

Abstract
Dynamic combinatorial chemistry (DCC) is a powerful tool to identify bioactive compounds. This efficient technique allows the target to select its own binders and circumvents the need for synthesis and biochemical evaluation of all individual derivatives. An ever‐increasing number of publications report the use of DCC on biologically relevant target proteins. This minireview complements previous reviews by focusing on the experimental protocol and giving detailed examples of essential steps and factors that need to be considered, such as protein stability, buffer composition and cosolvents.
Keywords: Target‐directed dynamic combinatorial chemistry, Protein stability, Hit identification, Medicinal chemistry, Biochemical activity
This minireview focuses on important aspects concerning the practical handling in protein‐templated dynamic combinatorial chemistry (ptDCC). The best biological and chemical conditions should be harmoniously combined in order to successfully apply ptDCC. Moreover, analytical techniques used to determine which compounds are favored are briefly discussed.
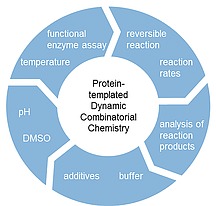
Introduction
Since its dawn more than two decades ago, combinatorial chemistry approaches1, 2, 3, 4, 5 have developed into target‐directed dynamic combinatorial chemistry (tdDCC) and have matured as a hit‐identification tool6, 7, 8, 9, 10, 11 A growing number of groups have shown the general applicability and scope of tdDCC for the identification of modulators of targets.6, 12, 13, 14, 15, 16, 17, 18, 19, 20 tdDCC refers to general pharmacologically relevant targets which next to proteins also include DNA and RNA, whereas protein‐templated DCC (ptDCC) only refers to proteins. Several reviews and book chapters on tdDCC have been published in recent years.21, 22, 23 This minireview covers our work on ptDCC and provides the key features of our protocol, explaining the essential steps in designing a successful ptDCC experiment.
Carefully chosen building blocks are connected in a reversible manner via covalent or noncovalent bonds to form a dynamic combinatorial library (DCL) (Figure 1). Biocompatibility, pH dependence, temperature, solubility and stability of the components are important factors, which should be taken into account. The ideal DCLs do not require cosolvents, however, it can occur that the formed products have a lower solubility than the building blocks and in order to keep all compounds in solution, a cosolvent such as dimethyl sulfoxide (DMSO) is commonly used. Precipitation of DCL components could lead to an undesired shift in the equilibrium. By contrast, a desired shift of the equilibrium can be obtained by the addition of an external stimulus, such as a protein target. There are in general two different approaches that can be followed in ptDCC: “adaptive DCC”, in which the target is present during the formation of the DCL and “pre‐equilibrated DCC”, in which the target is added after the DCL is established. An advantage of pre‐equilibrated DCC is that the exchange chemistry can be applied in conditions which are not tolerated by the protein. A disadvantage is that the screening step is performed under static conditions and no amplification effects can be observed since the protein does not alter the equilibrium.
Figure 1.
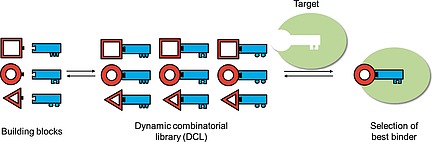
Schematic representation of target‐directed dynamic combinatorial chemistry.
In ptDCC, the member(s) of DCLs, which bind best will be amplified, leading to an increase in their concentration compared to a control reaction without the external stimulus. These binders can then be further evaluated for their biochemical properties.
To enable a comparative analysis of DCLs, a blank reaction, without the target, should be run concurrent with a templated reaction. Another approach of DCC is non‐comparative, in which the hits can be analyzed in complex with the target or after being released from the target. There are different techniques that can be used to analyze the DCLs: liquid and size‐exclusion chromatography coupled to mass spectrometry, NMR spectroscopy, fluorescence spectroscopy and X‐ray crystallography. Figure 2 illustrates the comparative approach vs. the non‐comparative approach, which can be adopted in DCC. The reaction mixture can be “frozen”, in order to prevent the library from re‐equilibrating during the analysis. In the case of acylhydrazone chemistry, this can be achieved by an increase in pH. Denaturation by heat, addition of a solvent or (ultra‐fast) centrifugation ensures that all binders are released from the protein before analysis.
Figure 2.

DCC approaches: comparative and non‐comparative. In the comparative approach the library in presence of a target is compared to the library in absence of the target. In the non‐comparative approaches, the hit–target complexes will be separated from the mixture and analyzed as a complex or as released hits. Adapted from Frei et al.21
1. Reversible Reaction Suitable for DCC
Only a limited number of reversible reactions have been used thus far, they are summarized in Scheme 1. One of the most frequently used reactions is the (acyl‐)hydrazone formation, which combines ketone or aldehyde building blocks with (acyl‐)hydrazides. This condensation reaction can take place in water, making it biocompatible.24 The synthesis of the building blocks is generally straightforward or they may be commercially available.
Scheme 1.
