

The innovation of new therapies to combat multidrug-resistant (MDR) bacteria is being outpaced by the continued rise of MDR bacterial infections. Of particular concern are hospital-acquired infections (HAIs) that are recalcitrant to antibiotic therapies. The Gram-positive intestinal pathobiont Enterococcus faecalis is associated with HAIs, and some strains are MDR. Therefore, novel strategies to control E. faecalis populations are needed.
Keywords: Enterococcus faecalis, CRISPR-Cas, antibiotic resistance, conjugation, plasmid, intestinal colonization, pheromone
ABSTRACT
The innovation of new therapies to combat multidrug-resistant (MDR) bacteria is being outpaced by the continued rise of MDR bacterial infections. Of particular concern are hospital-acquired infections (HAIs) that are recalcitrant to antibiotic therapies. The Gram-positive intestinal pathobiont Enterococcus faecalis is associated with HAIs, and some strains are MDR. Therefore, novel strategies to control E. faecalis populations are needed. We previously characterized an E. faecalis type II CRISPR-Cas system and demonstrated its utility in the sequence-specific removal of antibiotic resistance determinants. Here, we present work describing the adaption of this CRISPR-Cas system into a constitutively expressed module encoded on a pheromone-responsive conjugative plasmid that efficiently transfers to E. faecalis for the selective removal of antibiotic resistance genes. Using in vitro competition assays, we show that these CRISPR-Cas-encoding delivery plasmids, or CRISPR-Cas antimicrobials, can reduce the occurrence of antibiotic resistance in enterococcal populations in a sequence-specific manner. Furthermore, we demonstrate that deployment of CRISPR-Cas antimicrobials in the murine intestine reduces the occurrence of antibiotic-resistant E. faecalis by several orders of magnitude. Finally, we show that E. faecalis donor strains harboring CRISPR-Cas antimicrobials are immune to uptake of antibiotic resistance determinants in vivo. Our results demonstrate that conjugative delivery of CRISPR-Cas antimicrobials may be adaptable for future deployment from probiotic bacteria for exact targeting of defined MDR bacteria or for precision engineering of polymicrobial communities in the mammalian intestine.
INTRODUCTION
Disruption of the intestinal microbiota can predispose individuals to infection by opportunistic pathogens (1, 2). Antibiotics facilitate such disturbances, leading to the development of hospital-acquired infections (HAIs) (3, 4). Enterococcus faecalis, a normal constituent of the healthy human intestinal microbiota and historically used in probiotics and during food fermentation, is now a leading cause of HAIs (5–7). E. faecalis is an adept opportunist that can proliferate in the dysbiotic intestine following antibiotic perturbation (8). Antibiotic-resistant clinical isolates of E. faecalis typically possess expanded genomes compared to susceptible isolates due to the acquisition of mobile genetic elements (9, 10), and patients colonized with these multidrug-resistant (MDR) E. faecalis strains are at increased risk of acquiring bloodstream infections (11). Thus, there is a need for novel strategies to decolonize high-risk individuals of MDR E. faecalis (12).
CRISPR-Cas systems are protective barriers in prokaryotes that function in the adaptive immunity to mobile genetic elements (13–15). The well-studied type II CRISPR-Cas system consists of a DNA endonuclease (Cas9) that uses a programmable RNA guide to cleave a double-stranded DNA target sequence (16). Double-stranded DNA breaks can be lethal to bacteria (17); therefore, deploying type II CRISPR-Cas as a sequence-specific antimicrobial is an attractive alternative to conventional antibiotic therapy. Previous studies have explored this concept using phages to deliver engineered CRISPR systems targeting specific genes (18–21). Although effective, this method of delivery is likely to be challenging for many species of bacteria, including E. faecalis, due to phage receptor and other cell wall mutations that arise readily in response to lytic phage infection (22–25).
We previously demonstrated the utility of CRISPR-Cas9 as a sequence-specific in vitro antimicrobial in E. faecalis by delivering CRISPR guide sequences on mobilizable cloning vectors into E. faecalis cells that chromosomally encode cas9 (26). However, since MDR E. faecalis strains lack complete CRISPR-Cas systems, and specifically cas9 (27), efficient delivery of the entire CRISPR-Cas9 machinery to MDR E. faecalis would likely be required. An alternative method for the delivery of CRISPR-Cas is the use of pheromone-responsive plasmids (PRPs), which naturally achieve high rates of conjugation, have a narrow host range limited to E. faecalis, are capable of comprehensively infiltrating E. faecalis populations, and can disseminate within intestinal E. faecalis in the absence of antibiotic selection (28–31). The transmission of PRPs is tightly regulated, so the deployment of a PRP CRISPR-Cas antimicrobial could be conditionally tuned for precision targeting and delivery (32).
PRPs respond to pheromones (short peptides) that are secreted by plasmid-free E. faecalis cells (33). When donor cells detect excess pheromones, indicating the presence of plasmid-free cells in the vicinity (34), they synthesize an aggregation substance which facilitates contact between plasmid-bearing and plasmid-free cells (35). This enables the rapid and efficient transfer of PRPs from donor to recipient. In addition, many PRPs encode accessory genes such as antibiotic resistance genes and bacteriocins, small ribosomally synthesized antimicrobial proteins that target diverse bacteria and for which producing strains must encode immunity genes (36–38). Previous work demonstrated that the PRP pPD1 conjugated at high rates in the intestines of mice, and pPD1 transfer to plasmid-free E. faecalis was beneficial since recipients that failed to acquire the plasmid were killed by the pPD1-encoded bac-21 bacteriocin (29).
We recently discovered that E. faecalis cells possessing self-targeting CRISPR-Cas systems are able to transiently tolerate this self-targeting, albeit at a fitness cost, and that antibiotic selection determines the outcome of this conflict by selecting for CRISPR mutants or loss of the CRISPR-targeted gene (26, 39). We observed CRISPR-Cas self-targeting lethality when the expression of cas9 was increased using a constitutive promoter (40). In this study, we engineered pPD1 with a complete, constitutively expressed CRISPR-Cas9 targeting cassette, specific for the enterococcal antibiotic resistance genes ermB (encoding erythromycin resistance) and tetM (encoding tetracycline resistance). We chose pPD1 because it lacks natively encoded antibiotic resistance yet harbors the fitness-enhancing bac-21 bacteriocin (41, 42). Using this engineered PRP, we successfully depleted tetracycline and erythromycin resistance from E. faecalis populations in vitro. Crucially, this approach worked in the absence of externally applied selection for pPD1 in donor strains, showing practicality for its usage as a potential antimicrobial. Using an in vivo intestinal colonization model, we show that these constructs are conjugated to erythromycin-resistant recipient cells in vivo. Donors carrying the engineered PRP targeting ermB significantly reduced the prevalence of erythromycin-resistant intestinal E. faecalis, supporting the utility of the engineered PRP in mitigating MDR E. faecalis. Furthermore, we show that these donors are immune to the uptake of ermB-encoding PRPs harbored by recipient E. faecalis cells. This work is a first step in utilizing conjugative elements for specific delivery of CRISPR-Cas antimicrobials to precisely target antibiotic-resistant bacteria in the intestinal microbiota.
