

Abstract

An efficient and quick access toward a series of (E)-2-arylideneaminopyrroles 6 and to their benzyne-promoted aza-Diels–Alder cycloaddition products is provided. These products are three pyrrolo[2,3-c]isoquinolines 8a–c substituted in position 5 with different electron-acceptor (A) or electron-donor (D) aryl groups. Intermediates and products were obtained in good yields (up to 78 and 84%, respectively), and their structures were determined on the basis of NMR measurements and HRMS analysis. Photophysical properties of 8a–c were investigated, finding good Stokes shift in different solvents, but only the product 8c showed appreciable fluorescence intensity since its 5-aryl group (2,4-Cl2Ph) could favor the twisted intramolecular charge transfer effect. In addition, a riveting relationship between solvent viscosity and fluorescence intensity was found. Structures of 6 and 8 were studied and confirmed by single-crystal X-ray diffraction, observing that their electronic distributions effect the supramolecular assembly but with only long-distance hydrophobic interactions. A CE-B3LYP model was used to study the energetic topology and understand the crystal architecture of compounds as well as find a connection with both the synthetic and photophysical results.
Introduction
The development of simple and atom-economical approaches for the construction of fused N-heterocycles is very important because of their broad range of applications in medicinal chemistry and, nowadays, also in chemosensor design.1−3 In particular, isoquinoline derivatives are found in a plethora of natural products, pharmaceutical compounds, and organic materials.4−6 Although several syntheses of this scaffold have been developed, there still remains a great need to find efficient methodologies for its construction. Traditional synthesis of isoquinolines include the Bischler–Napieralski,7 Pictet–Spengler,8 and Pomeranz–Fritsch reactions.9 Likewise, the transition metal-catalyzed C–H or C–halogen bond activation and electrophile-triggered annulation reaction have enabled an orthogonal access to isoquinolines.10 However, these methods are often limited due to the use of special additives and/or expensive catalysts, which results in lower atom efficiency.11 In this sense, Maulide’s group reported a metal-free selective cycloaddition between ynamides and nitriles that allowed efficient access to isoquinolines.12 Another elegant approach is based on a metal-free annulation reaction between secondary eneamides and arynes.13
Regarding the synthesis of fused isoquinolines, the pyrrolo-fused derivatives have remained almost unexplored, possibly due to the dearth of direct approaches to obtain these scaffolds.14 Specifically, pyrrolo[2,3-c]isoquinolines synthesis is quite rare, but Biehl et al. reported the synthesis of derivative 2a by an acid-promoted cyclization of 4-formylmethylisoquinolin-3-amines 1.15 Likewise, Hajós developed a TBAF-promoted 5-endo-dig cyclization involving 4-ethynylisoquinoline-3-amines 3 as a viable route to derivative 2b (Scheme 1a).16 A few years ago, we reported the synthesis of the pyrrolo[2,3-c]isoquinoline 8a as a single example of cycloaddition between the (E)-arylideneaminopyrrole 6e with the aryne derived from 7 (Scheme 1b,i).17 More recently, we reported an efficient synthesis and significant structural studies by X-ray of a series of (E)-arylideneaminopyrroles 6a–c and their reduction products to secondary amines 9a–c via a two-step synthesis sequence (solvent-free condensation and reduction) starting from 5-amino-1-tert-butyl-1H-pyrrole-3-carbonitrile (4) and arylaldehydes 5 (Scheme 1b,ii).18
Scheme 1. (E)-Arylideneaminopyrroles 6a–c and Synthesis of Pyrrolo[2,3-c]isoquinolines 2 and 8a–c.

Inspired by these earlier studies along with our interest in the development of efficient protocols for the synthesis of fused N-heterocycles,2,19−23 we envisioned that using our established synthetic sequences17,18 might generate other (E)-arylideneaminopyrroles 6d–h, which would be suitable heterodienes for normal electron-demand aza-Diels–Alder cycloaddition with benzyne in situ generated from 7. Thus, reactions with three heterodienes substituted with different electron-acceptor (A) or electron-donor (D) groups led to the expected cycloadducts 10a–c that, with later in situ aromatization, provided pyrrolo[2,3-c]isoquinolines 8a–c in high atom economy and efficiency (Scheme 1c). We report herein a detailed study of the observed molecular conformations in the solid state of three (E)-arylideneaminopyrroles 6e–g and two pyrrolo[2,3-c]isoquinolines 8b and 8c discussed in terms of their molecular and supramolecular structures, including the interpretation of their Hirshfeld surface maps and energy frameworks. Considering the lack of short contacts in the solid state, the term “weak interactions” presents, in this work, a special meaning, leaving the interpretation of the intermolecular contacts necessarily in terms of energetic topology. In addition, a brief discussion about results of photophysical studies for compounds 8a–c is presented, which was carried out due to their fused nature25−27 and our continued interest in this important property for N-heterocyclic derivatives.24−27
It is important to note that, in the pyrroloisoquinoline moiety of 8a–c, an intramolecular charge transfer (ICT) process occurs due to the π-excedent character of the pyrrole ring versus isoquinoline ring (Figure 1a). In addition, since compounds 8a–c are substituted in position 5 with different aryl groups (A or D), there exist a direct influence over their molecular electronic distribution. Accordingly, photophysical properties of 8a–c were explored in order to establish the scope of the pyrrolo[2,3-c]isoquinoline core as a new organic fluorophore. Additionally, since the structures of aza-diene intermediates 6 and products 8 were studied by X-ray diffraction, these analyses were conveniently related to both the synthetic and photophysical results. For example, the product 8c (substituted with the 2,4-Cl2Ph group) showed a fully perpendicular conformation due to the steric effect that can occur between the Cl atom at position 2 of the aryl group18 and the fused moiety (Figure 1b). In fact, only 8c showed appreciable fluorescence intensity (FI) since, possibly, its aryl group favors a greater twisted intramolecular charge transfer (TICT) effect.3,27,28
Figure 1.

(a) Structure of pyrrolo[2,3-c]isoquinolines 8a–c. (b) Observed molecular conformation of 8c.
Results and Discussion
Some information about the approach used for the formation of (E)-arylideneaminopyrroles 6d–h and the synthesis of the pyrroloisoquinolines 8a–c was described in our previous works (Scheme 1b).17,18 In this work, we decided to explore the scope and limits of the solvent-free microwave-assisted synthesis of 6 with diverse arylaldehydes 5d–h and the 5-aminopyrrole 4 using our previously optimized conditions.18 Likewise, we carry out the synthesis of three pyrrolo[2,3-c]isoquinolines 8a–c substituted with different A or D groups by the aza-Diels–Alder cycloaddition between 6 and 2-(trimethylsilyl)phenyl triflate (7).17 In addition, we report results about the photophysical properties of the final products and a wide analysis by X-ray of some pyrrolylimines and pyrroloisoquinolines. In summary, the discussion of this article includes synthetic results on nine compounds distributed as follows: six novel compounds (6d, 6f, 6g, 8b, and 8c), three previously reported compounds by us (6b, 6e, and 8a), the photophysical study of the three fused compounds 8a–c and five novel crystal structures (6e–g and 8b and 8c) (Scheme 1c).
In this way, microwave-assisted reactions of 4 with 5d–h at 100 °C for 10 min under solvent-free conditions enabled the time-efficient formation of the (E)-arylideneaminopyrroles 6d–h in 67–78% yield (Table 1). This protocol was distinguished by its operational simplicity, broad substrate scope, and short reaction times, and no solvent, no transition metal catalyst, and no drying agent were used, rendering the experimental procedure user- and environment-friendly. The structures of pyrrolylimines 6d–h were confirmed by 1H NMR, 13C NMR, and HRMS measurements as well as by X-ray diffraction analysis (see Experimental Section and Figures S4–S19).
Table 1. Solvent-Free Microwave-Assisted Synthesis of (E)-Arylideneaminopyrroles 6d–ha.
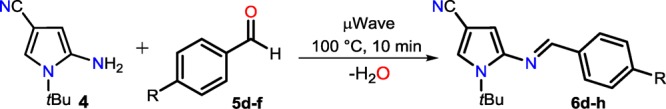
| entry | R | product | yield (%) |
|---|---|---|---|
| 1 | H | 6d | 70 |
| 2 | MeO | 6e | 67 |
| 3 | Ph2N | 6f | 68 |
| 4 | F | 6g | 78 |
| 5 | 4-BrC6H4 | 6h | 76 |
Equimolar quantities of reagents (2.0 mmol) under solvent-free conditions.
