

SUMMARY
Two types of monocytes, Ly6Chi and Ly6Clo, infiltrate the heart in murine experimental autoimmune myocarditis (EAM). We discovered a role for cardiac fibroblasts in facilitating monocyte-to-macrophage differentiation of both Ly6Chi and Ly6Clo cells, allowing these macrophages to perform divergent functions in myocarditis progression. During the acute phase of EAM, IL-17A is highly abundant. It signals through cardiac fibroblasts to attenuate efferocytosis of Ly6Chi monocyte-derived macrophages (MDMs) and simultaneously prevents Ly6Clo monocyte-to-macrophage differentiation. We demonstrated an inverse clinical correlation between heart IL-17A levels and efferocytic receptor expressions in humans with heart failure (HF). In the absence of IL-17A signaling, Ly6Chi MDMs act as robust phagocytes and are less proinflammatory, whereas Ly6Clo monocytes resume their differentiation into MHCII+ macrophages. We propose that MHCII+Ly6Clo MDMs are associated with the reduction of cardiac fibrosis and prevention of the myocarditis sequalae.
In Brief
Hou et al. show that cardiac fibroblasts facilitate infiltrating Ly6Chi and Ly6Clo monocytes to become macrophages. IL-17A trans-signaling through cardiac fibroblasts increases MerTK shedding and promotes a pro-inflammatory and pro-tissue remodeling gene expression profile in Ly6Chi monocyte-derived macrophages. Paradoxically, IL-17A signaling through cardiac fibroblasts can substantially inhibit Ly6Clo monocyte-to-macrophage differentiation.
Graphical Abstract
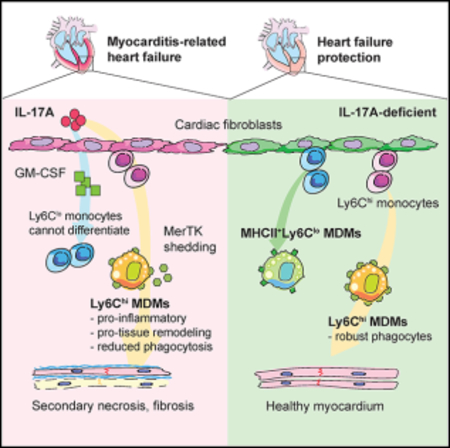
INTRODUCTION
Myocarditis remains a leading cause of heart failure (HF) in children and young adults (Caforio et al., 2013a; Dimas et al., 2009). A recent global estimate of myocarditis incidence is approximately 1.5-million cases annually (Kang and An, 2018). However, the actual myocarditis incidence could be significantly underestimated due to the heterogeneous clinical manifestations and a wide spectrum of disease presentations (Fung et al., 2016). Between 9% and 16% of patients with myocarditis develop dilated cardiomyopathy (DCM). Yet no biomarkers are currently available to identify myocarditis patients at risk of developing DCM (Fairweather et al., 2014; Hochholzer et al., 2010; Mason et al., 1995). To date, treatment of myocarditis and prevention of its sequelae, DCM, remain vital goals in the quest to reduce morbidity and mortality in patients.
In the experimental autoimmune myocarditis (EAM) model as well as human clinical studies, we and others have reported that monocytes and macrophages comprise approximately three-quarters of the infiltrating cells in the injured myocardium (Barin et al., 2012; Theaker et al., 1985). Cardiac tissue-resident macrophages originate from precursors that develop in the embryonic yolk sac and fetal liver and maintain locally as self-renewing populations to perform tissue-specific functions. After birth, a proportion of embryonically derived cardiacresident macrophages are progressively replaced by monocyte-derived macrophages (MDMs) that originate in the bone marrow (BM) (Epelman et al., 2014; Guilliams et al., 2014; Hashimoto et al., 2013; Molawi et al., 2014; Schulz et al., 2012; Yona et al., 2013). BM-derived circulating monocytes in mice consist of two subsets: the “classical” inflammatory Ly6Chi monocytes and the “non-classical” patrolling Ly6Clo monocytes. The former resemble human CD14hiCD16− and/or CD14+CD16+ monocytes, while the latter have similar characteristics to human CD14−CD16hi monocytes (Carlin et al., 2013; Geissmann et al., 2003; Gordon and Taylor, 2005; Ingersoll et al., 2010; Jakubzick et al., 2013; Shi and Pamer, 2011; Sunderkötter et al., 2004; Ziegler-Heitbrock, 2007). Cardiac injury triggers massive Ly6Chi monocytes mobilization, trafficking, and extravasation into the heart, where they promote inflammation associated with tissue repair and remodeling (Bronte and Pittet, 2013; Dutta et al., 2012; Leuschner et al., 2012; Robbins et al., 2012; Epelman et al., 2014; Heidt et al., 2014; Hilgendorf et al., 2014).
It is challenging to distinguish tissue-resident macrophages from recruited MDMs when they co-exist in the same inflammatory niche. Ly6Chi monocytes are known precursors to cardiac macrophages during cardiac injury (Epelman et al., 2014; Nahrendorf et al., 2007). However, the contribution of patrolling Ly6Clo monocytes to the macrophage pool is controversial. Ablation of Ly6Clo monocytes in a mouse model of myocardial infarction (MI) resulted in depletion of other monocyte and macrophage populations (Nahrendorf et al., 2007). Moreover, it is unclear whether the phenotype and function of either Ly6Chi or Ly6Clo MDMs differ from one another. The mechanism through which extrinsic stimuli influence monocytes survival, proliferation, differentiation, and function in the context of cardiac damage and inflammation is not understood.
We previously identified an important role of IL-17A in driving myocarditis progression to DCM (Baldeviano et al., 2010). IL-17A-deficient or IL-17 receptor alpha (IL17Ra)-deficient mice developed myocarditis with similar severity as wild-type (WT) animals. Remarkably, they have preserved cardiac functions and were protected from myocardial fibrosis and DCM (Baldeviano et al., 2010). We also reported that anti-IL-17A treatment during EAM onset can prevent DCM development in WT mice (Baldeviano et al., 2010). IL-17A signaling to cardiac fibroblasts results in the induction of granulocyte-macrophage colony-stimulating factor (GM-CSF) and CCL2, which drives Ly6Chi monocyte chemotaxis and accumulation in the heart, worsening DCM outcomes (Wu et al., 2014; Baldeviano et al., 2010). The pathogenic role of IL-17A in the development of cardiac fibrosis and HF has also been confirmed in MI (Chen et al., 2018).
To better understand the underlying mechanisms of how recruited monocytes drive DCM, we fate-mapped the accumulating monocyte subsets in the heart during myocardial inflammation. We found that IL-17A-activated cardiac fibroblasts significantly arrested Ly6Clo monocyte-to-macrophage differentiation and proliferation. We also discovered that IL-17A signaling through cardiac fibroblasts can downregulate efferocytosis receptors expressed by Ly6Chi MDMs, negatively affecting their phagocytic function during inflammation resolution. Furthermore, both intrinsic and extrinsic factors modulate distinct transcriptomic profiles of Ly6Chi and Ly6Clo MDMs, signifying their potential functional differences in the context of myocarditis. Our work provides evidence that cardiac fibroblasts play a decisive role in MDM ontogeny and function during cardiac injury. Our findings provide insights into monocyte and macrophage biology and their roles in the context of chronic cardiac inflammation.
RESULTS
Infiltrating Monocytes Contribute to Three Subsets of Macrophages during Myocarditis
We created parabiotic mice to elucidate the contribution of infiltrating monocytes to cardiac macrophage populations during myocarditis development. The circulations of CD45.1 and CD45.2 mice were surgically joined, and EAM was induced in CD45.1 mice 2 days prior to parabiosis (Figure 1A). On day 21 of EAM, we confirmed by histology that only CD45.1 parabionts developed severe myocardium infiltration (Figure 1B). Flow cytometric analysis showed a significantly elevated total number of infiltrating non-neutrophilic myeloid cells in the CD45.1 EAM hearts compared to their CD45.2 parabiotic partners (Figure 1C). We also observed that CD45.1 EAM hearts had more total F4/80hiCD64+ macrophages compared to non-EAM CD45.2 hearts, indicating cardiac macrophage expansion in response to inflammation (Figure 1D). We found a higher frequency of CD45.2+ MDMs in the CD45.1 EAM mice, compared to CD45.1+ MDMs in the CD45.2 non-EAM mice (Figures 1E and 1F; see the gating strategy in Figure S1A). However, a higher frequency of CD45.2+ MDMs in the CD45.1 EAM mice did not reflect the increase in infiltrating monocyte populations (Figure 1E). In addition, the heart Ly6Chi-to-Ly6Clo monocyte ratio between parabionts did not differ significantly (Figure 1G). Resident tissue macrophages can be further divided into three subsets, based on their expressions of CCR2 and MHCII (Epelman et al., 2014). We found that infiltrating CD45.2+ monocytes predominantly differentiated into CCR2+MHCII+ macrophages, though they possess the capacity to replenish all three macrophage subsets in the EAM hearts (Figures 1E and 1H). However, the heart-infiltrating CD45.1+ monocytes in the non-EAM CD45.2 mice differentiated only into MHCII+ macrophages (Figures 1E and 1H). Taken together, our data support the idea that during cardiac inflammation infiltrating monocytes readily differentiate into macrophages and these monocyte-derived cells can replenish all three macrophage subpopulations.
Figure 1. Infiltrating Monocytes Contribute to Three Subsets of Macrophages during Myocarditis.

(A) Schematics of parabiosis mice.
(B) Representative images of H&E-stained heart sections of the median CD45.1 (day 21 EAM) and CD45.2 (non-EAM) mice parabionts. Scale bars: 100 μm.
(C) Comparison of total grafted CD11b+Ly6G−myeloid cell counts between parabionts.
(D) Comparison of total cardiac macrophage counts between parabionts.
(E) Flow cytometry plots showing (top left) percentage of CD45.2+CD11b+Ly6G−Lineage (CD3e, B220, NKp46, CD90.2, and Ter119)− grafted cells infiltrating the CD45.1 EAM hearts; (top middle) percentage of grafted cells in the hearts that differentiated into macrophages or remained as monocytes; (top right) percentage of grafted MDM subsets; (bottom left) percentage of CD45.1+CD11b+Ly6G−Lineage grafted cells infiltrating the CD45.2 non-EAM hearts; (bottom middle) percentage of grafted cells in the hearts that differentiated into macrophages or remained as monocytes; and (bottom right) percentage of grafted MDM subsets.
(F) Percentages of grafted MDMs out of total number of grafted CD11b+Ly6G−Lineage− myeloid cells.
(G) Percentages of grafted Ly6Chi and Ly6Clo cells out of total number of grafted F4/80−CD64+ monocytes.
(H) Comparison of grafted MDM subsets defined by CCR2 and MHCII expressions between parabionts. Data are representative of two independent experiments with biological triplicates. n = 3.
(C, D, F, G) Groups were compared using Student’s t test. *p < 0.05. All data were presented as mean ± SD.
See also Figure S1.
During cardiac inflammation, mature Ly6Chi monocytes arise from common monocyte progenitors (cMoPs) in the BM before trafficking to the heart (Hettinger et al., 2013). We examined the mature monocyte population in the BM in response to cardiac inflammation (see the gating strategy in Figure S1B). We found a substantial presence of CD45.1+-grafted cells in the CD45.2 parabiont BM, whereas CD45.2+ monocytes were relatively rare in the CD45.1 EAM parabiont BM (Figures S1C and S1D). We speculated that this could be due to undifferentiated CD45.1+ Ly6Chi monocytes homing back to the CD45.1 BM in the absence of inflammation (Sunderkötter et al., 2004). Alternatively, a systemic response to adjuvants used for EAM induction in CD45.1+ parabionts could increase monocyte infiltration into non-EAM parabiont BM. Another possible explanation is that EAM BM is saturated with inflammatory cells, which prevented CD45.2+ monocytes from infiltrating CD45.1 BM. Nevertheless, the ratio of Ly6Chi-to-Ly6Clo monocytes was highly comparable between parabionts in the BM (Figure S1E). Collectively, the similarity of monocyte ratios in the BM between EAM and naive mice suggests the local cardiac inflammatory milieu is instrumental in monocyte-to-macrophage differentiation in the heart.
