

Abstract
Cancer cells have an increased demand for energy sources to support accelerated rates of growth. When nutrients become limiting, cancer cells may switch to nonconventional energy sources that are mobilized through nutrient scavenging pathways involving autophagy and the lysosome. Thus, several cancers are highly reliant on constitutive activation of these pathways to degrade and recycle cellular materials. Here, we focus on the MiT/TFE family of transcription factors, which control transcriptional programs for autophagy and lysosome biogenesis and have emerged as regulators of energy metabolism in cancer. These new findings complement earlier reports that chromosomal translocations and amplifications involving the MiT/TFE genes contribute to the etiology and pathophysiology of renal cell carcinoma, melanoma, and sarcoma, suggesting pleiotropic roles for these factors in a wider array of cancers. Understanding the interplay between the oncogenic and stress-adaptive roles of MiT/TFE factors could shed light on fundamental mechanisms of cellular homeostasis and point to new strategies for cancer treatment.
Keywords: TFEB, TFE3, MITF, lysosome, autophagy, mTORCl
INTRODUCTION
Lysosomes are membrane-bound organelles that function as the primary degradative compartment of eukaryotic cells through breakdown and recycling of a diverse array of intracellular and extracellular macromolecules (de Duve 2005). Several endocytic trafficking pathways including phagocytosis, macropinocytosis, clathrin- and caveolin-dependent endocytosis, and independent endocytosis import macromolecules from outside the cells and the plasma membrane to the lysosome for degradation (Conner & Schmid 2003, Di Fiore & von Zastrow2014, Goldstein & Brown 2015). Similarly, the self-catabolic process known as autophagy serves to capture and deliver diverse cytoplasmic content including lipid droplets, damaged or misfolded proteins, and organelles to the lysosome for elimination (Kaur & Debnath 2015, Mizushima & Komatsu 2011).
In addition to degradation and recycling of nutrients, the lysosome also plays a crucial role in the regulation of signaling pathways that control cellular anabolism. The mechanistic target of rapamycin complex 1 (mTORC1), a master regulator of organismal survival and growth, exerts its activity on the lysosomal surface, suggesting that the lysosome is at the interface between cellular catabolic and anabolic pathways (Sancak et al. 2010, Zoncu et al. 2011). Recent studies have implicated excessive lysosomal activity as a recurrent feature in cancer. First, cancer cells have higher metabolic demands than normal cells and thus may rely on induction of the autophagy-lysosome machinery for survival (Rabinowitz & White 2010). Second, cancer patients experience muscle atrophy—a syndrome known as cachexia—which is due to excessive muscle protein breakdown via not only activation of E3 ubiquitin ligases and upregulation of proteasome-mediated degradation but also induction of the autophagy-lysosome system (Bodine et al. 2001, Gomes et al. 2001, Sandri et al. 2016). Lastly, hyperactivation of mTORC1 signaling has largely been reported to promote cell growth in several malignancies (Ilagan & Manning 2016, Saxton & Sabatini 2017, Yecies & Manning 2011). The advantages and disadvantages of decreased and increased lysosomal function are outlined in Figure 1.
Figure 1.
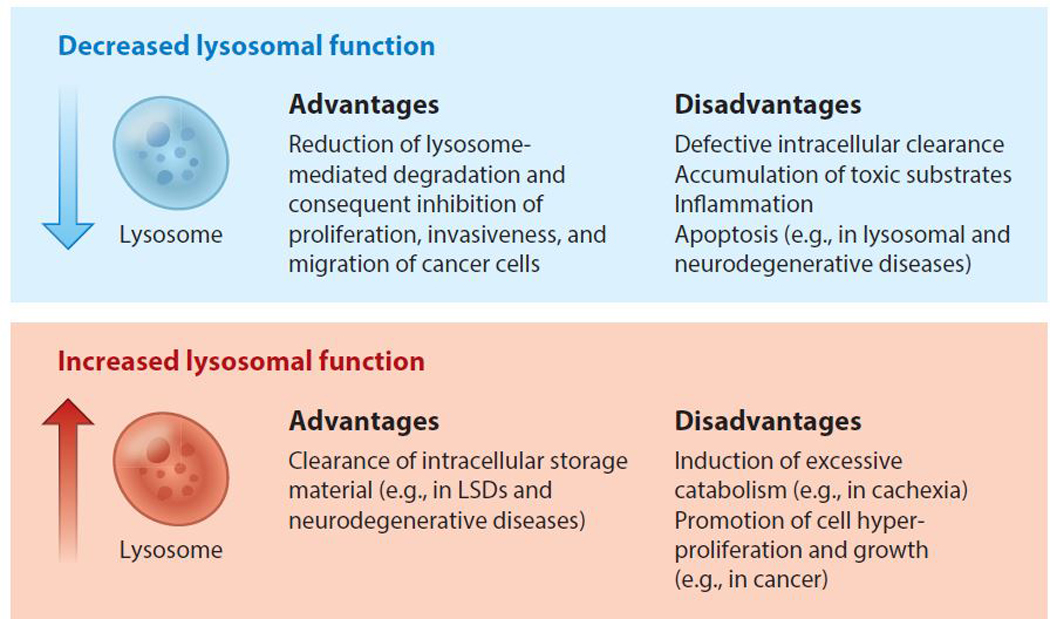
The advantages and disadvantages of decreased and increased lysosomal function for cell metabolism. A decrease in lysosomal function can be beneficial in pathological conditions such as cancer, in which lysosome-mediated degradation sustains high energy demands and promotes invasiveness and migration of malignant cells. However, defective lysosomal degradation leads to the accumulation of undigested material within the lysosomes of individuals affected by lysosomal storage diseases (LSDs) or common neurodegenerative diseases, such as Alzheimer’s, Parkinson’s, and Huntington’s disease. Conversely, enhancing lysosomal activity can be beneficial in diseases associated with dysfunctional lysosomes, but can be deleterious in conditions associated with excessive catabolism (such as cachexia) and in malignancies relying on lysosomal metabolism for cell growth.
Autophagy and Lysosome Dysfunction in Cancer
A hallmark of rapidly growing cancer cells is the ability to synthesize biomass at higher rates than normal cells (DeNicola & Cantley 2015, Lunt & Vander Heiden 2011, Piao & Amaravadi 2016, Rabinowitz & White 2010). During periods of growth when the supply of external nutrients is absent or limiting, lysosome-mediated degradation and recycling can contribute precursors to fuel the generation of new proteins, membrane lipids, DNA, and RNA. For example, fluctuations in in vivo microenvironment conditions due to poor vascularization can limit nutrient and oxygen availability to cancer cells, while infiltration of stromal and immune cells further competes with cancer cells for limited nutrient pools (Davidson et al. 2016, DeNicola & Cantley 2015, Lyssiotis & Kimmelman 2017, Perera & Bardeesy 2015). Under these conditions, tumor cell-associated activation of autophagy, which converges on the lysosome, aids in the generation of necessary building blocks through recycling of damaged or unnecessary cellular components to produce all classes of cellular macromolecules. For example, lung and pancreatic ductal adenocarcinoma (PDA) tumors appear to be reliant on constitutive activation of autophagy for supplying essential nutrients and for removing damaged mitochondria (Guo et al. 2011; Karsli-Uzunbas et al. 2014; Perera & Bardeesy 2015; Perera et al. 2015; Rao et al. 2014; Rebecca et al. 2017; Strohecker et al. 2013, White 2015; Yang et al. 2011, 2018). Several other cancers, including melanoma, breast, and prostate cancer, show context- and stage-specific reliance on autophagy during tumor initiation and progression (Huo et al. 2013, Lock et al. 2011, Santanam et al. 2016, Wei et al. 2011, Xie et al. 2015). Since the final steps of macropinocytosis and autophagy involve fusion with lysosomes for efficient degradation of cargo, these studies collectively highlight the importance of nutrient scavenging in cancer and establish lysosomal catabolism as essential for removing damaged organelles and supplying the building blocks for tumor growth (Guo et al. 2016).
Additional roles for autophagy in cancer promotion have also emerged that are distinct from its primary function in mediating autodigestion. These processes either do not require the formation of a double-membrane autophagosome or do not terminate at the lysosome. Such autophagy-related processes have been reviewed elsewhere, including secretion, LC3-associated phagocytosis, regulation ofinflammation, and immune signaling (Cadwell & Debnath2018, Heckmann et al. 2017, Levine & Deretic 2007, Monkkonen & Debnath 2018, Subramani & Malhotra 2013).
The MiT/TFE Family of Transcription Factors
The microphthalmia/transcription factor E (MiT/TFE) family of transcription factors (TFs) encodes four distinct genes: MITF, TFEB, TFE3,and TFEC (Hemesath et al. 1994). All family members share a common structure, consisting of a basic helix-loop-helix (bHLH) leucine zipper (LZ) dimerization motif, a transactivation domain, and an identical basic region required for DNA binding (Beckmann et al. 1990, Sato et al. 1997, Steingrímsson et al. 2004).
A new role for the MiT/TFE family proteins emerged in 2009 following the discovery that TFEB recognized and bound an E box-related consensus element found in the promoter region of many lysosomal genes, including those encoding hydrolases, lysosomal membrane permeases, andlysosome-associatedproteins (Napolitano & Ballabio 2016, Palmieri etal.2011, Sardiello et al. 2009, Settembre et al. 2013b). This element was termed the coordinated lysosomal expression and regulation (CLEAR) element and was required for TFEB-mediated induction of gene expression. In line with the high level of sequence conservation of the DNA-binding region among MiT/TFE family members, follow-up studies showed that MITF and TFE3 could also bind the CLEAR element and regulate lysosome biogenesis in several different cell types (Martina et al. 2014, Ploper et al. 2015).
