

Abstract
Type 1 and 2 diabetes mellitus are major medical epidemics affecting millions of patients worldwide. Diabetes mellitus is the leading cause of a form of chronic kidney disease known as Diabetic Kidney Disease (DKD), which is the most common cause of end-stage renal disease (ESRD). DKD is associated with significant changes in renal hemodynamics and electrolyte transport. Alterations in renal ion transport, triggered by pathophysiological conditions in diabetes, can exacerbate hypertension, accelerate renal injury, and are integral to the development of DKD. Renal ion transporters and electrolyte homeostasis play a fundamental role in functional changes and injury to the kidney during DKD. With the large number of ion transporters involved in DKD, understanding the roles of individual transporters as well as the complex cascades through which they interact is essential in the development of effective treatments for patients suffering from this disease. This chapter aims to gather current knowledge of the major renal ion transporters with altered expression and activity under diabetic conditions, and provide a comprehensive overview of their interactions and collective function in DKD.
Keywords: diabetic nephropathy, diabetic kidney disease, SGLT2, ENaC, TRPC6, TRPM6, NHE, KATP channel
1. INTRODUCTION
Diabetic Kidney Disease (DKD) is the primary contributor to the development of end stage renal disease (ESRD) and is associated with the onset of cardiovascular disease and stroke (Hagg et al., 2013; Maqbool, Cooper, & Jandeleit-Dahm, 2018; Umanath & Lewis, 2018). DKD is a subtype of chronic kidney disease (CKD) in which hyperglycemia, in combination with other manifestations of diabetes mellitus, leads to the development of severe renal complications. DKD is a steadily growing epidemic, with approximately 660,000 Americans diagnosed annually and Medicare expenditures in excess of $31 billion. In 2014, it was estimated that nearly 29 million Americans suffered from diabetes and an additional 86 million from prediabetes. In 2014, 44% of newly reported ESRD cases resulted from diabetes. The Center for Disease Control and Prevention predicted that diabetes could affect nearly 1 in 3 U.S. adults by 2050 if current trends continue. Worldwide, the number of diabetic patients is expected to increase to approximately 350 million by the year 2035, with more than 40% of these patients developing CKD (Gheith, Farouk, Nampoory, Halim, & Al-Otaibi, 2016; Pavkov, Collins, Coresh, & Nelson, 2018). With the increasing urgency of the health risk posed by diabetes-related renal complications, the development of effective therapies and strategies for prevention is paramount. However, the pathogenesis of DKD has not been fully elucidated. Renal ion transporters are central to the intricate pathophysiological mechanisms of DKD and its progression. This article will review research concerning the contribution of key renal transporters to the progression of DKD and assess their utility as candidate therapeutic targets.
2. DIABETIC KIDNEY DISEASE
In DKD, also known as diabetic nephropathy (DN), hyperglycemia overwhelms the kidney’s functionality, resulting in a breakdown of the glomerular filtration barrier (GFB) and overall dysfunction of the kidney. Hyperglycemia can lead to a variety of pathological cascades that affect ion transport in the kidney. The defining characteristics of DKD include a greater than 50% decline in glomerular filtration rate (GFR) over the course of the disease, microalbuminuria resulting from progressive GFB deterioration, and histological evidence of renal injury (interstitial fibrosis and glomerulosclerosis) (F. C. Brosius, 3rd et al., 2009; Schena & Gesualdo, 2005; Vallon & Komers, 2011). The substantial reduction in GFR associated with DKD is not observed during the initial stages of the disease. Patients initially exhibit hyperfiltration,an elevation in GFR, which gradually declines as DKD progresses over the course of 5 to 10 years. The initial hyperfiltration stage is an attempt by the kidneys to compensate for the apparent decrease in sodium delivery resulting from hyperglycemia. Once the tubuloglomerular feedback (TGF) system’s ability to compensate for increased sodium and glucose reabsorption reaches saturation, kidney function and GFR decline (F. C. Brosius, 3rd et al., 2009; Schena & Gesualdo, 2005; Tuttle, 2017; Vallon & Komers, 2011). The second hallmark of DKD, microalbuminuria, is caused by the breakdown of the GFB during disease progression. The GFB is comprised of podocytes and their foot processes (the slit diaphragm), the glomerular basement membrane, and endothelial cells. These components normally function together with a network of proteins to filter the contents of the glomerular capillaries and prevent the passage of substances larger than approximately 69 kDa (the approximate molecular weight of albumin). As renal function declines and glomerular hypertrophy ensues, the GFB deteriorates. As a result, albumin leaks from the capillaries and is excreted in the urine (Jefferson, Shankland, & Pichler, 2008). Renal histologic changes typical of DKD include cellular and tissue injury, mesangial matrix expansion, nodular glomerular lesions, arteriolar hyalinosis, thickening of glomerular basement membranes, and renal interstitial fibrosis.
