

Abstract
The primary objective of this study was to enhance the antitumor efficacy of a model cancer vaccine through co-delivery of pentaerythritol lipid A (PELA), an immunological adjuvant, and a model tumor antigen, ovalbumin (OVA), separately loaded into polyanhydride particles (PA). In vitro experiments showed that encapsulation of PELA into PA (PA-PELA) significantly enhanced its stimulatory capacity on dendritic cells as evidenced by increased levels of the cell surface costimulatory molecules, CD80/CD86. In vivo experiments showed that PA-PELA, in combination with OVA-loaded PA (PA-OVA), significantly expanded the OVA-specific CD8+ T lymphocyte population compared to PA-OVA alone. Furthermore, serum OVA-specific antibody titers of mice vaccinated with PA-OVA/PA-PELA displayed a significantly stronger shift toward a Th1-biased immune response compared to PA-OVA alone, as evidenced by the substantially higher IgG2C:IgG1 ratios achieved by the former. Analysis of E.G7-OVA tumor growth curves showed that mice vaccinated with PA-OVA/PA-PELA had the slowest average tumor growth rate.
Keywords: Pentaerythritol Lipid A, Toll-like receptor-4 agonist, immunologic adjuvants, polyanhydrides, tumor-specific immune responses, cancer vaccines
Graphical Abstract
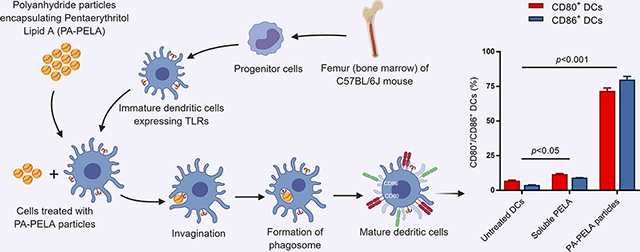
The study indicated that the immunostimulatory effect of PELA on DCs was significantly improved upon encapsulation into PA particles. The study also demonstrated that incorporation of PELA significantly enhanced the immunogenicity of PA particle-based cancer vaccines as indicated by significantly increased levels of OVA-specific CD8+ T cells and skewed the immune response toward a Th1 phenotype. Furthermore, the PELA containing formulation, when used in combination with PA-OVA, demonstrated a significant decrease in the rate of tumor progression when compared to PA-OVA alone. Taken together, these results emphasize the potential for PA-PELA to be utilized as an immunological adjuvant in cancer vaccines.
BACKGROUND
Cancer vaccines represent a promising approach to combating tumors and, in particular, their metastases without causing the deleterious side effects often associated with conventional chemotherapy1, 2. The primary goal of cancer vaccines is to harness the host’s own immune system to provide specific antitumor immunity capable of mediating tumor destruction and protecting against metastasis and tumor recurrence3–6. With steady growth in research toward finding effective approaches to treating cancer, immune-based therapies have undergone a considerable resurgence in the past decade1, 7, 8. Among all immunotherapies, cancer vaccines are the most common type currently being explored9. An essential component of a cancer vaccine is the tumor antigen(s) which can be delivered in a purified, tumor cell-derived (e.g., cell lysate) or encoded form10, 11. However, many tumor-associated antigens are poorly immunogenic, due primarily to tolerance mechanisms, and require an adjuvant and/or innate immune stimulus to promote their immunogenicity1, 12. In addition, the tumor microenvironment is known to be immunosuppressive13, 14. Thus, in order to enhance the potency of cancer vaccines, adjuvants capable of stimulating dendritic cell (DC) maturation, by binding to Toll-like receptors (TLRs), are often included in the formulation. These adjuvants can enhance tumor-specific adaptive immune responses12, 15–18.
From a research standpoint, available licensed adjuvants that can substantially enhance antitumor cytotoxic T cell activation are limited19, 20. Developing a vaccine formulation containing a TLR4 ligand has been a subject of wide research interest over the last few decades21. This has culminated in the generation of a number of prospective therapeutic cancer vaccines or prophylactic vaccines against cancer-causing viruses that have reached clinical trials or become FDA-approved, respectively22, 23. These vaccines contain monophosphoryl Lipid A (MPLA; a TLR4 agonist24) which is derived from Salmonella Minnesota. Recently, Oncothyreon Inc. (now known as Cascadian Therapeutics Inc.) has expanded the immunological adjuvant repertoire by synthesizing a more potent and safer version of lipid A (as compared to MPLA) known as pentaerythritol lipid A, or PET lipid A (PELA)25. PELA (C95H181N2O19P), a lipid A analog, is a hexa-acylated monosaccharide monophosphorylated ligand which is fully synthetic and thus is less subject to batch-to-batch variation and has greater quality control than MPLA26. PELA displays strong immunostimulatory (i.e., adjuvant) properties and can boost adaptive immunity by binding to TLR4 expressed on the surface of DCs26, 27. The acyl chains in PELA (Supplementary Material; Figure S1) are essential for binding to TLR4, which has been reported to trigger NF-κB and MAPK signaling pathways, involved in regulating and directing cellular immune responses28, 29. Due to the promising results that PELA has shown in preclinical studies, it has been advanced into clinical trials and incorporated into a liposomal formulation, ONT-10, which is a therapeutic cancer vaccine designed to treat cancers that express mucin-1, such as breast cancer30, 31.
Particulate delivery systems for cancer vaccine applications have been shown, in preclinical settings, to be an advantageous approach to delivering antigen and adjuvant compared to delivery in soluble form32–36. Not only does loading these components into particles results in protection from premature degradation, but particles also ensure more efficient delivery to DCs and sustained local availability for uptake by DCs32. In particular, polyanhydrides (PAs) have shown promise as biocompatible and biodegradable polymers37, 38 and have been used in some marketed controlled-release medical products such as Gliadel® (PA-based wafer containing carmustine for treating glioblastoma multiforme) and Septacin™ (PA-based beads loaded with gentamycin for treating osteomyelitis)39–41. PA and PELA have the potential to act at least additively in enhancing antigen-specific immune responses since both can bind to and activate TLR426, 27, 42. Since PA particles also offer the aforementioned advantages in addition to their adjuvanticity43–46, there is the possibility for synergistic enhancement of immune responses when used together as vaccines. In this study, the primary goal was to enhance the immune response and the antitumor efficacy of a previously reported PA-based model cancer vaccine, comprising of PA particles encapsulating ovalbumin (PA-OVA)47, by adding PA-PELA to the formulation. The encapsulation of PELA was found to enhance the stimulation of DCs and increase levels of antigen-specific CD8+ T cells. These enhancements in immunogenicity due to the combination of PA-OVA and PA-PELA were also reflected in a trend toward increased antitumor activity and decreased tumor progression in a prophylactic mouse tumor model, when compared to PA-OVA alone, suggesting the potential of PA-PELA adjuvants for use in cancer vaccines.