RESULTS
Delivery of CRISPR-Cas9 effectively removes antibiotic resistance in vitro by targeting plasmid-borne resistance genes.
To assess the ability of CRISPR-Cas9 to target and remove antibiotic resistance in E. faecalis, we modified the PRP pPD1 by introducing the cas9 gene and a CRISPR guide RNA under the control of the constitutive bacA promoter (Fig. 1A and Fig. S1 in the Supplemental Material). To facilitate detection of the plasmid, we included a chloramphenicol resistance marker (cat) for selection. We generated two pPD1 derivatives, referred to as pKH88[sp-tetM] and pKH88[sp-ermB], which carry guide RNAs that target the enterococcal tetM and ermB genes conferring tetracycline and erythromycin resistance, respectively. These plasmids retain pheromone response functions and bac-21 bacteriocin production (Fig. 1A).
FIG 1.

CRISPR-Cas engineered conjugative pPD1 derivative plasmids target and eliminate antibiotic resistance genes in in vitro coculture. (A) Schematic of the pPD1 derivative pKH88. pKH88 encodes cas9 and a CRISPR guide RNA under the control of the constitutive enterococcal bacA promoter. The guide RNA consists of a CRISPR repeat region and a unique spacer sequence with complementarity to the ermB gene (pKH88[sp-ermB]) of pAM771 or the tetM gene (pKH88[sp-tetM]) of pCF10. pKH88 derivatives encode genes necessary for conjugative transfer, chloramphenicol resistance (cat), and biosynthesis of the bacteriocin bac-21. (B) Cartoon depicting the delivery of pKH88 derivatives to target recipient E. faecalis cells. Abundance of erythromycin-resistant (C) or tetracycline-resistant (D) E. faecalis OG1SSp recipients following acquisition of pKH88[sp-ermB] or pKH88[sp-tetM]. Removal of erythromycin and tetracycline resistance from the transconjugant population depends on the introduction of a cognate spacer targeting pKH88 derivative. Markers: R, rifampin; F, fusidic acid; Cam, chloramphenicol; Abx, antibiotic resistance. Error bars represent the geometric mean plus or minus the geometric standard deviation. **, P = 0.006 (for passage 0 in C); **, P = 0.003 (for passage 1 in C); **, P = 0.009 (for passage 0 in D).
PRPs are vehicles for the transmission of antibiotic resistance genes in E. faecalis populations (30); thus, we first assessed the ability of our engineered pPD1 derivatives to eliminate antibiotic resistance genes present on other PRPs. We reasoned that bacteriocin production by pKH88 derivatives would promote maintenance of these plasmids in enterococcal populations. The donor strain used in these experiments was E. faecalis CK135, and the recipient was E. faecalis OG1SSp possessing either pAM771, which carries the erythromycin resistance gene ermB, or pCF10, which carries the tetracycline resistance gene tetM (Fig. 1B). Both CK135 and OG1SSp are isogenic derivatives of the human oral commensal isolate OG1 (43). We mixed donors and recipients and plated them on brain heart infusion (BHI) agar. After overnight growth we recovered the biofilm and enumerated CFU of the different cell types (passage 0 in Fig. 1C and D). We observed a significant reduction in the number of erythromycin- and tetracycline-resistant transconjugants only when recipient cells received a pKH88 derivative encoding a cognate CRISPR guide (Fig. 1C and D). Noncognate guides were ineffective at reducing antibiotic resistance. This loss of antibiotic resistance was maintained after growth of the community in fresh BHI broth overnight for the pKH88[sp-ermB] targeting group (passage 1 in Fig. 1C) and trended toward maintenance of antibiotic resistance loss for the pKH88[sp-tetM] targeting group (passage 1 in Fig. 1D).
Conjugation of pKH88[sp-ermB] to MDR E. faecalis resolves erythromycin resistance.
To determine whether the pKH88 plasmids would be effective against MDR E. faecalis, we used pKH88[sp-ermB] to target ermB in E. faecalis V583 (Fig. 2A), a vancomycin-resistant hospital-adapted isolate (44). V583 ermB is encoded on the plasmid pTEF1, and V583 does not encode a tetM gene (10, 45). Initially, we observed poor conjugation frequencies of pKH88 derivatives into V583 when using CK135 as the donor (Fig. S2A and B). E. faecalis V583 encodes a type IV restriction endonuclease that restricts entry of 5-methylcytosine-modified DNA and has been characterized as a barrier to DNA uptake in enterococci (40, 46, 47). We reasoned that this barrier could account for the reduced dissemination of pKH88 to E. faecalis V583. Therefore, we utilized the E. faecalis OG1 variant strain OG1RF deleted for a 5-methylcytosine DNA methyltransferase (ΔEfaRFI) as the donor strain. This strain cannot methylate plasmid DNA, and the plasmid will not be cleaved by the recipient cell following conjugation (47). We observed that delivery of pKH88[sp-ermB] from E. faecalis OG1RF(ΔEfaRFI) to E. faecalis V583 decreased total erythromycin resistance in the population (Fig. 2B). In addition, E. faecalis V583 did not tolerate CRISPR cleavage of ermB on pTEF1, indicated by a reduction in viable E. faecalis V583 cells (Fig. 2B), which is consistent with our previous findings (26). This was specific to ermB cleavage, as transconjugants that received the nonspecific targeting plasmid pKH88[sp-tetM] retained cell viability and erythromycin resistance (Fig. 2C).
FIG 2.