Hypertension is a frequent comorbidity in diabetic patients and is implicated in the progression of DKD (Fig 1). Hypertension is typically twice as common in patients with diabetes compared to the general population. Diabetes mellitus interacts synergistically with hypertension to promote kidney injury (Staruschenko, 2017). Therefore, the effects of both hyperglycemia and hypertension are required for kidney injury to occur in DKD (Z. Wang et al., 2017). For this reason, anti-hypertensive drugs remain the leading treatment for DKD. The development of hypertension in diabetes results from overactivation of the renin-angiotensin-aldosterone system (RAAS), upregulation of endothelin 1 (ET-1; a vasoconstrictor secreted by endothelial cells), overproduction of reactive oxygen species (ROS), downregulation of nitric oxide (NO; a vasodilator), as well as other aberrant signaling (Arora & Singh, 2013; Kohan, Rossi, Inscho, & Pollock, 2011; Patney, Chaudhary, & Whaley-Connell, 2018; Patney, Whaley-Connell, & Bakris, 2015).
Figure 1.
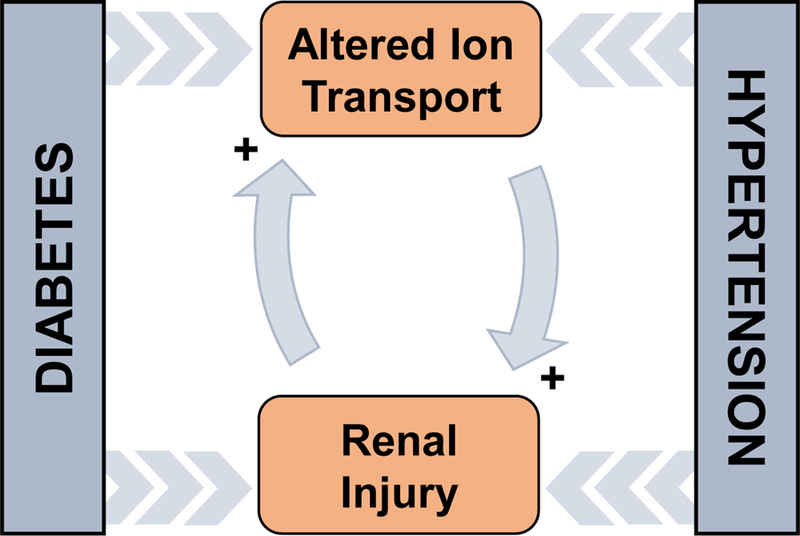
The positive feedback relationship between diabetes, hypertension, altered ion transport, and renal injury. This represents a simplified explanation for a very complex relationship that defines diabetic kidney disease (DKD).
In DKD, hyperglycemia leads to the activation of numerous pathways including RAAS, inflammatory cytokines, and oxidative stress cascades (Fig. 2), which ultimately result in the renal impairment characteristic of this disease. The RAAS is particularly important in the progression of DKD, given that the primary treatment for DKD is anti-RAAS drugs, such as angiotensin converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) (Anders, Huber, Isermann, & Schiffer, 2018; Umanath & Lewis, 2018). In response to hyperglycemic conditions, renal cells begin to secrete angiotensin II (Ang II), an integral member of RAAS. Intrarenal Ang II has been found to be substantially elevated when compared to circulating Ang II in DKD patients. This redistribution of Ang II, which likely has a considerable impact on renal ion transport, has been shown to have a causative influence on multiple distinctive features of DKD, including podocyte injury and apoptosis (Leehey, Singh, Alavi, & Singh, 2000; Vallon & Komers, 2011).
Figure 2.

Schematic for pathogenesis of diabetic kidney disease (DKD). Hyperglycemia and hyperinsulinemia induced by diabetes leads to the activation and expression of the different channels and transporters located along the various nephron segments. These changes are exercised either directly or indirectly upon the channel. The multitude of these channel alterations result in interstitial fibrosis, glomerulosclerosis, hypertrophy, breakdown of the glomerular filtration barrier (GFB) and albuminuria. The culmination of the damage to various portions of the nephron is the development of DKD. Abbreviations: calcium activated potassium channel 3.1 (KCa3.1); glucose transporters (GLUT1 and 2); sodium glucose cotransporter (SGLT1 and 2); large conductance Ca2+-activated K+ channel (BKCa); epithelial Na+ channel (ENaC); transient receptor potential canonical (TRPC) channel.