METHODS
Particle fabrication and characterization
PELA and OVA encapsulating PA particles were prepared using single and double emulsion solvent-evaporation techniques, respectively. Details of the fabrication and characterization of particles are provided in the Supplementary Material.
In vitro release kinetics
Release of OVA from PA particles:
Samples of PA particles encapsulating OVA (≈ 30 mg) were dispersed into 5 mL of phosphate buffered saline (PBS) (Sigma-Aldrich, St. Louis, MO) and incubated in the orbital incubator shaker (New Brunswick Scientific Co. Inc., Edison, NJ) set at 37°C and 300 rpm for one month. The amount of OVA released from particles into the PBS was measured at predetermined time intervals (1, 2, 4, 7, 10, 14, 20, and 30 days) and aliquots (0.5 mL) of the release medium were withdrawn and replaced by the same volume of fresh PBS at each time interval. Supernatants were stored at −20°C until OVA content was measured by the bicinchoninic acid protein assay (as described in the Supplementary Material). The experiment was performed in triplicate, and the results were expressed as the mean of cumulative OVA-release into PBS determined as a function of time ± standard deviation (SD).
Release of PELA from PA particles:
The release kinetics of PELA, which is poorly water-soluble as a result of its long hydrophobic acyl chains, was studied using PBS solution containing 1% v/v Tween-80 (Fisher Scientific, Fair Lawn, NJ). Tween-80, a nonionic surfactant, was added to the release medium to enhance PELA solubility and fulfill the sink conditions. Samples of PA particles (≈ 10 mg) were dispersed in 10 mL of PBS/Tween-80 solution and incubated in the orbital incubator shaker set at 37°C and 300 rpm for a period of one month. The amount of PELA released from particles was measured at predetermined time intervals (same as OVA-release time points), and aliquots (1 mL) of the release medium were withdrawn and replaced by the same volume of fresh PBS/Tween-80 solution at each time interval to maintain a constant volume of release medium. Samples were stored frozen at −20°C until PELA content was quantified by liquid chromatography-mass spectrometry (LC-MS) (as described in the Supplementary Material). The results were expressed as the mean of cumulative PELA release into PBS/Tween-80 determined as a function of time in three parallel experiments ± SD.
In vitro DC stimulation
In this study, the stimulatory effect of PELA encapsulated into particles and in its soluble form was assessed using DCs, which are professional antigen-presenting cells capable of efficiently priming naïve T cells48, 49. DCs were obtained from a C57BL/6J mouse through isolation of the bone marrow. Briefly, tibia and femur were extracted, and surrounding muscles were removed. This was followed by trimming both ends of the bone and flushing the media through the bone to collect the marrow. Primary cells were harvested and grown on Bacteriological Petri dishes in Roswell Park Memorial Institute medium (RPMI 1640) supplemented with 10 mM HEPES buffer, 1 mM sodium pyruvate, 0.1 mM minimal essential medium nonessential amino acids MEM-NEAA, 2 mM GlutaMAX (Life Technologies, Grand Island, NY), 50 mM 2-mercaptoethanol (Sigma-Aldrich), 50 ng/mL gentamicin sulfate (IBI Scientific, Peosta, IA), 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA), and 20 ng/mL of murine granulocyte-macrophage colony stimulating factor (PeproTech, Rocky Hill, NJ), and incubated in a well-controlled environment at 37°C with 5% CO2. Bone marrow-derived dendritic cells (BMDCs) were harvested at day 10 of culture, seeded in 12-well Cellstar plates (Greiner Bio-One, Germany) at a density of 3 × 105 cells/well, and incubated for 6 h. The cells were next stimulated by adding the treatments (1 and 3 μg PELA either encapsulated or soluble) and incubating for 24 h. After incubation with designated treatment, cells were flushed with existing media, collected and centrifuged for 5 min at 4°C using Eppendorf Centrifuge 5804-R (Eppendorf, Westbury, NY) set at 230 xg. Also, cell culture supernatants were collected to measure interleukin (IL-10 and IL12p70) concentrations using cytokine-specific mouse enzyme-linked immune-sorbent assay (ELISA) kits (Thermo Fisher Scientific, San Diego, CA), as per the manufacturer’s instructions. Cells collected by centrifugation were stained with an anti-CD11cfluorescein isothiocyanate (FITC) and either anti-CD80-phycoerythrin (PE) or anti-CD86-PE (eBioscience, San Diego, CA) using a standard direct immunofluorescence procedure. Controls involved staining DCs with FITC- or PE-conjugated isotype antibodies. Samples were run through a BD FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ) in triplicate, and data were analyzed with FlowJo software (Tree Star, Ashland, OR). Results were expressed as a percentage of CD80+/CD86+ DCs.
Animal studies
Mouse strains:
A murine tumor model was used for the evaluation of prophylactic cancer vaccine formulations. Wild-type C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME). Animals were maintained and preserved at the Medical Laboratories at the University of Iowa and kept on a daily 12 h light/12 h dark cycle. All animal experiments were performed in accordance with the University of Iowa guidelines for the care and use of laboratory animals. In all experiments, 6–8 week-old female mice were used. Mice were anesthetized via intraperitoneal injection of the ketamine xylazine mixture (dose: 87.5 mg/kg ketamine and 12.5 mg/kg xylazine) per mouse prior to vaccine administration or performing any other in vivo experiments.
