

SUMMARY
During autophagy, cargo recruitment and vesicle dynamics are driven by numerous receptors/adaptors that become tethered to the phagophore through interactions with lipidated ATG8/LC3 decorating the expanding membrane. Most currently described ATG8 binding proteins exploit a well-defined ATG8-interacting motif (AIM, or LC3-interacting region (LIR)) that contacts a hydrophobic patch on ATG8 known as the LIR/AIM-docking site (LDS). Here we describe a new class of ATG8 interactors that exploit ubiquitin-interacting motif (UIM)-like sequences for high-affinity binding to an alternative ATG8 interaction site. Assays with candidate UIM-containing proteins together with unbiased screens identified a large collection of UIM-based ATG8 interactors in plants, yeast and humans. Analysis of a subset also harboring UBX domains revealed a role for UIM-directed autophagy in clearing non-functional CDC48/p97 complexes, including some impaired in human disease. With this new class of adaptors/receptors, we greatly extend the reach of selective autophagy and identify new factors regulating autophagic vesicle dynamics.
Graphical Abstract
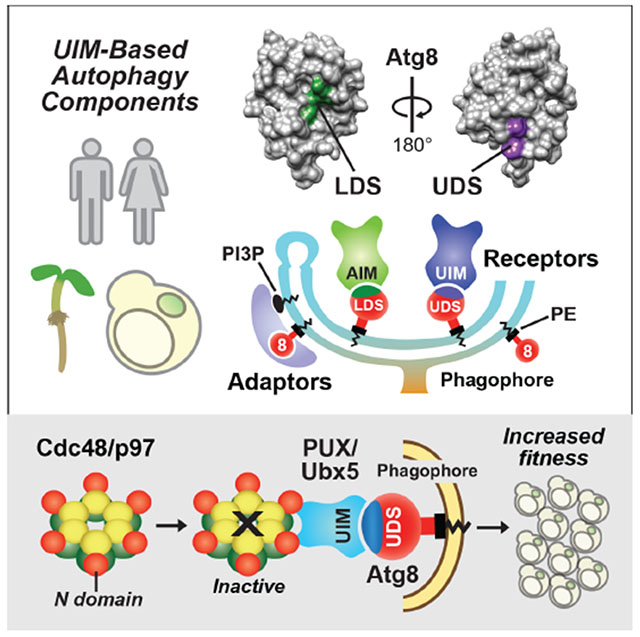
In Brief
Across organisms, the key autophagy protein, ATG8/LC3, binds a group of proteins on a site distinct from its classical interacting region, raising the possibility of dual binding interactions and identifying an array of unknown selective autophagic substrates, including some involved in human disease.
INTRODUCTION
All cellular organisms maintain internal homeostasis and avoid proteotoxic stress by engaging sophisticated catabolic pathways that identify and recycle unwanted or dysfunctional components. One dominant pathway in eukaryotes is macroautophagy (henceforth referred to as autophagy), which sequesters cytoplasmic material into vesicles for delivery to vacuoles (in plants and yeast) or lysosomes (in animals; Marshall and Vierstra, 2018; Levine and Kroemer, 2019). These vesicles emerge from the endoplasmic reticulum (ER) as a cup-shaped phagophore that eventually seals to generate a double membrane-bound autophagosome encapsulating substrates. Autophagosomes then fuse with the limiting membrane of the vacuole or lysosome to release the internal vesicle, called an autophagic body, into the lumen for degradation by resident hydrolases. As autophagy is essential for protecting against nutrient deprivation and often becomes mis-regulated in a variety of human diseases, its modulation has considerable therapeutic potential (Levine and Kroemer, 2019).
Central to autophagy is the ubiquitin-fold protein ATG8 that decorates the emerging phagophore and autophagosome following its modification with the lipid phosphatidylethanolamine (PE). Using an ATP-dependent conjugation cascade mechanistically analogous to ubiquitylation, mature ATG8 is activated by the E1 enzyme ATG7, transferred to the E2 enzyme ATG3, and finally conjugated to PE using a ligase complex containing an ATG12-ATG5 conjugate and ATG16 (Ohsumi, 2001; Marshall and Vierstra, 2018). Once inserted into autophagic membranes, lipidated ATG8 provides a docking platform for numerous autophagy adaptors and receptors that drive autophagic vesicle dynamics and cargo selection, respectively (Khaminets et al., 2016; Gatica et al., 2018; Marshall and Vierstra, 2018).
In the last few years, the list of known ATG8-binding proteins has grown substantially, including receptors for unwanted/dysfunctional mitochondria, peroxisomes, ER, lipid droplets, ribosomes, proteasomes and pathogens (Gatica et al., 2018). These interactors typically contain an ATG8-interacting motif (AIM, also known as an LC3-interacting region (LIR)) with the consensus sequence W/F/Y-X-X-L/I/V that tightly fits into two conserved hydrophobic pockets on ATG8 collectively termed the LIR/AIM docking site (LDS; Noda et al., 2008; 2010; Behrends et al., 2010). While most studies have focused on the AIM-LDS interface, possibilities for alternative ATG8 binding mechanisms have recently emerged, with several ATG8-interacting proteins now known to bind ATG8 in an AIM-independent manner (Behrends et al., 2010; Lin et al., 2013; Marshall et al., 2015). One example is Arabidopsis RPN10 that serves as the receptor for recruitment of inactive 26S proteasomes. Instead of using the canonical AIM sequence, RPN10 exploits an unrelated ubiquitin-interacting motif (UIM) for high-affinity binding to ATG8 (Marshall et al., 2015).
Under the premise that the UIM of RPN10 interacts with ATG8 through a novel contact that provides an alternative mechanism for tethering adaptors/receptors to autophagic membranes, we examined its interaction with ATG8 in detail. Remarkably, these studies led to the discovery of a previously unknown adaptor/receptor binding patch on ATG8, which we termed the UIM-docking site (UDS). By combining unbiased screens and candidate-based approaches to search for additional UIM-containing ATG8 interactors, we identified numerous Arabidopsis, yeast and human proteins that use this UIM-UDS interface. Detailed studies on a family of ATG8-interacting UIM- and UBX domain-containing proteins from Arabidopsis and yeast revealed that UIM-UDS binding facilitates the autophagic degradation of non-functional CDC48/p97 complexes, including mutant forms connected to several human pathologies (Tang and Xia, 2016). Through our discovery of the UIM-UDS interface and a large collection of adaptors and receptors that exploit this interaction, we greatly expand the reach of selective autophagy and identify new factors that might regulate autophagosome dynamics.
RESULTS
RPN10 Binds ATG8 at a Site Distinct from the LDS
Our discovery that the Arabidopsis proteaphagy receptor RPN10 uses its UIM2 sequence to bind ATG8 instead of the commonly employed AIM (Marshall et al., 2015) raised the possibility that a distinct surface on ATG8 other than the canonical LDS participates in this interaction. To test this possibility, we assayed by yeast two-hybrid (Y2H) pairwise interactions between RPN10 and representatives of the four Arabidopsis ATG8 sub-clades harboring alanine substitutions within the LDS (Y50A L51A in ATG8a; ΔLDS; Table S1). In all cases, the ΔLDS variants successfully bound to RPN10 but failed to interact with ATG1a, which is known to bind ATG8 via an AIM-LDS interface (Figures 1A and S1A; Suttangkakul et al., 2011). Because this interaction could be specific for proteaphagy receptors, we similarly tested binding of the yeast receptor Cue5 to Atg8 (Marshall et al., 2016), and found that the typical AIM-LDS interface is involved (Figures 1B and S1B). We thus speculated that UIM2 of Arabidopsis RPN10 connects with ATG8 via a novel interaction.
Figure 1. Arabidopsis RPN10 Interacts with ATG8 via a Novel Binding Site.

(A) Y2H assays reveal RPN10 binds ATG8a in an LDS-independent manner. Growth of cells on medium lacking leucine and tryptophan is shown in Figure S1 (also for panels (B) and (E)).
(B) Y2H assays reveal that the yeast proteaphagy receptor Cue5 binds Atg8 via the LDS.
(C) Identification of potential RPN10 binding sites in Arabidopsis ATG8a. Plant ATG8 sequences were aligned and the percent identity at each position calculated. Conserved, surfaced-exposed positions are underlined; the arrowhead indicates the ATG4 cleavage site.
(D) The 3-dimensional structure of yeast Atg8 (PDB file 3VXW), highlighted to locate the conserved surface patches identified in panel (C).
(E) Y2H assays identify a surface patch on Arabidopsis ATG8a that binds RPN10.
(F) In vitro binding assays confirm the UIM-UDS interface between RPN10 and ATG8e. Purified proteins were mixed and pulled down with Ni-NTA agarose beads. Input and bound proteins were visualized by immunoblotting with anti-ATG8 and anti-GST antibodies.
(G) BiFC assays show that RPN10 interacts with ATG8a at the UDS in planta. Fluorescence signals in N. benthamiana leaf cells were detected by confocal fluorescence microscopy. Scale bar, 10 μm.
(H) ATG8e can simultaneously interact with LDS- and UDS-binding partners. Purified proteins were mixed, pulled down with GST-Bind™ resin, and visualized as in panel (F).
Given that RPN10-ATG8 binding appears universal among plants (Marshall et al., 2015), we reasoned that a conserved surface outside of the LDS is involved. An alignment of 332 plant ATG8 sequences (Table S2; Kellner et al., 2017) revealed conserved clusters of amino acids (Figure 1C) that, when mapped onto the 3-dimensional structure of yeast Atg8, identified 8 additional surface patches besides the LDS as candidate UIM-binding sites (Figure 1D). When alanine substitutions of these sites were systematically tested, only one mutation cluster, centered on I77, F78 and V79, specifically blocked binding of ATG8 to RPN10 by Y2H (Figures 1E and S1C).
The importance of this patch (designated the UIM-docking site (UDS)) for binding RPN10 was confirmed using ΔUDS and ΔUIM mutants in in vitro pulldown, isothermal titration calorimetry (ITC) and in plants bimolecular fluorescence complementation (BiFC) assays. Whereas 6His-ATG8 beads readily pulled down GST-tagged RPN10, they did not pull down RPN10 bearing a ΔUIM2 mutant substituting the core Leu-Leu-Asp-Gln-Ala UIM sequence (Marshall et al., 2015), nor could the ΔUDS form of ATG8 pull down wild-type (WT) RPN10, showing that both UIM2 and the UDS were essential (Figure 1F). Similarly, while binding of RPN10 to ATG8 was clearly detected by ITC, this binding was absent when the UDS mutant of ATG8 was used (Figure S2G). By BiFC, we found that ATG8 and RPN10 bearing the N- and C-terminal halves of yellow fluorescent protein (YFP) in either orientation easily reconstituted fluorescence in Nicotians benthamiana (Figure 1G; Marshall et al., 2015); while cytoplasmic fluorescence was retained with the ΔLDS mutant form of ATG8, it was absent with the ΔUDS version (Figures 1G and S1D). Given the possibility that UDS mutations compromised binding of proteins to the LDS, we compared binding of the ΔLDS and ΔUDS mutants with RPN10 and DSK2, the latter of which uses an AIM for ATG8 binding (Nolan et al., 2017). Pulldown assays showed that neither the ΔLDS or ΔUDS variants impacted interaction of these ATG8-binding proteins with their corresponding non-mutated patch (Figure S1E).
The presence of separate binding sites on ATG8 suggested that a single ATG8 could dock UIM- and AIM-containing proteins simultaneously. Using a tripartite binding assay in which beads coated with GST-tagged RPN10 were used to pull down HA-tagged DSK2, we found that RPN10 could only pull down DSK2 when ATG8 was present (Figure 1H). Such dual binding raised the possibility that ATG8 can tether two adaptors/receptors simultaneously.
The UIM-UDS Interface is Widely Used by ATG8-interacting Proteins in Arabidopsis
To examine whether UIMs are employed by other autophagy adaptors/receptors, we tested by Y2H 13 Arabidopsis proteins predicted to contain one or more UIMs for binding to ATG8. Strikingly, three PLANT UBIQUITIN REGULATORY X DOMAIN (PUX) proteins were identified that displayed robust interactions (Figures 2A and S1G). All three (PUX7, PUX8 and PUX9) showed little preference for an ATG8 sub-clade, and equally bound ATG8a, ATG8e, ATG8f and ATG8i (Figures 2G and S1F). These PUX proteins are part of a 16-member family in Arabidopsis defined by a ubiquitin-like UBX domain, with some members also harboring ubiquitin-binding UBA domains and one or more UIMs (Figures S2A and S2B). Like relatives from yeast and animals, Arabidopsis PUX proteins bind CDC48 (p97 in humans) and play important roles in regulating the assembly and activity of this AAA-ATPase (Rancour et al., 2004; Gallois et al., 2013).
Figure 2. Multiple Arabidopsis UIM Proteins Bind ATG8 Through the UDS.

(A) A subset of Arabidopsis PUX proteins interact with ATG8a by Y2H. Growth of cells on medium lacking leucine and tryptophan is shown in Figure S1 (also for panels (B), (C) and (G)).
(B) Y2H assays reveal that the UIMs in Arabidopsis PUX7, 8, 9, and 13 are required for binding ATG8a at the UDS.
(C) Y2H assay reveal that components of the core autophagy pathway do not use the UDS interface to bind ATG8a.
(D) Quantification of the PUX8-ATG8e binding affinity. Various concentrations of 6His-ATG8e were incubated with 1 μM GST-PUX8 and pulled down with Ni-NTA beads. PUX8 remaining in the supernatant was quantified and expressed as a percentage of the input. Values represent the mean (±SD) from three independent biological replicates.
(E) ITC assays confirm the PUX8-ATG8e binding affinity. Shown are heats of injection (top panel and lower panel, squares) and the best fit to a single site binding model (solid lines) for interaction with WT (black) or ΔUDS (grey) forms of 6His-ATG8e. N and KD values represent the mean (±SD) from three independent biological replicates.
(F) BiFC assays in N. benthamiana leaf cells show that PUX proteins interact with ATG8a via their UIM sequences in planta. Scale bar, 10 μm.
(G) PUX7, 8, and 9 interact by Y2H with members of each Arabidopsis ATG8 sub-clade.
(H) Comparison of ATG8 isoform levels when expressed as Gal4 DNA-binding domain fusions in yeast.
(I) Screening of an Arabidopsis cDNA library by Y2H using WT, ΔLDS or ΔUDS forms of ATG8e identified multiple LDS- and UDS-binding proteins. A Venn diagram of the interaction categories is included; asterisks indicate proteins previously identified as ATG8 interactors.
(J) Dot blot binding assays confirm the candidate interactors identified in panel (I). Lysed E. coli cells expressing HA-tagged interactors were spotted onto nitrocellulose membrane and probed with WT, ΔLDS or ΔUDS forms of 6His-ATG8e. Binding of ATG8 variants and expression of the interactors were detected with anti-6His and anti-HA antibodies, respectively.
Asking whether other Arabidopsis PUX proteins not annotated to have UIMs might also bind ATG8, we tested the entire family by Y2H. Besides PUX7, PUX8 and PUX9, PUX13 also bound ATG8, thus identifying four isoforms with this activity (Figure S2C). Notably, when we re-analyzed the PUX13 sequence, a UIM-related sequence (residues 180-198) was discovered that might provide the ATG8-binding interface (Figure S2J).
To confirm that the UIMs in PUX7, PUX8, PUX9 and PUX13 were solely responsible for their binding to ATG8, we examined by Y2H deletion mutants individually missing the UBA and UBX domains, along with a set of substitutions impacting the UIMs (designated ΔUIM; Table S1), for binding to WT, ΔLDS or ΔUDS versions of ATG8. Collectively, we found that the PUX UIMs (and not the UBA or UBX domains) were essential for ATG8 binding, with the sole UIM in PUX7 and PUX13, and the N-terminal UIM (designated UIM1) in PUX8 and PUX9, providing the interfaces (Figures 2B and S1I). These binding activities were further supported by in vitro pulldown, ITC and BiFC assays (Figures 2D, 2E, 2F, S2D, S2E, S2F and S2G). By adjusting the pulldown conditions to provide quantitative equilibrium binding measurements, we determined a KD of between 3.3 and 7.4 μM for binding of the PUX proteins to ATG8e, which is comparable to the known affinity of Arabidopsis RPN10 and AIM-containing proteins for ATG8 (Marshall et al., 2015). This was further supported by ITC measurements, which detected a single binding site and confirmed the KD values to be in the low micromolar range (Figures 2E, S2F and S2G).
A number of proteins within the ATG system interact with ATG8, several of which use the AIM-LDS interface (Suttangkakul et al., 2011; Marshall and Vierstra, 2018). To test whether the others might alternatively engage the UDS, we examined their binding to WT, ΔLDS or ΔUDS versions of ATG8 by Y2H (Figures 2C and S1H). Whereas some indeed required the LDS for ATG8 binding (ATG1a, ATG5, ATG6, ATG12a, ATG16 and VPS34), none required the UDS. Interestingly, while binding of ATG3, ATG7 and ATG10 to ATG8 was expected to be independent of both the LDS and UDS, given that they thioester link to the C-terminus of ATG8, the independence of ATG9 and ATG11 implied that additional binding surface(s) exist.
Assuming that other Arabidopsis proteins besides PUX13 might bind ATG8 at the UDS but were not yet annotated to have a UIM, we performed non-biased screens of an Arabidopsis cDNA library using either WT, ΔLDS or ΔUDS variants of ATG8e as bait (Figure 2H). Overall, 112 ATG8e interactors were identified across the three Y2H screens (Table S3). These included 47 proteins that interacted with both WT and ΔUDS versions of ATG8e but not with the ΔLDS version, which we considered to be candidate LDS interactors, and 19 proteins that interacted with WT and ΔLDS versions of ATG8e but not with the ΔUDS version, which we considered to be candidate UDS interactors (Figure 2I). Importantly, the screen identified numerous ATG8-interacting proteins shown here and elsewhere to bind at the LDS or UDS. For the UDS, these included RPN10 (Marshall et al., 2015) and three of the four PUX proteins identified here by the candidate-based approach, while for the LDS these included two isoforms of ATG1, the adaptor protein SH3P2, and the cargo receptors ATI1/2, DSK2a and NBR1 (Suttangkakul et al., 2011 Nolan et al., 2017; Marshall and Vierstra, 2018).
To verify that these candidates indeed bound ATG8, we developed a dot blot assay to demonstrate binding in vitro (Figure 2J). Here, the prey cDNAs were excised from the Y2H vector and inserted into an IPTG-inducible vector in-frame with an N-terminal HA tag for expression in E. coli. Following induction, the bacterial cells were lysed, spotted onto a nitrocellulose membrane, and probed with purified 6His-ATG8e, either WT or harboring the ΔLDS or ΔUDS mutations, followed by immunoblot analysis with anti-6His antibodies to locate bound ATG8e. Probing the membranes with anti-HA antibodies confirmed adequate expression of each prey. All 66 of the candidate interactors (47 for the LDS and 19 for the UDS) bound WT 6His-ATG8e, and this binding was absent with either the ΔLDS or ΔUDS variants, thus confirming the specificity of the associations (Figure 2J). Both the LDS- and UDS-specific lists contained numerous proteins not previously known to be associated with ATG8 and/or autophagy that could function as specific cargo receptors, adaptors important for autophagic vesicle dynamics, or even cargo bearing their own ATG8-binding sites for self-recruitment. These included the clathrin adaptors AP19 and AP1M2/HAP13, the ATP synthase subunit ATPC2, the SWI/SNF complex subunit SWIB3, the ER-localized co-chaperone BAG7, the lipid body protein OLE1, plus a number of ion channels (KCO5/TPK5, VDAC2 and VDAC3) and transporter proteins (OPT7 and SWEET1; Figure 2I).
A Hydrophobic Patch Defines the UIM-binding UDS.