Cardiac Fibroblasts Facilitate Ly6Chi and Ly6Clo Monocyte-to-Macrophage Differentiation
The results from the parabiosis experiments suggested that MDMs in an inflamed cardiac environment are a highly heterogenous population. Whether macrophage heterogeneity is instigated by monocyte intrinsic differences and/or extrinsic environmental factors is not yet known. Cardiac fibroblasts play a key sentinel role during myocarditis as they produce myelotropic chemokines and cytokines such as GM-CSF and CCL2 (Baldeviano et al., 2010; Wu et al., 2014). This immune modulatory property of cardiac fibroblasts was also confirmed in MI and Kawasaki disease (Amoah et al., 2015; Chen et al., 2018). To examine whether Ly6Chi and Ly6Clo monocytes can differentiate into macrophages and whether cardiac fibroblasts facilitate this differentiation process, we established an in vitro co-culture system. We harvested primary cardiac fibroblasts from naive WT mice and co-cultured them with fluorescence-activated cell sorting (FACS)-sorted IL-17Ra−/− EAM splenic Ly6Chi or Ly6Clo monocytes for up to 160 h (Figure 2A; see the sorting strategy in Figure S2A). Due to the relative paucity of monocytes in blood, we used spleens as a surrogate source of monocytes. Flow cytometric analysis showed that approximately 80% of the Ly6Chi monocytes quickly differentiated to MDMs, while almost all Ly6Clo monocytes remained undifferentiated over the first 40 h of culture (Figures 2B and 2C). However, when we assessed their phenotypic changes at 160 h, approximately 30% of the Ly6Clo monocytes had become macrophages (Figures 2D and 2E). Neither Ly6Chi nor Ly6Clo monocytes were able to survival long term nor differentiate into macrophages in the absence of cardiac fibroblasts (Figures 2D and 2E). Ly6Chi and Ly6Clo MDMs were morphologically similar and resembled macrophages after 160 h of co-culture with cardiac fibroblasts (Figure 2F). To better understand how monocytes and cardiac fibroblasts interact in vitro, we used live-cell time-lapsed imaging to track carboxyfluorescein succinimidyl ester (CFSE)-labeled (CFSE+) Ly6Chi and Ly6Clo monocytes co-cultured with CFSE− cardiac fibroblasts. We found that CFSE+Ly6Chi monocytes started to cluster around cardiac fibroblasts as early as 9 h into co-culture. They adhered strongly to the cardiac fibroblasts and became static when adhesions formed (Figure 2G; Video S1). In contrast, CFSE+Ly6Clo monocytes were motile and they formed transient clusters around cardiac fibroblasts. Ly6Clo monocytes also lost CFSE at a higher rate, indicating faster proliferation when compared to Ly6Chi monocytes (Figure 2H; Video S2). Flow cytometric analysis confirmed that Ly6Clo monocytes indeed proliferate faster than Ly6Chi monocytes (Figures S2B and S2C). Next, we used transmission electron microscopy (EM) to examine whether MDMs and cardiac fibroblasts establish cell-to-cell connections, which would suggest the presence of molecular adhesion structures. Both Ly6Chi and Ly6Clo MDMs contained numerous cytoplasmic electron-lucent vesicles (Figures 2I and 2J). They each formed close cell-to-cell contact with cardiac fibroblasts (Figures 2K and 2L). Interestingly, these close cell-to-cell interactions are important for driving monocyte-to-macrophage differentiation. We demonstrated a significant reduction in monocyte-to-macrophage differentiation when monocyte-fibroblast contacts are inhibited by a transwell barrier (Figures S2D and S2E). We also showed that Ly6Chi monocytes induced cardiac fibroblasts to significantly upregulate Ccl2 mRNA levels compared to Ly6Clo monocytes, ensuring a feed-forward loop for a continuous supply of pro-inflammatory monocytes to accumulate in an inflamed microenvironment (Figure S2F). Taken together, Ly6Chi and Ly6Clo monocytes establish close physical contact with cardiac fibroblasts and these cell-to-cell interactions play a role in facilitating Ly6Chi and Ly6Clo monocytes survival and differentiation into macrophages.
Figure 2. Cardiac Fibroblasts Facilitate Ly6Chi and Ly6Clo Monocyte-to-Macrophage Differentiation.

(A) Schematics of the monocytes and cardiac fibroblasts co-culture system. Cardiac fibroblasts were harvested from WT naive mice, whereas monocytes weresorted from EAM IL-17Ra−/− mice.
(B) Flow cytometric analysis showing differentiation of viable Ly6Chi and Ly6Clo MDMs expressing F4/80hiCD64+ at 40 h.
(C) Frequency of macrophage differentiation at 40 h.
(D) Differentiation of viable Ly6Chi and Ly6Clo monocytes into MDMs at 160 h.
(E) Frequency of macrophage differentiation at 160 h.
(F) Giemsa staining of monocytes morphologies before culture and after 160 h co-culture with cardiac fibroblasts. Scale bars: (black) 8 μm.
(G and H) IncuCyte results showing CFSE+Ly6Chi (G) and CFSE+Ly6Clo (H) monocytes are in close contact with cardiac fibroblasts. Scale bars: (white) 50 μm.
(I–L) Representative EM images taken at 160 h of co-culture showing individual (I) Ly6Chi and (J) Ly6Clo MDMs and (K) Ly6Chi and (L) Ly6Clo MDMs interacting with cardiac fibroblasts. Scale bars: (white) 2 μm; (black) 2 μm.
(A–F) Data are representative of five independent experiments with technical triplicates. (C and E) n = 3. Groups were compared using one-way ANOVA followed by Tukey test. ****p < 0.0001. All data were presented as mean ± SD.
Inflammatory Monocytes Are the Main Precursors for MDMs during Myocarditis in Both Mice and Humans
We next sought to investigate whether both Ly6Chi and Ly6Clo monocytes could differentiate in vivo during EAM development. We FACS-sorted splenic CD45.2+Ly6Chi or Ly6Clo monocytes from WT mice on day 14 of EAM, and intracardially injected them into CD45.1 WT recipients at the peak of EAM (Figure 3A). Both Ly6Chi and Ly6Clo monocytes and MDMs of the donor origin persisted in the myocardium 40 h after the injection (Figures 3B and 3C). Approximately 50% of the injected Ly6Chi monocytes had differentiated into F4/80hiCD64+ macrophages (Figures 3D and 3E), whereas less than 1% of the Ly6Clo monocytes differentiated into macrophages (Figures 3D and 3F). We employed Barnes-Hut stochastic neighbor embedding (bhSNE) analysis to visualize monocyte-to-macrophage differentiation (Amir et al., 2013; Becher et al., 2014). We confirmed that CD45.2+Ly6Clo monocytes were mostly unable to differentiate into macrophages, while CD45.2+ Ly6Chi monocytes contributed significantly to the cardiac macrophage pool during EAM (Figures 3G and 3H). We also examined Ly6Chi and Ly6Clo monocytes differentiation at 160 h in vivo and found that very few injected monocytes were present in the myocardium, indicating a high local turnover rate. Notably, we did not find evidence suggesting Ly6Clo monocyte-to-macrophage differentiation (data not shown). Upon immunofluorescence examination of endomyocardial biopsy samples from a patient with giant cell myocarditis, we found the expression of CD68, a pan-macrophage marker, coincided with CD14 but not CD16. This suggests that the inflammatory CD14+CD16−/+ but not the patrolling CD14−CD16+ monocyte populations are the likely source of macrophages in giant cell myocarditis (Figures S3A and S3B). In parallel, we examined endomyocardial biopsies from three patients who had left ventricular assist devices implanted as a result of HF with various etiologies (Table S1). Flow cytometric analysis from all three patients indicated that CD14+CD68+, but not CD16+CD68+, macrophages, are present in the heart (Figure S3C). This further supports the hypothesis that CD14+CD16− monocytes are the main source of cardiac macrophages in human myocarditis. In conclusion, human CD14+ and mouse Ly6Chi monocyte subsets are the major contributors to the cardiac macrophage population during myocarditis development.
Figure 3. Inflammatory Monocytes Are the Main Precursors for MDMs during Myocarditis in Both Mice and Humans.

(A) Schematics of intracardiac injection of Ly6Chi or Ly6Clo CD45.2+ splenic monocytes into the hearts of CD45.1 day 21 EAM WT recipient mice.
(B and C) Gating of concatenated Ly6Chi (B) and Ly6Clo (C) donor cells from the total viable CD115+CD11b+ population.
(D) Percentages of injected Ly6Chi or Ly6Clo monocytes differentiated into macrophages.
(E and F) Flow cytometric analysis of the frequencies of Ly6Chi (E) and Ly6Clo (F) MDMs out of a viable CD45.2+CD115+CD11b+ population.
(G and H) F4/80 and CD64 expression intensities of Ly6Chi (G) and Ly6Clo (H) MDMs using a bh-SNE-dimensional reduction algorithm.
(D) Data are representative of three independent experiments. n = 3–4. Groups were compared using Student’s t test. **p < 0.01. All data were presented as mean ± SD.
IL-17A Signaling through Cardiac Fibroblasts Inhibits Ly6Clo Monocyte-to-Macrophage Differentiation and Ly6Clo Proliferation
EAM is a Th17-driven disease and IL-17A induces the production of myelotropic cytokines and chemokines by cardiac fibroblasts (Wu et al., 2014). The differentiation capacity of Ly6Clo monocytes in vivo appeared to be limited, and we therefore speculated that IL-17A plays a role in determining monocyte fate during EAM. Since monocytes require cardiac fibroblasts to differentiate into macrophages, we co-cultured FACS-sorted IL-17Ra−/− EAM Ly6Chi and Ly6Clo splenic monocytes with either cardiac fibroblasts alone or IL-17A-stimulated cardiac fibroblasts for 160 h. We found that IL-17A-stimulated cardiac fibroblasts significantly inhibited Ly6Clo monocyte-to-macrophage differentiation, while its effect on Ly6Chi monocyte differentiation was minimal (Figure 4A). Using CFSE labeling, we found that proliferation of Ly6Clo monocytes was almost completely inhibited by cardiac fibroblasts stimulated with IL-17A, whereas proliferation of Ly6Chi monocytes was minimally affected (Figure 4B). We described previously that cardiac fibroblasts are potent producers of GM-CSF upon IL-17A stimulation (Wu et al., 2014). We found that while treatment with recombinant GM-CSF only marginally suppressed Ly6Chi monocyte-to-macrophage differentiation (Figure 4C), it completely recapitulated the inhibitory effect of IL-17A through cardiac fibroblasts on Ly6Clo monocyte-to-macrophage differentiation (Figure 4D). IL-17A-induced inhibition of Ly6Clo monocyte-to-macrophage differentiation could be reversed with anti-GM-CSF treatment (Figure 4D). Similarly, GM-CSF did not change the proliferation of Ly6Chi monocytes and MDMs, while completely inhibiting proliferation of undifferentiated Ly6Clo monocytes (Figures 4E and 4F). Treatment of anti-GM-CSF to IL-17A-stimulated cardiac fibroblasts restored proliferation of Ly6Clo monocytes (Figure 4F). We further validated all of our major in vitro findings using splenic monocytes from WT animals and showed that monocytes responded to environmental cues similarly regardless of their sources (Figure S4). To summarize, GM-CSF acts as a downstream mediator of IL-17A signaling, through cardiac fibroblasts, that exhibits minor inhibitory effects on Ly6Chi monocyte differentiation and proliferation, but substantially inhibits Ly6Clo monocyte differentiation and proliferation in vitro.
Figure 4. IL-17A Signaling through Cardiac Fibroblasts Inhibits Ly6Clo Monocyte-to-Macrophage Differentiation and Ly6Clo Proliferation.

Naive WT cardiac fibroblasts were co-cultured with monocytes sorted from EAM IL-17Ra−/− mice. All co-cultured cells were assessed using flow cytometry at 160 h.
(A) Gating of concatenated Ly6Chi and Ly6Clo MDMs out of viable CD45+CD11b+.
(B) Histograms of mean CFSE fluorescent intensity (MFI) of viable Ly6Chi cells and Ly6Clo cells after co-culturing with either cardiac fibroblasts or IL-17A-treated cardiac fibroblasts.
(C and D) Percentages of Ly6Chi (C) and Ly6Clo (D) MDMs when co-cultured with cardiac fibroblasts only, IL-17A-stimulated cardiac fibroblasts, recombinant GM-CSF supplemented cardiac fibroblasts, and IL-17A-stimulated cardiac fibroblasts treated with anti-GM-CSF, respectively.
(E and F) Histograms of CFSE MFI showing viable Ly6Chi MDMs (E) and Ly6Clo monocytes (F) in conditions described in (C) and (D).
(C and D) Data are representative of three independent experiments with technical triplicates. n = 3. Groups were compared using one-way ANOVA followed by Dunnett test. *p < 0.05; ****p < 0.0001. All data were presented as mean ± SD.
See also Figure S4.
The Absence of IL-17A Signaling Enables Ly6Clo Monocyte-to-Macrophage Differentiation In vivo
We next aimed to test in vivo whether eliminating IL-17A signaling during EAM would enable Ly6Clo monocytes to undergo differentiation. To begin, we intracardially injected either FACS-sorted CD45.2+Ly6Chi or Ly6Clo monocytes into CD45.1 IL17Ra−/− recipients during the peak of EAM (Figure 5A). Both monocyte subsets were present in the myocardium 40 h post injection (Figures 5B and 5C; see the monocyte gating strategy in Figure S5A). Strikingly, while most of the Ly6Chi monocytes differentiated into macrophages, approximately 30% of the Ly6Clo monocytes also differentiated into macrophages (Figures 5D–5F). The bh-SNE algorithm further confirmed a non-negligible proportion of Ly6Clo MDMs (Figures 5G and 5H). In addition, we examined whether the Ly6Clo monocytes were able to traffic through blood and differentiate in the myocardium. We retro-orbitally injected FACS-sorted CD45.1+Ly6Clo monocytes into either CD45.2 WT or IL17Ra−/− recipients during the peak of EAM (Figure 5I). CD45.1+Ly6Clo cells were found in the myocardium 40 h post injection in both WT and IL-17Ra−/− mice compared to mice with no adoptive transferred cells (Figures 5J and 5K). Consistent to our finding, approximately 30% of the Ly6Clo monocytes had differentiated into macrophages in IL17Ra−/− recipients but not in WT (Figures 5L and 5M). Therefore, our results indicate that the absence of IL-17A signaling enables Ly6Clo monocyte-to-macrophage differentiation, again, underlining the inhibitory role of IL-17A on Ly6Clo monocyte-to-macrophage differentiation.