Vaccination and in vivo experiments schedule:
To test the in vivo efficacy of prepared formulations, mice were randomly divided into three groups and treated with subcutaneous (rear dorsal flank) injections of the following treatment groups: (I) naïve (i.e., unvaccinated), (II) PA-OVA, and (III) PA-OVA/PA-PELA. Prepared PA particles were dispersed in 1X Dulbecco’s phosphate-buffered saline (DPBS, pH 7.4) (Life Technologies) immediately prior to vaccination. Doses of 50 μg OVA and 20 μg PELA per mouse were regularly used during prime vaccination on the day (0) and booster vaccination a week later. These doses were based on a previous study using poly(D,L-lactide-co-glycolide) particles loaded with OVA and PELA where significant effects on OVA-specific CD8+ T cell responses were observed in vivo when doses of 60 and 18 μg were used, respectively50. It is well-established that encapsulation of antigen and/or adjuvant into particles enhances vaccine potency compared to delivery in soluble form35, 50, 51. Specifically, control groups such as soluble OVA alone and soluble OVA + soluble PELA have already been tested and have been shown to be poorly immunogenic compared to their particulate counterparts50. In addition, we have previously tested the in vivo efficacy of PA-OVA against soluble OVA and blank PA particles (i.e., containing no OVA), and the results indicated that 75% of mice survived in the PA–OVA group (at day 28 post-tumor challenge) while 0% of mice survived in the groups treated with either blank particles or soluble OVA52. Therefore, such controls were not included in this current animal study to align with ethical standards regarding minimizing the numbers of animals used in research experiments. On day 14 post-prime vaccination, tumor-specific CD8+ T lymphocyte levels (also known as cytotoxic T lymphocytes, CTLs) were measured in the peripheral blood harvested through submandibular bleeds. On day 28 post-prime vaccination, tumor-specific IgG1 and IgG2C antibody titers were measured in the serum harvested through submandibular bleeds. A week later, mice were challenged with tumor cells.
Cell-mediated immunity:
T cell receptors (TCR), expressed on CTLs, specifically recognize and bind to major histocompatibility complex (MHC) class I. MHC tetramer assay and direct immunofluorescence technique enable the direct detection and quantification of tumor-specific CTLs within blood samples. Using submandibular bleeding technique, approximately 200 μL of mouse peripheral blood was collected into tubes containing 3 mL of ACK (ammonium-chloride-potassium) lysing buffer, and the samples were incubated at room temperature for 5 min. After incubation, peripheral blood lymphocytes (PBLs) were washed twice with a complete medium using Eppendorf Centrifuge 5804-R set at 230 xg, 4°C, for 5 min. Then, PBLs were resuspended in 150 μL of ice-cold FACS (fluorescence-activated cell sorting) buffer and transferred to a v-bottom 96-well plate (Corning, Kennebunk, ME) incubated on ice. This was followed by centrifugation (at the same previous conditions), supernatants were discarded, and PBLs were resuspended in 50 μL of anti-mouse CD16/CD32 Fc receptor block (clone 93) (eBioscience) in FACS buffer and incubated for 15 min. Subsequently, 50 μL of H-2Kb SIINFEKL class I iTAg™ MHC tetramer (Kb-OVA257) la μL of a mixture of fluorescein isothiocyanate (FITC)-labeled rat anti-mouse CD8 and PE-Cy5-labeled hamster anti-mouse CD3 (eBioscience) antibodies in FACS buffer was added in the dark and incubated for 20 min. After incubation, PBLs were washed twice with FACS buffer to remove the unbounded antibodies. Subsequently, 100 μL of 1X Perm/wash buffer (BD Biosciences, San Jose, CA) was added, and this was immediately followed by centrifugation for 15 min at 660 xg and 4°C. Finally, PBLs were resuspended in FACS buffer, samples were acquired using BD FACScan flow cytometer, and data were analyzed with FlowJo software. Results were expressed as a percentage of total CD3+ CD8+ T lymphocytes in peripheral blood that were positive to tetramer staining assay.
Antibody-mediated immunity:
The titers of tumor-specific IgG antibodies, IgG1 and IgG2C, were measured using ELISA, as described previously47. In brief, mice were bled from the submandibular area, and to harvest sera, blood samples were incubated at room temperature for one hour. After incubation, blood clots were removed using clean tweezers, and the samples were centrifuged for 10 min using an Eppendorf Centrifuge 5804-R set at 3,000 xg and 4°C. Supernatants (sera) were collected and stored at −80°C until use. In the meanwhile, Immulon® 2HB flat-bottom microtiter 96-well plates (Thermo Fisher Scientific, Rochester, NY) were coated with 100 μL of PBS containing 0.5 μg OVA. Using OVA-coated plates and PBS containing 0.05% v/v Tween-20 (Sigma-Aldrich), sera samples were serially diluted and incubated overnight at room temperature. This was followed by incubation for 3 h at room temperature with either goat anti-mouse IgG1 (or goat anti-mouse IgG2C) antibody conjugated with alkaline phosphatase (Southern Biotech, Birmingham, AL). Subsequently, 100 μL of p-nitrophenyl phosphate (pNPP) in TRIS buffer (Sigma-Aldrich) was added in the dark. After 30 min, the absorbance was measured at 405 nm using a SpectraMax® Plus 384 microplate reader (Molecular Devices LLC, Sunnyvale, CA). To remove any proteins or antibodies that were not specifically bound, plates were washed three times with 150 μL of PBS/Tween-20 solution between all reagent addition steps. The reciprocal of mouse sera dilution (highest dilution at which the absorbance is three-times greater than those of negative control) was reported as serum antibody titers.
Tumor challenge:
Five weeks post-prime vaccination, all mice were subcutaneously challenged with 2 × 106 E.G7 cells (expressing OVA), purchased from American Type Culture Collection (ATCC®, Manassas, VA), suspended in 100 μL of sterile 1X DPBS. Cells were injected contralaterally to the vaccination site. Tumor progression was monitored regularly over time for the subsequent two months (using digital caliper), and tumor volumes were calculated as described in Equation 1. To minimize pain and discomfort, mice were euthanized when the tumor size exceeded 20 mm at the largest diameter or 10 mm in height.
| Equation 1: |
Statistical analysis
Data were initially analyzed by one-way analysis of variance (ANOVA) using F-test which was followed by a Tukey’s multiple comparison test to compare all pairs of treatments. To characterize the tumor progression and assess differences in the pattern of change over time in the mean tumor volumes between the two vaccinated groups (i.e., PA-OVA alone and PA-OVA/PA-PELA), longitudinal data of the tumor growth profiles of mice in both vaccinated groups were analyzed with a linear mixed-effects model using SAS PROC MIXED (SAS Institute Inc., Cary, NC). Initial analysis of survival data was performed by log-rank (Mantel-Cox) test using GraphPad-Prism 7 (GraphPad Software, La Jolla, CA). Using SAS 9.4, further statistical analysis was performed by pairwise comparisons and data were analyzed using the log-rank test (Tukey-Kramer adjusted). In all tests, differences were considered statistically significant when p<0.05.
RESULTS
Properties of PA particles
PA-OVA particles, prepared by the double emulsion method, had an average diameter of almost 1 μm while PA-PELA particles fabricated by the single emulsion method had an average diameter of nearly 0.5 μm (Table 1). All particles exhibited a narrow size distribution with average PDI values being < 0.2 (Table 1). In addition, all formulations possessed negatively charged surfaces (Table 1). PELA-PA particles were quantified using a validated LC-MS method (Supplementary Material; Figure S2), and it was found that the encapsulation efficiency of PELA was comparable to that of OVA (Table 1). Analysis of scanning electron microscope photomicrographs demonstrated that the particles were spherical in shape with smooth surfaces (Figure 1).