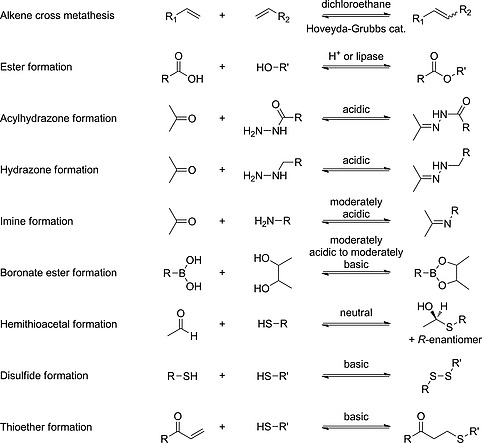
Reversible reactions used in target‐directed DCC to identify bioactive compounds. Adapted from Van der Vlag and Hirsch.23
At physiological conditions, neutral pH and room temperature, acylhydrazone formation and exchange are relatively slow. At acidic pH, the equilibrium is reached rapidly. However, Greaney and co‐workers have shown that the pH dependence can be influenced by the addition of a nucleophilic catalyst. They were able to reach equilibrium reasonably fast at a comparatively high pH of 6.2 by using aniline, as a nucleophilic catalyst.12 Previously Dawson and co‐workers have shown that aniline could serve as a catalyst for acylhydrazone formation and oxime ligation.25, 26 Derivatives of aniline, which bear substituents at the aryl ring, are even more effective catalysts.27
The acylhydrazone linkage is reversible but sufficiently stable to allow for direct analysis under acidic conditions and stable against hydrolysis at physiological pH values, allowing for the “freezing” of the reversible reaction upon increasing the pH.24
An overview of studies published over the past five years in the field of ptDCC is given in Table 1. It must be noted that much more work has been published applying DCC for the formation of diverse libraries in the drug‐discovery process. For example the coupling of DCC to DNA‐encoded libraries, creating so called DNA‐encoded dynamic combinatorial chemical libraries (EDCCLs). Iminobiotin and homotetrameric streptavidin were used as a model system to identify a bidentate protein/ligand interaction. The addition of an external stimulus, for example a target protein, can shift the thermodynamic equilibrium and hence a DNA amplification can be observed after sequencing28
Table 1.
Protein‐templated DCC studies reported over the past five years, in which a target was used as a template to influence the equilibrium. Therefore, only articles using an adaptive approach are listed, pre‐equilibrated DCC examples are omitted29, 30, 31 The table is adapted from Frei et al. and complemented32 a
| Target | Reversible reaction | Analysis | Library size | Equilibration time | Method applied for affinity measurement | Best affinity | Ref. |
|---|---|---|---|---|---|---|---|
| Wt Tau RNA | Disulfide | HPLC‐MS and NMR | 21 | 2 days | Fluorescence titration | EC 50 = 70 nm | Artigas et al. 201533 |
| HIV FSS RNA | Disulfide | MS | 12 | 4 days | n.a. | n.a. | McAnany et al. 201634 |
| Vascular endothelial growth factor receptor (VEGFR) 2 | Imine | HRMS | 297 | 24 h | In vitro activity against cancer cell lines | IC 50 = 2.4 µm | Yang et al. 201635 |
| Endothiapepsin | Acylhydrazone | HPLC‐MS | 90 | 20 h | Inhibition assay | IC 50 = 54.5 nm K i = 25.4 nm | Mondal et al. 201636 |
| FimH | Acylhydrazone | HPLC | 8 | 3 days | SPR | K D = 273 nm | Frei et al. 201737 |
| UDP‐galacto‐pyranose mutase | Acylhydrazone | HPLC | 11 | 24 h | Fluorescence‐based assay and MIC | K D = 3 µm mic = 26 µg mL–1 | Fu et al. 201738 |
| Myeloperoxidase (MPO) | Hydrazone | Activity assay | 6 | n.a. | in vivo activity assay | IC 50 = 79 nm | Soubhye et al. 201739 |
| ecFabH | Acylhydrazone | 19F‐NMR | 5 | 12 h | Enzymatic assay | IC 50 = 3 mm | Ektström et al. 201840 |
| Multi‐protein strategy on AlkB oxygenases: FTO, ALKBH3 and ALKBH5 | Acylhydrazone | DSF and HPLC | 10 | 5 h | HPLC‐based demethylase and DSF assays | IC 50 = 2.6 µm | Das et al. 201841 |
| Trypanosoma cruzi bromodomain‐containing (TcBDF3) | Acylhydrazone | HPLC‐MS | 30 | n.a. | DSF | IC 50 = 13–23 µm | García et al. 201842 |
| G‐Quadruplex DNA | Imine formation | HPLC and ESI‐MS | 10 | 24 h | n.a. | n.a. | Jana et al. 201943 |
DSF = differential scanning fluorimetry, HPLC = high‐pressure liquid chromatography, IC 50 = half maximal inhibitory concentration, ITC = isothermal titration calorimetry, K D = dissociation constant, K i = inhibition constant, MIC = minimum inhibitory concentration, MS = mass spectrometry, n.a. = not available, NMR = nuclear magnetic resonance, SPR = surface plasmon resonance.
2. A Closer Look on the Templating Protein
To obtain meaningful results from DCC experiments, the quality of the input template is critical. As the equilibrium of the library shifts by the templating effect of the added protein sample, it should consist of the target protein as close to its native state as possible. The quantity of the used template depends on the protein target, there are reported successful DCC projects with 0.1 to 1.5 equivalents of protein.29, 44 DCC experiments are also possible with a mixture of proteins, but a well‐defined sample eases up downstream data analysis and reduces the number of false positives for the desired target.41The condition of the protein sample depends on various variables. For DCC experiments the purity, concentration, tertiary and quaternary structure of the protein, additives and contaminations, as well as the pH‐value are of particular importance. During the experiment, which can take up to several days, protein degradation and precipitation could occur. The tests described herein should give an overview and help to choose suitable experimental conditions to plan new DCC experiments. In the next paragraphs, we will briefly discuss the influence of those factors and suitable analytical methods to monitor them.
2.1. Purity
In the case of a mixed or impure protein sample, there might be several templated reactions proceeding in parallel. It is impossible to differentiate between a small fraction of the sample showing a strong template effect and a large fraction of the protein pool showing only a weak amplification of a binder. This will result in overlapping data, which are difficult to analyze, and may result in false positives. We therefore recommend starting with the highest protein purity available.
