

Summary
Microrchidia (MORC) ATPases are critical for gene silencing and chromatin compaction in multiple eukaryotic systems but the mechanisms by which MORC proteins act are poorly understood. Here we apply a series of biochemical, single-molecule and cell-based imaging approaches to better understand the function of the C. elegans MORC-1 protein. We find that MORC-1 binds to DNA in a length-dependent but sequence non-specific manner, and compacts DNA by forming DNA loops. MORC-1 molecules diffuse along DNA, but become static as they grow into foci that are topologically entrapped on DNA. Consistent with the observed MORC-1 multimeric assemblies, MORC-1 forms nuclear puncta in cells, and can also form phase-separated droplets in vitro. We also demonstrate that MORC-1 compacts nucleosome templates. Together these results suggest that MORCs impact genome structure and gene silencing by forming multimeric assemblages to topologically entrap and progressively loop and compact chromatin.
Keywords: MORC, GHKL ATPases, genome organization, DNA binding protein, DNA compaction
Graphical Abstract
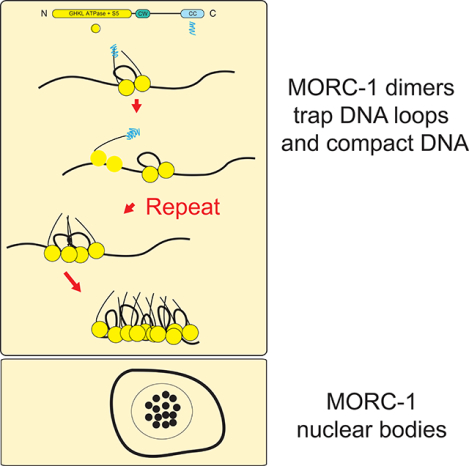
eTOC:
MORC proteins in plants and animals regulate compaction of chromatin, but the molecular mechanism is unknown. Kim et al. demonstrate that the C. elegans MORC-1 dimers can directly compact DNA by topologically entrapping DNA loops. Multiple MORC-1 dimers can also interact to form discrete foci of compacted DNA.
INTRODUCTION
MORC proteins are members of a highly conserved family of GHKL (Gyrase, HSP90, Histidine Kinase, MutL) type ATPases that are found in prokaryotic and eukaryotic organisms (Dong et al., 2018; Iyer et al., 2008). In Arabidopsis thaliana, MORC1 and MORC6 are required for silencing of DNA methylated genes and for large-scale compaction of pericentromeric heterochromatin regions (Moissiard et al., 2012). In mouse, MORC1 is involved in compacting and silencing transposons during male germline development (Pastor et al., 2014). In humans, MORC2 is part of the HUSH complex which is also implicated in transposon silencing (Douse et al., 2018; Tchasovnikarova et al., 2017). In Caenorhabditis elegans (C. elegans), MORC-1 is required for transgene silencing and for maintaining the transgenerational silencing of siRNA-targeted genes in the germline, and morc-1 mutants show visible genome decondensation (Weiser et al., 2017). Despite these genetic and genomic studies demonstrating a role of MORCs in chromatin compaction processes (Douse et al., 2018; Harris et al., 2016; Moissiard et al., 2014; Moissiard et al., 2012; Tchasovnikarova et al., 2017; Weiser et al., 2017), the molecular mechanisms by which MORCs act to compact the genome are not understood.
MORCs generally share a similar domain arrangement, possessing N-terminal GHKL ATPase and S5 domains and a large unstructured region followed by a C-terminal coiled coil domain that is thought to be important in homomer or heteromer formation (Douse et al., 2018; Iyer et al., 2008; Li et al., 2016; Mimura et al., 2010; Takahashi et al., 2007). The ATPase cassette adopts a prototypical GHKL ATPase (Bergerat) fold and dimerizes upon ATP binding, much like other GHKL family members such as bacterial Topoisomerase VIb (Corbett and Berger, 2005; Douse et al., 2018; Dutta and Inouye, 2000; Li et al., 2016). These properties have led to the proposal that MORCs might act as molecular clamps (Li et al., 2016; Mimura et al., 2010), however very little is known about how MORCs interact with DNA or with chromatin.
Here we demostrate that MORCs bind and compact DNA by progressively trapping DNA loops, resulting in large protein assemblies that are topologically trapped on DNA. Compaction in vitro does not require ATP, although it is stimulated by the addition of ATP and especially by addition of a non-hydrolyzable ATP analog. C. elegans MORC-1 is also found in nuclear bodies, and in vitro will undergo liquid-liquid phase separation. We also demonstrate that MORC-1 can compact DNA that has been assembled into chromatin. Together these observations provide mechanistic insight into how MORC family members function across eukaryotes to compact genomes and regulate gene silencing.
RESULTS
MORC-1 binds DNA with little sequence preference and prefers longer DNAs over shorter DNAs.
To study C. elegans MORC-1 in an in vitro reconstituted system, we purified bacterially expressed full length MORC-1, containing both the conserved N-terminal GHKL ATPase domain known to dimerize upon ATP binding and the C-terminal coiled coil domain thought to be important in multimerization (Figure S1A) (Harris et al., 2016; Inoue et al., 1999; Iyer et al., 2008; Li et al., 2016; Mimura et al., 2010; Moissiard et al., 2014; Moissiard et al., 2012; Tchasovnikarova et al., 2017). Native mass spectrometry analysis of the purified protein (Figure S1B) revealed it was well ordered and folded, and was predominantly in equilibrium as a monomer or dimer, with minor populations of trimers and tetramers (Figure S1C), consistent with the observation that plant and mammalian MORCs exist as homomers or heteromers in vivo (Harris et al., 2016; Mimura et al., 2010; Moissiard et al., 2014; Moissiard et al., 2012).
A. thaliana MORC1 was shown to bind DNA (Kang et al., 2012), and we therefore utilized gel shift assays to test the DNA binding activity of full length C. elegans MORC-1. MORC-1 bound radiolabeled 250 base pair (bp) double stranded DNA (Figure 1A). Both cold competitor DNA of the same sequence, or a 250 bp DNA with a different sequence competed equally for binding to the labeled probe, suggesting that MORC-1 has little sequence specificity (Figure 1B). However, a 50 bp DNA derived from the original 250 bp sequence was unable to efficiently compete for binding, suggesting that MORC-1 prefers longer DNAs over shorter DNAs (Figure 1B). To confirm this observation, we performed gel shift assays with a DNA ladder ranging from 100 to 1500 bp. At lower MORC-1 concentrations (< 400 nM), only longer DNAs (sizes larger than 1000 bp) were shifted, but at higher concentrations (> 800 nM) MoRc-1 could also shift shorter DNAs (Figure S1D). Therefore, MORC-1 is capable of binding both long and short DNAs, but exhibits a preference for longer DNAs under conditions where MORC-1 is limiting. In addition, the observation that MORC-1 could bind ladder DNAs derived from multiple plasmids of varying DNA sequences again suggests MORC-1 likely has little sequence preference, although we cannot rule out that MORC-1 may prefer particular DNA sequences not tested here.
Figure 1. MORC-1 is a DNA binding protein.

(A) Protein ([MORC-1] = 100 nM, 200 nM, 400 nM, 800 nM) was incubated with 32P labeled 250 bp DNA probe derived from λ-DNA and run on a 3% agarose gel, then dried before exposure to a phophor screen.
(B) 200 nM MORC-1 was added to 0.5 nM 32P labeled 250 bp λ-DNA that was premixed with cold 250 bp λ-DNA, 250 bp scrambled DNA, or 50 bp λ-DNA probe (1 nM, 10 nM, 20 nM, 50 nM) and processed as described above.
See also Figure S1.
MORC-1 robustly compacts DNA.
C. elegans morc-1 mutants exhibit X chromosome decondensation (Weiser et al., 2017). MORC mutants in other species also exhibit chromosome decompaction phenotypes (Moissiard et al., 2012). To directly test if MORC-1 might be able to compact DNA, we imaged flow-stretched DNAs tethered to a functionalized coverslip via a 3’ biotin neutravidin linkage and labeled at the 5’ end with a quantum dot (Figure 2A) (Graham et al., 2014; Kim and Loparo, 2016). Addition of MORC-1 into the flow cell induced DNA compaction, and the rate of compaction increased linearly with MORC-1 concentration (Figures 2B–2C). Compaction occurred in 150 mM NaCl, but did not occur at NaCl concentrations above 300 mM (Figure S2A). Importantly, compaction was largely reversible at a high salt concentration (500 mM NaCl) (Figure S2B), suggesting that the observed DNA compaction was not the result of non-specific protein aggregation.