Beyond I77, F78 and V79, the region surrounding the UDS in ATG8 includes a number of other conserved residues that could promote UIM binding. To better define the UDS and identify permissible residues, we analyzed the region in Arabidopsis ATG8a by saturating mutagenesis, followed by Y2H and dot blot binding assays with both RPN10 and PUX7. None of the mutations appeared to substantially impact ATG8 folding, as they all retained their ability to bind both ATG7 and DSK2 (Figure S3A). The results in Figures 3A and 3B showed remarkable consistency among binding partners and methods, with over 97% of the assays in agreement. They defined the UDS as a 4-residue region encompassing amino acids 77-80 that is enriched in hydrophobic residues and intolerant of charged amino acids. At its center is a phenylalanine, which could not be fully replaced by any other amino acid, and is thus considered essential (Figures 3A and 3B). Quantification of the interaction data and comparison with evolutionary mutation frequencies revealed a consensus motif for permitted residues of Ψ-F-Ψ-Ω/T, where Ψ and Ω represent small hydrophobic and aromatic residues, respectively (Figure 3C).
Figure 3. The UDS Contains a Conserved Phenylalanine Surrounded by Hydrophobic Residues.

(A and B) Amino acid sequence specificity of the ATG8 UDS for binding to UIMs. A panel of ATG8a mutants generated by saturating mutagenesis was tested for binding to Arabidopsis RPN10 (panel (A)) or PUX7 (panel (B)) by Y2H (left side) or dot blot binding assays (right side). Control interactions and expressions are shown in Figure S3.
(C) Quantification of the binding assays in panels (A) and (B), and comparison with expected substitution rates, identifies a core UDS sequence containing an invariant phenylalanine bracketed by hydrophobic amino acids. Ψ, small hydrophobic residues; Ω, aromatic residues.
(D) Sequence alignment of the UDS region from plant, fungi and animal ATG8 proteins. The core UDS is indicated by the solid red line.
(E) The 3-dimensional structure of yeast Atg8 (PDB file 3VXW) highlighting the opposed positions of the LDS (green) and UDS (purple).
When the UDS sequence was placed on an alignment of ATG8 proteins from a spectrum of eukaryotes, conservation of this hydrophobic patch was apparent (Figure 3D). Mapping this core region onto the 3-dimensional structure of yeast Atg8 showed that the UDS is located near the C-terminal glycine on the surface opposite to the LDS used by AIM-containing interactors (Figure 3E), thus explaining how AIM and UIM proteins could simultaneously bind.
UIM-UDS Interactions are Conserved in Yeast.
Both the conservation of the UDS and the widespread distribution of UIM-containing proteins outside of plants raised the possibility that the UIM-UDS interface is universally exploited. As a first test of this hypothesis, we examined by Y2H the seven yeast proteins annotated to contain a UIM for binding to Atg8 (Figures 4A and S4A). Positive interactions were seen for three proteins: Ent1 and Ent2, which are yeast Epsins that help generate membrane curvature during endocytosis (Wendland et al., 1999), and Ufo1, which is an F-Box ubiquitin ligase component (Kaplun et al., 2003). Focused Y2H assays of Ent1 and Ent2 harboring ENTH-domain deletions or ΔUIM mutations revealed that the N-terminal UIM (designated UIM1) is critical for binding the UDS of Atg8 (Figures 4B and S4B). Similar assays surprisingly failed to connect any of the three predicted UIMs in Ufo1 to Atg8 binding, thus leaving this interface unresolved (Figure S4C).
Figure 4. The UIM-UDS Interface Mediates ATG8 Binding in Yeast and Humans.

(A) A collection of yeast UIM proteins bind Atg8 by Y2H. Growth of cells on medium lacking leucine and tryptophan is shown in Figure S4 (also for panels (B), (C) and (E)).
(B) Y2H assays reveal that UIM regions in yeast Ent1 and Ent2 bind Atg8 at the UDS.
(C) Ubx5 interacts with the UDS of Atg8 via a UIM, from Y2H analysis of Ubx5 truncations (upper panel) or mutations (lower panel) paired with WT, ΔLDS or ΔUDS forms of Atg8.
(D) In vitro binding assays confirm the UIM-UDS interaction interface between Ubx5 and Atg8. Purified proteins were mixed and pulled down with Ni-NTA agarose beads. Input and bound proteins were visualized as in Figure 1F.
(E) Several human UIM-containing proteins interact with the ATG8 homologs MAP1LC3a and GABARAP by Y2H.
(F) Screening of a yeast cDNA library by Y2H using WT, ΔLDS or ΔUDS forms of yeast Atg8 identified multiple LDS- and UDS-binding proteins. A Venn diagram of the interaction categories is included; asterisks indicate proteins previously identified as Atg8 interactors.
(G) Dot blot binding assays confirm the candidate interactors identified in panel (F). The assay was performed as in Figure 2J.
Based on the confirmed interactions of Arabidopsis PUX proteins with ATG8, we considered it likely that one or more of the orthologous yeast Ubx proteins also bind Atg8 via the UIM-UDS interface. Testing all seven Ubx family members by Y2H revealed that only Ubx5 has affinity for Atg8 (Figures 4A and S4D), and that this binding requires the UDS (Figures 4C, 4D, S4E and S4F). Mapping of the UDS-binding site in Ubx5 localized the interacting region to residues 351-400 (Figures 4C and S4E), which we then found to contain a cryptic UIM at residues 360-379 (den Besten et al., 2012). Y2H and in vitro pulldown assays with substitution mutants confirmed that this patch is required (Figures 4C, 4D, and S4F).
To expand the list of UDS-interacting proteins in yeast, we next screened a cDNA library prepared from exponential phase cells, using WT, ΔLDS or ΔUDS versions of Atg8 as bait. From this screen, 92 interactors were identified (Table S3), including 38 candidate LDS interactors and 17 candidate UDS interactors (Figure 4F). The strength of the screen was again supported by the identification of numerous proteins previously shown to bind Atg8 (Figure 4F). Importantly, when assayed by in vitro dot blot binding, 51 of the 55 LDS or UDS interactors were confirmed (Figure 4G).
The UIM-UDS Interface Functions in Humans
As humans also express UIM proteins, we tested by Y2H whether they too might bind ATG8 via a UIM-UDS interface. Here, the MAP1LC3a and GABARAP isoforms, which represent the two main ATG8 sub-clades, were tested against 28 UIM candidates. Robust interactions were seen for six proteins (Figures 4E and S4G); of these, four (Epsin (EPN)-1, EPN2, EPN3 and Rabenosyn (RBSN)) interacted with both ATG8 isoforms, while two (Ataxin (ATXN)-3 and ATXN3L) interacted only with GABARAP (Figures 4E and S4G).
EPN1-3 are the human orthologs of yeast Ent1/2 (Ford et al., 2002), and are thus consistent with our prior identification of Ent1/2 as UDS interactors (Figures 4A, 4B, 4F and 4G). Like Ent1/2, they possess multiple UIMs; of the three predicted UIMs in EPN1, both UIM1 and UIM3 docked to MAP1LC3a and GABARAP, thus requiring mutation of each to prevent a Y2H interaction (Figure S4H). Similarly, elimination of both UIMs in EPN2 and EPN3 was needed to block MAP1LC3a and GABARAP binding (Figure S4H). ATXN3 and ATXN3L also contain at least two predicted UIMs; in both proteins, only the N-terminal UIM (UIM1) was needed for GABARAP binding (Figure S4I). RBSN contains a single predicted UIM, and its substitution blocked binding to both MAP1LC3a and GABARAP (Figure S4J). Together with our results from Arabidopsis and yeast, we propose that the UIM-UDS interface is a widely employed mechanism to dock autophagy adaptors/receptors to ATG8.
ATG8-interacting UIM Sequences have Diverse Amino Acid Properties.
UIMs in general are a 20-amino-acid stretch of considerable sequence diversity predicted to fold into an α-helix preceded by a short hairpin loop (Hofmann and Falquet, 2001). Using the same saturating mutagenesis approach as above for the UDS, we examined the UIM regions in Arabidopsis RPN10 (residues 282-301) and PUX7 (residues 328-347) for interaction with ATG8 by Y2H and dot blot binding assays (Figures 5A and 5B). The introduced mutations did not impact overall protein structure, as the mutant forms of RPN10 and PUX7 retained their ability to bind RPN9 and CDC48, respectively (Figures S3B and S3C), although local destabilization of the UIM α-helix could not be ruled out. As with the UDS, UIM mapping revealed remarkable consistency when comparing both binding partners and methods. Collectively, the assays defined a broad consensus sequence for ATG8 binding (Ψ-ζ-X-A-Ψ-X-X-S) that contains invariant alanine and serine residues, and with Ψ, ζ and X representing small hydrophobic residues, hydrophilic residues and any amino acid, respectively (Figures 5A, 5B and 5C). Alignment of the UIM regions demonstrated here to bind the UDS of ATG8 (17 UIMs from 14 proteins) confirmed this consensus sequence (Figure 5D), while visualization on a helical wheel indicated that the required residues cluster on one side of the predicted helix (Figure 5E).
Figure 5. Identification of a UIM Consensus Sequence that Binds the UDS.

(A and B) Amino acid sequence specificity of UIM-like sequences for binding the UDS. A panel of mutants generated by saturating mutagenesis of the Arabidopsis RPN10 (panel (A)) or PUX7 (panel (B)) UIMs was tested for binding to ATG8a by Y2H (top) or dot blot binding assays (bottom). Control interactions and expressions are shown in Figure S3.
(C) Quantification of the assays in panels (A) and (B), and comparison with expected substitution rates, identifies residues within the UIM important for ATG8a binding. Ψ, small hydrophobic residues; ζ, hydrophilic residues.
(D) Sequence alignment of ATG8-binding UIM sequences from Arabidopsis, yeast and humans. The core UIM is indicated by the solid red line; flanking residues are indicated by the dashed red line.
(E) Helical wheel representation of the core Arabidopsis ATG8-binding UIM, showing the positions of conserved residues in the predicted helical structure.
The ATG8-interacting PUX Proteins are Selective Autophagy Receptors for CDC48.
To demonstrate the significance of UIM-type autophagy adaptors/receptors, we focused on the Arabidopsis PUX proteins, given their potential to commit CDC48 to autophagy upon docking to ATG8 (Figures 2B and 2F). CDC48 assembles into a homo-hexameric ring that plays essential roles in protein quality control by using ATP hydrolysis to extract polypeptides from protein complexes or membranes for eventual turnover (Stach and Freemont, 2017), with its best understood function being the retrograde transport of proteins from the ER lumen during ER-associated protein degradation (ERAD; Ye et al., 2001; Marshall et al., 2008).
As a first step in connecting the ATG8-interacting PUX proteins and CDC48 to autophagy, we exploited an Arabidopsis line expressing a GFP-PUX7 fusion (Gallois et al., 2013) for interaction studies. Co-immunoprecipitation of GFP-PUX7 from seedling extracts with anti-GFP antibody beads detected four species (120, 95, 55 and 15 kDa) not found in extracts containing GFP alone (Figure S2H). Immunoblot analyses identified CDC48 and ATG8 in addition to GFP-PUX7, thus confirming that PUX7 can bind both in planta (Figure S2I). Mass spectrometric analysis confirmed CDC48 isoforms as the most abundant PUX7 interactors (Dataset S1).
To test whether both PUX7 and CDC48 are targets of autophagy, we subjected the GFP-PUX7 line, and a second line that expresses YFP-CDC48a (Park et al., 2008), to nitrogen starvation, and assayed each by the autophagy-specific GFP-release assay. As is widely exploited (Lu et al., 2014; Marshall and Vierstra, 2018), autophagic degradation of proteins tagged with GFP (or relatives such as YFP) invariably results in loss of the tagged protein concomitant with the appearance of free GFP, due to its relative stability inside vacuoles. This assay confirmed that both PUX7 and CDC48 are autophagy substrates, with accelerated release of free GFP/YFP from the GFP-PUX7 and YFP-CDC48a reporters being seen after starving WT seedlings for nitrogen, but not with similarly starved atg7-2, atg10-1 and atg13a-2 atg13b-2 seedlings missing key autophagy components (Figures 6A and 6B). Released GFP from GFP-PUX7 was seen even in well-fed WT seedlings, implying that PUX7 is continually turned over by autophagy (Figure 6A).
Figure 6. The Arabidopsis ATG8-interacting PUX Proteins are Autophagy Receptors for Dysfunctional CDC48 Complexes.

(A and B) PUX7 and CDC48a are degraded via autophagy upon nitrogen starvation. WT or autophagy-defective seedlings were starved for nitrogen for 16 hours, and release of free GFP or YFP from the GFP-PUX7 (panel (A)) or YFP-CDC48a (panel (B)) reporters was monitored by immunoblot analysis of total protein extracts with anti-GFP antibodies. Open and closed arrowheads locate fused and free GFP/YFP, respectively. Immunodetection of histone H3 was used to confirm near-equal protein loading.
(C and D) PUX7 and CDC48a are degraded via autophagy upon treatment with CB-5083. Seedlings were treated with or without 20 μM CB-5083 for 16 hours and GFP or YFP release was monitored as in panel (A).
(E and F) Inhibitor-induced autophagy of CDC48 requires PUX7, 8, 9 or 13. WT, atg7-2 or pux mutant seedlings were treated as in panel (D), and YFP release was monitored as in panel (B).
(G) Inhibited YFP-CDC48a accumulates in the vacuole in an autophagy- and PUX-dependent manner. Seedlings were treated as in panels (B) and (D), and imaged by confocal fluorescence microscopy. Scale bar, 10 μm. Nu, nucleus; Va, vacuole.
(H) CB-5083-induced vacuolar puncta containing YFP-CDC48a co-localize with the autophagic body marker mCherry-ATG8a. Seedlings were grown and analyzed as in panel (G).
(I) Homozygous pux-q plants are hypersensitive to CB-5083. WT or pux-q seedlings were grown for 10 days on GM medium containing either DMSO (control) or 2.5 μM CB-5083.
(J) Quantification of WT and pux-q seedling sensitivity to CB-5083; values represent the mean fresh weight (±SD) from three independent biological replicates.
(K and L) Mutations connected to human disease trigger PUX-dependent autophagy of Arabidopsis CDC48a. YFP-CDC48a variants were transiently expressed in protoplasts derived from WT, atg7-2 or pux-q plants, and YFP release was monitored as in panel (A).
(M) Mutations that trigger CDC48a degradation cluster within the N domain. The 3-dimensional structure of human CDC48 (p97; PDB file 5FTK) is shown, with residues identified in panel (K) highlighted in red.
(N) WT CDC48 is degraded by autophagy when associated with a mutant version. Seedlings expressing YFP-CDC48a were induced to express 6His-T7-CDC48a (either WT or DN mutant) in WT or atg7-2 backgrounds and harvested at the indicated times. YFP release was assayed as in panel (A), while immunodetection of the T7 tag confirmed transgene induction.
Beside nutritional stress, inhibitors often trigger autophagic turnover of proteins and their complexes, presumably as part of a quality control mechanism to purge non-functional species (Marshall et al., 2015; 2016). To test if inactivated CDC48 complexes become autophagy substrates, we examined whether a suite of CDC48 inhibitors designed to block the mammalian complex (CB-5083, DBeQ, ML240 and NMS-873), but not yet confirmed as effective in plants, could be exploited. As direct assays for CDC48 activity are not yet available in Arabidopsis, we indirectly monitored inhibitor efficacy by assessing the expression of the ER stress-responsive genes BIP1 and CRT1 that should be up-regulated upon inhibitor treatment if CDC48 activity and subsequent ERAD are compromised. Only CB-5083 robustly up-regulated these transcripts at micromolar concentrations (Figure S5A), while the other inhibitors were ineffective, most likely due to their insolubility in the Arabidopsis growth medium. A strong influence of CB-5083 on plant growth was also seen, with seedlings germinated on 10 μM CB-5083 barely emerging from the seed coat (Figure S5B).
Given the potency of CB-5083, we assayed whether it could stimulate autophagy of YFP-CDC48a in planta by the GFP-release assay. As shown in Figures 6C, 6D, S5C and S5D, CB-5083 treatment strongly induced the liberation of both free YFP from YFP-CDC48a and free GFP from GFP-PUX7 by a process requiring ATG7 and ATG10. CB-5083 also partially suppressed overall autophagic flux, as shown by the dampened release of free GFP from the GFP-ATG8a reporter upon nitrogen starvation following inhibitor treatment (Figures S5E and S5F). This effect was consistent with prior data from yeast and mammals showing that CDC48/p97 promotes autophagosome assembly (Krick et al., 2010).
Assuming that PUX7, PUX8, PUX9 and/or PUX13 are receptors for the autophagic clearance of inactive CDC48, we tested this genetically using a quadruple mutant missing all four isoforms. Likely null alleles for each locus that prevented accumulation of the full-length transcripts were identified in the T-DNA insertion collections (Figures S5G and S5H). Single homozygous pux7-2, pux8-4, pux9-4 and pux13-3 mutants, along with higher-order combinations, were fertile and phenotypically indistinguishable from their WT parents under well-fed conditions (Figure S5I). The effects of the mutants on turnover of inactive CDC48 were then tested by the GFP-release assay, using a quadruple mutant line lacking all four ATG8-interacting PUX proteins (designated pux-q) also expressing the YFP-CDC48a reporter. While the pux-q background had no effect on autophagic turnover of YFP-CDC48a upon nitrogen starvation, it effectively blocked this turnover when CDC48 was inhibited by CB-5083 (Figures 6E and 6F). None of the pux triple mutant combinations were as effective as pux-q in blocking YFP-CDC48a degradation, implying that all four isoforms work redundantly in clearing inactive CDC48 (Figure 6E). As one might predict, this failure to remove CDC48 made Arabidopsis hypersensitive to CB-5083, with pux-q seedling growth substantially repressed by increasing concentrations of the inhibitor as compared to WT (Figures 6I and 6J).
Confirmation that YFP-CDC48a was targeted to autophagy was provided by confocal fluorescence microscopy of root cells treated with CB-5083. Strong accumulation of YFP-labeled puncta in vacuoles was evident upon CB-5083 treatment of WT cells, but not in the pux-q mutant or the atg7-2 control, with cytoplasmic foci possibly representing aggregated YFP-CDC48a instead becoming visible (Figure 6G). By contrast, vacuolar YFP-CDC48a puncta accumulated in nitrogen-starved root cells either WT or harboring the pux-q combination, consistent with the PUX proteins recruiting only inactive CDC48 (Figure 6G). Co-localization of YFP-CDC48a with the autophagic reporter mCherry-ATG8a upon CB-5083 treatment demonstrated that these vacuolar puncta were indeed autophagic bodies (Figure 6H).
In addition to exploiting inhibitors to block CDC48, we also tested a suite of compromising amino acid substitutions that have been implicated in human disease (Figure S6; Tang and Xia, 2016). In total, 37 mutations, along with a dominant negative (DN) variant impaired in ATP hydrolysis (E308Q E581Q; Marshall et al., 2008; Park et al., 2008), were introduced into Arabidopsis CDC48a, and these variants were transiently expressed as YFP fusions in leaf mesophyll protoplasts. Autophagic degradation of the mutant proteins was then monitored by the YFP-release assay. WT YFP-CDC48a and several mutant forms accumulated to high levels in the protoplasts and did not display released free YFP (Figure 6K). By contrast, versions of YFP-CDC48a containing either the DN mutations or numerous other point mutations, including P141L, G160S, E189K, K389E and A442P, poorly accumulated, with free YFP instead being strongly detected (Figure 6K), indicating that these variants had become autophagy substrates. This scenario was confirmed by analyzing the fate of the YFP-CDC48a mutants in protoplasts generated from WT, atg7-2 and pux-q leaves. In each case, the release of free YFP was robust in WT, but was not evident in the atg7-2 and pux-q backgrounds (Figure 6L).
Mapping the effective mutations onto the 3-dimensional structure of human p97 revealed that they cluster within the N-terminal domain, which is responsible for co-factor binding and structural integrity of the complex (Stach and Freemont, 2017). Consequently, these mutations likely trigger autophagy by compromising the structure of the CDC48 hexamer (Figure 6M). To test this hypothesis, we generated Arabidopsis lines that accumulate YFP-CDC48a together with either WT or DN versions of 6His-T7-CDC48a whose expression was inducible by ethanol (Park et al., 2008), thus allowing us to test whether integration of impaired CDC48 into the hexamer would induce autophagy of a functional subunit. As shown by YFP-release assays, only upon expression of the DN form did WT YFP-CDC48a become an autophagy substrate (Figure 6N). Taken together, our Arabidopsis data identified an autophagic route of potential medical relevance that removes dysfunctional CDC48 complexes through dedicated PUX receptors and the UIM-UDS interface.
