Rab5A, Rab6A, Rab7, Rab8A, and Rab10). We used OPAL imaging to visualize anterograde Halo-NaV1.7 vesicles labeled with JF646-Halo; we used the far-red fluorophore to maximize the separation of the emission spectra from that of EGFP. Frames from a dual-color time-lapse movie demonstrate that Halo-NaV1.7 (magenta arrowheads) and EGFP-Rab6A (green arrowheads) cotraffic (white arrowheads) (Fig. 3A). The corresponding kymographs show that two vesicles, each containing Halo-NaV1.7 and EGFP-Rab6A, with the overlapping lines in the kymograph (white, merge) demonstrate comovement of the two proteins. This provides strong evidence that Halo-NaV1.7 and EGFP-Rab6A are packaged into the same vesicles. We sought to confirm that Rab6 is endogenously expressed in cultured DRG neurons using immunocytochemistry and observed strong labeling throughout the DRG soma and processes (fig. S6A), in contrast to the restricted perinuclear labeling seen in non-neuronal cells (fig. S6B).
Fig. 3. Anterograde NaV1.7 vesicles are Rab6A positive.
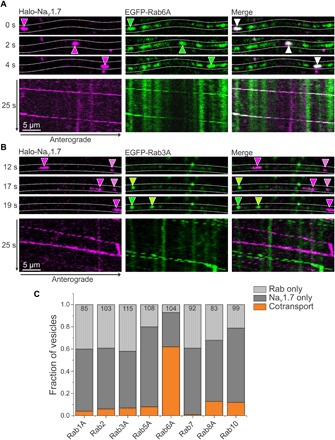
DRG neurons in MFCs were transfected with Halo-NaV1.7 and either EGFP-tagged Rab6A or Rab3A constructs. OPAL imaging was used to visualize anterograde Halo-NaV1.7 labeled with JF646-Halo, and two-color time-lapse imaging was performed. (A) Rab6A shows cotransport with Halo-NaV1.7. Selected frames from a time-lapse movie show the position of an anterograde vesicle containing Halo-NaV1.7 (magenta arrowheads) and EGFP-Rab6A (green arrowheads) over time. The overlay (white arrowheads) demonstrates comovement. The corresponding kymographs show a second vesicle containing both NaV1.7 and Rab6A as demonstrated by the overlapping lines on the kymograph. (B) Rab3A is an example of a Rab that does not cotransport with NaV1.7. The image sequence shows distinct NaV1.7 (magenta arrowheads) and Rab3A (green arrowheads), demonstrating independent movement. This can be visualized by distinct magenta and green lines on the kymograph. (C) DRG neurons in MFCs were transfected with NaV1.7 and either EGFP-tagged Rab1A, Rab2, Rab3A, Rab5A, Rab6A, Rab7, Rab8A, or Rab10. Anterograde NaV1.7 was labeled with JF646-Halo only in the somatic chamber, and two-color time-lapse imaging was performed. Kymographs were created for each Rab-NaV1.7 pair, and vesicle tracks were visually inspected and classified as Rab only, NaV1.7 only, or both. Rab6A shows a much higher association with NaV1.7 than any of the other Rabs investigated: Rab1A: 4%, 85 vesicles, 12 axons, 2 cultures; Rab2: 6%, 103 vesicles, 10 axons, 2 cultures; Rab3A: 7%, 115 vesicles, 8 axons, 2 cultures; Rab5A: 8%, 108 vesicles, 14 axons, 2 cultures; Rab6A: 62%, 104 vesicles, 10 axons, 4 cultures; Rab7A: 1%, 92 vesicles, 10 axons, 2 cultures; Rab8A: 13%, 83 vesicles, 10 axons, 2 cultures; Rab10: 12%, 99 vesicles, 12 axons, 2 cultures.
Rab3A is shown as an example of a Rab protein that does not cotraffic with NaV1.7. The time series of Halo-NaV1.7 (magenta arrowheads) and EGFP-Rab3A (green arrowheads) show distinct vesicles of each protein (Fig. 3B). This is emphasized in the kymographs where the separate magenta lines and green lines represent vesicles moving independently. We made similar kymographs for each tested Rab protein and Halo-NaV1.7 pairing and visually inspected kymographs for comovement. Since Rab1A, Rab2, Rab3A, Rab5, Rab7, Rab8, and Rab10 rarely colocalized with the channel (Fig. 3C), we focused on investigating cotrafficking of Halo-NaV1.7 and Rab6A. We observed a high likelihood that NaV1.7 and Rab6A traffic together, with 62% of vesicles carrying both NaV1.7 channels and Rab6A (64 of 104 vesicles from 10 axons, four independent cultures).
NaV1.7 current in DRG neurons is increased with IM
The studies described above demonstrate NaV1.7 surface distribution, mobility, and trafficking under normal conditions. It has been well established that up-regulation of NaV1.7 expression is involved in inflammatory pain (1, 19), so we next investigated how NaV1.7 trafficking and surface expression are altered after exposure to compounds known to be involved in the inflammatory pain response (IM). We first confirmed that DRG neurons treated with a cocktail of IM (31, 32) for 4 hours show the expected increase in current density. We recorded NaV1.7 current in the whole-cell voltage-clamp mode from NaV1.8-null DRG neurons expressing GFP-2A-NaV1.7R channels. Representative traces show that compared with control conditions (Fig. 4A), treatment with IM potentiates NaV1.7 currents (Fig. 4B). The average peak current amplitude was enhanced about twofold in IM-treated DRG neurons (Fig. 4C) (control: 23.1 ± 4.7 nA, n = 11; +IM: 45.3 ± 7.7 nA, n = 12; P < 0.05, two-sample t test). When currents were normalized to DRG neuron cell capacitance, the average peak current density of NaV1.7 in neurons treated with IM was more than twofold greater than control (Fig. 4D) (control: 1195 ± 246 pA/pF, n = 11; +IM: 2856 ± 526 pA/pF, n = 12; P < 0.05, two-sample t test).
Fig. 4. NaV1.7 current density increases following treatment with IM.
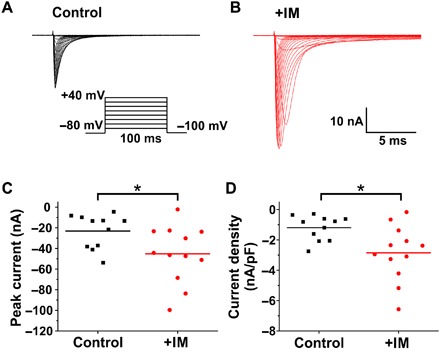
(A and B) Family of current traces evoked by 100-ms depolarizing voltage steps from −80 to +40 mV in 5-mV increments from a holding potential of −100 mV. Representative traces from NaV1.8-null DRG neurons expressing NaV1.7 channels in (A) control (black) and (B) IM-treated (IM; red) neurons. (C) Comparison of peak inward current between control (black) and IM-treated conditions (red). (D) Scatter plot showing enhanced current density in DRG neurons treated with IM. Current density was measured by normalizing maximal peak currents with cell capacitance. The bars indicate the means; *P < 0.05, two-sample t test.
Axonal trafficking of NaV1.7 is enhanced by IM
Reasoning that the increase in NaV1.7 current density in response to the 4-hour treatment of DRG neurons with IM most likely involves enhanced forward trafficking of NaV1.7, we used the Halo-NaV1.7 vesicular trafficking assay to investigate how IM affects anterograde axonal trafficking of NaV1.7. We cultured DRG neurons transfected with Halo-NaV1.7 in MFCs for 7 to 10 days and then incubated with IM in both the somatic and axonal chambers for 4 hours. We then imaged the neurons using OPAL imaging by labeling Halo-NaV1.7–expressing neurons in the somatic chamber with JF549-Halo and imaging in the axonal chamber. Incubation with IM markedly enhanced anterograde axonal trafficking of NaV1.7. Our observations indicate that exposure to IM triggers an increase in the number of NaV1.7-associated vesicles per axon, together with an increase in the number of channels per vesicle, and in the velocity of vesicle forward movement. Figure 5A shows representative images of Halo-NaV1.7 vesicles in axons under control conditions (left panels) and in axons after incubation with IM (right panels). The corresponding kymographs are shown below, representing vesicular movement over time (movie S2). After incubation with IM, there was a marked increase in the number of vesicles moving along each axon (Fig. 5B) (control: 12.7 ± 1.3 vesicles per axon per 60 s, n = 21 axons; +IM: 21.5 ± 1.5 vesicles per axon per 60 s, n = 20 axons; P ≤ 0.0001, two-sample t test). We observed that the fluorescence intensity of each vesicle appeared to be brighter after IM treatment. Thus, we estimated the number of labeled Halo-NaV1.7 channels per vesicle by measuring the fluorescence intensity of individual vesicles under both conditions. The median fluorescence intensity increased more than threefold after IM treatment (Fig. 5C) [control: 1840 arbitrary units (A.U.), n = 312 vesicles; +IM: 6220 A.U., n = 285 vesicles; P ≤ 0.0001, Mann-Whitney U test]. To determine approximately how many labeled channels were in each vesicle, we determined the fluorescence intensity of individual JF549-Halo molecules on a glass slide imaged under similar conditions (fig. S7). We used this to estimate that under control conditions, the majority of the vesicles contain only a single labeled Halo-NaV1.7 channel per vesicle, with the brightest vesicle containing an estimated nine channels. After treatment with IM, the median channel number per vesicle was 3, and the brightest vesicle contained an estimated 21 labeled channels.