Table 1:
Properties of polyanhydride particles encapsulating OVA or PELA.
| PA-OVA particles | PA-PELA particles | Empty PA particles | |
|---|---|---|---|
| Particle Size (nm) | 981 ± 18 | 486 ± 6 | 497 ± 5 |
| Polydispersity Index (PDI) | 0.15 ± 0.03 | 0.11 ± 0.04 | 0.11 ± 0.04 |
| Zeta Potential (mV) | −31.2 ± 4.5 | −19.8 ± 0.1 | −17.0 ± 0.2 |
| Loading Capacity (μg per mg of particles) | 7.7 ± 0.2 | 4.3 ± 0.2 | - |
| Encapsulation Efficiency (%) | 35.8 ± 1.1 | 35.6 ± 1.7 | - |
Fig. 1: Electron photomicrographs of polyanhydride particles.

(A) PA-OVA (scale bar is 1 μm); (B) PAPELA (scale bar is 0.5 μm); (C) PA empty particles (scale bar is 0.5 μm).
In vitro release kinetics of PA-OVA and PA-PELA particles
Upon dispersal of PA particles into release media, both OVA- and PELA-loaded PA particles demonstrated rapid short burst releases (Figure 2). A burst release was followed by a slower sustained release of the payloads over time. By day 30, OVA and PELA cumulative release from PA particles had reached 89% and 88%, respectively. Thus, it was found that the release of PELA exhibited a similar trend to the OVA cumulative release. Release profiles were also fit to a zero-order release kinetic model with burst effect, and the analysis revealed that the model adequately captured the data, and PA particles exhibited a steady increase in the release of cargo that approximated to zero-order release kinetics (Supplementary Material; Figure S3).
Fig. 2: Cumulative percentage release of OVA (●) and PELA (○) from polyanhydride particles over time.

Data are plotted as mean ± SD.
Evaluation of the effect of PA-PELA on the expression of CD80, CD86, IL-10, and IL-12 by BMDCs
BMDCs were harvested at day 10 of culture, at which point nearly 90% of the cells were CD11c positive as analyzed by the BD FACScan flow cytometer (data not shown). Prior to experimental assays, cell viability was tested by Trypan blue exclusion, and results indicated that the cell viability was greater than 95%. Results of surface staining of BMDCs revealed that PELA-PA particles promoted the upregulation of both CD80 and CD86, and this was significantly greater than untreated BMDCs, soluble PELA, PA particles alone, or a physical mixture of PA particles with PELA (PA + PELA), used at the equivalent doses (Figure 3.1). This clearly demonstrates that delivery of PELA in particulate form enhanced its costimulatory effect. In addition, the results showed that PA particles alone promoted the upregulation of both CD80 and CD86, which further demonstrates that PAs possess self-adjuvanting properties. Also, it was observed that the costimulatory effect of PAs was dose-dependent. The results also suggest that PELA and PAs worked synergistically and promoted stimulation of BMDCs, and the combination of these two is superior to PA or PELA alone. In addition, it was shown that while empty PA particles had little or no effect on both IL-12p70 and IL-10 secretion, PA-PELA had a significant impact on increasing the expression of both cytokines (compared to the same dose of soluble PELA). Interestingly, as the dose increased from 1 to 3 μg PELA (encapsulated in PA particles; PA-PELA), the relative levels of secretion of IL-10:IL-12p70 decreased significantly (t-test; p<0.0001) (Figure 3.2), suggesting that high doses of PA-PELA may preferentially favor the induction of Th1-type responses.
Fig. 3.1: Stimulatory effect of PELA on BMDCs (in vitro) via direct fluorescence staining.

BMDCs were stained with anti-CD80 and CD86. Representative histogram plots display relative surface expression intensity for CD80 and CD86 at 1 μg PELA. Data are plotted as percentage of CD80+/CD86+ DCs ± SD, n = 3. *p< 0.05, ***p<0.001.
Fig. 3.2: In vitro IL-10 and IL-12p70 production by BMDCs exposed to PA with or without PELA.

Levels of IL-10 and IL-12p70 were assessed in the supernatants by ELISA 24 h post incubation of BMDCs with the designated treatment. Results are plotted as mean ± SD, n = 3. *p< 0.05, ***p<0.001.
Assessment of Immunogenicity
Groups of immunocompetent mice were vaccinated (prime/boost on days 0/7) with a heterogeneous mix of PA particles separately loaded with OVA or PELA (PA-OVA/PA-PELA), or with a homogenous suspension of particles loaded with OVA alone (PA-OVA), and both humoral and cellular OVA-specific immune responses were assessed. Tetramer staining data showed that mice vaccinated with PA-OVA particles alone did not induce a significant increase in OVA-specific CD8+ T cell levels in the peripheral blood, however, when combined with PA-PELA particles, OVA-specific CD8+ T cell levels increased significantly compared to unvaccinated mice (Figure 4.1). In terms of humoral OVA-specific immune responses, co-delivery of PA-PELA with PA-OVA particles did not significantly enhance OVA-specific IgG1 antibody titers compared to when PA-OVA particles were administered alone (Figure 4.2A). However, co-delivery of PA-PELA with PA-OVA particles significantly improved the production of OVA-specific IgG2C compared to mice vaccinated with PA-OVA alone (Figure 4.2B). Also, the IgG2C:IgG1 ratio for mice immunized with PA-OVA/PA-PELA formulation was significantly higher compared to mice vaccinated with PA-OVA alone, indicating a correspondingly higher Th1-biased immune response (Figure 4.2C).
Fig. 4.1: Cell-mediated OVA-specific immune responses in mice vaccinated with different polyanhydride particle-based formulations containing OVA ± PELA.

Data are plotted as mean ± SEM, n = 10. **p<0.01, ***p<0.001.
Fig. 4.2: Humoral OVA-specific antibody responses in mice vaccinated with different polyanhydride particle-based formulations containing OVA ± PELA.

(A) Serum titers of OVA-specific IgG1; (B) Serum titers of OVA-specific IgG2C; (C) IgG2C:IgG1 ratio. Data are plotted as mean ± SEM, n = 10. ***p<0.001.