Continuing our study, we anticipated that aza-dienes 6 would be suitable candidates for cycloadditions with the aryne precursor 7, affording 1,2-dihydropyrrolo[2,3-c]isoquinolines 10, the oxidation of which would provide a direct route to the respective pyrroloisoquinolines 8. As a test reaction, we submitted a mixture of the aza-heterodiene 6a, the benzyne precursor 7, and cesium fluoride in acetonitrile at 70 °C for 24 h under oxidative conditions. The excess of MnO2 allowed an efficient in situ oxidation of the cycloadduct 10a to afford the heteroaromatic product 8a in 76% yield, though this cycloaddition was found to be unproductive at room temperature. Stimulated by this result, we investigated the scope of the reaction with other two aza-dienes under similar conditions (Scheme 2). In all cases, the reaction was found productive, affording compounds 8a–c in good yields. This aza-Diels–Alder/aromatization reaction sequence between electron-rich heterodienes and an aryne provided a time-efficient entry to functionalized fused isoquinolines (see Experimental Section). It is important to mention that an attempt was made to obtain a 5-(4-diphenylamino)phenyl derivative in order to find the best optical properties in this product due to the strong electron-donor character of its 5-aryl group,3,29 but the reaction did not proceed when the heterodiene 6f was used. Possibly, the stereoelectronic effects of the aryl group at the imine 6f decrease its reactivity with respect to the aza-Diels–Alder cycloaddition with benzyne.
Scheme 2. Synthesis of Pyrrolo[2,3-c]isoquinolines 8a–c via aza-Diels–Alder Reactions.

All reactions were performed with 1.0 equiv of heterodiene 6 and 1.2 equiv of the benzyne precursor 7 using CsF (2.4 equiv) and MnO2 (10 equiv) at 70 °C in ACN for 24 h.
Structures of products 8a–c were elucidated by 1H NMR, 13C NMR, and mass spectroscopy and, gratifyingly, also solved by single-crystal X-ray diffraction analysis. From crystallographic results, it was possible to observe a significant dihedral angle between the 5-aryl group and the heterocyclic core of products, which apparently have an important correlation with the observed fluorescence intensity in 8a–c (Figure 1 and Figure S19). The results and discussion of the photophysical studies are shown below and after X-ray diffraction studies are discussed.
Considering the molecular conformations observed and the special electronic properties of the fused products 8a–c, the three compounds were selected in order to carry out photophysical studies and thus establish their scope as novel fluorophores for possible uses in chemosensors. This study was done since compounds 8a–c have a fused structure substituted in position 5 with different electron-acceptor or electron-donor aryl groups (i.e., 8a 4-MeOPh, 8b Ph, and 8c 2,4-Cl2Ph), which is crucial in fluorescence phenomena that involve an intramolecular charge transfer (ICT) process, often observed in various N-heterocycles suitably substituted (see Figure 1). In addition, as it is observed from the crystallographic structures of 8b and 8c (perfectly extrapolated to 8a considering that no suitable crystals were obtained; Figure S19), the tendency in the orientation of aryl groups could favor a TICT phenomenon, presumably higher in 8c considering that it has a more pronounced dihedral angle due to its aryl group. The UV–vis and fluorescence emission spectra of 8a–c were achieved in cyclohexane (CH), tetrahydrofuran (THF), dichloromethane (DCM), N,N-dimethylformamide (DMF), and acetonitrile (ACN) (Figure 2 and Table 2).
Figure 2.

Absorption and emission spectra of compounds 8a–c in different solvents at 30 μM. (a, b) Compound 8a. (c, d) Compound 8b. (e, f) Compound 8c.
Table 2. Photophysical Properties of Compound 8a–c in Different Solventsa.

30 μM.
Relative quantum yields were obtained with anthracene (ϕF = 0.28 in ethanol) as the reference.
In general, UV–vis spectra of 8a–c show two bands around 255 and 345 nm with absorptivity coefficients ε ranging from 21,000 M–1 cm–1 (255 nm) to 8300 M–1 cm–1 (345 nm). When 8a–c are excited at their λab in an air-equilibrated solution at 298 K, they exhibit emission bands around 400 nm. The poor solvatochromic effect observed in both absorption and emission spectra and the short Stokes shift indicate that a small structural reorganization occur in these compounds when going from the ground to the excited state but without strong changes in the dipole moment. These properties indicate that there is no great ICT process in these compounds, which means that the absorption bands come essentially from n → π and π → π* transitions.30 As expected, the compound 8c exhibit higher fluorescence quantum yield (ϕF) versus 8a and 8b, which reflect an important effect of the dihedral angle (°) between the 5-aryl and heterocyclic moieties on the photophysical properties; thus, the 2,4-Cl2Ph group on 8c would favor a greater TICT effect, which is very sensitive to D–A efficacy, strength, and steric hindrance caused by substituents near the D–A junction,27,28 such as the Cl atom at position 2 of the 5-aryl group (see Figure 1b).
On the other hand, an interesting behavior was observed when high polar and viscous solvents were used. In all cases, no matter the 5-aryl group, the fluorescence intensity decreases dramatically when DMF was used as a solvent (e.g., for 8c, ϕF = 0.015 in CH, 0.010 in DCM, and 0.001 in DMF). This behavior reflects a high dependence of the FI with the dihedral angle between the 5-aryl group and the heterocyclic core. A possible explanation could be found in the relationship between the rotations of rotators in the molecule, which is gradually modified by changing the viscosity of the media, resulting in enhanced/decreased FI (i.e., increasing FI lifetime).31 Therefore, the λem has almost no change when the solvent polarity varies dramatically. Based on their structural and photophysical properties, the pyrrolo[2,3-c]isoquinolines 8 could be an interesting alternative for fluorescent molecular rotors (FMRs).32 Importantly, and due to the effect of the dihedral angle on the photophysical properties, corroborated by the crystal structure resolution, and the possibility of change the substituents in the periphery, these properties could be enhanced by an appropriate substitution pattern of this heterocyclic core.
Regarding the crystallographic discussion, since the structures of 6e–g and 8b and 8c were solved by single-crystal X-ray diffraction analysis,33 the molecular and supramolecular details of these five compounds were studied. The absence of short contacts to describe correctly the crystals made necessary a deeper analysis using Hirshfeld surfaces and CE-B3LYP model energies. In addition, we found an important connection of these analyses with both the synthetic and photophysical results. Crystal data details are summarized in Table 3 (see also Table S1). In general, the crystal structures of pyrrolylimines 6e–g show an interesting role of their aryl groups (4-MeOPh, 4-Ph2NPh, or 4-FPh) in the molecular conformations, showing different electronic distributions in the imine group (blue boxes in Figure 3a–c). Likewise, the molecular conformation observed in pyrrolo[2,3-c]isoquinolines 8a–c is a consequence of a stable orientation presented by the 5-aryl group, which is maintained once the compounds start the nucleation process in their trajectory to build the crystal (Figure 3d,e). As it is known, the molecular conformation plays an important role in features such as physical properties or biological activities.5,27
Table 3. Crystallographic Data of the Pyrrolylimines 6e–g and Pyrrolo[2,3-c]Isoquinolines 8b and 8c.
| compound | 6e | 6f | 6g | 8b | 8c |
|---|---|---|---|---|---|
| chemical formula | C17H19N3O | C28H26N4 | C16H16FN3 | C22H19N3 | C22H17Cl2N3 |
| Mr | 281.35 | 418.53 | 269.32 | 325.40 | 394.28 |
| crystal system/space group | triclinic/P1̅ | monoclinic/P21/c | monoclinic/P21/c | orthorhombic/Pna21 | triclinic/P1̅ |
| a, b, c (Å) | 6.5117(9), 10.8618(15), 12.3586(14) | 9.813(2), 25.305(4), 9.7316(11) | 8.8167(10), 17.559(2), 9.4619(9) | 8.8186(12), 10.9933(15), 18.078(2) | 8.4643(7), 9.8411(8), 12.2281(6) |
| α, β, γ (°) | 107.350(11), 96.512(11), 106.366(13) | 90.0, 101.981(14), 90.0 | 90.0, 90.43(1), 90.0 | 90.0, 90.0, 90.0 | 97.028(6), 98.958(6), 96.320(7) |
| V (Å3) | 781.70(19) | 2363.9(7) | 1464.8(3) | 1752.6(4) | 989.90(13) |
| Z | 2 | 4 | 4 | 4 | 2 |
| μ (mm–1) | 0.08 | 0.07 | 0.08 | 0.07 | 0.34 |
| crystal size (mm) | 0.31 × 0.25 × 0.19 | 0.23 × 0.17 × 0.14 | 0.21 × 0.15 × 0.15 | 0.21 × 0.16 × 0.15 | 0.25 × 0.20 × 0.19 |
Figure 3.