Figure 5. The Absence of IL-17A Signaling Enables Ly6Clo Monocyte-to-Macrophage Differentiation In vivo.

(A) Schematics of intracardiac injection of CD45.2+Ly6Chi or CD45.2+Ly6Clo monocytes into CD45.1 day 21 EAM IL-17Ra−/− recipient mice.
(B and C) Gating of concatenated Ly6Chi (B) and Ly6Clo (C) donor cells from the total viable CD115+CD11b+ population.
(D) Percentages of injected Ly6Chi or Ly6Clo MDMs.
(E and F) Frequencies of Ly6Chi MDMs (E) and Ly6Clo MDMs (F) out of the viable CD45.2+CD115+CD11b+ population.
(G and H) F4/80 and CD64 expression intensities of Ly6Chi (G) and Ly6Clo (H) MDMs using bh-SNE-dimensional reduction algorithm.
(I) Schematics of retro-orbital injection of CD45.1+Ly6Clo monocytes into CD45.2 day 21 EAM WT or IL-17Ra−/− recipient mice.
(J) Gating of concatenated Ly6Clo donor cells from the total viable CD115+CD11b+ population in WT and in IL-17Ra−/− recipient ice.
(K) CD45.1+ monocytes were gated based on no injection control CD45.2+ mice.
(L) Percentages of CD45.1+ Ly6Clo MDMs in the heart.
(M) Frequencies of Ly6Clo MDMs out of the viable CD45.1+CD115+CD11b+ population in WT and in IL-17Ra−/− recipient mice. Data are representative of two independent experiments with biological triplicates.
(D and L) n = 3. Groups were compared using Student’s t test. *p < 0.05, **p < 0.01. All data were presented as mean ± SD.
See also Figure S5.
Distinct Gene Expression Profiles in Ly6Chi and Ly6Clo MDM Subsets Differentiated In vitro in the Presence of Untreated or IL-17A-Treated Cardiac Fibroblasts
To explore the molecular differences between these MDM subpopulations, we next characterized the changes in gene expression that occurred during Ly6Clo and Ly6Chi monocyte-to-macrophage differentiation after being co-cultured with cardiac fibroblasts (conditions 1 and 2). In addition, we also examined the effect of IL-17A-stimulated cardiac fibroblasts on Ly6Chi MDMs (condition 3). Since there were very few Ly6Clo MDMs in the co-culture with IL-17A-stimulated cardiac fibroblasts, this condition was not included for this analysis. We performed microarray-based transcriptomic profiling of RNA isolated from FACS-sorted macrophages derived from the three conditions in triplicate (see the gating strategy in Figure S6A). Gene expression profiles of the three MDM subsets were evaluated by principal-component analysis (PCA) and hierarchical clustering analysis (Figures 6A and 6B). Both analyses demonstrated distinct gene expression profiles for the three MDM subsets, suggesting that both the microenvironment and monocyte intrinsic properties are determining factors. Differential gene expression analysis showed that consistent with both PCA and hierarchical clustering analysis, Ly6Clo MDMs versus Ly6Chi MDMs from IL-17A-treated cardiac fibroblast culture had the greatest number of differentially expressed genes (n = 1,057). Whereas Ly6Clo MDMs versus Ly6Chi MDMs from non-treated cardiac fibroblasts culture had the lowest number of differentially expressed genes (n = 644) (Figure S6B). We found that IL-17A signaling through cardiac fibroblasts significantly upregulated genes encoding inflammatory chemokines, cytokines, growth factors, Il6/Stat3, and the nuclear factor κB (NFκB) pathway in Ly6Chi MDMs (Figures 6C–6E). Moreover, IL-17A trans-signaling through cardiac fibroblasts upregulated genes in Ly6Chi MDMs known to promote tissue fibrosis such as Osm, as well as genes related to extracellular matrix degradation including Mmp9 and Timp1 (Figure 6F). In contrast, Ly6Clo MDMs uniquely upregulated genes associated with class II antigen processing, including Cd74 and H2-Ab1 (Figure 6D). Flow cytometric analysis showed that in vitro Ly6Clo MDMs indeed expressed MHCII, strongly suggesting their antigen presentation capabilities (Figure 6G). Ly6Clo MDMs expressed a significantly higher level of MHCII than Ly6Chi MDMs after being injected intracardially into EAM IL17Ra−/− recipients (Figure S6C). Furthermore, MHCII+Ly6Clo MDMs contribute to the increased proportion of MHCII+ macrophage in IL17Ra−/− mice during the resolution phase of EAM at day 28 (Figure 6H). We also validated that Ly6Clo MDMs do not carry a dendritic cell signature (Figure S6D). IL-17A signaling through cardiac fibroblasts significantly upregulated many genes encoding both M1 and M2 markers in the Ly6Chi MDMs, highlighting that the classic M1 and M2 paradigm cannot simply characterize macrophage activation complexity during disease states (Figure S6E) (Martinez and Gordon, 2014; Nahrendorf and Swirski, 2016). We conclude that IL-17A trans-signaling through cardiac fibroblasts promotes pro-inflammatory and pro-tissue remodeling characteristics in Ly6Chi MDMs. Interestingly, Ly6Clo MDMs upregulate genes associated with antigen presentation and have elevated surface expression of MHCII both in vitro and in vivo. Our findings suggest that Ly6Chi and Ly6Clo MDMs represent distinct subsets that have unique roles and functions in the inflamed heart.
Figure 6. Distinct Gene Expression Profiles in Ly6Chi and Ly6Clo MDM Subsets Differentiated In vitro in the Presence of Untreated or IL-17A-Treated Cardiac Fibroblasts.

(A) PCA analysis of microarray experiments. The principal components and their fraction of overall variability of the data (%) are shown on the x, y, and z axes.
(B) Supervised hierarchical clustering highlighting differential gene expression profiles among three groups of in vitro MDMs, using a threshold of 2-fold change and p value < 0.05. Sample number scheme is identical to the legend in all heatmaps below.
(C–F) Heatmaps showing relative fold changes in (C) genes associated with cytokines and growth factors, (D) NFκB pathway and antigen presentation, (E) chemokines and immune modulating activities, and (F) collagen production and matrix remodeling.
(G) Gating of MHCII+CD64+ macrophages derived from monocyte-fibroblast co-culture. Frequencies of MHCII+ subset out of total in vitro co-culture MDMs were assessed by flow cytometry.
(H) Frequencies of MHCII+ out of F4/80hiCD64+ macrophages were assessed by flow cytometry of the hearts of day 28 EAM mice.
(B–F) All genes displayed on the heatmaps have one-way ANOVA p value < 0.05 among groups compared. Data are representative of three independent experiments with technical triplicates. (G) n = 3. (H) n = 8 – 9. (G) Groups were compared using one-way ANOVA followed by Dunnett test. **p < 0.01. (H) Groups were compared using Student’s t test. **p < 0.01. All data were presented as mean ± SD.
See also Figure S6.
IL-17A Trans-Signaling through Cardiac Fibroblasts Downregulates MerTK Expression on Monocytes and MDMs
The clearance of apoptotic cells during inflammation resolution is critical for proper wound repair and tissue remodeling. The myeloid-epithelial-reproductive receptor tyrosine kinase (MerTK) is a major apoptotic cell receptor on macrophages, known to play a role in the clearance of dying cells, a process called efferocytosis (Scott et al., 2001; Zizzo et al., 2012). Efferocytosis is also known to induce an anti-inflammatory phenotype in macrophages and diminish proinflammatory cytokine release (Bosurgi et al., 2017; Camenisch et al., 1999; Healy et al., 2016). EM results showed engulfed apoptotic cells or large cell debris inside Ly6Chi MDMs (Figures 7A and 7B). Their phagocytic index was significantly elevated compared to the Ly6Clo MDMs derived from the co-culture (Figure 7C). This indicates that Ly6Chi MDMs are professional phagocytes. Consistent with our finding that IL-17A modulates the replenishment of the tissue macrophage pool with circulating monocytes, we observed an increase in the percentage of macrophages in IL-17Ra−/− EAM hearts compared to WT EAM hearts (Figure 7D; see the gating strategy in Figure S7A). In general, macrophages express considerably higher levels of MerTK than monocytes in the hearts (Figure S7B). We found that both monocytes and macrophages express higher levels of MerTK in IL17Ra−/− hearts compared to WT (Figures 7E and 7F). This suggests that IL-17A plays a role in downregulating MerTK expression on monocytes and macrophages. Furthermore, we found significantly less soluble Mer (sMer) present in IL17Ra−/− mice sera than WT animals, indicating that IL-17A might influence the rate at which MerTK is proteolytically cleaved from the surface of the cell (Figure 7G). In order to confirm that IL-17A signaling through cardiac fibroblasts was responsible for modulation of MerTK, we assessed MerTK expression in vitro. We confirmed that in vitro MerTK was predominantly expressed by Ly6Chi but not Ly6Clo monocytes and MDMs (Figure 7H). Moreover, IL-17A signaling through cardiac fibroblasts resulted in a significant downregulation of MerTK expression on Ly6Chi monocytes and MDMs (Figure 7H). In addition, we observed that sMer was significantly elevated in the co-culture supernatants when IL-17A signaling was present (Figure 7I). Our findings suggest that IL-17A-stimulated cardiac fibroblasts can suppress macrophages’ phagocytic function by promoting MerTK cleavage, resulting in reduced surface MerTK expression. Our results also suggest that Ly6Chi MDMs in WT EAM hearts have compromised phagocytic activities during heart inflammation. We retroorbitally injected fluorescein isothiocyanate (FITC)-conjugated latex beads in both IL17Ra−/− and WT EAM mice. We found significantly more FITC+ macrophages in the hearts of IL17Ra−/− compared to WT, indicating enhanced phagocytic activity in an IL-17A-signaling-deficient environment (Figures 7J and 7K). Human myocarditis patients were reported to have high serum IL-17A, as well as an abundant presence of Th17 cells in their hearts and blood (Myers et al., 2016). Indeed, we found higher frequencies of IL-17A expressing infiltrating leukocytes in the hearts of myocarditis patients compared to patients with ischemic cardiomyopathy (Figures S7C and S7D; Tables S2 and S3). We therefore hypothesized that myocarditis patients could have lower MerTK expression in their cardiac myeloid population. In a cohort of HF patients, we found a significant reduction in myeloid MerTK expression in patients with myocarditis as compared to those with ischemic HF, similar to our finding in a murine model (Figure 7L; Tables S2 and S3). In conclusion, IL-17A signals through cardiac fibroblasts to promote MerTK cleavage on Ly6Chi MDMs, thus compromising the processes of efferocytosis. With reduced phagocytic ability as a result of surface MerTK modulation, we predict that macrophages are less efficient in clearing apoptotic cells efficiently and more likely to exhibit a proinflammatory phenotype, which in turn could promote irreversible cardiac remodeling and fibrosis.
Figure 7. IL-17A Trans-Signaling through Cardiac Fibroblasts Downregulates MerTK expression on Monocytes and MDMs.

(A and B) Representative EM images showing (A) apoptotic cells or apoptotic cellular debris internalized by Ly6Chi MDMs (arrowheads) and (B) engulfed cellular debris were largely absent in Ly6Clo MDMs. Scale bars: 2 μm.
(C) Macrophage phagocytic index was calculated using the following formula: (number of engulfed apoptotic cells/total number of macrophages) × (number of macrophages with engulfed apoptotic cells/total number of macrophages) × 100.
(D–G) Hearts from day 21 EAM mice.
(D) Frequencies of F4/80hiCD64+ macrophages out of viable CD45+Ly6G–CD11b+ cells were assessed by flow cytometry.
(E and F) MerTK MFI of F4/80hiCD64+ macrophages (E) and F4/80–CD64+ monocytes (F) in the hearts.
(G) ELISA showing soluble Mer (sMer) in WT and IL-17Ra−/− EAM mice sera.
(H and I) Cardiac fibroblasts were harvested from WT naive mice, whereas monocytes were sorted from EAM IL-17Ra−/− mice.
(H) MerTK MFI of Ly6Chi or Ly6Clo monocytes and MDMs in vitro after 160 h post-co-culture with cardiac fibroblasts stimulated with or without IL-17A.
(I) sMer detected in supernatants of the monocyte-fibroblast co-culture by ELISA.
(J) Flow cytometric analysis of the frequencies of FITC+F4/80hiCD64+ macrophages in the myocardium of WT, IL-17Ra−/−, and non-treated controls.