Fig. 5. Anterograde NaV1.7 vesicular trafficking is enhanced by IM.
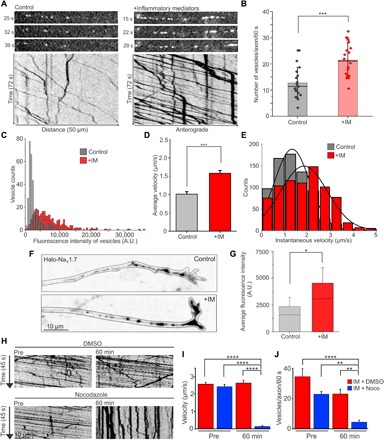
DRG neurons expressing Halo-NaV1.7 were cultured in MFCs for 7 to 10 days. Treatment dishes were exposed to IM in both the somatic and axonal chambers for 4 to 5 hours before imaging. Anterograde vesicles labeled with JF549-Halo were visualized via OPAL imaging. (A) Representative frames from time-lapse movies showing Halo-NaV1.7 vesicles traveling along axons from control dishes (left) or dishes incubated with IM (right). Below are the corresponding kymographs showing vesicle movement over time. The images were processed and contrasted identically. (B) The number of vesicles moving along each axon per minute was significantly enhanced after incubation with IM (control: 12.7 ± 1.3 vesicles per axon per 60 s, n = 21 axons; +IM: 21.5 ± 1.5 vesicles per axon per 60 s, n = 20 axons; ***P ≤ 0.0001, two-sample t test). (C) The fluorescence intensity was measured for individual vesicles under either control or +IM conditions. The median fluorescence intensity of vesicles was significantly enhanced after incubation with IM (control: 1840 A.U., n = 312 vesicles; +IM: 6220 A.U., n = 285 vesicles; P ≤ 0.0001, Mann-Whitney U test). (D) Vesicle tracks from kymographs were analyzed to measure the average velocity, including stops and pauses. The average vesicle velocity was significantly faster after incubation with IM (control: 1.01 ± 0.03 μm/s, n = 330 vesicles; +IM: 1.58 ± 0.04 μm/s, n = 532 vesicles; ***P ≤ 0.0001, Mann-Whitney U test). (E) Vesicle tracks were broken into segments that could be fit with a straight line, representing movement of a consistent velocity or instantaneous velocity. Stops or pauses were excluded. The histogram shows the counts for the number of line segments of each velocity under control (gray) and +IM (red) conditions. (F) Compressed z-stacks (inverted signal) showing accumulation of Halo-NaV1.7 vesicles at the distal axon under control (top) and +IM (bottom) conditions. (G) Quantitation of fluorescence intensity from the distal 60 μm of axons. The fluorescence intensity was significantly greater in axons from the +IM condition versus control (control: 1825 ± 223 A.U., n = 28 axons; +IM: 4535 ± 770 A.U., n = 27 axons; *P ≤ 0.0014, Mann-Whitney U test). (H) Kymographs generated from an axon from the +IM condition before and 60 min after either DMSO or Noco treatment. (I) Average vesicle velocities before and after DMSO or Noco treatment. ****P < 0.0001, two-way ANOVA with Tukey’s multiple comparisons test. (J) The number of vesicles with a velocity >0.1 μm/s within a 30-μm segment of axon per 60 s. ****P < 0.0001, **P < 0.003, two-way ANOVA with Tukey’s multiple comparisons test.
We used the entirety of the vesicle tracks from representative kymographs to calculate the average velocity of each vesicle, including pauses and stops. This analysis showed a considerable increase in the average vesicle velocities after treatment with IM (Fig. 5D) (control: 1.01 ± 0.03 μm/s, n = 330 vesicles; +IM: 1.58 ± 0.04 μm/s, n = 532 vesicles; P ≤ 0.0001, Mann-Whitney U test). Changes in overall vesicle velocities can reflect changes in vesicle speed or changes in the number or length of stops and pauses. To further investigate the vesicle behaviors that resulted in increased overall velocities, we measured instantaneous velocities and run lengths. We determined instantaneous velocities by breaking vesicle runs into sections with consistent slopes and by excluding stops or pauses. The histogram of instantaneous velocities for the control condition was fit with a peak around 1.3 ± 0.75 μm/s, while the velocities for the +IM condition was fit with a peak around 1.8 ± 1.01 μm/s (Fig. 5E). To measure run length, we used the method described by Lim et al. (33), whereby we summed the total distance traveled for all clearly visible vesicles in the kymographs and then divided by the number of stops or pauses observed. Control conditions showed an average run length of 13.0 μm (123 stops over 1598.3 μm; 126 vesicles from 21 axons), while the +IM condition showed an average run length of 25.6 μm (95 stops over 2433.8 μm; 207 vesicles from 20 axons). Thus, both vesicle velocities and run lengths are increased in the presence of IM.
Since vesicles accumulate at the distal axons under control conditions, we next investigated whether the channels also target to distal axons under IM conditions. We observed the accumulation of vesicles at the distal axonal ends and did not observe vesicle accumulation at other sites along the axon within the axonal chamber in treated cultures. We measured the fluorescence signal intensity of accumulated Halo-NaV1.7 at the distal axon from compressed confocal z-stacks of the distal 60 μm of the axon (Fig. 5F). We found that the fluorescence signal intensity under the IM conditions was more than double than that under control conditions (control: 1825 ± 223 A.U., n = 28 axons; +IM: 4535 ± 770 A.U., n = 27 axons; P ≤ 0.0014, Mann-Whitney U test) (Fig. 5G). This suggests that the majority of the additional anterograde NaV1.7 vesicles visualized using OPAL imaging under IM conditions are targeted to the distal axon. We also investigated whether NaV1.7 and Rab6A still associate after treatment with IM and found that, under these conditions, more than 70% of NaV1.7 vesicles associate with Rab6A in cotrafficking studies (87 of 111 vesicles from 12 axons, three independent cultures).
Next, we investigated whether the up-regulated trafficking after inflammatory treatment is microtubule dependent by imaging IM-treated axons before and after the addition of Noco (Fig. 5H). Similar to the results seen under control conditions (Fig. 2, D to F), the average velocity of vesicles within the Noco-treated axons was significantly reduced from a high baseline velocity (pre-Noco: 2.41 ± 0.12 μm/s, n = 109 vesicles, seven axons; post-Noco: 0.12 ± 0.04 μm/s, n = 68 vesicles, seven axons; P ≤ 0.0001, two-way ANOVA with Tukey’s multiple comparisons test), while DMSO-treated axons displayed stable velocity (pre-DMSO: 2.56 ± 0.10 μm/s, n = 146 vesicles, seven axons; post-DMSO: 2.6 ± 0.15 μm/s, n = 113 vesicles, seven axons) (Fig. 5I). The number of forward-moving vesicles (with an anterograde velocity >0.1 μm/s) was also significantly inhibited by Noco (pre-Noco: 23.14 ± 3.10 moving vesicles per axon per 60 s, n = 7 axons; post-Noco: 4.29 ± 1.33 moving vesicles per axon per 60 s, n = 7 axons; P = 0.0024, two-way ANOVA with Tukey’s multiple comparisons test), while DMSO-treated axons continued to have forward trafficking (pre-DMSO: 34.62 ± 5.32 moving vesicles per axon per 60 s, n = 7 axons; post-DMSO: 22.86 ± 1.87 moving vesicles per axon per 60 s, n = 7 axons; P ≤ 0.08, two-way ANOVA with Tukey’s multiple comparisons test) (Fig. 5J). Together, this suggests that in both control and up-regulated inflammatory state, trafficking of Halo-NaV1.7–containing vesicles is microtubule dependent.