Figure 2. MORC-1 compacts DNA using a loop trapping mechanism.

(A) A schematic of the flow stretched DNA assay.
(B) A representative kymograph demonstrating compaction of a quantum dot labelled λ-DNA over time. Scale bars represent 10 seconds and 5 μm.
(C) Effect of ATP on MORC-1 compaction. Data was calculated from 48 different experiments, with a total of 1199 different trajectories. n=85, 48, 98, 122, 66, 84, 57, 97 (without ATP), and n=66, 94, 79, 52, 61, 76, 47, 67 (with 2 mM ATP). Error bars: SE.
(D) AMP-PNP further stimulates DNA compaction. Compaction rates were from two independent experiments for each condition. Error bars represent the SE. n=45, 46, and 56 for experiments with no nucleotide, 2 mM ATP, and 2 mM AMP-PNP, respectively.
(E) Left: schematic of DNA motion capture assay depicting the location of the five EcoRI binding sites. Middle: Plotted trajectories of location of EcoRI-E111Q conjugated quantum dots over time. Below: representative kymograph. [MORC-1] = 40 nM, [ATP] =1 mM.
See also Figures S2 and S3, Video S1.
MORCs are ATPases, and we therefore assayed the effect of adding ATP to the compaction assays. While the addition of ATP was not strictly required for compaction, the addition of ATP stimulated the rate of compaction by ~30% (Figures 2C and S2C–S2D; Supplemental document S1). Moreover, the non-hydrolysable ATP analog adenylyl-imidodiphosphate (AMP-PNP) further stimulated DNA compaction by approximately two-fold relative to ATP (Figure 2D). ATP binding is known to induce GHKL head dimerization (Corbett and Berger, 2005; Douse et al., 2018; Li et al., 2016), and thus this observation suggests that it is ATP binding, rather than hydrolysis, that enables MORC-1 to most efficiently compact DNA.
MORC-1 uses a loop trapping mechanism to compact DNA.
We used a DNA motion capture assay to analyze the mechanism of MORC-1-mediated DNA compaction by following how segments along the DNA length were condensed (Graham et al., 2014; Kim and Loparo, 2016). λ-DNA was labeled at its five EcoRI binding sites by quantum-dot labeled catalytically inactive EcoRI (EcoRIE111Q) (Figure 2E). Compaction initiated from the free end of the DNA and moved sequentially down to the tether point (Figure 2E), consistent with either a DNA loop-trapping mechanism as demonstrated for bacterial Spo0J (ParB), or with a loop extrusion mechanism as shown for yeast condensin (Ganji et al., 2018; Graham et al., 2014). Proteins that compact DNA using either loop forming mechanism are sensitive to the force applied to DNA by flow, and therefore preferentially initiate compaction from the free end of the DNA, which experiences substantially lower drag force compared to the DNA segments closer to the tether point (Graham et al., 2014; Kim and Loparo, 2016).
To directly test whether MORC-1 extrudes DNA loops, we tethered both ends of λ-DNA, allowing for slack in the DNA, and then flowed MORC-1 and ATP into the flow cell orthogonal to the direction of DNA tethering. DNA was imaged using the intercalating DNA stain SYTOX Orange. DNA compaction events occurred on both single and double tethered DNA in the same field of view. Singly tethered DNA was compacted down to the tether point, while doubly tethered became taut over time, showing that MORC-1 could compact both singly and doubly tethered DNAs. In contrast to what was previously shown for condensin that acts via a loop extrusion mechanism (Ganji et al., 2018), we did not see evidence for extruded DNA loops on MORC-1 compacted doubly tethered DNA (Figure S3; Video S1). The absence of extruded loops implies that MORC-1 uses a loop trapping mechanism rather than a loop extrusion mechanism to condense DNA. This interpretation is also consistent with the observation that MORC-1 can compact DNA without the addition of ATP, since loop extrusion is an active ATP-requiring process driven by directional motor activity (Ganji et al., 2018).
MORC-1 forms discrete foci as it compacts DNA.
To investigate MORC-1 behavior as it compacts DNA, we non-specifically labeled MORC-1 with Cy3, at an average of ~1 Cy3 per MORC-1 dimer. Fluorophore labeling did not perturb the ability of MORC-1 to compact DNA (Figure 3A). At concentrations below 2 nM, where the majority of DNA-bound proteins were single MORC-1 dimers, we observed MORC-1 diffusing along DNA (Figure S4A). However, DNA compaction was not observed. Addition of higher concentrations of labeled MORC-1 (> 10 nM) to the flow cell resulted in progressive DNA compaction down to the tether point, along with the formation of static MORC-1 foci on the DNA that grew brighter with time (Figures 3A and S4B; Video S2). Additionally, the number of foci per substrate increased as a function of MORC-1 concentration (Figure S4C). We frequently observed collision events between mobile/static MORC-1 proteins, suggesting that the growth of static foci was in part due to the addition of additional DNA bound MORC-1 proteins (Video S3). However, the relatively fast growth of MORC-1 foci suggests that additional MORC-1 proteins were likely also added to the growing foci from the population in solution. As DNA compaction proceeded, static MORC-1 foci came into close proximity and merged into larger foci (Figure 3A; Video S2). There was a linear relationship between the concentration of MORC-1 and the average number of MORC-1 molecules in each body, as estimated by Cy3 fluorescence (Pearson Correlation coefficient: > 0.99) (Figures 3B and S4B–S4D). These results mirror the linear relationship between MORC-1 concentration and DNA compaction rate, and are consistent with a model in which additional MORC-1 molecules added to existing foci contribute to further DNA compaction. Additionally, the growth of existing MORC-1 foci suggests that initial MORC-1 binding events seed additional MORC-1 binding in part via protein-protein interactions.
Figure 3. MORC-1 forms bodies that grow in size with concentration.

(A) A representative kymograph showing Cy3 labeled MORC-1 cluster formation.
(B) At varying MORC-1 concentrations, foci intensities were quantified at a fixed time point (140 seconds after flowing in sample) and presented as box plots. The number of clusters was n=21, 34, 55, and 34 for [MORC-1] = 2, 5, 10, and 40 nM, respectively. The red line inside each box corresponds to the median value while the bottom and top edges of each box correspond to the 25th and 75th percentiles, respectively. A Black dotted line connecting nearby median values was added for visual guidance and showed displayed a strong linear correlation (Pearson correlation coefficient: r = 0.99996).
MORC-1 forms discrete nuclear foci in vivo and forms phase separated droplets in vitro.
The MORC-1 foci observed on DNA during compaction are reminiscent of in vivo nuclear bodies observed with plant MORCs (Harris et al., 2016; Moissiard et al., 2012). C. elegans MORC-1 was previously observed to be localized adjacent to dense areas of heterochromatin (Weiser et al., 2017). We used Structured Illumination Microscopy (SIM) to visualize the nuclear distribution of C-terminal 3xFLAG tagged MORC-1 expressed under its endogenous promoter in vivo (Weiser et al., 2017). We observed MORC-1 punctate bodies in both mitotically dividing germline stem cells as well as in gonadal nuclei undergoing meiosis (Figure S5A). Consistent with previously published data in adult intestinal nuclei, MORC-1 bodies were mostly adjacent to, but not overlapping with, the most DAPI-dense areas corresponding to heterochromatin (Figure S5A) (Weiser et al., 2017). Imaging these same cell types at higher resolution using stochastic optical reconstruction microscopy (STORM) (Köhler et al., 2017) showed that, in addition to large MORC-1 puncta, a series of smaller bodies could also be seen throughout the nucleus (Fig. 4A). These data are also consistent with observations that MORCs in multiple species form punctate bodies in vivo (Harris et al., 2016; Mimura et al., 2010; Moissiard et al., 2012). Together with the MORC-1 foci formation on flow stretched DNAs, these results suggest that MORC-1 acts in part via higher order oligomerization to cause DNA compaction.
Figure 4. MORC-1 is localized to bodies in vivo and forms condensates in vitro.

(A) MORC-1 are found in nuclear bodies of varying sizes in vivo. MORC-1 mitotic bodies (n=96) are on average (mean) 229 nm; MORC-1 pachytene bodies (n=53) are on average (mean) 170 nm. Scale bar represents 5 μm. (B) MORC-1 undergoes liquid-liquid phase separation. Unlabeled protein was buffer exchanged into 75 mM NaCl and mixed with labeled protein to achieve a final population of 1–2% labeling. Single color droplets were pre-formed by adding a final concentration of 10% PEG 3350. To image dual color droplets, pre-formed droplets were mixed together and incubated at room temperature before imaging. To image dual color MORC-1 on DNA, Cy5-MORC-1 was added to Cy3-MORC-1 preincubated with DNA. White represents areas of signal overlap. Scale bars represent 5 μm.