In addition to regulating lysosomal genes, TFEB was shown to control the expression of several genes involved in autophagy, including those associated with autophagosome initiation (BECN1, WIPI1, ATG9B, and NRBF2), elongation (GABARAP, MAP1LC3B, and ATG5), substrate capture (SQSTM1), and autophagosome trafficking and fusion with lysosomes (UVRAG and RAB7), which harbor CLEAR elements in their promoters (Palmieri et al. 2011, Settembre et al. 2011). Thus, TFEB is a true master regulator of cellular catabolism, capable of controlling the cell’s ability to select and capture substrates via autophagy and to degrade them via the lysosome.
This procatabolic activity of MiT/TFE factors occurs in most cells and has tissue- and organ- specific roles. For example, in the liver, in response to fasting, TFEB promotes autophagy and lipid catabolism via activation of a gene expression program that includes the master metabolic TF PGC1a, as well as many of its downstream genes involved in fatty acid oxidation and mitochondrial biogenesis (Settembre & Ballabio 2014, Settembre et al. 2013a). Liver-specific deletion of Tfeb rendered mice hypersensitive to the effects of a high-fat diet, whereas its overexpression promoted resistance to lipid accumulation in an autophagy-dependent manner. The regulatory action of TFEB in lipid metabolism is also conserved in Caenorhabditis elegans (O’Rourke & Ruvkun 2013, Settembre et al. 2013a). In addition, muscle-specific TFEB deletion impairs the cell’s ability to adapt energy metabolism to physical exercise (Mansueto et al. 2017). These studies provided strong evidence that regulation of the autophagy-lysosome system may be a key aspect of metabolic adaptation, both at the cellular and at the organismal level.
Regulation of MiT/TFE Factors
The importance of lysosomal function for maintaining cellular and tissue health suggested that there must be mechanisms in place by which cells can rapidly and adaptively boost lysosome number or activity in response to cellular need. Interestingly, in the presence of nutrients, TFEB was shown to be in the cytoplasm but rapidly translocates into the nucleus in response to nutrient starvation (Settembre et al. 2011). This observation was followed by the key discovery that pathways involved in nutrient sensing and growth control regulate the MiT/TFE factors (Martina et al. 2012, 2014; Peña-Llopis et al. 2011; Roczniak-Ferguson et al. 2012; Settembre et al. 2012). This process is primarily regulated by mTORC1, which, once activated by nutrients, phosphorylates TFEB at critical serine residues, promoting its cytoplasmic localization (Martina et al. 2012, Roczniak-Ferguson et al. 2012, Settembre et al. 2012). Conversely, nutrient depletion inhibits mTORC1 and limits TFEB phosphorylation. Dephosphorylation of TFEB is mediated by the phosphatase calcineurin (CaN), which is activated following TRPML1-mediated lysosomal calcium release (Medina et al. 2015) and enables TFEB to translocate into the nucleus. Physical exercise also activates CaN, thus linking TFEB-mediated autophagy induction to the health benefits of exercise (Mansueto et al. 2017, Medina et al. 2015).
Along with mTORC1, other growth-regulating kinases such as MAPK kinase (MEK)/ extracellular signal-regulated kinase (ERK) and glycogen synthase kinase 3 (GSK3) also affect MiT/TFE nuclear localization (Marchand et al. 2015, Ploper et al. 2015, Settembre et al. 2011). Notably, acute inhibition of mTORC1 via catalytic inhibitors completely abolished TFEB phosphorylation, leading to its rapid nuclear translocation, suggesting that mTORC1 is predominant with respect to other kinases in regulating TFEB activity (Settembre et al. 2012). Follow-up studies in several cellular systems showed that the TFEB serine residues phosphorylated by mTORCl are conserved in MITF and TFE3, whose subcellular localization in response to nutrient starvation and mTORCl inhibition is regulated in a similar way (Martina et al. 2014). Thus, in normal cells, anabolic (mTORCl signaling) and catabolic (MiT/TFE activation) pathways are mutually exclusive.
Recent studies revealed that MiT/TFE proteins, which are substrates of mTORCl, are in turn able to reciprocally regulate mTORCl activity, thus identifying an MiT/TFE-mTORCl feedback loop (Di Malta et al. 20l7). Mechanistically, MiT/TFE factors strongly induce the expression of RRAGD, the gene encoding RagD, one of the four mammalian Ras-related small GTP-binding proteins called Rag GTPases (Kim et al. 2008, Sancak et al. 2008). Unlike Ras-related and most other small G proteins, Rags exist as heterodimers, where the highly similar RagA and RagB bind to either RagC or RagD (Schürmann et al. l995, Sekiguchi et al. 200l). In response to amino acids, Rags switch to an active conformation in which RagA or B is GTP loaded and RagC or D is GDP loaded. Active Rag GTPases subsequently facilitate mTORCl relocalization from the cytoplasm to the lysosomal surface, which is necessary for mTORCl activation (Buerger et al. 2006; Kim et al. 2008; Sancak et al. 2008, 20l0). Consequently, transcriptional induction of RagD promotes Rag assembly on the lysosomal surface, enabling mTORCl recruitment once nutrients become available. Thus, MiT/TFE TFs and mTORCl are involved in a feedback loop by which mTORCl inhibits TFEB nuclear localization and function, and in turn, TFEB regulates mTORCl lysosomal recruitment and activity through the RagD GTPase (Di Malta et al. 20l7). Figure 2 summarizes the regulation of MiT/TFE factors and their downstream pathways.
Figure 2.
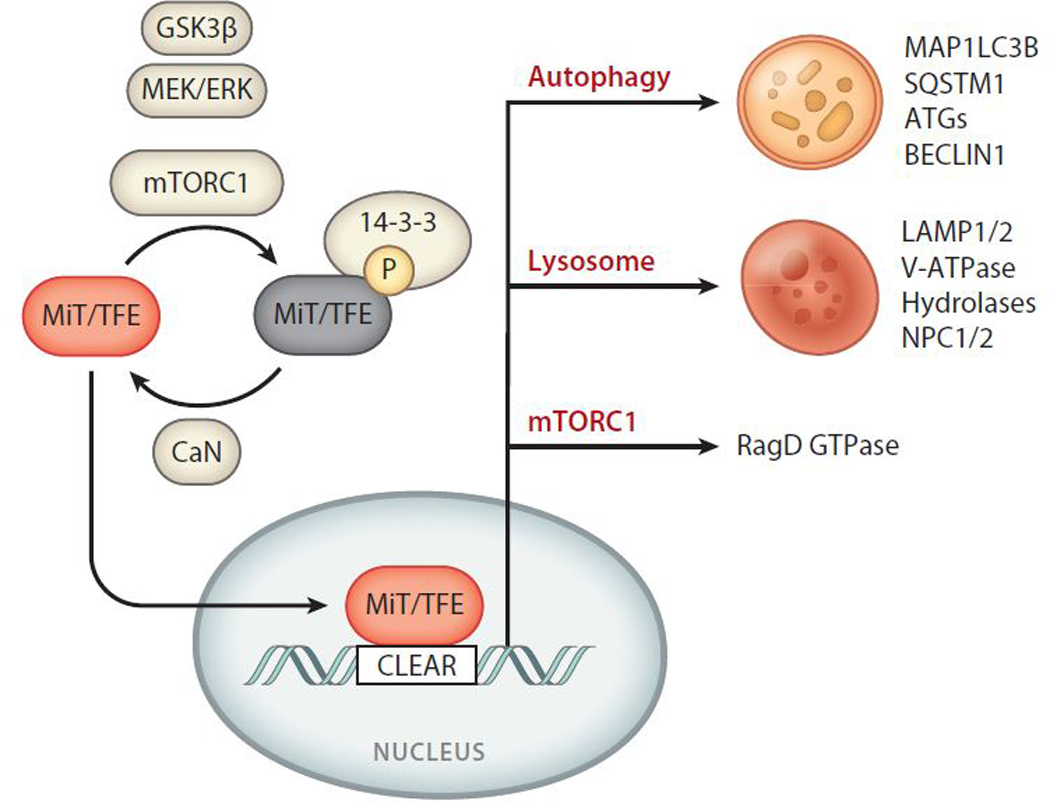
Regulation and downstream targets of MiT/TFE transcription factors. MiT/TFE proteins are negatively regulated through phosphorylation of conserved serine residues by mTORCl as well as GSK3 β and ERK, leading to 14–3-3 binding and cytoplasmic retention. Dephosphorylation by CaN enables the nuclear translocation of MiT/TFE proteins and consequent binding to CLEAR sequences present in their target genes. Pathways and cellular processes regulated by MiT/TFE factors include autophagy, lysosomal biogenesis, and mTORCl signaling through upregulation of RagD GTPase. Abbreviations: ATGs, autophagy-related proteins; CaN, calcineurin; CLEAR, coordinated lysosomal expression and regulation; ERK, extracellular signal-regulated kinase; GSK3 β, glycogen synthase kinase 3 beta; MEK, MAPK kinase; MiT/TFE, microphthalmia/transcription factor E; mTORCl, mechanistic target ofrapamycin complex 1.