(K) Percentages of FITC+F4/80hiCD64+ macrophages out of viable CD45+Ly6G–CD11b+ cells.
(L) MerTK MFI in patients with either myocarditis or ischemic cardiomyopathy.
(D–G) Data are representative of five independent experiments. n = 8 – 9. (H and I) Data are representative of three independent experiments with technical triplicates. n = 3. (J and K) Data are representative of two independent experiments. n = 3. (C–G, I, K, and L) Groups were compared using Student’s t test. *p < 0.05; **p < 0.01. (H) Groups were compared using one-way ANOVA followed by Dunnett test. **p < 0.01; ****p < 0.0001. All data were presented as mean ± SD.
DISCUSSION
Monocytes and macrophages are key effector cells in the injured myocardium during human myocarditis and EAM (Barin et al., 2012; Theaker et al., 1985). Although both Ly6Chi and Ly6Clo monocytes infiltrate the myocardium during EAM, we found that an excessive accumulation of Ly6Chi rather than Ly6Clo monocytes in the heart leads to adverse cardiac remodeling and the development of DCM (Wu et al., 2014). However, it was unknown whether both infiltrating Ly6Chi and Ly6Clo monocytes contribute to the adverse cardiac tissue remodeling during development of EAM. Moreover, the role of the local pro-inflammatory cardiac microenvironment in influencing Ly6Chi and Ly6Clo monocyte fate and function was not well understood. We used three fate-mapping strategies to evaluate the phenotypic and functional changes of monocytes in the heart during myocarditis: parabiosis, adoptive transfer of monocytes, and an in vitro co-culture system.
Under steady state, adult cardiac tissue macrophages in mice are of mixed embryonic and hematopoietic origins (Epelman et al., 2014; Hashimoto et al., 2013; Hoeffel et al., 2015; Schulz et al., 2012; Sheng et al., 2015). In cases of aging or perturbed homeostasis, a greater proportion of embryonic-derived macrophages are replaced by infiltrating monocytes, changing the landscape of macrophage dynamics in the heart. (Epelman et al., 2014; Heidt et al., 2014; Molawi et al., 2014). Our parabiosis fate-mapping results reveal that monocytes extravasate into hearts during EAM and readily differentiate into macrophages, which contributes to the increase in the cardiac macrophage number during EAM. It was previously reported that cardiac CCR2− macrophages, including MHCII+ and MHCII− populations, are largely embryonically derived, whereas cardiac CCR2+ macrophages represent a hematopoietically derived lineage at steady state (Epelman et al., 2014; Lavine et al., 2014). Embryonically derived macrophages are shown to have regenerative and reparative properties (Aurora et al., 2014; Lavine et al., 2014), whereas CCR2+ macrophages are thought to initiate inflammation and contribute to adverse cardiac remodeling (Bajpai et al., 2018; Li et al., 2016). We report here that infiltrating monocytes predominantly contribute to the CCR2+MHCII+ macrophage compartment. Albeit to a lesser extent, they are also able to differentiate into CCR2−MHCII+ macrophages, with very few becoming CCR2−MHCII− macrophages in the chronically inflamed heart.
Ly6Chi monocytes are known to replenish the macrophage pool during cardiac injury; however, the contribution of Ly6Clo monocytes to the cardiac macrophage pool has not been previously described (Bronte and Pittet, 2013; Dutta et al., 2012; Leuschner et al., 2012; Robbins et al., 2012; Epelman et al., 2014; Heidt et al., 2014; Hilgendorf et al., 2014; Nahrendorf et al., 2007). Here, we show that both Ly6Chi and Ly6Clo monocytes have the capability to differentiate into macrophages, a process driven by direct contact with cardiac fibroblasts. Previously, we demonstrated that IL-17A can stimulate cardiac fibroblasts to produce significant amounts of GM-CSF (Wu et al., 2014). Evidence from EAM, Kawasaki syndrome, and the mouse model of MI show that cardiac fibroblasts are potent producers of GM-CSF (Anzai et al., 2017; Chen et al., 2018; Stock et al., 2016; Wu et al., 2014). An accumulating body of literature indicates that GM-CSF plays a key role in signaling myelopoiesis in response to tissue injury. GM-CSF also induces a pathogenic transcriptional signature in pro-inflammatory monocytes propelling the inflammatory cascade, resulting in further tissue damage (Anzai et al., 2017; Croxford et al., 2015; Wu et al., 2014). Here, we show a profound inhibitory role of IL-17A as it works in trans through cardiac fibroblastderived GM-CSF on Ly6Clo monocyte-to-macrophage differentiation in vitro. Other studies have implemented Ly6Clo latex bead labeling methods to track Ly6Clo monocytes in vivo (Potteaux et al., 2011; Tacke et al., 2007). However, due to the inadequate labeling efficiency in the EAM model, we were unable to distinguish and quantify Ly6Clo MDMs in EAM hearts using this method (data not shown). Alternatively, we used both intracardial and retro-orbital adoptive transfer methods and confirmed in vivo the inhibitory effect of IL-17A signaling on Ly6Clo monocyte-to-macrophage differentiation. We hypothesize that these Ly6Clo MDMs have distinct functions in myocarditis compared to Ly6Chi MDMs.
Gene expression profiling results substantiate that Ly6Chi and Ly6Clo MDMs represent distinct cell types, possibly driven partly by intrinsic properties. Recent studies have demonstrated that Ly6Chi and Ly6Clo monocytes are phenotypically heterogenous populations. Intrinsic properties such as transcription factors regulate Ly6Chi and Ly6Clo monocyte fate and function during steady state or tissue injury (Menezes et al., 2016; Yanez et al., 2017; Ikeda et al., 2018; Satoh et al., 2017; Mildner et al., 2017). However, we found that IL-17A signaling through cardiac fibroblasts accentuates a pro-inflammatory and pro-tissue remodeling gene profile in Ly6Chi MDMs. These macrophages upregulate genes such as Il6, Mmp9, Timp1, and Osm, all of which have been implicated in cardiac dysfunction and maladaptive cardiac remodeling (Hansson et al., 2011; Heymans et al., 2005; Hirota et al., 2004; Kubin et al., 2011; Kubota et al., 2000; Morishita et al., 2017). Unexpectedly, we found that Ly6Clo MDMs are enriched with genes associated with class II antigen processing. We confirm that Ly6Clo MDMs express higher levels of MHCII than their Ly6Chi counterparts in vitro. IL17Ra−/− mice develop EAM similarly to their WT counterparts, but are protected from DCM, cardiac fibrosis, and dysfunction (Baldeviano et al., 2010). In vivo, IL17Ra−/− EAM hearts contain more MHCII+ macrophages than WT EAM controls during disease resolution, coinciding with DCM protection in IL17Ra−/− mice. The anti-inflammatory and proangiogenic properties of Ly6Clo monocytes were previously investigated, and their roles in the resolution of cardiac inflammation were frequently highlighted (Hilgendorf et al., 2014; Nahrendorf et al., 2007; Wu et al., 2014). This suggests that MHCII+Ly6Clo MDMs might be beneficial in disease resolution and contribute to the protection of IL17Ra−/− mice from DCM and HF. The functions of Ly6Clo MDMs were placed under scrutiny recently. It has been implicated that they are the precursors for wound-healing macrophages in a soft tissue injury model (Olingy et al., 2017). Paradoxically, they were also shown to give rise to inflammatory macrophages in an autoimmune arthritis model (Misharin et al., 2014). We offer clues that Ly6Clo MDMs are a non-inflammatory and non-tissue-remodeling population. The increased presence of these macrophages and an increased Ly6Clo-to-Ly6Chi ratio in EAM myocardium correlate with DCM protection in IL17Ra−/− mice (Wu et al., 2014). Although phagocytic activity of Ly6Clo MDMs is unremarkable, their potential antigen presentation capability predicts their immune regulatory role in the heart. Whether MHCII+Ly6Clo MDMs play a role in effector and/or memory T cell or regulatory T cell activation in the heart requires further examination. Recent evidence indicated that MHCII+ macrophages loaded with cardiac myosin peptide are not effective in stimulating the proliferation of autoreactive CD4+ T cells (Anzai et al., 2019). However, the beneficial effects of macrophages and regulatory T cell interactions during inflammation resolution and tissue regeneration have been reported (Panduro et al., 2018; Proto et al., 2018). Currently, it remains unfeasible to selectively deplete Ly6Clo monocytes in mice. However, further experiments utilizing Nr4a1−/−, CX3CR1−/−, or S1PR5−/− mice with reduced Ly6Clo monocytes can help to better assess the specific functional role of Ly6Clo monocytes-derived macrophage in the context of myocarditis (Debien et al., 2013; Hanna et al., 2011; Landsman et al., 2009).
Macrophage function can be shaped by both progenitor origin and tissue microenvironment (Amit et al., 2016; Lavine et al., 2014). We tested the hypothesis that upon IL-17A stimulation, cardiac fibroblasts play a role in modulating macrophage function. Recent data directly link the level of efferocytosis receptors on monocytes and macrophages to phagocytosis-dependent wound healing and restoration of organ function (Foo et al., 2005; Whelan et al., 2010; Howangyin et al., 2016; Wan et al., 2013). Uncontrolled cellular necrosis and apoptosis during cardiac injury are integral components that contribute to adverse tissue healing and remodeling. Inefficient phagocytic clearance can also lead to exposure of self-antigens, which contribute further to autoimmune reactivity (Wermeling et al., 2009). One of these efferocytosis receptors, MerTK, has been shown to be upregulated on phagocytic cells during inflammation and plays an essential role in apoptotic cell recognition and clearance (Chen et al., 1997; Hilliard et al., 2014; Scott et al., 2001; Seitz et al., 2007). Moreover, efferocytosis triggers a shift in macrophage activation, making them phenotypically less proinflammatory (Camenisch et al., 1999; Fadok et al., 1998; Healy et al., 2016). Notably, our in vivo and in vitro results suggest that IL-17A signaling through cardiac fibroblasts leads to MerTK shedding and the release of sMer, which significantly reduces surface MerTK expression on Ly6Chi monocytes and MDMs. Cardiomyocytes have been shown to induce shedding of macrophage MerTK to suppress phagocytosis (Zhang et al., 2015). This highlights another mechanism by which IL-17A through cardiac fibroblasts can induce MerTK shedding and, in turn, contributes to cardiac pathology in autoimmune myocarditis. Moreover, we showed that myeloid MerTK was significantly lower in human myocarditis patients when compared to ischemic patients. These findings warrant future studies to determine whether sMer can be used as a biomarker in clinical settings to differentiate patients with myocarditis from those with other types of cardiac diseases.
Taken together, we underscore the fates of Ly6Chi and Ly6Clo monocyte subsets as a result of changes in the cardiac microenvironment. We demonstrate how the local cardiac milieu instructs cardiac fibroblasts to facilitate monocyte differentiation and proliferation as well as to regulate the phenotype and function of monocytes and MDMs. Monocytes are versatile immune cells playing a multi-faceted role in a wide range of inflammatory disorders (Arnold et al., 2007; Misharin et al., 2017; Mishra et al., 2012; Nahrendorf et al., 2007; Olingy et al., 2017; Rahman et al., 2017). Our findings have broader implications for inflammatory diseases in the heart and other organs. We recently reported that IL-17A signaling to cardiac fibroblasts is associated with severe fibrosis and post-infarct death (Chen et al., 2018), which demonstrates a potential parallel concept that warrants future investigation. Ultimately, we anticipate that further studies of macrophage-cardiac fibroblast interactions will provide valuable insights into the development of targeted therapies that prevent deleterious inflammatory responses.
STAR⋆METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniela Ciháková (cihakova@jhmi.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients
Left ventricle apex endomyocardial biopsies were obtained from patients with end stage heart failure (AHA stage D) of various etiologies. These patients were undergoing either an implantation of Left Ventricular Assist Device (LVAD), or an orthotopic heart transplant after LVAD explant at the Texas Heart Institute. Details including patients’ age and gender are summarized in Table S1 – 3. Informed consent was obtained from human subjects and the study protocol was approved by the Committee for the Protection of Human Subjects (University of Texas Health Science Center at Houston. IRB #HSC-MS-05–0074). Samples were properly preserved in cryovials embedded in liquid nitrogen (−190°C) and then kept at −80°C in the tissue bank at the Texas Heart Institute, Houston, TX as previously described (Razeghi et al., 2001). Aliquots of the samples were shipped frozen and processed in house. Reported diagnoses corresponded to clinical charts, based on standard histology (H&E), clinical presentation, hemodynamic parameters, and routine clinical biochemical and serology parameters. Patient’s information of all samples was processed with random non-linked code relabeling in a database at the time of preservation. Furthermore, for each sample, a separate random non-linked code was assigned for the analysis process. Separately, one paraffin embedded, virus negative, acute giant cell myocarditis left ventricle transmural endomyocardial biopsy sample in Figures S3A and S3B were provided by Dr. Karin Klingel. The sample was taken for routine diagnostic purposes to identify infectious agents in the myocardium as described before (Zuern et al., 2013). Informed consent was obtained and the study was approved by local ethic committee (Project-No.253/2009BO2). Diagnosis of giant cell myocarditis was based on established criteria (Caforio et al., 2013b).