(Bottom) Structures of (a) 6e, (b) 6f, (c) 6g, (d) 8b, and (e) 8c showing anisotropic displacement ellipsoids at the 30% probability level and highlighting the azomethine fragment in blue (6e–g) and the C–C bond length distance (8b and 8c). (Top) Dihedral angles between the least-squares mean planes made by the pyrrolic (red plane) and aryl (blue plane) rings (6e–g) and the pyrroloisoquinolinic core (red plane) and aryl ring (blue plane) (8b and 8c).
In this sense, the aryl group effect in 6e–g is noticed in the dihedral angles between the least-squares mean planes made by the pyrrole and benzene rings. In 6e, the dihedral angle along the azomethine fragment (C—N=C—C, Figure 3a) is −179.92(14)°, and the bond distance between the C atom of the pyrrolic ring and the N atom of the imine group has a value of 1.386(2) Å. This evidences a greater resonance effect in 6e as result of the electron-donor character of its MeO group, a structural property not seen in 6f and 6g (conformational effects) where the bond lengths are 1.398(3) and 1.395(3) Å, respectively. These findings suggest that groups of proper electronic nature could regulate the resonance properties in 6e–g. Considering that the same interactions are detected in all the molecules (C–H···N between the tBu group and the N atom of the azomethine group), the diverse conformations seem to be independent of other factors (i.e., intramolecular hydrogen bonds). Regarding 8a–c, its 5-aryl group is connected to the pyrroloisoquinolinic core by sp2 carbon atoms generating bond lengths of 1.487(5) and 1.492(3) Å for 8b and 8c, respectively, which suggests distances deviated from expected values for free rotational bonds as a consequence of hybridization effects (Figure 3d,e; see also Scheme 2). The two conformations for these compounds are characterized by dihedral angles observed in Figure 3d,e.
In the supramolecular assembly of 6e–g, no classic hydrogen bonds were found, and only weak hydrogen interactions were detected based on the intermolecular H···(O, N) lengths. In 6e, pairs of inversion-related molecules are connected by two equivalent weak C–H···Oi (2.65 Å; (i) 1 – x, 1 – y, 1 – z) hydrogen interactions, involving one methyl in the tBu group (Figure 4a). These pairs are the slabs of infinite chains running along the [−111] direction and connected by weaker C–H···Nii (2.78 Å; (ii) 1 – x, 2 – y, 2 – z) interactions, involving the CN and MeO groups. The three-dimensional (3D) assembly is completed by C–H···Oiii (2.76 Å; (iii) 2 – x, 2 – y, 1 – z) hydrogen interactions, joining molecules by the connection between tBu and MeO groups in adjacent molecules along the a axis (Figure 4a). In 6f, the formation of weak C–H···Ni,ii interactions involving the tBu (2.71 Å; (i) x, 3/2 – y, −1/2 + z) and triphenylamine (TPA) (2.72 Å; (ii) 1 – x, −1/2 + y, 3/2 – z) groups builds the solid forming chains running along the [010] direction. These chains are further connected in the three dimensions by the same sort of C–H···N contacts forming a framework that seems solely dependent on these connections (Figure 4b). In 6g, the presence of a F atom over the aryl group allows the connection of pairs of inversion-related molecules (by C–H···Fi; 2.72 Å; (i) 1 + x, y, 1 + z) connected by two equivalent C–H···Nii (2.68 Å; (ii) 1 – x, 1 – y, 2 – z) hydrogen interactions. As it was observed in 6e and 6f, the CN group interacts with the azomethine (in C–H···Nii) and tBu groups, C–H···Niii (2.72 Å; (iii) 2 – x, 1 – y, 2 – z), forming bifurcated hydrogen interactions building (010) sheets, which are further connected in the [010] direction by van der Waals forces (Figure 4c).
Figure 4.

Crystal structures of (a) 6e, (b) 6f, and (c) 6g showing the C–H···O, C–H···N, and C–H···F hydrogen-bond interactions. The (i), (ii), and (iii) codes serve as a reference with the description in the text.
Hirshfeld (HF) surface analysis mapped over dnorm (analysis of the contact distances di and de from the HF surface to the nearest atom inside and outside, respectively) was performed in order to investigate these contacts further. In 6e, the bright red spots over dnorm corresponds exclusively to the C–H···O (2.65 Å; 1 – x, 1 – y, 1 – z) hydrogen interactions showing that C–H···N interactions are not relevant (Figure S20a). On the other hand, C–H···N interactions are clearly, but weakly, observed in the HF surface for 6f, showing that, in this crystal, they are “close contacts” compared with the remaining interactions. Additional C–H···π interactions (3.38 Å) were detected between the tBu group and one Ph group of 6f (Figure S20b). In 6g, the red spots are showing mainly the C–H···N contacts, leaving other contacts with nearly nonobservable spots; including C–H···F (Figure S20c). In the 2D fingerprint plots, H···N/N···H contacts comprise 12–17% of the total HF surface in 6e–g, which is not the highest effect to the crystal structure. In all compounds, no close (<2.6 Å) contacts are observed, showing the absence of strong hydrogen bonds in the blue/white regions on the HF surfaces. The nonbonded H···H (40–57%) contacts suggest a principal role of long-range hydrophobic interaction in the formation of the crystals.
Energy framework representations of the intermolecular interactions were computed using the CE-B3LYP model, which serves to interpret the topological architecture of the supramolecular features from an energetic perspective, considering the difficulty to interpret the formation of the crystal based on long hydrogen contacts (Figure 5). The strongest interaction in 6e is not related to the closest C–H···O (2.65 Å; 1 – x, 1 – y, 1 – z) hydrogen contact as expected from the atom–atom analysis. Instead, the strongest contacts form chains of molecules connected by total interaction energies of −36.6 kJ mol–1 along the [100] direction connected between them by interaction energies of −37.9 and −32.1 kJ mol–1 along the [011] direction with contributions from electrostatic and dispersion energies in similar magnitudes (Figure 5a and Table S2). The energy framework describes an anisotropic topology, showing that the crystal is formed mainly by dispersion forces along the a axis. These chains of molecules are connected between them by a combination of electrostatic (only important in the connection of the chains along [011] by the azomethine fragments, red framework) and dispersion contributions forming the 3D structure (Figure 5a). The topology is characterized by the formation of layers stacked along the [01-1] direction.
Figure 5.

Description of molecular close contacts and energy framework diagrams for electrostatic (red) and dispersion (green) contributions to the total interaction energies (blue) in compounds (a) 6e, (b) 6f, and (c) 6g.
The interaction energies for selected molecular pairs in the first coordination sphere around the asymmetric unit for 6e–g are summarized in Table S2. As it is observed, the dispersion terms contribute in high proportion to the formation of the crystals, explaining the absence of short hydrogen contacts due to the electronic distributions that do not allow a great nucleophilicity over O and N atoms and the absence of acidic hydrogen atoms. Additionally, the highest total energies are observed in 6f, which is related to the highest thermal stability compared with 6e and 6g, considering that their melting points are 142–143, 181–182, and 138–139 °C for 6e–g, respectively. As it was observed previously, these framework energies are comparable with the experimental sublimation enthalpies.34
As observed in 6e–g, the hydrogen C–H···N interactions involving the nitrile group are weak, with distances greater than 2.6 Å rather difficult to detect without using Hirshfeld surfaces. In 8b, one possibility for such contacts is observed in Figure 6a. However, this weak C–H···Ni contact has a length of 2.92 Å ((i) 1/2 + x, 3/2 – y, z), leaving this interaction as not relevant. The planar conformation on the pyrroloisoquinolinic core allows the formation of π···πii (3.740(2) Å; (ii) 1/2 + x, 3/2 – y, z) stacking contacts (Figure 6a). These interactions form chains of molecules along the [100] direction. Hirshfeld surface shows a complete lack of short contacts, leaving the map without red spots and only blue regions, suggesting that the crystal is formed only by long-distance hydrophobic interactions despite the presence of potential donor/acceptor regions in the molecule as it is observed in the electrostatic potential map in Figure S21a.