Role of MiT/TFE Proteins as Oncogenes
MiT/TFE proteins have established roles in promoting tumorigenesis (Haq & Fisher 2011, Kauffman et al. 2014). Genomic amplifications of MITF are found in 5–20% of melanomas (Cancer Genome Atlas Res. Netw. 2015), while translocations and rearrangements of TFE3 and TFEB are found in pediatric renal cell carcinoma (RCC) and alveolar soft part sarcoma (ASPS) (Argani et al. 2001, Ramphal et al. 2006). In addition, a recent study showed upregulated expression of MiT/TFE factors in PDA (Perera et al. 2015).
RENAL CELL CARCINOMA
RCC is the most common form of kidney cancer, and it includes multiple histopathologically distinct subtypes that originate from the renal tubular epithelium (Linehan et al. 2010). RCCs include clear cell carcinoma (65–70%), papillary RCC (15–20%), and chromophobe RCC (5–10%) (Amin et al. 2002). Several mutations in genes involved in metabolism (VHL, SDHB, SDHC, SDHD, and FH), mTOR signaling (FLCN, TSC1, TSC2, and PTEN) (Linehan & Ricketts 2013), tyrosine kinase receptor-mediated signaling (MET) (Schmidt et al. 1997), and Polycomb repressor complex proteins (BAP1) (Farley et al. 2013, Peña-Llopis et al. 2012) are associated with hereditary RCC.
Approximately 5% of sporadic RCC tumors define a rare subgroup termed translocation-RCC (tRCC), which involves MiT/TFE genes (Figure 3). These chromosomal abnormalities include translocations and rearrangements involving TFE3 located centromerically on the short arm of the X chromosome (Xp11) and, less commonly, TFEB (Bruder et al. 2004, Cancer Genome Atlas Res. Netw. 2013). Translocations involving TFE3 were first discovered in the mid-1990s (Shipley et al. 1995, Sidhar et al. 1996, Weterman et al. 1996) and are among the earliest reported cancer- associated gene fusions. Unlike the common histopathological subtypes of RCC, tRCC is most common in children, representing 20–50% of all pediatric RCC cases (Kauffman et al. 2014). The morphologic spectrum of tRCC is diverse and has potential overlap with common RCC subtypes such as clear cell and papillary RCC. tRCC-associated TFE3 gene fusions can occur with several partners including PRCC, ASPSCR1, SFPQ, NONO, and CLTC (Kauffman et al. 2014, Linehan et al. 2010). ASPSCR1-TFE3 and SFPQ-TFE3 fusions are not restricted to RCC and were originally identified in ASPS (Argani et al. 2001) and in a subset of benign tumors known as perivascular epithelioid cell neoplasms (Tanaka et al. 2009), respectively. For each of these fusion proteins, rearrangement occurs within an intron of either gene partner, but the specific intron involved can vary between patients, resulting in different messenger RNA (mRNA) isoforms consisting of the N-terminal portion of the fusion partner linked to C-terminal coding exons of TFE3. TFE3 fusion proteins vary considerably in size—47.8 kDa for PRCC-TFE3 and up to 136.1 kDa for CLTC-TFE3—however, the fusion proteins always retain a 280-amino acid C-terminal portion, harboring the bHLH-LZ dimerization, DNA-binding domain, and putative nuclear localization signal of TFE3 (Kauffman et al. 2014).
Figure 3.
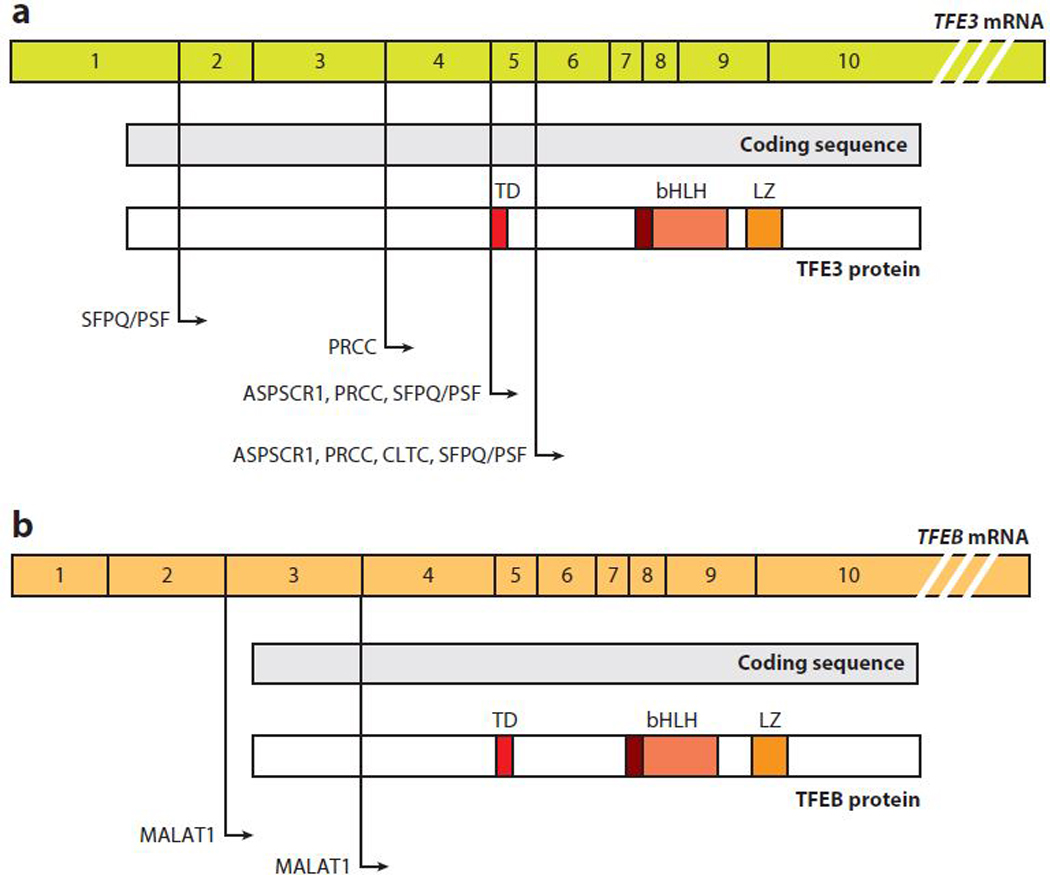
TFE3 and TFEB gene fusions. Exons, coding sequences, and functional domains of (a) TFE3 and (b) TFEB. Positions of known fusions with the indicated genes are shown with lines and arrows. Abbreviations: bHLH, basic helix-loop-helix domain; LZ, leucine zipper domain; mRNA, messenger RNA; TD, transactivation domain.
TFEB-associated tRCCs are less common than TFE3 tRCCs and result from a gene fusion between TFEB on chromosome 6p21 and the metastasis-associated lung adenocarcinoma transcript 1 gene known as MALAT1 on chromosome 11q12 (Davis et al. 2003). MALAT1-TFEB fusion breakpoints generally are located before the start codon in exon 3 of the TFEB coding sequence, thus resulting in full retention of TFEB under the control of the strong MALAT1 promoter. TFEB-tRCC patients usually have a better prognosis than TFE3-tRCC patients. More recently, a comprehensive genomic analysis of 161 primary papillary RCCs led to the identification of novel fusion partners for both TFE3 and TFEB, including RBM10 and DVL2 for TFE3 and COL21A1 and CADM2 for TFEB (Cancer Genome Atlas Res. Netw. 2016).
The mechanisms mediating the oncogenic effects of TFE fusions in RCC remain unclear. It has been postulated that chimeric proteins resulting from TFE3 fusions retain some activity of the fusion partner (Kauffman et al. 2014). For example, most of the TFE3 fusion partners appear to have regulatory roles in either mRNA splicing (NONO, SFPQ, and PRCC) or mitosis (PRCC and CLTC) that could promote cellular transformation. For instance, fusion of TFE3 with genes associated with mRNA splicing may ensure constitutive nuclear localization of the fusion protein. An alternative hypothesis is the so-called dysregulated activity model, according to which the fusion leads to upregulation of oncogenic activity already present in the wild-type protein. Consistent with this idea, all TFE3 fusion partners display constitutively active gene promoters, and therefore, TFE3 fusion proteins are usually expressed at much higher levels than wild-type TFE3 (Argani et al. 2003b; Clark et al. 1997; Weterman et al. 1996, 2000). This is also the case for MALAT1-TFEB, since MALAT1 provides a much stronger promoter without changing the protein-coding sequence of TFEB (Kuiper et al. 2003). No MITF-associated fusions have been reported as causative of RCC; however, a specific MITF germline gain-of-function mutation (E318K) confers an increased risk for developing RCC or melanoma (Bertolotto et al. 2011).
Several patient tumor-derived cell line models of tRCC (Shipley et al. 1995, Sidhar et al. 1996, Weterman et al. 1996) have been developed to study key features of the disease. More recently, a conditional, kidney-specific, TFEB-overexpressing mouse line was generated, which displayed phenotypic features commonly observed in patients with tRCC such as cysts, clear cells, fibrosis, and multilayered basement membranes (Calcagni et al. 2016). Starting from postnatal day 30 (P30), this mouse model also developed papillary carcinoma that was eventually associated with hepatic metastases from P90. Thus, this mouse line well recapitulates the human disease, further supporting the hypothesis that tRCC tumorigenesis is mediated by increased activity ofMiT/TFE genes. Furthermore, these mice represent a valuable tool for studying the pathogenic mechanisms underlying tRCC and a model for testing new therapeutic strategies.