2.2. Stability
Not only the initial state, but also the stability of the templating protein during the reaction should be checked by preliminary tests before conducting a DCC experiment. The time span over which a DCC experiment, pre‐equilibrated or adaptive, is monitored can vary. It depends on the reaction rate and concentration and should ideally be monitored until the library reaches an equilibrium state. Usually, the DCL reaches a new equilibrium within the first few days, depending on the reversible reaction and conditions used (Table 1). However, if the protein is stable for longer periods of time, longer equilibration times are possible, for example up to 20 days for the very stable protease endothiapepsin (see Section 2.4).24
It is important that the protein is not precipitating or degrading during the experiment. Precipitation of the protein will remove the template from the solution. Denaturation of the template will lead to entirely new templates, which would affect the equilibrium state of the DCL. This can lead to random and irreproducible amplification of compounds by the unordered protein and a decrease of initially already amplified best binders of the native template. If the protein target is labile, it is therefore necessary to follow the reaction over time to identify the temporary, templated equilibrium of the DCC library. In this, compounds amplified by the native state of the template can be found.
Eventually, after prolonged incubation times, nearly every protein will degrade and, by this, change the equilibrium of the DCL again. Compounds amplified in this step should be disregarded, as they were not templated by the native protein. Observation of the DCC experiment for longer timeframes than the template's stability under the specific conditions should therefore be avoided.
2.3. Buffer and pH
When choosing a buffer for DCC experiments, several different requirements have to be met. Attention should be paid to possible side reactions with the DCL or chelation effects. For example, Tris buffer could form imines with aldehyde building blocks, which might influence the formation of the DCL. Some stabilization of the protein is beneficial, but strong interactions of the buffer with the target protein should also be avoided, for instance, a phosphate buffer for a phosphate binding protein. The phosphate could compete with possible binders; possible effects of competition are discussed in more detail in Section 2.5. So far, in most cases common buffer systems have been used, which are shown in Table 2 and Scheme 2. The choice of buffer is, however, not limited to the established systems.
Table 2.
Buffers commonly used in different DCC reactions. *Tris buffer requires special attention
Scheme 2.
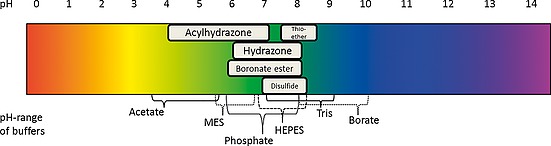
Example of possible buffers and the pH ranges of reactions used in DCC experiments.
For many protein targets, the stability at room temperature and the optimal buffer conditions are not known. We therefore recommend determining these conditions prior to performing DCC experiments. As several interdependent factors, like pH, buffer, ionic strength and ions influence the stability of a protein, it is difficult to suggest a stepwise flow scheme for the determination of the ideal buffer composition for a given protein.28 Not only the protein but also the exchange chemistry might be affected significantly by varying these parameters. We propose to first measure the effect of pH, buffer and ionic strength over a wide range in parallel. Afterwards, a small selection (2 to 5) of the most stabilizing combinations can be evaluated for their long‐term effect on the protein. Subsequently, the best condition will then be used to determine the influence of DMSO (Section 2.6) and, if of interest, additives (Section 2.5). The selection of the initial buffers could be broadened, in case no suitable condition was found.
Two or more buffers should be screened per pH value to distinguish the influence of the buffer component and the pH value on the stability of the protein. It is also possible to use a so‐called “superbuffer”,52 a mix of three or more buffer components, enabling the adjustment of a wide pH range, without changing the buffer composition or concentration.
The effect of the buffer components on a protein can be measured in a straightforward way, by determining the melting point of the target protein via a thermal‐shift assay/differential scanning fluorimetry (TSA/DSF).28 In this method, the protein is incubated together with a lipophilic dye, for example sypro orange. The dye shows an increase in fluorescence after binding to the hydrophobic parts of a protein. These are often located at the inside of a protein and become exposed during temperature‐induced unfolding/melting. The temperature–dependent increase in fluorescence can be measured in a RT‐PCR apparatus and yields the T m of the protein.
Other methods, like DSC, ITC and CD (differential scanning calorimetry, isothermal titration calorimetry and circular dichroism spectroscopy) and the determination of melting points by CD could also be used to gain information on the interaction and possible stabilization of the protein with its buffer, but require a high amount of protein and/or long measurement time. The TSA, however, offers high throughput and a short assay time, together with already several published or commercially available kits in 96‐well format.53, 54 These kits were originally intended to screen for optimal crystallization conditions and cover several stability‐influencing conditions. When performing DCC experiments, the design of an individual 96‐well plate layout, tailored to the buffers and conditions compatible with the planned DCC reaction, might be useful. This is a short time investment, which might pay off quickly in the future, if ptDCC is used on several different targets.
After a DCC–compatible, stabilizing buffer condition has been identified, the protein should be checked for its long‐term stability. To check for cleavage of the protein backbone an analysis by SDS‐PAGE is of sufficient sensitivity (Figure 3). To determine if the protein folding is affected, TSA is again the method of choice, since the signal directly depends on the unfolding process of the protein. With prolonged degradation, the melting point decreases slightly. As a secondary effect of the degradation, the fluorescence curve can show bi‐ and multi‐phasic melting points and an overall decrease in signal intensity and resolution. A fully denatured enzyme will just show a decreasing fluorescence signal with no peak from protein unfolding. As controls, a fresh and a heat‐treated sample of the target protein should be included in the experiment.
Figure 3.
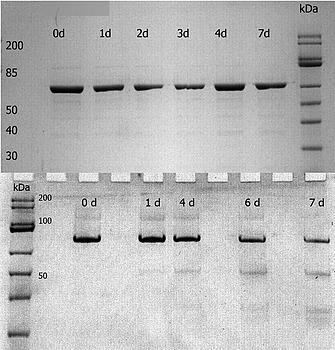
12 % SDS‐PAGE of different homologues of the enzyme 5‐deoxyxylulose 5‐phosphate synthase (DXS) after incubation at RT. The protein on the upper gel shows no sign of degradation. The second protein, shown on the lower gel, shows signs of degradation, starting already at day one with a very faint band around 50 kDa. From day 6 on a decrease of the main protein band also becomes clear. In the top left corner, a gel‐label was removed using image processing software.