This introduction to DKD represents only a glimpse of the complexity involved in the development and progression of this disease. The variation in renal ion transporter expression and activity and their intersecting pathways, signaling cascades, and feedback loops both respond and contribute to pathophysiological states during the progression of diabetes to DKD. These pathological states, including hyperglycemia, hypertension, and dysfunctional insulin signaling substantially dysregulate renal ion transport and electrolyte homeostasis, accelerating renal injury characteristic of DKD. The intricacy of DKD makes it particularly difficult to fully understand all details involved in its progression.
3. GLUCOSE TRANSPORTERS
3.1. Sodium glucose cotransporters
The kidneys play a major role in glucose regulation in humans, and are responsible for reabsorbing 99% of plasma glucose. Glucose reabsorption in the kidney occurs via sodium-glucose transporter 2 (SGLT2) in the early proximal tubule (PT) and to a lesser extent via sodium-glucose transporter 1 (SGLT1) in the late PT. Several SGLT1, SGLT2 or dual SGLT1/SGLT2 inhibitors have been shown to lower blood glucose by preventing glucose reabsorption at the PT, and act as effective antiglycemic drugs that may have utility in the treatment or prevention of DKD (Fig. 3) (Rieg & Vallon, 2018). These inhibitors are well described and have been marketed for treatment of type 2 diabetes mellitus (T2DM).
Figure 3.

Tubular view of glucose transport within the proximal tubule of the kidneys. A. Healthy proximal tubule segments and normal function of sodium glucose cotransporters (SGLT1 and SGLT2) and their glucose transporter (GLUT1 and GLUT2) counterparts. B. The altered function of SGLT and GLUT transporters under DKD conditions. C. Examples of the inhibitors specific for SGLT2, SGLT1, and dual inhibitors for both transporters and their general effects under DKD conditions.
Members of the SGLT family of glucose transporters are involved in the re-uptake of glucose across the apical cell membrane. Their structure is composed of 14 transmembrane helices (Deng & Yan, 2016). There are 6 members of the SGLT family (SGLT1–6), but only SGLT1 and SGLT2 are well-characterized and have been shown to be highly expressed in the kidney (Harada & Inagaki, 2012; Poulsen, Fenton, & Rieg, 2015). Although SGLT1 primarily functions within the small intestine, it does contribute to the maintenance of normal glucose balance in the kidney (Tahrani, Barnett, & Bailey, 2013); whereas SGLT2 is predominantly responsible for glucose uptake within the kidney. SGLT2 is located in the brush border of segments 1 and 2 of the PT and is responsible for approximately 90% of the glucose reabsorption in this part of the nephron. The remaining 10% is reabsorbed via SGLT1 in late segments of the PT (Hediger & Rhoads, 1994; Poulsen et al., 2015). The driving force for these cotransporters is the active movement of sodium via the sodium potassium ATPase (Na+/K+ ATPase) causing reuptake of glucose by the cell. In a study of these two cotransporters, it was found that only 1 sodium ion is required by SGLT2 for the reabsorption of 1 glucose molecule through the transporter. In contrast, SGLT1 requires 2 Na+ ions to be moved for each glucose molecule absorbed, Km ~0.4 mM. Normally, SGLT2 (Km ≤ 6 mM) works at 50% capacity, only becoming fully saturated at a glucose level greater than and/or equal to 35 mM (E. Ferrannini & Solini, 2012; Ghezzi, Loo, & Wright, 2018; Harada & Inagaki, 2012; Hummel et al., 2011; Szablewski, 2017). Together with Na+/K+ ATPase, SGLT1 and SGLT2 make up the first stage of glucose transport and prevent excessive loss of glucose in the urine. Powell and colleagues confirmed the important role of the SGLTs in glucose transport showing that in a double knockout mouse model, the absence of both SGLT1 and SGLT2 resulted in the excretion of the entire filtered load of glucose (Ghezzi et al., 2018; Powell, DaCosta, et al., 2013). Given these transporters’ substantial impact on overall glucose homeostasis, their implication in the development diabetes mellitus and its progression to DKD is unsurprising.