Figure 6.

Crystal structures of (a) 8b and (b, c) 8c showing the C–H···N and C–H···π hydrogen-bond interactions and π···π stacking contacts. The (i), (ii) and (iii) codes serve as reference with the description in the text.
In 8c, the nitrile group has bifurcated weak C–H···N contacts with the pyrrolici and 2,4-dichlorophenyli rings (2.74 Å, (i) −x, −y, 1 – z), forming chains of molecules along [100] (Figure 6b). C–H···π interactions play an important role in the crystal with contacts involving also the pyrrolicii and 2,4-dichloreophenyliii rings as hydrogen acceptors (2.73 Å, (ii) 1 + x, y, z; 2.91 Å, (iii) 2 – x, 1 – y, 1 – z) (Figure 6c). Hirshfeld surface analysis shows that the C–H···π interaction involving the pyrrolicii ring also has another connotation. The H···N distance has a value 2.75 Å, and the C–H···N angle is 159°, indicating an interaction between the H atom and the nonhybridized 2p atomic orbital in the pyrrolic N atom that in fact is perpendicular to the plane of the ring and is part of the π cloud. This bifurcated interaction, C–H···π and C–H···N, is observed as a doublet spot over the HF surface (Figure S21b).
Another interesting interaction is detected in the HF surface involving a Me group from the tBu group and the C atom placed between the substituent halogens in the aryl group of 8c. Due to the geometry (H···C distance of 2.81 Å and C–H···C angle of 156°), this contact could be understood as a C–H···C interaction, which is not frequently reported (Figure S21b). The contact is possible due to the sp2 hybridization of the C atom that allows the interaction of the H atom with the perpendicular pure 2p orbital overlapped in the π cloud. The 2D fingerprints show that, essentially, the crystal in 8b is mainly built by hydrophobic interactions with H···H contacts comprising 56% of the total HF surface (Figure S21a). Despite the suggestion of other interactions, such as H···N, they showed to be very large in 8b and thus irrelevant in the supramolecular assembly. In 8c, the 2D fingerprint plot shows that H···N interactions comprise 13% of the HF surface (Figure S21b). In this case, the H···H contacts are less relevant (35%) compared with 8b, suggesting a less hydrophobic character.
In order to obtain a better understanding of the crystal packing, the CE-B3LYP model was used to investigate the supramolecular assembly in terms of energy frameworks (Table S3). In 8b, the strongest pairwise interactions have a total energy value −60.9 kJ mol–1, forming chains of molecules along the [100] direction interacting by the fused heterocyclic moiety in a π···π stacking disposition (Figure 7a). Considering the high stabilization in this direction, it is possible to assume that the short distance among adjacent centroids (5.72 Å) is due to dispersion forces. In fact, this term contributes mainly to this interaction, showing that these forces are responsible for assembling along the a axis instead of particular atom–atom contacts. The second strongest interactions connect these chains along the [001] direction with −22.7 kJ mol–1 as the total energy, which is also represented in higher proportion by the dispersion term. The energy frameworks show a curious topology. Electrostatic forces act to keep the molecules assembled along the a axis (Figure 7a). Observing the closeness of neighboring molecules due to dispersion forces along this [100] direction, the electrostatic term rises due to the short distance between heterocyclic-fused rings where some degree of electronic polarization exists. However, the dispersion forces are the most important interactions in the crystal, forming (001) sheets stacked along the c axis and building a 3D structure by less energetic interactions giving an anisotropic tendency.
Figure 7.

Description of molecular close contacts and energy framework diagrams for electrostatic (red) and dispersion (green) contributions to the total interaction energies (blue) in compounds (a) 8b and (b) 8c.
In 8c, the strongest pairwise interactions occur between the nearest (7.39 Å) molecules involving the pyrroloisoquinolinic moieties oriented in a parallel form, allowing a total energy value of −62.9 kJ mol–1 (Figure 7b and Table S3). Similar to 8b, the dispersion term contributes in higher proportion to the interaction, showing the importance of the π cloud in the pyrroloisoquinolinic core to build “strong” dispersion attractions in this sort of structures. Other molecules are connected by total energies such as −38.9 and −31.8 kJ mol–1 in which the dispersion term always represents the most important interaction. The energy frameworks show similar electrostatic interactions as observed in 8b, forming chains along the [100] direction with a curious zigzag topology joining the fused heterocyclic rings. The dispersion-cylinder magnitudes show an isotropic structure built by the strongest interactions forming a sort of compacted scaffolding structure. Between the strong pillars, other diagonal cylinders of low magnitude are present acting between halogen atoms, which does not mean an actual absence but a low energetic contact by a possible repulsion between close electron shells (Figure 7b) compared with 8b, suggesting a less hydrophobic character.
In order to have a deeper insight into the electronic properties for the studied compounds and achieve a better connection of the experimental results, the energy levels of the electron-donor HOMO (highest occupied molecular orbital) and electron-acceptor LUMO (lowest unoccupied molecular orbital) for aza-dienes 6c,186e–g, and products 8b and 8c were computed (Figure 8). These calculations were carried out through the STO-3G basis set at the Hartree–Fock level of theory implemented in CrystalExplorer35 using the crystallographic information files (.cif) obtained from the X-ray diffraction measurements. In the analyzed aza-dienes, the electron densities in the frontier molecular orbitals (FMO) are mostly localized over the whole of the molecules (except the tBu group), especially over the pyrrolylimine and aryl fragments (Figure 8a).
Figure 8.

Computed energy levels HOMO and LUMO for aza-dienes (a) 6c and 6e–g and (b) products 8b and 8c using the crystallographic information files (.cif) by the STO-3G basis set at the Hartree–Fock level of theory.
The HOMO–LUMO energy gaps suggest that 6g and 6f are the less and more reactive molecules toward electrophilic reagents, respectively. These results are consistent with the strong electron-donor character of the 5-aryl group in 6f,3,28 though FMO can be affected by the solvent, which could shift the electron density in the FMO from one fragment to another.36 Remarkably, the HOMO in 6f has a poor contribution (low electronic density) in the aza-dienic moiety versus the other imines analyzed (6c, 6e, and 6g), which could explain the poor reactivity of 6f (mentioned above) with respect to the aza-Diels–Alder reaction with benzyne (yellow circle in Figure 8a). In 8b and 8c, the electron densities are mostly localized over the pyrroloisoquinolinic fragment; however, the HOMO in 8c present lower electronic density over the 2,4-dichlorophenyl group as a consequence of the marked dihedral angle (75.01(8)°) between this aryl group with the pyrroloisoquinolinic fragment, as mentioned earlier in the crystallographic results. Consequently, the charge transfer from the pyrroloisoquinolinic core to 2,4-dichlorophenyl group could be better observed upon photoexcitation of 8c (yellow circle in Figure 8b).37 These findings confirm that the fluorescence phenomenon of 8c is governed by a TICT mechanism.3,27,28
Conclusions
In summary, we have established a general synthetic access to various (E)-2-arylideneaminopyrroles 6, which served as key intermediates of some pyrrolo[2,3-c]isoquinolines 8a–c through the aryne aza-Diels–Alder cycloaddition. Both intermediates and final products were obtained in good yields (up to 78 and 84%, respectively), their structures were determined on the basis of NMR measurements and HRMS analysis, and pleasantly, the structures were confirmed by single-crystal X-ray diffraction analyses. The fused N-heterocycles 8a–c are substituted in position 5 with aryl groups of different electronic nature; thus, their photophysical properties were investigated, finding absorption bands with a non-ICT character and a fluorescence intensity sensible to the dihedral angle between the 5-aryl group and the heterocyclic core. Interestingly, we find a relationship between viscosity and the solvent with the fluorescence intensity of these kinds of compounds, this property could be very useful for fluorescence molecular rotor (FMR) applications. On the other hand, the crystallographic results show that all the interactions found in 6e–g and 8b and 8c are weak, and van der Waals forces seem to be the main responsible for the solid formation. The analysis of individual interatomic interactions and the difficult search for hydrogen bonds did not allow a rational description of the crystal formation. However, using the CE-B3LYP model, in addition to studying the energetic topology and understanding the crystal architecture of compounds, we managed to find an important connection with both the synthetic and photophysical results.