The tendency of a protein to precipitate is concentration‐dependent. Because of this, the assays determining the protein stability should be performed with the same protein concentration that is intended to be used in the DCC experiment. If this is not possible, due to limited protein availability, the first experiments might be done with less protein. However, at least for the chosen final condition, the stability assessment should be repeated with the protein concentration that will be used in the DCC experiments.
2.4. Functional Enzyme Assay
For enzymatic protein targets, a functional assay can be used instead of TSA and PAGE measurements for the assessment of long‐term stability. The analysis of activity data of a functional assay to determine the best experimental conditions of the DCC experiments leaves less room for interpretation than the analysis of the results of a melting‐point analysis. Therefore, if a functional assay is available, and the enzyme is showing catalytic activity in the desired pH range of the DCC reaction, the activity assay should be the method of choice.
In a previous study from 2014, we could monitor the activity of the target protein endothiapepsin by performing a fluorescence‐based assay (Figure 4). The pH optimum of endothiapepsin is 4.5, and the enzymatic activity was not affected even after 20 days incubation at RT and a pH of 4.6. Considering this high stability, no buffer optimization was needed.24
Figure 4.
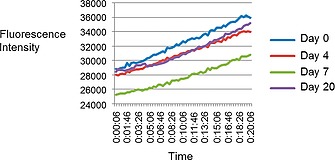
Activity of endothiapepsin, a pepsin‐like aspartic protease, in a fluorescence‐based assay at different time intervals of incubation at room temperature. Adapted from Mondal et al.24
2.5. Additives and Contaminations
During the purification, the protein might be in contact with different buffers and conditions. Some of the buffer components might remain bound to the protein, even after buffer exchange. These contaminants might influence the experiment. It is therefore recommended to critically evaluate the composition of the protein sample. Not only should the protein storage buffer be evaluated, but also the origin of the sample.
Common substances that could be found in protein samples are for example imidazole as a leftover from an IMAC (immobilized metal affinity chromatography) purification step. Protein samples are often supplemented with reducing agents like 2‐ME, DTT or TCEP (2‐Mercaptoethanol, Dithiothreitol or Tris(2‐carboxyethyl)phosphine) in concentrations up to 10 mm to keep the protein in a reducing environment. If disulfide formation is the reversible reaction of choice, the final reducing agent concentration should be evaluated to make sure that the formation of disulfide bonds is not inhibited.
The effect of additives and contaminations is related to the volume of the protein sample used in the individual DCC experiments. This not only determines the final concentration of protein, but also the concentrations of the contaminants. If the batch‐to‐batch concentration of the protein varies and its volume is adjusted to reach the same final concentration in the DCC experiment, it should be noted that the concentrations and effects of the additives in the DCC experiment might vary.
Compounds that remain in the protein sample can have an influence on the DCC reaction or on the target protein. One should critically check every buffer component on possible interference with the planned exchange chemistry. Performing a control experiment with all buffer components, in the absence of protein, can assure that no side reactions are taking place. If the exact composition of the protein sample is unknown, a small volume of the buffer might be obtained by concentration of the protein using an ultrafiltration device and using the flow‐through for the control experiment.
Some agents used during protein purification, such as cryoprotectants like glycerol or detergents like Tween, will interact in a non‐specific way with the protein surface. From our experience, if there is no hint that they might affect the experiment, leftover cryoprotectants and detergents can be tolerated. Special care should be taken if cofactors, coenzymes or ions are supplemented during the purification process to stabilize the enzyme. The same holds true for buffer components structurally related to those supplements. Everything that binds to the targeted binding pocket is competing with the DCC library. If a natural, tight binding cofactor is present during the experiment, it could prevent the building blocks from binding and therefore also inhibit their amplification. However, the use of tight binders can be beneficial in control experiments. If a compound with a known binding site is inhibiting the formation of some previously observed binders this can be taken as a hint that the templated binders are targeting the same protein pocket.
2.6. DMSO
Addition of a small percentage of DMSO to the reaction solution is a common practice in the design of enzymatic assays to improve the solubility of hydrophobic compounds. For biochemical assays, DMSO concentrations up to 10 % are regularly used.55
In DCC experiments, the building blocks of the library are typically dissolved in DMSO stock solutions to enable the easy assembly of a library. Depending on the library composition and number of compounds used, the final DMSO concentration would vary. To keep the reaction conditions comparable, we recommend adding DMSO up to a concentration that can be kept constant for all experiments of a project. This fixed concentration should be evaluated and chosen beforehand, to ensure the protein tolerates it.
DMSO has a very broad range of effects on proteins, it can even decrease the solubility and induce precipitation.56 Both, rate acceleration, as well as inhibition of the enzyme‐catalyzed reaction by DMSO have been observed. An influence of already low percentages of DMSO on the enzymatic activity often hints to DMSO acting as an unspecific effector, interacting with the active site of the enzyme.57 If the enzymatic activity is reduced by DMSO at higher concentrations (>10 % DMSO ), it is often by influencing the overall protein conformation by displacing water molecules bound to the surface and unfolding the protein.58 On the other hand, there are DMSO‐tolerant enzymes known which show activity up to 80 % DMSO.57 Enzyme activity assays are the method of choice to estimate the effect of DMSO on an enzyme. If no activity assay is available, the effect of DMSO could also be measured using TSA, however, interactions with the active site are difficult to detect with this method. We often observe a small effect on the T m of a protein, but a strong effect on the enzymatic activity. Taken together, the DMSO concentration has several effects on the protein structure. The benefits of DMSO addition need to be weighed against the risk of creating an artificial enzymatic fold, which could amplify compounds that would not bind under native conditions. Therefore, the DMSO concentration should be as low as possible, in our lab up to 5 % are regularly used.