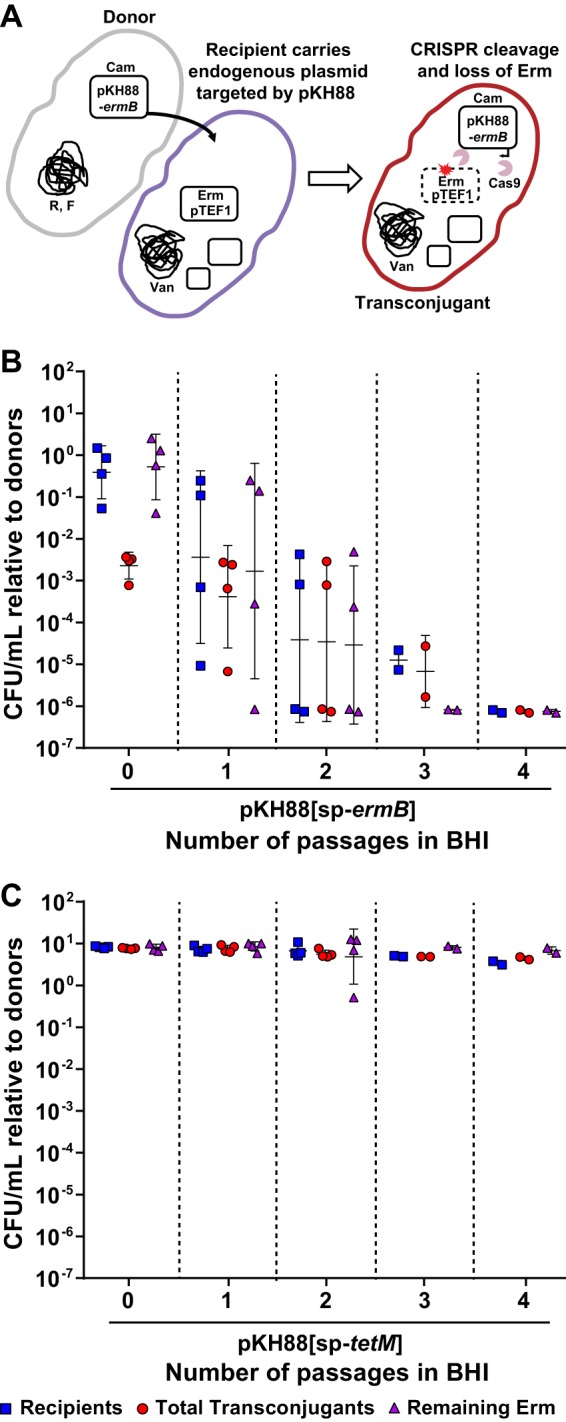
Engineered CRISPR-Cas targeting plasmid specifically reduces drug resistance in MDR E. faecalis. (A) Cartoon depicting the transfer, targeting, and removal of erythromycin resistance from MDR E. faecalis V583. Conjugation of E. faecalis V583 with pKH88[sp-ermB] (B) but not pKH88[sp-tetM] (C) reduces erythromycin-resistant E. faecalis V583 over the course of four serial passages in BHI broth. Markers: Erm, erythromycin; Van, vancomycin; R, rifampin; F, fusidic acid; S, streptomycin; Sp, spectinomycin; Cam, chloramphenicol. Error bars represent the geometric mean plus or minus the geometric standard deviation.
CRISPR-Cas9 selectively depletes erythromycin resistance within enterococcal populations in the dysbiotic murine intestine.
We next evaluated the efficacy of pKH88[sp-ermB] for the removal of erythromycin resistance using a mouse model of E. faecalis intestinal colonization. Mice were administered an antibiotic cocktail in their drinking water for 7 days. Antibiotic water was removed for 24 hours, and the mice were gavaged with E. faecalis OG1SSp(pAM771) recipients, modeling a bloom of antibiotic-resistant E. faecalis in the intestine. After an additional 24 hours, mice were gavaged with donor E. faecalis CK135 carrying either pKH88[sp-ermB] (targeting) or pKH88[sp-tetM] (nontargeting), and fecal samples were collected over the course of 23 days. A schematic of this experiment is depicted in Fig. S3. Whereas E. faecalis CK135 donors colonized the intestine to a sustained high density, E. faecalis OG1SSp(pAM771) recipient cells gradually declined in abundance over time (Fig. S4A and B). We attribute this gradual reduction in the recipient population to competitive killing by bacteriocin-producing donors, as previously described for pPD1-carrying E. faecalis (29), and/or competition from the reemerging native microbiota that was initially displaced by the antibiotic treatment. Robust numbers of pKH88-containing transconjugants (i.e., chloramphenicol-resistant OG1SSp) appeared 2 days postcolonization of the donor strains for both the pKH88[sp-ermB] and pKH88[sp-tetM] groups (Fig. S4C). However, the initial frequency of transconjugants per recipient reached only ∼10−5, indicating that overall conjugation frequency was relatively low (Fig. 3A). This was followed by a gradual increase in the transconjugant to recipient ratio, approaching 1:1 (Fig. 3A) as the total recipient population decreased over time (Fig. S4A). This suggests that the initial transconjugant population remains fixed due to a pKH88 bacteriocin-mediated fitness advantage upon restoration of the indigenous microbiota.
FIG 3.

Saturation of the E. faecalis OG1SSp(pAM771) recipient population by pKH88 is time-dependent. The ratio of transconjugants to recipients was calculated for both the pKH88[sp-ermB] targeting group (ermB) and the pKH88[sp-tetM] nontargeting group (tetM) for 23 days of cocolonization in antibiotic-treated (A) and gnotobiotic (B) mice. Error bars represent the geometric mean plus or minus the geometric standard deviation.
Although there appeared to be a trend of reduced erythromycin resistance within the recipient population for the pKH88[sp-ermB] group, no statistically significant reduction in the number of erythromycin-resistant recipients compared to the total recipient population was observed in the pKH88[sp-ermB] targeting group (Fig. 4A). Similarly, no significant difference in the erythromycin-resistant recipients compared to total recipients was observed for the pKH88[sp-tetM] nontargeting group (Fig. 4B). However, to our surprise, the difference in erythromycin-resistant recipients between the pKH88[sp-ermB] and pKH88[sp-tetM] groups was considerably less than the difference between the total erythromycin-resistant intestinal E. faecalis, which includes both donors and recipients. The magnitude of this difference was ∼3 log by the end of the experiment (Fig. 5A). This indicated that there was likely a substantial amount of conjugation by pAM771 into the donor population. Indeed, in as little as 2 days, donors acquired erythromycin resistance, but only in the pKH88[sp-tetM] nontargeting group (Fig. 5B). These data indicate that E. faecalis CK135(pKH88[sp-tetM]) donors are susceptible to counterconjugation of pAM771 from recipients but that CK135(pKH88[sp-ermB]) donors are immune, presumably due to CRISPR-Cas targeting of ermB on pAM771 upon counterconjugation. We conclude that our CRISPR antimicrobial dramatically reduces the prevalence of a targeted conjugative plasmid within the input E. faecalis population, likely through a multifactorial mechanism involving a bacteriocin-dependent competitive advantage, cleavage of the targeted plasmid, and prevention of counterplasmid acquisition. This in turn substantially impacts the distribution of drug resistance, as mice colonized with pKH88[sp-ermB] donors had 0.1% of the total levels of erythromycin-resistant E. faecalis possessed by mice colonized with pKH88[sp-tetM] donors.
FIG 4.

Assessment of erythromycin-resistant transconjugants from pKH88[sp-ermB] targeting and pKH88[sp-tetM] nontargeting populations in the murine intestine. (A) Antibiotic-treated mice cocolonized with E. faecalis OG1SSp(pAM771) and CK135(pKH88[sp-ermB]) (targeting group). (B) Antibiotic-treated mice cocolonized with E. faecalis OG1SSp(pAM771) and CK135(pKH88[sp-tetM]) (nontargeting group). The total OG1SSp(pAM771) recipient population and recipients that are erythromycin resistant were enumerated by selective plating of mouse feces over the course or 23 days of cocolonization. The solid horizontal line indicates the limit of detection. Markers: S, streptomycin; Sp, spectinomycin; Erm, erythromycin resistance. Error bars represent the geometric mean plus or minus the geometric standard deviation.
FIG 5.