Experimental Section
General Information
All reagents were purchased from commercial sources and used without further purification, unless otherwise noted. All starting materials were weighed and handled in air at room temperature. The reactions were monitored by TLC and visualized by UV (254 nm). Column and flash chromatography was performed on silica gel (230–400 mesh or 70–230 mesh, respectively). All reactions under microwave irradiation were performed using a sealed reaction vessel (10 mL, max pressure = 300 psi) containing a Teflon-coated stirring bar (obtained from CEM). Microwave-assisted reactions were performed in a CEM Discover focused microwave (ν = 2.45 GHz) reactor equipped with a built-in pressure measurement sensor and a vertically focused IR temperature sensor; controlled temperature, power, and time settings were used for all reactions. NMR spectra were recorded at 400 MHz (1H) and 100 MHz (13C) at 298 K. NMR spectroscopic data were recorded in CDCl3 and DMSO-d6 using, as internal standards, the residual nondeuterated signal for 1H NMR and the deuterated solvent signal for 13C NMR spectroscopy. DEPT spectra were used for the assignment of carbon signals. Chemical shifts (δ) are given in ppm and coupling constants (J) are given in Hz. The following abbreviations are used for multiplicities: s = singlet, d = doublet, t = triplet, and m = multiplet. Melting points were collected using a Stuart SMP10 melting point apparatus, and the acquired data are uncorrected. High-resolution mass spectra (HRMS) were recorded using an Agilent Technologies Q-TOF 6520 spectrometer by electrospray ionization (ESI). 5-Amino-1-(tert-butyl)-1H-pyrrole-3-carbonitrile 4 was prepared using a known procedure.38 The electronic absorption and fluorescence emission spectra were recorded in quartz cuvettes having a path length of 1 cm. UV–vis and emission measurements were achieved at room temperature (20 °C). For fluorescence measurements, both the excitation and the emission slit widths were 5 nm.
Synthesis and Characterization
General Procedure for the Synthesis of (E)-5-(Arylideneamino)-1-tert-butyl-1H-pyrrole-3-carbonitriles 6d–h (Scheme 1b)
A 10.0 mL sealable oven-dried tubular reaction vessel was charged with 5-amino-1-(tert-butyl)-1H-pyrrole-3-carbonitrile (4; 327 mg, 2.0 mmol) and arylaldehydes 5d–h (2.0 mmol). The resulting mixture was subjected to microwave under solvent-free conditions at 100 °C and maintained this temperature for 40 min, after which the reaction mixture was cooled to 50 °C by airflow. The resulting crude product was purified by flash chromatography on silica gel using a mixture of n-pentane/CH2Cl2 (1:1, v/v) as an eluent to give the expected N-pyrrolylimines 6d–h in good yields. Crystals suitable for X-ray structure analysis were obtained from methanol by slow evaporation of the solution at room temperature.
(E)-5-(Benzylideneamino)-1-(tert-butyl)-1H-pyrrole-3-carbonitrile (6d)
Following the general procedure in the reaction with benzaldehyde (5d; 205 μL, 2.0 mmol), the imine 6d was obtained as a yellow solid (352 mg, 70%). mp 86–87 °C. Recrystallization of 6d from methanol afforded crystalline yellow prisms suitable for X-ray diffraction analysis. 1H NMR (CDCl3, 400 MHz): δ 1.72 (s, 9H), 6.42 (d, J = 1.9 Hz, 1H), 7.27 (d, J = 1.9 Hz, 1H), 7.46–7.49 (m, 3H), 7.83–7.86 (m, 2H), 8.46 (s, 1H) ppm. 13C{1H} NMR (CDCl3, 101 MHz): δ 30.1 (CH3), 58.4 (C), 90.2 (C), 98.2 (CH), 117.0 (C), 124.9 (CH), 128.4 (CH), 128.8 (CH), 131.2 (CH), 136.2 (C), 143.0 (C), 155.1 (CH) ppm. HRMS (ESI+): calcd for C16H18N3+ 252.1495 [M + H]+; found, 252.1494.
(E)-1-(tert-Butyl)-5-((4-methoxybenzylidene)amino)-1H-pyrrole-3-carbonitrile (6e)
Following the general procedure in the reaction with 4-methoxybenzaldehyde (5e; 250 μL, 2.1 mmol), the imine 6e was obtained as a yellow solid (377 mg, 67%). mp 142–143 °C (lit.17 mp 143–144 °C). Recrystallization of 6e from methanol afforded crystalline yellow prisms suitable for X-ray diffraction analysis. 1H NMR (CDCl3, 400 MHz): δ 1.71 (s, 9H), 3.88 (s, 3H), 6.34 (d, J = 1.8 Hz, 1H), 6.98 (d, J = 8.8 Hz, 2H), 7.24 (d, J = 1.8 Hz, 1H), 7.79 (d, J = 8.8 Hz, 2H), 8.38 (s, 1H) ppm. 13C{1H} NMR (CDCl3, 101 MHz): δ 30.0 (CH3), 55.4 (CH3), 58.2 (C), 89.9 (C), 97.6 (CH), 114.3 (CH), 117.2 (C), 124.5 (CH), 129.2 (C), 130.2 (CH), 143.5 (C), 154.8 (CH), 162.2 (C) ppm. These NMR data matched the previously reported data.17
(E)-1-(tert-Butyl)-5-((4-(diphenylamino)benzylidene)amino)-1H-pyrrole-3-carbonitrile (6f)
Following the general procedure in the reaction with 4-(diphenylamino)benzaldehyde (5f; 303 mg, 1.1 mmol), the imine 6f was obtained as a yellow solid (570 mg, 68%). mp 181–182 °C. Recrystallization of 6f from methanol afforded crystalline yellow prisms suitable for X-ray diffraction analysis. 1H NMR (CDCl3, 400 MHz): δ 1.70 (s, 9H), 6.33 (d, J = 1.8 Hz, 1H), 7.06 (d, J = 8.7 Hz, 2H), 7.11 (t, J = 7.5 Hz, 2H), 7.15 (d, J = 7.8 Hz, 4H), 7.24 (d, J = 1.8 Hz, 1H), 7.31 (t, J = 7.8 Hz, 4H), 7.67 (d, J = 8.7 Hz, 2H), 8.35 (s, 1H) ppm. 13C{1H} NMR (CDCl3, 101 MHz): δ 30.0 (CH3), 58.2 (C), 89.9 (C), 97.5 (CH), 117.3 (C), 121.2 (CH), 124.1 (CH), 124.4 (CH), 125.5 (CH), 129.3 (C), 129.5 (CH), 129.5 (CH), 143.6 (C), 146.8 (C), 150.7 (C), 154.5 (CH) ppm. HRMS (ESI+): calcd for C28H27N4+ 419.2230 [M + H]+; found, 419.2226.
(E)-1-(tert-Butyl)-5-((4-fluorobenzylidene)amino)-1H-pyrrole-3-carbonitrile (6g)
Following the general procedure in the reaction with 4-fluorobenzaldehyde (5g; 204 μL, 1.9 mmol), the imine 6g was obtained as a yellow solid (400 mg, 78%). mp 138–139 °C. Recrystallization of 6g from methanol afforded crystalline yellow prisms suitable for X-ray diffraction analysis. 1H NMR (CDCl3, 400 MHz): δ 1.71 (s, 9H), 6.40 (d, J = 2.0 Hz, 1H), 7.15 (t, J = 8.7 Hz, 2H), 7.27 (d, J = 2.0 Hz, 1H), 7.82–7.85 (m, 2H), 8.42 (s, 1H) ppm. 13C{1H} NMR (CDCl3, 101 MHz): δ 30.1 (CH3), 58.3 (C), 90.2 (C), 98.2 (CH), 116.0 (CH, d, JC–F = 22.1 Hz), 117.0 (C), 124.9 (CH), 130.3 (CH, d, JC–F = 8.8 Hz), 132.6 (C), 142.8 (C), 153.7 (CH), 164.6 (C, d, JC–F = 252.4 Hz) ppm. HRMS (ESI+): calcd for C16H17FN3+ 270.1401 [M + H]+; found, 270.1400.