2.7. Temperature
To speed up the rate at which the DCL reaches equilibrium, the experiments are normally performed at room temperature. For labile proteins, a lower reaction temperature may be necessary, which can improve the stability of the proteins. At the same time, the equilibration rate is decreased, leading to a prolonged incubation time. The optimal temperature for protein stability in DCC could vary from enzyme to enzyme and thus needs to be evaluated in each individual case but room temperature is used in most cases.
3. Setting up a ptDCC Experiment
When crystal structures are available, or even co‐crystal structures, a structure‐based approach can be undertaken to design promising building blocks. In this case, also non‐binders could be designed as control elements, which are not supposed to emerge as hits. The type of reversible linkage should be carefully selected because it influences the molecular recognition by the target. For example, the acylhydrazone linkage resembles the amide functionality and features hydrogen‐bond donors and acceptors. We showed that by combining DCC with de novo structure‐based design, the risks associated with this attractive approach are reduced.24
3.1. Formation of the DCLs
The building blocks might have to be dissolved in DMSO, allowing them as well as the formed products to stay soluble in the final mixture. In principle, they could also be dissolved in the desired buffer, which would be most ideal. In 2014, we coupled DCC to saturation‐transfer difference (STD) NMR spectroscopy, which requires lower concentrations of protein than a general DCC experiment (Table 3). STD‐NMR spectroscopy enables selection of the binders from the DCL, since the intensity of these signals is stronger due to a more efficient saturation transfer. As a result, STD‐NMR spectra cannot be used to determine concentrations of DCL members and therefore amplification cannot be calculated. In follow‐up experiments, it is possible to determine the KD value of a ligand via STD‐NMR or other biophysical assays.59
Table 3.
General protocol for DCC and protocol for DCC coupled to 1H‐STD‐NMR. * Aniline or another nucleophilic catalyst could be added when required. ** In a control experiment, no protein is added. *** Buffer conditions to guarantee protein stability should be determined a priori
| Final concentration in general DCC | Final concentration used in DCC coupled to 1H‐STD‐NMR24 | |
|---|---|---|
| Aldehyde | 0.1 mm | 0.4 mm |
| Hydrazide | 0.1–0.3 mm | 1 mm (for each of the five hydrazides) |
| DMSO | 5–10 % | 5–10 % |
| Aniline* | 10 mm | – |
| Protein** | 10–100 µm | 4 µm |
| Buffer*** | 0.1 m | Ammonium acetate in D2O (0.1 m, pH 4.6) |
| pH* | Acidic–neutral | pH 4.6 |
The ratio of hydrazides vs. aldehydes should allow for the formation of all possible products, therefore at least one equivalent of each hydrazide per aldehyde should be used. For example, if three aldehydes are used then at least three equivalents of each hydrazide should be added, making sure that there is an excess of hydrazides. When required, a nucleophilic catalyst like aniline could be added. The most frequently used concentration of DMSO lies around 5–10 %.
Control experiments should be considered, which should clarify where binding of molecules to the protein occurs and if it is specific or unspecific. This could for example be performed by the addition of a known inhibitor. If the previously observed amplification is not observedany longer, then the hit compounds are competitive binders. Based on the work of Danieli et al., B. Ernst and co‐workers propose that the use of bovine serum albumin (BSA), as a negative control template for which no amplification is expected since the binding pocket is different, is not a good control since it could influence the library composition, whilst the use of a competitive inhibitor is better. BSA has been used in DCC to show that the applied library only gives hits with the real target and that BSA would yield the same result as the blank.32, 60 BSA is commonly known for its stability and was thought not to interfere with biological reactions, however recently DCC experiments have even been used to target BSA.61
3.2. Analysis of the DCLs
Different techniques such as fluorescence‐polarization, SPR, ITC, MST, STD‐NMR, crystallography and others can be used to evaluate and possibly optimize obtained hits. We and Rademann and co‐workers have reviewed the analytical methods used in protein‐templated dynamic combinatorial chemistry to detect hit compounds.23, 62
A commonly applied method to analyze DCC experiments is the recording of HPLC‐MS chromatograms of the libraries. As an illustrative example of the comparative approach, we drew HPLC chromatograms of a blank library and a target library (Figure 5). When we compare both chromatograms, we see that peak number five has increased in the library containing the target, whereas peaks three and six have decreased. The total amount of building blocks stays the same, only the equilibrium can be shifted towards one or more products.
Figure 5.
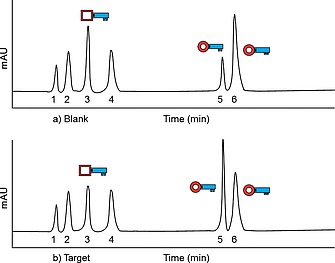
Schematic example of HPLC chromatograms: (a) blank library chromatogram, (b) target library chromatogram.
In order to accurately determine the amplification or decrease of peaks, their relative peak areas (RPA) should be compared. The fictional RPAs of both chromatograms in Figure 5 are given in Table 4. The amplification factor in percentage can be calculated by Equation (1), where the amplification factor in “fold” is given by Equation (2). Using these two equations, the product at peak five has increased by 100 % or twofold. Frei et al. report on a particularly thorough analysis of a DCL using the lectin FimH as a target, using HPLC analysis with an optimized DCC protocol.37
| (1) |
| (2) |
Table 4.