IM enhance delivery of NaV1.7 channels to the plasma membrane at the distal axon
Incubation with IM causes increased current density and enhanced vesicular trafficking to the distal axon, implying that the enhanced NaV1.7 vesicular delivery results in additional channel delivery to the neuronal plasma membrane. To test this hypothesis, we compared the expression of Venus-NaV1.7-BAD at distal axons under control conditions or after treatment with IM. We obtained confocal z-stacks for the Venus signal (total protein) and SA-CF640 (surface) labeling from the distal 45 μm of axons. Figure 6A shows representative axons from each group. As seen from the images and the line profiles (Fig. 6B), NaV1.7 localizes into distinct nanoclusters along the axon under both conditions, suggesting that the overall distribution of channels on the plasma membrane is not rearranged in the presence of IM. However, when measuring total fluorescence intensity in the distal axon, the average fluorescence intensities of both the Venus signal (control: 317.4 ± 26.9 A.U., n = 28 axons; +IM: 463.7 ± 46.9 A.U., n = 26 axons; P = 0.01, two-sample t test) (Fig. 6C) and surface labeling (Fig. 6D) (control: 192.3 ± 17.4 A.U., n = 28 axons; +IM: 367.2 ± 44.5 A.U., n = 26 axons; P ≤ 0.001, Mann-Whitney U test) are significantly increased after exposure to IM. This is consistent with enhanced vesicular trafficking resulting in enhanced surface delivery of NaV1.7 to the distal axonal membrane. We next used single-particle tracking to determine whether the mobility of individual channels is altered after IM. We still observed two populations of NaV1.7 channels, those with low mobility and a second population with higher mobility. Again, single-particle tracks were grouped by low diffusivity (<0.004 μm2/s) or higher diffusivity (>0.004 μm2/s) using ad hoc thresholds. Low-diffusivity channels (black traces) showed diffusion coefficients of 0.0010 ± 0.001 μm2/s (mean ± SD; 353 trajectories), while channels with higher diffusivity (blue traces) showed diffusion coefficients of 0.0122 ± 0.0086 μm2/s (mean ± SD; 128 trajectories) (Fig. 6E). Together, these data suggest that IM enhance vesicular trafficking and membrane delivery of NaV1.7 channels to the neuronal plasma membrane but do not obviously alter the overall surface distribution or mobility of channels on the distal axon.
Fig. 6. Enhanced surface expression of NaV1.7 channels after incubation with IM.
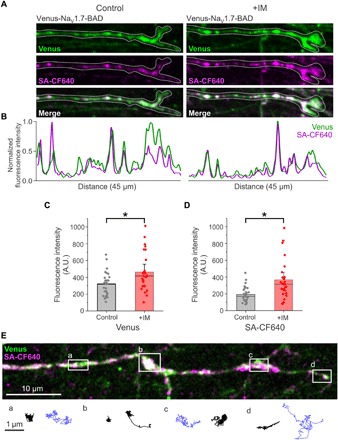
DRG neurons expressing Venus-NaV1.7-BAD were cultured in MFCs for 7 to 10 days. Treatment dishes were exposed to IM in both the somatic and axonal chambers for 4 to 5 hours before imaging. (A) Compressed z-stacks of distal axons under control conditions (left) and after exposure to IM (right). Under both conditions, nanoclusters can be seen along the distal axon with both the Venus signal (green) and CF640 surface labeling (magenta). (B) Line profile of the axons in (A) showing fluorescence intensity normalized to the maximum fluorescence intensity, with peaks representing nanoclusters of NaV1.7. (C and D) Quantification of the fluorescence intensity of the Venus signal (C) and CF640 surface labeling (D) shows significantly more surface labeling at distal axons after incubation with IM versus control conditions. *P< 0.01, two-sample t test (C), Mann-Whitney U test (D). (E) Single-molecule tracks of individual surface labeled Venus-NaV1.7-BAD channels on a distal axon. A compressed z-stack is used to demonstrate the overall morphology of a distal axon expressing Venus-NaV1.7-BAD where the overlay between the Venus signal (green) and surface labeling (magenta) appears as white. The white dots along the axon represent the location of axonal NaV1.7 nanoclusters. The lines below the picture represent the trajectories of single molecules over time, tracked from time-lapse movies. Black lines represent molecule tracks with relatively low mobility, while blue lines represent molecule tracks with higher mobility. Tracks are representative of single-molecule behaviors from the regions indicated by the white boxes (boxes a to d).
DISCUSSION
We demonstrate here real-time dynamic regulation of trafficking and surface delivery of the NaV1.7 sodium channel in the distal axonal membrane of DRG neurons. We show that NaV1.7 channels are transported to distal axons via microtubule-dependent trafficking in vesicles that appear to be enriched for Rab6A and localize to nanoclusters at the membrane surface in the distal axonal terminus. Single-particle tracking shows low mobility of channels within these nanoclusters and greater mobility outside them. We also demonstrated substantial enhancement of channel vesicular delivery to distal axons and increased number of channels within the plasma membrane upon treatment of DRG neurons with a cocktail of IM.
A major challenge for neurons, especially those with very long axons such as primary afferents, is the need to deliver membrane proteins to distant compartments in a precise and timely manner. Local protein synthesis of axonal mRNA has been suggested as a strategy for NaV delivery to distal axons. Although limited evidence supports local translation of ion channels in DRG axons, including NaV1.8, but not NaV1.7 (34, 35) or NaV1.6 channels (36), these studies do not preclude channel biogenesis in the soma and transport to axons. Three pathways have been suggested to deliver membrane proteins from the soma to the distal axons: (i) polarized direct delivery from the trans-Golgi network, (ii) nonpolarized delivery and subsequent selective endocytosis or retention, and (iii) transcytosis via delivery to the soma, retrieval in endosomes, and then polarized delivery (37). Previous strategies investigated these using GFP-tagged full-length NaV channels or reporter proteins with partial channel fragments, which carry potential targeting motifs (18, 25). However, only a few moving vesicles have been visualized using this strategy, which limits the conclusions that can be made based on these data. Using the Halo-NaV1.7 construct and selective labeling in the MFC somatic compartment, we developed the OPAL imaging technique, whereby we were able to visualize extensive anterograde vesicular trafficking of NaV1.7 for a long distance in sensory neurons to distal axonal ends with remarkable resolution and show that vesicles newly traveling from the somatic chamber preferentially accumulate near the end of the distal axon. While we do not directly demonstrate that these vesicles deliver their cargo to the neuronal surface, we have observed increased surface channels in the same region of the axon, which suggests that this vesicular trafficking does translate into channel insertion in the plasma membrane. Thus, our data provide evidence supporting polarized delivery of NaV1.7 channels to distal sensory axons. These studies used the OPAL imaging technique to selectively label and investigate trafficking behavior of anterograde vesicles.
Because of the background fluorescence of most labeling techniques, it is difficult to investigate the behavior of low-expressing molecules in live neurons. However, by selecting for a subpopulation of proteins, namely, those actively transported from one chamber to the other, proteins with low numbers can be visualized in an environment that is virtually free of background noise. The development of the highly sensitive OPAL imaging assay provided the signal-to-noise ratio necessary for tracking individual Halo-NaV1.7 vesicles in real time, which is not possible through previous imaging methods. Furthermore, we have achieved single-molecule resolution, enabling the use of this technique to observe proteins with low expression levels; we could observe vesicles containing even a single tagged channel (Fig. 5C). The high resolution of this technique can be used to investigate long-distance axonal transport of many proteins including other NaV channels as well as other ion channels, vesicular proteins, and motor proteins involved in vesicular trafficking in either the anterograde or the retrograde direction. We have used the HaloTag technology, but by combining this with SNAP-tag, FLAG-tag, or other labeling techniques, it will be possible to investigate cotrafficking of multiple proteins simultaneously with single-molecule resolution. We observed that surface NaV1.7, a threshold channel, accumulates within nanoclusters along the axonal membrane and estimated that the majority of these nanoclusters contain 10 to 15 channels; however, some contained as few as 3 or as many as 108 channels per nanocluster. These numbers are intended as approximations of the number of channels per nanocluster, as there are several factors that contribute to uncertainties about the absolute number of channels, including the presence of endogenous channels and ambiguities about labeling efficiency.
The formation of nanoclusters of NaV1.7 channels in the axonal compartment devoid of Schwann cells suggests that this organization is axon intrinsic, which might be analogous to the axon-intrinsic property of AIS formation (38). We show that NaV1.7 channels within the nanoclusters display low mobility, suggesting that they might be tethered to the cytoskeleton. In contrast, channels outside of the nanoclusters are highly mobile, which could be important to cover a larger membrane area with fewer channels. A similar single-particle behavior was observed for NaV1.6 channels on the soma of hippocampal neurons (23), where channels within nanoclusters demonstrate low mobility, while channels outside the nanoclusters show higher mobility. We speculate that the accumulation of NaV1.7 channels in nanoclusters at the surface of distal axonal ends provides sites of action potential generation, and the mobile channels outside the nanoclusters boost response to local depolarization in a larger membrane area. In this study, we used a spinning disk confocal microscope to perform the single-particle tracking, requiring slow imaging rates. More detailed studies of channel diffusion will require the development of novel imaging or labeling strategies.
Since most nociceptors in DRG neurons give rise to unmyelinated axons with high input resistance, even a small concentration of channels can generate membrane depolarization and are, thus, functionally important. Freeze-fracture electron microscopy has previously shown particles, presumed to be NaV channels, accumulated in small clusters in unmyelinated segments of the axons of retinal ganglion neurons (39), suggesting a possible role in facilitating impulse conduction in these unmyelinated segments. Computer modeling studies have also suggested that clustering of NaV channels along C fibers enables microsaltatory conduction (40). We have previously shown in direct patch-clamp recordings of distal axon ends of cultured small DRG neurons, 650 to 720 μm from the soma, a TTX-S conductance that boosts subthreshold depolarization and a combination of TTX-S and tetrodotoxin-resistant (TTX-R) currents that activate sequentially, as predicted, supporting action potentials (41). The specific isoform(s) that contribute to the TTX-S conductance at the distal axon ends is not known. Our data indicate that NaV1.7 channels could contribute to the initiation and propagation of action potentials in these unmyelinated fibers and could be the NaV isoform that boosts subthreshold receptor potentials because of their demonstrable ability to produce a large ramp current (42).