ALVEOLAR SOFT PART SARCOMA
ASPS is a rare malignant tumor generally arising in the skeletal muscle of the lower limbs in adolescents and young adults or in the head and neck in children. Accounting for approximately 1% of all soft tissue sarcomas, all ASPS tumors harbor a characteristic chromosomal translocation resulting in oncogenic fusion or rearrangement of TFE3 with ASPSCR1 on chromosome 17 (Argani et al. 2001). First identified in ASPS, ASPSCR1-TFE3 fusions were later discovered in a subset of RCC. The ASPSCR1-TFE3 fusion replaces the N-terminal portion of TFE3 with ASPSCR1 but retains the TFE3 DNA-binding domain. There are two forms of the ASPSCR1-TFE3 chimera, involving an in-frame fusion of ASPSCR1 to either exon 6 of TFE3 (type 1) or exon 5 (type 2), which retains the transactivation domain of TFE3 (Figure 3). Similar to tRCC-specific TFE3 fusion proteins, ASPSCR1-TFE3 has a predominant nuclear localization (Argani et al. 2003a) and is a stronger transcriptional activator than the native TFE3.
ASPS tumors display characteristic alveolar histology and an abundant vascular network. Despite being a slow-growing tumor, ASPS can metastasize to the lung and brain. Several angiogenesis-associated genes are found to be upregulated in ASPS, and patients respond to anti-angiogenesis inhibitors (Azizi et al. 2006, Ghose et al. 2012, Stacchiotti et al. 2009). However, only a few angiogenesis-associated genes appear to be direct transcriptional targets of ASPSCR1- TFE3, as determined by chromatin immunoprecipitation analysis (Kobos et al. 2013), suggesting that additional molecular pathways or microenvironment factors may contribute to ASPS pathophysiology. Two studies have developed in vivo mouse models to study the pathophysiology of ASPS. Goodwin et al. (2014) developed a tamoxifen-inducible conditional ASPSCR1-TFE3 (type 2 variant) mouse model that developed spatially restricted tumors within the intracranial vault, but no tumors in the skeletal muscle—the most common site of human ASPS. A further limitation of this model was the lack of vascular invasion and metastasis, often seen in human ASPS. Nevertheless, mouse intracranial tumors in this GEM (genetically engineered mouse) model displayed characteristic histological features and overlapping transcriptional profiles with human ASPS. Interestingly, the development of ASPS-like tumors within the cranial vault was proposed to be associated with a preference for high environmental lactate, which was shown to be taken up by tumor cells and to promote proliferation, highlighting a previously unrecognized environmental factor that contributes to ASPS development.
An additional ex vivo model of ASPS developed by Tanaka et al. (2017) used primary mouse embryonic osteochondrogenic progenitors (eMCs), which were in vitro transduced with ASPSCR1- TFE3 followed by subcutaneous injection. These xenografts displayed characteristic histological features of human ASPS and, importantly, showed prominent angiogenesis; over 50% metastasized to the lung. Interestingly, abundant mitochondria and lysosomes were observed in ASPS xenografts, and gene set enrichment analysis of microarray data generated from ASPS xenografts and ASPSCR1-TFE3-expressing eMCs showed a statistically significant upregulation of lysosome and autophagy signatures. Thus, the cell of origin and the tissue microenvironment likely have complementary roles in ASPS initiation and progression.
MELANOMA
Melanoma is an aggressive type of cancer that arises within any anatomic territory occupied by melanocytes. The most common type, the cutaneous melanoma, develops from epidermal melanocytes of the skin. In rare cases, cutaneous melanoma can also originate from melanocytes located in the choroidal layer of the eye or in the mucosa of respiratory, gastrointestinal, or genitourinary systems (Tsao et al. 2012). Approximately 50% of melanoma cases harbor a V600E activating mutation in the BRAF kinase, resulting in hyperactivation of the MAPK signaling pathway (Davies et al. 2002, Hodis et al. 2012). Amplification of the MITF gene was detected in 5–20% of melanoma cases, and these tumors appear to have better prognosis (Cancer Genome Atlas Res. Netw. 2015).
MITF is a melanocyte lineage determinant, and if overexpressed, together with S0X10 and PAX3, it can directly reprogram human and mouse fibroblasts into functional melanocytes (Tachibana et al. 1996, R. Yang et al. 2014). MITF-associated tumors are usually caused by gene amplifications; however, in rarer cases, somatic mutations of MITF have also been reported. These mutations are generally located in the MITF transactivation domain, suggesting that increased transcriptional activity of MITF is sufficient to trigger tumorigenesis (Cronin et al. 2009). In addition, two independent studies identified a rare oncogenic MITFE318K mutation in patients with familial melanoma and a small fraction of sporadic cases (Bertolotto et al. 2011, Yokoyama et al. 2011). Interestingly, these patients are also predisposed to developing RCC (Bertolotto et al. 2011). The MITFE318K mutation occurs at a site previously described as a target of MITF SUMOylation (addition of a small ubiquitin-like modifier) (Miller et al. 2005) and represents a gain of function of MITF activity.
It has been proposed that the levels of MITF can fluctuate within a tumor to potentially influence activation of diverse cellular programs and response to therapy (Ennen et al. 2015, Hoek et al. 2008, Konieczkowski et al. 2014, Müller et al. 2014, Tirosh et al. 2016). High expression of MITF in melanoma cells is associated with differentiation and proliferation, while low levels favor stem cell-like, invasive potential and intrinsic resistance to multiple targeted therapies. Fine-tuning of MITF levels and activity in melanoma likely involves the integration of microenvironmental cues, epigenetic states, and activities of upstream signaling pathways.
PANCREATIC DUCTAL ADENOCARCINOMA
PDA is among the most lethal of cancers with a five-year survival rate of only 6% following initial diagnosis (Maitra & Hruban 2008, Ryan et al. 2014, Ying et al. 2016). Activating mutations in the KRAS oncogene occur in more than 90% of patients with PDA and are early initiating events in this disease, while frequent inactivating mutations in TRP53, CDKN2A,and SMAD4 occur during later stages of malignant progression. Constitutive activation of autophagy and increased rates of macropinocytosis, which is responsible for the bulk uptake of extracellular material, most notably serum albumin, help to maintain metabolic homeostasis in PDA cells and tumors (Commisso et al. 2013; Davidson et al. 2017; Kamphorst et al. 2013,2015; Palm et al. 2015; Yang et al. 2011; A. Yang et al. 2014). Recently, increased mRNA and protein expression of MITF, TFE3, and TFEB was detected in PDA cell lines and patient tumors (Perera et al. 2015). Importantly, prominent nuclear staining of these TFs was observed in PDA, similar to that seen in RCC and ASPS. Functionally, MiT/TFE factors were shown to control autophagy in PDA and promoted an increase in lysosome biogenesis (discussed below).
DOWNSTREAM MECHANISMS IMPLICATED IN THE PATHOGENESIS OF MiT/TFE-DEPENDENT TUMORS
The ability of MiT/TFE factors to regulate lysosomal biogenesis and mTORC1 signaling suggests that these pathways may be dysregulated in MiT/TFE-associated malignancies. In addition, co-occurring genetic alterations and tissue-specific microenvironmental factors likely influence tumor progression in the different tumor types associated with MiT/TFE dysregulation. Here we describe established and emerging roles of MiT/TFE proteins in the regulation of pro- tumorigenic processes in cancer.
Autophagy and mTORC1 Signaling
Constitutive activation of autophagy is a hallmark of PDA (Yang et al. 2011). In cell lines and patient-derived PDA cultures, MiT/TFE proteins were found to be upregulated and required for maintaining high levels of autophagy and lysosome gene expression (Perera et al. 2015). Accordingly, PDA cells display a 12-fold increase in lysosome biogenesis. Importantly, MiT/TFE proteins bypass mTORC1-mediated surveillance and are constitutively localized in the nucleus of PDA cells regardless of nutrient availability. Boosted lysosome-mediated catabolic activity in PDA tumors was required for maintenance of intracellular amino acid levels. Importantly, inactivation of MiT/TFE proteins in PDA cells results in downregulation of autophagy and lysosome genes, defective lysosomal function, and autophagic flux, as well as decreased degradation of macropinocytosis-derived proteins, culminating in the inhibition of tumor growth both in vitro and in vivo.
PDA cells and tumors also show high rates of macropinocytosis, which mediates uptake of extracellular serum albumin and potentially other external fuel sources. Carbon tracing of 13C- labeled albumin taken up via macropinocytosis in PDA cell lines and in vivo tumors revealed specific labeling of multiple metabolite species, indicating that lysosome-mediated digestion of albumin is followed by the utilization of the resulting free amino acids in the cytoplasm (Commisso et al. 2013, Davidson et al. 2017, Kamphorst et al. 2015). Collectively, these data show that by governing both autophagic flux and lysosomal catabolism, the MiT/TFE proteins support an integrated cellular clearance program that enables efficient processing of cargo from autophagy as well as macropinocytosis.
More recently, the discovery that MiT/TFE factors are positive regulators of mTORC1 signaling has led to the identification of a novel pathogenetic mechanism prevalent among MiT/TFE- associated malignancies (Di Malta et al. 2017). Specifically, tumors associated with hyperactivation of MiT/TFE factors (melanoma, tRCC, and PDA) display constitutive induction of RRAGD and, to a lesser extent, RRAGC transcripts. For example, analysis of patient metastatic melanoma TCGA (The Cancer Genome Atlas) data and human melanoma cell lines identified a correlation between MITF and RRAGD gene expression levels. Similarly, increased RRAGD transcript levels were detected in kidney tissue and primary kidney cells from a mouse model of RCC driven by TFEB overexpression (TFEB-RCC). As Rag GTPases are responsible for mTORC1 lysosomal recruitment and activation, increased RagD levels in tumors correlated with hyperactivation of mTORC1 signaling and proliferation. Importantly, treatment of TFEB-RCC mice with an mTOR inhibitor or silencing of RRAGD in an MITF-dependent melanoma cell line suppressed proliferation or reduced in vivo tumor growth, respectively. Together, these findings highlight RRAGD as a new transcriptional target associated with the pathogenesis of MiT/TFE-dependent tumors (Di Malta et al. 2017).