Example of relative peak areas (RPA) obtained from HPLC chromatograms from Figure 4
| Peak number | Relative peak area in blank [%] | Relative peak area in target [%] | Amplification in % | Amplification in “fold” |
|---|---|---|---|---|
| 1 | 10 | 10 | – | 1 |
| 2 | 15 | 15 | – | 1 |
| 3 | 20 | 16 | –20 % | 0.8 |
| 4 | 16 | 16 | – | 1 |
| 5 | 12 | 24 | 100 % | 2 |
| 6 | 27 | 19 | –30 % | 0.7 |
| Total | 100 % | 100 % |
3.3. DCL Analyzed with STD‐NMR spectroscopy
Inspired by the work of Ramström and co‐workers,20 we analyzed the formed DCLs by STD‐NMR spectroscopy (Scheme 3). Five sub‐libraries enabled a clear analysis. As a control with a known binder we used saquinavir (Ki = 48 nm), a potent peptidic inhibitor, to differentiate specific from nonspecific binding. Each sub‐library contained all five hydrazides and one of the aldehyde building blocks and was allowed to equilibrate for 24 hours before adding the target. By analyzing the imine‐type proton signals of the acylhydrazone products in the 1H‐STD‐NMR spectra (Figure 6), we identified in total eight binders. To confirm the results from STD‐NMR, we performed an enzyme‐inhibition assay and showed that the hits were inhibitors with IC 50 values ranging from 12.8 µm to 365 µm. The high hit rate in this report may be a result of the powerful and synergistic combination of de novo structure‐based drug design and DCC. In addition, it is due to use of five sublibraries in which the best binder of each library is detected, whereas in a regular ptDCC setup only the overall best binders will be discovered. In STD‐NMR the protein is used as a tool to analyze the library, whereas in a ptDCC experiment the protein influences the equilibrium and hence the concentrations
Scheme 3.
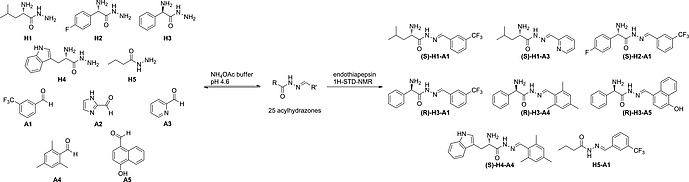
Formation of dynamic combinatorial library and enzymatic selection of the best binders by 1H‐STD‐NMR analysis. Adapted from Mondal et al.24
Figure 6.
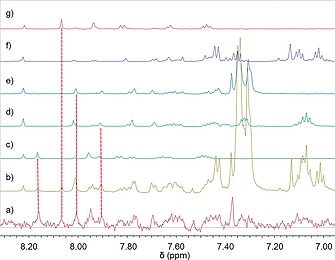
DCL generated from H1–5 + A4: (aromatic region) a) 1H‐STD‐NMR spectrum of H1–5 + A4, b) 1H‐NMR spectrum of H1–5 + A4, c) 1H‐NMR spectrum of H3+A4, d) 1H‐NMR spectrum of H4+A4 (2 singlets correspond to the E/Z isomers), e) H1+A4, f) H2+H4 and g) H5+A4. Adapted from Mondal et al.24
4.4. How to proceed after obtaining hits
Having obtained a validated hit, identified by de novo structure‐based drug design in combination with DCC and STD‐NMR, we have used a structure‐based design approach to improve the molecular recognition by the target.63 In this specific case, we were fortunate to have an X‐ray crystal structure of the target endothiapepsin in complex with the hit. If this is not the case, optimization is still possible, relying on structure–activity relationships.
Conclusions
There are a number of steps, which should be carefully taken into account, in order to obtain active hits by DCC. If information on the target is available, e.g. a crystal‐structure, one could consider a structure‐based design when choosing the building blocks. The type of reversible linkage to be used can be chosen at this stage. Conditions necessary for the equilibration to take place should be compatible with the target. After establishing conditions, which will ensure the target remains folded, the actual DCC experiment can be started. To do so, stock solutions of building blocks, catalyst and protein should be prepared. The formed DCLs can be analyzed by different techniques such as STD‐NMR or HPLC‐MS. Compounds that have been selected by the target, and their biochemical properties should be evaluated and possibly optimized in further studies.
Acknowledgements
Funding from Netherlands Organisation for Scientific Research (VIDI grant: 723.014.008; LIFT grant: 731.015.414) and from the Helmholtz Association's Initiative and Networking Fund is gratefully acknowledged. We thank Dr. Ravindra Jumde for fruitful discussions regarding this manuscript.
Biographies
Alwin M. Hartman studied Chemistry at the University of Groningen. In his Master's research, he synthesized inhibitors of the aspartic protease endothiapepsin in the Hirsch group. In September 2015, he started his PhD research in the same group, focussing on new applications of dynamic combinatorial chemistry to medicinal chemistry and chemical biology.

Robin M. Gierse studied Biochemistry at the University of Greifswald. He obtained his M.Sc. with a thesis on the synthesis of crosslink‐active microRNAs in the bio‐organic chemistry lab of Prof. S. Müller. Subsequently, he worked at the company Enzymicals as a junior scientist. He joined the Hirsch group in the fall of 2016 as a PhD student. His research focuses on the development of novel anti‐infectives and includes molecular and structural biology as well as computational drug design.