(E)-5-((4-Bromobenzylidene)amino)-1-(tert-butyl)-1H-pyrrole-3-carbonitrile (6h)
Following the general procedure in the reaction with 4-bromobenzaldehyde (5h; 352 mg, 1.90 mmol), the imine 6h was obtained as a yellow solid (477 mg, 76%). mp 156–157 °C. 1H NMR (CDCl3, 400 MHz): δ 1.71 (s, 9H), 6.43 (d, J = 1.8 Hz, 1H), 7.28 (d, J = 1.8 Hz, 1H), 7.60 (d, J = 8.6 Hz, 2H), 7.70 (d, J = 8.6 Hz, 2H), 8.40 (s, 1H) ppm. 13C{1H} NMR (CDCl3, 101 MHz): δ 30.1 (CH3), 58.4 (C), 90.4 (C), 98.5 (CH), 116.9 (C), 125.2 (CH), 125.7 (C), 129.7 (CH), 132.1 (CH), 135.1 (C), 142.6 (C), 153.6 (CH) ppm. HRMS (ESI+): calcd for C16H1779BrN3+ 330.0600 [M + H]+; found, 330.0599.
General Procedure for the Synthesis of 3-(tert-Butyl)-5-aryl-3H-pyrrolo[2,3-c]isoquinoline-1-carbonitriles 8a–c (Scheme 1b)
A sealable (Teflon screw cap) oven-dried tubular reaction vessel was charged with the appropriate arylideneaminopyrrole 6 (0.30 mmol), CsF (109 mg, 0.72 mmol), and manganese dioxide (263 mg, 3.0) and subjected to high vacuum for 20 min. The mixture was placed under an argon atmosphere, and anhydrous acetonitrile (2.0 mL) was added. To this stirred solution kept at room temperature was added 2-(trimethylsilyl)phenyl triflate (7; 87 μL, 0.36 mmol) in one portion, the reaction vessel was sealed, and the resulting reaction mixture was stirred at 70 °C for 24 h. The product reaction mixture was diluted with EtOAc and water. The organic layer was separated, and the aqueous layer was extracted twice with EtOAc. The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum to give the crude product. The resulting crude product was purified by flash chromatography on silica gel using a mixture of n-pentane/EtOAc (50:1, v/v) as an eluent to give the pyrrolo[2,3-c]isoquinolines 8a–c. Crystals suitable for X-ray structural analyses were obtained from methanol by slow evaporation of the solution at room temperature.
3-(tert-Butyl)-5-(4-methoxyphenyl)-3H-pyrrolo[2,3-c]isoquinoline-1-carbonitrile (8a)
Following the general procedure in the reaction with the imine 6e (87 mg, 0.31 mmol), the compound 8a was obtained as a white solid (80 mg, 73%). mp 190–191 °C (lit.17 190–191 °C). 1H NMR (CDCl3, 400 MHz): δ 1.89 (s, 9H), 3.93 (s, 3H), 7.09 (d, J = 8.4 Hz, 2H), 7.51 (t, J = 7.3 Hz, 1H), 7.71 (d, J = 8.3 Hz, 2H), 7.79 (dd, J = 7.2 Hz, 1H), 7.85 (s, 1H), 8.25 (d, J = 8.5 Hz, 1H), 8.78 (d, J = 8.3 Hz, 1H) ppm. 13C{1H} NMR (CDCl3, 101 MHz): δ 29.5 (CH3), 55.4 (CH3), 55.7 (C), 82.8 (C), 113.3 (C), 113.7 (CH), 117.7 (C), 122.4 (CH), 123.2 (C), 125.2 (CH), 128.7 (CH), 130.2 (CH), 130.3 (CH), 131.6 (CH), 131.7 (C), 132.6 (C), 142.1 (C), 155.4 (C), 159.9 (C) ppm. HRMS (ESI+): calcd for C23H22N3O+ 356.1757 [M + H]+; found, 356.1759. These NMR data matched the previously reported data.17
3-(tert-Butyl)-5-phenyl-3H-pyrrolo[2,3-c]isoquinoline-1-carbonitrile (8b)
Following the general procedure in the reaction with the imine 6d (73 mg, 0.29 mmol), the compound 8b was obtained as a white solid (72 mg, 76%). mp 186–187 °C. Recrystallization of 8b from methanol afforded crystalline yellow prisms suitable for X-ray diffraction analysis. 1H NMR (CDCl3, 400 MHz): δ 1.89 (s, 9H), 7.49–7.58 (m, 4H), 7.75 (d, J = 8.2 Hz, 2H), 7.79 (dd, J = 7.1 Hz, 1H), 7.87 (s, 1H), 8.21 (d, J = 8.4 Hz, 1H), 8.79 (d, J = 8.2 Hz, 1H) ppm. 13C{1H} NMR (CDCl3, 101 MHz): δ 29.5 (CH3), 58.8 (C), 82.9 (C), 113.6 (C), 117.6 (C), 122.4 (CH), 123.2 (C), 125.3 (CH), 128.2 (CH), 128.4 (CH), 128.6 (CH), 130.3 (CH), 130.3 (CH), 130.5 (CH), 131.6 (C), 140.1 (C), 142.0 (C), 155.7 (C) ppm. HRMS (ESI+): calcd for C22H20N3+ 326.1652 [M + H]+; found, 326.1656.
3-(tert-Butyl)-5-(2,4-dichlorophenyl)-3H-pyrrolo[2,3-c]isoquinoline-1-carbonitrile (8c)
Following the general procedure in the reaction with the imine 6c (89 mg, 0.28 mmol), the compound 8c was obtained as a white solid (93 mg, 84%). mp 176–174 °C. Recrystallization of 8c from methanol afforded crystalline yellow prisms suitable for X-ray diffraction analysis. 1H NMR (CDCl3, 400 MHz): δ 1.86 (s, 9H), 7.41–7.44 (m, 2H), 7.50 (t, J = 7.0 Hz, 1H), 7.61 (s, 1H), 7.70 (d, J = 8.3 Hz, 1H), 7.81 (t, J = 7.0 Hz, 1H), 7.90 (s, 1H), 8.80 (d, J = 8.3 Hz, 1H) ppm. 13C{1H} NMR (CDCl3, 101 MHz): δ 29.5 (CH3), 59.0 (C), 82.2 (C), 114.4 (C), 117.3 (C), 122.5 (CH), 123.5 (C), 125.7 (CH), 127.0 (CH), 127.9 (CH), 129.8 (CH), 130.6 (CH), 130.8 (CH), 131.2 (C), 132.5 (CH), 134.7 (C), 135.0 (C), 137.2 (C), 141.6 (C), 152.2 (C) ppm. HRMS (ESI+): calcd for C22H1835Cl2N3+ 394.0872 [M + H]+; found, 394.0869.
Photophysical Properties
UV–Vis Absorption and Fluorescence Studies
The solvatochromic studies of the compounds 8a–c were carried out from 50 μM stock solutions in cyclohexane (CH), tetrahydrofuran (THF), dichloromethane (DCM), dimethylformamide (DMF), and acetonitrile (ACN). The relative quantum yields were obtained using anthracene (ϕF = 0.28 in ethanol at 340 nm) as the reference and calculated according to the following equation24−27
where x and st indicate the sample and standard solution, respectively, ϕF is the quantum yield, F is the integrated area of the emission, A is the absorbance at the excitation wavelength, and η is the index of refraction of the solvents.