Interestingly, elevated RRAGD levels were also identified among a cohort of 332 genes identified as direct targets of ASPSCR1-TFE3 in ASPS. Kobos et al. (2013) performed transcript tome analysis in cells expressing the ASPSCR1-TFE3 fusion protein and in ASPS tumor samples. Among the differentially upregulated genes were several autophagy and lysosome genes, as well as RRAGD. Further studies will be necessary to unravel the contribution of autophagy and lysosome function and RRAGD upregulation in ASPS and to evaluate mTORC1 signaling activation and its role in the growth of this cancer, as well as in additional cancer types.
Angiogenesis.
Angiogenesis is a physiological process through which new capillaries grow from preexisting blood vessels; it is associated with tumor invasiveness and metastasis (De Palma et al. 2017, Potente et al. 2011). A role for MiT/TFE factors in the regulation of angiogenesis was first hypothesized following the observation that Tfeb knockout mice die prenatally due to a defect in placental vascularization (Steingrimsson et al. 1998). Arecent study by Fan et al. (2018) showedno significant differences between control mice and mice overexpressing Tfeb in endothelial cells under baseline conditions. However, transgenic mice presented enhanced blood flow during recovery from ischemia, which was mediated by increased expression of proangiogenic functions, including enhanced migration, enhanced tube formation, and inhibited apoptosis, whereas endothelial-specific Tfeb conditional knockout mice displayed the opposite phenotype. These effects required functional AMPKa and autophagy, as inhibition of these pathways blunted Tfeb-mediated proangiogenic functions. Interestingly, ischemia increased Tfeb expression levels, while treatment with proangiogenic stimuli promoted its nuclear localization, suggesting that TFEB may function as a homeostatic regulator of angiogenesis.
In addition to TFEB, MITF has also been found to positively regulate angiogenesis. This occurs through direct transcriptional control of hypoxia inducible factor 1 (HIF1 a) (Busca et al. 2005). HIF1a in turn induces the expression of several genes implicated in the response to hypoxia, including VEGF—the prototypic angiogenic factor. Moreover, MITF regulates the expression of the antiapoptotic gene BCL2 (McGill et al. 2002), which increases VEGF levels by both inducing HIF1a expression (Trisciuoglio et al. 2011) and enhancing the transcriptional activity of STAT3 (Kaneko et al. 2007). A direct role for TFE3 in the regulation of angiogenesis has not been described. However, ASPSCR1-TFE3-driven ASPS is a highly vascularized tumor, which responds to antiangiogenic therapy (Lazar et al. 2007, Zhou et al. 2017). While several angiogenesis-associated genes were shown to be upregulated in ASPS and are unique to this sarcoma (Lazar et al. 2007), only a few appear to be direct targets of ASPSCR1-TFE3 (Kobos et al. 2013). ASPSCR1- TFE3-mediated regulation of receptor tyrosine kinase-dependent proangiogenic pathways may provide an indirect mechanism for activation of angiogenesis downstream of TFE3 fusion proteins in ASPS and tRCC tumors.
Dysregulation of WNT signaling.
Dysregulation of WNT signaling has been reported in RCC, but the relevance of this alteration in disease progression has remained elusive (Xu et al. 2016). The recent generation of a Tfeb conditional transgenic model, which closely recapitulates human RCC, has helped to dissect the signaling pathways associated with tRCC (Calcagnì et al. 2016). Transcriptomic analysis of kidney tumors from these mice revealed a significant induction of Wnt pathway components (Axin2, Fzd3, Rnf146, and Kdm6a) and direct target genes (Ccnd1 and Myc). Additionally, increased protein levels of β -catenin and inactivation of Gsk3β were observed in several tumors. Accordingly, treatment with Wnt pathway inhibitors significantly attenuated tumor growth, suggesting that targeting WNT signaling could be a therapeutic approach in TFE tRCC (Calcagni et al. 2016).
Lysosomal exocytosis.
Tumor cells must break down basement membranes and extracellular matrices to invade surrounding tissues prior to metastasizing to secondary organ sites (Eddy et al. 2017). Lysosomal exocytosis—a process whereby lysosomes dock, subsequently fuse with the plasma membrane, and secrete their contents into the extracellular space (Cheng et al. 2014, Medina et al. 2011, Polishchuk et al. 2014)—mediates plasma membrane repair upon mechanical injury (Andrews et al. 2014, Jimenez & Perez 2017) and remodeling of the extracellular matrix. This pathway has an important role in several physiological processes, including bone resorption by osteoclasts, organ development, and branching morphogenesis (Naegeli et al. 2017, Settembre et al. 2013b). Lysosomal exocytosis can be triggered by changes in cellular calcium levels (Cheng et al. 2015, Reddy et al. 2001). Increased expression of TFEB has been shown to promote lysosomal exocytosis through transcriptional regulation of the lysosomal Ca2+ transporter TRPML1.
Cancer cells can also breach local basement membranes via lysosomal exocytosis to transmigrate to other tissues (Olson & Joyce 2015). For example, the extralysosomal functions of the cysteine cathepsin family of lysosomal proteases include the degradation and turnover of the extracellular matrix; activation and processing of growth factors, cytokines, and chemokines; and shedding of cell-cell adhesion molecules, all of which promote tissue invasion and metastases. It remains to be determined whether cancers harboring increased MiT/TFE-dependent lysosome activity utilize this pathway to promote invasion and metastasis through lysosomal exocytosis and the release of luminal proteases.
Cell fate determination.
Master TFs often control the specification of tissue lineage, the establishment of cell types, and their differentiation status within a given organ. Evidence in support of MiT/TFE factors controlling lineage commitment and differentiation of normal cells in several organs (Ferron et al. 2013, Hershey & Fisher 2004, Leeman et al. 2018, Shibahara et al. 2000) suggests that these factors may also regulate cell fate in cancer. For instance, MITF is a key determinant of melanocyte differentiation and regulates the expression of numerous genes associated with pigmentation and melanocyte differentiation (Steingrimsson et al. 2004).
Differentiation and maintenance of several additional cell types in the body, including the retinal pigment epithelium (RPE) (Shibahara et al. 2000), mast cells (Kitamura et al. 2002, Qi et al. 2013, Steingrímsson et al. 1994), and osteoclasts (Ferron et al. 2013, Hershey & Fisher 2004, Steingrimsson et al. 2002), are dependent on MiT/TFE factors. A common theme among these cell types is a role for lysosomes in executing their normal functions: phagocytosis and the degradation of photoreceptor outer segment membranes by the RPE; the release of granules containing cytokines, histamines, and proteases in mast cells; and bone resorption in osteoclasts.
THERAPEUTIC IMPLICATIONS
The diverse roles of MiT/TFE proteins in promoting tumorigenesis make these TFs and their downstream pathways important targets for generating anticancer agents. To date, several studies using in vitro and in vivo tumor models have shown that targeting autophagy and lysosome function significantly inhibits tumor growth (White 2015). For example, over 40 currently ongoing clinical trials incorporate the lysosomal inhibitor hydroxychloroquine (HCQ) in the treatment of a diverse array of tumor types (Perera & Bardeesy 2015). Most recently, treatment of 31 preoperative PDA patients with HCQ and gemcitabine resulted in a significant increase in overall survival (Boone et al. 2015). Importantly, increased LC3II staining in isolated peripheral blood mononuclear cells posttreatment provided clear evidence of autophagy inhibition in vivo in responding patients. Results from ongoing trials of HCQ in combination with chemotherapy (gemcitabine/Abraxane® and FOLFIRINOX) in PDA are highly anticipated. The success of autophagy-lysosome inhibition against an array of tumor types including glioblastoma, myeloma, prostate cancer, and breast cancer highlights the broader importance of this pathway in promoting tumorigenesis (Mahalingam et al. 20l4, Rangwala et al. 20l4, Vogl et al. 20l4). It remains to be determined whether MiT/TFE proteins function in these cancer settings and whether their activation status represents a biomarker of response.
A further implication of recent studies is that MiT/TFE factors can function upstream of mTORCl and stimulate its activity through a feedback mechanism (Di Malta et al. 20l7). Accordingly, mTORCl pathway activation was shown to be higher in tRCC than in non-tRCC (Argani et al. 20l0). Several clinical trials incorporating mTOR inhibitors as both first- and second-line therapies are underway in RCCs associated with poor prognosis (Sánchez-Gastaldo et al. 20l7). These treatments have shown significant improvement in overall survival. However, the presence of an MiT/TFE feedback mechanism may confer resistance to mTOR inhibitors in some settings. Thus, simultaneous suppression of several pathways downstream of MiT/TFE factors with next-generation inhibitors, such as the recently described mTOR RapaLink compound (Rodrik- Outmezguine et al. 20l6), and more potent lysosome inhibitors (McAfee et al. 20l2, Rebecca et al. 20l7), could confer enhanced efficacy in MiT/TFE-dependent cancers.