Anna Hirsch read Natural Sciences at the University of Cambridge and developed the double conjugate addition of dithiols to propargylic carbonyl systems in the group of Prof. Steven V. Ley. She received her Ph.D. with Prof. François Diederich from ETH Zurich in 2008 on de novo design and synthesis of the first inhibitors of an anti‐infective target. After a postdoc in the group of Prof. Jean‐Marie Lehn in Strasbourg, she took up a position as assistant professor at the Stratingh Institute for Chemistry at the University of Groningen in 2010 and was promoted to associate professor in 2015. In 2017, she became head of the department for drug design and optimization at the HIPS. Her work focuses on rational approaches to drug design, including structure‐ and fragment‐based drug design in combination with dynamic combinatorial chemistry and kinetic target‐guided synthesis, focusing on anti‐infective targets

References
- 1. Lam K. S., Salmon S. E., Hersh E. M., Hruby V. J., Kazmierski W. M. and Knapp R. J., Nature, 1991, 354, 82–84. [DOI] [PubMed] [Google Scholar]
- 2. Houghten R. A., Pinilla C., Blondelle S. E., Appel J. R., Dooley C. T. and Cuervo J. H., Nature, 1991, 354, 84–86. [DOI] [PubMed] [Google Scholar]
- 3. Houghten R. A., Proc. Natl. Acad. Sci. USA, 1985, 82, 5131–5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Geysen H. M., Meloen R. H. and Barteling S. J., Proc. Natl. Acad. Sci. USA 1984, 81, 3998 LP‐4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Frank R., Heikens W., Heisterberg‐Moutsis G. and Blöcker H., Nucl. Acid. Res., 1983, 11, 4365–4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huc I. and Lehn J.‐M., Proc. Natl. Acad. Sci. USA, 1997, 94, 2106–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ohlmeyer M. H., Swanson R. N., Dillard L. W., Reader J. C., Asouline G., Kobayashi R., Wigler M. and Still W. C., Proc. Natl. Acad. Sci. USA 1993, 90, 10922 LP1‐0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rowan S. J., Lukeman P. S., Reynolds D. J. and Sanders J. K. M., New J. Chem., 1998, 22, 1015–1018. [Google Scholar]
- 9. Polyakov V. A., Nelen M. I., Nazarpack‐Kandlousy N., Ryabov A. D. and Eliseev A. V., J. Phys. Org. Chem., 1999, 12, 357–363. [Google Scholar]
- 10. Ganesan A., Angew. Chem. Int. Ed., 1998, 37, 2828–2831; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 1998, 110, 2989. [Google Scholar]
- 11. Karan C. and Miller B. L., Drug Discovery Today, 2000, 5, 67–75. [DOI] [PubMed] [Google Scholar]
- 12. Bhat V. T., Caniard A. M., Luksch T., Brenk R., Campopiano D. J. and Greaney M. F., Nat. Chem., 2010, 2, 490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fang Z., He W., Li X., Li Z., Chen B., Ouyang P. and Guo K., Bioorg. Med. Chem. Lett., 2013, 23, 5174–5177. [DOI] [PubMed] [Google Scholar]
- 14. Demetriades M., Leung I. K. H., Chowdhury R., Chan M. C., McDonough M. A., Yeoh K. K., Tian Y.‐M., Claridge T. D. W., Ratcliffe P. J., Woon E. C. Y., et al., Angew. Chem. Int. Ed., 2012, 51, 6672–6675; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2012, 124, 6776. [Google Scholar]
- 15. Woon E. C. Y., Demetriades M., Bagg E. A. L., Aik W., Krylova S. M., Ma J. H. Y., Chan M., Walport L. J., Wegman D. W., Dack K. N., et al., J. Med. Chem., 2012, 55, 2173–2184. [DOI] [PubMed] [Google Scholar]
- 16. Sakai S., Shigemasa Y. and Sasaki T., Tetrahedron Lett., 1997, 38, 8145–8148. [Google Scholar]
- 17. Lins R. J., Flitsch S. L., Turner N. J., Irving E. and Brown S. A., Angew. Chem. Int. Ed., 2002, 41, 3405–3407; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2002, 114, 3555. [Google Scholar]
- 18. Lins R. J., Flitsch S. L., Turner N. J., Irving E. and Brown S. A., Tetrahedron, 2004, 60, 771–780. [Google Scholar]
- 19. Shi B., Stevenson R., Campopiano D. J. and Greaney M. F., J. Am. Chem. Soc., 2006, 128, 8459–8467. [DOI] [PubMed] [Google Scholar]
- 20. Caraballo R., Dong H., Ribeiro J. P., Jiménez‐Barbero J. and Ramström O., Angew. Chem. Int. Ed., 2010, 49, 589–593; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2010, 122, 599. [Google Scholar]
- 21. Frei P., Hevey R. and Ernst B., Chem. Eur. J., 2019, 25, 60–73. [DOI] [PubMed] [Google Scholar]
- 22. Mondal M. and Hirsch A. K. H., Chem. Soc. Rev., 2015, 44, 2455–2488. [DOI] [PubMed] [Google Scholar]
- 23. Van der Vlag R., Hirsch A. K. H., in: Compr. Supramol. Chem. 2, Elsevier, 2017, pp. 487–509. [Google Scholar]
- 24. Mondal M., Radeva N., Köster H., Park A., Potamitis C., Zervou M., Klebe G. and Hirsch A. K. H., Angew. Chem. Int. Ed., 2014, 53, 3259–3263; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2014, 126, 3324. [Google Scholar]
- 25. Dirksen A., Dirksen S., Hackeng T. M. and Dawson P. E., J. Am. Chem. Soc., 2006, 128, 15602–15603. [DOI] [PubMed] [Google Scholar]
- 26. Dirksen A., Hackeng T. M. and Dawson P. E., Angew. Chem. Int. Ed., 2006, 45, 7581–7584; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2006, 118, 7743. [Google Scholar]