See also Figure S5.
Some proteins found in nuclear bodies like the heterochromatic protein HP1 have properties of phase separated condensates (Alberti et al., 2019; Boija et al., 2018; Larson et al., 2017; Sabari et al., 2018; Strom et al., 2017). MORC-1 contains a long predicted intrinsically disordered region (IDR) as determined by IUPred2A, and IDRs are known to be able to mediate phase separation (Figure S1A) (Lin et al., 2015; Mészáros et al., 2018). We therefore tested whether purified MORC-1 protein may be able to form phase separated droplets in vitro. Using both confocal and differential interference contrast (DIC) microscopy, we observed MORC-1 phase separated droplets at 75 mM NaCl in the presence of 10% polyethylene glycol 3350 (PEG 3350), a crowding agent that can mimic a crowded cellular environment (Lin et al., 2015; Protter et al., 2018) (Figure 4B). We then tested for droplet fusion to confirm that these were phase-separated droplets (Lin et al., 2015; Protter et al., 2018). Cy3 or Cy5-labeled MORC-1 droplets were pre-formed in 10% PEG 3350, then mixed and observed to form droplets containing both Cy3 and Cy5 dye (Figure 4B). Furthermore, addition of λ-DNA to MORC-1 in 75 mM NaCl without any PEG crowding agent caused MORC-1 to coalesce into fiber-like conformations that are also likely representative of a phase separated state (Figure 4B) (Lin et al., 2015). Thus MORC-1 is able to form phase separated droplets in vitro.
A subset of proteins found in cellular bodies with properties of phase transitioned condensates show dispersion in vivo after exposure to 1,6 hexanediol, a solvent that can interfere with weak hydrophobic interactions (Alberti et al., 2019; Boija et al., 2018; Larson et al., 2017; Sabari et al., 2018; Strom et al., 2017). We thus subjected the same mitotic and meiotic germ cells to 5% 1,6 hexanediol treatment and quantified whether the MORC-1 bodies remained punctate or became diffuse upon treatment. We found that the proportion of nuclei containing puntate MORC-1 bodies remained largely unchanged upon 5% 1,6 hexanediol treatment, even though we observed that the peripheral localization of chromatin became disorganized and the ribonucleoprotein bodies that represented P granules became largely dispersed in the presence of 1,6 hexanediol (Updike et al., 2011; Wheeler et al., 2016) (Figure S5B). Thus, while the in vivo MORC-1 bodies may represent intracellular condensates, these bodies are likely held together by additional complex interactions other than weak hydrophobic interactions (Kroschwald et al., 2015; Kroschwald et al., 2017).
MORC-1 topologically entraps DNA
The observation that MORC-1 nuclear bodies were largely 1,6 hexanediol resistant in vivo suggested that like HP1, these MORC-1 bodies may engage chromatin to produce a more metastable phase separated state in vivo. The presence of additional scaffolding materials like nucleic acids can impact the physical properties of nuclear bodies (Kroschwald et al., 2015; Larson and Narlikar, 2018; Shin and Brangwynne, 2017; Strom et al., 2017; Wheeler et al., 2016). In addition, our in vitro phase transition experiments showed that MORC-1 formed granular fibrils upon incubation with lambda DNA (Figure 4B). Thus, a possible explanation for MORC-1 body resistance to 1,6 hexanediol in vivo is that it is stably engaged with DNA (Alberti et al., 2019; Larson and Narlikar). MORC ATPases have also been proposed to act like other GHKLs that can gate and encircle their substrates, and we therefore sought to test whether MORC-1 could topologically entrap DNA using single molecule experiments (Corbett and Berger, 2005; Douse et al., 2018; Li et al., 2016; Mimura et al., 2010).
We observed that pre-formed, static Cy3-labeled MORC-1 bodies on flow stretched DNAs without quantum dots became mobile when subjected to a high salt wash (500 mM NaCl). Rather than slowly disintegrating, they were frequently observed to move to the free end of DNA as a single unit before disengaging from the DNA and traveling in the direction of buffer flow (Video S4). This implies that MORC-1 may be topologically entrapping DNA, and thus needs to slide off the free end to be released. To test this hypothesis, we attached quantum dots to the free end of DNAs to determine whether this blocked the dissociation of MORC-1 foci. Indeed, quantum dot-labeled DNAs retained MORC-1 foci at a much higher rate (82% retained, n=28) than unlabeled DNAs (16% retained, n=32) (Figure 5; Video S5, Video S6). In addition, MORC-1 foci often accumulated at the free end when quantum dots were present (Figure 5). Static MORC foci could similarly block the movement of mobile foci (Figure S6A; Video S7). These observations are consistent with MORC-1 topologically entrapping DNA.
Figure 5. MORC-1 foci topologically entrap DNA.

Representative kymographs of Cy3-MORC-1 foci movement on quantum dot labeled DNA (top panel, [MORC-1] = 5 nM, [ATP] = 2 mM) and on bare DNA (bottom panel, [MORC-1] = 10 nM, [ATP] = 2 mM). Schematics below the kymographs illustrate the experiment. MORC-1 is applied to the flow cell and allowed to form foci, then subjected to a 500 mM NaCl and 2 mM MgCl2 wash in the presence of 2 mM ATP. Scale bars represent 20 seconds and 5 μm. SYTOX Orange stained DNA is shown to the right of the kymographs to indicate approximate location of foci on the DNA. Note that the quantum dot is not visible any of the displayed spectral channels. In the top panel, the top focus (magenta asterisk) contains approximately 2 MORC-1s and the bottom focus (lavender asterisk) contains approximately 7 MORC-1s. In the bottom panel, the time gap between the two events as marked by the dashed line is 7.2 seconds. The top focus (orange asterisk) has approximately 7 MORC-1s and the bottom (peach asterisk) has approximately 13 MORC-1s. See also Figure S6, and Videos S4, S5, and S6.
To further confirm topological entrapment, we incubated 8xHis-tagged MBP-MORC-1 with plasmid DNAs of various topologies (supercoiled, open circular, or linear), captured MORC-1-DNA complexes with nickel resin, and then washed with either a low salt or high salt wash (Cuylen et al., 2011; Kanno et al., 2015; Murayama and Uhlmann, 2013). MORC-1 remained bound to all forms of DNA after low salt washes (150 mM NaCl) (Figures 1A and 6A–6B). However, upon high salt washes, MORC-1 retained open circular and supercoiled forms of DNA but did not retain significant amounts of linear DNA (Figures 6A–6B). This occurred without added ATP, and addition of ATP or AMP-PNP had only minor effects on retention preferences (Figures S6B–S6E). We also confirmed the preference of MORC-1s for retention of circular DNA by first incubating MORC-1 with nicked circular plasmid DNA, and then linearizing the plasmid via restriction enzyme digest. We found that MORC-1 retained the circular plasmid but lost the linearized DNA (Figure 6B). Together, these results indicate that MORC-1, like eukaryotic SMC proteins (Cuylen et al., 2011; Kanno et al., 2015; Murayama and Uhlmann, 2013), are able to topologically entrap DNAs.
Figure 6. MORC-1 selectively retains circular DNA after high salt washes.

(A) MORC-1 was incubated with DNA of varying topologies and precipitated with magnetic resin, then washed twice before proteinase K elution. Retained DNA is visualized by resolving samples on an 1% 1x TAE agarose gel and post-stained with ethidium bromide. (B) 400 nM MORC-1 was incubated with 50 ng open circular DNA that was made by Nt.BsmAI digestion and precipitated with magnetic resin, then washed twice with a high salt buffer and once with Restriction Enzyme (RE) buffer before digesting with restriction enzymes to alter the topology. The sample was then washed using low or high salt, then eluted using proteinase K. Retained DNA is visualized by resolving samples on an 1% 1x TAE agarose gel and post-stained with ethidium bromide. Note that PstI digestion was not complete and there were trace amounts of open circular plasmid remaining. However, MORC-1 washed with high salt only retains the open circular form and not the linearized material.
See also Figure S6.
MORC-1 can compact nucleosomal DNA.