E. faecalis CK135 pKH88[sp-ermB] donors are immune to acquisition of erythromycin resistance from recipients in the murine intestine. (A) Total intestinal load of erythromycin resistance among input E. faecalis donors and recipients from antibiotic-treated mice. Erythromycin resistance is acquired by donors only in the pKH88[sp-tetM] nontargeting group in both antibiotic-treated (B) and gnotobiotic (C) mice. The solid horizontal line indicates the limit of detection. Markers: Erm, erythromycin. Error bars represent the geometric mean plus or minus the geometric standard deviation. ***, P = 0.0003; ****, P < 0.0001.
We next assessed whether the microbial complexity of the intestinal environment plays a role in the efficacy of the CRISPR-Cas antimicrobial, since curtailing antibiotic therapy spontaneously restores the microbiota in the antibiotic dysbiosis model (48). We first colonized germfree mice with E. faecalis OG1SSp(pAM771) recipient cells and 24 hours later introduced donor E. faecalis CK135 carrying either pKH88[sp-ermB] or pKH88[sp-tetM]. Gnotobiotic mice maintained a high intestinal burden of both donors and recipients for the entire 23 days, in contrast to the antibiotic dysbiosis model, where the recipient population was reduced over time (Fig. S5A and B and S4A). Despite sustained high-density colonization of both donors and recipients, the total number of transconjugants was similar to that observed in the antibiotic dysbiosis model (Fig. S5C and S4C), but the transconjugant per recipient ratio remained stably low at 10−5 (Fig. 3B). Sustained high recipient density indicated that the apparent competitive advantage possessed by pKH88-containing donors via bac-21 does not manifest in gnotobiotic mice. These data suggest that even with prolonged high densities of both donors and recipients in the absence of microbial complexity, there may be biogeographical barriers that restrict the dissemination of pKH88 derivatives. This also suggests that the competitive advantage possessed by the donor is most prevalent in the presence of the microbiota, and therefore some degree of microbial complexity is likely necessary for the functionality of our CRISPR-Cas antimicrobial. Interestingly, similar to observations in the antibiotic dysbiosis model, pKH88 donors acquired erythromycin resistance, but only in the pKH88[sp-tetM] nontargeting group (Fig. 5C).
These results demonstrate that our engineered CRISPR-Cas antimicrobial is a robust barrier to in vivo plasmid acquisition and reduces the in vivo prevalence of a specifically targeted drug resistance trait. From a technological viewpoint, this is the ideal situation of a prospective “probiotic” donor—a strain that can effectively disseminate a CRISPR targeting plasmid while being protected from acquisition of undesirable traits.
CRISPR-Cas antimicrobial prevents antibiotic-mediated intestinal expansion of transconjugants.
A hallmark of enterococcal intestinal colonization is its ability to expand its population upon antibiotic perturbation of the indigenous microbiota (4, 25). This antibiotic-mediated expansion is attributed to both intrinsic and acquired antibiotic resistance. To determine the ability of our CRISPR-Cas antimicrobial to minimize the outgrowth of erythromycin-resistant E. faecalis in the intestine following antibiotic therapy, we utilized both our antibiotic dysbiosis and gnotobiotic mouse models. We treated the mice 27 days after cocolonization with a single 40-μg dose of oral erythromycin and determined the abundance of erythromycin-resistant transconjugants and total erythromycin-resistant E. faecalis. Prior to antibiotic treatment, there were no erythromycin-resistant transconjugants recovered from mice receiving donors carrying the targeting plasmid pKH88[sp-ermB], whereas two of eight mice receiving donors carrying the nontargeting plasmid pKH88[sp-tetM] had recoverable erythromycin-resistant transconjugants (Fig. 6). Upon oral erythromycin treatment, there was an ∼4 log expansion of erythromycin-resistant transconjugants in the pKH88[sp-tetM] nontargeting group, and no erythromycin-resistant transconjugants emerged in the mice colonized with E. faecalis CK135 pKH88[sp-ermB] donors (Fig. 6). This suggests that the perceived absence of erythromycin-resistant transconjugants in six of the eight mice in the pKH88[sp-tetM] nontargeting group was due to their abundance being below the limit of detection prior to antibiotic treatment. Additionally, the total number of erythromycin-resistant E. faecalis in the pKH88[sp-tetM] nontargeting group was significantly higher than the pKH88[sp-ermB] targeting group (Fig. 6). Together, these data indicate that transconjugants receiving pKH88[sp-ermB] were depleted of erythromycin resistance and that pKH88[sp-ermB] can significantly reduce the overall total erythromycin resistance within intestinal E. faecalis, pre- and post-antibiotic therapy. Interestingly, any remaining viable transconjugants in the pKH88[sp-ermB]-targeting group were impaired in their ability to increase in cell density compared to mice receiving pKH88[sp-tetM] nontargeting donors (Fig. 6), further indicating that targeted E. faecalis cells fail to expand their population due to loss of pAM771-encoded erythromycin resistance.
FIG 6.

Transconjugants that receive pKH88[sp-ermB] do not bloom in the intestines of dysbiotic mice following oral erythromycin treatment. Twenty-seven days after cocolonization with E. faecalis OG1SSp(pAM771) and CK135(pKH88[sp-ermB]) (targeting group) or with E. faecalis OG1SSp(pAM771) and CK135(pKH88[sp-tetM]) (nontargeting group), mice received a single 40-μg dose of oral erythromycin. The numbers of recipients, transconjugants, and erythromycin-resistant transconjugants were enumerated from fecal pellets before and after oral erythromycin treatment. The solid horizontal line indicates the limit of detection. Markers: Erm, erythromycin; S, streptomycin; Sp, spectinomycin; Cam, chloramphenicol. Error bars represent the geometric mean plus or minus the geometric standard deviation. *, P = 0.01; **, P = 0.004.
In gnotobiotic mice, following oral erythromycin treatment, we again did not recover any erythromycin-resistant transconjugants in the pKH88[sp-ermB]-targeting group (Fig. S6). Interestingly, in gnotobiotic mice, we did not observe outgrowth of erythromycin-resistant transconjugants in the pKH88[sp-tetM] nontargeting group, even though we did not recover any erythromycin-resistant transconjugants in the pKH88[sp-ermB]-targeting group, similar to the antibiotic dysbiosis model (Fig. S6). We attribute this to the high level of both donors and recipients in the gnotobiotic intestine that were unaffected by oral erythromycin treatment (Fig. S5A and B). Intestinal colonization saturation by donors and recipients may be spatially restricting, thus promoting colonization resistance against transconjugant expansion upon erythromycin treatment in the pKH88[sp-tetM] nontargeting group.