Mice
IL-17Ra–/– mice on BALB/c background were provided by Amgen Inc. (Thousand Oaks, CA) and Dr. J. Kolls (Children’s Hospital, University of Pittsburgh Medical Center, Pittsburgh, PA) (Wu et al., 2014; Yu et al., 2008). CByJ.SJL(B6)-Ptprca/J (CD45.1) mice (006584) and WT BALB/cJ (000651) mice were purchased from The Jackson Laboratory. IL-17Ra–/– mice were crossed to CByJ.SJL(B6)-Ptprca/J (CD45.1) mice and bred to homozygosity at both loci. All mice were housed and maintained in the Johns Hopkins University School of Medicine specific pathogen-free vivarium. 6 – 10 weeks old healthy naive male mice were randomly selected as the subjects of all of our studies. All experiments involving animals were in compliance with the Animal Welfare Act and strictly followed the Guide for the Care and Use of Laboratory Animals. The Animal Care and Use Committee of The Johns Hopkins University has approved all procedures and protocols used in this study.
Primary Adult Cardiac Fibroblasts Isolation and Culture
50U heparin was injected i.p. into 6 – 10-week-old male BALB/cJ naive mice prior to sacrifice. Hearts were cannulated through the aorta and perfused for 3 minutes with 37°C perfusion buffer at 4 mL/minute: 7.03 g/L NaCl, 1.1 g/L KCl, 0.082 g/L KH2PO4, 0.085 g/L Na2HPO4, 0.144 g/L MgSO4, 2.38 g/L HEPES, 0.39 g/L NaHCO3, 1 g/L glucose, 3.74 g/L Taurine, 1 g/L 2,3-Butanedione monoxime (all Sigma), and for 8 min with Collagenase II and Protease XIV (Worthington) and 0.03M CaCl2. Hearts were cut into small pieces and cells separated by gently pipetting for 3 minutes or until no large tissue pieces were observed. Cells were filtered through a 100 mm filter and washed in DMEM (GIBCO). Cells were plated in DMEM with 20% FBS (GE Healthcare Life Sciences), nonessential amino acids (Sigma), Penicillin/Streptomycin, 2 mM L-Glutamine, and 25mM HEPES (all Quality Biological). Cells were incubated in a humidified 5% CO2 incubator at 37°C from here on. Non-adherent cells were washed away after 1 hour. Fibroblasts from second passage were used in experiments. The purity of cardiac fibroblast cultures was confirmed by qPCR and flow cytometry. Cells and supernatants were harvested at the indicated time points after addition of recombinant mouse cytokine IL-17A at 50 ng/mL or recombinant mouse GM-CSF at 50ng/mL (Peprotech). LEAF™ Purified anti-GM-CSF at 50μg/mL was used (MP1–22E9, BioLegend). Monocytes and fibroblasts separation in the co-culture was achieved by 0.4 μM transwell inserts (Corning).
METHOD DETAILS
Parabiosis Surgery
Pairs of mice were anesthetized by isoflurane inhalation, 4.0–5.0% v/v induction (Baxter). Fur was removed thoroughly from the entire flank region using clippers. Anesthesia in surgical pairs was maintained with intramuscular injections of ketamine (80 mg/kg) and xylazine (16 mg/kg). The mice were laid supine and the site was disinfected with betadine followed by 70% EtOH. We administered buprenorphine (0.1 mg/kg) intraperitoneally for the initial analgesia, and at 12 hours postoperatively. Longitudinal incisions were made through the skin starting from the elbow joint and extended down to the knee joint. Non-absorbable 4–0 interrupted sutures were placed around the knee and elbow joints. Pairs where attached from the elbow joints first. To increase skin anastomosis, we used a continuous 5–0 absorbable vicryl sutures through the muscular layer and connect the pairs further, before attaching the knee joints. Surgical stapler was used to connect the skins of the pairs. Baytril was used upon completion of the procedure. Animals were provided with moistened chow and gel food diet supplement every other day until sacrifice at 19 to 20 days after parabiosis.
EAM Induction
To induce EAM, we injected mice with 125 μg myosin heavy chain α peptide MyHCα614–629 (Ac-SLKLMATLFSTYASAD; Genscript) (Pummerer et al., 1996) emulsified in CFA (Sigma-Aldrich) supplemented with 5mg/mL heat-killed Mycobacterium tuberculosis strain H37Ra (Difco) on days 0 and 7. On the first day of immunization, mice also received a dose of 500 ng pertussis toxins intraperitoneally (List Biological Laboratories) (Ciháková et al., 2004).
EAM Histopathology Assessments
Myocarditis severity was evaluated by histology on days 21. Heart tissues were fixed in SafeFix solution (Thermo Fisher Scientific), embedded and cut into 5 μm serial sections. Sections were stained with H&E and ventricular inflammation was scored via a microscope by two independent blinded investigators and averaged using the following criteria for hematopoietic infiltrates: grade 0, no inflammation; grade 1, < 10% of the heart section is involved; grade 2, 10%–30%; grade 3, 30%–50%; grade 4, 50%–90%; grade 5, > 90% (Ciháková et al., 2004).
Isolation of Splenic Ly6Chi and Ly6Clo Monocytes
Spleens from WT or IL-17Ra–/– BALB/c mice on day 14 of EAM were dissected and mechanically disrupted. Cells were filtered through 40 mm cell strainers and washed. Histopaque 1119 and 1077 (Sigma) were used to isolate mononuclear cells. Anti-Ly6G MicroBead kit (Miltenyi Biotech) was used to deplete Ly6G+ cells, and CD11b MicroBead kit (Miltenyi Biotech) was used to positively enrich CD11b+ cells. Cells were sorted on an AriaIIu cell sorter. Sorted cells from IL-17Ra–/– BALB/c mice spleens were used for in vitro co-culture with primary cardiac fibroblasts, major outcomes were validated with sorted cells from WT BALB/c mice spleens co-cultured with cardiac fibroblasts. Sorted cells from WT BALB/c mice spleens were used in adoptive transfer experiments (see below).
Cell Staining and Light Microscopy
Modified Giemsa staining was achieved by using Differential Quik Stain Kit, following the manufacturer’s instructions (Polysciences). Cell staining images were acquired on Olympus BX43 microscope with a camera (DP72) using cellSens Standard software (version 1.4.1; Olympus).
IncuCyte ZOOM Imaging
Once the monocyte-fibroblast co-cultures were established, the plates were placed into the IncuCyte ZOOM™ (Essen Bioscience) apparatus and images of cells were recorded with 20 × magnification undisturbed every 1 hour for a total duration of 160 hours. Cardiac fibroblasts alone were used as controls and triplicated wells were included in all data acquisitions.
Transmission Electron Microscopy
Co-cultured cells were washed with 1 × PBS and fixed in 2.5% glutaraldehyde dissolved in 0.1 M Na cacodylate. Samples were then processed by the Johns Hopkins Microscope Facility (Diny et al., 2017) before examination using the Philips CM120 transmission electron microscope. Images were captured using Advanced Microscopy Techniques V602 software. The phagocytic index was calculated according to the following formula: phagocytic index = (total number of engulfed cells/total number of counted macrophages) × (number of macrophages containing engulfed cells/total number of counted macrophages) × 100. Approximately 60 total macrophages derived from each monocyte subset were counted, numbers were acquired from two separate assessments.
Immunofluorescence Microscopy
Human paraffin-embedded giant cell myocarditis biopsy sample slides were processed using xylene and rehydrated with decreasing concentration of ethyl alcohol and rinsed with distilled water. After blocking with 1% BSA and 0.1% tween-20 in 1 × PBS, tissues were incubated with anti-CD14-biotin sheep antibodies (R & D systems) and anti-CD68 mouse antibodies (Abcam). We then used NL-637 Streptavidin and donkey NL-557 anti-mouse IgG (R & D systems) as secondary antibodies diluted in 0.05% Evans Blue for counterstaining. DAPI was used for staining nuclei. Finally, Sudan Black was used to suppress auto-fluorescence as a result of tissue fixation and paraffin treatment. Images were acquired using Zeiss Axio Imager.A2 microscope with AxioCam MRm at 20 × magnification. We used AxioVision Rel. 4.8 software to acquire images and Adobe Photoshop CC to process images.
Intracardiac Injection
WT or IL-17Ra–/– BALB/c mice on day 21 of EAM were depilated and anesthetized with 3.5% isoflurane (Baxter). The mice were subsequently endotracheally intubated, 100% oxygen and 2% isoflurane were provided to the animals throughout the operation by mechanical ventilation (Model 845, Harvard Apparatus). Pre-operational analgesics (0.05 mg/kg Buprenorphine, Reckitt Benckiser) and paralytics (1 mg/kg Succinylcholine, Henry Schein) were administered prior to operation. Mice were subjected to a thoracotomy, typically around the 4th or 5th intercostal space to expose the heart ventricles. Roughly 1.5 – 2 × 105 cells were injected with a 29G 29G ½ insulin syringe (BD) into 2 – 3 ventricular locations. Mice were placed under the heat lamp to recover post-surgery, post-operational analgesics (0.05 mg/kg Buprenorphine, Reckitt Benckiser) were administered. Mice were sacrifice at 40 hours or at 160 hours post-surgery to assess cell transfer outcomes.
Retro-Orbital Injection of Monocytes
WT or IL-17Ra–/– BALB/c mice on day 21 of EAM were depilated and anesthetized with 100% oxygen and 3.5% isoflurane (Baxter). Mice were subjected to an injection into the ophthalmic venous sinus. Roughly 4 – 5 × 105 FACS sorted monocytes were injected with a 29G ½ insulin syringe (BD).
Retro-Orbital Injection of Liposomes and Latex Beads
We administered a single treatment of 10 μl Clodrosome (clodronate loaded liposomes; Encapsula Nano Sciences) per gram of animal weight seventeen hours prior to latex microsphere labeling to better visualize FITC+ macrophages in vivo. 0.5-μm FITC-conjugated (yellow gold) plain microspheres (2.5% solids [wt/vol]; Polysciences, Inc.) were diluted 1:25 in 1XPBS. We administered 250 μl of the diluted latex beads to examine macrophage phagocytic activity in vivo. Hearts were harvested 24 hours post bead administration.
Quantitative Reverse Transcription PCR
Cell mRNAs were extracted in TRIzol (Invitrogen), and reverse transcribed using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Target cDNAs were amplified with Power SYBR Green PCR Master Mix (Bio-Rad) and real time cycle thresholds were detected via MyiQ2 themocycler running on an iQ5 software (Bio-Rad). Target genes fold induction were calculated using the 2−ΔΔCt method by normalizing cycle thresholds to the Hprt housekeeping gene and medium controls (Livak and Schmittgen, 2001). Verified Ccl2 primer sequences were acquired from the PrimerBank (Harvard Medical School, Massachusetts General Hospital and The Center for Computational and Integrative Biology), and commercially synthesized (Integrated DNA Technologies). Ccl2 forward primer sequence (5′ to 3′): TTAAAAACCTGGATCGGAACCAA, and reverse primer sequence (5′ to 3′): GCATTAGCTTCAGATTTACGGGT.
Flow Cytometry Analysis and Barnes-Hut Stochastic Neighbor Embedding Analysis
Hearts of naive or EAM mice were perfused through the ventricles with 1 × PBS for 3 minutes. GentleMACS C Tubes were used to mechanically disassociate the tissue according to manufacturer’s instructions (Miltenyi Biotech). We used 3000 U Collagenase II and 300 U DNase I (Worthington) dissolved in 5mL HBSS to digest tissue and obtain single cell suspension. Splenocytes, blood, and BM cells (femurs and tibias) were isolated by mechanical disruption, followed by red blood cell lysis using ACK buffer (ThermoFisher). Heart samples were filtered through 40 μm filters, whereas the other tissues were filtered through 70 μm filters. Single-cell suspensions were stained with LIVE/DEAD Fixable Far Red Dead Cell Stain Kit (ThermoFisher), washed, FcγRII/III blocked with a-CD16/CD32, and stained with fluorochrome-conjugated antibodies (eBioscience, BioLegend, BD PharMingen). Alternatively, we used APC Annexin V Apoptosis Detection Kit with 7-AAD (BioLegend) instead of LIVE/DEAD. CellTrace™ CFSE Kit was used to track cell proliferation both in vivo and in vitro (ThermoFisher). Samples were acquired on a BD LSRII or LSRFortessa 4-laser cytometers running FACSDiva 6.0 (BD Immunocytometry) and analyzed using FlowJo 10.4 software. We ran an interactive visualization tool called cyt in MATLAB to analyze high-dimensional flow cytometry data (Becher et al., 2014; Amir et al., 2013). bh-SNE is an unsupervised non-linear dimensionality reduction embedding technique (Amir et al., 2013).