3.2. SGLT1 and SGLT2 in DKD
In treating diabetes mellitus, controlling blood glucose levels is imperative to prevent disease progression. The tubules reabsorb glucose at a maximal rate of approximately 375 mg/min after which their capacity for transport is saturated. In diabetes, plasma glucose increases to levels of hyperglycemia, bombarding the TGF system and the macula densa with overwhelming quantities of glucose. The overloading of this system causes hyperfiltration, which as previously mentioned, ultimately leads to glucose excretion in the urine and DKD (Abdul-Ghani, Norton, & DeFronzo, 2015; Brenner, 1983; Farber, Berger, & Earle, 1951; Ruggenenti et al., 2012; Vallon et al., 2011). SGLT transporters, especially SGLT2, have been targeted by multiple therapeutics in attempt to regulate hyperglycemia in diabetes. The goal of SGLT inhibitors is to increase the urinary output of glucose, thereby decreasing circulating glucose content and minimizing damage (Spatola, Finazzi, Angelini, Dauriz, & Badalamenti, 2018). Vallon et al. found that administering an SGLT2 inhibitor (empagliflozin) to the Akita mouse, a model of type 1 diabetes mellitus (T1DM), prevented hyperfiltration and reduced kidney hypertrophy and albumin excretion in the early stages of DKD. In the T2DN rat, a model of type 2 diabetic nephropathy (DN), the progression of DKD was slowed using a SGLT2 inhibitor which led to an appreciable reduction in GFR, glomerulosclerosis, tubulointerstitial fibrosis, and proteinuria. Similar results were seen in a mouse model of type 2 DKD with SGLT2 inhibition causing a reduction in mesangial expansion and expression of inflammatory markers (Gembardt et al., 2014; Koepsell, 2017; Kojima, Williams, Takahashi, Miyata, & Roman, 2013). Interestingly, insulin receptor deletion significantly reduced SGLT2 expression and increased urinary glucose excretion and urine flow (Nizar, Shepard, Vo, & Bhalla, 2018).
Unlike T1DM where insulin treatments are greatly beneficial and contribute to an improved quality of life, insulin treatment in T2DM is not as effective. These patients are, to some extent, insulin resistant. For T2DM patients, research has been focused on therapies targeting glucose transporters, specifically SGLT2 because the bulk of glucose re-uptake occurs through this transporter (Defronzo, 2009; Tahrani et al., 2013). Additionally, this cotransporter is primarily located in the kidney which minimizes off target effects of globally inhibiting SGLT2 (Koepsell, 2017). Patients with T2DM have similar renal protein expression of SGLT1 and SGLT2 compared to normoglycemic patients, although Norton and colleagues found that SGLT1 and SGLT2 mRNA expression levels in T2DM are higher than in normoglycemic patients. SGLT2 inhibitors reduce glucose reabsorption in the PT, ultimately resulting in improved kidney function (Sano, Takei, Shiraishi, & Suzuki, 2016; Zou, Zhou, & Xu, 2017). SGLT2 inhibitors also result in increased sodium delivery to the macula densa, which activates the TGF system to constrict the afferent arteriole and decrease GFR (Cherney et al., 2014; Vallon, Blantz, & Thomson, 2003; Zou et al., 2017). In the late stages of T2DM and DKD, the SGLT2 inhibitor canagliflozin was found to improve glycemic control and reduce albuminuria. Kohan et al. found that dapagliflozin, another SGLT2 inhibitor, reduced the body weight and blood pressure of T2DM patients without the change in glycemic control that Yale et al. observed (Kohan, Fioretto, Tang, & List, 2014; Yale et al., 2013; Zou et al., 2017). Similar results were found by other groups showing multiple benefits of SGLT2 inhibitors in diabetes and DKD, including improvements to renal oxygenation, natriuresis, and oxidative stress (Dekkers, Gansevoort, & Heerspink, 2018; Tanaka et al., 2018; X. X. Wang et al., 2017). SGLT2 expression and/or activity has been found to be upregulated in both type 1 and 2 diabetes, with an increase in the maximum glucose transport of approximately 20%. Administration of these inhibitors decreases this maximum capacity by roughly 30 to 50% (DeFronzo et al., 2013; Gallo, Wright, & Vallon, 2015). In addition, a study by Ferrannini et al found that empagliflozin improved pancreatic beta cell function and insulin sensitivity in T2DM patients (E. Ferrannini et al., 2014). Inhibitors of SGLT2 also reduced urate concentrations in the blood, which may contribute to the protective effects of these inhibitors in the progression of DKD (Ficociello et al., 2010; Haring et al., 2014; Jabbour, Hardy, Sugg, & Parikh, 2014; Koepsell, 2017; Kovacs et al., 2014; Rosenstock et al., 2014; Wilding et al., 2012). Figure 3 summarizes the effects of hyperglycemia on these transporters and the effects of their various inhibitors in DKD.