Ubx5 is a UIM-type Autophagy Receptor for Cdc48 in Yeast.
Given that Ubx5 is the likely yeast ortholog of the ATG8-interacting PUX proteins, we examined whether it could target dysfunctional yeast Cdc48 for autophagy. We first assessed whether yeast Cdc48 is an autophagy substrate, using the YFP-release assay with cells expressing YFP-Cdc48 and starved for nitrogen. As shown in Figure 7A, yeast degrades Cdc48 upon nitrogen stress via an autophagic route analogous to that in Arabidopsis, requiring Atg7 and Atg13, but not Ubx5 or the alternative autophagy receptor Cue5. Confocal fluorescence microscopy then demonstrated the autophagic deposition of YFP-Cdc48 into vacuoles. Whereas cells grown in nitrogen-rich medium localized the YFP signal to the cytoplasm and nucleus, it moved to the vacuole upon nitrogen starvation via a process dependent on Atg7 but not Ubx5 (Figure 7B).
Figure 7. Ubx5 is an Autophagy Receptor for Dysfunctional Cdc48 Complexes in Yeast.

(A) Yeast Cdc48 is degraded via autophagy upon nitrogen starvation. WT or mutant yeast cells expressing YFP-CDC48 were starved for nitrogen for the indicated periods of time, and release of free YFP was monitored as in Figure 6A.
(B) Cdc48 accumulates in yeast vacuoles in an autophagy-dependent manner upon nitrogen starvation. Cells were treated as in panel (A) for 24 hours and imaged by confocal fluorescence microscopy. Scale bar, 2 μm. Nu, nucleus; Va, vacuole.
(C) Mutant forms of Cdc48 are degraded in an autophagy- and Ubx5-dependent manner. Cells expressing YFP-CDC48 variants in the indicated backgrounds were grown in medium containing galactose for 16 hours, and YFP release was monitored as in panel (A).
(D) Autophagic degradation of Cdc48 mutants requires the UIM and UBX domain of Ubx5. Cells were grown as in panel (C), and YFP release was monitored as in panel (A).
(E) Mutant Cdc48 accumulates in yeast vacuoles in an autophagy- and Ubx5-dependent manner. Cells were grown as in panel (C) and imaged as in panel (B). Ag, aggregate.
(F, G and H) Yeast cell growth is hypersensitive to expression of Cdc48 mutants in the absence of Ubx5. Cells were switched to medium containing galactose, and growth was monitored by measuring culture density at OD600 (panels (F) and (G)), or by spotting 5-fold serial dilutions onto synthetic dropout medium and incubating at 30°C for 36 hours (panel (H)).
(I and J) Yeast cells unable to degrade Cdc48 are hypersensitive to ER stress. Cells were grown as in panel (F) in the presence or absence of 1.5 mM DTT or 200 ng/ml tunicamycin (Tm), and growth monitored as in panels (G) and (H), with OD600 values in the presence of each chemical normalized to the value in its absence.
In panels (G) and (J), letters indicate values that are significantly different from one another and the control (p < 0.05).
To examine whether non-functional Cdc48 is similarly degraded by autophagy, we attempted to find conditions that would inactivate this ATPase. None of the CDC48/p97 inhibitors tested above (CB-5083, DBeQ, ML240 and NMS-873) appeared effective in yeast, based on their failure to activate ER stress-responsive genes (DER1 and ERO1), inhibit cell growth, or even impact autophagic flux as assayed with the engineered Pho8Δ60 reporter assay that quantifies autophagic transport (Noda et al., 1995), even when applied to the Δerg6 or Δpdr5 backgrounds that might facilitate drug uptake (Figures S7A, S7B, S7C and S7D).
As an alternative, we tested mutations that might trigger autophagic turnover of Cdc48 like that seen in Arabidopsis. Because Cdc48 is necessary for autophagosome assembly in yeast (Krick et al., 2010), the mutations were introduced into a YFP-CDC48 reporter under the control of the galactose-inducible GAL1 promoter in cells retaining their functional copy of CDC48. In this way, expression of the mutant YFP-Cdc48 variants would only mildly impair bulk autophagic flux, in contrast to the dramatic reduction seen when the mutants replaced the genomic locus (Figures S7E and S7F). We first tested YFP-Cdc48 harboring the temperature-sensitive cdc48-3 and cdc48-6 alleles, which contain P257L R387K and P257L A540T mutations, respectively (Ruggiano et al., 2016). However, even prolonged incubation of YFP-cdc48-3 and YFP-cdc48-6 cells for 48 hours at 37°C did not yield high leve ls of free YFP, indicating that these thermosensitive variants were only weakly targeted to autophagy (Figure S7G).
Finally, we tested three mutations shown to trigger robust autophagic turnover of Arabidopsis CDC48a (Figure 6K; P147L, E195K and K396E in yeast). While their expression only minimally impacted autophagic flux (Figure S7H), each strongly induced autophagic breakdown of YFP-Cdc48 based on the YFP-release assay, via a process requiring Ubx5 and the core autophagy components Atg7 and Atg8, but not Cue5 (Figures 7C and S7I). To confirm that Ubx5 was required and acted through its UIM, we introduced HA-tagged WT Ubx5, or mutants eliminating the UBA, UIM or UBX domains, into Δubx5 cells expressing the YFP-Cdc48 mutants. Whereas free YFP was released in WT cells and in Δubx5 mutants complemented with full-length Ubx5, it was absent when Ubx5 variants lacking the UIM or UBX domain were used instead (Figure 7D).
The UIM of Ubx5 was expected to bind Atg8 at the UDS and, as confirmation, we found that free YFP was released from the YFP-Cdc48 mutants in Δatg8 cells complemented with HA-tagged WT Atg8, but not when the ΔUDS variant was expressed instead (Figure S7I). Given the remote possibility that the ΔUDS mutant slowed YFP-Cdc48 turnover by discouraging Atg8 lipidation, we assayed for this adduct by subjecting total protein extracts from nitrogen-starved cells to SDS-PAGE in the presence of 6 M urea, which permits separation of the free and lipidated forms of Atg8 (Krick et al., 2010), with the identity of Atg8-PE then confirmed by its loss upon treatment of the extracts with phospholipase D (Figure S7J). However, as shown in Figures S7J and S7K, lipidated Atg8 accumulated to near normal levels in cells expressing the ΔUDS mutant.
The autophagic transport of the P147L, E195K and K396E variants of YFP-Cdc48 was additionally confirmed by confocal fluorescence microscopy; non-mutated YFP-Cdc48 remained in the cytoplasm and nucleus, while the YFP-Cdc48 mutants mainly localized to the vacuole in WT cells (Figure 7E). As expected for an autophagy target requiring Ubx5, this vacuolar movement was blocked in Δatg7 and Δubx5 cells, with these cells instead accumulating the YFP-Cdc48 signal in cytoplasmic puncta (Figure 7E). These puncta resembled the foci seen for YFP-CDC48a in Arabidopsis, and with other autophagic targets (Marshall et al., 2016; Gatica et al., 2018), suggesting that they represent mis-folded Cdc48 aggregates awaiting autophagy, although their degradation was independent of the aggrephagy receptor Cue5 (Figure 7C; Lu et al., 2014).
We predicted that this failure to degrade inactive Cdc48 would have physiological consequences in yeast, as it does in Arabidopsis. To test this notion, we compared growth rates of WT and Δubx5 strains expressing either unaltered YFP-Cdc48 or the three mutant forms prone to autophagy. Whereas the Δubx5 mutation did not compromise growth of cells expressing WT YFP-Cdc48, growth was substantially slowed when the mutant variants were expressed (Figures 7F, 7G, and 7H). Each mutant was modestly toxic to WT cells, which could reflect their ability to interfere with the Cdc48 hexamer, but became strongly toxic when the nonfunctional complexes could no longer be cleared by Ubx5-mediated autophagy.
Reasoning that the stabilization of non-functional Cdc48 in Δubx5 cells should compromise ERAD and protein homeostasis, we examined the sensitivity of these cells to the ER stress agents DTT and tunicamycin. While growth of WT cells was modestly impaired by both treatments, the Δubx5 cells were more sensitive (Figures 7I and 7J). Although this growth suppression was alleviated by complementation with HA-tagged Ubx5, it was not alleviated with the ΔUIM or ΔUBX versions (Figures 7I and 7J). These results are consistent with Ubx5 helping direct autophagic degradation of Cdc48 complexes that become non-functional during ER stress, using its UBX domain to bind Cdc48 and its UIM to bind the UDS of Atg8.
DISCUSSION
As our understanding of autophagy has progressed, one focus has been to define the array of ATG8-binding proteins that help generate and transport autophagic vesicles and recruit appropriate substrates. While these adaptors/receptors were thought to be limited to the collection of AIM/LIR-containing proteins that bind the LDS on ATG8 (Noda et al., 2008; 2010), we discovered that a second, conserved ATG8-binding mechanism exists that uses a UIM-UDS interface, thus greatly expanding the number of components involved in autophagy. As an example, our studies on the Arabidopsis and yeast PUX/Ubx5 family showed that the UIM-UDS contact is critical for clearing non-functional CDC48 complexes. As CDC48/p97 is essential for ERAD and other protein quality control pathways relevant to several human pathologies, this turnover has strong medical implications (Ye et al., 2001; Tang and Xia, 2016).
The UIM-binding UDS sequence within ATG8 is highly conserved, with only a few differences among species. While our saturating mutagenesis approach identified a clear Ψ-F-Ψ-Ω/T consensus motif for residues permitted within the UDS, it should be noted that, like the LDS, adjacent residues within the tertiary structure of ATG8 also likely contribute. By contrast, a clear description of the UIM subtypes that bind ATG8 as opposed to other ubiquitin-fold proteins remains elusive. In fact, we emphasize that many currently annotated UIM-containing proteins do not bind ATG8, at least by Y2H (10 of 14, 4 of 8, and 22 of 28 tested from Arabidopsis, yeast and humans, respectively), whereas some without a predicted UIM did, demonstrating that the presence of a UIM does not a priori confer ATG8 binding and should be confirmed empirically. While our ATG8 binding assays generated a list of permissible residues within the UIM, the limited number of known ATG8-binding UIMs across species (17 to date) prevented us developing a robust consensus sequence beyond the internal Ψ-ζ-X-A-Ψ-X-X-S motif. Clearly, 3-dimensional structures of one or more UIM-UDS interfaces are now needed to fully resolve this interaction.
Our combined ATG8 interactomes identified 204 ATG8-binding proteins in plants and yeast, including over 40 proteins that utilize the UDS. While some were described previously, a majority had no prior connections to autophagy, and likely represent new autophagy adaptors, receptors, or even cargo that bind ATG8 directly. Our identification of the four Arabidopsis PUX proteins and yeast Ubx5 as bona fide autophagy receptors expands the reach of selective autophagy in these organisms. The PUX proteins join ATI1/2, DSK2, NBR1/Joka2, ORM1/2, RPN10 and TSPO as the only autophagic cargo receptors thus far described in plants (Nolan et al., 2017; Marshall and Vierstra, 2018; Yang et al., 2019).
Many proteins regulating vesicle dynamics were also found to bind ATG8, particularly in yeast; these likely modulate autophagosome expansion, closure, and trafficking to the vacuole. Examples include eight SNAREs, three Rab GTPases, three p24 complex components, four clathrin adaptors, and the UDS-binding Epsins Ent1 and Ent2 (plus their human counterparts EPN1-3), suggesting an intimate cross-talk between the autophagic and vesicular trafficking machineries mediated directly by Atg8. Surprisingly, several ATG8 interactors also influence ubiquitylation, including the human ATXN3 and ATXN3L deubiquitylases, the Ufo1 F-Box component of a yeast SCF ubiquitin ligase, and two potentially orthologous ubiquitin conjugating enzymes, Arabidopsis UBC19 and yeast Ubc1, further connecting ubiquitin to autophagy (Khaminets et al., 2016). It should also be noted that several ATG8-interacting proteins required neither the LDS or UDS for binding, implying that additional interactions interfaces exist between ATG8 and its partners.
CDC48/p97 is an essential, highly abundant segregase that uses the energy from ATP hydrolysis to mechanically extract proteins from membranes or multi-subunit complexes (Stach and Freemont, 2017). As such, it plays crucial roles in, among others, ERAD, ribosome-mediated quality control, chromatin re-modeling, extraction of membrane-bound transcription factors, capping of 20S proteasomes, and autophagic clearance of stress granules and ruptured lysosomes (Ye et al., 2001; Marshall et al., 2008; Park et al., 2008; Krick et al., 2012; den Besten et al., 2012; Stach and Freemont, 2017). Regulating the activity and abundance of CDC48/p97 is therefore likely critical for maintaining proteostasis. While little is currently known about post-transcriptional mechanisms that might control CDC48/p97, our results with the PUX proteins in Arabidopsis and Ubx5 in yeast place autophagy as a crucial regulator. In fact, Arabidopsis pux-q seedlings unable to degrade CDC48 are hypersensitive to the CB-5803 inhibitor, while yeast cells missing Ubx5 or expressing only the ΔUDS variant of Atg8 have strongly impaired growth and are hypersensitive to ER stress.
Two autophagic routes were identified for CDC48 turnover, promoted by either nitrogen starvation or inactivation. The former requires signaling from the ATG1 kinase complex and is independent of the PUX/Ubx proteins, while the latter is independent of ATG1 but dependent on these receptors. Thus, as with proteaphagy (Marshall et al., 2015; 2016), cells have the capacity to separately adjust CDC48 degradation in response to nutritional cues or upon recognition of non-functional complexes. Whether there is a selective receptor for the former route, or whether ubiquitylation acts a signal for the latter, remains unknown.
Importantly, mutations in CDC48/p97 have been implicated in several human diseases, including inclusion body myopathy with early-onset Paget’s disease of bone and frontotemporal dementia (IBMPFD), familial amyotrophic lateral sclerosis (FALS), and Charcot-Marie-Tooth disease (Tang and Xia, 2016). We found here that numerous mutations associated with these pathologies trigger autophagic clearance of CDC48 in both Arabidopsis and yeast (e.g., P141L, G160S, E189K, K389E and A442P), implying that this recycling influences disease onset and/or severity. Here, two scenarios are possible: either accelerated degradation of functional CDC48/p97 in complex with mutant forms triggers disease symptoms due to CDC48 insufficiency, or the known age-associated decline in autophagic capacity (Levine and Kroemer, 2019) allows the cytotoxic accumulation of impaired complexes. Our observation that a dominant-negative mutant of CDC48 stimulates the degradation of presumably functional subunits implies that hexameric complexes are degraded in toto, even though some subunits might retain normal activity. Given that the PUX proteins and Ubx5 bind functional CDC48 (Rancour et al., 2004; Gallois et al., 2013), a plausible model is that inhibition or mutation of CDC48 causes a conformational change that increases the accessibility and/or affinity of bound PUX/Ubx5 for ATG8. Taken together, novel curative strategies for CDC48/p97-associated disorders might be possible by manipulating its autophagic clearance, possibly involving the human orthologs of PUX/Ubx5.
In conclusion, we discovered a novel UIM-UDS interface within the autophagy system that is exploited by a large, heretofore unknown collection of adaptors and receptors to bind ATG8. This discovery should greatly expand the complexity of the autophagy system and the pathways involved in selective cargo recruitment beyond the well-described AIM-LDS interface (Noda et al., 2008; 2010). Additionally, the discovery that CDC48/p97 is targeted to autophagy by a collection of UIM- and UBX domain-containing proteins that recruit non-functional complexes to ATG8-decorated autophagic vesicles demonstrates the importance of the UIM-UDS interface for multiple facets of eukaryotic proteostasis and human health.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Richard D. Vierstra (rdvierstra@wustl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Plant Materials and Growth Conditions
All Arabidopsis thaliana plants were the Columbia-0 (Col-0) ecotype, except for the SAIL T-DNA insertion lines, which were the Columbia-3 (Col-3) ecotype, and the FLAG T-DNA insertion lines and the 35S::GFP and 35S::GFP-PUX7 lines, which were the Wassilewskija (Ws) ecotype. Details of all T-DNA insertion mutants and stable transgenic lines are provided in Table S4. Sterilized seeds (obtained via vapor-phase or liquid-phase sterilization) were vernalized at 4°C for 3 to 4 days and germinated on solid GM medium (3.2 g/l Gamborg’s B5 basal medium with minimal organics, 1% (w/v) sucrose, 0.05% (w/v) 2-(N-morpholino)ethanesulfonic acid (MES; pH 5.7), 0.7% (w/v) agar) at 21 to 23°C under a long-day photoperiod (16 hours light/8 hours darkness) with a light intensity of 75 to 100 μmol/m2/sec and a relative humidity of 40 to 50%.
For selection and maintenance of transgenic plants, agar plates were supplemented with appropriate combinations of antibiotics or herbicide (50 μg/ml kanamycin, 25 μg/ml hygromycin B, 7.5 μg/ml sulphadiazine or 10 μg/ml BASTA). For growth assays, plates were supplemented with the indicated concentrations of CB-5083 (1-(4-(benzylamino)-7,8-dihydro-5H-pyrano-(4,3-d)-pyrimidin-2-yl)-2-methyl-1H-indole-4-carboxamide), DBeQ (N2,N4-dibenzylquinazoline-2,4-diamine), ML240 ((2-(2-aminobenzimidazol-1-yl)-N-benzyl-8-methoxyquinazolin-4-amine) or NMS-873 (3-(3-(cyclopentylsulphanyl)-5-((3-methyl-4-(4-methylsulfonylphenyl)-phenoxy)-methyl)-1,2,4-triazol-4-yl)-pyridine) and grown horizontally or vertically for 10 or 7 days, respectively. When required, seedlings were transferred to soil (mixed in a 1:1 ratio with organic Coco Coir planting mixture, supplemented before use with 2 g/l Peters 20-20-20 fertilizer, 80 mg/l Ca(NO3)2 and 80 mg/l MgSO4) after 2 to 3 weeks, and again grown at 21 to 23°C under a long-day photoperiod. Plants for protoplast preparation were grown as above, except under a short-day photoperiod (8 hours light/16 hours darkness).
For chemical treatments, seedlings were grown in either 2 ml or 50 ml cultures of liquid GM medium (as above, but without agar) at 21 to 23°C under continuous light with gentle shaking (90 rpm). On the sixth day, fresh medium was supplemented with the indicated concentrations of CB-5083, DBeQ, ML240, NMS-873 and/or MG132 ((N-benzyloxycarbonyl)-leucinyl-leucinyl-leucinal) for the indicated times, with equivalent volumes of DMSO added to the controls. For nitrogen starvation, seedlings were grown in liquid MS medium (4.4 g/l Murashige and Skoog basal medium, 1% (w/v) sucrose, 0.05% (w/v) MES (pH 5.7)) before transfer to fresh MS medium or MS medium lacking nitrogen (Murashige and Skoog basal salt micronutrient solution supplemented with 3 mM CaCl2, 1.5 mM MgSO4, 1.25 mM KH2PO4, 5 mM KCl, 1% (w/v) sucrose, 0.05% (w/v) MES (pH 5.7)) for the indicated times. Both control and treated seedlings were washed three times in fresh medium prior to treatment. For induction of 6His-T7-CDC48a variants (Park et al., 2008), fresh medium was supplemented with 2% ethanol, again for the indicated times. Following all treatments, tissue was harvested, immediately frozen in liquid nitrogen, and stored at −80°C before analysis.
Tobacco (Nicotiana benthamiana) plants for Agrobacterium tumefaciens-mediated leaf infiltration were sown directly onto soil (pre-treated as described above), vernalized at 4°C for 4 to 5 days, and grown at 21 to 23°C under a 12 hour light/12 hour darkness photoperiod, with a light intensity of 75 to 100 μmol/m2/sec and relative humidity of 60 to 75%. Plants were refertilized once a week.