Because MORCs likely act on chromatin in vivo, we tested whether MORC-1 could compact a chromatinized template in our flow-based single-molecule experiments. We assembled nucleosomes on λ DNA with a single biotinylated end using HeLa core histones, the histone chaperone NAP1, and ATP-utilizing chromatin assembly and remodeling factor protein complex (ACF). The nucleosomal template was then tethered to the flow cell via the biotin linkage and visualized with SYTOX Orange staining. We observed that the length of the chromatinized substrates (mode = 2.86 μm) was approximately 3.5 fold shorter than mock-treated unassembled λ DNA (mode = 10.08 μm) (Figure 7A). This degree of DNA condensation is consistent with a previous report that showed that DNA with repeated positioning sequences with nucleosomes was lengthened by approximately 3.6 fold upon removal of the histone octamers (Brower-Toland et al., 2002). Introduction of 75 nM MORC-1 into the flow cell resulted in robust compaction (Figures 7B–7C). Notably, independent of initial substrate length and thus the amount of nucleosomal compaction, the chromatinized substrates compacted to less than 1 micron in length, demonstrating that MORC-1 can efficiently compact nucleosomal arrays (Figure 7C), although the mean compaction rate (43.5 nm/sec at 75 nM MORC-1 with 2 mM ATP, n=113) was 6~8-fold lower than with bare lambda DNA.
Figure 7. MORC-1 compacts chromatin.

(A) Distribution of substrate lengths under flow. Mode of DNA substrates (n=301) = 10.03 μm; Mode of chromatin substrates (n=415) = 2.84 μm. Substrates were flowed into the flow cell and subsequently visualized with 3 μM SYTOX Orange. (B) Kymograph depicting MORC-1 mediated compaction of a chromatinized template. Scale bars represent 7 μm and 3 seconds, respectively. [MORC-1] = 75 nM. (C) MORC-1 compacts chromatinized substrates of varying lengths. [MORC-1] = 75 nM was flowed into the flow cell and compaction was allowed to proceed. Upper panel; scatter plot of individual trajectories plotted by the initial and final lengths. Bottom panel; projection of the scatter plot, binning the trajectories by final DNA length.
DISCUSSION
Here we demonstrate that MORC-1 can bind to, compact, and topologically entrap DNA. From single molecule flow stretched DNA experiments, we observed that after MORC-1 binds DNA, it seeds the formation of foci containing multiple MORC-1 proteins, which mirrors the MORC-1 nuclear puncta seen in vivo. MORC-1 is also able to compact DNA that has been assembled into nucleosome arrays.
The finding that MORC-1 compaction activity is stimulated by AMP-PNP more than by ATP is consistent with a model in which ATP binding by MORC-1, rather than ATP hydrolysis, promotes a conformation that can loop and compact DNA. MORC family members are known to dimerize through their N-terminal ATPase domain upon ATP binding (Douse et al., 2018; Li et al., 2016), and MORC-1 bound to AMP-PNP should therefore be constitutively dimerized at its N terminus. AMP-PNP likely promotes MORC-1 stability on DNA because it disfavors the opening of the ATPase dimers, preventing topologically entrapped DNA from dissociating from MORC-1. It was recently reported that mutations in human MORC2 that create constitutive dimerization within the N-terminal ATPase domain were also more efficient than wild-type at silencing a transgene reporter, whereas mutations that disrupted ATP binding or dimer formation could not re-establish silencing (Douse et al., 2018). Together with our data, this supports a model in which MORCs utilize ATP metabolism to stimulate the conformational changes that regulate their stability and turnover on DNA.
MORC-1 demonstrates a preference for binding long DNAs over shorter DNAs. This preference can be explained by our observation that MORC-1 dimers can diffuse along DNA at low concentrations, and forms assemblages of multiple MORC-1 molecules on DNA at higher concentrations. Thus longer DNA substrates are able to, on average, load more MORC-1 molecules. The observation that MORC dimers can freely diffuse, but then become static before growing into larger foci suggests that a conformational change occurs, likely coupled with initial DNA loop trapping, at the time that MORC-1 becomes statically localized on DNA. We propose that this conformational change also stimulates the interaction of MORC-1 with other freely diffusing MORC-1 molecules, recruiting them to static MORC-1 foci to initiate further DNA compaction. Both DNA compaction and the number of MORC-1 molecules per body scale linearly with MORC-1 concentration, suggesting an ordered process in which additional MORC-1 molecules recruited to static foci contribute to additional steps of DNA looping and compaction. This process eventually leads to assemblages of MORC-1 molecules that stably compact DNA.
Though MORC-1 preferred longer over shorter DNAs, it appeared to display little preference for particular sequences, as it was able to bind to different DNA templates, including a DNA ladder composed of many different sequences. It therefore seems unlikely that MORC-1 would use DNA sequence as the basis for selecting its chromatin targets in vivo, which is consistent with the view that MORCs in different organisms act as epigenetic regulators that silence targets such as transposons that are composed of a diverse array of sequences (Moissiard et al., 2012; Pastor et al., 2014). It seems more likely that MORCs are guided to their chromatin targets by other proteins or by epigenetic marks. In this regard, one well understood example is mouse MORC3 which contains a CW domain that binds to H3K4 trimethylation in vitro, and which shows co-localization with H3K4 trimethylation in vivo (Li et al., 2016). C. elegans MORC-1 also contains a CW domain, but it is currently unknown whether it serves to bind methylated histones.
We observed that MORC-1 formed nuclear foci in vivo, and that the MORC-1 protein contains a large intrinsically disordered region and could form phase separated droplets in vitro. Plant MORCs also form prominent nuclear bodies in vivo (Harris et al., 2016; Moissiard et al., 2012), and also contain predicted intrinsically disordered regions, suggesting that these features may be generally important for the function of eukaryotic MORC. Several chromatin regulators such as HP1 and Mediator have been shown to assemble into condensates to create an epigenetic nuclear microenvironment that enforces a particular epigenetic state (Boija et al., 2018; Larson et al., 2017; Sabari et al., 2018; Shin et al., 2018; Strom et al., 2017). Similarly, we propose that MORC-1 may first bind, topologically entrap, and locally compact its chromatin targets, but also form intracellular condensates to concentrate MORC-1 target loci into the observed in vivo MORC-1 bodies within nuclei.
In summary, the results of this study suggest that MORC-1 acts via a mechanism to trap a loop of DNA and then multimerize upon DNA binding to form topologically entrapped foci to cause stable chromatin compaction. Given that eukaryotic MORC proteins are highly conserved and evolved from ancient prokaryotic restriction-modification systems (Iyer et al., 2008), it is likely that other plant and animal MORCs use a similar mechanism to enforce chromatin compaction.
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Steve E. Jacobsen (jacobsen@ucla.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C. elegans were maintained using standard procedures at 20°C and fed OP50 E. coli. The morc-1∷3xflag strain used in this study is listed in the Key Resource Table.
METHOD DETAILS
Cloning and Purification of 8x-His-MBP-CeMORC
C. elegans MORC-1 coding sequence (CDS) was cloned into the pRSF vector with an 8xHis MBP tag at the N terminus using In-Fusion cloning (Takara 638920). Plasmid was transformed into BL-21 (DE3) Rosetta2 cells (Novagen) and grown at 37°C, induced with 0.1 mM isopropyl p-D-1-thiogalactopyranoside and moved to 16°C for overnight expression. Cells were harvested then lysed by sonication in 20 mM Tris-Cl pH 7.5, 500 mM NaCl, 10 mM imidazole, 5 mM DTT, and protease inhibitors. Lysate was clarified by centrifugation and supernatant was applied to Ni-NTA agarose (Qiagen). After binding, resin was washed with 20 mM Tris-Cl pH 7.5, 500 mM NaCl, 30 mM imidazole, 5 mM DTT, then treated with at least 2 units of apyrase (NEB M0398L) prior to elution except for the experiment shown in Supplementary Fig. 2. Protein was eluted from the resin then dialyzed against 20 mM Tris pH 7.5, 150 mM NaCl, 5 mM DTT, 10% glycerol. Sample was then subjected to size exclusion chromatography (Superdex 200) in dialysis buffer. Fractions were pooled and concentrated in an Amicon Ultra-15 (Millipore) for storage.
Gel shift assays
Probe was 32P labeled with T4 PNK (NEB) and used at a final concentration of 0.45 nM unless otherwise indicated. Protein and probe were incubated at 23°C for 30 minutes in 20 mM Tris pH 7.5, 50 mM NaCl, 1 mM DTT, 1 mM MgCl2, 0.1 mg/ml BSA, then resolved on a 3% 0.5X TBE agarose gel and visualized by exposure to a phosphorscreen unless otherwise indicated. All images were collected with a Typhoon Imager 9000 (GE Amersham). For the ladder gel shift, a 100 bp double stranded DNA ladder (NEB N3231S) was used as substrate, and visualized by SyBR gold staining (Invitrogen S11494).