DISCUSSION
This study presents a strategy for the delivery of a CRISPR-based antimicrobial that targets antibiotic-resistant E. faecalis. We show that this conjugative antimicrobial significantly reduces antibiotic resistance from in vitro and in vivo enterococcal communities in the absence of externally applied selection for donor strains. Our approach for the delivery of a CRISPR-Cas antimicrobial differs from studies which employed either transformable plasmids or phages for the recognition and delivery to target cells (18–21). Although these studies provided the foundation for the use of CRISPR antimicrobial activity against bacteria, limitations were noted. In the case of phage-mediated CRISPR antimicrobial dissemination, this technique suffered from incomplete phage dissemination. This scenario is expected due to selective pressures imposed on bacteria by phages during infection, including receptor mutations (22–25). Such issues have recently been addressed using multiphage delivery systems or by coopting phage particles for the delivery of CRISPR-Cas targeting modules embedded within mobilizable genome islands (49, 50). Although the use of a phage-based delivery system for E. faecalis should be possible, enterococcal phage biology is understudied, and limited information exists concerning phage host ranges and genetics. Therefore, we chose to engineer a PRP for the dissemination of an E. faecalis-specific CRISPR-Cas antimicrobial for several reasons. First, the conditions governing PRP regulation have been extensively studied. Second, PRPs are specific for E. faecalis. Third, PRPs achieve high conjugation rates and can be genetically modified to diversify host tropism by altering pheromone responsiveness. Fourth, PRPs are stably maintained and disseminate in the absence of selection (51, 52).
The intestine is a reservoir for MDR E. faecalis, and E. faecalis infections often follow exposure to broad-spectrum antibiotics. We applied our CRISPR-Cas antimicrobial method for the removal of an antibiotic resistance trait from intestinal E. faecalis. The mouse models of intestinal colonization used were designed to mimic enterococcal overgrowth in the human intestine following antibiotic therapy (3, 4). Antibiotics disrupt the intestinal microbiota, allowing antibiotic-resistant enterococci to become the dominant colonizer. In our study, we used E. faecalis donor strains for the conjugative delivery of CRISPR-Cas to remove erythromycin resistance from an abundant intestinal E. faecalis target population. The conjugative transfer of an ermB-specific CRISPR-Cas targeting system into recipient E. faecalis cells showed little to no reduction in erythromycin resistance from the total intestinal E. faecalis recipient population. However, within the recipient population that did receive the CRISPR targeting plasmid carrying a cognate spacer, all transconjugants were fully depleted of erythromycin resistance. This indicates that, although the frequency of conjugation was low compared to the total available recipient population and not as robust as in vitro conjugation frequencies, the specificity of our CRIPSR-Cas targeting system in the intestine was absolute. If successful conjugation occurs, there is complete loss of erythromycin resistance. In addition, we observed that donors carrying a cognate CRISPR antimicrobial plasmid were protected from acquisition of erythromycin resistance from recipients. This is an important feature of our deliverable system since the transmission of undesirable target traits to donor cells would impede the usefulness of such a delivery strategy within complex microbial environments. The discovery that E. faecalis donor cells carrying the CRISPR-Cas antimicrobial are immunized against the acquisition of antibiotic resistance suggests that donors could be used as a probiotic. These probiotic E. faecalis cells would be protected from the acquisition of antibiotic resistance traits and could preemptively occupy the intestine to outcompete or inhibit invading multidrug-resistant strains.
There are several reasons why intestinal conjugation frequency might be suboptimal in our study. First, our analysis measured transconjugants from fecal samples, not the intestinal mucosa. The spatial distribution of donor and recipient E. faecalis cells colonizing the intestinal mucosal layer is unknown, and evidence suggests that conjugation frequency could be impacted by the regional location of E. faecalis intestinal colonization (28). Second, pKH88 derivatives require recipient-derived cPD1 pheromone to induce conjugation (53). We know little about the kinetics of pheromone signaling in the intestine. Low rates of conjugation could reflect low local concentrations of cPD1 in the vicinity of donors, resulting in few donors that effectively conjugate, thereby minimizing conjugation frequencies. Alternatively, excess donors can inhibit conjugation to recipient cells (54). This is due to the production of inhibitory self-signaling peptides produced by the donors that compete with recipient pheromones and reduce conjugation frequencies. Exploring the efficiency of conjugation upon altering donor-to-recipient ratios could lead to more permissive conjugation in the intestine. Another possibility is that conjugation in the intestine may be more efficient at a precise density of donors, recipients, or both, which may require future optimization for effective plasmid delivery.
Moving forward, improvements to our current system are justified if an engineered conjugative CRISPR antimicrobial is to be clinically effective for precision removal of antibiotic resistance genes in E. faecalis or other MDR bacteria. Modifications include multiplexing CRISPR guide RNAs to target different resistance genes and/or the same genes at multiple sites, as well as increasing the overall conjugation frequency by altering PRP regulation, dosing of donor strains, or the plasmid replicon itself. Theoretically, resistance to bac-21 could compromise the efficacy of our system, and future iterations should utilize alternative bacteriocins as well as bacteriocin-free constructs. In this study, we focused on targeting antibiotic resistance that resides on plasmids. However, many E. faecalis antibiotic resistance genes are chromosomally encoded. Future iterations of this system should also be engineered for the specific targeting of chromosomal antibiotic resistance genes. Recently we showed that CRISPR-Cas targeting of the vanB locus on the E. faecalis V583 chromosome is lethal (40). Targeting chromosomal antibiotic resistance genes could have the added feature of bacterial lethality. Such a strategy may be preferable depending on whether the intended use of the CRISPR-Cas antimicrobial is for reshaping the microbiota or for targeting drug-resistant strains during infection.
Our model system has relied on laboratory-domesticated strains of E. faecalis OG1 (43); it will be imperative to determine the efficacy of this system against more “wild” clinical isolates, which are the intended targets of such a therapy. For safe and effective delivery of the CRISPR antimicrobial in the clinic, the use of an attenuated E. faecalis strain or a related member of the Lactobacillales approved for probiotic use would be essential. Finally, this system could be adapted for the targeted removal of antibiotic resistance from bacteria other than E. faecalis. Gram-positive conjugative plasmids like pIP501, which belongs to the rep1 family, are broadly disseminated conjugative plasmids (51, 52, 55). pIP501 is transferrable among enterococci, other lactic acid bacteria, staphylococci, and clostridia (55). The modification of a promiscuous mobilizable plasmid such as pIP501 with CRISPR-Cas antimicrobial function could expand the capabilities of our current system by facilitating broad targeting of multiple Gram-positive pathogens.
MATERIALS AND METHODS
Bacterial culture methods and molecular biology techniques.
Escherichia coli cultures were incubated with shaking at 220 rpm at 37°C in lysogeny broth (LB). E. faecalis cultures were grown in brain heart infusion (BHI) broth at 37°C without shaking. PCR was performed using Taq polymerase (New England Biolabs) and Q5 DNA polymerase (New England Biolabs) for molecular cloning. The PureLink PCR purification kit (Invitrogen) was used to purify DNA fragments, and the GeneJet plasmid purification kit (Thermo Fisher) was used for plasmid purification. Primers were obtained from Sigma-Aldrich. Sanger sequencing was performed at the Massachusetts General Hospital DNA Core Facility. E. coli EC1000 was used for routine plasmid propagation (56). E. faecalis and E. coli competent cells were prepared as described previously (26). Genomic DNA was extracted using the Mo Bio microbial DNA isolation kit (Qiagen). Antibiotics were used at the following concentrations: chloramphenicol, 15 μg/ml; streptomycin, 500 μg/ml; spectinomycin, 500 μg/ml; vancomycin, 10 μg/ml; erythromycin, 50 μg/ml; rifampin, 50 μg/ml; fusidic acid, 25 μg/ml; tetracycline, 10 μg/ml.