Yeast Strains and Manipulations
Unless otherwise stated, all yeast (Saccharomyces cerevisiae) manipulations were performed according to standard protocols (as described in Marshall et al., 2016). Details of all strains used in this study are given in Table S5. Strains BY4741 and BY4742 were used as WT controls. Deletion strains (Δatg7, Δatg13, Δcue5, Δhac1, Δpdr5 and Δubx5) were obtained from the yeast knockout collection (Dharmacon) and cultured on YPDA medium containing 200 μg/ml Geneticin®, except for the Δerg6 deletion, which was grown on YPDA medium containing 200 μg/ml hygromycin B (Marshall et al., 2016). All genomic deletions were confirmed by PCR genotyping (using primer pairs A + B, A + KanB, C + D, KanC + D, and A + D for each deletion collection strain; details of all oligonucleotide primers are provided in Table S6).
Yeast strains expressing YFP-CDC48 from the genomic CDC48 locus in WT or deletion backgrounds were generated by standard PCR-based homologous recombination, with the coding sequence for EYFP (amplified from pAG424-GAL-EYFP-ccdB (Addgene, product number 14343)) integrated following the initiator methionine. Strains in which the endogenous ATG8 locus was replaced by ATG8ΔLDS or ATG8ΔUDS variants were generated in the same way. Correct integration was confirmed by sequencing of the genomic region and, where appropriate, confocal fluorescence microscopy (see below). Plasmid-based expression of YFP-CDC48, HA-ATG8 or HA-UBX5 variants was driven from the pAG423-GAL-EYFP-ccdB or pAG425-GPD-ccdB vectors (Addgene, product numbers 14341 and 14154, respectively; see below for cloning details). The sequence for the HA-tag on the N-terminus of ATG8 or UBX5 was incorporated into the appropriate PCR amplification primer. Transformation with the indicated plasmids was performed using the standard lithium acetate procedure, and transformed cells were cultured on synthetic dropout medium lacking leucine or histidine as required.
For time course experiments, 15 ml liquid cultures in YPDA medium were grown overnight at 30°C with vigorous shaking, diluted to an OD600 of 0.1 in 15 ml, then grown for an additional 2 to 3 hours until an OD600 of approximately 0.5 was reached. To initiate nitrogen starvation, cells were harvested by centrifugation at 1,000 x g for 2 minutes, washed twice in sterile distilled H2O, and re-suspended in synthetic dropout medium lacking nitrogen (0.17% yeast nitrogen base without amino acids and ammonium sulfate, 2% glucose), followed by continued incubation at 30°C. For expression of YFP-CDC48 variants from the GAL1 promoter, cells were harvested and washed as above then re-suspended in YPDA medium containing 2% galactose instead of 2% glucose, followed by continued incubation at 30°C. Cell aliquots corresponding to 1.5 OD units were collected at the indicated times, pelleted by centrifugation at 5,000 x g for 1 minute, washed once in sterile distilled H2O, pelleted again, and immediately frozen in liquid nitrogen. For treatment with CB-5083, DBeQ, ML240 or NMS-873, cultures were grown and diluted as above before the inhibitor was added to the indicated concentration (with equivalent volumes of DMSO for the controls).
METHOD DETAILS
Gene Cloning and Site-directed Mutagenesis
To clone required coding sequences, total RNA was first extracted from Arabidopsis seedlings or yeast cells and then converted into cDNA. Arabidopsis RNA was extracted from 50 to 100 mg of 7-day-old liquid grown seedlings using the RNeasy® plant mini kit (QIAGEN). To obtain total yeast RNA, the cell walls of 2 x 107 freshly harvested BY4741 cells (taken from a 15 ml culture grown in YPDA) were digested with 100 U of lyticase (from Arthrobacter luteus) in 100 μl buffer Y1 (1 M sorbitol, 100 mM EDTA, 0.1% (v/v) β-mercaptoethanol (pH 7.4)) for 1 hour at 30°C, and RNA was then extracted using the RNeasy® mini kit (QIAGEN). For human RNA, a commercially available batch of total HeLa cell RNA (Clontech, product number 636543) was used. In all cases, the RNA concentration was determined using a NanoDrop™ 1000 spectrophotometer (Thermo Fisher Scientific), 1 μg of total RNA was treated with DNase I (Invitrogen), and RNA integrity was assessed via a combination of OD260/OD280 and OD260/OD230 measurements, and by analyzing samples on a denaturing formaldehyde agarose gel. RNA was converted into cDNA using the Superscript® III first-strand synthesis system (Invitrogen) and oligo(dT)20 primers.
For yeast two-hybrid, bimolecular fluorescence complementation, and recombinant protein expression in E. coli or yeast, coding sequences amplified from cDNA by PCR were first recombined into pDONR™221 via the Gateway® BP clonase™ II reaction (Invitrogen). When required, site-directed mutagenesis was performed at this step using the QuikChange™ II site-directed mutagenesis kit (Agilent Technologies). Mutation of individual or small groups of adjacent residues was performed using single primer pairs, while mutations of larger groups of residues were introduced in two stages. For large-scale saturating mutagenesis, degenerate primers were used in which the three bases of the relevant codon were permitted to be any of A, C, G or T. Details of all ΔLDS, ΔUDS and ΔUIM mutations are given in Table S1. Sequence-confirmed genes were recombined in-frame with appropriate Gateway®-compatible destination vectors via the Gateway® LR clonase™ II reaction (Invitrogen; see relevant sections above and below). To prepare plasmids for cDNA library screening or transient expression in Arabidopsis mesophyll protoplasts, standard restriction enzyme-based cloning was used (see relevant sections below).
Immunological Techniques
Frozen Arabidopsis tissue samples were homogenized in 3 volumes of protein extraction buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 2 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), 50 µM MG132, 1X plant protease inhibitor cocktail) and clarified by centrifugation at 16,000 x g for 5 minutes at 4°C. The supernatant was then mixed with 0.25 volumes of 5X SDS-PAGE sample buffer (200 mM Tris-HCl (pH 6.8), 25% (v/v) glycerol, 10% (w/v) SDS, 10% (v/v) β-mercaptoethanol, 0.005% (w/v) bromophenol blue). Total protein extracts from yeast were usually obtained by re-suspending harvested cells in 500 μl of lysis buffer (0.2 N NaOH, 1% β-mercaptoethanol), followed by precipitation of proteins with 50 μl of 50% trichloroacetic acid. Proteins were collected by centrifugation at 16,000 x g for 5 minutes at 4°0, washed once with 1 ml of ice-cold acetone, and re-suspended in 150 μl 2X SDS-PAGE sample buffer (80 mM Tris-HCl (pH 6.8), 10% (v/v) glycerol, 4% (w/v) SDS, 4% (v/v) β-mercaptoethanol, 0.002% (w/v) bromophenol blue).
For analysis of Atg8 lipidation, yeast cell walls were first digested with 200 U of lyticase in 100 μl 1X PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4). Resulting spheroplasts were harvested by centrifugation at 100 x g for 5 minutes at 4°0, then lysed by resuspension in yeast homogenization buffer (50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM PMSF, 10 mM iodoacetamide, 1% (v/v) Triton X-100) followed by vigorous mixing with a vortex. Extracts were clarified by centrifugation at 16,000 x g for 5 minutes at 4°C, and the supernatant was incubated for 1 hour at 37°C in the presence or absence of 250 U phospholipase D (Enzo Life Sciences), then mixed with 0.25 volumes of 5X SDS-PAGE sample buffer. All protein samples were heated at 95°C for 5 minutes and then subjected to SDS-PAGE analysis, with 10 to 20 μl of each sample being run on linear gels containing between 10 and 14% acrylamide, as appropriate. To enable separation of Atg8 and Atg8-PE, gels were supplemented with 6 M urea.
For direct protein visualization, gels were subjected to staining with Coomassie Brilliant Blue R-250 or silver nitrate, exactly as previously described (Marshall et al., 2015; 2016). For immunoblot analyses, proteins separated by SDS-PAGE were electrophoretically transferred onto Immobilon®-P polyvinylidene difluoride (PVDF) membrane (Millipore) for 16 hours at 80 mA, and the membrane was blocked for at least 60 minutes with a 10% (w/v) non-fat dry milk solution in 1X PBS, which was first filtered through two layers of Miracloth. All incubations were performed at room temperature. The membrane was incubated with primary antibody solution (in 1% (w/v) non-fat dry milk solution in PBS) for 60 minutes, before being washed once with PBS, once with PBST (PBS containing 0.1% Triton X-100), and once with PBS for 10 minutes each. The membrane was re-blocked with 10% (w/v) non-fat dry milk solution in PBS for 30 minutes, incubated for 60 minutes with secondary antibody solution (in 1% (w/v) non-fat dry milk solution in PBS), then washed again as above. Details of all primary and secondary antibodies are given in the Key Resources Table. Blots were developed using the SuperSignal® West Pico Plus Chemiluminescent Substrate or the SuperSignal® West Femto Maximum Sensitivity Substrate (both from Thermo Fisher Scientific). Densitometric quantifications were performed using TotalLab™ software (Non-linear Dynamics), with at least three different exposures of the same blot being quantified to ensure that the exposure level was within the linear range of the film.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chicken polyclonal anti-CDC48a | Rancour et al., 2004 | N/A |
| Mouse monoclonal anti-GFP | Sigma-Aldrich | Cat# 11814460001; RRID: AB_390913 |
| Mouse monoclonal anti-GST | Santa Cruz Biotechnology | Cat# SC138; RRID: AB_627677 |
| Mouse monoclonal anti-HA | Covance | Cat# MMS-101R; RRID: AB_2314672 |
| Mouse monoclonal anti-6His | Covance | Cat# MMS-156P; RRID: AB_2565061 |
| Rabbit polyclonal anti-ATG8a | Thompson et al., 2005 | N/A |
| Rabbit polyclonal anti-Gal4 BD | Sigma-Aldrich | Cat# G3042; RRID: 439688 |
| Rabbit polyclonal anti-H3 | Abcam | Cat# AB1791; RRID: AB_302613 |
| Goat anti-mouse HRP conjugate | Seracare | Cat# 074-1806; RRID: AB_2307348 |
| Goat anti-rabbit HRP conjugate | Seracare | Cat# 074-1506; RRID: AB_2721169 |
| Rabbit anti-chicken IgY HRP conjugate | Rancour et al., 2004 | N/A |
| Bacterial and Virus Strains | ||
| Agrobacterium tumefaciens strain GV3101 | Lab stock | N/A |
| Escherichia coli strain BL21(DE3) pLysS | Promega | Cat# L1195 |
| Escherichia coli strain BNN132 | Elledge et al., 1991 | N/A |
| Escherichia coli strain DH5α | Lab stock | N/A |
| Biological Samples | ||
| Human HeLa cell total RNA | Clontech | Cat# 636543 |
| Arabidopsis thaliana universal normalized cDNA library | Clontech | Cat# 630487 |
| Saccharomyces cerevisiae cDNA library | Elledge et al., 1991 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Acetosyringone | Sigma-Aldrich | Cat# D134406 |
| 3-amino-1,2,4-triazole | Sigma-Aldrich | Cat# A8056 |
| Bovine serum albumin | Affymetrix | Cat# AAJ10857 |
| CB-5083 | Selleckchem | Cat# S8101 |
| Cellulase “Onozuka” R-10 | Yakult Pharmaceutical Industries | N/A |
| 2-chloroacetamide | Sigma-Aldrich | Cat# C0267 |
| Concanavalin A | Sigma-Aldrich | Cat# C2010 |
| DBeQ | Selleckchem | Cat# S7199 |
| Dithiothreitol | Sigma-Aldrich | Cat# D9779 |
| DNase I, RNase-free | Thermo Fisher Scientific | Cat# EN0521 |
| FastDigest™ restriction enzymes (BamHI; BglII; EcoRV; NcoI; SalI; SfiI; XbaI; XhoI) | Thermo Fisher Scientific | Cat# FD0054; FD0084; FD0304; FD0574; FD0644; FD1824; FD0684; FD0694 |
| Influenza haemagglutinin (HA) peptide | Sigma-Aldrich | Cat# I2149 |
| Isopropyl-β-D-thiogalactopyranoside | Research Products International | Cat# I56000 |
| Klenow fragment | Thermo Fisher Scientific | Cat# EP0051 |
| Lyticase from Arthrobacter luteus | Sigma-Aldrich | Cat# L4025 |
| Macerozyme R-10 | Yakult Pharmaceutical Industries | N/A |
| MG132 | Selleckchem | Cat# S2619 |
| ML240 | Sigma-Aldrich | Cat# SML1071 |
| NMS-873 | Selleckchem | Cat# S7285 |
| Phenylmethylsulphonyl fluoride | Sigma-Aldrich | Cat# P7626 |
| Phospholipase D from Streptomyces chromofuscus | Enzo Life Sciences | Cat# BML-SE301-0025 |
| Polyethylene glycol 4000 | Sigma-Aldrich | Cat# 81240 |
| p-nitrophenol | Sigma-Aldrich | Cat# 1048 |
| p-nitrophenyl phosphate disodium salt hexahydrate | Sigma-Aldrich | Cat# N4645 |
| Sequencing grade modified porcine trypsin | Promega | Cat# V5111 |
| Silver nitrate | Sigma-Aldrich | Cat# S8157 |
| Sodium thiosulphate pentahydrate | Sigma-Aldrich | Cat# 217247 |
| Trifluoroacetic acid | Sigma-Aldrich | Cat# 302031 |
| Tunicamycin | Sigma-Aldrich | Cat# T7765 |
| Urea | Thermo Fisher Scientific | Cat# BP169-212 |
| Critical Commercial Assays | ||
| LightCycler® 480 SYBR Green I master mix | Roche Diagnostics | Cat# 04707516001 |
| Pierce™ BCA protein assay kit | Thermo Fisher Scientific | Cat# 23225 |
| ProQuest™ two-hybrid system | Thermo Fisher Scientific | Cat# PQ1000101 |
| QIAprep® spin miniprep kit | QIAGEN | Cat# 27106 |
| QuikChange™ II site-directed mutagenesis kit | Agilent Technologies | Cat# 200524 |
| RNeasy® mini kit | QIAGEN | Cat# 74106 |
| RNeasy® plant mini kit | QIAGEN | Cat# 74904 |
| SuperScript® III first strand synthesis system | Thermo Fisher Scientific | Cat# 18080051 |
| SuperSignal® West Femto maximum sensitivity substrate | Thermo Fisher Scientific | Cat# 34096 |
| SuperSignal® West Pico Plus chemiluminescent substrate | Thermo Fisher Scientific | Cat# 34578 |
| Zymoprep™-96 yeast plasmid miniprep kit | Zymo Research | Cat# D2006 |
| Deposited Data | ||
| Mass spectrometric analysis of GFP-PUX7 immunoprecipitations | ProteomeXchange consortium | PXD011397 |
| Experimental Models: Organisms/Strains | ||
| Arabidopsis thaliana wild type ecotypes (Col-0; Col-3; Ws) | Arabidopsis Biological Resource Center | Cat# CS60000; CS908; CS1602 |
| Arabidopsis thaliana transgenic plant lines | Thompson et al., 2005; Park et al., 2008; Suttangkakul et al., 2011; Gallois et al., 2013 | See Table S4 for details |
| Arabidopsis thaliana T-DNA insertion mutants | Arabidopsis Biological Resource Center; Versailles Arabidopsis Stock Center; GABI-Kat collection | See Table S4 for details |
| Nicotiana benthamiana plants | Michael Dyer (Washington University in St. Louis) | N/A |
| Saccharomyces cerevisiae wild type strains (BY4741; BY4742; DF5; SEY6210; W303-1B) | Daniel Finley (Harvard Medical School); Audrey P. Gasch (University of Wisconsin); Mark Hochstrasser (Yale University) | See Table S5 for details |
| Saccharomyces cerevisiae gene knockout collection strains (multiple) | Dharmacon | See Table S5 for details |
| Saccharomyces cerevisiae strains cdc48-3; cdc48-6 | Pedro Carvalho (Centre de Regulació Genòmica) | See Table S5 for details |
| Saccharomyces cerevisiae strain PHO8Δ60 | Noda et al., 1995 | See Table S5 for details |
| Saccharomyces cerevisiae strain YFP-CDC48 | This study | See Table S5 for details |
| Oligonucleotides | ||
| Oligonucleotide primers | Integrated DNA Technologies | See Table S6 for details |
| Recombinant DNA | ||
| pBlueScript II SK(+) | Agilent Technologies | Cat# 212205 |
| pDHA | Marshall et al., 2008 | N/A |
| pDONR™221 | Thermo Fisher Scientific | Cat# 12536017 |
| pET17-b | EMD Millipore | Cat# 69663 |
| pGEX-4T-1 | GE Healthcare | Cat# 28-9545-49 |
| pGBKT7 | Clontech | Cat# 630443 |
| pSITE-N-EYFP-C1; pSITE-C-EYFP-C1 | Arabidopsis Biological Resource Center | Cat# CD3-1648; CD3-1649 |
| Escherichia coli Gateway® destination vectors (pDEST™14; pDEST™15; pDEST™17) | Thermo Fisher Scientific | Cat# 11801016; 11802014; 11803012; |
| Saccharomyces cerevisiae Gateway® destination vectors (pAG423-GAL-EYFP-ccdB; pAG424-GAL-EYFP-ccdB; pAG425-GPD-ccdB) | Addgene | Cat# 14341; 14343; 14154 |
| Software and Algorithms | ||
| Adobe Illustrator CC; Adobe Photoshop CC | Adobe Systems | N/A |
| BoxShade version 3.21 | ExPASy Bioinformatics Resource Portal | www.ch.embnet.org/software/BOX_form.html |
| Clustal Omega | European Bioinformatics Institute | www.clustal.org/omega |
| eulerAPE version 3 | University of Kent | www.eulerdiagrams.org/eulerAPE |
| FigTree version 1.4.2 | University of Edinburgh | www.tree.bio.ed.ac.uk/software/figtree |
| ImageQuant™ version 5.2 | GE Healthcare | N/A |
| Jalview version 2 | University of Dundee | www.jalview.org |
| MrBayes version 3.2.2 | Ronquist and Huelsenbeck, 2003 | www.mrbayes.sourceforge.net |
| Nikon Elements Viewer | Nikon Imaging Software | https://www.nikoninstruments.com/products/software |
| Origin® version 7.0 | MicroCal | N/A |
| Proteome Discoverer™ version 2.0.0.802 | Thermo Fisher Scientific | N/A |
| ProtParam | ExPASy Bioinformatics Resource Portal | www.web.expasy.org/protparam |
| T-DNA Express | Salk Institute Genomic Analysis Laboratory | www.signal.salk.edu/cgi-bin/tdnaexpress |
| TotalLab™ Quant | Non-linear Dynamics | www.totallab.com/home/cliqs |
| UCSF Chimera | University of California, San Francisco | www.cgl.ucsf.edu/chimera |
| Other | ||
| Amicon® Ultra-15 centrifugal filter units | EMD Millipore | Cat# UFC901024 |
| Bond Elut™ OMIX C18 pipette tips | Agilent Technologies | Cat# A57003100 |
| BugBuster® master mix | EMD Millipore | Cat# 71456 |
| EZview™ red anti-HA affinity gel | Sigma-Aldrich | Cat# E6779 |
| Gamborg’s B5 basal medium with minimal organics | Sigma-Aldrich | Cat# G5893 |
| Gateway® BP clonase™ II/LR clonase™ II enzyme mix | Thermo Fisher Scientific | Cat# 11789; 11791 |
| GFP-Trap®_A beads | ChromoTek | Cat# GTA-20 |
| GST-Bind™ resin | EMD Millipore | Cat# 70541 |
| Hybond™-C Extra nitrocellulose membrane | GE Healthcare | Cat# RPN303E |
| Immobilon®-P PVDF transfer membrane | EMD Millipore | Cat# IPVH00010 |
| Murashige and Skoog basal salts with macronutrients and micronutrients | Caisson Labs | Cat# MSP01 |
| Murashige and Skoog basal salt micronutrient solution | Sigma-Aldrich | Cat# M0529 |
| Nickel-nitrilotriacetic acid agarose beads | QIAGEN | Cat# 30230 |
| Plant protease inhibitor cocktail | Sigma-Aldrich | Cat# P9599 |
| Yeast complete supplement mixture lacking histidine, leucine and tryptophan | MP Biomedicals | Cat# 114530522 |
| Yeast complete supplement mixture lacking adenine, histidine, leucine and tryptophan | MP Biomedicals | Cat# 114540422 |
| Yeast nitrogen base without amino acids and ammonium sulphate | Sigma-Aldrich | Cat# Y1251 |
To assay 6His-ATG8e/Atg8 binding via dot blots, 1 ml cultures of E. coli strain BL21(DE3) pLysS (Promega) expressing the indicated genes from either pDEST™15 (Thermo Fisher Scientific) or pET17-b (Novagen; see below for growth and induction conditions) were harvested by centrifugation at 5,000 x g for 5 minutes at 4°C, frozen in liquid nitrogen, a nd lysed in 100 μl BugBuster® Master Mix (EMD4 Biosciences). An appropriate volume of cell lysate (normalized for expression of the GST- or HA-tagged protein; typically between 1 and 10 μl was then spotted onto Hybond™-C Extra nitrocellulose membrane (GE Healthcare). The membrane was blocked with 3% (w/v) bovine serum albumin (BSA) in 1X TBS (10 mM Tris-HCl (pH 7.3), 125 mM NaCl), then incubated with 500 nM purified 6His-ATG8e or 6His-Atg8 variants (see below for purification details) in 10 ml of binding buffer (150 mM Tris-HCl (pH 7.4), 50 mM NaCl, 5 mM MgCl2, 1 mM DTT, 5% (v/v) glycerol, 1% (w/v) BSA, 0.01% (v/v) Triton X-100) for 60 minutes with gentle rotation. All incubations were performed at 4°C. The membranes were then washed once with TBST (TBS plus 0.1% (w/v) BSA, 0.1% (w/v) SDS, 0.1% (v/v) Triton X-100) and twice with TBS for 10 minutes each, incubated with primary anti-6His antibody (in 1% (w/v) BSA in TBS) for 60 minutes, washed as above, re-blocked with 3% (w/v) BSA in TBS for 30 minutes, incubated for 60 minutes with secondary goat anti-rabbit antibody conjugated to horseradish peroxidase (in 1% (w/v) BSA in TBS), then washed again and developed as above.