Native mass spectrometry data collection
Protein samples (~0.5–1 μM) were buffer exchanged into 150 mM ammonium acetate, pH 7.5 using MicroBioSpin6 columns (Bio-Rad 7326221). Native mass spectrometry experiments were carried out on an Exactive Plus EMR mass spectrometer (Thermo Fisher Scientific), which was calibrated in the extended mass range with 5 mg/mL CsI prepared in water. Samples were sprayed from borosilicate capillaries (NanoES spray capillaries, borosilicate; Thermo Fisher Scientific) using a flow rate of 5–40 μL/min, and analyzed in positive ion mode. Experimental parameters were optimized for each run but were generally: spray voltages, 0.8–1.5 kV; injection flatapole, 5; interflatapole lens, 5; bent flatapole, 5; transfer multipole, +4 to −4; C-trap entrance lens, −10 to +10; source DC offset, 25 V; fragmentation collision energy, 20–150 and collision-induced dissociation, 5–150; injection times, 50–200 μs; trapping gas pressure, 7.5; resolution, 17,500 arbitrary units; mass range, 500–20,000 m/z; capillary temperature, 250 °C; S-lens RF value was set to 200 V; microscan s, 10; and automatic gain control was set to 1e6.
DNA substrate preparation
DNA substrates were prepared as previously described (Kim and Loparo, 2016). Briefly, to prepare a DNA substrate with a biotin at one end and a quantum dot at the other end, a biotinylated BL2-oligo (5’-ggg cgg cga cct/BioTEG/-3’) and an oligo with digoxigenin (5’-agg tcg ccg ccc aaa aaa aaa aaa/Digoxigenin/-3’) was annealed to each cos site of genomic bacteriophage λ-DNA (NEB N3013S). The biotin at one end of the substrate was used for surface-tethering of the DNA to a neutravidin coated coverslip while digoxigenin was used for labeling the DNA with anti-digoxigenin antibody-conjugated quantum dots (Thermo Fisher). The DNA substrate used in DNA motion capture experiments was constructed by annealing the BL1 oligo (5’-agg tcg ccg ccc/BiotinTEG/-3’) to λ-DNA.
Single-molecule flow-stretching assays
Coverglass passivation and flow cell construction and were performed as previously described (Kim and Loparo, 2016). For this study coverslips were functionalized with 4% biotinylated PEG. A home-built total internal reflection fluorescence (TIRF) microscope was used as described in Graham et al. (Graham et al., 2017). DNA flow stretching experiments were performed by first preincubating the λ-DNA substrate with a biotin and a digoxigenin at each end with anti-digoxigenin antibody-conjugated quantum dot at room temperature (anti-digoxigenin antibody: Roche 11 214 667 001, Qdot 605 antibody conjugation kit: Thermo Fisher Scientific Q22001MP). 0.25 mg/mL neutravidin was applied to the flow cell and unbound neutravidin was washed from the flow cell after 5~10 minutes by EcoRI binding buffer (EBB buffer: 10 mM Tris, pH 8.0, 150 mM NaCl, 10 mM MgCl2) that contains 0.2 mg/mL BSA. Quantum-dot labeled biotinylated λ-DNA was flowed into the flow cell at a rate of 20–30 μL/min, and unbound excess DNA was washed away using MORC-1 sample buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 2 mM MgCl2). MORC-1 stock was diluted to the desired concentration by adding MORC-1 sample buffer either in the absence or presence of 2 mM ATP, and then flowed into the flow cell at a rate of 50 μL/min. The movies of flow-stretching assays were recorded using MicroManager (Edelstein et al., 2014).
DNA motion capture assay
The assay was performed as previously described (Kim and Loparo, 2016; Kim and Loparo, 2018). Briefly, catalytically-inactive EcoRI (6xHis-EcoRI E111Q; See Graham et al. 2014) was incubated at room temperature for 30 minutes with anti-His-tag antibody-conjugated quantum dot 605 (Thermo Fisher Scientific Q22001MP) with a two-to-one ratio in 20 mM Tris, pH 7.5, 100 mM NaCl, and 0.1 mg/mL bovine serum albumin. Then, the mixture was incubated for 30 minutes at room temperature with λ-DNA where one end was biotinylated through a biotin oligo (BL1: 5’-agg tcg ccg ccc/BiotinTEG/-3’). In parallel a surface-passivated flow cell was treated with 0.25 mg/mL neutravidin for 5~10 minutes and unbound excess neutravidin was washed out with EcoRI binding buffer (EBB buffer) that contained 0.2 mg/mL BSA. Subsequent steps were identical to those described above in the section entitled “Single-molecule flow-stretching assays” with the exception of site-specifically quantum-dot labeled λ-DNA.
Double-tethered DNA assay
A flow cell was constructed by sandwiching a cross-shaped (1.8 mm and 2.5 mm widths, respectively) channel made out of double-sided tape (0.12 mm thickness) between a passivated coverglass and a quartz slide. PE60 tubes were used as inlet and outlet tubing for each channel. 0.25 mg/ml of neutravidin (Thermo Scientific 31000) was added to the flow cell using a gel loading pipet tip. After 6–7 minutes of incubation, EBB buffer (10 mM Tris pH 8.0, 150 mM NaCl, 10 mM MgCl2, 0.2 mg/ml bovine serum albumin) was flowed in using a syringe pump to wash away excess neutravidin. 180 μL of the 400-fold diluted BL1-DNA substrate was flowed in at a rate of 30 μL/min and allowed to incubate for 2 minutes in the absence of flow. Unbound DNAs were then washed away by flowing in 140 μL of MORC sample buffer at a rate of 100 μL/min. In order to tether the other end of DNA to the flow cell, 380 μL of 100 nM BL2-oligo was flowed in at 30 μL/min, and 120 μL of MORC-1 sample buffer was immediately flowed in at 100 μL/min to remove unbound BL2-oligo. DNA compaction was observed by adding 80 nM MORC-1 and 6 nM SYTOX-Orange to the flow cell using the channel perpendicular to DNA tethering in the presence of 2 mM ATP.
Chromatin assembly and compaction assay
Chromatin compaction assays were imaged on a total internal reflection fluorescence microscope (Olympus). Biotinylated DNA was chromatinized using the Active Motif chromatin assembly kit (Active Motif 53500). Assembly was checked by MNAse digestion. Substrate was diluted into EBB+BSA and introduced into the flow cell using the parameters described above. 75 nM MORC-1 was diluted into MORC sample buffer as described above supplemented with 2 mM ATP, and introduced into the flow cell also using parameters described above, and visualized by 3 nM SYTOX Orange staining. Chromatin compaction movies were recorded.
Circular DNA binding assay
Unless otherwise noted, 2.4 μM protein was incubated for 30 minutes at room temperature with a pBR322 derivative of varying topologies in 20 mM Tris, pH 7.5, 150 mM NaCl, 1mM DTT). Topology was altered by restriction enzyme digest. Protein was precipitated using Ni-NTA magnetic agarose (Qiagen 36113), then washed 2x with either a high salt (600 mM NaCl) or a low salt buffer (150 mM NaCl). Reactions were quenched with a stop buffer (20 mM Tris pH 8, 2% SDS, 1 mM EDTA) and samples were eluted by proteinase K digestion. For the re-linearization assay, 400 nM MORC-1 was bound to 50 ng total pBR322 pre-modified with Nt.BsmAI. Protein was precipitated as described above, then washed with 20 mM Tris pH 7.5, 500 mM NaCl, 1 mM DTT, then buffer exchanged into a Restriction Enzyme (RE) buffer (New England Biosciences CutSmart Buffer, B7204) and digested for 30 minutes room temperature, then washed again with either a low salt buffer (150 mM NaCl) or a high salt buffer (500 mM NaCl.) For assays with nucleotide, all buffers were supplemented with 4 mM MgCl2. Reactions were loaded on a 1% 1xTAE gel and run overnight at room temperature, then post stained using ethidium bromide, and imaged using a Gel Logic 212 Pro (Carestream).
Protein labeling with fluorescent dyes
Proteins were incubated with NHS-Cy3 or NHS-Cy5 dye (Lumiprobe 11020, 13020) at 4°C. Labeled protein was separated from free dye using Biospin 6 columns (Biorad). Cy-dye labeling efficiency was quantified using a Nanodrop; final concentrations were determined by averaging three readings.