Evaluation of tumor progression and survival
Five weeks post-prime vaccination, groups of mice were challenged with a lethal dose of E.G7 cells, and tumor growth and survival were subsequently recorded. As expected, naïve mice had tumors that grew rapidly compared to vaccinated mice, and four mice from the control group were euthanized on day 16 post-tumor challenge as their tumors already reached a predetermined size limit (Figure 5.1). In contrast, mice vaccinated with either PA-OVA alone or PA-OVA/PA-PELA had tumor volumes that were, on average, significantly smaller than those of unvaccinated mice (as tested on day 16 using ANOVA, p<0.001). In addition, it was observed that 60% of mice treated with PA-OVA/PA-PELA were tumor free on day 16 post tumor challenge whereas all mice in the other two groups had detectable tumors. Also, longitudinal data of the tumor growth of mice in both vaccinated groups (i.e., PA-OVA alone and PA-OVA/PA-PELA) were analyzed using a linear mixed-effects model. The analysis revealed that the combination of PA-OVA and PA-PELA caused a significant reduction in the tumor progression compared to mice administered with PA-OVA alone (p<0.001). Co-delivery of PA-PELA and PA-OVA particle formulations also marginally extended the median survival time (MST = 35 days) when compared to the delivery of PA-OVA particles alone (MST = 31.5 days), albeit not significantly (Figure 5.2). Survival analysis also revealed that both vaccine strategies resulted in a statistically significant extended survival compared to the naïve control group (MST = 18 days) (p<0.001).
Figure 5.

1 Prophylactic antitumor effect of vaccinating mice with different polyanhydride particle-based formulations containing OVA ± PELA. Each curve represents the tumor growth for each individual mouse except in the average tumor growth graph where the average tumor volume (mean ± SEM) is reported; naïve (●); PA-OVA (■); PA-OVA/PA-PELA (□). Numbers in parentheses represent the percent of mice with complete tumor regressions. In the average tumor growth graph, # indicates the statistical difference between vaccinated groups and naïve control group at day 16 post-tumor challenge (ANOVA, p<0.001) while ★ refers to the statistical difference between the two vaccinated groups (longitudinal data analysis with the linear mixed-effects model, p<0.001). 2 Survival curve of mice bearing E.G7-OVA tumors. Prior to tumor challenge, mice were vaccinated with the indicated formulation; naïve (●); PA-OVA (■); PA-OVA/PA-PELA (□). The dotted line represents the median (50%) survival while # indicates the statistical difference between vaccinated groups and naïve control group (p<0.001).
DISCUSSION
We have previously demonstrated that PA particles loaded with OVA were capable of protecting against challenges with OVA-expressing tumor cells when the particles were used as prophylactic vaccines in immunocompetent mice47. Here, we attempted to further improve the immunogenicity of OVA-loaded PA particles through co-delivery of the encapsulated TLR-4 agonist, PA-PELA. It has been previously shown by others and us that providing TLR agonists in a particulate form, using different polymers, generates more potent vaccines as opposed to when soluble TLR agonists are used35, 50, 53. Thus, side-by-side comparisons of two formulations were carried out. One treatment formulation involved PA-OVA alone, a homologous suspension of OVA-loaded PA particles. The second treatment formulation involved a heterologous blend of PA-OVA and PA-PELA. In the particle preparation process, PELA and OVA were independently encapsulated in order to maximize the loading for each component and to ensure that the dosage ratio of OVA:PELA delivered in vivo was similar to a previous study involving a different polymer50. While some studies have shown that co-loading can be advantageous in terms of stimulating strong immune responses, we and others have found that simultaneous delivery of the antigen and adjuvant using independently loaded particles results in similar enhancement of immune responses17, 50, 54, 55. This is because separately encapsulated antigen and adjuvant will not necessarily remain segregated in vivo as both types of particle formulations could be internalized by the same DCs, which generally have the capacity to internalize more than one particle at a time56. Specifically, our group has previously demonstrated that co-loading PELA and OVA into the same poly(lactide-co-glycolide)-based particles generated similar cellular immune responses and provided similar tumor protection to independently loaded particles50. Analysis of in vitro release profiles for both PA-OVA and PA-PELA demonstrated an initial burst release followed by sustained delivery of both antigen and adjuvant over time (up to 30 days), implying that the payloads were homogeneously distributed throughout the particles. Interestingly, the release kinetics of each of the payloads were similar despite their distinct chemical properties (e.g., PELA is a much more hydrophobic molecule than OVA). Thus, it appears that the release kinetics of these payloads were mainly determined by polymer degradation rate although the presence of proteins and enzymes in vivo may also have the potential to modulate the release profile. This observation can be explained by the fact that PA particles predominately degrade through surface erosion which make them appropriate biomaterials for sustained release of payloads57–59. The sustained release witnessed may be advantageous in terms of in vivo vaccine applications for at least two reasons. The first is that, in the 1–2 days subsequent to vaccination, the payload still present inside the particles is likely to be substantial and therefore capable of exerting significant downstream consequences, upon uptake by DCs, in terms of DC activation and immune response stimulation. The second reason is that those particles potentially not taken up by DCs in the initial 1–2 days may act as a depot for both PELA and OVA for a period of days to weeks, which may be beneficial in terms of generating OVA-specific adaptive immune responses. Recently, we reported on the uptake of PA particles, and the results demonstrated that PA particles are readily and efficiently internalized by BMDCs56. Also, it is of note that particle size plays a crucial role in determining their ultimate fate and the magnitude of the immune response32, 60. Generally, larger particles (> 100–200 nm) remain at the vaccine injection site and require uptake by migratory DCs in order to be delivered to the local draining lymph node while smaller particles (< 100–200 nm) can potentially travel independently to the draining lymph node where they can be taken up by the resident dendritic cells61, 62. Since the size of both particle formulations (PA-OVA and PA-PELA) is above that threshold, it would be expected that particles would remain in situ until being internalized by peripheral DCs.
In vitro studies on DC activation by PA-PELA were performed to test if this formulation had the capacity to further enhance, over soluble PELA, the costimulatory potential of DCs by inducing their maturation as defined by the up-regulation of CD80 and CD8663. Generally, naïve T cells require costimulatory signals (in addition to the engagement of the T cell receptor) in order to become activated into an effector phenotype capable of proliferation and imparting function in an antigen-specific manner. Two well-studied costimulatory surface proteins are CD80 and CD8663. Our in vitro studies indicated that the expression of CD80 and CD86 were significantly upregulated on the surface of DCs when the cells were cultured with PA-PELA compared to when DCs were cultured with soluble PELA alone or soluble PELA plus empty PA particles. This further supports the observation that the encapsulation of PELA into particles provides additional advantages compared to soluble counterparts. Interestingly, incubation of DCs with PA-PELA resulted in a synergistic increase in expression levels of the costimulatory proteins compared to when both components were added independently to the same culture, where an additive increase in expression was observed (Figure 3.1). Finally, it was found that empty PA particles per se induced up-regulation of CD80 and CD86 on DCs, further supporting the body of literature on the adjuvanticity of PA particles43–46.