Yeast Two-Hybrid Assays
Assays for direct protein-protein interaction by Y2H were performed using the ProQuest™ two-hybrid system (Thermo Fisher Scientific). Sequence-confirmed coding sequences cloned into pDONR™221 as above were recombined in-frame with either the GAL4 activation domain or GAL4 DNA-binding domain in the pDEST™22 or pDEST™32 vectors (Thermo Fisher Scientific), respectively, via the Gateway® LR clonase™ II reaction. Pairwise combinations of coding sequences in pDEST™22 and pDEST™32 (or the empty vectors as controls) were then co-transformed into yeast strain MaV203. Cells transformed with both plasmids were selected for by growth for 2 days at 30°C on synthetic dropo ut medium lacking leucine and tryptophan. Protein-protein interactions were then identified by growing for 2 days at 30°C on synthetic dropout medium lacking leucine, tryptophan and histidine, and containing 25 mM 3-amino-1,2,4-triazole (3-AT), with at least four individual colonies tested for each interaction pair. To confirm interactions, single colonies were diluted in sterile H2O to an OD600 of 0.1, and 5 μl was spotted onto both types of selective medium and again grown for 2 days at 30°C.
The following interaction pairs were included as positive controls in the Y2H assays: Arabidopsis ATG1a/ATG8, ATG7/ATG8, CDC48a/PUX1-16 and RPN9a/RPN10; yeast Atg1/Atg8, Atg7/Atg8, Cdc48/Ubx1-7, Dsk2/Cue5, Pan1/Ent1-2 and Skp1/Ufo1; and human ATG7/GABARAP, ATG7/MAP1LC3a, EPS15/EPN1-3, KLF5/ATXN3-3L and VPS45/RBSN.
Bimolecular Fluorescence Complementation
In planta protein-protein interactions were assayed using bimolecular fluorescence complementation in N. benthamiana leaves. Sequence-confirmed coding sequences cloned into pDONR™221 as above were recombined in-frame with the N- or C-terminal halves of EYFP in the pSITE-N-EYFP-C1 or pSITE-C-EYFP-C1 vectors (ABRC stock numbers CD3-1648 and CD3-1649, respectively) via the Gateway® LR clonase™ II reaction, with expression driven by the cauliflower mosaic virus (CaMV) 35S promoter. The resulting plasmids were introduced into A. tumefaciens strain GV3101, overnight cultures of which were re-suspended in 5 ml infiltration buffer (10 mM MgCl2, 10 mM MES (pH 5.7), 100 μM 3′,5′-dimethoxy-4′-hydroxyacetophenone (acetosyringone)), incubated at room temperature for 8 to 12 hours, and then used for direct infiltration of 4 to 6-week-old N. benthamiana leaves. Leaf sections of approximately 2 mm x 2 mm were excised 36 to 48 hours after infiltration and visualized by confocal fluorescence microscopy as described below.
Confocal Laser Scanning Microscopy
Arabidopsis seedlings expressing GFP, YFP or mCherry reporters were grown in 2 ml liquid GM medium at 21 to 23°C under continuous light for 4 days with gentle shaking (90 rpm), then transferred to fresh medium either containing or lacking nitrogen and/or supplemented with 20 μM CB-5083, with an equivalent volume of DMSO added to the controls. Following incubation for 16 hours, root cells within the lower elongation zone were visualized with a Nikon A1+ confocal laser scanning microscope, using 20X or 40X oil objectives (numerical apertures 0.75 and 1.30, respectively). Excitation was performed at 488 nm and 543 nm, and emission was collected between 500 and 530 nm or 565 and 615 nm, for GFP/YFP and mCherry signals, respectively. Identical settings were used for imaging of N. benthamiana leaf sections subjected to A. tumefaciens-mediated infiltration as described above.
For imaging of yeast, cells expressing YFP reporters grown in YPDA medium (containing 2% glucose or 2% galactose as required), or subjected to nitrogen starvation as described above, were visualized using a 100X oil objective (numerical aperture 1.46) with the same settings as above. To prevent cell movement, all cover slips were washed in 1 N NaOH, rinsed with sterile distilled H2O, and coated with a 2 mg/ml solution of concanavalin A for 10 minutes. The slips were then air-dried, rinsed with sterile distilled H2O, left to dry again, and stored for up to 2 months before use. To avoid auto-fluorescence from the YPDA medium, cells were first pelleted by centrifugation at 1,000 x g for 1 minute, and then re-suspended in synthetic dropout medium lacking appropriate amino acids prior to imaging.
All confocal images were scanned in single-track mode, except for co-localization studies, where the GFP and mCherry signals were detected simultaneously in multi-track mode. Images were processed using Elements Viewer (Nikon Imaging Software) and/or Adobe Photoshop CC, before conversion to TIFF files for use in the figures. Within each figure, all images (including negative controls) were captured using identical microscope settings, with the exception of bright field images, where channel gain was adjusted to provide uniform exposure between images.
Recombinant Protein Expression and Purification
Sequence-confirmed coding sequences cloned into pDONR™221 as above were recombined in-frame with either no tag, a glutathione S-transferase (GST) tag, or a 6His tag in the pDEST™14, pDEST™15 or pDEST™17 vectors (Thermo Fisher Scientific), respectively, via the Gateway® LR clonase™ II reaction. The sequence for the HA-tag at the N-terminus of DSK2a was incorporated into the appropriate PCR amplification primer. The GST protein alone, encoded in the unmodified pGEX-4T-1 plasmid (GE Healthcare), was used as a control for the GST fusions. All proteins were expressed in E. coli strain BL21(DE3) pLysS. The cells were cultured at 37°C in 800 ml LB medium to an OD 600 of between 0.6 and 0.8, followed by a 4 hour induction with 1 mM isopropyl-β-D-thiogalactopyranoside at either 30 or 37°C. Cell s were harvested by centrifugation at 5,000 x g for 20 minutes at 4°C, frozen in liquid nitrogen, and lysed in two rounds of 10 ml BugBuster® Master Mix (EMD4 Biosciences).
GST or GST fusion proteins were affinity purified using GST-Bind™ Resin (EMD4 Biosciences). Bacterial cell lysates were incubated with 1 ml PBS-washed GST-Bind™ Resin for 1 hour at 4°C with continual rotation, and then applied to a 30 ml EconoPac® chromatography column (Bio-Rad). After 5 minutes, the flow-through was allowed to run out, and the beads were washed twice with PBS containing 1 M NaCl, and once with PBS containing 2 M NaCl. Bound proteins were eluted by incubating the beads with 10 ml of 25 mM 3-(N-morpholino)propanesulphonic acid (MOPS)-KOH (pH 7.5) containing 10 mM reduced glutathione (GSH), and the eluants were dialyzed overnight at 4°C against 25 mM MOPS-KOH (pH 7.5), before being concentrated 10-fold using an Amicon® Ultra-15 centrifugal filter unit with a 10 kDa cut-off limit (Millipore).
Proteins tagged with 6His were affinity purified using nickel-nitrilotriacetic acid (Ni-NTA) agarose beads (QIAGEN). Bacterial cell lysates in BugBuster® Master Mix containing 10 mM imidazole were applied three times to either 1 or 3 ml of PBS-washed Ni-NTA beads in a 30 ml EconoPac® chromatography column at 4°C. For in vitro binding assays, the beads were washed once with NaH2PO4 wash buffer (50 mM NaH2PO4 (pH 7.8), 300 mM NaCl, 20 mM imidazole), once with Tris-40 buffer (50 mM Tris-HCl (pH 7.8), 300 mM NaCl, 40 mM imidazole), and once with Tris-60 buffer (50 mM Tris-HCl (pH 7.8), 300 mM NaCl, 60 mM imidazole). Bound proteins were then eluted with 2 or 6 ml of Tris elution buffer (50 mM Tris-HCl (pH 7.8), 100 mM NaCl, 300 mM imidazole). For isothermal titration calorimetry (ITC) experiments, the beads were washed once with NaP-20 buffer (20 mM Na2HPO4, 5 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, 500 mM urea, 2% glycerol), once with NaP-40 buffer (20 mM Na2HPO4, 5 mM NaH2PO4, 300 mM NaCl, 40 mM imidazole, 500 mM urea, 2% glycerol), and once with NaP-60 buffer (20 mM Na2HPO4, 5 mM NaH2PO4, 300 mM NaCl, 60 mM imidazole, 500 mM urea, 2% glycerol). Bound proteins were then eluted with 5 ml of NaP elution buffer (20 mM Na2HPO4, 5 mM NaH2PO4, 150 mM NaCl, 300 mM imidazole, 500 mM urea, 2% glycerol). All eluants were dialyzed overnight at 4°C to remove the imidazole.
The HA-DSK2a protein was purified using EZview™ red anti-HA affinity gel (Sigma-Aldrich). The bacterial cell lysate was incubated with 100 μl PBS-washed anti-HA antibody beads for 1 hour at 4°C with continual rotation, then applied to a 12 ml PolyPrep® chromatography column (Bio-Rad). After 5 minutes the flow-through was allowed to run out, and the beads were washed three times with PBS containing 1 M NaCl. Bound protein was eluted by incubating the beads for 30 minutes at 4°C with 300 μl of PBS containing 500 ng/µl of the HA peptide (YPYDVPDYA; Sigma-Aldrich), and the eluant was dialyzed overnight at 4°C against PBS. In all cases, protein concentration was determined using the Pierce™ BCA protein assay kit (Thermo Fisher Scientific). For ITC experiments, protein concentrations were additionally confirmed using OD280 measurements on a DU® 800 spectrophotometer (Beckman Coulter), using extinction co-efficients calculated in ProtParam (www.web.expasy.org/protparam).
Quantitative in vitro Binding Assays
For testing of protein-protein interactions in vitro, full-length proteins were used, except for Arabidopsis ATG8e and yeast Atg8, where the processed forms (i.e., amino acids 1 to 118 and 1 to 116, respectively) were used, and RPN10, where only the C-terminal portion (i.e., amino acids 201 to 386) was used, due to the insolubility of recombinant full-length GST-RPN10. To assay direct binding between 6His-ATG8e/Atg8 and their potential interactors, equal amounts (5 μg) of both proteins were combined in 500 μl of binding buffer (150 mM Tris-HCl (pH 7.4), 300 mM NaCl, 5 mM MgCl2, 1 mM DTT, 5% (v/v) glycerol, 0.01% (v/v) Triton X-100). Binding was allowed to proceed for 2 hours at 4°C, before the a ddition of 100 μl Ni-NTA agarose beads pre-equilibrated with binding buffer. Samples were incubated for a further 2 hours at 4°0 with continual rotation, and the beads were then pelleted by centrifugation at 3,000 x g for 1 minute at 4°C. The beads were washed 5 times with binding buffer, and bound proteins were eluted into 100 μl of 2X SDS-PAGE sample buffer by heating at 95°0 for 5 minutes. Samples were analyzed by SDS-PAGE followed by immunoblotting with appropriate antibodies as described above. Three-way binding assays to confirm the simultaneous interaction of Arabidopsis ATG8e with DSK2a and RPN10 were performed as above with equal amounts (2 μg) of the three interactors, except that the binding buffer contained 150 mM NaCl, and GST-Bind™ Resin was used to perform the pulldowns.
To determine the affinity of the interactions between Arabidopsis ATG8e and the four PUX proteins, quantitative equilibrium binding assays were performed as previously described (Marshall et al., 2015), with minor modifications. Varying concentrations of purified 6His-ATG8e (from 0 to 40 µM) were incubated with 1 µM purified GST-PUX7, GST-PUX8, GST-PUX9 or GST-PUX13 in 100 μl of binding buffer. Binding was allowed to proceed as above and, after pelleting of the Ni-NTA agarose beads, 10 μl of supernatant was analyzed by SDS-PAGE followed by immunoblotting with anti-ATG8 or anti-GST antibodies. Densitometric quantification of the PUX protein bands was performed as above, and the amount remaining in the supernatant when 6His-ATG8e was absent was set at 100% (i.e., 0% bound). The percentage of PUX protein bound was then plotted against the concentration of 6His-ATG8e, with the KD being the concentration of 6His-ATG8e at which 50% of the GST-PUX protein was bound.
ITC experiments were conducted using a MicroCal™ VP-ITC instrument (Malvern Panalytical) at 15°C, with a syringe protein concentration of 100 μM, a cell protein concentration of 10 μM, and a stirring speed of 300 rpm. Thermodynamic analyses were performed by titrations of RPN10, PUX7, PUX8, PUX9 or PUX13 (syringe proteins) into ATG8e (cell protein), with an initial injection of 2 μl followed by 21 consecutive injections of 12 μl, each separated by a time interval of 360 seconds. The syringe samples were also titrated to a cell filled only with buffer, and the obtained reference data were used for subtracting the heat of dilution. The raw titration data (excluding the initial 2 μl injection) were integrated and fitted to a single-site binding model using MicroCal™ Origin software version 7.0, and the resulting curve was used to determine the stoichiometry of binding (N), the dissociation constant (KD), and the change in enthalpy (ΔH).
Arabidopsis and Yeast cDNA Library Screens
Coding sequences of the processed forms of Arabidopsis ATG8e and yeast ATG8 were PCR-amplified from cDNA using primers encoding XhoI and NcoI restriction enzyme sites at the 5′ end and a BamHI restriction enzyme site at the 3′ end. Amplified PCR products were digested with FastDigest™ XhoI and BamHI (Thermo Fisher Scientific) and ligated into pBlueScript II (Agilent Technologies) cut with the same enzymes. Site-directed mutagenesis to obtain the ΔLDS and ΔUDS mutations was then performed using the QuikChange™ II site-directed mutagenesis kit. Sequence-confirmed genes were excised by digestion with FastDigest™ NcoI and BamHI (Thermo Fisher Scientific) and ligated into pGBKT7 (Clontech) cut with the same enzymes. Resulting clones were then transformed into yeast strain AH109 (Clontech).
Strains expressing the above bait plasmids were either mated to strain Y187 that had been transformed with an Arabidopsis cDNA library, or subsequently transformed with 500 μg of a yeast cDNA library. The commercially available Arabidopsis cDNA library in pGADT7-RecAB was constructed from a mixture of poly(A)-containing mRNAs isolated from 11 tissues chosen to represent a broad range of expressed genes, and was normalized to reduce the copy number of abundant cDNA derived from highly represented mRNAs (Clontech, product number 630487). A yeast cDNA library in the pACT2 vector, derived via automatic sub-cloning from bacteriophage λ-YES in E. coli strain BNN132 (Elledge et al., 1991), was generously provided by Stephen J. Elledge (Harvard Medical School).
Positive yeast clones expressing putative interacting partners were identified by growth at 30°C for up to 12 days on synthetic dropout medi um lacking adenine, histidine, leucine and tryptophan. Prey plasmids were then extracted from the resulting colonies using the Zymoprep™-96 yeast plasmid miniprep kit (Zymo Research), transformed into E. coli strain DH5α, and extracted again using the QIAprep® spin miniprep kit (QIAGEN). The prey cDNAs were then identified by DNA sequencing. To confirm the interacting pairs, bait and prey plasmids were individually transformed into strain MaV203, and interactions re-tested by standard Y2H as described above. Venn diagrams displaying the interactors were produced using eulerAPE (www.eulerdiagrams.org/eulerAPE), and homology identification was performed by reciprocal BLASTn searches (www.ncbi.nlm.nih.gov/blast).
To confirm the positive Y2H interactions via dot blot assays, cDNA inserts were excised from the prey plasmids and ligated into an E. coli expression vector. Arabidopsis inserts were excised by digestion with FastDigest™ SfiI (Thermo Fisher Scientific), while yeast inserts were excised by digestion with appropriate restriction enzymes that cut either side of the insert within the multiple-cloning site (FastDigest™ BglII (Thermo Fisher Scientific) was the preferred option, followed by FastDigest™ XhoI). In all cases, the resulting sticky ends were converted to blunt ends using Klenow fragment (Thermo Fisher Scientific), and the product was ligated into pET17-b (Novagen) digested with FastDigest™ EcoRV (Thermo Fisher Scientific). With the exception of yeast cDNAs excised with BglII, ligation occurred into a version of pET17-b pre-engineered to encode an N-terminal HA tag between the NheI and EcoRI restriction sites. Sequence-confirmed clones were transformed into E. coli strain BL21(DE3) pLysS, and proteins were expressed in 1 ml cultures. Cell growth, induction, lysis, and subsequent dot blot binding assays were performed as described above.
Immunprecipitation of GFP-PUX7
Arabidopsis seedlings of the indicated genotype (WT (Ws), 35S::GFP or 35S::GFP-PUX7) were grown in 50 ml liquid GM medium as above, with ~100 mg of dry seeds used per culture, resulting in ~5 g of fresh weight tissue. Frozen tissue was ground to a fine powder at liquid N2 temperatures, and proteins were extracted on ice for 20 minutes with 1.5 volumes of extraction buffer (50 mM HEPES (pH 7.5), 150 mM NaCl, 10 mM MgCl2, 10% (v/v) glycerol, 2% (w/v) polyvinylpyrrolidone, 5 mM DTT, 2 mM PMSF, 0.1% (v/v) Triton X-100, 1X plant protease inhibitor cocktail). Extracts were filtered through two layers of Miracloth, clarified at 30,000 x g for 20 minutes at 4°C, and the resulting supernatan ts were immediately applied three times at 4°0 over a 12 ml PolyPrep® chromatography column containing 100 μl (equal to a 50 μl bead volume) of GFP-Trap®_A beads (ChromoTek) pre-equilibrated in extraction buffer. The column was washed five times with wash buffer (50 mM HEPES (pH 7.5), 150 mM NaCl, 10 mM MgCl2, 10% (v/v) glycerol, 2 mM DTT), and remaining bound proteins were eluted with 100 μl of 200 mM glycine-HCl (pH 2.5) and immediately neutralized with 20 μl of 1 M Tris-HCl (pH 8.0). Samples of the crude extract, flow through (both diluted 1/10), third wash step and elution were analyzed by SDS-PAGE followed by silver staining or immunoblotting with appropriate antibodies (as above). Alternatively, elution fractions were subjected to tandem mass spectrometry (see below).