Refinement
Crystal data, data collection, and structure refinement details are summarized in Table S1. Single crystals of the compounds were obtained by recrystallization from methanol and mounted in oil. The X-ray diffraction intensity data were measured at room temperature (298(2) K) with an Agilent SuperNova, Dual, Cu at zero, Atlas four-circle diffractometer equipped with a CCD plate detector. In the data acquisition, a Mo microfocus sealed tube was used (λ = 0.71073 Å). The collected frames were integrated with the CrysAlisPro software package (Agilent Technologies, version 1.171.37.35). Data were corrected for the absorption effect using the same CrysAlisPro software package by the empirical absorption correction using spherical harmonics implemented in the SCALE3 ABSPACK scaling algorithm. The structure was solved using an iterative algorithm39 and subsequently completed by a difference Fourier map. All the hydrogen atoms, with H-atom parameters constrained during refinements, were placed in calculated positions (C–H = 0.93–0.96 Å) and included as riding contributions with isotropic displacement parameters set at 1.2–1.5 times the Ueq value of the parent atom. The crystal structures were refined using the program SHELXL2014.40 The graphic material was prepared using the software Mercury 3.8.41 In the structural calculations, the software PLATON was used.42
Computational Methods
Hirshfeld (HF) surface analysis43 mapped over dnorm (analysis of the contact distances di and de from the HF surface to the nearest atom inside and outside, respectively) was calculated in order to investigate the close contacts further which are observed as bright red spots over dnorm. Electrostatic potentials were calculated using TONTO, a Fortran-based object-oriented system for quantum chemistry and crystallography,44,45 and subsequently mapped over the HF surface using the STO-3G basis set at the Hartree–Fock level of theory over the range of ±0.14 a.u. Energy frameworks describing the intermolecular interaction energies were computed based on CE-B3LYP interaction energies (kJ mol–1), which use B3LYP/6-31G(d,p) molecular wave functions calculated applying the crystal symmetry obtained from X-ray results, which include electrostatic (Eele), polarization (Epol), dispersion (Edis), and exchange-repulsion (Erep) terms.46 Computed energies between molecular pairs are represented using cylinders joining the centroids (centers of mass) of the molecules with a radius proportional to the magnitude of the interaction. The calculations were performed using the models implemented in CrystalExplorer.35
Acknowledgments
We thank the Department of Chemistry and Vicerrectoría de Investigaciones at Universidad de los Andes for financial support. We also acknowledge Edwin Guevara (Universidad de los Andes) for acquiring the mass spectra. J.-C.C. thanks the Universidad Pedagógica y Tecnológica de Colombia UPTC for financial support. A.T. acknowledges COLCIENCIAS for a postdoctoral fellowship (Con. 784 of 2017). Y.C. and J.R. thank Aix-Marseille Université and the Centre National de la Recherche Scientifique (CNRS). M.A.M. thanks the “Fondo de Apoyo para Profesores Asistentes” of the Universidad de los Andes, Bogotá (Colombia).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b02043.
Copies of NMR spectra (1H and 13C{1H}) for imines 6d–h and products 8a–c and details of X-ray crystallography (tables and graphic material) (PDF)
Accession Codes
CCDC 1959987 (6e), 1959991 (6f), 1959989 (6g), 1959988 (8b), and 1959990 (8c) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif or by emailing data_request@ccdc.cam.ac.uk or contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
This paper was published ASAP on October 9, 2019, with incorrect accession codes for the crystallographic data for the compounds. The corrected version was reposted on October 22, 2019.
Supplementary Material
References
- Eftekhari-Sis B.; Zirak M. Chemistry of α-Oxoesters: A Powerful Tool for the Synthesis of Heterocycles. Chem. Rev 2015, 115, 151–264. 10.1021/cr5004216. [DOI] [PubMed] [Google Scholar]
- Castillo J.-C.; Portilla J. Recent advances in the synthesis of new pyrazole derivatives. Targets Heterocycl. Syst. 2018, 22, 194–223. 10.17374/targets.2019.22.194. [DOI] [Google Scholar]
- Mako T. L.; Racicot J. M.; Levine M. Supramolecular Luminescent Sensors. Chem. Rev. 2019, 119, 322–477. 10.1021/acs.chemrev.8b00260. [DOI] [PubMed] [Google Scholar]
- Chrzanowska M.; Grajewska A.; Rozwadowska M. D Asymmetric Synthesis of Isoquinoline Alkaloids: 2004–2015. Chem. Rev. 2016, 116, 12369–12465. 10.1021/acs.chemrev.6b00315. [DOI] [PubMed] [Google Scholar]
- Buskes M. J.; Harvey K. L.; Prinz B.; Crabb B. S.; Gilson P. R.; Wilson D. J. D.; Abbott B. M. Exploration of 3-Methylisoquinoline-4-carbonitriles as Protein Kinase A Inhibitors of Plasmodium falciparum. Bioorg. Med. Chem. 2016, 24, 2389–2396. 10.1016/j.bmc.2016.03.048. [DOI] [PubMed] [Google Scholar]
- Guo T.; Yu L.; Zhao B.; Li Y.; Tao Y.; Yang W.; Hou Q.; Wu H.; Cao Y. Highly Efficient, Red-Emitting Hyperbranched Polymers Utilizing a Phenyl-Isoquinoline Iridium Complex as the Core. Macromol. Chem. Phys. 2012, 213, 820–828. 10.1002/macp.201100676. [DOI] [Google Scholar]
- Awuah E.; Capretta A. Strategies and Synthetic Methods Directed Toward the Preparation of Libraries of Substituted Isoquinolines. J. Org. Chem. 2010, 75, 5627–5634. 10.1021/jo100980p. [DOI] [PubMed] [Google Scholar]
- Seayad J.; Seayad A. M.; List B. Catalytic Asymmetric Pictet–Spengler Reaction. J. Am. Chem. Soc. 2006, 128, 1086–1087. 10.1021/ja057444l. [DOI] [PubMed] [Google Scholar]
- Birch A. J.; Jackson A. H.; Shannon P. V. R. A New Modification of the Pomeranz–Fritsch Isoquinoline Synthesis. J. Chem. Soc., Perkin Trans. 1 1974, 0, 2185–2190. 10.1039/P19740002185. [DOI] [Google Scholar]
- Zhao D.; Lied F.; Glorius F. Rh(III)-Catalyzed C–H Functionalization/Aromatization Cascade with 1,3-Dienes: A Redox-Neutral and Regioselective Access to Isoquinolines. Chem. Sci. 2014, 5, 2869–2873. 10.1039/c4sc00628c. [DOI] [Google Scholar]
- Qi L.; Hu K.; Yu S.; Zhu J.; Cheng T.; Wang X.; Chen J.; Wu H. Tandem Addition/Cyclization for Access to Isoquinolines and Isoquinolones via Catalytic Carbopalladation of Nitriles. Org. Lett. 2017, 19, 218–221. 10.1021/acs.orglett.6b03499. [DOI] [PubMed] [Google Scholar]
- Xie L.-G.; Niyomchon S.; Mota A. J.; González L.; Maulide N. Metal-Free Intermolecular Formal Cycloadditions Enable an Orthogonal Access to Nitrogen Heterocycles. Nat. Commun 2016, 7, 10914. 10.1038/ncomms10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore C. D.; Allan K. M.; Stoltz B. M. Orthogonal Synthesis of Indolines and Isoquinolines via Aryne Annulation. J. Am. Chem. Soc. 2008, 130, 1558–1559. 10.1021/ja0780582. [DOI] [PubMed] [Google Scholar]
- Sun L.-L.; Liao Z.-Y.; Tang R.-Y.; Deng C.-L.; Zhang X.-G. Palladium and Copper Cocatalyzed Tandem N–H/C–H Bond Functionalization: Synthesis of CF3-Containing Indolo- and Pyrrolo[2,1-a]isoquinolines. J. Org. Chem. 2012, 77, 2850–2856. 10.1021/jo3000404. [DOI] [PubMed] [Google Scholar]