The above in vitro demonstration of potent DC activation by PA-PELA spurred further studies in vivo to assess the immunostimulatory potential of these particles when used in combination with PAOVA as a cancer vaccine. It was shown that the combination of PA-PELA and PA-OVA induced a modest yet significant increase in OVA-specific CD8+ T cell levels in the peripheral blood of vaccinated mice within two weeks of the priming vaccination (Figure 4.1). Significant increases were also seen in the titers of OVA-specific IgG2C antibodies (four weeks after prime), indicating a push, albeit marginal, toward a Th1-type immune response. This was in contrast to when PA-OVA was used alone as a vaccine and demonstrated no significant increase in either of the aforementioned parameters when compared to mice receiving no vaccination. When tested for the capacity of the combination of PA-PELA and PAOVA to protect against a subsequent tumor challenge, a significant increase in antitumor activity (as assessed through average tumor volume measurements) was observed compared to mice vaccinated with PA-OVA alone (Figure 5.1). The reasons for the lack of a significant increase in the survival study are at this stage unknown but may stem from insufficient DC stimulation subsequently resulting in insufficient CD8+ T cell activation. As mentioned above, the observed increases in OVA-specific T cells in the peripheral blood of mice immunized with PA-PELA and PA-OVA, while being significant when compared to mice vaccinated with PA-OVA alone, were marginal and perhaps the protective response may not have benefited from higher levels of OVA-specific CD8+ T cells being generated. It is possible that insufficient stimulation of Th1-leaning cytokine production, such as IL-12, or alternatively an inappropriate ratio of Th1:Th2 cytokines hindered the expansion and/or function of the OVA-specific CD8+ T cells. Our ELISA studies, where both IL-12p70 and IL-10 production by DCs co-cultured in vitro with PA-PELA were measured, revealed that the ratio of production levels of these cytokines was dose-dependent where 1 μg of PELA (encapsulated in PA particles; PA-PELA) cultured with DCs resulted in an approximate 1.3:1 ratio of IL-10:IL-12 while 3 μg of PELA (PA-PELA) resulted in a 0.5:1 ratio (Figure 3.2). Therefore, at higher doses of PA-PELA, a greater skewing toward a Th1-type response was observed. IL-12 has been shown to enhance CD8+ T cell responses while IL-10 has often been associated with abrogation of Th1 responses64. We, therefore, expect that increased amounts of PA-PELA or both PA-PELA and PA-OVA would increase the magnitude of the antigen-specific immune response stimulated even further.
Immunosuppressive strategies recruited by tumors and inadequate specificity of existing cancer treatments are among the major limitations to the current therapies, and the vast majority of approved immunotherapies utilize systemic delivery of cells or immunomodulators which makes addressing these hurdles more challenging65. Biomaterials such as PAs offer a unique platform to address these challenges by harnessing the advantages of therapeutic targeting, co-delivery, sustained release, activation/maturation of DCs, and promotion of CTLs. Enhancing the immune responses against tumors through particle-based delivery of TLR(s) can further improve the clinical impact of biomaterials in cancer immunotherapy. In previously published studies, we reported that the TLR-9 agonist CpG ODN was prone to hinder rather than improve the immunogenicity and antitumor activity of PA particle-based vaccines47. In contrast, in this study, it was demonstrated that the TLR4 agonist (PELA) when encapsulated in PA particles, significantly improved OVA-specific CD8+ T cell responses, a result potentially attributable to the effect observed in vitro where DCs were induced to express significantly higher amounts of CD80/CD86 and IL-12p70. This improvement in the immunogenicity may explain the trend toward slower tumor progression and extended survival. Further studies on the role of increased doses of PA-PELA in vivo on the antitumor potential of the formulation need to be performed. Also, the vaccine formulation will need to be tested in a therapeutic setting and using a more clinically relevant tumor model, and in the presence and absence of immune checkpoint inhibitors.
Supplementary Material
Acknowledgments
We acknowledge support from the Nanovaccine Institute, the Vlasta Klima Balloun Faculty Chair, the National Institutes of Health (1U01CA213862–01A1 and P30 CA086862) and the Lyle and Sharon Bighley Chair of Pharmaceutical Sciences.
Abbreviations:
- ANOVA
one-way analysis of variance
- BMDCs
bone marrow-derived dendritic cells
- CTLs
cytotoxic T lymphocytes
- DCs
dendritic cells
- DPBS
Dulbecco’s phosphate-buffered saline
- ELISA
enzyme-linked immune-sorbent assay
- FDA
food and drug administration
- FITC
fluorescein isothiocyanate
- IL
interleukin
- LC-MS
liquid chromatography-mass spectrometry
- MAPK
mitogen-activated protein kinase
- MPLA
monophosphoryl lipid A
- MST
median survival time
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- OVA
ovalbumin
- PA
polyanhydride
- PBS
phosphate buffered saline
- PE
phycoerythrin
- PELA
pentaerythritol lipid A
- SD
standard deviation
- TLRs
Toll-like receptors
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest:
The authors have declared that no competing interest exists.