Our data show the association of a NaV isoform and a Rab protein in moving vesicles. Rab proteins are small cytoplasmic GTPases that regulate multiple aspects of vesicular trafficking, with mutations or dysfunction in Rabs causing many human disorders (28). We observed that >70% of NaV1.7-carrying vesicles contain the Rab6A, which has been shown to regulate anterograde transport of dense core vesicles (DCV) that carry neuropeptides (43). Whether Rab6A-DCV codeliver neuropeptides and NaV1.7 channels together is not known. By contrast, we observed little or no cotrafficking of NaV1.7 with Rab1A, Rab2, Rab3A, Rab5A, Rab7, Rab8A, and Rab10. Thus, NaV1.7 sorting into Rab6A vesicles appears to be selective. The molecular determinants in NaV1.7 that underlie the selective sorting that we observed in this study are not known, but the assay that we developed here will allow us to identify them in future studies.
Vesicular transport of neuronal membrane proteins in axons involves kinesin motor proteins (KIF) along microtubules and nonmuscle myosin motor proteins (myh) along actin filaments. Using Noco to destabilize microtubules, we confirm that the trafficking of NaV1.7 is microtubule dependent both in control conditions and in the inflammatory state. The speed of anterograde long-distance vesicular transport under control conditions in our system fits a single-mode broad distribution with an average rate of 1 μm/s. This speed is consistent with the involvement of kinesin motors that are generally thought to move at speeds of 1 to 2 μm/s (44), although studies have also reported kinesin speeds of 0.5 to 0.8 μm/s for kinesin-1 and 2 to 3 μm/s for kinesin-3 (33, 43). Kinesin-1 has been shown to play an important role in the dynamic regulation of a-amino-3-hydoxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors in Caenorhabditis elegans axons (45). A member of the kinesin-1 family (KIF5B) interacts with NaV1.8 in DRG neurons, but it does not interact with NaV1.7 in biochemical assays (17, 46). While there is no direct evidence for the involvement of KIF5B in NaV1.7 axonal vesicular trafficking, other kinesin motors could be involved. It is interesting that Rab6A (47, 48) and NaV1.7 (46) interact with nonmuscle myosins MYH9 and MYH10, which suggests that myosins might be involved in the exit of these vesicles from the trans-Golgi system, or for local trafficking at the growth cone where there is a high density of actin filaments.
Taking full advantage of the power of live-cell imaging and the single-molecule resolution achieved using OPAL imaging, we report dynamic changes to channel trafficking and surface delivery in response to IM in real time. Treatment with IM for 4 hours causes a twofold increase in NaV1.7 current density in DRG neurons measured by whole-cell voltage-clamp recording and nearly a twofold increase in the number of surface channels in distal axons, albeit in a different culture system. This IM-induced increase in surface channels is paralleled by increased vesicular trafficking to the distal axon where we measured a more than threefold increase in the average fluorescence intensity of NaV1.7-carrying vesicles. We estimate that, under control conditions, most of the vesicles contained only one labeled channel. While we cannot be certain about the absolute number of NaV1.7 channels per vesicle due to the presence of endogenous channels and uncertainty about labeling efficiency, these data suggest presence of only small numbers of channels per vesicle. The enhanced trafficking in response to IM occurred within 4 hours, a time frame that could include de novo synthesis of sodium channels and mobilization of a metabolically stable channel cytoplasmic pool (49). A shorter time course series is needed to distinguish between the contributions of these two mechanisms.
Perhaps of special interest was the increase in the velocity of the vesicles in IM-treated cultures. While neuronal activity increased the delivery of AMPA receptors to synapses in C. elegans in a kinesin-1–dependent manner, the velocity of the transport was not altered (50). The average transport rate of NaV1.7 under control conditions (1.01 μm/s) is consistent with other reports of transport rates for long-range axonal transport of DCVs; however, we observed a marked increase in vesicular velocities (1.6 μm/s) upon treatment with IM. Studies in both mouse (43) and Drosophila (33) suggest that both the slower kinesin-1 (0.5 to 0.8 μm/s) and faster kinesin-3 (2 to 3 μm/s) motor proteins can be attached to DCVs. Thus, one explanation for our data could be a shift in the contributions between two kinesin motors that might be present in NaV1.7-carrying vesicles. This idea is also supported by our observation of increased run lengths after incubation with IM since different kinesins have different processivity, for example, kinesin-3 is 10-fold more processive than kinesin-1 (51). However, we cannot rule out a posttranslational modulation of one kinesin isoform after treatment with IM, which might account for the increase in speed and run length that we observed. Another recent study showed that AMPA receptor trafficking is up-regulated, both in terms of vesicle number and speed, in response to a chemical long-term potentiation protocol (52). Together with the present study, these findings support the conclusion that neurons can modulate the trafficking of key proteins such as voltage- and ligand-gated channels in response to relevant stimuli.
In conclusion, this study provides the first real-time characterization of the dynamics of NaV channel trafficking in sensory neurons, mostly unmyelinated C fibers. We provide evidence for vesicular trafficking of NaV1.7 channels that shows selective sorting into Rab6A vesicles, and the accumulation of channels within nanoclusters at the distal end of axons. We also provide evidence for enhanced channel trafficking and insertion in distal membranes upon treatment of cultures with IM, thus mimicking an inflammatory response. We are cognizant of limitations of our studies, foremost among them being the use of an in vitro system combined with overexpression of tagged proteins, time in culture of 7 to 10 days, and the short length of axons in MFCs, which may not recapitulate the in vivo situation. However, to date, this is the only viable approach to investigating real-time dynamics of NaV channels in live neurons. By design, our studies have focused on anterograde trafficking of vesicles carrying NaV1.7 channels. Future investigations will explore retrograde trafficking and protein recycling using the OPAL method with labeling occurring in the distal axonal compartment, and may reveal the involvement of different motor and Rab proteins.
In conclusion, our results demonstrate with single-molecule resolution the live visualization of NaV1.7 channels in DRG neurons including long-distance microtubule-dependent vesicular transport in Rab6A-containing vesicles, and reveal the presence of nanoclusters that contain a median of 12.5 channels at the plasma membrane on axon termini. We also demonstrate that IM trigger an increase in the number of NaV1.7-carrying vesicles per axon together with a threefold increase in the median number of NaV1.7 channels per vesicle and a ~50% increase in forward velocity. This remarkable enhancement of NaV1.7 vesicular trafficking and surface delivery under conditions that mimic a disease state provides new insights into the contribution of NaV1.7 to inflammatory pain. Further development of this system will permit future studies that can unravel molecular pathways that regulate channel sorting, trafficking, and polarized distribution in multiple types of neurons under a variety of conditions.
MATERIALS AND METHODS
DNA constructs
The parent plasmid was previously described (53). Briefly, human NaV1.7 plasmid was rendered TTX-R by substituting amino acid Tyr362 with serine (Y362S) using QuikChange Lightning site-directed mutagenesis (Agilent Technologies). Subsequently, a modified plasmid (GFP-2A-hNaV1.7R) was made by fusing EGFP and a “StopGo” 2A linker in-frame with the N-terminus of the channel as described previously (54), which was used for patch-clamp recording.
hNaV1.7R was modified for cell surface tracking and cytoplasmic trafficking. Briefly, hNaV1.7R was tagged with an in-frame fusion of the fluorescent protein Venus (239 amino acids) via a 7–amino acid linker (SGLRSAT) on the N-terminus and a 50–amino acid insertion that includes the 17–amino acid BAD (biotin-accepting lysine: GLNDIFEAQKIEWH) after position 1529 of the channel in the extracellular linker of domain IV between S1 and S2 using Mega mutagenesis. This insertion was previously used to tag NaV1.6 channels (25). The BAD is recognized by a bacterial biotin ligase (BirA) that is cotransfected with the channel, which biotinylates the lysine residue in the BAD sequence (underlined), allowing for specific labeling of surface channels via streptavidin-conjugated fluorophores (20).
Halo-NaV1.7 was created in two steps. First, the transmembrane segment of the sodium channel β4 subunit was fused to the N-terminus of hNaV1.7R to produce a channel with 25 transmembrane segments. Second, we replaced the immunoglobulin G domain of the β4 subunit with the HaloTag enzyme (gift from D. Toomre, Yale University). The final construct topology was in order from the N-terminus: 1– to 30–amino acid β4 signal peptide, 3× myc tag (EQKLISEEDL), HaloTag enzyme (297 amino acids), 3× hemagglutinin tag (YPYDVPDYA), 21–amino acid transmembrane segment (β4 163 to 183), 7–amino acid linker (SGLRSAT), hNaV1.7.
The EGFP-Rab1A (plasmid #49467, Addgene), EGFP-Rab2 (#49541), EGFP-Rab3A (#49542), EGFP-Rab5 (#49888), EGFP-Rab6A (#49469), EGFP-Rab8 (#49543), and EGFP-Rab10 (#49472) were obtained from Addgene as a gift from M. Scidmore (55); GFP-Rab7 (plasmid #12605, Addgene) was a gift from R. Pagano (56).