Microarray
Ly6Chi and Ly6Clo in vitro co-culture derived macrophages in triplicate were FACS sorted and cells were lysed using RLT buffer (QIAGEN). RNA was extracted using RNeasy micro kit. RNA samples were labeled using Thermal Fisher 3′ IVT Pico Reagent kit (Affymetrix) according to manufacturer’s guidelines and probed using the Affymetrix Mouse Clariom S Array. RNA concentration and integrity were determined with Agilent Bioanalyzer Pico Chip.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
GraphPad Prism 7 software was used for statistical analysis. Statistical analysis details are described in the figure legends. To determine if datasets show a normal distribution, either the D’Agostino-Pearson or the Shapiro-Wilk normality test was used. P values were considered statistically significant at p < 0.05. All data were presented as mean ± SD.
DATA AND CODE AVAILABILITY
Microarray Gene Expression Analysis
Raw data generated from Clariom S Arrays were processed using Affymetrix Expression Console Software. CEL files containing feature intensity values were converted into summarized expression values by Partek Genomic Suite Software including background adjustment, quantile normalization and summarization across all chips. All samples passed QC thresholds for hybridization, labeling and the expression of spiked in controls. The variances, both between and within groups, in log2 transformed expression values were analyzed by one-way ANOVA. PCA graph was generated using Partek Genomic Suite Software, heatmaps were generated using TIBCO Spotfire Software. The complete normalized dataset is available on GEO: GSE118861.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BUV 395 Hamster anti-Mouse CD11c (clone HL3) | BD Horizon | Cat# 564080; RRID:AB_2738580 |
| BUV 395 Hamster IgG1, λ1 Isotype Control (clone G235–2356) | BD Horizon | Cat# 564075 |
| BV 421 anti-Mouse CCr2 (clone SA203G11) | BioLegend | Cat# 150605; RRID:AB_2571913 |
| BV 605 anti-Mouse CD115 (clone AFS98) | BioLegend | Cat# 135517; RRID:AB_2562760 |
| PE anti-Mouse MerTK (Clone 2B10C42) | BioLegend | Cat# 151506; RRID:AB_2617037 |
| PE Rat IgG2a Isotype Control (Clone RTK2758) | BioLegend | Cat# 400508; RRID:AB_326530 |
| PerCP/Cy5.5 anti-Mouse/Human CD11b (clone M1/70) | BioLegend | Cat# 101228; RRID:AB_893232 |
| APC eflour 780 anti-Mouse Ly6C (clone HK1.4) | Invitrogen | Cat# 47–5932–82; RRID:AB_2573992 |
| PE Cy7 anti-Mouse F4/80 (clone BM8) | BioLegend | Cat# 123114; RRID:AB_893478 |
| PE Texas Red anti-Mouse CD45 | Invitrogen | Cat# MCD4517; RRID:AB_10392557 |
| PE anti-Mouse CD11c (clone N418) | BioLegend | Cat# 117307; RRID:AB_313776 |
| PE Armenian Hamster IgG1 (clone HTK888) | BioLegend | Cat# 400907; RRID:AB_326593 |
| BV 711 anti-Mouse CD64 (clone X45–5/7.1) | BioLegend | Cat# 139311; RRID:AB_2563846 |
| BV785 anti-Mouse Ly6G (clone 1A8) | BioLegend | Cat# 127645; RRID:AB_2566317 |
| FITC anti-Mouse Ly6G (clone 1A8) | BioLegend | Cat# 127606; RRID:AB_1236494 |
| FITC anti-Mouse NKp46 (clone 29A1.4) | BioLegend | Cat# 137606; RRID:AB_2298210 |
| FITC anti-Mouse CD49b clone DX5 | BioLegend | Cat# 108906; RRID:AB_313413 |
| FITC anti-Mouse CD19 (clone 65) | BioLegend | Cat# 115506; RRID:AB_313641 |
| FITC anti-Mouse TER119 (clone TER-119) | BioLegend | Cat# 116206; RRID:AB_313707 |
| FITC anti-Mouse CD3e (clone 145–2C11) | BioLegend | Cat# 100306; RRID:AB_312671 |
| FITC anti-Mouse TCRβ chain (Clone H57–597) | BioLegend | Cat# 109206; RRID:AB_313429 |
| FITC anti-Mouse CD11c (clone N418) | BioLegend | Cat# 117306; RRID:AB_313775 |
| Alexa Flour 700 anti-Mouse I-A/I-E (clone M5/114.15.2) | BioLegend | Cat# 107622; RRID:AB_493727 |
| BV 605 rat IgG2a, kappa Isotype control (Clone RTK2758) | BioLegend | Cat# 400539; RRID:AB_11126979 |
| APC anti-mouse NK-46 (clone 29A1.4) | BioLegend | Cat# 137607; RRID:AB_10612749 |
| APC anti-mouse B220 (clone RA3–6B2) | BioLegend | Cat# 103211; RRID:AB_312996 |
| APC anti-mouse CD90.2 (clone 53–2.1) | BioLegend | Cat# 140311; RRID:AB_10645337 |
| APC anti-mouse CD3 (clone 17A2) | BioLegend | Cat# 100236; RRID:AB_2561456 |
| APC anti-mouse CD19 (clone 65) | BioLegend | Cat# 115512; RRID:AB_313647 |
| Pacific Blue anti-mouse F4/80 | Invitrogen | Cat# MF48028; RRID:AB_10373419 |
| PE Dazzle 594 CD45.1 (clone A20) | BioLegend | Cat# 110748; RRID:AB_2564295 |
| APC CD45.1 (clone A20) | BioLegend | Cat# 110714; RRID:AB_313503 |
| PE/Dazzle 594 anti-mouse CD45.2 (Clone 104) | BioLegend | Cat# 109845; RRID:AB_2564176 |
| APC anti- mouse CD45.2 (clone 104) | BioLegend | Cat# 109813; RRID:AB_389210 |
| BV711 anti- mouse IgG1 Isotype control (clone MOPC-21) | BioLegend | Cat# 400167; RRID:AB_11218607 |
| PE Cy7 Rat IgG2a Isotype control (clone RTK2758) | BioLegend | Cat# 400522; RRID:AB_326542 |
| PE anti- mouse CD135 (clone A2F10) | BioLegend | Cat# 135305; RRID:AB_1877218 |
| PE Rat IgG2b Isotype control (clone eB149/10H5) | eBioscience | Cat# 12–4031–81; RRID:AB_470041 |
| Pacific Blue anti- mouse Ly6G (clone 1A8) | BioLegend | Cat# 127612; RRID:AB_2251161 |
| Pacific Blue anti-human/mouse CD11b (clone M1/70) | BioLegend | Cat# 101224; RRID:AB_755986 |
| BV421 anti-human/mouse CD11b (clone M1/70) | BioLegend | Cat# 101235; RRID:AB_10897942 |
| BUV 395 Rat anti-mouse CD117 (clone 2B8) | BD Horizon | Cat# 564011; RRID:AB_2738541 |
| BV650 anti-mouse Ly6G (clone 1A8) | Biolegend | Cat# 127641; RRID:AB_2565881 |
| BV785 anti-mouse CD45.2 (clone 104) | Biolegend | Cat# 109839; RRID:AB_2562604 |
| Donkey NL-557 anti-mouse IgG | R&D Systems | Cat# NL007; RRID:AB_663768 |
| Northern Lights 637 Streptavidin | R&D Systems | Cat# NL998; RRID:AB_10175723 |
| Goat anti-Rabbit IgG HNL | Abcam | Cat# AB150077; RRID:AB_2630356 |
| Human CD14 Biotinylated Antibody | R&D Systems | Cat# BAF383; RRID:AB_356435 |
| Recombinant CD16 antibody | Abcam | Cat# AB109223; RRID:AB_10863447 |
| Anti-CD68 antibody | Abcam | Cat# AB955; RRID:AB_ 307338 |
| LEAF Purified anti-mouse GM-CSF antibody (clone MP1–22E9) | BioLegend | Cat# 505408; RRID:AB_315384 |
| PerCP/Cy5.5 anti-human CD14 (clone 36D3) | BioLegend | Cat# 367110; RRID:AB_2566712 |
| PE-TR anti-human CD16 (clone 3G8) | ThermoFisher | Cat# MHCD1617; RRID:AB_1464937 |
| PE anti-human MerTK (clone 590H11G1E3) | BioLegend | Cat# 367608; RRID:AB_2566401 |
| BV711 anti-mouse/human CD11b (clone M1/70) | BioLegend | Cat# 101242; RRID:AB_2563310 |
| APC anti-human CD68 (Clone Y1/82a) | BioLegend | Cat# 333810; RRID:AB_2275735 |
| BUV395 anti-mouse/human CD45 (clone HI30) | BD Biosciences | Cat# 563791; RRID:AB_2744400 |
| APC Annexin V apoptosis Detection Kit with 7-AAD | BioLegend | Cat# 640930 |
| Biological Samples | ||
| Endomyocardial Biopsies | University of Texas Health Science Center | https://www.texasheart.org/; https://www.uth.edu/ |
| Giant Cell Endomyocardial Biopsies Sample | University of Tubingen | https://uni-tuebingen.de/en/university.html |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 0.05% Trypsin-EDTA (1x) | Life Technologies | Cat#25300–054 |
| MyHCa614–629 | GenScript | Cat#639666–1 |
| Protease, Type XIV: Bacterial, From Streptomyces griseus | Sigma-Aldrich | Cat#P5147–5G |
| Deoxyribonuclease I | Worthington | Cat#LS002139 |
| Collagenase II | Worthington | Cat#LS004177 |
| Pertussis Toxin from Bordetella pertussis | List Biological Laboratories, Inc. | Cat#180, #181 |
| Mycobacterium Tuberculosis Des. H37 Ra | BD | 231141 |
| L-Glutamine | Corning Inc. | Cat#25–005-CI |
| Freund’s Adjuvant Complete | Sigma | Cat#F5881–10X10ML |
| Sodium Pyruvate | Sigma | Cat#S8636 |
| Antibiotic antimycotic solution (100x) | Corning Inc. | Cat#30–004-CI |
| CellStripper | Corning Inc. | Cat#25–05-CI |
| 1M HEPES Buffer pH7.3 | Quality Biological | Cat#118–089–721 |
| Histopaque-1119 | Sigma | Cat#11191–6X100ML |
| HIstopaque-1077 | Sigma | Cat#10771–6X100ML |
| Bovine Serum Albumin | Sigma | Cat#A3608–100G |
| ACK Lysing Buffer | Quality Biological | Cat#118–156–721 |
| MEM NON-Essential Amino Acid solution 100x | Sigma-Aldrich | Cat# M7145 |
| Lysis Buffer RLT Plus | QIAGEN | Cat#1053393 |
| EDTA | Corning Inc. | Cat#46–034-CI |
| Fluoresbrite Plain YG 0.5 Micron Microspheres (2.5%Solids Latex) | Polysciences, Inc. | Cat#17152 |
| Differential Quik Stain Solution C Xanthene Dye | Polysciences, Inc. | Cat#24606C-250 |
| Differential Quik Stain Solution A Fixative | Polysciences, Inc. | Cat#24606A-250 |
| Differential Quik Stain Solution B Blue Dye | Polysciences, Inc. | Cat#24606B-250 |
| Liposomal Clodronate | Clodrosome | Cat#CLD-8909 |
| DMEM (1X) | GIBCO | Cat#11995–065 |
| Anti-Ly6G Microbead Kit mouse | Miltenyi Biotec | Cat#130–092–332 |
| Anti-CD11b Microbead Kit mouse/human | Miltenyi Biotec | Cat#130–049–601 |
| TRIzol reagent | ThermoFisher | Cat#15596026 |
| Heat Inactivated Fetal Bovine Serum | ThermoFisher | Cat#A3840001 |
| Recombinant Murine GM-CSF | Peprotech | Cat#315–03 |
| DAPI Solution (1mg/ml) | ThermoFisher | Cat#62248 |
| SafeFix II All-Purpose Fixative | ThermoFisher | Cat#23–042600 |
| Evans Blue | Sigma | Cat#E21129 |
| Sudan Black B | Sigma | Cat#199664–25G |
| BD Cytofix Fixation Buffer | BD Biosciences | Cat# 554655 |
| CountBright absolute counting beads | Invitrogen | Cat# C36850 |
| Anti-MoCD-16/CD32 | eBioscience | Cat#14–0161–86 |
| Forane Liquid for inhalation 100ml | Baxter | Cat#1001936040 |
| Buprenorphine (1mg/ml) | ZooPharm | Cat#BZ8069317 |
| Succinyl Choline (20mg/ml) | Hospira, Inc. | Cat# 81–081-EV |
| High Capacity cDNA reverse Transcription Kit | FisherScientific | Cat#4368814 |
| SYBR Green PCR Master Mix | BioRad | Cat#4367659 |
| CFSE CellTrace Cell Proliferation Kit | Invitrogen | Cat# C34554 |
| D-Glucose Anhydrous | Amresco | Cat3 0188–1KG |
| Magnesium Sulfate | Amresco | Cat# E797–500G |
| Potassium Chloride | Sigma | Cat# 60130–250G |
| Taurine | Sigma | Cat3T0625–100G |
| Tween 200 | Sigma | Cat3 P1379–500ML |
| Sodium Chloride | Fisher BioReagents | Cat#M-11615 |
| Sodium Bicarbonate | Amresco | Cat# 0865–1KG |
| Sodium phosphate dibasic heptahydrate, ACS regent | Sigma | Cat#S9390–500G |
| Potassium Phosphate monobasic | Sigma | Cat#P9791–100G |
| Heparin sodium salt from porcine intestinal mucosa | Sigma | Cat#H3393–10Ku |
| Triton X-100 | Sigma | Cat#T9284–100ML |
| 2,3-butanedione | Sigma | Cat# B0753–100G |
| RNeasy Micro Kit | QIAGEN | Cat#74004 |
| GeneChip 3′ IVT Pico Kit | ThermoFisher | Cat#902789 |
| Live/Dead Fixable Far Red Dead Cell Stain Kit | Invitrogen | Cat# L10120 |
| Live/Dead Fixable Aqua Dead Cell Stain Kit | ThermoFisher | Cat# L34966 |
| Critical Commercial Assays | ||
| Mouse MER ELISA Kit | Abcam | Cat#AB210572 |
| Human MER ELISA Kit | Abcam | Cat#AB119604 |
| Clariom S Assay, mouse | ThermoFisher | Cat#902930 |
| Deposited Data | ||
| Raw and analyzed microarray data | This paper | GEO: GSE118861 |
| Experimental Models: Organisms/Strains | ||
| Mouse: BALB/cJ | The Jackson Laboratory | Cat# 000651 |
| Mouse: CByJ.SJL(B6)-Ptprca/J | The Jackson Laboratory | Cat# 006584 |
| Mouse: IL-17Ra−/− (BALB/c) | Amgen Inc.; Children’s Hospital University of Pittsburg Medical Center (Dr. J Kolls) | Yu et al., 2008; Wu et al., 2014 |
| Mouse: IL-17Ra−/− (CByJ.SJL(B6)-Ptprca/J) | This Paper | N/a |
| Oligonucleotides | ||
| Ccl2 forward primer (5′ to 3′): TTAAAAACCTGGATCGGAACCAA | This paper | N/A |
| Ccl2 reverse primer (5′ to 3′): GCATTAGCTTCAGATTTACGGGT | This paper | N/A |
| Hprt forward primer (5′ to 3′): TCAGTCAACGGGGGACATAAA | This paper | N/A |
| Hprt reverse primer (5′ to 3′): GGGGCTGTACTGCTTAACCAG | This paper | N/A |
| Software and Algorithms | ||
| FlowJo | FlowJo | RRID:SCR_008520 |
| Cyt | Amir et al., 2013; Becher et al., 2014 | https://systemsbiology.columbia.edu/center-for-computational-biology-and-bioinformatics-c2b2 |
| MATLAB | MATLAB | RRID:SCR_001622 |
| PRISM | PRISM | RRID:SCR_005375 |
| Partek Genomics Suite | Partek Genomics Suite | RRID:SCR_011860 |
| Spotfire | TIBCO | RRID:SCR_008858 |
Highlights.