- Rao U. N.; Han X.; Biehl E. R. Preparation of 3H-pyrrolo[2,3-c]isoquinolines and 3H-pyrrolo[2,3-c][2,6]- and 3H-pyrrolo[2,3-c][1,7]naphthyridines. ARKIVOC 2002, x, 61–66. [Google Scholar]
- Dixit B.; Balog J.; Riedl Z.; Drahos L.; Hajós G. New Approach for the Synthesis of 3H-pyrrolo[2,3-c]isoquinoline Derivatives. Tetrahedron 2012, 68, 3560–3565. 10.1016/j.tet.2012.03.011. [DOI] [Google Scholar]
- Castillo J.-C.; Quiroga J.; Abonia R.; Rodriguez J.; Coquerel Y. The Aryne aza-Diels–Alder Reaction: Flexible Syntheses of Isoquinolines. Org. Lett. 2015, 17, 3374–3377. 10.1021/acs.orglett.5b01704. [DOI] [PubMed] [Google Scholar]
- Macías M. A.; Castillo J.-C.; Portilla J. A Series of (E)-5-(Arylideneamino)-1-tert-Butyl-1H-pyrrole-3-carbonitriles and Their Reduction Products to Secondary Amines: Syntheses and X-Ray Structural Studies. Acta Cryst. 2018, C74, 82–93. 10.1107/S2053229617017260. [DOI] [PubMed] [Google Scholar]
- Portilla J.; Quiroga J.; Abonía R.; Insuasty B.; Nogueras M.; Cobo J.; Mata E. Solution-phase and solid-phase synthesis of 1-pyrazol-3-ylbenzimidazoles. Synthesis 2008, 2008, 387–394. 10.1055/s-2008-1032030. [DOI] [Google Scholar]
- Castillo J.-C.; Quiroga J.; Abonia R.; Rodriguez J.; Coquerel Y. Pseudo-Multicomponent Reactions of Arynes with N-Aryl Imines. J. Org. Chem. 2015, 80, 9767–9773. 10.1021/acs.joc.5b01564. [DOI] [PubMed] [Google Scholar]
- Castillo J.-C.; Rosero H.-A.; Portilla J. Simple Access Toward 3-Halo- and 3-Nitro-pyrazolo[1,5-a]pyrimidines Through a One-Pot Sequence. RSC Adv. 2017, 7, 28483–28488. 10.1039/C7RA04336H. [DOI] [Google Scholar]
- Charris-Molina A.; Castillo J.-C.; Macías M.; Portilla J. One-Step Synthesis of Fully Functionalized Pyrazolo[3,4-b]pyridines via Isobenzofuranone Ring Opening. J. Org. Chem. 2017, 82, 12674–12681. 10.1021/acs.joc.7b02471. [DOI] [PubMed] [Google Scholar]
- Orrego-Hernández J.; Lizarazo C.; Cobo J.; Portilla J. Pyrazolo-fused 4-azafluorenones as key reagents for the synthesis of fluorescent dicyanovinylidene-substituted derivatives. RSC Adv 2019, 9, 27318–27323. 10.1039/C9RA04682H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrego-Hernández J.; Portilla J. Synthesis of Dicyanovinyl-Substituted 1-(2-Pyridyl)pyrazoles: Design of a Fluorescent Chemosensor for Selective Recognition of Cyanide. J. Org. Chem. 2017, 82, 13376–13385. 10.1021/acs.joc.7b02460. [DOI] [PubMed] [Google Scholar]
- Castillo J.-C.; Tigreros A.; Portilla J. 3-Formylpyrazolo[1,5-a]pyrimidines as Key Intermediates for the Preparation of Functional Fluorophores. J. Org. Chem. 2018, 83, 10887–10897. 10.1021/acs.joc.8b01571. [DOI] [PubMed] [Google Scholar]
- Tigreros A.; Rosero H.-A.; Castillo J.-C.; Portilla J. Integrated Pyrazolo[1,5-a]pyrimidine-Hemicyanine System as a Colorimetric and Fluorometric Chemosensor for Cyanide Recognition in Water. Talanta 2019, 196, 395–401. 10.1016/j.talanta.2018.12.100. [DOI] [PubMed] [Google Scholar]
- García M.; Romero I.; Portilla J. Synthesis of Fluorescent 1,7-Dipyridyl-bis-pyrazolo[3,4-b:4′,3′-e]pyridines: Design of Reversible Chemosensors for Nanomolar Detection of Cu2+. ACS Omega 2019, 4, 6757–6768. 10.1021/acsomega.9b00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.; Liu W.; Ge J.; Zhang H.; Wang P. New Sensing Mechanisms for Design of Fluorescent Chemosensors Emerging in Recent Years. Chem. Soc. Rev. 2011, 40, 3483–3495. 10.1039/c0cs00224k. [DOI] [PubMed] [Google Scholar]
- Peckus D.; Matulaitis T.; Franckevičius M.; Mimaitė V.; Tamulevičius T.; Simokaitienė J.; Volyniuk D.; Gulbinas V.; Tamulevičius S.; Gražulevičius J. V. Twisted Intramolecular Charge Transfer States in Trinary Star-Shaped Triphenylamine-Based Compounds. J. Phys. Chem. A 2018, 122, 3218–3226. 10.1021/acs.jpca.8b00981. [DOI] [PubMed] [Google Scholar]
- Tigreros A.; Ortiz A.; Insuasty B. Effect of π-conjugated linkage on photophysical properties: Acetylene linker as the better connection group for highly solvatochromic probes. Dyes Pigm. 2014, 111, 45–51. 10.1016/j.dyepig.2014.05.035. [DOI] [Google Scholar]
- Yang Z.; Cao J.; He Y.; Yang J. H.; Kim T.; Peng X.; Kim J. S. Macro–/micro-environment-sensitive chemosensing and biological imaging. Chem. Soc. Rev. 2014, 43, 4563–4601. [DOI] [PubMed] [Google Scholar]
- Lee S.-C.; Heo J.; Woo H. C.; Lee J.-A.; Seo Y. H.; Lee C.-L.; Kim S.; Kwon O.-P. Fluorescent Molecular Rotors for Viscosity Sensors. Chem. – Eur. J. 2018, 24, 13706–13718. 10.1002/chem.201801389. [DOI] [PubMed] [Google Scholar]
- CCDC 1817750 (6e), 1817752 (6f), 1899534 (6g), 1817752 (8b), and 1899534 (8c) contain the supplementary crystallographic data for this paper.
- Thomas S. P.; Spackman P. R.; Jayatilaka D.; Spackman M. A. Accurate Lattice Energies for Molecular Crystals from Experimental Crystal Structures. J. Chem. Theory Comput. 2018, 14, 1614–1623. 10.1021/acs.jctc.7b01200. [DOI] [PubMed] [Google Scholar]
- Turner M. J.; McKinnon J. J.; Wolff S. K.; Grimwood D. J.; Spackman P. R.; Jayatilaka D.; Spackman M. A. (2017). CrystalExplorer17. University of Western Australia. http://hirshfeldsurface.net/. [Google Scholar]
- Saha S.; Quiney H. M. Solvent effects on the excited state characteristics of adenine–thymine base pairs. RSC Adv 2017, 7, 33426–33440. 10.1039/C7RA03244G. [DOI] [Google Scholar]
- Misra R.; Bhttacharyya S. P.. Intramolecular Charge Transfer: Theory and Applications. 2018. First Ed. Wiley-VCH Verlag GmbH & Co. KGaA. p: 16–17. [Google Scholar]
- Allegretti M.; Anacardio R.; Cesta M. C.; Curti R.; Mantovanini M.; Nano G.; Topai A.; Zampella G. A Practical Synthesis of 7-Azaindolylcarboxy-endo-tropanamide (DF 1012). Org. Process Res. Dev. 2003, 7, 209–213. 10.1021/op025570t. [DOI] [Google Scholar]
- Palatinus L.; Chapuis G. SUPERFLIP - a computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. 10.1107/S0021889807029238. [DOI] [Google Scholar]
- Sheldrick G. M. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrae C. F.; Bruno I. J.; Chisholm J. A.; Edgington P. R.; McCabe P.; Pidcock E.; Rodriguez-Monge L.; Taylor R.; van de Streek J.; Wood P. A. Mercury CSD 2.0 - new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. 10.1107/S0021889807067908. [DOI] [Google Scholar]
- Spek A. L. PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 9–18. 10.1107/S2053229614024929. [DOI] [PubMed] [Google Scholar]
- Spackman M. A.; Jayatilaka D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. 10.1039/B818330A. [DOI] [Google Scholar]
- Jayatilaka D.; Grimwood D. J.; Lee A.; Lemay A.; Russel A. J.; Taylor C.; Wolff S. K.; Cassam-Chenai P.; Whitton A. (2005). TONTO. http://hirshfeldsurface.net/.
- Spackman M. A.; McKinnon J. J.; Jayatilaka D. Electrostatic potentials mapped on Hirshfeld surfaces provide direct insight into intermolecular interactions in crystals. CrystEngComm 2008, 10, 377–388. 10.1039/B715227B. [DOI] [Google Scholar]
- Mackenzie C. F.; Spackman P. R.; Jayatilaka D.; Spackman M. A. CrystalExplorer model energies and energy frameworks: extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ. 2017, 4, 575–587. 10.1107/S205225251700848X. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.