CONCLUSIONS
Cancer-associated alterations of MiT/TFE genes in subsets of RCC, melanoma, and ASPS first established these genes as bona fide oncogenes. As outlined here, MiT/TFE proteins are linked to several protumorigenic pathways, which may be activated in a context-dependent manner based on tissue lineage, co-occurring genetic alterations, and microenvironmental conditions. The recent discovery that MiT/TFE proteins regulate autophagy, lysosome biogenesis, and mTORCl activation suggests that these TFs may be broadly implicated in a wider array of cancers than previously anticipated. Further defining the full cohort of interacting partners and transcriptional targets of MiT/TFE proteins may help pinpoint the specific MiT/TFE-dependent gene programs activated in different tissues and states. Similarly, continued characterization of upstream signaling cascades that control MiT/TFE levels, stability, localization, and activity may inform the generation of novel therapeutic strategies to switch off MiT/TFE in cancer cells.
ACKNOWLEDGMENTS
We thank Roberto Zoncu for critical reading of the manuscript. R.M.P. is supported by an NIH (National Institutes of Health) Director’s New Innovator Award (DP2CA2l6364), the Damon Runyon-Rachleff Innovation Award, and the AACR (American Association for Cancer Research) Pancreatic Cancer Action Network Career Development Award. C.D.M. is supported by the University of Naples Federico II under the STAR Program. A.B. is supported by the Telethon Foundation, the European Research Council (ERC) advanced grant ERC-LYSOSOMICS, NIH R0l-NS078072, and AIRC IG 20l5 ID l7639. We apologize to colleagues whose work we were not able to cite due to space limitations.
Footnotes
DISCLOSURE STATEMENT
A.B. is a cofounder of Casma Therapeutics.
LITERATURE CITED
- Amin MB, Amin MB, Tamboli P, Javidan J, Stricker H, et al. 2002. Prognostic impact of histologic subtyping of adult renal epithelial neoplasms: an experience of 405 cases. Am. J. Surg. Pathol. 26(3):281–91 [DOI] [PubMed] [Google Scholar]
- Andrews NW, de Almeida PE, Corrotte M. 2014. Damage control: cellular mechanisms of plasma membrane repair. Trends Cell Bio.24(12):734–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argani P, Antonescu CR, Illei PB, Lui MY, Timmons CF, et al. 2001. Primary renal neoplasms with the ASPL-TFE3 gene fusion of alveolar soft part sarcoma. Am. J. Pathol. 159(1):179–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argani P, Hicks J, De Marzo AM, Albadine R, Illei PB, et al. 2010. Xp11 translocation renal cell carcinoma (RCC): extended immunohistochemical profile emphasizing novel RCC markers. Am. J. Surg. Pathol. 34(9):1295–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argani P, Lal P, Hutchinson B, Lui MY, Reuter VE, Ladanyi M. 2003a. Aberrant nuclear immunoreactivity for TFE3 in neoplasms with TFE3 gene fusions: a sensitive and specific immunohistochemical assay. Am.J. Surg. Pathol. 27(6):750–61 [DOI] [PubMed] [Google Scholar]
- Argani P, Lui MY, Couturier J, Bouvier R, Fournet J-C, Ladanyi M. 2003b. A novel CLTC-TFE3 gene fusion in pediatric renal adenocarcinoma with t(X;17)(p11.2;q23). Oncogene 22(34):5374–78 [DOI] [PubMed] [Google Scholar]
- Azizi AA, Haberler C, Czech T, Gupper A, Prayer D, et al. 2006. Vascular-endothelial-growth-factor (VEGF) expression and possible response to angiogenesis inhibitor bevacizumab in metastatic alveolar soft part sarcoma. Lancet Oncol.7(6):521–23 [DOI] [PubMed] [Google Scholar]
- Beckmann H, Su LK, Kadesch T. 1990. TFE3: a helix-loop-helix protein that activates transcription through the immunoglobulin enhancer |J.E3 motif. Genes Dev. 4(2):167–79 [DOI] [PubMed] [Google Scholar]
- Bertolotto C, Lesueur F, Giuliano S, Strub T, de Lichy M, et al. 2011. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 480(7375):94–98 [DOI] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, et al. 2001. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294(5547):1704–8 [DOI] [PubMed] [Google Scholar]
- Boone BA, Bahary N, Zureikat AH, Moser AJ, Normolle DP, et al. 2015. Safety and biologic response of pre-operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma. Ann. Surg. Oncol. 22(13):4402–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruder E, Passera O, Harms D, Leuschner I, Ladanyi M, et al. 2004. Morphologic and molecular characterization of renal cell carcinoma in children and young adults. Am. J. Surg. Pathol. 28(9):1117–32 [DOI] [PubMed] [Google Scholar]
- Buerger C, DeVries B, Stambolic V. 2006. Localization of Rheb to the endomembrane is critical for its signaling function. Biochem. Biophys. Res. Commun. 344(3):869–80 [DOI] [PubMed] [Google Scholar]
- Buscà R, Berra E, Gaggioli C, Khaled M, Bille K, et al. 2005. Hypoxia-inducible factor 1a is a new target of microphthalmia-associated transcription factor (MITF) in melanoma cells. J. Cell Biol. 170(1):49–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell K, Debnath J. 2018. Beyond self-eating: the control of nonautophagic functions and signaling pathways by autophagy-related proteins. J. Cell Biol. 217(3):813–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcagnì A, Kors L, Verschuren E, De Cegli R, Zampelli N, et al. 2016. Modelling TFE renal cell carcinoma in mice reveals a critical role of WNT signaling. eLife 5:e17047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Res. Netw. 2013. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499(7456):43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Res. Netw. 2015. Genomic classification of cutaneous melanoma. Cell 161(7):1681–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Res. Netw. 2016. Comprehensive molecular characterization of papillary renal-cell carcinoma. N. Engl. J. Med. 374(2):135–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Zhang X, Gao Q, Ali Samie M, Azar M, et al. 2014. The intracellular Ca2+ channel MCOLN1 is required for sarcolemma repair to prevent muscular dystrophy. Nat. Med. 20(10):1187–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Zhang X, Yu L, Xu H. 2015. Calcium signaling in membrane repair. Semin. Cell Dev. Biol. 45:24–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J, Lu YJ, Sidhar SK, Parker C, Gill S, et al. 1997. Fusion of splicing factor genes PSF and NonO (p54nrb) to the TFE3 gene in papillary renal cell carcinoma. Oncogene 15(18):2233–39 [DOI] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, et al. 2013. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497(7451):633–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner SD, Schmid SL. 2003. Regulated portals of entry into the cell. Nature 422(6927):37–44 [DOI] [PubMed] [Google Scholar]
- Cronin JC, Wunderlich J, Loftus SK, Prickett TD, Wei X, et al. 2009. Frequent mutations in the MITF pathway in melanoma. Pigment Cell Melanoma Res.22(4):435–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Jonas O, Keibler MA, Hou HW, Luengo A, et al. 2017. Direct evidence for cancer-cell- autonomous extracellular protein catabolism in pancreatic tumors. Nat. Med. 23(2):235–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, et al. 2016. Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. CellMetab. 23(3):517–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, et al. 2002. Mutations of the BRAF gene in human cancer. Nature 417(6892):949–54 [DOI] [PubMed] [Google Scholar]
- Davis IJ, Hsi B-L, Arroyo JD, Vargas SO, Yeh YA, et al. 2003. Cloning of an Alpha-TFEB fusion in renal tumors harboring the t(6;11)(p21;q13) chromosome translocation. PNAS 100(10):6051–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Duve C 2005. The lysosome turns fifty. Nat. Cell Biol. 7(9):847–49 [DOI] [PubMed] [Google Scholar]
- De Palma M, Biziato D, Petrova TV. 2017. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 17(8):457–74 [DOI] [PubMed] [Google Scholar]
- DeNicola GM, Cantley LC. 2015. Cancer’s fuel choice: new flavors for a picky eater. Mol. Cell. 60(4):514–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fiore PP, von Zastrow M. 2014. Endocytosis, signaling, and beyond. Cold Spring Harb. Perspect. Biol. 6(8):a016865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Malta C, Siciliano D, Calcagni A, Monfregola J, Punzi S, et al. 2017. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science 356(6343):1188–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy RJ, Weidmann MD, Sharma VP, Condeelis JS. 2017. Tumor cell invadopodia: invasive protrusions that orchestrate metastasis. Trends CellBiol.27(8):595–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennen M, Keime C, Kobi D, Mengus G, Lipsker D, et al. 2015. Single-cell gene expression signatures reveal melanoma cell heterogeneity. Oncogene 34(25):3251–63 [DOI] [PubMed] [Google Scholar]
- Fan Y, Lu H, Liang W, Garcia-Barrio MT, Guo Y, et al. 2018. Endothelial TFEB (transcription factor EB) positively regulates postischemic angiogenesis. Circ. Res. 122(7):945–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley MN, Schmidt LS, Mester JL, Pena-Llopis S, Pavia-Jimenez A, et al. 2013. A novel germline mutation in BAP1 predisposes to familial clear-cell renal cell carcinoma. Mol. CancerRes. 11(9):1061–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron M, Settembre C, Shimazu J, Lacombe J, Kato S,et al. 2013. ARANKL-PKCβ-TFEB signaling cascade is necessary for lysosomal biogenesis in osteoclasts. Genes Dev.27(8):955–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose A, Tariq Z, Veltri S. 2012. Treatment of multidrug resistant advanced alveolar soft part sarcoma with sunitinib. Am.J. Ther. 19(1):e56–58 [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. 2015. A century of cholesterol and coronaries: from plaques to genes to statins. Cell 161(1):161–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. 2001. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. PNAS 98(25):14440–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin ML, Jin H, Straessler K, Smith-Fry K, Zhu J-F, et al. 2014. Modeling alveolar soft part sarcomage- nesis in the mouse: a role for lactate in the tumor microenvironment. Cancer Cell 26(6):851–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Chen H-Y, Mathew R, Fan J, Strohecker AM, et al. 2011. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25(5):460–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Teng X, Laddha SV, Ma S, Van Nostrand SC, et al. 2016. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev.30(15): 1704–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq R, Fisher DE. 2011. Biology and clinical relevance of the micropthalmia family of transcription factors in human cancer. J. Clin. Oncol. 29(25):3474–82 [DOI] [PubMed] [Google Scholar]