Cardiac fibroblasts facilitate differentiation of Ly6Chi and Ly6Clo monocytes
IL-17A trans-signaling inhibits Ly6Clo monocyte-to-macrophage differentiation
IL-17A trans-signaling increases MerTK shedding on monocyte-derived macrophages
IL-17A trans-signaling promotes monocyte-derived macrophages to be proinflammatory
ACKNOWLEDGMENTS
The authors would like to extend their gratitude to Dr. Noel Rose for his kind sponsorship and mentorship, Amgen Inc. (Thousand Oaks, CA) and Dr. J. Kolls (Children’s Hospital, University of Pittsburgh Medical Center, Pittsburgh, PA) for IL-17Ra−/− mice, Xiaoling Zhang and Jessica Gucwa at the Johns Hopkins Flow Cytometry Cores, Leonard Marque and Firozeh Dastani at the Johns Hopkins animal resources, Julie Schaub for mouse colony management, Karen Fox-Talbot for human sample immunohistochemistry work, Barbara Smith at the Johns Hopkins SOM microscopy facility, and Haiping Hao and C. Conover Talbot Jr. at the Johns Hopkins Deep Sequencing and Microarray Core Facility.
This work was supported by NIH/NHLBI grants R01HL118183 and R01HL136586, an American Heart Association (AHA) AWRP Winter 2017 Grant-in-Aid (17GRNT33700274), AHA 2019 Transformational Project Award 19TPA34910007, and the Matthew Poyner MVP Memorial Myocarditis Research Fund to D.C.; the Johns Hopkins Autoimmune Disease Research Center O’Leary-Wilson Fellowship, the Johns Hopkins Bloomberg School of Public Health Richard J. and Margaret Conn Himelfarb Student Support fund, and a Katherine E. Welsh Fellowship to X.H.; a Myocarditis Foundation Postdoctoral Fellowship (90072351) to G.C.; an AHA Postdoctoral Fellowship (16POST31330012) to W.B.-B.; a predoctoral fellowship to H.S.C. (16PRE31170040) and N.L.D. (15PRE25400010); as well as AARDA awards to W.B.-B., a Gilead Research Scholar grant and AARDA award to J.G.B., and Deutsche Forschungsgemeinschaft grant KL595/2-3 to K.K.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.06.007.
REFERENCES
- Amir AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, and Pe’er D (2013). viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 31, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit I, Winter DR, and Jung S (2016). The role of the local environment and epigenetics in shaping macrophage identity and their effect on tissue homeostasis. Nat. Immunol. 17, 18–25. [DOI] [PubMed] [Google Scholar]
- Amoah BP, Yang H, Zhang P, Su Z, and Xu H (2015). Immunopathogenesis of Myocarditis: The Interplay Between Cardiac Fibroblast Cells, Dendritic Cells, Macrophages and CD4+ T Cells. Scand. J. Immunol. 82, 1–9. [DOI] [PubMed] [Google Scholar]
- Anzai A, Choi JL, He S, Fenn AM, Nairz M, Rattik S, McAlpine CS, Mindur JE, Chan CT, Iwamoto Y, et al. (2017). The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. J. Exp. Med. 214, 3293–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzai A, Mindur JE, Halle L, Sano S, Choi JL, He S, McAlpine CS, Chan CT, Kahles F, Valet C, et al. (2019). Self-reactive CD4+ IL-3+ T cells amplify autoimmune inflammation in myocarditis by inciting monocyte chemotaxis. J. Exp. Med. 216, 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, and Chazaud B (2007). Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 204, 1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, Sadek HA, and Olson EN (2014). Macrophages are required for neonatal heart regeneration. J. Clin. Invest. 124, 1382–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M, Epelman S, Kreisel D, Liu Y, Itoh A, et al. (2018). The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 24, 1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldeviano GC, Barin JG, Talor MV, Srinivasan S, Bedja D, Zheng D, Gabrielson K, Iwakura Y, Rose NR, and Cihakova D (2010). Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ. Res. 106, 1646–1655. [DOI] [PubMed] [Google Scholar]
- Barin JG, Rose NR, and Ciháková D (2012). Macrophage diversity in cardiac inflammation: a review. Immunobiology 217, 468–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher B, Schlitzer A, Chen J, Mair F, Sumatoh HR, Teng KW, Low D, Ruedl C, Riccardi-Castagnoli P, Poidinger M, et al. (2014). High-dimensional analysis of the murine myeloid cell system. Nat. Immunol. 15, 1181–1189. [DOI] [PubMed] [Google Scholar]
- Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, Weinstein JS, Licona-Limon P, Schmid ET, Pelorosso F, et al. (2017). Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 356, 1072–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronte V, and Pittet MJ (2013). The spleen in local and systemic regulation of immunity. Immunity 39, 806–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caforio AL, Marcolongo R, Jahns R, Fu M, Felix SB, and Iliceto S (2013a). Immune-mediated and autoimmune myocarditis: clinical presentation, diagnosis and management. Heart Fail. Rev. 18, 715–732. [DOI] [PubMed] [Google Scholar]
- Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, Fu M, Helio T, Heymans S, Jahns R, et al. ; European Society of Cardiology Working Group on Myocardial and Pericardial Diseases (2013b). Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 34, 2636–2648. [DOI] [PubMed] [Google Scholar]
- Camenisch TD, Koller BH, Earp HS, and Matsushima GK (1999). A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J. Immunol. 162, 3498–3503. [PubMed] [Google Scholar]
- Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, Hedrick CC, Cook HT, Diebold S, and Geissmann F (2013). Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell 153, 362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Bracamonte-Baran W, Diny NL, Hou X, Talor MV, Fu K, Liu Y, Davogustto G, Vasquez H, Taegtmeyer H, et al. (2018). Sca-1(+) cardiac fibroblasts promote development of heart failure. Eur. J. Immunol. 48, 1522–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Carey K, and Godowski PJ (1997). Identification of Gas6 as a ligand for Mer, a neural cell adhesion molecule related receptor tyrosine kinase implicated in cellular transformation. Oncogene 14, 2033–2039. [DOI] [PubMed] [Google Scholar]
- Ciháková D, Sharma RB, Fairweather D, Afanasyeva M, and Rose NR (2004). Animal models for autoimmune myocarditis and autoimmune thyroiditis. Methods Mol. Med. 102, 175–193. [DOI] [PubMed] [Google Scholar]
- Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, Clausen BE, Jung S, Greter M, and Becher B (2015). The Cytokine GM-CSF Drives the Inflammatory Signature of CCR2+ Monocytes and Licenses Autoimmunity. Immunity 43, 502–514. [DOI] [PubMed] [Google Scholar]
- Debien E, Mayol K, Biajoux V, Daussy C, De Aguero MG, Taillardet M, Dagany N, Brinza L, Henry T, Dubois B, et al. (2013). S1PR5 is pivotal for the homeostasis of patrolling monocytes. Eur. J. Immunol. 43, 1667–1675. [DOI] [PubMed] [Google Scholar]
- Dimas VV, Denfield SW, Friedman RA, Cannon BC, Kim JJ, Smith EO, Clunie SK, Price JF, Towbin JA, Dreyer WJ, and Kertesz NJ (2009). Frequency of cardiac death in children with idiopathic dilated cardiomyopathy. Am. J. Cardiol. 104, 1574–1577. [DOI] [PubMed] [Google Scholar]
- Diny NL, Baldeviano GC, Talor MV, Barin JG, Ong S, Bedja D, Hays AG, Gilotra NA, Coppens I, Rose NR, and Ciháková D (2017). Eosinophil-derived IL-4 drives progression of myocarditis to inflammatory dilated cardiomyopathy. J. Exp. Med. 214, 943–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, Iwamoto Y, Thompson B, Carlson AL, Heidt T, et al. (2012). Myocardial infarction accelerates atherosclerosis. Nature 487, 325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, et al. (2014). Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40, 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, and Henson PM (1998). Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 101, 890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairweather D, Guilarte TR, and Cooper LT Jr. (2014). Biomarker and more: can translocator protein 18 kDa predict recovery from brain injury and myocarditis? Biomarkers Med. 8, 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo RS, Mani K, and Kitsis RN (2005). Death begets failure in the heart. J. Clin. Invest. 115, 565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung G, Luo H, Qiu Y, Yang D, and McManus B (2016). Myocarditis. Circ. Res. 118, 496–514. [DOI] [PubMed] [Google Scholar]
- Geissmann F, Jung S, and Littman DR (2003). Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19, 71–82. [DOI] [PubMed] [Google Scholar]
- Gordon S, and Taylor PR (2005). Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5, 953–964. [DOI] [PubMed] [Google Scholar]
- Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, and Yona S (2014). Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat. Rev. Immunol. 14, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, Geissmann F, and Hedrick CC (2011). The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat. Immunol. 12, 778–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson J, Vasan RS, Ärnlöv J, Ingelsson E, Lind L, Larsson A, Michaëlsson K, and Sundström J (2011). Biomarkers of extracellular matrix metabolism (MMP-9 and TIMP-1) and risk of stroke, myocardial infarction, and cause-specific mortality: cohort study. PLoS ONE 6, e16185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, et al. (2013). Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38, 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy LM, Perron G, Won SY, Michell-Robinson MA, Rezk A, Ludwin SK, Moore CS, Hall JA, Bar-Or A, and Antel JP (2016). MerTK Is a Functional Regulator of Myelin Phagocytosis by Human Myeloid Cells. J. Immunol. 196, 3375–3384. [DOI] [PubMed] [Google Scholar]
- Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, Sun Y, Da Silva N, Panizzi P, van der Laan AM, et al. (2014). Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 115, 284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettinger J, Richards DM, Hansson J, Barra MM, Joschko AC, Krijgsveld J, and Feuerer M (2013). Origin of monocytes and macrophages in a committed progenitor. Nat. Immunol. 14, 821–830. [DOI] [PubMed] [Google Scholar]
- Heymans S, Schroen B, Vermeersch P, Milting H, Gao F, Kassner A, Gillijns H, Herijgers P, Flameng W, Carmeliet P, et al. (2005). Increased cardiac expression of tissue inhibitor of metalloproteinase-1 and tissue inhibitor of metalloproteinase-2 is related to cardiac fibrosis and dysfunction in the chronic pressure-overloaded human heart. Circulation 112, 1136–1144. [DOI] [PubMed] [Google Scholar]
- Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, Iwamoto Y, Liao R, Zirlik A, Scherer-Crosbie M, et al. (2014). Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res. 114, 1611–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliard BA, Zizzo G, Ulas M, Linan MK, Schreiter J, and Cohen PL (2014). Increased expression of Mer tyrosine kinase in circulating dendritic cells and monocytes of lupus patients: correlations with plasma interferon activity and steroid therapy. Arthritis Res. Ther. 16, R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota H, Izumi M, Hamaguchi T, Sugiyama S, Murakami E, Kunisada K, Fujio Y, Oshima Y, Nakaoka Y, and Yamauchi-Takihara K (2004). Circulating interleukin-6 family cytokines and their receptors in patients with congestive heart failure. Heart Vessels 19, 237–241. [DOI] [PubMed] [Google Scholar]
- Hochholzer W, Morrow DA, and Giugliano RP (2010). Novel biomarkers in cardiovascular disease: update 2010. Am. Heart J. 160, 583–594. [DOI] [PubMed] [Google Scholar]
- Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, Beaudin AE, Lum J, Low I, Forsberg EC, et al. (2015). C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 42, 665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howangyin KY, Zlatanova I, Pinto C, Ngkelo A, Cochain C, Rouanet M, Vilar J, Lemitre M, Stockmann C, Fleischmann BK, et al. (2016). Myeloid-Epithelial-Reproductive Receptor Tyrosine Kinase and Milk Fat Globule Epidermal Growth Factor 8 Coordinately Improve Remodeling After Myocardial Infarction via Local Delivery of Vascular Endothelial Growth Factor. Circulation 133, 826–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda N, Asano K, Kikuchi K, Uchida Y, Ikegami H, Takagi R, Yotsumoto S, Shibuya T, Makino-Okamura C, Fukuyama H, et al. (2018). Emergence of immunoregulatory Ym1+Ly6Chi monocytes during recovery phase of tissue injury. Sci. Immunol. 3, eaat0207. [DOI] [PubMed] [Google Scholar]
- Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, Lang R, Haniffa M, Collin M, Tacke F, et al. (2010). Comparison of gene expression profiles between human and mouse monocyte subsets. Blood 115, e10–e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, Ivanov S, Duan Q, Bala S, Condon T, et al. (2013). Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity 39, 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M, and An J (2018). Viral Myocarditis (StatPearls). [PubMed] [Google Scholar]
- Kubin T, Pöling J, Kostin S, Gajawada P, Hein S, Rees W, Wietelmann A, Tanaka M, Lörchner H, Schimanski S, et al. (2011). Oncostatin M is a major mediator of cardiomyocyte dedifferentiation and remodeling. Cell Stem Cell 9, 420–432. [DOI] [PubMed] [Google Scholar]
- Kubota T, Miyagishima M, Alvarez RJ, Kormos R, Rosenblum WD, Demetris AJ, Semigran MJ, Dec GW, Holubkov R, McTiernan CF, et al. (2000). Expression of proinflammatory cytokines in the failing human heart: comparison of recent-onset and end-stage congestive heart failure. J. Heart Lung Transplant. 19, 819–824. [DOI] [PubMed] [Google Scholar]
- Landsman L, Bar-On L, Zernecke A, Kim KW, Krauthgamer R, Shagdarsuren E, Lira SA, Weissman IL, Weber C, and Jung S (2009). CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood 113, 963–972. [DOI] [PubMed] [Google Scholar]
- Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, Ornitz DM, Randolph GJ, and Mann DL (2014). Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 111, 16029–16034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuschner F, Rauch PJ, Ueno T, Gorbatov R, Marinelli B, Lee WW, Dutta P, Wei Y, Robbins C, Iwamoto Y, et al. (2012). Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J. Exp. Med. 209, 123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Hsiao HM, Higashikubo R, Saunders BT, Bharat A, Goldstein DR, Krupnick AS, Gelman AE, Lavine KJ, and Kreisel D (2016). Heart-resident CCR2+ macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight 1, 87315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Martinez FO, and Gordon S (2014). The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JW, O’Connell JB, Herskowitz A, Rose NR, McManus BM, Billingham ME, and Moon TE; The Myocarditis Treatment Trial Investigators (1995). A clinical trial of immunosuppressive therapy for myocarditis. N. Engl. J. Med. 333, 269–275. [DOI] [PubMed] [Google Scholar]
- Menezes S, Melandri D, Anselmi G, Perchet T, Loschko J, Dubrot J, Patel R, Gautier EL, Hugues S, Longhi MP, et al. (2016). The Heterogeneity of Ly6Chi Monocytes Controls Their Differentiation into iNOS+ Macrophages or Monocyte-Derived Dendritic Cells. Immunity 45, 1205–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schonheit J, Giladi A, David E, Lara-Astiaso D, Lorenzo-Vivas E, Paul F, Chappell-Maor L, Priller J, Leutz A, et al. (2017). Genomic Characterization of Murine Monocytes Reveals C/EBPbeta Transcription Factor Dependence of Ly6C(−) Cells. Immunity 46, 849–862.e7. [DOI] [PubMed] [Google Scholar]
- Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK 3rd, Berdnikovs S, Filer A, Clark AR, Buckley CD, et al. (2014). Nonclassical Ly6C(−) monocytes drive the development of inflammatory arthritis in mice. Cell Rep. 9, 591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, Chen CI, Anekalla KR, Joshi N, Williams KJN, et al. (2017). Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 214, 2387–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra MK, Wang J, Silva C, Mack M, and Yong VW (2012). Kinetics of proinflammatory monocytes in a model of multiple sclerosis and its perturbation by laquinimod. Am. J. Pathol. 181, 642–651. [DOI] [PubMed] [Google Scholar]
- Molawi K, Wolf Y, Kandalla PK, Favret J, Hagemeyer N, Frenzel K, Pinto AR, Klapproth K, Henri S, Malissen B, et al. (2014). Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 211, 2151–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita T, Uzui H, Mitsuke Y, Amaya N, Kaseno K, Ishida K, Fukuoka Y, Ikeda H, Tama N, Yamazaki T, et al. (2017). Association between matrix metalloproteinase-9 and worsening heart failure events in patients with chronic heart failure. ESC Heart Fail. 4, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers JM, Cooper LT, Kem DC, Stavrakis S, Kosanke SD, Shevach EM, Fairweather D, Stoner JA, Cox CJ, and Cunningham MW (2016). Cardiac myosin-Th17 responses promote heart failure in human myocarditis. JCI Insight 1, 85851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahrendorf M, and Swirski FK (2016). Abandoning M1/M2 for a Network Model of Macrophage Function. Circ. Res. 119, 414–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, and Pittet MJ (2007). The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 204, 3037–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olingy CE, San Emeterio CL, Ogle ME, Krieger JR, Bruce AC, Pfau DD, Jordan BT, Peirce SM, and Botchwey EA (2017). Non-classical monocytes are biased progenitors of wound healing macrophages during soft tissue injury. Sci. Rep. 7, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panduro M, Benoist C, and Mathis D (2018). Treg cells limit IFN-g production to control macrophage accrual and phenotype during skeletal muscle regeneration. Proc. Natl. Acad. Sci. USA 115, E2585–E2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, Thomas MJ, Sorci-Thomas MG, and Randolph GJ (2011). Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe−/− mice during disease regression. J. Clin. Invest. 121, 2025–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proto JD, Doran AC, Gusarova G, Yurdagul A Jr., Sozen E, Subramanian M, Islam MN, Rymond CC, Du J, Hook, et al. (2018). Regulatory T Cells Promote Macrophage Efferocytosis during Inflammation Resolution. Immunity 49, 666–677.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pummerer CL, Luze K, Grässl G, Bachmaier K, Offner F, Burrell SK, Lenz DM, Zamborelli TJ, Penninger JM, and Neu N (1996). Identification of cardiac myosin peptides capable of inducing autoimmune myocarditis in BALB/c mice. J. Clin. Invest. 97, 2057–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, Gusarova V, Gromada J, Weinstock A, Moore KJ, et al. (2017). Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J. Clin. Invest. 127, 2904–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, and Taegtmeyer H (2001). Metabolic gene expression in fetal and failing human heart. Circulation 104, 2923–2931. [DOI] [PubMed] [Google Scholar]
- Robbins CS, Chudnovskiy A, Rauch PJ, Figueiredo JL, Iwamoto Y, Gorbatov R, Etzrodt M, Weber GF, Ueno T, van Rooijen N, et al. (2012). Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation 125, 364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Nakagawa K, Sugihara F, Kuwahara R, Ashihara M, Yamane F, Minowa Y, Fukushima K, Ebina I, Yoshioka Y, et al. (2017). Identification of an atypical monocyte and committed progenitor involved in fibrosis. Nature 541, 96–101. [DOI] [PubMed] [Google Scholar]
- Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, et al. (2012). A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90. [DOI] [PubMed] [Google Scholar]
- Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, and Matsushima GK (2001). Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411, 207–211. [DOI] [PubMed] [Google Scholar]
- Seitz HM, Camenisch TD, Lemke G, Earp HS, and Matsushima GK (2007). Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J. Immunol. 178, 5635–5642. [DOI] [PubMed] [Google Scholar]
- Sheng J, Ruedl C, and Karjalainen K (2015). Most Tissue-Resident Macrophages Except Microglia Are Derived from Fetal Hematopoietic Stem Cells. Immunity 43, 382–393. [DOI] [PubMed] [Google Scholar]
- Shi C, and Pamer EG (2011). Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 11, 762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock AT, Hansen JA, Sleeman MA, McKenzie BS, and Wicks IP (2016). GM-CSF primes cardiac inflammation in a mouse model of Kawasaki disease. J. Exp. Med. 213, 1983–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunderkötter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA, and Leenen PJ (2004). Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J. Immunol. 172, 4410–4417. [DOI] [PubMed] [Google Scholar]
- Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, et al. (2007). Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Invest. 117, 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theaker JM, Gatter KC, Heryet A, Evans DJ, and McGee JO (1985). Giant cell myocarditis: evidence for the macrophage origin of the giant cells. J. Clin. Pathol. 38, 160–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan E, Yeap XY, Dehn S, Terry R, Novak M, Zhang S, Iwata S, Han X, Homma S, Drosatos K, et al. (2013). Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ. Res. 113, 1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wermeling F, Karlsson MC, and McGaha TL (2009). An anatomical view on macrophages in tolerance. Autoimmun. Rev. 9, 49–52. [DOI] [PubMed] [Google Scholar]
- Whelan RS, Kaplinskiy V, and Kitsis RN (2010). Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu. Rev. Physiol. 72, 19–44. [DOI] [PubMed] [Google Scholar]
- Wu L, Ong S, Talor MV, Barin JG, Baldeviano GC, Kass DA, Bedja D, Zhang H, Sheikh A, Margolick JB, et al. (2014). Cardiac fibroblasts mediate IL-17A-driven inflammatory dilated cardiomyopathy. J. Exp. Med. 211, 1449–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanez A, Coetzee SG, Olsson A, Muench DE, Berman BP, Hazelett DJ, Salomonis N, Grimes HL, and Goodridge HS (2017). Granulocyte-Monocyte Progenitors and Monocyte-Dendritic Cell Progenitors Independently Produce Functionally Distinct Monocytes. Immunity 47, 890–902.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JJ, Ruddy MJ, Conti HR, Boonanantanasarn K, and Gaffen SL (2008). The interleukin-17 receptor plays a gender-dependent role in host protection against Porphyromonas gingivalis-induced periodontal bone loss. Infect. Immun. 76, 4206–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Yeap XY, Grigoryeva L, Dehn S, DeBerge M, Tye M, Rostlund E, Schrijvers D, Zhang ZJ, Sumagin R, et al. (2015). Cardiomyocytes induce macrophage receptor shedding to suppress phagocytosis. J. Mol. Cell. Cardiol. 87, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler-Heitbrock L (2007). The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J. Leukoc. Biol. 81, 584–592. [DOI] [PubMed] [Google Scholar]
- Zizzo G, Hilliard BA, Monestier M, and Cohen PL (2012). Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J. Immunol. 189, 3508–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuern CS, Müller KA, Seizer P, Geisler T, Banya W, Klingel K, Kandolf R, Bauer A, Gawaz M, and May AE (2013). Cyclophilin A predicts clinical outcome in patients with congestive heart failure undergoing endomyocardial biopsy. Eur. J. Heart Fail. 15, 176–184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microarray Gene Expression Analysis
Raw data generated from Clariom S Arrays were processed using Affymetrix Expression Console Software. CEL files containing feature intensity values were converted into summarized expression values by Partek Genomic Suite Software including background adjustment, quantile normalization and summarization across all chips. All samples passed QC thresholds for hybridization, labeling and the expression of spiked in controls. The variances, both between and within groups, in log2 transformed expression values were analyzed by one-way ANOVA. PCA graph was generated using Partek Genomic Suite Software, heatmaps were generated using TIBCO Spotfire Software. The complete normalized dataset is available on GEO: GSE118861.