Liquid droplet sample preparation and imaging
Samples were buffer exchanged using Micro Bio-spin P-30 gel columns (Biorad 732–6223) into 20 mM Tris pH 7.5, 150 mM NaCl, 5mM DTT. Concentration was measured three times using a NanoDrop spectrophotometer (Thermo Fisher). Protein was diluted to the 75 mM NaCl. Final amounts: 10% PEG or 500 ng total, or 0.8 nM final, λ-DNA (NEB N3011S) with a final unlabeled protein concentration of 4.3 uM with 1–2% Cy-labeled protein acting as tracer. Final reaction volume was 20 μL. Samples were imaged using a Zeiss LSM 880 with spectral imaging at 63X magnification. Signals from Cy3 and Cy5 were linear unmixed in Zen 9.0 (Zeiss).
Gonad dissection and immunostaining for SIM
Gravid adult C. elegans were dissected in egg buffer (25 mM HEPES pH 7.4, 118 mM NaCl, 48 mM KCl, 2 mM EDTA, 0.5 mM EGTA), containing 15 mM sodium azide and 0.1% Tween-20. Samples were fixed in 1% formaldeyhyde in egg buffer for 10 seconds followed by freeze-cracking in liquid nitrogen, 1 min methanol fixation at −20°C, and three washes in PBS T. Slides were then incubated with primary mouse anti-FLAG antibody (1:100, Sigma F1804) in a humid chamber overnight at 4°C or 2 h at room temperature. Slides were washed three times in PBST and then incubated with secondary antibody (1:300, Invitrogen AlexaFluor 555 goat anti-mouse) in a humid chamber for 2 h at room temperature. Slides were washed three times in PBST, stained with 0.5 μg/mL DAPI in PBST for 15 min, washed in PBST, and mounted with Vectashield (Vectorlabs H-1000).
Structured illumination and image processing
Structural Illumination Microscopy (SIM) was performed with Zeiss ELYRA PS.1 system (Carl Zeiss, Germany) equipped with a 100x/1.46 oil immersion objective and a PCO Edge 4.2 camera. Excitation wavelengths used were 405 nm and 561 nm. Images were acquired after 5 rotations and 5 standard shifts of the grating pattern. Image processing was carried out using Zen 2.1 SP3 software. For wild type, n (mitotic)=11, n (pachytene)=12; For RNAi, n (mitotic)= 3, n (pachytene)=4.
STORM imaging
Immunofluorescence of extruded gonads was performed as previously described with minor modifications (Köhler et al., 2017). Briefly, germline extrusion was performed in egg buffer (25 mM HEPES pH 7.4, 118 mM NaCl, 48 mM KCl, 2 mM EDTA, 0.5 mM EGTA), containing 30 mM sodium azide and 0.1% Tween-20. Samples were fixed in 1% formaldeyhyde in egg buffer for 10 seconds followed by a 1 min methanol fixation at −20°C. Primary mouse anti-FLAG antibody used was 1:100 (Sigma F1804) in normal goat serum and PBST and the secondary antibody was used at 1:100 (Invitrogen AlexaFluor 647 goat antimouse) in PBST. All washes and staining were performed in suspension. Germlines were mounted in imaging buffer (Tris·HCl, pH 7.5, containing 100 mM cysteamine, 5% glucose, 0.8 mg/mL glucose oxidase, and 40 μg/mL catalase) and imaged immediately. At least n (mitotic) =8 germlines and n (pachytene)=11 germlines were imaged. Scale bar represents 5 μm.
STORM image processing
STORM was performed with a Zeiss ELYRA PS.1 system (Carl Zeiss, Germany) equipped with a 100x/1.46 oil immersion objective and a PCO Edge 4.2 camera. The 647-nm laser was used to excite fluorescence from antibody-labeled proteins after photobleaching them into the dark state. The 405-nm laser was adjusted during image acquisition. Images were acquired with 43.55 ms exposure time per frame for >13,000 frames. Events that emit fewer than 500 photons or that were not resolved to within 50 nm were discarded. Image processing was performed using Zen 2.1 SP3 software.
Hexanediol treatment and imaging
Gravid adult C. elegans were dissected in egg buffer (118 mM NaCl, 48 mM KCl, 2 mM EDTA, 0.5 mM EGTA, 25 mM HEPES [pH 7.4]), containing 15 mM sodium azide and 0.1% Tween-20. Samples were incubated briefly in either 1x egg buffer or 5% 1,6-hexanediol in egg buffer then fixed in 1% formaldeyhyde in egg buffer for 10 sec followed by a 1 min methanol fixation at −20°C. Primary mouse anti-FLAG antibody (Sigma F1804) was used at 1:100 and rabbit anti-CSR-1 was used at 1:200 in normal goat serum and PBST. The secondary antibody was used at 1:300 (Invitrogen AlexaFluor 555 goat antimouse and 488 goat anti-rabbit) in PBST. All washes and staining were performed in suspension. Germlines were stained with DAPI (0.5 Mg/mL) then mounted with Vectashield (Vectorlabs H-1000). Images were acquired on a Zeiss LSM700 confocal microscope at 63x magnification. Image processing was performed using Zen SP5 software. Experiments were performed in biological triplicate. A total of n=144 nuclei were quantified in 0% 1,6-hexanediol, and n=72 nuclei were quantified in 5% 1,6-hexanediol.
QUANTIFICATION AND STATISTICAL ANALYSIS
Native mass spectrometry data analysis
MagTran (Zhang and Marshall, 1998) and PeakSeeker (Lu et al., 2015) were used to process collected spectra. The m/z values were converted to charge state envelopes. The zero-charge state of each species was calculated from the charge state envelopes. Primary sequences were input into ExPASy ProtParam (web.expasy.org/protparam) to calculate average protein masses.
Quantification of flow-stretching assays
DNA compaction trajectories were determined by fitting the positions of quantum dots using Gaussian fitting with custom-written MATLAB (MathWorks) codes that have provided in the previous publication (Kim and Loparo, 2016). From the trajectories, the positions and time points of the compaction initiation and completion were determined and its compaction rates were quantified.
Chromatin compaction data analysis
MORC-1 mediated chromatin compaction was determined by measuring the change in the flow stretched DNA length using custom-written MATLAB (MathWorks) code (Song et al., 2016). Briefly, to improve the signal-to-noise ratio, five frames (corresponding to 0.5 s of the movie) were averaged. A region of interest (ROI) containing fluorescence signal was determined manually for each DNA molecule. For each averaged image, the pixels in the ROI were summed along the short axis of the DNA molecule to generate a fluorescence intensity line profile. The position of the free DNA end was then determined in each averaged image by applying a threshold of 50% of the maximum intensity after subtraction of the baseline; linear interpolation was used to determine this position to sub-pixel resolution. A similar procedure was followed to determine the position of the tethered end of the DNA, except that all line profiles were averaged and the position was determined from the resulting averaged profiles. Rates of compaction were determined from DNA length trajectories as described for quantum dot labeled DNAs.
STORM data analysis
The longest diagonal distance of each MORC-1 punctate was measured by ImageJ (Schneider et al., 2012). Ninety six and fifty two random but discrete puncta of mitotic and meiotic puncta were measured respectively.
Hexanediol data analysis
Quantification was performed in a binary fashion. Based upon the red signal (anti-FLAG), MORC-1 was categorized as either punctate (1) or diffuse (0) as determined by eye. To remove bias, in each germline, the six most proximal pachytene nuclei were used for quantification. The same nuclei were used for P granule quantification for consistency, which was also done in a binary fashion. If one P granule remained, that nucleus was designated as “P granules present.”
Quantification of circular DNA binding assay
Quantification was performed using Fiji (Schindelin et al., 2012). Intensities were averaged if there were replicates within the experiment, and normalized to the value obtained for open circular.
Supplementary Material
Supplemental Document 1 | Raw analysis data for MORC-1 compaction rates (Related to Figure 2).
The Excel document contains raw analysis data for MORC-1 compaction rates. Each tab represents the MORC-1 concentration, and it contains results both in the absence and presence of ATP. In the Summary tab, the information on the compaction rate fold increases in the presence of ATP is included as well.
Video S1 | MORC-1 does not extrude DNA loops. (Related to Figure 2 and S3).
Both ends of bacteriophage lambda DNA were tethered on the microfluidic flow cell with slack. Subsequent addition of 80 nM MORC-1 and 2 mM ATP led to DNA compaction. While the two DNAs became taut, no extruded loops were observed. Dimension: 18.7 μm Ü 8.3 μm. Movie speed: 6x real-time.