Strain and plasmid construction.
All strains used in this study can be found in Table S1. The donor strain used for competition experiments was a rifampin and fusidic acid-resistant derivative of CK135 (57). All plasmids used in this study can be found in Table S2. Fragments obtained from the plasmids pHA101 (58), pPD1 (24), PbacA-pG19, pKH12 (26), and pGR tetM/ermB (40) were amplified using Q5 DNA polymerase and assembled using NEB HiFi DNA assembly master mix (New England Biolabs). For simplicity, assembly of the CRISPR-targeting construct (PbacA-cas9-cat-guide RNA) was divided into two steps. First, PbacAcas9 was introduced into pPD1 using homologous recombination. Then the cat gene from pKH12 and the PbacA-driven CRISPR guide RNA were integrated downstream of PbacA-cas9. Following the generation of this construct, we discovered that the first integration site obstructed the pheromone response, and therefore the entire engineered region (PbacA-cas9-cat-guide RNA) was amplified as one fragment and integrated into native pPD1 between open reading frames (ORFs) ppd4 and ppd5 using the integration plasmid pCOP88 (Fig. S1). The modified pPD1 derivatives are designated pKH88[sp-tetM] and pKH88[sp-ermB], which target tetM or ermB, respectively. Primer sequences are listed in Table S3.
In vitro competition assays.
Overnight cultures of donors and recipients were pelleted, resuspended in fresh BHI broth, and cultured for 1.5 hours. Donors and recipients were mixed in a volume ratio of 1:9, and the mixtures were spread on BHI agar. Following overnight incubation, lawns were scraped into sterile phosphate-buffered saline (PBS), and serial dilutions (10 μl each) were spotted on BHI plates supplemented with antibiotics to enumerate the CFU of donors, recipients, and transconjugants as well as the total CFU of tetracycline- and erythromycin-resistant cells. For individual passages, the recovered conjugation mixture was diluted 1:1,000 in fresh BHI broth without selection and incubated overnight, after which the constituents of the mixture were again enumerated.
Mouse models of E. faecalis colonization.
All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Colorado School of Medicine (protocol number 00253).
For the antibiotic dysbiosis model, 7 days prior to colonization, 6- to 8-week-old C57BL6/J mice were gavaged with 150 μl of an antibiotic cocktail (streptomycin, 1 mg/ml; gentamicin, 1 mg/ml; erythromycin, 200 μg/ml) and given a water bottle ad libitum with the same antibiotic cocktail for 6 days following the initial gavage. Twenty-four hours prior to colonization with recipient E. faecalis, antibiotic water was removed and replaced with standard sterile reverse osmosis water. Donor and recipient strains were cultured overnight in BHI, and mice were gavaged with ∼109 CFU in 100 μl of sterile PBS. All mice were first gavaged with E. faecalis OG1SSp(pAM771). After 24 hours, mice (n = 13; 7 female [F] and 6 male [M]) were gavaged with CK135(pKH88[sp-ermB]), while the second group (n = 12; 5 F and 7 M) were gavaged with CK135(pKH88[sp-tetM]). Fecal samples were collected at the designated time points, resuspended in 1 ml of sterile PBS, and plated on BHI agar supplemented with rifampin, fusidic acid, erythromycin, chloramphenicol, streptomycin, and spectinomycin in combinations that would select for the desired populations and using the concentrations described above. After 27 days, 8 mice (4 F and 4 M) from the pKH88[sp-ermB] targeting group and 7 mice (3 F and 4 M) from the pKH88[sp-tetM] nontargeting group were gavaged with erythromycin (40 μg). Fecal samples were collected from all mice preerythromycin gavage. Posterythromycin gavage, fecal samples were collected from all 8 mice from the pKH88[sp-ermB] targeting group and from 6 of 7 mice from the pKH88[sp-tetM] nontargeting group due to the inability of a single mouse from this group to defecate.
All germfree mice were gavaged with E. faecalis OG1SSp(pAM771). Twenty-four hours later, these gnotobiotic mice were gavaged with E. faecalis CK135(pKH88[sp-ermB]) (n = 8; 4 F and 4 M) or E. faecalis CK135(pKH88[sp-tetM]) (n = 8; 4 F and 4 M). Fecal samples were collected at the designated time points, and donors, recipients, and transconjugants were enumerated on selective medium as described above. After 27 days, all 8 gnotobiotic mice from each group were gavaged with 40 μg of erythromycin.
Statistical analysis.
GraphPad-Prism version 8.2.0 was used to determine statistical significance, and the following tests were employed based on the experimental setup. For in vitro competition assays, a log normal distribution was assumed and a Student’s t test was performed. For intestinal colonization studies, due to a higher degree of variation in the colonization kinetics between individual mice, a normal distribution was not assumed, and thus, a nonparametric Mann-Whitney test was performed. Values below the limit of detection for in vivo animal experiments were represented as constant (one-half of the value of the limit of detection). The limit of detection for all animal experiments was 100 CFU per gram of feces. P values indicate the following: *, 0.01 to 0.05; **, 0.001 to <0.01; ***, 0.0001 to <0.001; ****, <0.0001.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants R01AI116610 (K.L.P.), R01AI141479 (B.A.D.), and K01DK102436 (B.A.D.).
We thank members of the Palmer and Duerkop labs for critical feedback on the manuscript. We thank Christopher Kristich for providing E. faecalis CK135(pPD1).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01454-19.