- Heckmann BL, Boada-Romero E, Cunha LD, Magne J, Green DR. 2017. LC3-associated phagocytosis and inflammation. J. Mol. Biol. 429(23):3561–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemesath TJ, Steingrimsson E, McGill G, Hansen MJ, Vaught J, et al. 1994. Microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 8(22):2770–80 [DOI] [PubMed] [Google Scholar]
- Hershey CL, Fisher DE. 2004. Mitfand Tfe3: members of a b-HLH-ZIP transcription factor family essential for osteoclast development and function. Bone 34(4):689–96 [DOI] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, et al. 2012. A landscape of driver mutations in melanoma. Cell 150(2):251–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek KS, Eichhoff OM, Schlegel NC, Döbbeling U, Kobert N, et al. 2008. In vivo switching of human melanoma cells between proliferative and invasive states. CancerRes. 68(3):650–56 [DOI] [PubMed] [Google Scholar]
- Huo Y, Cai H, Teplova I, Bowman-Colin C, Chen G, et al. 2013. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov. 3(8):894–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilagan E, Manning BD. 2016. Emerging role of mTOR in the response to cancer therapeutics. Trends Cancer 2(5):24l–5l [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez AJ, Perez F. 2017. Plasma membrane repair: the adaptable cell life-insurance. Curr. Opin. Cell Biol. 47:99–l07 [DOI] [PubMed] [Google Scholar]
- Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, et al. 20l3. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. PNAS ll0(22):8882–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, et al. 20l5. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 75(3):544—53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko T, Zhang Z, Mantellini MG, Karl E, Zeitlin B, et al. 2007. Bcl-2 orchestrates a cross-talk between endothelial and tumor cells that promotes tumor growth. Cancer Res. 67(20):9685–93 [DOI] [PubMed] [Google Scholar]
- Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, et al. 20l4. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 4(8):9l4–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman EC, Ricketts CJ, Rais-Bahrami S, Yang Y, Merino MJ, et al. 20l4. Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat. Rev. Urol. ll(8):465–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur J, Debnath J. 20l5. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. l6(8):46l–72 [DOI] [PubMed] [Google Scholar]
- Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan K-L. 2008. Regulation of TORCl by Rag GTPases in nutrient response. Nat. Cell Biol. l0(8):93 5–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura Y, Morii E, Jippo T, Ito A. 2002. Regulation of mast cell phenotype by MITF. Int. Arch. Allergy Immunol. l27(2):l06–9 [DOI] [PubMed] [Google Scholar]
- Kobos R, Nagai M, Tsuda M, Merl MY, Saito T, et al. 20l3. Combining integrated genomics and functional genomics to dissect the biologyofa cancer-associated, aberrant transcription factor, the ASPSCRl-TFE3 fusion oncoprotein. J. Pathol. 229(5):743–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, et al. 20l4. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov.4(7): 816–2 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper RP, Schepens M, Thijssen J, van Asseldonk M, van den Berg E, et al. 2003. Upregulation of the transcription factor TFEB in t(6;ll)(p2l;ql3)-positive renal cell carcinomas due to promoter substitution. Hum. Mol. Genet. l2(l4):l66l–69 [DOI] [PubMed] [Google Scholar]
- Lazar AJF, Das P, Tuvin D, Korchin B, Zhu Q, et al. 2007. Angiogenesis-promoting gene patterns in alveolar soft part sarcoma. Clin. Cancer Res. l3(24):73l4–2l [DOI] [PubMed] [Google Scholar]
- Leeman DS, Hebestreit K, Ruetz T, Webb AE, McKay A, et al. 20l8. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science 359(638l):l277–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Deretic V. 2007. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 7(l0):767–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan WM, Ricketts CJ. 20l3. The metabolic basis of kidney cancer. Semin. Cancer Biol. 23(l):46–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan WM, Srinivasan R, Schmidt LS. 20l0. The genetic basis of kidney cancer: a metabolic disease. Nat. Rev. Urol. 7(5):277–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock R, Roy S, Kenific CM, Su JS, Salas E, et al. 20ll. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell. 22(2):l65–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG. 20ll. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27:44l–64 [DOI] [PubMed] [Google Scholar]
- Lyssiotis CA, Kimmelman AC. 20l7. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 27(ll):863–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, et al. 20l4. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy l0(8):l403–l4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra A, Hruban RH. 2008. Pancreatic cancer. Annu. Rev. Pathol. 3:l57–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansueto G, Armani A, Viscomi C, D’Orsi L, De Cegli R, et al. 2017. Transcription factor EB controls metabolic flexibility during exercise. Cell Metab. 25(1):182–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand B, Arsenault D, Raymond-Fleury A, Boisvert F-M, Boucher M- J. 2015. Glycogen synthase kinase-3 (GSK3) inhibition induces prosurvival autophagic signals in human pancreatic cancer cells. J. Biol. Chem. 290(9):5592–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Chen Y, Gucek M, Puertollano R. 2012. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8(6):903–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Diab HI, Lishu L, Jeong-A L, Patange S, et al. 2014. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. Signal. 7(309):ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAfee Q, Zhang Z, Samanta A, Levi SM, Ma X-H, et al. 2012. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. PNAS 109(21):8253–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill GG, Horstmann M, Widlund HR, Du J,Motyckova G, et al. 2002. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell 109(6):707–18 [DOI] [PubMed] [Google Scholar]
- Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, et al. 2015. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 17(3):288–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, et al. 2011. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell. 21(3):421–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AJ, Levy C, Davis IJ, Razin E, Fisher DE. 2005. Sumoylation ofMITF and its related family members TFE3 and TFEB. J. Biol. Chem. 280(1):146–55 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M. 2011. Autophagy: renovation of cells and tissues. Cell 147(4):728–41 [DOI] [PubMed] [Google Scholar]
- Monkkonen T, Debnath J. 2018. Inflammatory signaling cascades and autophagy in cancer. Autophagy 14(2):190–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller J, Krijgsman O, Tsoi J, Robert L, Hugo W, et al. 2014. LowMITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 5:5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naegeli KM, Hastie E, Garde A, Wang Z, Keeley DP, et al. 2017. Cell invasion in vivo via rapid exocytosis of a transient lysosome-derived membrane domain. Dev. Cell. 43(4):403–17.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano G, Ballabio A. 2016. TFEB at a glance. J. Cell Sci. 129(13):2475–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson OC, Joyce JA. 2015. Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 15(12):712–29 [DOI] [PubMed] [Google Scholar]
- O’Rourke EJ, Ruvkun G. 2013MXL-3 andHLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat. Cell Biol. 15(6):668–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, et al. 2015. The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell 162(2):259–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri M, Impey S, Kang H,di Ronza A, Pelz C, et al. 2011. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 20(19):3852–66 [DOI] [PubMed] [Google Scholar]
- Pena-Llopis S, Vega-Rubin-de-Celis S, Liao A, Leng N, Pavia-Jimenez A, et al. 2012. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 44(7):751–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, Wolff NC, Tran TAT, et al. 2011. Regulation of TFEB and V-ATPases by mTORC1. EMBOJ. 30(16):3242–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Bardeesy N. 2015. Pancreatic cancer metabolism: breaking it down to build it back up. Cancer Discov.5(12):1247–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, et al. 2015. Transcriptional control of autophagy- lysosome function drives pancreatic cancer metabolism. Nature 524(7565):361–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao S,Amaravadi RK. 2016. Targeting the lysosome in cancer. Ann. N.Y. Acad. Sci. 1371(1):45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploper D, Taelman VF, Robert L, Perez BS, Titz B, et al. 2015. MITF drives endolysosomal biogenesis and potentiates Wnt signaling in melanoma cells. PNAS 112(5):E420–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polishchuk EV, Concilli M, Iacobacci S, Chesi G, Pastore N, et al. 2014. Wilson disease protein ATP7B utilizes lysosomal exocytosis to maintain copper homeostasis. Dev. Cell. 29(6):686–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potente M, Gerhardt H, Carmeliet P. 2011. Basic and therapeutic aspects of angiogenesis. Cell 146(6):873–87 [DOI] [PubMed] [Google Scholar]
- Qi X, Hong J, Chaves L, Zhuang Y, Chen Y, et al. 2013. Antagonistic regulation by the transcription factors C/EBP a and MITF specifies basophil and mast cell fates. Immunity 39(1):97–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JD, White E. 2010. Autophagy and metabolism. Science 330(6009):1344–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramphal R, Pappo A, Zielenska M, Grant R, Ngan B-Y. 2006. Pediatric renal cell carcinoma: clinical, pathologic, and molecular abnormalities associated with the members of the MiT transcription factor family. Am.J. Clin. Pathol. 126(3):349–64 [DOI] [PubMed] [Google Scholar]