Video S2 | MORC-1 forms foci on DNA. (Related to Figures 3 and S4, Video S3).
Cy3-labeled MORC-1 foci appear on a flow stretched DNA and grow in size over time. As DNA compaction proceeds, they eventually merge into one large focus. [Cy3-MORC-1] = 40 nM, [ATP] = 2 mM, Flow direction: upward, Movie speed: 4x real-time.
Video S3 | Formation and growth of MORC-1 foci. (Related to Figures 3 and S4, Video S2).
Diffusing MORC-1 molecules can form static foci. Collisions between sliding molecules and static foci lead to apparent growth of foci, although free MORC-1 molecules are also likely recruited from solution. Note that collisions among foci are also involved in the growth of foci. [Cy3-MORC-1] = 10 nM, [ATP] = 2 mM, Flow direction: upward, Speed: 20 frames per second = 4x-real time.
Video S4 | Static MORC-1 foci are dislodged upon high salt wash. (Related to Figure 5).
Cy3-labeled MORC-1 foci were pre-formed on flow stretched DNA, then subjected to a high salt wash (500 mM NaCl, 2 mM MgCl2) in the presence of 2 mM ATP. The high salt wash causes static MORC-1 foci to quickly move along the DNA in the flow direction and appears to cause them to slide off the DNA. Flow stretched DNAs are not quantum dot labeled in this example. [Cy3-MORC-1] = 10 nM, [ATP] = 2 mM, Flow direction: upward, Movie speed: 3x real-time.
Video S5 | MORC-1 foci are retained on DNA end labeled with a QDot. (Related to Figure 5).
As in Video S4 MORC-1 foci were pre-formed on DNA and then subjected to high salt wash. The presence of a quantum dot at the DNA ends blocks dissociation of MORC-1 foci. Note that the quantum dot is not visible in this spectral channel. Movie speed: Flow stretched DNAs are quantum dot labeled in this example. [Cy3-MORC-1] = 5 nM, [ATP] = 2 mM, Flow direction: upward, 20 frames per second = 4x real-time.
Video S6 | MORC-1 foci are not retained on DNA without QDots. (Related to Figure 5).
Pre-formed foci slide along and dissociate from DNA upon high salt wash. [Cy3-MORC-1] = 10 nM, [ATP] = 2 mM, Flow direction: upward, Flow stretched DNAs are not quantum dot labeled in this example. 20 frames per second = 4x-real time.
Video S7 | Static MORC-1 foci act as a roadblock to diffusing foci. (Related to Figure 5, Figure 6, and Figure S6).
In this movie, a static MORC-1 focus is capable of preventing a mobile focus from dissociating from the free end of DNA. Flow stretched DNAs are not quantum dot labeled in this example. [Cy3-MORC-1] = 10 nM, [ATP] = 2 mM, Flow direction: upward, 20 frames per second = 4x-real time.
Highlights:
C. elegans MORC-1 traps DNA loops
Recruitment of additional MORC-1s cause further loop trapping and DNA compaction
MORC-1 assemblages become topologically entrapped on DNA
MORC-1 forms discrete foci in vivo and can phase transition in vitro
ACKNOWLEDGMENTS
We thank Michael Carey, Reid Johnson, Geeta Narlikar, Sridhar Mandali, and Elizabeth Thrall for discussion and advice, Sean Carney for assistance with reagent preparation, and Alison Frand and Sophie Katz for guidance and assistance with microscopy. The supercoiled pBR322 derivative was a gift from Sridhar Mandali and Reid Johnson. The MBP vector was a gift from Andrew Ah Young. This work was supported by National Institutes of Health (NIH) Grant 1R35GM130272 and a grant from the W. M. Keck Foundation (S.E.J); NIH R01 GM114054 (J.J.L) and a John and Virginia Kaneb Fellowship (J.J.L); the University of Texas Rio Grande Valley Start-up Grant and the University of Texas System’s Rising STARs Award (H.J.K.); NIH grant P41GM103481 and a University of California, San Francisco Research Resource Program (RRP) Shared Equipment Award (A.L.B.) for the purchase of the Exactive EMR mass spectrometer; a Ruth L. Kirschstein National Research Service Award GM007185 and a University of California, Los Angeles Dissertation Year Fellowship (L.Y); a NIH R01 GM129301 (J.K.K). S.E.J is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA AND CODE AVAILABILITY
All codes are available upon request. Raw data have been deposited to Mendeley Data (http://dx.doi.org/10.17632/876gx33wm2.1).
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alberti S, Gladfelter A, and Mittag T (2019). Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 176, 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM, et al. (2018). Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 175, 1842–1855.e1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower-Toland BD, Smith CL, Yeh RC, Lis JT, Peterson CL, and Wang MD (2002). Mechanical disruption of individual nucleosomes reveals a reversible multistage release of DNA. Proceedings of the National Academy of Sciences 99, 1960–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett KD, and Berger JM (2005). Structural Dissection of ATP Turnover in the Prototypical GHL ATPase TopoVI. Structure 13, 873–882. [DOI] [PubMed] [Google Scholar]
- Cuylen S, Metz J, and Haering CH (2011). Condensin structures chromosomal DNA through topological links. Nature Structural & Molecular Biology 18, 894–901. [DOI] [PubMed] [Google Scholar]
- Dong W, Vannozzi A, Chen F, Hu Y, Chen Z, Zhang L, and Pritham E (2018). MORC Domain Definition and Evolutionary Analysis of the MORC Gene Family in Green Plants. Genome Biology and Evolution 10, 1730–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douse CH, Bloor S, Liu Y, Shamin M, Tchasovnikarova IA, Timms RT, Lehner PJ, and Modis Y (2018). Neuropathic MORC2 mutations perturb GHKL ATPase dimerization dynamics and epigenetic silencing by multiple structural mechanisms. Nature communications 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R, and Inouye M (2000). GHKL, an emergent ATPase/kinase superfamily. Trends in Biochemical Sciences 25, 24–28. [DOI] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, and Stuurman N (2014). Advanced methods of microscope control using μManager software. Journal of Biological Methods 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganji M, Shaltiel IA, Bisht S, Kim E, Kalichava A, Haering CH, and Dekker C (2018). Real-time imaging of DNA loop extrusion by condensin. Science 360, 102–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TG, Wang X, Song D, Etson CM, van Oijen AM, Rudner DZ, and Loparo JJ (2014). ParB spreading requires DNA bridging. Genes & development 28, 1228–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TGW, Walter JC, and Loparo JJ (2017). Ensemble and Single-Molecule Analysis of Non-Homologous End Joining in Frog Egg Extracts In DNA Repair Enzymes: Cell, Molecular, and Chemical Biology, Eichman BF, ed. (Academic Press; ), pp. 233–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CJ, Husmann D, Liu W, Kasmi FE, Wang H, Papikian A, Pastor WA, Moissiard G, Vashisht AA, Dangl JL, et al. (2016). Arabidopsis AtMORC4 and AtMORC7 Form Nuclear Bodies and Repress a Large Number of Protein-Coding Genes. PLOS Genetics 12, e1005998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue N, Hess KD, Moreadith RW, Richardson LL, Handel MA, Watson ML, and Zinn AR (1999). New gene family defined by MORC, a nuclear protein required for mouse spermatogenesis. Human Molecular Genetics 8, 1201–1207. [DOI] [PubMed] [Google Scholar]
- Iyer LM, Abhiman S, and Aravind L (2008). MutL homologs in restriction-modification systems and the origin of eukaryotic MORC ATPases. Biology direct 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H-G, Woo Choi H, von Einem S, Manosalva P, Ehlers K, Liu P-P, Buxa SV, Moreau M, Mang H-G, Kachroo P, et al. (2012). CRT1 is a nuclear-translocated MORC endonuclease that participates in multiple levels of plant immunity. Nature communications 3, 1297. [DOI] [PubMed] [Google Scholar]
- Kanno T, Berta, Davide G, and Sjögren C (2015). The Smc5/6 Complex Is an ATP-Dependent Intermolecular DNA Linker. Cell Reports 12, 1471–1482. [DOI] [PubMed] [Google Scholar]
- Kim H, and Loparo JJ (2016). Multistep assembly of DNA condensation clusters by SMC. Nature communications 7, 10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, and Loparo JJ (2018). Observing Bacterial Chromatin Protein-DNA Interactions by Combining DNA Flow-Stretching with Single-Molecule Imaging. In Bacterial Chromatin, pp. 277–299. [DOI] [PubMed] [Google Scholar]