Tandem Mass Spectrometry
Protein samples from the GFP immunoprecipitation experiments (80 μl of the 120 μl elution volume, from three independent biological replicates) were vacuum dried in a SpeedVac™ centrifugal evaporator (Savant Instruments, model number SUC100H) to a final volume of approximately 20 μl and denatured in 8 M urea, 25 mM (NH4)HCO3 in a total volume of 300 µl. Proteins were then reduced with 10 mM DTT at room temperature for 1 hour, and alkylated in the dark in the presence of 50 mM 2-chloroacetamide at room temperature for a further 1 hour. Excess alkylating agent was quenched with 50 mM DTT for 5 minutes at room temperature, and the samples were diluted with 1.2 ml of 25 mM (NH4)HCO3 to reduce the urea concentration to below 1.5 M. Proteolytic digestion was then initiated by adding 1 μg of sequencing grade modified porcine trypsin (Promega), and the samples were incubated overnight at 37°C. Peptides were vacuum dried as above to a final volume of approximately 300 μl, acidified with 10% trifluoroacetic acid (TFA) until the pH was less than 3.0, and desalted and concentrated on a 100 μl Bond Elut™ OMIX C18 pipette tip (Agilent Technologies). The peptides were eluted in 50 μl of 75% acetonitrile, 0.1% acetic acid, then lyophilized and re-suspended in 17 μl 5% acetonitrile, 0.1% formic acid.
Nano-scale liquid chromatography (LC) separation of tryptic peptides was performed on a Dionex Ultimate™ 3000 Rapid Separation LC system (Thermo Fisher Scientific). 6.8 μl of each protein digest was loaded onto a 20 μl nanoViper™ sample loop (Thermo Fisher Scientific) and separated on a C18 analytical column (Acclaim® PepMap™ RSLC C18 column, 2 μm particle size, 100 Å pore size, 75 μm x 25 cm (Thermo Fisher Scientific)) by the application of a linear 2 hour gradient from 4.0% to 36.0% acetonitrile in 0.1% formic acid, with a column flow rate set to 250 nl/min. Analysis of the eluted tryptic peptides was performed online using a Q Exactive™ Plus mass spectrometer (Thermo Fisher Scientific) possessing a Nanospray Flex™ ion source (Thermo Fisher Scientific) fitted with a stainless steel nano-bore emitter operated in positive electro-spray ionization (ESI) mode at a capillary voltage of 1.9 kV. Data-dependent acquisition of full MS scans within a mass range of 380-1500 m/z was performed at a resolution of 70,000, with the automatic gain control (AGC) target set to 3.0 x 106, and the maximum fill time set to 200 msec. High energy collision-induced dissociation (HCD) fragmentation of the top 8 most intense peaks was performed with a normalized collision energy of 28, an intensity threshold of 1.3 x 104 counts, and an isolation window of 3.0 m/z, excluding precursors that had an unassigned, +1, +7 or +8 charge state. MS/MS scans were conducted at a resolution of 17,500, with an AGC target of 2.0 x 105 and a maximum fill time of 300 msec. Dynamic exclusion was performed with a repeat count of 2 and an exclusion duration of 30 seconds, while the minimum MS ion count for triggering MS/MS was set to 4.0 x 103 counts.
The resulting MS/MS spectra were analyzed using Proteome Discoverer™ (version 2.0.0.802; Thermo Fisher Scientific), which was set up to search the Arabidopsis Col-0 proteome database, as downloaded from the Arabidopsis Information Resource (www.arabidopsis.org; ID number TAIR10_pep_20101214). Peptides were assigned using SEQUEST HT, with search parameters set to assume the digestion enzyme trypsin with a maximum of 1 missed cleavage, a minimum peptide length of 6, precursor mass tolerances of 10 ppm, and fragment mass tolerances of 0.02 Da. Carbamidomethylation of cysteine was specified as a static modification, while oxidation of methionine and N-terminal acetylation were specified as dynamic modifications. The target false discovery rate (FDR) of 0.01 (strict) was used as validation for peptide-spectral matches (PSMs) and peptides. Proteins that contained similar peptides, and which could not be differentiated based on the MS/MS analysis alone, were grouped to satisfy the principles of parsimony. All samples were injected in duplicate, with the resulting values combined for the final analysis.
Quantitative Real-Time PCR Analysis
Total RNA extraction from Arabidopsis seedlings or yeast cells treated with or without the indicated concentrations of CDC48/p97 inhibitors, and its subsequent conversion into cDNA, were performed as described above. Quantitative real-time PCR of ER stress-responsive transcripts (BIP1 and CRT1 for Arabidopsis, and DER1 and ERO1 for yeast) was then performed on three independent biological replicates using a LightCycler® 480 machine (Roche Diagnostics) and LightCycler® 480 SYBR Green I master mix (Roche Diagnostics), with three technical replicates for each reaction. In all cases, the amplification factor of the primer pair was experimentally determined to be between 1.90 and 2.10, with the gradient of the standard curve being between −3.59 and −3.10, and the R2 value being greater than 0.975.
For each PCR reaction, 5 μl of cDNA (diluted 1/30 following first-strand synthesis) was amplified in a 20 μl reaction volume also containing 10 μl SYBR Green I master mix, 3 μl sterile H2O, and 1 μl each of 10 μM forward and reverse primers. Reaction parameters were 95°C for 5 minutes, 45 cycles of 95°C for 10 seconds, 55°C for 10 seconds and 72°C for 30 seconds, followed by a melting curve program and cooling at 40°C for 2 minutes. Fluorescence data were collected at the end of each 72°C extension step, a nd continuously during the melting curve program. Relative transcript abundance of target genes was determined using the comparative threshold cycle method, using as internal controls the reference genes ACT2 and PP2A for Arabidopsis, and ALG9 and TFC1 for yeast. All data were normalized to untreated Col-0 seedlings or BY4741 cells for Arabidopsis and yeast, respectively.
Identification of Arabidopsis T-DNA Insertion Mutants
T-DNA insertion mutants were identified in the SALK T-DNA Express database (www.signal.salk.edu/cgi-bin/tdnaexpress) and obtained from the Arabidopsis Biological Resource Center (Ohio State University, USA), the Versailles Arabidopsis Stock Center (Institut Jean-Pierre Bourgin, France) or the GABI-Kat collection (Universität Bielefeld, Germany). All insertions were confirmed by genomic PCR using 5' and 3' gene-specific primers (LP and RP, respectively) in conjunction with an appropriate T-DNA left border-specific primer (BP). Details of all oligonucleotide primers are provided in Table S6. Before analysis, each mutant was backcrossed at least twice to its WT ecotype and then selfed to obtain homozygous progeny. Higher order mutants were generated by multiple rounds of standard introgression and selfing.
Transient Expression in Arabidopsis Protoplasts
The coding sequence of Arabidopsis CDC48a was PCR-amplified from Col-0 cDNA using primers encoding SalI and XbaI restriction enzyme sites at the 5′ end and a SalI restriction enzyme site at the 3' end. The amplified PCR product was digested with FastDigest™ SalI (Thermo Fisher Scientific) and ligated into pBlueScript II cut with the same enzyme. An internal XbaI site within CDC48a was then mutated synonymously using the QuikChange™ II site-directed mutagenesis kit, and the sequence-confirmed gene was excised by digestion with FastDigest™ XbaI and SalI and ligated into pDHA (Marshall et al., 2008) cut with the same enzymes, for transient expression driven by the CaMV 35S promoter. Further site-directed mutagenesis to insert coding substitutions associated with various human diseases (Tang and Xia, 2016) was then performed.
Arabidopsis mesophyll protoplasts from Col-0, atg7-2 or pux-q leaves were prepared and transfected as previously described (Marshall et al., 2008), with minor modifications. Leaves from 4 to 5-week-old soil-grown plants were scarified on their underside using a sterilized scalpel blade and incubated in 1X enzyme solution (1.5% (w/v) cellulase “Onozuka” R-10, 0.4% (w/v) macerozyme R-10 (both from Yakult Pharmaceutical Industries), 400 mM mannitol, 20 mM KCl, 20 mM MES (pH 5.7)) for 6 hours in the dark. Protoplasts were released by gentle swirling, and the protoplast/enzyme mixture was diluted with 1 volume of W5 buffer (150 mM NaCl, 125 mM CaCl2, 5 mM KCl, 4 mM MES (pH 5.7)) and filtered through a 75 μm nylon mesh to remove remaining leaf debris. Protoplasts were collected by centrifugation at 100 x g for 5 minutes, washed once in 20 ml W5 buffer, incubated on ice for 30 minutes, resuspended in MMG buffer (400 mM mannitol, 15 mM MgCl2, 4 mM MES (pH 5.7)) at a concentration of 107 cells/ml, and then incubated in the dark for 30 minutes.
Protoplasts were transfected by mixing 500 μl of cells with 20 μg plasmid DNA, followed by drop-wise addition of 500 μl PEG buffer (40% polyethylene glycol 4000, 400 mM mannitol, 100 mM CaCl2) with gentle mixing. Protoplasts were then incubated for a further 30 minutes at room temperature, 1 ml of W5 solution was added drop-wise and gently mixed, and the cells were collected by centrifugation at 100 x g for 5 minutes, followed by re-suspension in 1 ml WI buffer (400 mM mannitol, 20 mM KCl, 4 mM MES (pH 5.7)). The cells were then incubated in the dark for 16 hours to permit gene expression, collected by centrifugation at 100 x g for 5 minutes, and the cell pellets were frozen on dry ice. Cells were homogenized by addition of 300 μl homogenization buffer (150 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1.5% (v/v) Triton X-100, 1X plant protease inhibitor cocktail) followed by vigorous mixing with a vortex, clarified twice by centrifugation at 16,000 x g for 5 minutes at 4°C, and the supernatant was made 1X with 5X SDS-PAGE sample buffer. Samples were then heated at 95°C for 5 minutes before analysis by SDS-PAGE and immunoblot as above.
Yeast Growth and Viability Assays
Cells of the indicated genotypes were grown in 15 ml synthetic dropout medium lacking appropriate amino acids and containing 2% glucose as the carbon source. Following 12 hours overnight growth, cultures were diluted to an OD600 of 0.2 in 50 ml synthetic dropout medium lacking appropriate amino acids and containing either 2% glucose or 2% galactose as the carbon source. Growth was then monitored by measurement of OD600 values in the presence or absence of 1.5 mM DTT or 200 ng/ml tunicamycin (both from Sigma-Aldrich) using a Smartspec™ 3000 UV/Vis spectrophotometer (Bio-Rad), or by growth of cells on solid synthetic dropout medium lacking appropriate amino acids and containing glucose or galactose as indicated. Susceptibility to DTT or tunicamycin was determined by normalizing the OD600 value of each strain in the presence of the chemical to growth in their absence. For growth on solid medium, cells were re-suspended in liquid synthetic dropout medium lacking appropriate amino acids, and containing glucose or galactose as the carbon source, to an OD600 of 1.0, then subjected to a series of 5-fold dilutions, before 5 μl of each dilution was spotted onto the same medium containing or lacking 1.5 mM DTT or 200 ng/ml tunicamycin. Cells were then grown for 48 hours at 30°C prior to imaging.
Pho8Δ60 Enzyme Assays
The Pho8Δ60 assay for quantitative measurement of yeast autophagic flux was performed essentially as described (Noda et al., 1995), using the spectrophotometric assay with minor modifications. Strains TN124 or RSM294-302 were grown in 250 ml cultures with an appropriate carbon source, subjected to nitrogen starvation, and aliquots corresponding to 5.0 OD units were collected at the indicated time points, all as described above. Cell pellets were resuspended in 500 μl lysis buffer (20 mM piperazine-N,N'-bis-2-ethanesulfonic acid (PIPES)-KOH (pH 8.5), 50 mM KCl, 100 mM KCH3COO, 10 mM MgSO4, 10 μM ZnSO4, 0.5% (v/v) Triton X-100, 1 mM PMSF) and lysed by the addition of acid-washed glass beads to a volume of approximately 200 μl, followed by vigorous mixing with a vortex for a total of 5 minutes at 4°C (10 rounds of mixing for 30 seconds, then resting on ice for 30 seconds). Non-lysed cells and insoluble debris were pelleted by centrifugation at 16,000 x g for 5 minutes at 4°C, and the supernatant was collected for subsequent analysis.
The total protein concentration of each sample was determined by Pierce™ BCA protein assay kit, and equal amounts of protein (20 μg) were assayed for alkaline phosphatase activity. Protein samples in a volume of 100 μl were mixed with 400 μl pre-warmed assay buffer (250 mM Tris-HCl (pH 8.5), 10 mM MgSO4, 10 μM ZnSO4, 1.5 mM p-nitrophenyl phosphate, 1% (v/v) Triton X-100) and incubated for 10 minutes at 37°C. Reactions were quenched by addition of 500 μl 1 M glycine-KOH (pH 11.0), and the absorbance of p-nitrophenol at 400 nm was measured spectrophotometrically, as above. Following subtraction of the appropriate enzyme and substrate only controls, specific alkaline phosphatase activity was calculated from a p-nitrophenol standard curve. Three technical replicates were performed for each sample, and the data from three independent biological replicates was averaged and normalized to the activity observed at the 0 hour time point.
Sequence Alignments and Phylogenetic Analysis
Predicted full-length nucleotide and protein sequences were obtained from appropriate databases (for full details see Tables S1 and S2), while domain positions were predicted by the SMART (www.smart.embl-heidelberg.de) and PFAM (www.pfam.sanger.ac.uk) databases. Progressive alignments of predicted full-length amino acid sequences were performed using Clustal Omega (www.clustal.org/omega) with the default settings and, following minor manual editing, final alignments were displayed with BoxShade version 3.2.3 (www.ch.embnet.org/software/BOX_form.html). Species abbreviations used are: At, Arabidopsis thaliana; Bd, Brachypodium distachyon; Ce, Caenorhabditis elegans; Dm, Drosophila melanogastep Gm, Glycine max; Hs, Homo sapiens; Os, Oryza sativa; Pp, Physcomitrella patens; Pt, Populus trichocarpa; Sc, Saccharomyces cerevisiae; Sm, Selaginella moellendorffii; Vv, Vitis vinifera; Zm, Zea mays. Sequence conservation of the plant ATG8 isoforms was calculated and displayed using JalView version 2 (www.jalview.org).
To determine UDS and UIM consensus sequences, the interaction strength of each point mutation variant of the ATG8a UDS and RPN10 and PUX7 UIMs was determined based on its mean interaction signal, as quantified using ImageQuant version 5.2 (GE Healthcare); the variant was considered a true interactor when both Y2H and dot blot binding signals were above the background level. The mean BLOSUM62 value for each site within the UDS or UIMs was then calculated to give the complete set of mutations that yielded true interaction variants. Since a higher BLOSUM62 value implies stronger conservation of the mutation, mean BLOSUM62 values were applied to quantify the evolutionary relevance of the complete set of mutations for each site within or surrounding the UDS and UIMs. The stretch of amino acids with high positive mean BLOSUM62 values were defined as the consensus region for each domain.
Bayesian phylogenetic analyses of the full-length Arabidopsis PUX proteins was performed with MrBayes version 3.2.2 (Ronquist and Huelsenbeck, 2003), using the General Time Reversible evolutionary model with the mixed amino acid model and γ-distributed rate variation with a proportion of invariable sites. The software was run for 1,000,000 generations, with a sampling frequency of every 1,000 generations. The first 250,000 generations (25%) were discarded as “burn-in” after checking that the log likelihood values had plateaued and that the potential scale reduction factor was close to 1.000. The resulting consensus tree was displayed using FigTree version 1.4.2 (www.tree.bio.ed.ac.uk/software/figtree).
Protein Structure Modeling
The 3-dimensional positions of potential UIM-binding sites in Arabidopsis ATG8, and of mutated residues within CDC48/p97 that trigger its autophagic degradation, were mapped with UCSF Chimera (Pettersen et al., 2004), using as templates the structures 3VXW and 5FTK, respectively, from the Protein Data Bank (www.rcsb.org/pdb).
QUANTIFICATION AND STATISTICAL ANALYSIS
All datasets were statistically analyzed using one-way analysis of variance (ANOVA), followed by Tukey’s post-hoc tests to identify significantly different data points. Significance was defined as p < 0.05 or p < 0.01. Statistical details for each experiment can be found in the Figure Legends. At least three biological replicates were performed in all cases, unless otherwise noted. Due to the nature of the experiments, no methods were employed to determine strategies for sample randomization, stratification, and/or size estimation.
DATA AND SOFTWARE AVAILABILITY
Datasets generated from cDNA library screens are provided in Table S3. The mass spectrometry-based proteomics data have been deposited to the ProteomeXchange Consortium (www.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD011397. All accession numbers for genes and proteins used in this study are provided in Table S1.
Supplementary Material
Dataset S1: Mass Spectrometric Analysis of GFP-PUX7 Co-immunoprecipitations; Related to Figure 6.
Figure S4. Control Y2H assays used to identify and map UIM-containing ATG8-interacting proteins from yeast and humans. Related to Figure 4.
(A, B, D, E, F and G) Growth of transformed yeast cells on synthetic dropout medium lacking leucine and tryptophan from the experiments in Figures 4A, 4B, 4C and 4E, confirming that all plasmid combinations are able to support growth on non-selective medium.
(C) Yeast Ufo1 does not use a UIM to interact with Atg8. Various Ufo1 combinations, either full length, missing the FBX domain, or with mutations in one or more UIM sequences, were tested for interaction with WT, ΔLDS or ΔUDS forms of Atg8. Details of all mutations are given in Table S1. Shown are cells grown on selective and non-selective medium, as in Figure S2C.
(H, I and J) The UIM regions in human ATXN3, ATXN3L, EPN1, EPN2, EPN3 and RBSN are required for binding to MAP1LC3a and/or GABARAP at the UDS. Various combinations of the above proteins, either WT or with mutations in one or more of their UIM sequences, were tested for interaction with WT, ΔLDS or ΔUDS forms of MAP1LC3a or GABARAP. Shown are cells grown on both selective and non-selective medium, as in panel (C).
Figure S5. Identification of p97 inhibitors effective in plants and assembly of a quadruple mutant eliminating the four Arabidopsis ATG8-interacting PUX proteins. Related to Figure 6.
(A) The human CDC48/p97 inhibitor CB-5083 induces ER stress in Arabidopsis. Total RNA was extracted from 7-day-old WT seedlings treated with the given concentrations of CB-5083, DBeQ, ML240 or NMS-873 for 16 hours, and converted to first-strand cDNA. Relative transcript abundance of the ER stress-responsive genes BIP1 and CRT1 was determined by quantitative real-time PCR, using the ACT2 and PP2A genes as internal reference standards. All data points were normalized to untreated seedlings; the bars represent mean (±SD) from three independent biological replicates, each with three technical replicates.
(B) CB-5083 treatment impairs Arabidopsis seedling growth. WT seedlings were grown for 7 days on solid GM medium containing the indicated concentrations of CB-5083, DBeQ, ML240 or NMS-873.
(C and D) CB-5083 stimulates the autophagic turnover of both GFP-PUX7 (panel (C)) and YFP-CDC48a (panel (D)), as determined by the GFP/YFP-release assay. Seedlings were grown for 6 days and then switched from nutrient-rich medium to either medium lacking nitrogen or medium containing 20 μM of CB-5083, DBeQ, ML240 or NMS-873 for 16 hours. Total protein extracts were assayed for GFP or YFP release as in Figure 6A.
(E) CB-5083 treatment reduces bulk autophagic flux in Arabidopsis. Seedlings expressing GFP-ATG8a were grown for 6 days, treated with 20 μM CB-5083, DBeQ, ML240 or NMS-873 for 16 hours, then switched from nutrient-rich medium to medium lacking nitrogen for an additional 16 hours. Total protein extracts were assayed for GFP release as in panel (C).