Although SGLT2 is the primary target for treatment of T2DM, it has been proposed that SGLT1 expression increases as a result of SGLT2 inhibition. As SGLT2 becomes overloaded and is inhibited, saturating glucose transport in the early PT, there is an increased need for SGLT1 contribution to handle the increase in glucose delivery to the later segment of the PT. Also, it has been hypothesized that SGLT2 inhibitors are less effective in T2DM patients with renal impairments, and SGLT1 inhibitors have been found to improve glycemic control in these cases (Gorboulev et al., 2012; J. J. Liu, Lee, & DeFronzo, 2012; Spatola et al., 2018; Yale et al., 2013). SGLT1 inhibitors also improve glucose homeostasis by exerting substantial effects on the gastrointestinal system, increasing the release of glucagon-like peptide-1 (GLP-1) and reducing the absorption of glucose in the gut (P. Song, Onishi, Koepsell, & Vallon, 2016).
Although broadly beneficial for the treatment of diabetes, there are also complications associated with SGLT inhibitors. (G. Ferrannini & Ryden, 2018; Lupsa & Inzucchi, 2018). Adverse effects of SGLT2 blockers include genital mycotic and urinary tract infections. Euglycemic diabetic ketoacidosis has been found in some cases, likely due to increases in glucagon secretion and stimulation of lipolysis and ketogenesis. Furthermore, SGLT2 inhibitor monotherapy seems to be ineffective at maintaining long-term control of hyperglycemia in some cases of DKD (Hershon, 2016; Zou et al., 2017). Combined inhibitors of SGLT1 and SGLT2, such as sotagliflozin, have been shown to effectively reduce glucose and insulin levels in the plasma of T2DM patients (Powell, Smith, et al., 2013; Zambrowicz et al., 2012). Recent studies also revealed beneficial effects of sotagliflozin in combination with insulin treatment in patients with T1DM (Garg et al., 2017). SGLT2 inhibitors have also shown to be effective in combination with RAAS blockers by reducing cardiovascular events, albuminuria, hyperfiltration, and blood pressure in DKD (Bautista et al., 2004; Kojima et al., 2015; Kojima et al., 2013; Zou et al., 2017). Combining SGLT2 inhibitors with other targets of the glycemic control pathway, specifically dipeptidyl peptidase-4 (DPP-4) inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists, has also shown to be beneficial in treating DKD. GLP-1 receptor agonists can potentially counteract increases in glucagon secretion caused by SGLT2 inhibitors; whereas DPP-4 inhibitors combined with SGLT2 inhibitors showed reduced hypoglycemia, albuminuria, hyperglycemia, and blood pressure in DKD (DeFronzo et al., 2015; Scheen & Delanaye, 2018; Secrest, Udell, & Filion, 2017).
3.3. GLUT glucose transporters
In addition to the sodium glucose cotransporters, glucose transporters (GLUTs) are also vital for proper glucose homeostasis, and therefore have implications in the development of DM and DKD. GLUTs along with SGLTs are members of the major facilitator superfamily (S. S. Pao, Paulsen, & Saier, 1998; Thorens & Mueckler, 2010). GLUTs facilitate the energy independent movement of glucose down its electrochemical gradient. They are expressed in every cell in the body and are essential for energy metabolism. There are 17 GLUT proteins in the SLC2 family, which are further divided into three classes based on structure. Class I contains GLUT1–4, Class II contains GLUT5, 7, 9 and 11, and Class III contains GLUT6, 8, 10, 12 as well as the H+/myo-inositol transporter (HMIT). Expression patterns, regulation, and properties of GLUTs are tissue specific. During different disease stages, GLUT expression levels tend to vary as well (Szablewski, 2017). This review will focus on specific GLUTs with prominent effects in the kidney.