REFERENCES
- 1.Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB and Wang XY. Therapeutic cancer vaccines: past, present, and future. Adv Cancer Res. 2013; 119: 421–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Emens LA. Cancer vaccines: on the threshold of success. Expert Opin Emerg Drugs. 2008; 13: 295–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Burg SH, Arens R, Ossendorp F, van Hall T and Melief CJ. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer. 2016; 16: 219–33. [DOI] [PubMed] [Google Scholar]
- 4.Papaioannou NE, Beniata OV, Vitsos P, Tsitsilonis O and Samara P. Harnessing the immune system to improve cancer therapy. Ann Transl Med. 2016; 4: 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palucka K, Ueno H, Fay J and Banchereau J. Harnessing dendritic cells to generate cancer vaccines. Ann N Y Acad Sci. 2009; 1174: 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenblatt J, Stone RM, Uhl L, Neuberg D, Joyce R, Levine JD, et al. Individualized vaccination of AML patients in remission is associated with induction of antileukemia immunity and prolonged remissions. Sci Transl Med. 2016; 8: 368ra171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finn OJ. Vaccines for cancer prevention: a practical and feasible approach to the cancer epidemic. Cancer Immunol Res. 2014; 2: 708–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melief CJ, van Hall T, Arens R, Ossendorp F and van der Burg SH. Therapeutic cancer vaccines. J Clin Invest. 2015; 125: 3401–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bot A, Obrocea M and Marincola FM. Cancer Vaccines: From Research to Clinical Practice. CRC Press, 2015. [Google Scholar]
- 10.Even-Desrumeaux K, Baty D and Chames P. State of the art in tumor antigen and biomarker discovery. Cancers (Basel). 2011; 3: 2554–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buonaguro L, Petrizzo A, Tornesello ML and Buonaguro FM. Translating tumor antigens into cancer vaccines. Clin Vaccine Immunol. 2011; 18: 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coffman RL, Sher A and Seder RA. Vaccine adjuvants: putting innate immunity to work. Immunity. 2010; 33: 492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim R, Emi M and Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007; 121: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015; 35 Suppl: S185–98. [DOI] [PubMed] [Google Scholar]
- 15.Marciani DJ. Vaccine adjuvants: role and mechanisms of action in vaccine immunogenicity. Drug Discov Today. 2003; 8: 934–43. [DOI] [PubMed] [Google Scholar]
- 16.Awate S, Babiuk LA and Mutwiri G. Mechanisms of action of adjuvants. Front Immunol. 2013; 4: 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ilyinskii PO, Roy CJ, O’Neil CP, Browning EA, Pittet LA, Altreuter DH, et al. Adjuvant-carrying synthetic vaccine particles augment the immune response to encapsulated antigen and exhibit strong local immune activation without inducing systemic cytokine release. Vaccine. 2014; 32: 2882–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaurav M, Minz S, Sahu K, Kumar M, Madan J and Pandey RS. Nanoparticulate mediated transcutaneous immunization: Myth or reality. Nanomedicine. 2016; 12: 1063–81. [DOI] [PubMed] [Google Scholar]
- 19.Lee S and Nguyen MT. Recent advances of vaccine adjuvants for infectious diseases. Immune Netw. 2015; 15: 51–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hogenesch H Mechanism of immunopotentiation and safety of aluminum adjuvants. Front Immunol. 2012; 3: 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reed SG, Hsu FC, Carter D and Orr MT. The science of vaccine adjuvants: advances in TLR4 ligand adjuvants. Curr Opin Immunol. 2016; 41: 85–90. [DOI] [PubMed] [Google Scholar]
- 22.Ozao-Choy J, Lee DJ and Faries MB. Melanoma vaccines: mixed past, promising future. Surg Clin North Am. 2014; 94: 1017–30, viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tritto E, Mosca F and De Gregorio E. Mechanism of action of licensed vaccine adjuvants. Vaccine. 2009; 27: 3331–4. [DOI] [PubMed] [Google Scholar]
- 24.Bazzill JD, Stronsky SM, Kalinyak LC, Ochyl LJ, Steffens JT, van Tongeren SA, et al. Vaccine nanoparticles displaying recombinant Ebola virus glycoprotein for induction of potent antibody and polyfunctional T cell responses. Nanomedicine. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.PET Lipid A Vaccine Adjuvant and Related Technologies. Seattle, WA: Oncothyreon, 2014. [Google Scholar]
- 26.Jiang Z-H, Budzynski WA, Skeels LN, Krantz MJ and Koganty RR. Novel lipid A mimetics derived from pentaerythritol: synthesis and their potent agonistic activity. Tetrahedron. 2002; 58: 8833–42. [Google Scholar]
- 27.Pestano LA, Christian B, Koppenol S, Millard J, Christianson G, Klucher K, et al. ONT-10, a liposomal vaccine targeting hypoglycosylated MUC1, induces a potent cellular and humoral response and suppresses the growth of MUC1 expressing tumors. Cancer Research. 2011; 71: 762-. [Google Scholar]
- 28.Carpenter S and O’Neill LA. Recent insights into the structure of Toll-like receptors and post-translational modifications of their associated signalling proteins. Biochem J. 2009; 422: 1–10. [DOI] [PubMed] [Google Scholar]
- 29.Kuper C, Beck FX and Neuhofer W. Toll-like receptor 4 activates NF-kappaB and MAP kinase pathways to regulate expression of proinflammatory COX-2 in renal medullary collecting duct cells. Am J Physiol Renal Physiol. 2012; 302: F38–46. [DOI] [PubMed] [Google Scholar]
- 30.Nemunaitis J, Bedell C, Klucher K, Vo A and Whiting S. Phase 1 dose escalation of ONT-10, a therapeutic MUC1 vaccine, in patients with advanced cancer. Journal for ImmunoTherapy of Cancer. 2013; 1: 240. [Google Scholar]
- 31.Nemunaitis JJ, Adams N, Bedell C, Klucher K, Taylor J and Whiting SH. Tolerability, humoral immune response, and disease control in phase 1 patients receiving ONT-10, a MUC1 liposomal vaccine. Journal of Clinical Oncology. 2014; 32: 3091. [Google Scholar]
- 32.Suliman SO, Wafa EI, Geary SM and Salem AK. Polymeric Particles as Cancer Vaccine Vectors. American Pharmaceutical Review. 2019; 22: 32–9. [Google Scholar]
- 33.de Barros CM, Wafa EI, Chitphet K, Ahmed K, Geary SM and Salem AK. Production of Adjuvant-Loaded Biodegradable Particles for Use in Cancer Vaccines. Methods Mol Biol. 2017; 1494: 201–13. [DOI] [PubMed] [Google Scholar]
- 34.Fontana F, Liu D, Hirvonen J and Santos HA. Delivery of therapeutics with nanoparticles: what’s new in cancer immunotherapy? Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2017; 9. [DOI] [PubMed] [Google Scholar]