REFERENCES
- 1.Round JL, Mazmanian SK. 2009. The gut microbiome shapes intestinal immune responses during health and disease. Nat Rev Immunol 9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y-J, Li S, Gan R-Y, Zhou T, Xu D-P, Li H-B. 2015. Impacts of gut bacteria on human health and diseases. Int J Mol Sci 16:7493–7519. doi: 10.3390/ijms16047493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donskey CJ, Chowdhry TK, Hecker MT, Hoyen CK, Hanrahan JA, Hujer AM, Hutton-Thomas RA, Whalen CC, Bonomo RA, Rice LB. 2000. Effect of antibiotic therapy on the density of vancomycin-resistant enterococci in the stool of colonized patients. N Engl J Med 343:1925–1932. doi: 10.1056/NEJM200012283432604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, Viale A, Socci ND, van den Brink MRM, Kamboj M, Pamer EG. 2010. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Invest 120:4332–4341. doi: 10.1172/JCI43918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foulquié Moreno MR, Sarantinopoulos P, Tsakalidou E, De Vuyst L. 2006. The role and application of enterococci in food and health. Int J Food Microbiol 106:1–24. doi: 10.1016/j.ijfoodmicro.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 6.Franz C, Huch M, Abriouel H, Holzapfel W, Gálvez A. 2011. Enterococci as probiotics and their implications in food safety. Int J Food Microbiol 151:125–140. doi: 10.1016/j.ijfoodmicro.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 7.Lebreton F, Willems RJL, Gilmore MS. 2014. Enterococcus diversity, origins in nature, and gut colonization In Gilmore MS, Clewell DB, Ike Y, Shankar N (ed), Enterococci: from commensals to leading causes of drug resistant infection, Massachusetts Eye and Ear Infirmary, Boston, MA. [PubMed] [Google Scholar]
- 8.Weiner LM, Webb AK, Limbago B, Dudeck MA, Patel J, Kallen AJ, Edwards JR, Sievert DM. 2016. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2011-2014. Infect Control Hosp Epidemiol 37:1288–1301. doi: 10.1017/ice.2016.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim S-K, Tanimoto K, Tomita H, Ike Y. 2006. Pheromone-responsive conjugative vancomycin resistance plasmids in Enterococcus faecalis isolates from humans and chicken feces. Appl Environ Microbiol 72:6544–6553. doi: 10.1128/AEM.00749-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paulsen IT, Banerjei L, Myers GSA, Nelson KE, Seshadri R, Read TD, Fouts DE, Eisen JA, Gill SR, Heidelberg JF, Tettelin H, Dodson RJ, Umayam L, Brinkac L, Beanan M, Daugherty S, DeBoy RT, Durkin S, Kolonay J, Madupu R, Nelson W, Vamathevan J, Tran B, Upton J, Hansen T, Shetty J, Khouri H, Utterback T, Radune D, Ketchum KA, Dougherty BA, Fraser CM. 2003. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299:2071–2074. doi: 10.1126/science.1080613. [DOI] [PubMed] [Google Scholar]
- 11.Alevizakos M, Gaitanidis A, Nasioudis D, Tori K, Flokas ME, Mylonakis E. 2017. Colonization with vancomycin-resistant enterococci and risk for bloodstream infection among patients with malignancy: a systematic review and meta-analysis. Open Forum Infect Dis 4:ofw246. doi: 10.1093/ofid/ofw246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Septimus EJ, Schweizer ML. 2016. Decolonization in prevention of health care-associated infections. Clin Microbiol Rev 29:201–222. doi: 10.1128/CMR.00049-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 14.Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadan AH, Moineau S. 2010. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 15.Marraffini LA, Sontheimer EJ. 2008. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322:1843–1845. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. 2014. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. 2013. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bikard D, Euler C, Jiang W, Nussenzweig PM, Goldberg GW, Duportet X, Fischetti VA, Marraffini LA. 2014. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat Biotechnol 32:1146–1150. doi: 10.1038/nbt.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Citorik RJ, Mimee M, Lu TK. 2014. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat Biotechnol 32:1141–1145. doi: 10.1038/nbt.3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomaa AA, Klumpe HE, Luo ML, Selle K, Barrangou R, Beisel L. 2014. Programmable removal of bacterial strains by use of genome-targeting CRISPR-Cas systems. mBio 5:1–9. doi: 10.1128/mBio.00928-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park JY, Moon BY, Park JW, Thornton JA, Park YH, Seo KS. 2017. Genetic engineering of a temperate phage-based delivery system for CRISPR/Cas9 antimicrobials against Staphylococcus aureus. Sci Rep 7:44929. doi: 10.1038/srep44929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duerkop BA, Huo W, Bhardwaj P, Palmer KL, Hooper LV. 2016. Molecular basis for lytic bacteriophage resistance in enterococci. mBio 7:1–12. doi: 10.1128/mBio.01304-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lossouarn J, Briet A, Moncaut E, Furlan S, Bouteau A, Son O, Leroy M, DuBow MS, Lecointe F, Serror P, Petit MA. 2019. Enterococcus faecalis countermeasures defeat a virulent Picovirinae bacteriophage. Viruses 11:48. doi: 10.3390/v11010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho K, Huo W, Pas S, Dao R, Palmer KL. 2018. Loss-of-function mutations in epaR confer resistance to phiNPV1 infection in Enterococcus faecalis OG1RF. Antimicrob Agents Chemother 62:e00758-18. doi: 10.1128/AAC.00758-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chatterjee A, Johnson CN, Luong P, Hullahalli K, McBride SW, Schubert AM, Palmer KL, Carlson PE Jr, Duerkop BA. 2019. Bacteriophage resistance alters antibiotic-mediated intestinal expansion of enterococci. Infect Immun 87:e00085-19. doi: 10.1128/IAI.00085-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hullahalli K, Rodrigues M, Palmer KL. 2017. Exploiting CRISPR-Cas to manipulate Enterococcus faecalis populations. Elife 6:e26664. doi: 10.7554/eLife.26664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palmer KL, Gilmore MS. 2010. Multidrug-resistant enterococci lack CRISPR-cas. mBio 1:e00227-10. doi: 10.1128/mBio.00227-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirt H, Greenwood-Quaintance KE, Karau MJ, Till LM, Kashyap PC, Patel R, Dunny GM. 2018. Enterococcus faecalis sex pheromone cCF10 enhances conjugative plasmid transfer in vivo. mBio 9:e00037-18. doi: 10.1128/mBio.00037-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kommineni S, Bretl DJ, Lam V, Chakraborty R, Hayward M, Simpson P, Cao Y, Bousounis P, Kristich CJ, Salzman NH. 2015. Bacteriocin production augments niche competition by enterococci in the mammalian gastrointestinal tract. Nature 526:719–722. doi: 10.1038/nature15524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Licht TR, Laugesen D, Jensen LB, Jacobsen BL. 2002. Transfer of the pheromone-inducible plasmid pCF10 among Enterococcus faecalis microorganisms colonizing the intestine of mini-pigs. Appl Environ Microbiol 68:187. doi: 10.1128/AEM.68.1.187-193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price VJ, McBride SW, Hullahalli K, Chatterjee A, Duerkop BA, Palmer KL. 2019. Enterococcus faecalis CRISPR-Cas is a robust barrier to conjugative antibiotic resistance dissemination in the murine intestine. mSphere 4:e00464-19. doi: 10.1128/mSphere.00464-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kozlowicz BK, Dworkin M, Dunny GM. 2006. Pheromone-inducible conjugation in Enterococcus faecalis: a model for the evolution of biological complexity? Int J Med Microbiol 296:141–147. doi: 10.1016/j.ijmm.2006.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dunny GM, Brown BL, Clewell DB. 1978. Induced cell aggregation and mating in Streptococcus faecalis: evidence for a bacterial sex pheromone. Proc Natl Acad Sci U S A 75:3479–3483. doi: 10.1073/pnas.75.7.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chandler JR, Flynn AR, Bryan EM, Dunny GM. 2005. Specific control of endogenous cCF10 pheromone by a conserved domain of the pCF10-encoded regulatory protein PrgY in Enterococcus faecalis. J Bacteriol 187:4830–4843. doi: 10.1128/JB.187.14.4830-4843.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clewell DB. 1993. Bacterial sex pheromone-induced plasmid transfer. Cell 73:9–12. doi: 10.1016/0092-8674(93)90153-h. [DOI] [PubMed] [Google Scholar]