Primary rat and mouse DRG neuron culture and transfection
Animal studies followed a protocol approved by the Veterans Administration Connecticut Healthcare System Institutional Animal Care and Use Committee. DRG neurons were isolated from 2- to 4-day-old Sprague-Dawley rat pups for imaging studies and from homozygous NaV1.8-cre mice (4 to 8 weeks of age) that lack functional endogenous NaV1.8 channels for voltage-clamp recordings using previously described protocols that permit NaV1.7 currents to be recorded in isolation within a DRG neuron background (57). Briefly, dissected DRGs were first incubated at 37°C for 20 min in complete saline solution (CSS) [137 mM NaCl, 5.3 mM KCl, 1 mM MgCl2, 25 mM sorbitol, 3 mM CaCl2, and 10 mM Hepes (pH 7.2), adjusted with NaOH], supplemented with 0.6 mM EDTA and collagenase A (1.5 mg/ml; Roche) for rat DRG, or Liberase TM (0.5 U/ml; Sigma-Aldrich, St. Louis, MO) for mouse DRG. Rat DRGs were then incubated for 20 min at 37°C in CSS containing collagenase D (1.5 mg/ml; Roche), 0.6 mM EDTA, and papain (30 U/ml; Worthington Biochemical), while mouse DRGs were incubated for 15 min at 37°C in CSS containing Liberase TL (0.5 U/ml; Sigma-Aldrich, St. Louis, MO), 0.6 mM EDTA, and papain (30 U/ml). DRGs were centrifuged and triturated in 1 ml of DRG culture medium [Dulbecco’s modified Eagle’s medium (DMEM)/F12 (1:1) with penicillin (100 U/ml), streptomycin (0.1 mg/ml; Life Technologies, Grand Island, NY), 2 mM l-glutamine, and 10% fetal bovine serum (Hyclone, Logan, UT) containing bovine serum albumin (1.5 mg/ml; low endotoxin, Sigma-Aldrich) and trypsin inhibitor (1.5 mg/ml; Sigma-Aldrich)]. For rat DRG neurons, the cell suspension was filtered through a 70-μm nylon mesh cell strainer (Becton Dickinson) to remove debris, and the mesh was then washed once with 1 ml of DRG culture medium.
Transfection of DRG neurons was performed as previously described (57). Briefly, DRG neurons were pelleted (200 g, 3 min) and gently resuspended with 20 μl of Nucleofector solution, then the cell suspension was mixed with DNA (either 2 μg of Venus-NaV1.7-BAD + 0.5 μg of BirA, 2 μg of Halo-NaV1.7 channel DNA, or 0.1 μg of EGFP-Rab + 2 μg of Halo-NaV1.7 channel DNA for imaging studies, or 2.5 μg of GFP-2A-hNaV1.7R for voltage-clamp recordings), and were transfected using Nucleofector IIS Electroporator (Lonza, Walkersville, MD) using protocol SCN-BNP 6 and Amaxa SCN Nucleofector reagents (VSPI-1003, Lonza). After electroporation, 100 μl of calcium-free DMEM (37°C) was added, and cells were incubated at 37°C for 5 min in a 95% air/5% CO2 (v/v) incubator to allow neurons to recover. The cell mixture was then diluted with DRG medium containing bovine serum albumin (1.5 mg/ml) and trypsin inhibitor (1.5 mg/ml). Rat DRGs were carefully seeded onto 35-mm glass bottom dishes coated with poly-d-lysine (MatTek) or onto the somatic chamber of MFCs, and mouse DRGs were seeded onto poly-d-lysine/laminin–coated coverslips (Corning, Discovery Labware, Bedford, MA). DRGs cultured on coverslips were incubated at 37°C for 45 min to allow DRG neurons to attach. Then, DRG medium was added to each well to a final volume of 1.5 ml (rat) or 1.0 ml (mouse). DRG neurons were maintained at 37°C in a 95% air/5% CO2 (v/v) incubator before use.
Microfluidic chambers
MFCs (RD450, two-chamber 450-μm groove, Xona Microfluidics, Temecula, CA) were modified using a razor blade to expose the region between the reservoirs on either side (fig. S8). This was performed to (i) ensure that cells were plated immediately adjacent to the microgroove barriers and (ii) allow access to the cells for the labeling and washing steps in the imaging experiments. MFCs were placed on 50-mm glass bottom dishes (P50G-1.55-30-F, MatTek) that were coated with poly-l-lysine (0.5 mg/ml) overnight at 37°C. The glass surface was washed twice with sterile double-distilled water and then air dried under the hood. The dishes were then coated with laminin (10 mg/ml) for at least 2 hours at 37°C, excess laminin was aspirated, and the dishes were dried under the hood before MFCs were adhered. Transfected DRG neuron suspension was applied in the soma chamber containing DRG medium with growth factors (50 ng/mg) [nerve growth factor (NGF) and glial cell line–derived neurotrophic factor (GDNF) from PeproTech], and 4× growth factors (200 ng/mg) were added to the axonal chamber. Medium was changed to serum-free medium in both chambers after 24 hours (Neurobasal medium supplemented with 2% B27, 1% penicillin/streptomycin, same 1:4 ratio of NGF, GDNF in soma and axonal chambers), and 1 μM uridine/5-fluoro-2-deoxyuridine was added to inhibit the growth of fibroblasts and glia. During the 7 to 10 days before imaging, volumes were kept slightly higher (~50 μl) in the somatic chamber as recommended by the manufacturer to fluidically isolate the chambers and maintain the high concentration of growth factors in the axonal chamber.
Patch-clamp recordings
Validation of biophysical properties of the tagged channel was performed in HEK293 cells. HEK293 cells were transiently transfected with Venus-BAD-hNaV1.7R, Halo-hNaV1.7R, or GFP-2A-hNaV1.7R plasmids (0.5 μg per well) using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) and were maintained under standard culture conditions (37°C with 5% CO2) in DMEM/F12 medium supplemented with 10% fetal bovine serum and 1% penicillin. Voltage-clamp recordings were obtained at room temperature (RT; 20° ± 1°C) using an Axon MultiClamp 700B amplifier (Molecular Devices, USA). Data were digitized via an analog-to-digital converter Digidata 1440A (Molecular Devices, USA). The data were acquired at 100 kHz and filtered at 10 kHz. Patch pipettes were fabricated from borosilicate glass (World Precision Instruments) using a P-97 puller (Sutter Instruments) and fire polished for a resistance of <1.5 megohms when filled with internal solutions. The pipette internal solution contained 140 mM CsF, 10 mM NaCl, 1 mM EGTA, 10 mM Hepes, and 10 mM dextrose (pH 7.30) with CsOH (adjusted to 310 mosmol/liter with mannitol). The extracellular bath solution contained 140 mM NaCl, 3 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM Hepes, 10 mM dextrose (pH 7.30) with NaOH (adjusted to 320 mosmol/liter with mannitol). Cells were held at −120 mV, except for recovery from inactivation protocols where they were held at −90 mV. Conductance was calculated as G = I/(Vm − ENa), normalized to the maximum conductance and fit with the Boltzmann equation y = A2 + (A1 − A2)/(1 + exp.((x − x0)/dx)), where A1 and A2 represent 0 and 100% current availability, V is membrane voltage, V1/2 is voltage when reaction rate is half maximal, and k is slope coefficient. Data were analyzed using pClamp 10.6 (Molecular Devices), Origin 2018 (OriginLab Co.), and SPSS (IBM SPSS Statistics) and presented as means ± SEM. Statistical significance for multigroup comparisons was determined by one-way ANOVA and one-way ANOVA with repeated measures, as specifically indicated.
Whole-cell patch-clamp recordings were also obtained from DRG neurons from NaV1.8-null mice that were transfected with GFP-2A-hNaV1.7R under control conditions and after treatment with an IM cocktail (described in detail below). Voltage-clamp recording data from small (<25 μm) DRG neurons with robust green fluorescence were acquired with an EPC10-double amplifier (HEKA Instruments) at RT (20° ± 1°C). The extracellular solution contained 70 mM NaCl, 70 mM choline chloride, 3 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM Hepes, 5 mM CsCl, 20 mM TEA-Cl, 1 mM 4-AP, and 0.1 mM CdCl2 (pH 7.3), adjusted with NaOH (320 mOsm adjusted with dextrose). TTX (500 nM) was included in the bath to block endogenous TTX-S sodium currents. Patch pipettes were fabricated from borosilicate glass using a P-97 puller and fire polished to have a resistance between 0.7 and 1.2 megohms when filled with internal pipette solution. Pipette solution contained 140 mM CsF, 10 mM NaCl, 1 mM EGTA, 10 mM Hepes, and 10 mM dextrose (pH 7.3), adjusted with CsOH (310 mOsm adjusted with dextrose). Series resistance compensation (85 to 90%) was applied to minimize voltage errors. Recordings were sampled at 50 kHz through a low-pass Bessel filter of 2.9 kHz. After achieving whole-cell configuration, a 5-min delay was applied to allow adequate time for the pipette solution and cytoplasmic milieu to equilibrate. The electrophysiological data were acquired using Patchmaster (HEKA Instruments), analyzed using Fitmaster (HEKA Instruments), Excel, and Origin 8.5 (Northampton, MA) software, and presented as means ± SEM. Statistical significance was determined by unpaired Student’s t test.