- 35.Geary SM, Hu Q, Joshi VB, Bowden NB and Salem AK. Diaminosulfide based polymer microparticles as cancer vaccine delivery systems. J Control Release. 2015; 220: 682–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huntimer LM, Ross KA, Darling RJ, Winterwood NE, Boggiatto P, Narasimhan B, et al. Polyanhydride nanovaccine platform enhances antigen-specific cytotoxic T cell responses. TECHNOLOGY. 2014; 02: 171–5. [Google Scholar]
- 37.Huntimer L, Ramer-Tait AE, Petersen LK, Ross KA, Walz KA, Wang C, et al. Evaluation of biocompatibility and administration site reactogenicity of polyanhydride-particle-based platform for vaccine delivery. Adv Healthc Mater. 2013; 2: 369–78. [DOI] [PubMed] [Google Scholar]
- 38.Brenza TM, Ghaisas S, Ramirez JEV, Harischandra D, Anantharam V, Kalyanaraman B, et al. Neuronal protection against oxidative insult by polyanhydride nanoparticle-based mitochondria-targeted antioxidant therapy. Nanomedicine. 2017; 13: 809–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perry J, Chambers A, Spithoff K and Laperriere N. Gliadel wafers in the treatment of malignant glioma: a systematic review. Curr Oncol. 2007; 14: 189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li LC, Deng J and Stephens D. Polyanhydride implant for antibiotic delivery--from the bench to the clinic. Adv Drug Deliv Rev. 2002; 54: 963–86. [DOI] [PubMed] [Google Scholar]
- 41.Jain JP, Modi S, Domb AJ and Kumar N. Role of polyanhydrides as localized drug carriers. J Control Release. 2005; 103: 541–63. [DOI] [PubMed] [Google Scholar]
- 42.Tamayo I, Irache JM, Mansilla C, Ochoa-Reparaz J, Lasarte JJ and Gamazo C. Poly(anhydride) nanoparticles act as active Th1 adjuvants through Toll-like receptor exploitation. Clin Vaccine Immunol. 2010; 17: 1356–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kipper MJ, Wilson JH, Wannemuehler MJ and Narasimhan B. Single dose vaccine based on biodegradable polyanhydride microspheres can modulate immune response mechanism. J Biomed Mater Res A. 2006; 76: 798–810. [DOI] [PubMed] [Google Scholar]
- 44.Huntimer L, Wilson Welder JH, Ross K, Carrillo-Conde B, Pruisner L, Wang C, et al. Single immunization with a suboptimal antigen dose encapsulated into polyanhydride microparticles promotes high titer and avid antibody responses. J Biomed Mater Res B Appl Biomater. 2013; 101: 91–8. [DOI] [PubMed] [Google Scholar]
- 45.Phanse Y, Carrillo-Conde BR, Ramer-Tait AE, Broderick S, Kong CS, Rajan K, et al. A systems approach to designing next generation vaccines: combining alpha-galactose modified antigens with nanoparticle platforms. Sci Rep. 2014; 4: 3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ulery BD, Petersen LK, Phanse Y, Kong CS, Broderick SR, Kumar D, et al. Rational design of pathogen-mimicking amphiphilic materials as nanoadjuvants. Sci Rep. 2011; 1: 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wafa EI, Geary SM, Goodman JT, Narasimhan B and Salem AK. The effect of polyanhydride chemistry in particle-based cancer vaccines on the magnitude of the anti-tumor immune response. Acta Biomater. 2017; 50: 417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yewdall AW, Drutman SB, Jinwala F, Bahjat KS and Bhardwaj N. CD8+ T cell priming by dendritic cell vaccines requires antigen transfer to endogenous antigen presenting cells. PLoS One. 2010; 5: e11144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neek M, Kim TI and Wang SW. Protein-based nanoparticles in cancer vaccine development. Nanomedicine. 2019; 15: 164–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahmed KK, Geary SM and Salem AK. Development and Evaluation of Biodegradable Particles Coloaded With Antigen and the Toll-Like Receptor Agonist, Pentaerythritol Lipid A, as a Cancer Vaccine. J Pharm Sci. 2016; 105: 1173–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Snapper CM. Distinct Immunologic Properties of Soluble Versus Particulate Antigens. Front Immunol. 2018; 9: 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Joshi VB, Geary SM, Carrillo-Conde BR, Narasimhan B and Salem AK. Characterizing the antitumor response in mice treated with antigen-loaded polyanhydride microparticles. Acta Biomater. 2013; 9: 5583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Standley SM, Mende I, Goh SL, Kwon YJ, Beaudette TT, Engleman EG, et al. Incorporation of CpG oligonucleotide ligand into protein-loaded particle vaccines promotes antigen-specific CD8 T-cell immunity. Bioconjug Chem. 2007; 18: 77–83. [DOI] [PubMed] [Google Scholar]
- 54.Kazzaz J, Singh M, Ugozzoli M, Chesko J, Soenawan E and O’Hagan DT. Encapsulation of the immune potentiators MPL and RC529 in PLG microparticles enhances their potency. J Control Release. 2006; 110: 566–73. [DOI] [PubMed] [Google Scholar]
- 55.Malyala P, Chesko J, Ugozzoli M, Goodsell A, Zhou F, Vajdy M, et al. The potency of the adjuvant, CpG oligos, is enhanced by encapsulation in PLG microparticles. J Pharm Sci. 2008; 97: 1155–64. [DOI] [PubMed] [Google Scholar]
- 56.Wafa EI, Geary SM, Ross KA, Goodman JT, Narasimhan B and Salem AK. Single dose of polyanhydride particle-based vaccine generates potent antigen-specific antitumor immune responses. J Pharmacol Exp Ther. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Katti DS, Lakshmi S, Langer R and Laurencin CT. Toxicity, biodegradation and elimination of polyanhydrides. Adv Drug Deliv Rev. 2002; 54: 933–61. [DOI] [PubMed] [Google Scholar]
- 58.Shen E, Kipper MJ, Dziadul B, Lim MK and Narasimhan B. Mechanistic relationships between polymer microstructure and drug release kinetics in bioerodible polyanhydrides. J Control Release. 2002; 82: 115–25. [DOI] [PubMed] [Google Scholar]
- 59.Haughney SL, Ross KA, Boggiatto PM, Wannemuehler MJ and Narasimhan B. Effect of nanovaccine chemistry on humoral immune response kinetics and maturation. Nanoscale. 2014; 6: 13770–8. [DOI] [PubMed] [Google Scholar]
- 60.Joshi VB, Geary SM and Salem AK. Biodegradable particles as vaccine delivery systems: size matters. AAPS J. 2013; 15: 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O’Neil CP, et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol. 2007; 25: 1159–64. [DOI] [PubMed] [Google Scholar]
- 62.Rao DA, Forrest ML, Alani AW, Kwon GS and Robinson JR. Biodegradable PLGA based nanoparticles for sustained regional lymphatic drug delivery. J Pharm Sci. 2010; 99: 2018–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Acuto O and Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. 2003; 3: 939–51. [DOI] [PubMed] [Google Scholar]
- 64.Starbeck-Miller GR, Xue HH and Harty JT. IL-12 and type I interferon prolong the division of activated CD8 T cells by maintaining high-affinity IL-2 signaling in vivo. J Exp Med. 2014; 211: 105–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gammon JM, Dold NM and Jewell CM. Improving the clinical impact of biomaterials in cancer immunotherapy. Oncotarget. 2016; 7: 15421–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.