- 36.Ike Y, Clewell DB, Segarra RA, Gilmore MS. 1990. Genetic analysis of the pAD1 hemolysin/bacteriocin determinant in Enterococcus faecalis: Tn917 insertional mutagenesis and cloning. J Bacteriol 172:155–163. doi: 10.1128/jb.172.1.155-163.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomita H, Fujimoto S, Tanimoto K, Ike Y. 1997. Cloning and genetic and sequence analyses of the bacteriocin 21 determinant encoded on the Enterococcus faecalis pheromone-responsive conjugative plasmid pPD1. J Bacteriol 179:7843–7855. doi: 10.1128/jb.179.24.7843-7855.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ness IF, Diep DB, Ike Y. 2014. Enterococcal bacteriocins and antimicrobial proteins that contribute to niche control In Gilmore MS, Clewell DB, Ike Y, Shankar N (ed), Enterococci: from commensals to leading causes of drug resistant infection, Massachusetts Eye and Ear Infirmary, Boston, MA. [PubMed] [Google Scholar]
- 39.Price VJ, Huo W, Sharifi A, Palmer KL. 2016. CRISPR-Cas and restriction-modification act additively against conjugative antibiotic resistance plasmid transfer in Enterococcus faecalis. mSphere 1:e00064-16. doi: 10.1128/mSphere.00064-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hullahalli K, Rodrigues M, Nguyen UT, Palmer KL. 2018. An attenuated CRISPR-Cas system in Enterococcus faecalis permits DNA acquisition. mBio 9:e00414-18. doi: 10.1128/mBio.00414-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clewell DB. 1981. Plasmids, drug resistance, and gene transfer in the genus Streptococcus. Microbiol Rev 45:409–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fujimoto S, Tomita H, Wakamatsu E, Tanimoto K, Ike Y. 1995. Physical mapping of the conjugative bacteriocin plasmid pPD1 of Enterococcus faecalis and identification of the determinant related to the pheromone response. J Bacteriol 177:5574–5581. doi: 10.1128/jb.177.19.5574-5581.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gold OG, Jordan HV, van Houte J. 1975. The prevalence of enterococci in the human mouth and their pathogenicity in animal models. Arch Oral Biol 20:473–477. doi: 10.1016/0003-9969(75)90236-8. [DOI] [PubMed] [Google Scholar]
- 44.Sahm DF, Kissinger J, Gilmore MS, Murray PR, Mulder R, Solliday J, Clarke B. 1989. In vitro susceptibility studies of vancomycin-resistant Enterococcus faecalis. Antimicrob Agents Chemother 33:1588–1591. doi: 10.1128/AAC.33.9.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McBride SM, Fischetti VA, Leblanc DJ, Moellering RC, Gilmore MS. 2007. Genetic diversity among Enterococcus faecalis. PLoS One 2:e582. doi: 10.1371/journal.pone.0000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huo W, Adams HM, Trejo C, Badia R, Palmer KL. 2018. A type I restriction-modification system associated with Enterococcus faecium subspecies separation. Appl Environ Microbiol 85:e02174-18. doi: 10.1128/AEM.02174-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huo W, Adams HM, Zhang MQ, Palmer KL. 2015. Genome modification in Enterococcus faecalis OG1RF assessed by bisulfite sequencing and single-molecule real-time sequencing. J Bacteriol 197:1939–1951. doi: 10.1128/JB.00130-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suez J, Zmora N, Zilberman-Schapira G, Mor U, Dori-Bachash M, Bashiardes S, Zur M, Regev-Lehavi D, Ben-Zeev Brik R, Federici S, Horn M, Cohen Y, Moor AE, Zeevi D, Korem T, Kotler E, Harmelin A, Itzkovitz S, Maharshak N, Shibolet O, Pevsner-Fischer M, Shapiro H, Sharon I, Halpern Z, Segal E, Elinav E. 2018. Post-antibiotic gut mucosal microbiome reconstitution is impaired by probiotics and improved by autologous FMT. Cell 174:1406–1423e16. doi: 10.1016/j.cell.2018.08.047. [DOI] [PubMed] [Google Scholar]
- 49.Yosef I, Manor M, Kiro R, Qimron U. 2015. Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc Natl Acad Sci U S A 112:7267–7272. doi: 10.1073/pnas.1500107112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ram G, Ross HF, Novick RP, Rodriguez-Pagan I, Jiang D. 2018. Conversion of staphylococcal pathogenicity islands to CRISPR-carrying antibacterial agents that cure infections in mice. Nat Biotechnol 36:971–976. doi: 10.1038/nbt.4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clewell DB, Weaver KE, Dunny GM, Coque TM, Francia MV, Hayes F. 2014. Extrachromosomal and mobile elements in enterococci: transmission, maintenance, and epidemiology In Gilmore MS, Clewell DB, Ike Y, Shankar N (ed), Enterococci: from commensals to leading causes of drug resistant infection, Massachusetts Eye and Ear Infirmary, Boston, MA. [PubMed] [Google Scholar]
- 52.Jensen LB, Garcia-Migura L, Valenzuela AJ, Lohr M, Hasman H, Aarestrup FM. 2010. A classification system for plasmids from enterococci and other Gram-positive bacteria. J Microbiol Methods 80:25–43. doi: 10.1016/j.mimet.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 53.Nakayama J, Takanami Y, Horii T, Sakuda S, Suzuki A. 1998. Molecular mechanism of peptide-specific pheromone signaling in Enterococcus faecalis: functions of pheromone receptor TraA and pheromone-binding protein TraC encoded by plasmid pPD1. J Bacteriol 180:449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bandyopadhyay A, O’Brien S, Frank KL, Dunny GM, Hu W-S. 2016. Antagonistic donor density effect conserved in multiple enterococcal conjugative plasmids. Appl Environ Microbiol 82:4537–4545. doi: 10.1128/AEM.00363-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kohler V, Vaishampayan A, Grohmann E. 2018. Broad-host-range Inc18 plasmids: occurrence, spread and transfer mechanisms. Plasmid 99:11–21. doi: 10.1016/j.plasmid.2018.06.001. [DOI] [PubMed] [Google Scholar]
- 56.Leenhouts K, Buist G, Bolhuis A, Ten Berge A, Kiel J, Mierau I, Dabrowska M, Venema G, Kok J. 1996. A general system for generating unlabelled gene replacements in bacterial chromosomes. Mol Gen Genet 253:217–224. doi: 10.1007/s004380050315. [DOI] [PubMed] [Google Scholar]
- 57.Kristich CJ, Little JL. 2012. Mutations in the beta subunit of RNA polymerase alter intrinsic cephalosporin resistance in enterococci. Antimicrob Agents Chemother 56:2022–2027. doi: 10.1128/AAC.06077-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bhardwaj P, Ziegler E, Palmer KL. 2016. Chlorhexidine induces VanA-type vancomycin resistance genes in enterococci. Antimicrob Agents Chemother 60:2209–2221. doi: 10.1128/AAC.02595-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.