Immunocytochemistry
DIV7 DRG neurons grown in an MFC were fixed for 15 min with 4% paraformaldehyde (PFA) [20% PFA aqueous solution (Electron Microscopy Sciences, Hatfield, PA) diluted in neuronal imaging saline (NIS)]. Cells were incubated with blocking solution (2% bovine serum albumin, 4% donkey serum, and 0.1% Triton X-100) for 10 min. Primary antibodies were chicken anti-peripherin (catalog: PER, Aves Labs) and rabbit anti-PGP9.5 (catalog: ab15503, Abcam), diluted 1:1000 in blocking solution, and incubated overnight at 4°C. Secondary antibodies were donkey anti-chicken Alexa Fluor 549 and donkey anti-rabbit Alexa Fluor 488, diluted 1:1000, and incubated at RT for 3 hours.
DRG neurons grown in 35-mm glass bottom dishes (MatTek) were fixed, permeabilized, and blocked as above. Rabbit anti-Rab6 primary antibody (catalog: ab95954, Abcam), diluted 1:200 in blocking solution, was incubated for 2 hours at RT. Anti-rabbit Alexa Fluor 549 was diluted 1:500 and incubated at RT for 1 hour.
Inflammatory mediators
IM cocktail (31, 32) was prepared in advance as a stock solution and diluted to a final concentration containing (all from Sigma-Aldrich) bradykinin (1 μM), PGE-2 (10 μM), histamine (10 μM), 5-HT (10 μM), and adenosine 5′-triphosphate (15 μM). The IM cocktail was stored in aliquots and frozen until use. Stock solution was first mixed with a portion of the medium removed from the culture dish or slide before mixing the diluted IM back into the original well, thus preventing exposure of the cells to high concentrations of the compounds. The neurons were then incubated for 4 hours before imaging or recording.
Imaging system
Images were acquired using an Andor Dragonfly spinning disk confocal microscope built on a Nikon Eclipse Ti fluorescence microscope. Images were collected using an Andor iXon Ultra 888 electron multiplying charge-coupled device camera through either an SF Apo TIRF 100× [numerical aperture (NA), 1.49] or Plan Apo Lambda 60× (NA, 1.4) oil objective. The system includes an Andor Integrated Laser Engine containing 100-mW 405-nm, 150-mW 488-nm, 150-mW 561-nm, and 140-mW 637-nm solid-state lasers. The Nikon Perfect-Focus system was used to maintain focus in the z plane during time-lapse imaging.
Live-cell imaging and surface labeling
All experiments were performed at 37°C using a stage incubator (Tokai Hit, Shizuoka, Japan). Neurons were kept in DRG-NIS containing 136 mM NaCl, 3 mM KCl, 1 mM MgSO4, 2.5 mM CaCl2, 0.15 mM NaH2PO4, 0.1 mM ascorbic acid, 20 mM Hepes, and 8 mM dextrose (pH 7.35) with NaOH (adjusted to 320 mosmol/liter) during incubation and imaging. Surface labeling of biotinylated channels was performed as previously described (25, 58). Briefly, neurons cotransfected with Venus-NaV1.7-BAD and BirA (to biotinylate the lysine residue in the BAD) were plated on 35-mm glass bottom dishes or MFCs. Neurons were washed with DRG-NIS to remove medium that may contain biotin and decrease efficacy of labeling and then incubated with 1.2 nM streptavidin-conjugated fluorophore Streptavidin-CF640 (SA-CF640) (Biotium) or streptavidin-conjugated Alexa Fluor 568 (SA-568) (Thermo Fisher Scientific) for 20 min to label the biotinylated surface channels. Cells were washed three times with DRG-NIS and then imaged for up to 1 hour after labeling.
OPAL imaging
An optical pulse-chase technique was used to selectively label anterograde vesicles containing Halo-NaV1.7 during long-distance axonal transport. DRG neurons transfected with Halo-NaV1.7 were cultured in two-chamber MFCs for 7 to 10 days to allow the axons to extend through into the axonal chamber. Neurons were rinsed with DRG-NIS, and then JF549-Halo (100 nm) [gift from L. D. Lavis and J. B. Grimm (Janelia)] (27) was added to the somatic chamber only. To fluidically isolate the somatic chamber, the volume of DRG-NIS added to the axonal chamber was slightly greater (~50 μl) than the total volume in the somatic chamber to fluidically isolate the chambers and prevent any passive flow of dye through the microfluidic channels. Neurons were incubated for 20 min at 37°C, and then the somatic chamber was washed three times with DRG-NIS to remove excess HaloTag ligand. Dishes were imaged on the Dragonfly spinning disk confocal microscope for up to 1 hour after labeling. Time-lapse images of labeled vesicles were acquired (250 frames at 3 Hz). Acquisition conditions were 50% 561-nm laser power, 100-ms exposures, and 2× frame averaging.
Nocodazole
Baseline time-lapse images of labeled vesicles in distal axons were acquired, with or without IM treatment. Subsequently, Noco (20 μM; Millipore-Sigma) was added to the distal chamber. The same axons were then imaged again 1 hour after the addition of Noco. The IM-treated cultures were maintained in IM containing DRG-NIS during the hour of imaging.
Kymograph analysis of vesicles
Vesicle movement was analyzed using KymographClear and KymographDirect (59). Time-lapse movies were loaded into ImageJ, and the KymographClear toolset was used to create kymographs from sections of axons. Axons chosen for analysis were isolated from other axons and had multiple clearly visible vesicles that move through the region during the image sequence. Representative axons were chosen from each field of view, with six to eight axons analyzed from each culture. At least three independent cultures were analyzed for each condition. Vesicle tracks were manually traced in ImageJ, and the tracks were recorded using KymographClear. The kymograph and manual traces were loaded into KymographDirect, where information on average fluorescence intensities and velocities was obtained for each vesicle track. Data were imported into Origin for statistical analysis.
Cotrafficking with Rab GTPases
Neurons were transfected with one of the EGFP-Rab constructs (100 ng) and Halo-NaV1.7 (2 μg) and plated in the MFCs as described above. Neurons were cultured for 7 to 10 days to allow the axons to grow through the microgroove barriers. Neurons were then labeled with 100 nM HaloTag ligand on the somatic side only to allow visualization of Halo-NaV1.7–containing vesicles as described above. JF646-Halo was used for labeling Halo-NaV1.7 in these experiments to eliminate any chance of spectral overlap between the EGFP signal of the Rabs and the HaloTag ligand signal. Image sequences were obtained on the Dragonfly spinning disk by sequentially acquiring images alternating between the green (488 nm) and far red (637 nm) channels, 100-ms exposures, and 2× frame averaging for 100 frames. Kymographs were created as described above. Vesicle tracks were visually inspected and classified as Rab only, NaV1.7 only, or both. Axons were used for analysis only if the Rab signal showed distinct vesicular movement. It was then confirmed that the axon contained at least one clearly visible moving Halo-NaV1.7 vesicle.
Single-particle tracking of NaV1.7 in the plasma membrane
DIV7 to DIV10 DRG neurons expressing Venus-NaV1.7-BAD or Halo-NaV1.7 in MFCs were surface labeled with SA-CF640 or Halo ligand JF549i (100 nM for 15 min) [gift from L. D. Lavis and J. B. Grimm (Janelia)]. Time-lapse images were taken of axons in a single z plane. Image sequences were acquired on the spinning disk using the 100× objective, 2 pixel by 2 pixel binning, and 4× frame averaging to obtain an imaging rate of 2.5 Hz. These imaging conditions were used instead of TIRF microscopy as typically used for single-particle tracking because the majority of DRG neurons and axons do not adhere tightly enough to the glass to fall within the TIRF field, even when using substrates such as laminin, especially in older cultures.
A confocal z-stack was first acquired, and then an image sequence of surface-labeled proteins was taken for single-particle tracking. Segments of image sequences with low particle density after some photobleaching had occurred were used for tracking to ensure that individual particles could be visualized. Image sequences were processed in ImageJ. Average background values were subtracted, and a 0.6 Gaussian blur filter was applied to each sequence to enhance the automatic tracking. The MATLAB code uTrack (24) was used to automatically track particle movement over time. Only particles tracked for 50 frames or more were used for analysis. Mean squared displacements and diffusion coefficients were calculated using custom MATLAB codes as previously described (23).
Counting number of channels in nanoclusters
DIV7 to DIV10 DRG neurons expressing Venus-NaV1.7-BAD in MFCs were surface labeled with SA-CF640. Time-lapse images were taken of axons in a single z plane. Single molecules were identified as particles moving with Brownian-like diffusion. The summed fluorescence intensity was measured for 20 single molecules from each image series to ensure that imaging conditions were identical to the nanoclusters analyzed. The summed fluorescence intensity of nanoclusters within the field of view was divided by the average intensity for the single molecules to approximate the number of channels per nanocluster (fig. S4). Both the single molecule and nanocluster measurements were background subtracted.