3.4. GLUTs in DKD
GLUTs 1, 2, 4, 5, 8–10, and 12 all function in different segments of the kidney to facilitate glucose transport (Chin et al., 1997; C. Heilig et al., 1995; C. W. Heilig, Brosius, & Cunningham, 2006; Mather & Pollock, 2011). GLUT1 has been found to be upregulated in the renal cortex in diabetes as well as in glomerular hypertension. Similar to SGLT1 in the PT, GLUT1 is a low capacity glucose transporter in the glomerulus with a high affinity for glucose (C. W. Heilig, Brosius, & Henry, 1997). Wang et al. showed that when GLUT1 is overexpressed in glomerular mesangial cells of the C57BL6 mouse (a relatively DKD resistant strain), the glomerulus develops damage like that of the glomerulosclerosis typically seen in DKD. More interestingly, these mice were not hyperglycemic or hypertensive during overexpression, insinuating that GLUT1 plays a potential role in the development of glomerulosclerosis in DKD (Y. Wang et al., 2010). GLUT1 is also expressed in the podocytes of the glomerulus, where it is widely localized to both the apical and basolateral membrane, within vesicles in the cytoplasm and plasma membrane of the foot processes. GLUT1 plays an important role in the proper function of podocytes in the GFB, which impacts overall kidney function (F. C. Brosius, 3rd, Briggs, Marcus, Barac-Nieto, & Charron, 1992; F. C. Brosius & Heilig, 2005; Coward et al., 2005; Jefferson et al., 2008; Wasik & Lehtonen, 2018). GLUT1 is activated in models of streptozotocin (STZ) induced T1DM as well as in mouse models of T2DM (Chen, Heilig, Brosius, & Heilig, 2003; D’Agord Schaan et al., 2001). Although GLUT1 overexpression in mesangial cells leads to the further development of DKD, its overexpression in podocytes appears to be protective in the progression of DKD (Y. Wang et al., 2010; Wasik & Lehtonen, 2018; Zhang et al., 2010). Several studies also implied that genetic variations in GLUT1 results in a genetic predisposition for DKD (F. C. Brosius & Heilig, 2005; Grzeszczak et al., 2001; Hodgkinson, Millward, & Demaine, 2001; Hsu et al., 2011; Z. H. Liu, Guan, Chen, & Li, 1999; Ng et al., 2002; Tarnow, Grarup, Hansen, Parving, & Pedersen, 2001; Vaulont & Kahn, 1994).
In the later stages of glucose reuptake by the kidney, GLUT2 is the primary transporter responsible for the basolateral movement of glucose on the brush border of the PT (Ghezzi et al., 2018; Mather & Pollock, 2011). In the diabetic kidney, it has been found that there is an increase in GLUT2 expression in the PT. For example, Chin et al. found that GLUT2 mRNA expression in the PT was increased in a T1DM animal model (Chin et al., 1997). Kamran et al. found that GLUT2 is overexpressed in both STZ-treated Sprague Dawley rats and diabetic Zucker rats (Kamran, Peterson, & Dominguez, 1997). Marks et al. found similar results in the PT of STZ-treated rats (Marks, Carvou, Debnam, Srai, & Unwin, 2003). GLUT1 and 2 work within the PT together with SGLT1 and 2 (Fig. 3). However, SGLT1 and 2 work more closely through GLUT2, high capacity and low glucose affinity, to handle the bulk of glucose transport in the PT (Ghezzi et al., 2018; Mather & Pollock, 2011). In contrast to SGLTs, finding specific inhibitors of GLUT2 has proven challenging considering the close homology between members of the GLUT family (Ghezzi et al., 2018; Yan, 2015).
In addition to GLUT1 and 2, GLUTs 4, 5, 8, 9, and 10 have also been detected in the kidney. In podocytes, GLUT4 is located on both the apical and basolateral membranes and within intracellular vesicles in the podocyte foot processes. GLUT4 is also expressed in mesangial cells, whereas GLUT8 is only expressed in podocytes (F. C. Brosius, 3rd et al., 1992; F. C. Brosius & Heilig, 2005; C. W. Heilig et al., 2006; Mather & Pollock, 2011). GLUT4 and 8 are considered to be insulin responsive transporters, another key function linking them to DKD. It was found that GLUT8 mRNA and protein levels are regulated by plasma glucose levels in both normal conditions and in cases of diabetes, including insulin resistant forms of diabetes. GLUT4 relocation to the plasma membrane has been found to be induced by the insulin-stimulated increase in phosphoinositide 3-kinase (PI3K)/AKT pathway (F. C. Brosius & Heilig, 2005; Marcus et al., 1994; Schiffer et al., 2005; Wasik & Lehtonen, 2018). In a model of STZ-induced T1DM, GLUT4 expression in glomeruli was reduced (Marcus et al., 1994). Additionally, Coward et al. found that GLUT4 redistributes to the basal membrane in podocytes in a T2DM model. This suggests that despite a marked reduction in GLUT4 expression, functionality may not be affected during DKD progression (Coward et al., 2005). Additional studies have found that GLUT1 and 4 are responsive to insulin in the podocyte specifically, being stimulated in some fashion by insulin in these cells. Insulin causes activation of GLUT4 and its translocation from the perinuclear and cytosolic vesicular structures to the plasma membrane of the podocyte. Additional studies of GLUT4 deficient mice with DKD demonstrate that reduced GLUT4 activity protects podocytes from DKD by reducing mechanistic target of rapamycin (mTOR) activity (Coward et al., 2005; Guzman et al., 2014; Wasik & Lehtonen, 2018).