(F) Quantification of the ratio of free GFP to GFP-ATG8a from the experiment shown in panel (E) and two additional independent biological replicates. Bars represent the mean (±SD) from three independent biological replicates; letters indicate values statistically significantly different from one another and the control (p < 0.05).
(G) Diagram of the PUX7, PUX8, PUX9 and PUX13 genes. The boxes represent coding regions (colored) or predicted untranslated regions (white), while lines represent introns. The green, red and blue boxes identify UBA domains, UIMs and UBX domains, respectively. Positions of the T-DNA insertions are indicated by the red triangles; sites of amino acid sequence disruption are shown in Figure S2D. Positions of oligonucleotide primers used for reverse transcription (RT)-PCR in panel (H) are indicated by the half arrowheads.
(H) RT-PCR analysis of the PUX7, PUX8, PUX9 and PUX13 transcripts in the indicated T-DNA insertions. Total RNA was extracted from 7-day-old WT or homozygous mutant seedlings, converted to first-strand cDNA, and subjected to RT-PCR using the primer pairs shown in panel (G). Amplification of ACT2 was included to confirm analysis of equal amounts of cDNA.
(I) Phenotypes of individual and higher order pux mutant plants. Plants with the indicated pux genotypes were grown on soil for 3 weeks under a long-day photoperiod.
Figure S6. p97 amino acids associated with human disease are conserved in Arabidopsis CDC48. Related to Figure 6.
Sequence alignment of CDC48 proteins from Arabidopsis thaliana, Saccharomyces cerevisiae and humans. Identical amino acids (55% threshold value) are shown with a black background; similar amino acids are shown with a grey background. Positions of the N-terminal domain and the D1 and D2 ATPase domains are indicated by green, red and blue lines, respectively, while the C-terminal HbYX motif that docks onto the 20S proteasome is indicated with a yellow line. Conserved residues implicated in human disease are indicated by purple arrowheads; residues critical for ATP binding or hydrolysis are indicated by the red and brown asterisks, respectively. Amino acid numbers are indicated on the right; accession numbers for these proteins are listed in Table S1.
Figure S7. Human p97 inhibitors do not impact yeast Cdc48, but genetic Cdc48 mutants can impact autophagic flux. Related to Figure 7.
(A) Human p97 inhibitors do not induce ER stress in yeast. Total RNA was extracted from WT cells following 16 hour incubation with the given concentrations of CB-5083, DBeQ, ML240 or NMS-873, and converted to first-strand cDNA. The relative transcript abundance of the ER stress-responsive genes DER1 and ERO1 was determined by quantitative real-time PCR, using the ALG9 and TFC1 genes as internal reference standards. All data points were normalized to untreated cells. Similar results were obtained with Δerg6 and Δpdr5 cells.
(B) Yeast growth is not impacted by p97 inhibitors. Cells of the indicated genotype were treated for 16 hours with or without 50 μM MG132 or 2 mM CB-5083, DBeQ, ML240 or NMS-873, and equal numbers of cells were subjected to 5-fold serial dilutions, spotted onto synthetic dropout medium also containing the inhibitors, and incubated at 30°C for 36 hours. The treatment with MG132 was included as a positive control for the Δerg6 and Δpdr5 deletions.
(C) Bulk autophagic flux in yeast is not impacted by p97 inhibitors, as measured by assay of the Pho8Δ60 reporter. Cells of the indicated genotypes expressing PHO8Δ60 were treated with or without 2 mM CB-5083, DBeQ, ML240 or NMS-873, or switched to growth at 37Ό (for cdc48-6 cells only) for 16 hours, then subjected to nitrogen starvation for 0 or 8 hours. All data points were normalized to those obtained after 0 hours nitrogen starvation.
(D) Yeast Cdc48 is not subject to autophagy following treatment with human p97 inhibitors, as determined by the YFP release assay. Release of free YFP from the YFP-Cdc48 reporter upon nitrogen starvation or treatment with 2 mM CB-5083, DBeQ, ML240 or NMS-873 for 16 hours was monitored in WT, Δerg6 or Δpdr5 cells by immunoblot analysis of total protein extracts with anti-GFP antibodies, as in Figure 7A.
(E) Mutations in genomic CDC48 impact bulk autophagic flux as induced by nitrogen starvation. Cells of the indicated genotypes expressing PHO8Δ60 were grown at 30 or 37°C for 8 hours, then subjected to nitrogen starvation for 0 or 8 hours, as in panel (C).
(F) Mutant versions of Cdc48 ectopically expressed from a galactose-inducible promoter in WT cells have less impact on autophagic flux than when they replace the genomic locus. Cells expressing PHO8Δ60 were transformed with plasmids encoding YFP-CDC48, YFP-cdc48-3 or YFP-cdc48-6 under control of the GAL1 promoter. Cells were then grown at either 30 or 37Ό in medium containing glucose (−) or galactose (+) for 8 hours, and subjected to nitrogen starvation for 0 or 8 hours, as in panel (C).
(G) The cdc48-3 and cdc48-6 proteins are poor autophagy substrate, as determined by the YFP release assay. WT cells were transformed with the same plasmids as in panel (F), grown at 30°C, then switched to growth at 30 or 37°C in medi um containing galactose for 48 hours. Total protein extracts were assayed for YFP release by immunoblot analysis as in panel (D).
(H) Ectopic expression of Cdc48 variants containing mutations in conserved disease-associated amino acids only mildly impacts bulk autophagic flux. WT cells expressing PHO8Δ60 were transformed with plasmids encoding YFP-CDC48 or the P147L, E195K or K396E variants under control of the GAL1 promoter. Cells were grown in medium containing glucose (−) or galactose (+) for 8 hours, and then subjected to nitrogen starvation for 0 or 8 hours, as in panel (C).
(I) Autophagic degradation of Cdc48 mutants requires the UDS of Atg8. WT or Δatg8 cells expressing YFP-CDC48 mutants, with or without rescue with HA-tagged WT Atg8 or the ΔLDS or ΔUDS variants, were grown in medium containing galactose for 16 hours, and YFP release was monitored as in panel (D).
(J) The UDS is not required for Atg8 lipidation. Cells expressing WT Atg8 or the ΔLDS or ΔUDS variants were grown in nutrient-rich medium then subjected to nitrogen starvation for 2 hours. Total cell extracts were treated for 1 hour with or without phospholipase D, separated by SDS-PAGE in the presence of 6 M urea, and subjected to immunoblot analysis with anti-ATG8a antibodies.
(K) Quantification of the ratio of Atg8 to Atg8-PE from the experiment shown in panel (J) and two additional independent biological replicates.
In each of panels (A), (C), (E), (F), (H) and (K), bars represent the mean (±SD) from three independent biological replicates, each comprised of three technical replicates (except panel (K)). Asterisks indicate data points that are statistically significantly different to the control (p < 0.05).
Table S1: Accession Numbers of Genes and Proteins Used in This Study, and Associated Named Mutations; Related to Figures 1, 2 and 4, and STAR Methods.
Table S2: List and Alignment of Plant ATG8 Nucleotide and Amino Acid Sequences Used in This Study; Related to Figure 1.
Table S3: Results of Arabidopsis thaliana and Saccharomyces cerevisiae cDNA Library Screens; Related to Figures 2 and 4.
Table S5: Saccharomyces cerevisiae Strains Used in This Study; Related to STAR Methods.
Table S6: Oligonucleotide Primers Used in This Study; Related to STAR Methods.
Figure S1. Control Y2H, BiFC and in vitro binding assays used to map the RPN10 binding site on ATG8. Related to Figures 1 and 2.
(A, B and C) Growth of transformed yeast cells on synthetic dropout medium lacking leucine and tryptophan from the Y2H assays in Figures 1A, 1B and 1E, confirming that all plasmid combinations are able to support growth on non-selective medium.
(D) BiFC assays showing that the NYFP- or CYFP-tagged RPN10 and ATG8a constructs do not produce a fluorescence signal due to auto-activation. N. benthamiana leaf epidermal cells were co-infiltrated with the indicated plasmid combinations, and fluorescence signals were detected by confocal fluorescence microscopy 36 hours following infiltration. Merged bright field (BF) and fluorescence images are shown. Scale bar, 10 μm.
(E) Mutation of the ATG8e LDS or UDS does not impair binding of RPN10 or DSK2a to the alternate site. Equal amounts (2 μg) of purified protein were incubated together and pulled down with Ni-NTA agarose beads. Input and bound proteins were then visualized by immunoblotting with anti-ATG8 and anti-GST antibodies.
(F, G, H and I) Growth of transformed yeast cells on synthetic dropout medium lacking leucine and tryptophan from the experiments in Figures 2A, 2B, 2C and 2G, confirming that all plasmid combinations are able to support growth on non-selective medium.
Figure S2. Four Arabidopsis PUX proteins interact with both ATG8 and CDC48. Related to Figures 2 and 6.
(A) Box diagrams of the 16-member Arabidopsis UBX domain-containing family. Positions of the UBA, PUB, SEP and UBX domains are indicated by the green, purple, orange and blue boxes, respectively, while the UIMs are highlighted in red. The amino acid length of each polypeptide is shown on the right. Accession numbers for the given proteins are listed in Table S1.
(B) Phylogenetic analysis of the Arabidopsis PUX protein family. The full-length predicted amino acid sequences of the 16 Arabidopsis PUX proteins were aligned in Clustal Omega, subjected to Bayesian phylogenetic analysis with MrBayes version 3.2, and the resulting consensus tree displayed using FigTree. ATG8-interacting PUX proteins are highlighted in red; the scale bar represents substitutions per site.
(C) Y2H assays testing interactions between ATG8a and the 16 Arabidopsis PUX proteins. Full-length proteins fused with either the GAL4 activating (AD) or DNA-binding (BD) domains at their N-termini were co-expressed in the indicated pair-wise orientations. Shown are cells grown on medium lacking leucine and tryptophan (left panel), or leucine, tryptophan and histidine and containing 25 mM 3-amino-1,2,4-triazole (3-AT; right panel).
(D and E) Quantification of the binding affinity between PUX7, PUX9 or PUX13 and ATG8e. Various concentrations of 6His-ATG8e were incubated with 1 μM GST-tagged PUX protein and pulled down with Ni-NTA beads. PUX protein remaining in the supernatant was then quantified by immunoblotting with anti-GST antibodies and expressed as a percentage of the input. Values represent the mean (±SD) from three independent biological replicates.
(F) Purity of the proteins used for isothermal titration calorimetry analysis. The indicated proteins were expressed and purified from E. coli by Ni-NTA affinity chromatography, and equal amounts of protein (2 μg) were subjected to SDS-PAGE and stained for protein with Coomassie Brilliant Blue.
(G) Quantification of the binding affinity between RPN10, PUX7, PUX9 or PUX13 and ATG8e, as determined by isothermal titration calorimetry (ITC). Top panels show heat differences upon successive injections of 6His-tagged RPN10 or PUX protein into WT 6His-ATG8e, while lower panels show integrated heats of injection (squares) and the best fit to a single site binding model (solid lines) for interaction with WT (black) or ΔUDS (grey) forms of ATG8e. The N and KD values represent the mean (±SD) from three independent biological replicates.
(H and I) CDC48 and ATG8 co-immunoprecipitate together with GFP-PUX7 from Arabidopsis seedlings. Total protein extracts from 7-day-old WT, GFP or GFP-PUX7 seedlings were incubated with GFP-Trap®_A beads, washed, and eluted at low pH. Fractions were subjected to SDS-PAGE and either stained for protein with silver, or immunoblotted with anti-CDC48, anti-GFP or anti-ATG8 antibodies.
(J) Sequence alignment of all ATG8-interacting PUX proteins from Arabidopsis. Identical amino acids (50% threshold value) are shown with a black background; similar amino acids are shown with a grey background. Positions of the UBA and UBX domains and the UIMs are indicated by green, blue and red lines, respectively. The amino acids mutated to abolish UIM binding activity are indicated by red asterisks; the positions of T-DNA insertions analyzed in Figures S5G and S5H are shown by purple arrowheads. Amino acid numbers are indicated on the right.
Figure S3. Control Y2H and dot blot binding assays used to determine the requirements for UIM-UDS interaction. Related to Figures 3 and 5.
(A) Saturating mutagenesis of the Arabidopsis ATG8a UDS does not impact its interaction with ATG7 or DSK2a, or its expression in E. coli. The panel of mutants employed in Figures 3A and 3B were tested for their ability to interact with ATG7 by Y2H (upper left panel), with DSK2a by Y2H and in vitro dot blot binding assay (lower panels), or to accumulate in E. coli, as detected by immunoblot of lysed cell extracts with anti-ATG8a antibodies (upper right panel).
(B and C) Saturating mutagenesis of the Arabidopsis RPN10 and PUX7 UIMs does not impact their interaction with RPN9a and CDC48a, respectively, or their expression in E. coli. The panel of mutants employed in Figures 5A and 5B were tested for their ability to interact with RPN9a or CDC48a by Y2H (upper panel), or to accumulate in E. coli, as detected by immunoblot of lysed cell extracts with anti-GST antibodies (lower panel).
HIGHLIGHTS.
A new binding site for autophagy adaptors and receptors was discovered on ATG8.
This site engages UIM-like sequences rather than the canonical ATG8-interacting motif.
UIM-type autophagy adaptors/receptors can be found in plants, yeast and humans.
UIM-containing UBX domain proteins mediate autophagic clearance of CDC48/p97.
ACKNOWLEDGEMENTS
We wish to thank Sebastian Y. Bednarek, Pedro Carvalho, Stephen J. Elledge, Jean-Luc Gallois, Daniel J. Klionsky, and Mamie Z. Li for generously sharing reagents, and Robert C. Augustine, E. Sethe Burgie, Ryan Calcutt, Michael Dyer, Faqiang Li, Katrice E. McLoughlin, Samantha K. Powers and Joseph M. Walker for advice and technical support. Work in the Vierstra lab was supported by grants from the National Institutes of Health, National Institute of General Medical Science (R01-GM124452-01A1) and the National Science Foundation, Plant Genome Research Program (IOS-1339325). Work in the Hua lab was supported by an Ohio University Start-Up grant (SU-1007172) and a National Science Foundation CAREER award (MCB-1750361).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Bechtold N, Ellis J, and Pelletier G (1993). In plants Agrobacterium-mediated gene transfer by infiltration of adult Arabidopsis thaliana plants. C. R. Acad. Sci 316, 1194–1199. [Google Scholar]
- Behrends C, Sowa ME, Gygi SP, and Harper JW (2010). Network organization of the human autophagy system. Nature 466, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davis A, Cost GJ, Caputo E, Li J, Hieter P, and Boeke JD (1998). Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132. [DOI] [PubMed] [Google Scholar]
- Clough SJ, and Bent AF (1998). Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743. [DOI] [PubMed] [Google Scholar]
- Curtis MD, and Grossniklaus U (2003). A Gateway® cloning vector set for high-throughput functional analysis of genes in planta. Plant Physiol 133, 462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler SR, Ehrhardt DW, Griffitts JS, and Somerville CR (2000). A set of random GFP::cDNA fusions enables visualization of subcellular structures in cells of Arabidopsis at high frequency. Proc. Natl. Acad. Sci. USA 97, 3718–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Besten W, Verma R, Kleiger G, Oania RS, and Deshaies RJ (2012). NEDD8 links cullin-RING ubiquitin ligase function to the p97 pathway. Nat. Struct. Mol. Biol 19, 511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge SJ, Mulligan JT, Ramer SW, Spottswood M, and Davis RW (1991). λ-YES: a multi-functional cDNA expression vector for the isolation of genes by complementation of yeast and Escherichia coli mutations. Proc. Natl. Acad. Sci. USA 88, 1731–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley D, Ozkaynak E, and Varshavsky A (1987). The yeast polyubiquitin gene is essential for resistance to high temperatures, starvation, and other stresses. Cell 48, 1035–1046. [DOI] [PubMed] [Google Scholar]
- Ford MG, Mills IG, Peter BJ, Vallis Y, Praefcke GJ, Evans PR, and McMahon HT (2002). Curvature of clathrin-coated pits driven by Epsin. Nature 419, 361–366. [DOI] [PubMed] [Google Scholar]
- Gallois JL, Drouaud J, Lecureuil A, Guyon-Debast A, Bonhomme S, and Guerche P (2013). Functional characterization of the plant ubiquitin regulatory X (UBX) domain-containing protein PUX7 in Arabidopsis thaliana. Gene 526, 299–308. [DOI] [PubMed] [Google Scholar]
- Gatica D, Lahiri V, and Klionsky DJ (2018). Cargo recognition and degradation by selective autophagy. Nat. Cell Biol 20, 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajdukiewicz P, Svab Z, and Maliga P (1994). The small, versatile pPZP family of Agrobacterium binary vectors for plant transformation. Plant Mol. Biol 25, 989–994. [DOI] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, and Elledge SJ (1993). The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75, 805–816. [DOI] [PubMed] [Google Scholar]
- Hofius D, Schultz-Larsen T, Joensen J, Tsitsigiannis DI, Petersen NH, Mattsson O, J0rgensen LB, Jones JDG, Mundy J, and Petersen M (2009). Autophagic components contribute to hypersensitive cell death in Arabidopsis. Cell 137, 773–783. [DOI] [PubMed] [Google Scholar]
- Hofmann K, and Falquet L (2001). A ubiquitin-interacting motif conserved in components of the proteasomal and lysosomal protein degradation systems. Trends Biochem. Sci 26, 347–350. [DOI] [PubMed] [Google Scholar]
- Kaplun L, Ivantsiv Y, Bakhrat A, and Raveh D (2003). DNA damage response-mediated degradation of HO endonuclease via the ubiquitin system involves its nuclear export. J. Biol. Chem 278, 48727–48734. [DOI] [PubMed] [Google Scholar]
- Karimi M, Inze D, and Depicker A (2002). Gateway® vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci 7, 193–195. [DOI] [PubMed] [Google Scholar]
- Kellner R, De La Concepcion JC, Maqbool A, Kamoun S, and Dagdas YF (2017). ATG8 expansion: a driver of selective autophagy diversification. Trends Plant Sci 22, 204–214. [DOI] [PubMed] [Google Scholar]
- Khaminets A, Behl C, and Dikic I (2016). Ubiquitin-dependent and -independent signals in selective autophagy. Trends Cell Biol 26, 6–16. [DOI] [PubMed] [Google Scholar]
- Krick R, Bremer S, Welter E, Schlotterhose P, Muehe Y, Eskelinen EL, and Thumm M (2010). Cdc48/p97 and Shp1/p47 regulate autophagosome biogenesis in concert with ubiquitin-like Atg8. J. Cell Biol 190, 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, and Kroemer G (2019). Biological functions of autophagy genes: a disease perspective. Cell 176, 11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Yang P, Huang X, Zhang H, Lu Q, and Zhang H (2013). The scaffold protein EPG-7 links cargo receptor complexes with the autophagic assembly machinery. J. Cell Biol 201, 113–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Psakhye I, and Jentsch S (2014). Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 158, 549–563. [DOI] [PubMed] [Google Scholar]
- Marshall RS, Jolliffe NA, Ceriotti A, Snowden CJ, Lord JM, Frigerio L, and Roberts LM (2008). The role of CDC48 in the retro-translocation of non-ubiquitinated toxin substrates in plant cells. J. Biol. Chem 283, 15869–115877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall RS, Li F, Gemperline DC, Book AJ, and Vierstra RD (2015). Autophagic degradation of the 26S proteasome is mediated by the dual ATG8/ubiquitin receptor RPN10 in Arabidopsis. Mol. Cell 58, 1053–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall RS, McLoughlin F, and Vierstra RD (2016). Autophagic turnover of inactive 26S proteasomes in yeast is directed by the ubiquitin receptor Cue5 and the Hsp42 chaperone. Cell Rep 16, 1717–1732. [DOI] [PubMed] [Google Scholar]
- Marshall RS, and Vierstra RD (2018). Autophagy: the master of bulk and selective recycling. Annu. Rev. Plant Biol 69, 173–208. [DOI] [PubMed] [Google Scholar]
- Noda T, Matsuura A, Wada Y, and Ohsumi Y (1995). A novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun 210, 126–132. [DOI] [PubMed] [Google Scholar]
- Noda NN, Kumeta H, Nakatogawa H, Satoo K, Adachi W, Ishii J, Fujioka Y, Ohsumi Y, and Inagaki F (2008). Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells 13, 1211–1218. [DOI] [PubMed] [Google Scholar]
- Noda NN, Ohsumi Y, and Inagaki F (2010). An Atg8-family interacting motif crucial for selective autophagy. FEBS Lett 584, 1379–1385. [DOI] [PubMed] [Google Scholar]
- Nolan TM, Brennan B, Yang M, Chen J, Zhang M, Li Z, Wang X, Bassham DC, Walley J, and Yin Y (2017). Selective autophagy of BES1 mediated by DSK2 balances plant growth and survival. Dev. Cell 41, 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsumi Y (2001). Molecular dissection of autophagy: two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol 2, 211–216. [DOI] [PubMed] [Google Scholar]
- O’Malley RC, Barragan CC, and Ecker JR (2015). A user’s guide to the Arabidopsis T-DNA insertion mutant collections. Methods Mol. Biol 1284, 323–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Rancour DM, and Bednarek SY (2008). In planta analysis of the cell cycle-dependent localization of AtCDC48a and its critical roles in cell division, expansion and differentiation. Plant Physiol 148, 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips AR, Suttangkakul A, and Vierstra RD (2008). The ATG12-conjugating enzyme ATG10 is essential for autophagic vesicle formation in Arabidopsis thaliana. Genetics 178, 1339–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancour DM, Park S, Knight SD, and Bednarek SY (2004). Plant UBX domain-containing protein 1, PUX1, regulates the oligomeric structure and activity of Arabidopsis CDC48. J. Biol. Chem 279, 54264–54274. [DOI] [PubMed] [Google Scholar]
- Robinson JS, Klionsky DJ, Banta L, and Emr SD (1988). Protein sorting in Saccharomyces cerevisiae: isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol. Cell. Biol 8, 4936–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, and Huelsenbeck JP (2003). Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. [DOI] [PubMed] [Google Scholar]
- Ruggiano A, Mora G, Buxo L, and Carvalho P (2016). Spatial control of lipid droplet proteins by the ERAD ubiquitin ligase Doa10. EMBO J 35, 1644–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stach L, and Freemont PS (2017). The AAA-ATPase p97: a cellular multi-tool. Biochem. J 474, 2953–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttangkakul A, Li F, Chung T, and Vierstra RD (2011). The ATG1/ATG13 protein kinase complex is both a regulator and a target of autophagic recycling in Arabidopsis. Plant Cell 23, 3761–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang WK, and Xia D (2016). Mutations in the human AAA chaperone p97 and related diseases. Front. Mol. Biosci 3, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas BJ, and Rothstein R (1989). Elevated recombination rates in transcriptionally active DNA. Cell 56, 619–630. [DOI] [PubMed] [Google Scholar]
- Thompson AR, Doelling JH, Suttangkakul A, and Vierstra RD (2005). Autophagic nutrient recycling in Arabidopsis directed by the ATG8 and ATG12 conjugation pathways. Plant Physiol 138, 2097–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Brachmann RK, Fattaey A, Harlow E, and Boeke JD (1996). Reverse two-hybrid and one-hybrid systems to detect dissociation of protein-protein and DNA-protein interactions. Proc. Natl. Acad. Sci. USA 93, 10315–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland B, Steece KE, and Emr SD (1999). Yeast Epsins contain an essential N-terminal ENTH domain, bind clathrin, and are required for endocytosis. EMBO J 18, 4383–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Bentio R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Diet-rich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M’Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Véronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, and Davis RW (1999). Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906. [DOI] [PubMed] [Google Scholar]
- Yang F, Kimberlin AN, Elowsky CG, Liu Y, Gonzalez-Solis A, Cahoon EB, and Alfano JR (2019). A plant immune receptor degraded by selective autophagy. Mol. Plant 12, 113–123. [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, and Rapoport TA (2001). The AAA-ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414, 652–656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dataset S1: Mass Spectrometric Analysis of GFP-PUX7 Co-immunoprecipitations; Related to Figure 6.