Image analysis and statistical analysis
Images were processed with either ImageJ or Volocity. For figures with multiple panels, matched panels were processed and contrasted identically. Statistical analysis was performed using Origin or Prism, and specific tests are identified in the results. Data are presented as means ± SE unless otherwise stated, and statistical significance was considered at P < 0.05.
Supplementary Material
Acknowledgments
We thank M. Tamkun, C. Benson, and M. Estacion for the helpful discussions. We thank D. Krapf and D. Akin for writing codes for the data analysis. Funding: This work was supported by Merit Review Award B9253-C from the U.S. Department of Veterans Affairs Rehabilitation Research and Development Service (S.G.W. and S.D.D.-H.). The Center for Neuroscience and Regeneration Research is a collaboration of the Paralyzed Veterans of America with Yale University. G.P.H. was supported by the NIH/NIGMS Medical Training Program T32GM007205. Author contributions: S.D.D.-H., S.G.W., E.J.A., and G.P.H. designed the experiments. E.J.A., G.P.H., M.A.M., B.S.T., and T.A. performed the experiments. F.B.D.-H. and S.L. provided technical support, reagents, and materials. S.D.D.-H., S.G.W., and E.J.A. wrote the manuscript. S.D.D.-H., S.G.W., E.J.A., G.P.H., M.A.M., B.S.T., and T.A. revised and edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax4755/DC1
Fig. S1. Venus-NaV1.7-BAD and Halo-NaV1.7 display WT currents and express in cultured DRG neurons (related to Figs. 1A and 2A).
Fig. S2. Venus-NaV1.7-BAD surface labeling does not display bleed-through, and DRG neurons display autofluorescence (related to Fig. 1).
Fig. S3. Venus-NaV1.7-BAD and Halo-NaV1.7 nanoclusters are visible in DRG axons (related to Fig. 1).
Fig. S4. Methods for estimating number of channels per nanocluster (related to Fig. 1G).
Fig. S5. Retrograde, but not anterograde, vesicles can be visualized using the fluorescent protein signal (related to Fig. 2).
Fig. S6. Rab6 expression in cultured DRG neurons (related to Fig. 3).
Fig. S7. Fluorescence intensity for individual JF549-Halo molecules (related to Fig. 5).
Fig. S8. Modification of MFCs (related to Materials and Methods).
Movie S1. Anterograde trafficking of Halo-NaV1.7.
Movie S2. Anterograde trafficking of Halo-NaV1.7 under control conditions and after exposure to IM.
REFERENCES AND NOTES
- 1.Bennett D. L., Clark A. J., Huang J., Waxman S. G., Dib-Hajj S. D., The role of voltage-gated sodium channels in pain signaling. Physiol. Rev. 99, 1079–1151 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Dib-Hajj S. D., Waxman S. G., Sodium channels in human pain disorders: Genetics and pharmacogenomics. Annu. Rev. Neurosci. 42, 87–106 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Persson A.-K., Black J. A., Gasser A., Fischer T. Z., Waxman S. G., Sodium-calcium exchanger and multiple sodium channel isoforms in intra-epidermal nerve terminals. Mol. Pain 6, 84 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Black J. A., Frézel N., Dib-Hajj S. D., Waxman S. G., Expression of Nav1.7 in DRG neurons extends from peripheral terminals in the skin to central preterminal branches and terminals in the dorsal horn. Mol. Pain 8, 82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L., Huang J., Zhao P., Persson A.-K., Dib-Hajj F. B., Cheng X., Tan A., Waxman S. G., Dib-Hajj S. D., Conditional knockout of NaV1.6 in adult mice ameliorates neuropathic pain. Sci. Rep. 8, 3845 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shields S. D., Cheng X., Üçeyler N., Sommer C., Dib-Hajj S. D., Waxman S. G., Sodium channel Nav1.7 is essential for lowering heat pain threshold after burn injury. J. Neurosci. 32, 10819–10832 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vasylyev D. V., Han C., Zhao P., Dib-Hajj S., Waxman S. G., Dynamic-clamp analysis of wild-type hNav1.7 and erythromelalgia mutant channel L858H. J. Neurophysiol. 111, 1429–1443 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Alexandrou A. J., Brown A. R., Chapman M. L., Estacion M., Turner J., Mis M. A., Wilbrey A., Payne E. C., Gutteridge A., Cox P. J., Doyle R., Printzenhoff D., Lin Z., Marron B. E., West C., Swain N. A., Storer R. I., Stupple P. A., Castle N. A., Hounshell J. A., Rivara M., Randall A., Dib-Hajj S. D., Krafte D., Waxman S. G., Patel M. K., Butt R. P., Stevens E. B., Subtype-selective small molecule inhibitors reveal a fundamental role for Nav1.7 in nociceptor electrogenesis, axonal conduction and presynaptic release. PLOS ONE 11, e0152405 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minett M. S., Nassar M. A., Clark A. K., Passmore G., Dickenson A. H., Wang F., Malcangio M., Wood J. N., Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat. Commun. 3, 791 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Black J. A., Nikolajsen L., Kroner K., Jensen T. S., Waxman S. G., Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann. Neurol. 64, 644–653 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Persson A. K., Gasser A., Black J. A., Waxman S. G., Nav1.7 accumulates and co-localizes with phosphorylated ERK1/2 within transected axons in early experimental neuromas. Exp. Neurol. 230, 273–279 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Devor M., Wall P. D., Catalan N., Systemic lidocaine silences ectopic neuroma and DRG discharge without blocking nerve conduction. Pain 48, 261–268 (1992). [DOI] [PubMed] [Google Scholar]
- 13.Gould H. J. III, England J. D., Soignier R. D., Nolan P., Minor L. D., Liu Z. P., Levinson S. R., Paul D., Ibuprofen blocks changes in Nav 1.7 and 1.8 sodium channels associated with complete freund's adjuvant-induced inflammation in rat. J. Pain 5, 270–280 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Liang L., Fan L., Tao B., Yaster M., Tao Y.-X., Protein kinase B/Akt is required for complete Freund’s adjuvant-induced upregulation of Nav1.7 and Nav1.8 in primary sensory neurons. J. Pain 14, 638–647 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bi R. Y., Ding Y., Gan Y. H., Non-steroidal anti-inflammatory drugs attenuate hyperalgesia and block upregulation of trigeminal ganglionic sodium channel 1.7 after induction of temporomandibular joint inflammation in rats. Chin. J. Dent. Res. 19, 35–42 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Strickland I. T., Martindale J. C., Woodhams P. L., Reeve A. J., Chessell I. P., McQueen D. S., Changes in the expression of Nav1.7, Nav1.8 and Nav1.9 in a distinct population of dorsal root ganglia innervating the rat knee joint in a model of chronic inflammatory joint pain. Eur. J. Pain 12, 564–572 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Su Y. Y., Ye M., Li L., Liu C., Pan J., Liu W. W., Jiang Y., Jiang X. Y., Zhang X., Shu Y., Bao L., KIF5B promotes the forward transport and axonal function of the voltage-gated sodium channel Nav18. J. Neurosci. 33, 17884–17896 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barry J., Gu Y., Jukkola P., O'Neill B., Gu H., Mohler P. J., Rajamani K. T., Gu C., Ankyrin-G directly binds to kinesin-1 to transport voltage-gated Na+ channels into axons. Dev. Cell 28, 117–131 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dib-Hajj S. D., Cummins T. R., Black J. A., Waxman S. G., Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 33, 325–347 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Chen I., Howarth M., Lin W., Ting A. Y., Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat. Methods 2, 99–104 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Dzhashiashvili Y., Zhang Y., Galinska J., Lam I., Grumet M., Salzer J. L., Nodes of Ranvier and axon initial segments are ankyrin G-dependent domains that assemble by distinct mechanisms. J. Cell Biol. 177, 857–870 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kole M. H., Ilschner S. U., Kampa B. M., Williams S. R., Ruben P. C., Stuart G. J., Action potential generation requires a high sodium channel density in the axon initial segment. Nat. Neurosci. 