- 27. Crisalli P. and Kool E. T., J. Org. Chem., 2013, 78, 1184–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reddavide F. V., Lin W., Lehnert S. and Zhang Y., Angew. Chem. Int. Ed., 2015, 54, 7924–7928; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2015, 127, 8035. [Google Scholar]
- 29. Monjas L., Swier L. J. Y. M., Setyawati I., Slotboom D. J. and Hirsch A. K. H., ChemMedChem, 2017, 12, 1693–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kern F. T. and Wanner K. T., ChemMedChem, 2015, 10, 396–410. [DOI] [PubMed] [Google Scholar]
- 31. Kern F. and Wanner K. T., Bioorg. Med. Chem., 2019, 27, 1232–1245. [DOI] [PubMed] [Google Scholar]
- 32.see ref.21
- 33. Artigas G., López‐Senín P., González C., Escaja N. and Marchán V., Org. Biomol. Chem., 2015, 13, 452–464. [DOI] [PubMed] [Google Scholar]
- 34. McAnany J. D., Reichert J. P. and Miller B. L., Bioorg. Med. Chem., 2016, 24, 3940–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang Z., Fang Z., He W., Wang Z., Gan H., Tian Q. and Guo K., Bioorg. Med. Chem. Lett., 2016, 26, 1671–1674. [DOI] [PubMed] [Google Scholar]
- 36. Mondal M., Radeva N., Fanlo‐Virgós H., Otto S., Klebe G. and Hirsch A. K. H., Angew. Chem. Int. Ed., 2016, 55, 9422–9426; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem., 2016, 128, 9569. [Google Scholar]
- 37. Frei P., Pang L., Silbermann M., Eris D., Mühlethaler T., Schwardt O. and Ernst B., Chem. Eur. J., 2017, 23, 11570–11577. [DOI] [PubMed] [Google Scholar]
- 38. Fu J., Fu H., Dieu M., Halloum I., Kremer L., Xia Y., Pan W. and Vincent S. P., Chem. Commun., 2017, 53, 10632–10635. [DOI] [PubMed] [Google Scholar]
- 39. Soubhye J., Gelbcke M., Van Antwerpen P., Dufrasne F., Boufadi M. Y., Nève J., Furtmüller P. G., Obinger C., Zouaoui Boudjeltia K. and Meyer F., ACS Med. Chem. Lett., 2017, 8, 206–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ekström A. G., Wang J. T., Bella J. and Campopiano D. J., Org. Biomol. Chem., 2018, 16, 8144–8149. [DOI] [PubMed] [Google Scholar]
- 41. Das M., Yang T., Dong J., Prasetya F., Xie Y., Wong K. H. Q., Cheong A. and Woon E. C. Y., Chem. Asian J., 2018, 13, 2854–2867. [DOI] [PubMed] [Google Scholar]
- 42. García P., Alonso V. L., Serra E., Escalante A. M. and Furlan R. L. E., ACS Med. Chem. Lett., 2018, 9, 1002–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jana S., Panda D., Saha P., Pantos̨ G. D. and Dash J., J. Med. Chem., 2019, 62, 762–773. [DOI] [PubMed] [Google Scholar]
- 44. Clipson A. J., Bhat V. T., McNae I., Caniard A. M., Campopiano D. J. and Greaney M. F., Chem. Eur. J., 2012, 18, 10562–10570. [DOI] [PubMed] [Google Scholar]
- 45. Sindelar M. and Wanner K. T., ChemMedChem, 2012, 7, 1678–1690. [DOI] [PubMed] [Google Scholar]
- 46. Nguyen R. and Huc I., Chem. Commun., 2003, 942–943. [DOI] [PubMed] [Google Scholar]
- 47. Erlanson D. A., Braisted A. C., Raphael D. R., Randal M., Stroud R. M., Gordon E. M. and Wells J. A., Proc. Natl. Acad. Sci. USA, 2000, 97, 9367–9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ludlow R. F. and Otto S., J. Am. Chem. Soc., 2008, 130, 12218–12219. [DOI] [PubMed] [Google Scholar]
- 49. Shi B. and Greaney M. F., Chem. Commun., 2005, 886–888. [DOI] [PubMed] [Google Scholar]
- 50.see ref.14
- 51. Leung I. K. H., Brown T., Schofield C. J. and Claridge T. D. W., Medchemcomm, 2011, 2, 390–395. [Google Scholar]
- 52. Newman J., Acta Crystallogr., Sect. D, 2004, 60, 610–612. [DOI] [PubMed] [Google Scholar]
- 53. Reinhard L., Mayerhofer H., Geerlof A., Mueller‐Dieckmann J. and Weiss M. S., Acta Crystallogr., Sect. F Struct. Biol. Cryst. Commun., 2013, 69, 209–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Niesen F. H., Berglund H. and Vedadi M., Nat. Protoc., 2007, 2, 2212–2221. [DOI] [PubMed] [Google Scholar]
- 55.G. S. Sittampalam, N. P. Coussens, K. Brimacomber, A. Grossman, M. Arkin, D. Auld, C. Austin, J. Baell, B. Bejcek, J. M. M, Caaveiro, et al. 2004.
- 56. Arakawa T., Kita Y. and Timasheff S. N., Biophys. Chem., 2007, 131, 62–70. [DOI] [PubMed] [Google Scholar]
- 57. Rammler D. H., Ann. N. Y. Acad. Sci., 1967, 141, 291–299. [DOI] [PubMed] [Google Scholar]
- 58. Jackson M. and Mantsch H. H., Biochim. Biophys. Acta ‐ Protein Struct. Mol. Enzymol., 1991, 1078, 231–235. [DOI] [PubMed] [Google Scholar]
- 59. Viegas A., Manso J., Nobrega F. L. and Cabrita E. J., J. Chem. Educ., 2011, 88, 990–994. [Google Scholar]
- 60. Danieli B., Giardini A., Lesma G., Passarella D., Peretto B., Sacchetti A., Silvani A., Pratesi G. and Zunino F., J. Org. Chem., 2006, 71, 2848–2853. [DOI] [PubMed] [Google Scholar]
- 61. Qiu C., Fang Z., Zhao L., He W., Yang Z., Liu C. and Guo K., React. Chem. Eng., 2019, 4, 658–662. [Google Scholar]
- 62. Jaegle M., Wong E. L., Tauber C., Nawrotzky E., Arkona C. and Rademann J., Angew. Chem. Int. Ed., 2017, 56, 7358–7378; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem., 2017, 129, 7464. [Google Scholar]
- 63. Hartman A. M., Mondal M., Radeva N., Klebe G. and Hirsch A. K., Int. J. Mol. Sci. 2015, 16, 10.3390/ijms160819184 [DOI] [PMC free article] [PubMed] [Google Scholar]