In addition to GLUT expression in glomeruli and the PT, Linden et al. found that GLUT12 is located within the distal tubule and collecting duct (CD) of the nephron. GLUT12 protein was found to be predominantly located in the cytoplasm and apical membranes of these segments. Using STZ-treated Ren-2 transgenic rats as a model of DKD, they saw an increase in both GLUT12 and GLUT1, indicating that both GLUT transporters have some involvement in the progression of this disease (Linden et al., 2006; Mather & Pollock, 2011). Expression levels for GLUT3 were also reported, but protein localization has not been determined (C. Heilig et al., 1995). GLUT5, which has been proposed to be fructose specific, has shown increased mRNA expression in the PT during chronic T1DM induced by STZ. Thus, GLUTs, together with SGLTs, play critical roles in glucose transport and contribute towards DKD progression.
4. SODIUM TRANSPORTERS
Sodium absorption in the kidney is precisely regulated and controlled by numerous physiological mechanisms. Under pathological conditions such as diabetes mellitus, Na+ transport is significantly altered. Eriguchi et al. recently performed a sodium transporter profile immunoblot analysis in wild type mice where T1DM was induced by injection of STZ. After 6 months of STZ injections, the authors found no significant changes in total Na+/H+ exchanger (NHE) and Na+-K+−2Cl− cotransporter (NKCC) expression. However, NKCC2 phosphorylation was significantly increased in diabetic mice compared with nondiabetic controls. Similarly, analysis of distal tubule transporters revealed increased expression in both total and phosphorylated sodium-chloride cotransporter (NCC), and in subunits of the epithelial Na+ channel (ENaC). Diabetic animals exhibited elevations in total α- and β-ENaC as well as cleaved forms of α- and γ-ENaC (Eriguchi et al., 2018). These data demonstrate that STZ-mediated hypoinsulinemia and hyperglycemia cause upregulation of most major sodium transporters. This section will delineate additional details about specific sodium transporters, and their potential contributions to DKD.
4.1. ENaC
Expressed primarily in principal cells of the distal nephron, ENaC plays a central role in maintaining salt and water homeostasis, regulating extracellular fluid volume, controlling blood pressure, and overall renal function (Hanukoglu & Hanukoglu, 2016; Kleyman, Kashlan, & Hughey, 2018; Pavlov & Staruschenko, 2017; Staruschenko, 2012). Diabetes and DKD have been associated with increased ENaC activity and expression, which may reflect or contribute to the pathophysiology of DKD. Studies in humans and animals present multiple mechanisms by which the diabetic state can elicit changes in ENaC, which interfere with renal blood pressure control, exacerbate hypertension, and thereby contribute to the progression of DKD.
ENaC subunits are located on the apical membrane of principal cells in the aldosterone-sensitive distal nephron where they are tightly controlled by various hormones and mediate fine-tuning of sodium absorption in the kidney (Staruschenko, 2012). We and others have shown that insulin augments ENaC expression and activity (Gonzalez-Rodriguez, Gaeggeler, & Rossier, 2007; Ilatovskaya, Levchenko, Brands, Pavlov, & Staruschenko, 2015; Mansley et al., 2016; A. C. Pao, 2016; Pavlov et al., 2013; Tiwari, Nordquist, Halagappa, & Ecelbarger, 2007). As an example, single-channel analysis in freshly isolated, split-open tubules demonstrated that ENaC activity was acutely activated by insulin. Moreover, insulin receptor knockout mice have significantly lower activity compared to their wild-type littermates (Pavlov et al., 2013). Interestingly, high fat-fed mice had no increase in ENaC activity (Nizar et al., 2016). Recent studies by Irsik et al. (Irsik, Blazer-Yost, Staruschenko, & Brands, 2017; Irsik & Brands, 2018) have utilized a sophisticated insulin-clamping technique in rats, which allowed them to test the role of daily variations in insulin on sodium excretion. They found that rats whose insulin was clamped to prevent increases in response to carbohydrate showed elevated sodium excretion over the first 4 hours post carbohydrate administration (Irsik & Brands, 2018).