11, 178–186 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Akin E. J., Solé L., Johnson B., Beheiry M. E., Masson J. B., Krapf D., Tamkun M. M., Single-molecule imaging of Nav1.6 on the surface of hippocampal neurons reveals somatic nanoclusters. Biophys. J. 111, 1235–1247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaqaman K., Loerke D., Mettlen M., Kuwata H., Grinstein S., Schmid S. L., Danuser G., Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods 5, 695–702 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akin E. J., Solé L., Dib-Hajj S. D., Waxman S. G., Tamkun M. M., Preferential targeting of Nav1.6 voltage-gated Na+ channels to the axon initial segment during development. PLOS ONE 10, e0124397 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakada C., Ritchie K., Oba Y., Nakamura M., Hotta Y., Iino R., Kasai R. S., Yamaguchi K., Fujiwara T., Kusumi A., Accumulation of anchored proteins forms membrane diffusion barriers during neuronal polarization. Nat. Cell Biol. 5, 626–632 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Grimm J. B., English B. P., Chen J., Slaughter J. P., Zhang Z., Revyakin A., Patel R., Macklin J. J., Normanno D., Singer R. H., Lionnet T., Lavis L. D., A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat Methods 12, 244–250 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banworth M. J., Li G., Consequences of Rab GTPase dysfunction in genetic or acquired human diseases. Small GTPases 9, 158–181 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mis M. A., Rogers M. F., Jeffries A. R., Wilbrey A. L., Chen L., Yang Y., Dib-Hajj S., Waxman S. G., Stevens E. B., Randall A. D., Differential aging-related changes in neurophysiology and gene expression in IB4-positive and IB4-negative nociceptive neurons. Aging Cell 17, e12795 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Usoskin D., Furlan A., Islam S., Abdo H., Lönnerberg P., Lou D., Hjerling-Leffler J., Haeggström J., Kharchenko O., Kharchenko P. V., Linnarsson S., Ernfors P., Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 18, 145–153 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Hockley J. R., Boundouki G., Cibert-Goton V., McGuire C., Yip P. K., Chan C., Tranter M., Wood J. N., Nassar M. A., Blackshaw L. A., Aziz Q., Michael G. J., Baker M. D., Winchester W. J., Knowles C. H., Bulmer D. C., Multiple roles for Nav1.9 in the activation of visceral afferents by noxious inflammatory, mechanical, and human disease-derived stimuli. Pain 155, 1962–1975 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan Z. Y., Piekarz A. D., Priest B. T., Knopp K. L., Krajewski J. L., McDermott J. S., Nisenbaum E. S., Cummins T. R., Tetrodotoxin-resistant sodium channels in sensory neurons generate slow resurgent currents that are enhanced by inflammatory mediators. J. Neurosci. 34, 7190–7197 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lim A., Rechtsteiner A., Saxton W. M., Two kinesins drive anterograde neuropeptide transport. Mol. Biol. Cell 28, 3542–3553 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruangsri S., et al. , Relationship of axonal voltage-gated sodium channel 1.8 (NaV1.8) mRNA accumulation to sciatic nerve injury-induced painful neuropathy in rats. J. Biol. Chem. 286, 39836–39847 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thakor D. K., et al. , Increased peripheral nerve excitability and local NaV1.8 mRNA up-regulation in painful neuropathy. Mol. Pain 5, 14 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalez C., Cánovas J., Fresno J., Couve E., Court F. A., Couve A., Axons provide the secretory machinery for trafficking of voltage-gated sodium channels in peripheral nerve. Proc. Natl. Acad. Sci. U.S.A. 113, 1823–1828 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lasiecka Z. M., Winckler B., Mechanisms of polarized membrane trafficking in neurons—Focusing in on endosomes. Mol. Cell. Neurosci. 48, 278–287 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rasband M. N., The axon initial segment and the maintenance of neuronal polarity. Nat. Rev. Neurosci. 11, 552–562 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Black J. A., Waxman S. G., Foster R. E., Spatial heterogeneity of the axolemma of non-myelinated fibers in the optic disc of the adult rat: Freeze-fracture observations. Cell Tissue Res. 224, 239–246 (1982). [DOI] [PubMed] [Google Scholar]
- 40.Neishabouri A., Faisal A. A., Saltatory conduction in unmyelinated axons: Clustering of Na+ channels on lipid rafts enables micro-saltatory conduction in C-fibers. Front. Neuroanat. 8, 109 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vasylyev D. V., Waxman S. G., Membrane properties and electrogenesis in the distal axons of small dorsal root ganglion neurons in vitro. J. Neurophysiol. 108, 729–740 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Cummins T. R., Howe J. R., Waxman S. G., Slow closed-state inactivation: A novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J. Neurosci. 18, 9607–9619 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gumy L. F., Katrukha E. A., Grigoriev I., Jaarsma D., Kapitein L. C., Akhmanova A., Hoogenraad C. C., MAP2 defines a pre-axonal filtering zone to regulate KIF1- versus KIF5-dependent Cargo transport in sensory neurons. Neuron 94, 347–362.e7 (2017). [DOI] [PubMed] [Google Scholar]
- 44.R. Milo, R. Philips, Cell Biology by the numbers (Garland Science, 2016). [Google Scholar]
- 45.Hoerndli F. J., Maxfield D. A., Brockie P. J., Mellem J. E., Jensen E., Wang R., Madsen D. M., Maricq A. V., Kinesin-1 regulates synaptic strength by mediating the delivery, removal and redistribution of AMPA receptors. Neuron 80, 1421–1437 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dash B., Han C., Waxman S. G., Dib-Hajj S. D., Nonmuscle myosin II isoforms interact with sodium channel alpha subunits. Mol. Pain 14, 1744806918788638 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wakana Y., van Galen J., Meissner F., Scarpa M., Polishchuk R. S., Mann M., Malhotra V., A new class of carriers that transport selective cargo from the trans Golgi network to the cell surface. EMBO J. 31, 3976–3990 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wandinger-Ness A., Zerial M., Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb. Perspect. Biol. 6, a022616 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmidt J. W., Catterall W. A., Biosynthesis and processing of the α subunit of the voltage-sensitive sodium channel in rat brain neurons. Cell 46, 437–444 (1986). [DOI] [PubMed] [Google Scholar]
- 50.Hoerndli F. J., Wang R., Mellem J. E., Kallarackal A., Brockie P. J., Thacker C., Madsen D. M., Maricq A. V., Neuronal activity and CaMKII regulate kinesin-mediated transport of synaptic AMPARs. Neuron 86, 457–474 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scarabelli G., Soppina V., Yao X.-Q., Atherton J., Moores C. A., Verhey K. J., Grant B. J., Mapping the processivity determinants of the kinesin-3 motor domain. Biophys. J. 109, 1537–1540 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hangen E., Cordelières F. P., Petersen J. D., Choquet D., Coussen F., Neuronal activity and intracellular calcium levels regulate intracellular transport of newly synthesized AMPAR. Cell Rep. 24, 1001–1012.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herzog R. I., Cummins T. R., Ghassemi F., Dib-Hajj S. D., Waxman S. G., Distinct repriming and closed-state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons. J. Physiol. 551, 741–750 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang Y., Huang J., Mis M. A., Estacion M., Macala L., Shah P., Schulman B. R., Horton D. B., Dib-Hajj S. D., Waxman S. G., Nav1.7-A1632G mutation from a family with inherited erythromelalgia: Enhanced firing of dorsal root ganglia neurons evoked by thermal stimuli. J. Neurosci. 36, 7511–7522 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rzomp K. A., Scholtes L. D., Briggs B. J., Whittaker G. R., Scidmore M. A., Rab GTPases are recruited to chlamydial inclusions in both a species-dependent and species-independent manner. Infect. Immun. 71, 5855–5870 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Choudhury A., Dominguez M., Puri V., Sharma D. K., Narita K., Wheatley C. L., Marks D. L., Pagano R. E., Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J. Clin. Invest. 109, 1541–1550 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dib-Hajj S. D., Choi J. S., Macala L. J., Tyrrell L., Black J. A., Cummins T. R., Waxman S. G., Transfection of rat or mouse neurons by biolistics or electroporation. Nat. Protoc. 4, 1118–1126 (2009). [DOI] [PubMed] [Google Scholar]
- 58.Deutsch E., Weigel A. V., Akin E. J., Fox P., Hansen G., Haberkorn C. J., Loftus R., Krapf D., Tamkun M. M., Kv2.1 cell surface clusters are insertion platforms for ion channel delivery to the plasma membrane. Mol. Biol. Cell 23, 2917–2929 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mangeol P., Prevo B., Peterman E. J., KymographClear and KymographDirect: Two tools for the automated quantitative analysis of molecular and cellular dynamics using kymographs. Mol. Biol. Cell 27, 1948–1957 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax4755/DC1
Fig. S1. Venus-NaV1.7-BAD and Halo-NaV1.7 display WT currents and express in cultured DRG neurons (related to Figs. 1A and 2A).
Fig. S2. Venus-NaV1.7-BAD surface labeling does not display bleed-through, and DRG neurons display autofluorescence (related to Fig. 1).
Fig. S3. Venus-NaV1.7-BAD and Halo-NaV1.7 nanoclusters are visible in DRG axons (related to Fig. 1).
Fig. S4. Methods for estimating number of channels per nanocluster (related to Fig. 1G).
Fig. S5. Retrograde, but not anterograde, vesicles can be visualized using the fluorescent protein signal (related to Fig. 2).
Fig. S6. Rab6 expression in cultured DRG neurons (related to Fig. 3).
Fig. S7. Fluorescence intensity for individual JF549-Halo molecules (related to Fig. 5).
Fig. S8. Modification of MFCs (related to Materials and Methods).
Movie S1. Anterograde trafficking of Halo-NaV1.7.
Movie S2. Anterograde trafficking of Halo-NaV1.7 under control conditions and after exposure to IM.