- Rangwala R, Leone R, Chang YC, Fecher LA, Schuchter LM, et al. 2014. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 10(8):1369–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, et al. 2014. A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 5:3056. [DOI] [PubMed] [Google Scholar]
- Rebecca VW, Nicastri MC, McLaughlin N, Fennelly C, McAfee Q, et al. 2017. A unified approach to targeting the lysosome’s degradative and growth signaling roles. Cancer Discov. 7(11):1266–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A, Caler EV, Andrews NW. 2001. Plasma membrane repair is mediated by Ca2+-regulated exocytosis oflysosomes. Cell 106(2):157–69 [DOI] [PubMed] [Google Scholar]
- Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, et al. 2012. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 5(228):ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrik-Outmezguine VS, Okaniwa M, Yao Z, Novotny CJ, McWhirter C, et al. 2016. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 534(7606):272–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan DP, Hong TS, Bardeesy N. 2014. Pancreatic adenocarcinoma. N. Engl. J. Med. 371(11):1039–49 [DOI] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. 2010. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141(2):290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, et al. 2008. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320(5882):1496–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Gastaldo A, Kempf E, González Del Alba A, Duran I. 2017. Systemic treatment ofrenal cell cancer: a comprehensive review. Cancer Treat. Rev. 60:77–89 [DOI] [PubMed] [Google Scholar]
- Sandri M 2016. Protein breakdown in cancer cachexia. Semin. Cell Dev. Biol. 54:11–19 [DOI] [PubMed] [Google Scholar]
- Santanam U, Banach-Petrosky W, Abate-Shen C, Shen MM, White E, DiPaola RS. 2016. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 30(4):399–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, et al. 2009. A gene network regulating lysosomal biogenesis and function. Science 325(5939):473–77 [DOI] [PubMed] [Google Scholar]
- Sato S, Roberts K, Gambino G, Cook A, Kouzarides T, Goding CR. 1997. CBP/p300 as a co-factor for the Microphthalmia transcription factor. Oncogene 14(25):3083–92 [DOI] [PubMed] [Google Scholar]
- Saxton RA, Sabatini DM. 2017. mTOR signaling in growth, metabolism, and disease. Cell 169(2):361–71 [DOI] [PubMed] [Google Scholar]
- Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, et al. 1997. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 16(1):68–73 [DOI] [PubMed] [Google Scholar]
- Schürmann A, Brauers A, Massmann S, Becker W, Joost HG. 1995. Cloning of a novel family of mammalian GTP-binding proteins (RagA, RagBs, RagB1) with remote similarity to the Ras-related GTPases. J. Biol. Chem. 270(48):28982–88 [DOI] [PubMed] [Google Scholar]
- Sekiguchi T, Hirose E, Nakashima N, Ii M, Nishimoto T. 2001. Novel G proteins, Rag C and Rag D, interact with GTP-binding proteins, Rag A and Rag B. J. Biol. Chem. 276(10):7246–57 [DOI] [PubMed] [Google Scholar]
- Settembre C, Ballabio A. 2014. Lysosome: regulator of lipid degradation pathways. Trends Cell Bio/.24(12):743–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, et al. 2013a. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 15(6):647–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, et al. 2011. TFEB links autophagy to lysosomal biogenesis. Science 332(6036):1429–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Medina DL, Ballabio A. 2013b. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14(5):283–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, et al. 2012. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB: self-regulation of the lysosome via mTOR and TFEB. EMBOJ. 31(5):1095–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibahara S, Yasumoto K, Amae S, Udono T, Watanabe K, et al. 2000. Regulation of pigment cell-specific gene expression byMITF. Pigment Cell Res. 13(Suppl. 8):98–102 [DOI] [PubMed] [Google Scholar]
- Shipley JM, Birdsall S, Clark J, Crew J, Gill S, et al. 1995. Mapping the X chromosome breakpoint in two papillary renal cell carcinoma cell lines with a t(X;1)(p11.2;q21.2) and the first report of a female case. Cytogenet. Cell Genet. 71(3):280–84 [DOI] [PubMed] [Google Scholar]
- Sidhar SK, Clark J, Gill S,Hamoudi R, Crew AJ,et al. 1996. The t(X;1)(p11.2;q21.2) translocation in papillary renal cell carcinoma fuses a novel gene PRCC to the TFE3 transcription factor gene. Hum. Mol. Genet. 5(9):1333–38 [DOI] [PubMed] [Google Scholar]
- Stacchiotti S, Tamborini E, Marrari A, Brich S, Rota SA, et al. 2009. Response to sunitinib malate in advanced alveolar soft part sarcoma. Clin. Cancer Res. 15(3):1096–1104 [DOI] [PubMed] [Google Scholar]
- Steingrímsson E, Copeland NG, Jenkins NA. 2004. Melanocytes and the microphthalmia transcription factor network. Annu. Rev. Genet. 38:365–411 [DOI] [PubMed] [Google Scholar]
- Steingrímsson E, Moore KJ, Lamoreux ML, Ferré-D’Amaré AR, Burley SK, et al. 1994. Molecular basis of mouse microphthalmia (mi) mutations helps explain their developmental and phenotypic consequences. Nat. Genet. 8(3):256–63 [DOI] [PubMed] [Google Scholar]
- Steingrímsson E, Tessarollo L, Pathak B, Hou L, Arnheiter H, et al. 2002. Mitf and Tfe3, two members of the Mitf-Tfe family of bHLH-Zip transcription factors, have important but functionally redundant roles in osteoclast development. PNAS 99(7):4477–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steingrímsson E, Tessarollo L, Reid SW, Jenkins NA, Copeland NG. 1998. The bHLH-Zip transcription factor Tfeb is essential for placental vascularization. Development 125(23):4607–16 [DOI] [PubMed] [Google Scholar]
- Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ,et al. 2013. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 3(11):1272–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramani S,Malhotra V. 2013. Non-autophagic roles of autophagy-related proteins. EMBO Rep.14(2):143–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Takeda K, Nobukuni Y, Urabe K, Long JE, et al. 1996. Ectopic expression of MITF, a gene for Waardenburg syndrome type 2, converts fibroblasts to cells with melanocyte characteristics. Nat. Genet. 14(1):50—54 [DOI] [PubMed] [Google Scholar]
- Tanaka M, Homme M, Yamazaki Y, Shimizu R, Takazawa Y, Nakamura T. 2017. Modeling alveolar soft part sarcoma unveils novel mechanisms of metastasis. Cancer Res. 77(4):897–907 [DOI] [PubMed] [Google Scholar]
- Tanaka M, Kato K, Gomi K, Matsumoto M, Kudo H, et al. 2009. Perivascular epithelioid cell tumor with SFPQ/PSF-TFE3 gene fusion in a patient with advanced neuroblastoma. Am.J. Surg. Pathol. 33(9):1416–20 [DOI] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, et al. 2016. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352(6282):189–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trisciuoglio D, Gabellini C, Desideri M, Ragazzoni Y, De Luca T, et al. 2011. Involvement of BH4 domain of bcl-2 in the regulation of HIF-1-mediated VEGF expression in hypoxic tumor cells. Cell Death Differ. 18(6):1024–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao H, Chin L, Garraway LA, Fisher DE. 2012. Melanoma: from mutations to medicine. Genes Dev. 26(11):1131–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl DT, Stadtmauer EA, Tan K- S, Heitjan DF, Davis LE, et al. 2014. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 10(8):1380—90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Wei S, Gan B,Peng X,Zou W, Guan J-L. 2011. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev.25(14):1510–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weterman MA, Wilbrink M, Geurts van Kessel A. 1996. Fusion of the transcription factor TFE3 gene to a novel gene, PRCC, in t(X;1)(p11;q21)-positive papillary renal cell carcinomas. PNAS 93(26):15294–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weterman MJ, van Groningen JJ, Jansen A, van Kessel AG. 2000. Nuclear localization and transactivating capacities of the papillary renal cell carcinoma-associated TFE3 and PRCC (fusion) proteins. Oncogene 19(1):69–74 [DOI] [PubMed] [Google Scholar]
- White E 2015. The role for autophagy in cancer. J. Clin. Investig. 125(1):42–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Koh JY, Price S, White E, Mehnert JM. 2015. Atg7 overcomes senescence and promotes growth of BrafV600E-driven melanoma. Cancer Discov. 5(4):410–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Krause M, Samoylenko A, Vainio S. 2016. Wnt signaling in renal cell carcinoma. Cancers 8(6):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, et al. 2018. Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov.8(3):276–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, et al. 2014. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov.4(8):905–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Zheng Y, Li L, Liu S, Burrows M, et al. 2014. Direct conversion of mouse and human fibroblasts to functional melanocytes by defined factors. Nat. Commun. 5:5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang X, Contino G, Liesa M, Sahin E, et al. 2011. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25(7):717–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yecies JL, Manning BD. 2011. mTOR links oncogenic signaling to tumor cell metabolism. J. Mol. Med. 89(3):221–28 [DOI] [PubMed] [Google Scholar]
- Ying H, Dey P, Yao W, Kimmelman AC, Draetta GF, et al. 2016. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev.3 0(4): 3 5 5–8 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama S, Woods SL, Boyle GM, Aoude LG, MacGregor S, et al. 2011. A novel recurrent mutation in MITFpredisposes to familial and sporadic melanoma. Nature 480(7375):99–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Tang F, Wang Y,Min L, Luo Y, et al. 2017. Advanced alveolar soft part sarcoma responds to apatinib. Oncotarget 8(30):50314—22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. 2011. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 334(6056):678–83 [DOI] [PMC free article] [PubMed] [Google Scholar]