Figure S4. Control Y2H assays used to identify and map UIM-containing ATG8-interacting proteins from yeast and humans. Related to Figure 4.
(A, B, D, E, F and G) Growth of transformed yeast cells on synthetic dropout medium lacking leucine and tryptophan from the experiments in Figures 4A, 4B, 4C and 4E, confirming that all plasmid combinations are able to support growth on non-selective medium.
(C) Yeast Ufo1 does not use a UIM to interact with Atg8. Various Ufo1 combinations, either full length, missing the FBX domain, or with mutations in one or more UIM sequences, were tested for interaction with WT, ΔLDS or ΔUDS forms of Atg8. Details of all mutations are given in Table S1. Shown are cells grown on selective and non-selective medium, as in Figure S2C.
(H, I and J) The UIM regions in human ATXN3, ATXN3L, EPN1, EPN2, EPN3 and RBSN are required for binding to MAP1LC3a and/or GABARAP at the UDS. Various combinations of the above proteins, either WT or with mutations in one or more of their UIM sequences, were tested for interaction with WT, ΔLDS or ΔUDS forms of MAP1LC3a or GABARAP. Shown are cells grown on both selective and non-selective medium, as in panel (C).
Figure S5. Identification of p97 inhibitors effective in plants and assembly of a quadruple mutant eliminating the four Arabidopsis ATG8-interacting PUX proteins. Related to Figure 6.
(A) The human CDC48/p97 inhibitor CB-5083 induces ER stress in Arabidopsis. Total RNA was extracted from 7-day-old WT seedlings treated with the given concentrations of CB-5083, DBeQ, ML240 or NMS-873 for 16 hours, and converted to first-strand cDNA. Relative transcript abundance of the ER stress-responsive genes BIP1 and CRT1 was determined by quantitative real-time PCR, using the ACT2 and PP2A genes as internal reference standards. All data points were normalized to untreated seedlings; the bars represent mean (±SD) from three independent biological replicates, each with three technical replicates.
(B) CB-5083 treatment impairs Arabidopsis seedling growth. WT seedlings were grown for 7 days on solid GM medium containing the indicated concentrations of CB-5083, DBeQ, ML240 or NMS-873.
(C and D) CB-5083 stimulates the autophagic turnover of both GFP-PUX7 (panel (C)) and YFP-CDC48a (panel (D)), as determined by the GFP/YFP-release assay. Seedlings were grown for 6 days and then switched from nutrient-rich medium to either medium lacking nitrogen or medium containing 20 μM of CB-5083, DBeQ, ML240 or NMS-873 for 16 hours. Total protein extracts were assayed for GFP or YFP release as in Figure 6A.
(E) CB-5083 treatment reduces bulk autophagic flux in Arabidopsis. Seedlings expressing GFP-ATG8a were grown for 6 days, treated with 20 μM CB-5083, DBeQ, ML240 or NMS-873 for 16 hours, then switched from nutrient-rich medium to medium lacking nitrogen for an additional 16 hours. Total protein extracts were assayed for GFP release as in panel (C).
(F) Quantification of the ratio of free GFP to GFP-ATG8a from the experiment shown in panel (E) and two additional independent biological replicates. Bars represent the mean (±SD) from three independent biological replicates; letters indicate values statistically significantly different from one another and the control (p < 0.05).
(G) Diagram of the PUX7, PUX8, PUX9 and PUX13 genes. The boxes represent coding regions (colored) or predicted untranslated regions (white), while lines represent introns. The green, red and blue boxes identify UBA domains, UIMs and UBX domains, respectively. Positions of the T-DNA insertions are indicated by the red triangles; sites of amino acid sequence disruption are shown in Figure S2D. Positions of oligonucleotide primers used for reverse transcription (RT)-PCR in panel (H) are indicated by the half arrowheads.
(H) RT-PCR analysis of the PUX7, PUX8, PUX9 and PUX13 transcripts in the indicated T-DNA insertions. Total RNA was extracted from 7-day-old WT or homozygous mutant seedlings, converted to first-strand cDNA, and subjected to RT-PCR using the primer pairs shown in panel (G). Amplification of ACT2 was included to confirm analysis of equal amounts of cDNA.
(I) Phenotypes of individual and higher order pux mutant plants. Plants with the indicated pux genotypes were grown on soil for 3 weeks under a long-day photoperiod.
Figure S6. p97 amino acids associated with human disease are conserved in Arabidopsis CDC48. Related to Figure 6.
Sequence alignment of CDC48 proteins from Arabidopsis thaliana, Saccharomyces cerevisiae and humans. Identical amino acids (55% threshold value) are shown with a black background; similar amino acids are shown with a grey background. Positions of the N-terminal domain and the D1 and D2 ATPase domains are indicated by green, red and blue lines, respectively, while the C-terminal HbYX motif that docks onto the 20S proteasome is indicated with a yellow line. Conserved residues implicated in human disease are indicated by purple arrowheads; residues critical for ATP binding or hydrolysis are indicated by the red and brown asterisks, respectively. Amino acid numbers are indicated on the right; accession numbers for these proteins are listed in Table S1.
Figure S7. Human p97 inhibitors do not impact yeast Cdc48, but genetic Cdc48 mutants can impact autophagic flux. Related to Figure 7.
(A) Human p97 inhibitors do not induce ER stress in yeast. Total RNA was extracted from WT cells following 16 hour incubation with the given concentrations of CB-5083, DBeQ, ML240 or NMS-873, and converted to first-strand cDNA. The relative transcript abundance of the ER stress-responsive genes DER1 and ERO1 was determined by quantitative real-time PCR, using the ALG9 and TFC1 genes as internal reference standards. All data points were normalized to untreated cells. Similar results were obtained with Δerg6 and Δpdr5 cells.
(B) Yeast growth is not impacted by p97 inhibitors. Cells of the indicated genotype were treated for 16 hours with or without 50 μM MG132 or 2 mM CB-5083, DBeQ, ML240 or NMS-873, and equal numbers of cells were subjected to 5-fold serial dilutions, spotted onto synthetic dropout medium also containing the inhibitors, and incubated at 30°C for 36 hours. The treatment with MG132 was included as a positive control for the Δerg6 and Δpdr5 deletions.
(C) Bulk autophagic flux in yeast is not impacted by p97 inhibitors, as measured by assay of the Pho8Δ60 reporter. Cells of the indicated genotypes expressing PHO8Δ60 were treated with or without 2 mM CB-5083, DBeQ, ML240 or NMS-873, or switched to growth at 37Ό (for cdc48-6 cells only) for 16 hours, then subjected to nitrogen starvation for 0 or 8 hours. All data points were normalized to those obtained after 0 hours nitrogen starvation.
(D) Yeast Cdc48 is not subject to autophagy following treatment with human p97 inhibitors, as determined by the YFP release assay. Release of free YFP from the YFP-Cdc48 reporter upon nitrogen starvation or treatment with 2 mM CB-5083, DBeQ, ML240 or NMS-873 for 16 hours was monitored in WT, Δerg6 or Δpdr5 cells by immunoblot analysis of total protein extracts with anti-GFP antibodies, as in Figure 7A.
(E) Mutations in genomic CDC48 impact bulk autophagic flux as induced by nitrogen starvation. Cells of the indicated genotypes expressing PHO8Δ60 were grown at 30 or 37°C for 8 hours, then subjected to nitrogen starvation for 0 or 8 hours, as in panel (C).
(F) Mutant versions of Cdc48 ectopically expressed from a galactose-inducible promoter in WT cells have less impact on autophagic flux than when they replace the genomic locus. Cells expressing PHO8Δ60 were transformed with plasmids encoding YFP-CDC48, YFP-cdc48-3 or YFP-cdc48-6 under control of the GAL1 promoter. Cells were then grown at either 30 or 37Ό in medium containing glucose (−) or galactose (+) for 8 hours, and subjected to nitrogen starvation for 0 or 8 hours, as in panel (C).
(G) The cdc48-3 and cdc48-6 proteins are poor autophagy substrate, as determined by the YFP release assay. WT cells were transformed with the same plasmids as in panel (F), grown at 30°C, then switched to growth at 30 or 37°C in medi um containing galactose for 48 hours. Total protein extracts were assayed for YFP release by immunoblot analysis as in panel (D).
(H) Ectopic expression of Cdc48 variants containing mutations in conserved disease-associated amino acids only mildly impacts bulk autophagic flux. WT cells expressing PHO8Δ60 were transformed with plasmids encoding YFP-CDC48 or the P147L, E195K or K396E variants under control of the GAL1 promoter. Cells were grown in medium containing glucose (−) or galactose (+) for 8 hours, and then subjected to nitrogen starvation for 0 or 8 hours, as in panel (C).
(I) Autophagic degradation of Cdc48 mutants requires the UDS of Atg8. WT or Δatg8 cells expressing YFP-CDC48 mutants, with or without rescue with HA-tagged WT Atg8 or the ΔLDS or ΔUDS variants, were grown in medium containing galactose for 16 hours, and YFP release was monitored as in panel (D).
(J) The UDS is not required for Atg8 lipidation. Cells expressing WT Atg8 or the ΔLDS or ΔUDS variants were grown in nutrient-rich medium then subjected to nitrogen starvation for 2 hours. Total cell extracts were treated for 1 hour with or without phospholipase D, separated by SDS-PAGE in the presence of 6 M urea, and subjected to immunoblot analysis with anti-ATG8a antibodies.
(K) Quantification of the ratio of Atg8 to Atg8-PE from the experiment shown in panel (J) and two additional independent biological replicates.
In each of panels (A), (C), (E), (F), (H) and (K), bars represent the mean (±SD) from three independent biological replicates, each comprised of three technical replicates (except panel (K)). Asterisks indicate data points that are statistically significantly different to the control (p < 0.05).
Table S1: Accession Numbers of Genes and Proteins Used in This Study, and Associated Named Mutations; Related to Figures 1, 2 and 4, and STAR Methods.
Table S2: List and Alignment of Plant ATG8 Nucleotide and Amino Acid Sequences Used in This Study; Related to Figure 1.
Table S3: Results of Arabidopsis thaliana and Saccharomyces cerevisiae cDNA Library Screens; Related to Figures 2 and 4.
Table S5: Saccharomyces cerevisiae Strains Used in This Study; Related to STAR Methods.
Table S6: Oligonucleotide Primers Used in This Study; Related to STAR Methods.
Figure S1. Control Y2H, BiFC and in vitro binding assays used to map the RPN10 binding site on ATG8. Related to Figures 1 and 2.
(A, B and C) Growth of transformed yeast cells on synthetic dropout medium lacking leucine and tryptophan from the Y2H assays in Figures 1A, 1B and 1E, confirming that all plasmid combinations are able to support growth on non-selective medium.
(D) BiFC assays showing that the NYFP- or CYFP-tagged RPN10 and ATG8a constructs do not produce a fluorescence signal due to auto-activation. N. benthamiana leaf epidermal cells were co-infiltrated with the indicated plasmid combinations, and fluorescence signals were detected by confocal fluorescence microscopy 36 hours following infiltration. Merged bright field (BF) and fluorescence images are shown. Scale bar, 10 μm.
(E) Mutation of the ATG8e LDS or UDS does not impair binding of RPN10 or DSK2a to the alternate site. Equal amounts (2 μg) of purified protein were incubated together and pulled down with Ni-NTA agarose beads. Input and bound proteins were then visualized by immunoblotting with anti-ATG8 and anti-GST antibodies.
(F, G, H and I) Growth of transformed yeast cells on synthetic dropout medium lacking leucine and tryptophan from the experiments in Figures 2A, 2B, 2C and 2G, confirming that all plasmid combinations are able to support growth on non-selective medium.
Figure S2. Four Arabidopsis PUX proteins interact with both ATG8 and CDC48. Related to Figures 2 and 6.
(A) Box diagrams of the 16-member Arabidopsis UBX domain-containing family. Positions of the UBA, PUB, SEP and UBX domains are indicated by the green, purple, orange and blue boxes, respectively, while the UIMs are highlighted in red. The amino acid length of each polypeptide is shown on the right. Accession numbers for the given proteins are listed in Table S1.
(B) Phylogenetic analysis of the Arabidopsis PUX protein family. The full-length predicted amino acid sequences of the 16 Arabidopsis PUX proteins were aligned in Clustal Omega, subjected to Bayesian phylogenetic analysis with MrBayes version 3.2, and the resulting consensus tree displayed using FigTree. ATG8-interacting PUX proteins are highlighted in red; the scale bar represents substitutions per site.
(C) Y2H assays testing interactions between ATG8a and the 16 Arabidopsis PUX proteins. Full-length proteins fused with either the GAL4 activating (AD) or DNA-binding (BD) domains at their N-termini were co-expressed in the indicated pair-wise orientations. Shown are cells grown on medium lacking leucine and tryptophan (left panel), or leucine, tryptophan and histidine and containing 25 mM 3-amino-1,2,4-triazole (3-AT; right panel).
(D and E) Quantification of the binding affinity between PUX7, PUX9 or PUX13 and ATG8e. Various concentrations of 6His-ATG8e were incubated with 1 μM GST-tagged PUX protein and pulled down with Ni-NTA beads. PUX protein remaining in the supernatant was then quantified by immunoblotting with anti-GST antibodies and expressed as a percentage of the input. Values represent the mean (±SD) from three independent biological replicates.
(F) Purity of the proteins used for isothermal titration calorimetry analysis. The indicated proteins were expressed and purified from E. coli by Ni-NTA affinity chromatography, and equal amounts of protein (2 μg) were subjected to SDS-PAGE and stained for protein with Coomassie Brilliant Blue.
(G) Quantification of the binding affinity between RPN10, PUX7, PUX9 or PUX13 and ATG8e, as determined by isothermal titration calorimetry (ITC). Top panels show heat differences upon successive injections of 6His-tagged RPN10 or PUX protein into WT 6His-ATG8e, while lower panels show integrated heats of injection (squares) and the best fit to a single site binding model (solid lines) for interaction with WT (black) or ΔUDS (grey) forms of ATG8e. The N and KD values represent the mean (±SD) from three independent biological replicates.
(H and I) CDC48 and ATG8 co-immunoprecipitate together with GFP-PUX7 from Arabidopsis seedlings. Total protein extracts from 7-day-old WT, GFP or GFP-PUX7 seedlings were incubated with GFP-Trap®_A beads, washed, and eluted at low pH. Fractions were subjected to SDS-PAGE and either stained for protein with silver, or immunoblotted with anti-CDC48, anti-GFP or anti-ATG8 antibodies.
(J) Sequence alignment of all ATG8-interacting PUX proteins from Arabidopsis. Identical amino acids (50% threshold value) are shown with a black background; similar amino acids are shown with a grey background. Positions of the UBA and UBX domains and the UIMs are indicated by green, blue and red lines, respectively. The amino acids mutated to abolish UIM binding activity are indicated by red asterisks; the positions of T-DNA insertions analyzed in Figures S5G and S5H are shown by purple arrowheads. Amino acid numbers are indicated on the right.
Figure S3. Control Y2H and dot blot binding assays used to determine the requirements for UIM-UDS interaction. Related to Figures 3 and 5.
(A) Saturating mutagenesis of the Arabidopsis ATG8a UDS does not impact its interaction with ATG7 or DSK2a, or its expression in E. coli. The panel of mutants employed in Figures 3A and 3B were tested for their ability to interact with ATG7 by Y2H (upper left panel), with DSK2a by Y2H and in vitro dot blot binding assay (lower panels), or to accumulate in E. coli, as detected by immunoblot of lysed cell extracts with anti-ATG8a antibodies (upper right panel).
(B and C) Saturating mutagenesis of the Arabidopsis RPN10 and PUX7 UIMs does not impact their interaction with RPN9a and CDC48a, respectively, or their expression in E. coli. The panel of mutants employed in Figures 5A and 5B were tested for their ability to interact with RPN9a or CDC48a by Y2H (upper panel), or to accumulate in E. coli, as detected by immunoblot of lysed cell extracts with anti-GST antibodies (lower panel).
Data Availability Statement
Datasets generated from cDNA library screens are provided in Table S3. The mass spectrometry-based proteomics data have been deposited to the ProteomeXchange Consortium (www.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD011397. All accession numbers for genes and proteins used in this study are provided in Table S1.