- Köhler S, Wojcik M, Xu K, and Dernburg AF (2017). Superresolution microscopy reveals the three-dimensional organization of meiotic chromosome axes in intact Caenorhabditis elegans tissue. Proc Natl Acad Sci USA 114, E4734–E4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroschwald S, Maharana S, Mateju D, Malinovska L, Nuske E, Poser I, Richter D, and Alberti S (2015). Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. eLife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, and Narlikar GJ (2017). Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature 547, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AG, and Narlikar GJ (2018). The Role of Phase Separation in Heterochromatin Formation, Function, and Regulation. Biochemistry 57, 2540–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Yen L, Pastor WA, Johnston JB, Du J, Shew CJ, Liu W, Ho J, Stender B, Clark AT., et al. (2016). Mouse MORC3 is a GHKL ATPase that localizes to H3K4me3 marked chromatin. Proceedings of the National Academy of Sciences of the United States of America 113, E5108–5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Protter DSW, Rosen MK, and Parker R (2015). Formation and Maturation of Phase Separated Liquid Droplets by RNA Binding Proteins. Molecular cell 60, 208–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Trnka MJ, Roh S-H, Robinson PJJ, Shiau C, Fujimori DG, Chiu W, Burlingame AL, and Guan S (2015). Improved Peak Detection and Deconvolution of Native Electrospray Mass Spectra from Large Protein Complexes. Journal of The American Society for Mass Spectrometry 26, 2141–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mészáros B, Erdős G, and Dosztányi Z (2018). IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Research 46, W329–W337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimura Y, Takahashi K, Kawata K, Akazawa T, and Inoue N (2010). Two-step colocalization of MORC3 with PML nuclear bodies. Journal of cell science 123, 2014–2024. [DOI] [PubMed] [Google Scholar]
- Moissiard G, Bischof S, Husmann D, Pastor WA, Hale CJ, Yen L, Stroud H, Papikian A, Vashisht AA, Wohlschlegel JA, et al. (2014). Transcriptional gene silencing by Arabidopsis microrchidia homologues involves the formation of heteromers. Proceedings of the National Academy of Sciences of the United States of America 111, 7474–7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moissiard G, Cokus SJ, Cary J, Feng S, Billi AC, Stroud H, Husmann D, Zhan Y, Lajoie BR, McCord RP, et al. (2012). MORC family ATPases required for heterochromatin condensation and gene silencing. Science 336, 1448–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama Y, and Uhlmann F (2013). Biochemical reconstitution of topological DNA binding by the cohesin ring. Nature 505, 367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Stroud H, Nee K, Liu W, Pezic D, Manakov S, Lee SA, Moissiard G, Zamudio N, Bourc’his D, et al. (2014). MORC1 represses transposable elements in the mouse male germline. Nature communications 5, 5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protter DSW, Rao BS, Van Treeck B, Lin Y, Mizoue L, Rosen MK, and Parker R (2018). Intrinsically Disordered Regions Can Contribute Promiscuous Interactions to RNP Granule Assembly. Cell reports 22, 1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nature Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nature Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y, and Brangwynne CP (2017). Liquid phase condensation in cell physiology and disease. Science 357. [DOI] [PubMed] [Google Scholar]
- Shin Y, Chang Y-C, Lee DSW, Berry J, Sanders DW, Ronceray P, Wingreen NS, Haataja M, and Brangwynne CP (2018). Liquid Nuclear Condensates Mechanically Sense and Restructure the Genome. Cell 175, 1481–1491.e1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song D, Graham TGW, and Loparo JJ (2016). A general approach to visualize protein binding and DNA conformation without protein labelling. Nature communications 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, and Karpen GH (2017). Phase separation drives heterochromatin domain formation. Nature 547, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yoshida N, Murakami N, Kawata K, Ishizaki H, Tanaka-Okamoto M, Miyoshi J, Zinn AR, Shime H, and Inoue N (2007). Dynamic regulation of p53 subnuclear localization and senescence by MORC3. Molecular biology of the cell 18, 1701–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchasovnikarova IA, Timms RT, Douse CH, Roberts RC, Dougan G, Kingston RE, Modis Y, and Lehner PJ (2017). Hyper-activation of HUSH complex function by Charcot-Marie-Tooth disease mutation in MORC2. Nat Genet 49, 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Updike DL, Hachey SJ, Kreher J, and Strome S (2011). P granules extend the nuclear pore complex environment in theC. elegansgerm line. The Journal of Cell Biology 192, 939–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser NE, Yang DX, Feng S, Kalinava N, Brown KC, Khanikar J, Freeberg MA, Snyder MJ, Csankovszki G, Chan RC, et al. (2017). MORC-1 Integrates Nuclear RNAi and Transgenerational Chromatin Architecture to Promote Germline Immortality. Developmental cell 41, 408–423.e407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler JR, Matheny T, Jain S, Abrisch R, and Parker R (2016). Distinct stages in stress granule assembly and disassembly. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, and Marshall AG (1998). A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. Journal of the American Society for Mass Spectrometry 9, 225–233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Document 1 | Raw analysis data for MORC-1 compaction rates (Related to Figure 2).
The Excel document contains raw analysis data for MORC-1 compaction rates. Each tab represents the MORC-1 concentration, and it contains results both in the absence and presence of ATP. In the Summary tab, the information on the compaction rate fold increases in the presence of ATP is included as well.
Video S1 | MORC-1 does not extrude DNA loops. (Related to Figure 2 and S3).
Both ends of bacteriophage lambda DNA were tethered on the microfluidic flow cell with slack. Subsequent addition of 80 nM MORC-1 and 2 mM ATP led to DNA compaction. While the two DNAs became taut, no extruded loops were observed. Dimension: 18.7 μm Ü 8.3 μm. Movie speed: 6x real-time.
Video S2 | MORC-1 forms foci on DNA. (Related to Figures 3 and S4, Video S3).
Cy3-labeled MORC-1 foci appear on a flow stretched DNA and grow in size over time. As DNA compaction proceeds, they eventually merge into one large focus. [Cy3-MORC-1] = 40 nM, [ATP] = 2 mM, Flow direction: upward, Movie speed: 4x real-time.
Video S3 | Formation and growth of MORC-1 foci. (Related to Figures 3 and S4, Video S2).
Diffusing MORC-1 molecules can form static foci. Collisions between sliding molecules and static foci lead to apparent growth of foci, although free MORC-1 molecules are also likely recruited from solution. Note that collisions among foci are also involved in the growth of foci. [Cy3-MORC-1] = 10 nM, [ATP] = 2 mM, Flow direction: upward, Speed: 20 frames per second = 4x-real time.
Video S4 | Static MORC-1 foci are dislodged upon high salt wash. (Related to Figure 5).
Cy3-labeled MORC-1 foci were pre-formed on flow stretched DNA, then subjected to a high salt wash (500 mM NaCl, 2 mM MgCl2) in the presence of 2 mM ATP. The high salt wash causes static MORC-1 foci to quickly move along the DNA in the flow direction and appears to cause them to slide off the DNA. Flow stretched DNAs are not quantum dot labeled in this example. [Cy3-MORC-1] = 10 nM, [ATP] = 2 mM, Flow direction: upward, Movie speed: 3x real-time.
Video S5 | MORC-1 foci are retained on DNA end labeled with a QDot. (Related to Figure 5).
As in Video S4 MORC-1 foci were pre-formed on DNA and then subjected to high salt wash. The presence of a quantum dot at the DNA ends blocks dissociation of MORC-1 foci. Note that the quantum dot is not visible in this spectral channel. Movie speed: Flow stretched DNAs are quantum dot labeled in this example. [Cy3-MORC-1] = 5 nM, [ATP] = 2 mM, Flow direction: upward, 20 frames per second = 4x real-time.
Video S6 | MORC-1 foci are not retained on DNA without QDots. (Related to Figure 5).
Pre-formed foci slide along and dissociate from DNA upon high salt wash. [Cy3-MORC-1] = 10 nM, [ATP] = 2 mM, Flow direction: upward, Flow stretched DNAs are not quantum dot labeled in this example. 20 frames per second = 4x-real time.
Video S7 | Static MORC-1 foci act as a roadblock to diffusing foci. (Related to Figure 5, Figure 6, and Figure S6).
In this movie, a static MORC-1 focus is capable of preventing a mobile focus from dissociating from the free end of DNA. Flow stretched DNAs are not quantum dot labeled in this example. [Cy3-MORC-1] = 10 nM, [ATP] = 2 mM, Flow direction: upward, 20 frames per second = 4x-real time.