One proposed mechanism suggests that ENaC involvement with DKD is inexorably linked to the serum and glucocorticoid-regulated kinase (SGK1) protein. SGK1 is stimulated by insulin, which causes more ENaC to be translocated to the membrane (through the Nedd4–2 signaling pathway) increasing sodium reabsorption from the tubule. This may lead to excess renal sodium retention, hypertension, and ultimately renal damage associated with DKD (McCormick, Bhalla, Pao, & Pearce, 2005; Pearce et al., 2015). In vitro studies found that both ENaC and SGK1 are up-regulated by high levels of extracellular glucose (Hills, Bland, Bennett, Ronco, & Squires, 2006). It has been well established that the over-activity of ENaC can result in hypertension, and increased ENaC expression has been identified in animal models of both type 1 and type 2 diabetes (C. T. Chang et al., 2007). In a rat model of STZ-induced T1DM, increased glucose was correlated with upregulation of all three ENaC subunits, attributed to elevations in aldosterone and vasopressin (J. Song, Knepper, Verbalis, & Ecelbarger, 2003). Another proposed mechanism of ENaC increases in DKD involves the serine protease, plasmin (Kleyman et al., 2018; Ray et al., 2018). Urinary plasmin has been found to be elevated in human subjects with DKD as well as in the puromycin aminonucleoside rat model of nephrotic syndrome. Dysfunction of the GFB in DKD causes plasmin to be filtered to the tubules where it activates ENaC and increases sodium reabsorption (Svenningsen, Skott, & Jensen, 2012). In a study of patients with T2DM, microalbuminuria, a hallmark of GFB breakdown, is associated with increased aberrant filtration of plasmin. This surge of filtered plasmin was shown to be sufficient to increase the open probability for ENaC, and was proposed as a possible mechanism contributing to hypertension in diabetes (Buhl et al., 2014). Clinical studies have also found that amiloride, an ENaC blocker, may be protective in DKD as it significantly increased sodium excretion, and reduced blood pressure, albuminuria, and plasmin in urine of diabetic patients (Andersen et al., 2015). Recently a pilot randomized cross-over study comparing the effects of daily administration of either oral amiloride or the NCC inhibitor, hydrochlorothiazide (HCTZ), to patients with type 2 diabetes and proteinuria revealed similar effects with both drugs resulting in reduced systolic blood pressure (Unruh et al., 2017).
It is widely accepted that oxidative stress plays a central role in diabetes-induced renal injury. Prolonged hyperglycemia causes excess glucose to contact and react with proteins and lipids resulting in advanced glycation end-products (AGEs), which are known to cause multiple complications in diabetic patients and are implicated in DKD. The role of AGEs in DKD may be especially important to understand as they are capable of having substantial effects, including oxidative stress, that persists long term even after blood glucose control is regained in the patient (Singh, Bali, Singh, & Jaggi, 2014). AGEs have been shown to be upregulated in diabetic subjects with hypertension, with an especially pronounced elevation in the distal nephron where ENaC is highly expressed (Schleicher, Wagner, & Nerlich, 1997). When applied to cultured tubular epithelial cells in concentrations comparable to what occurs in diabetes, AGEs increased ENaC mRNA and protein and stimulated ENaC activity by inhibiting catalase and increasing intracellular ROS production (Q. Wang et al., 2015). The effect on ENaC activity persisted for more than 72 hours after removal of AGEs. This sustained ENaC elevation may be key to understanding why DKD often continues to progress despite adequate glucose control and provide key insights necessary for the development of more effective treatments. From these studies, it is evident that diabetes creates pathophysiological conditions that affect ENaC via multiple pathways, causing a sustained increase in activity or expression, ultimately resulting in blood pressure elevation (Fig. 4). As hypertension is one of the most important risk factors in the progression from diabetes to DKD, ENaC is a critical mechanistic and potential therapeutic target in DKD research.
Figure 4.
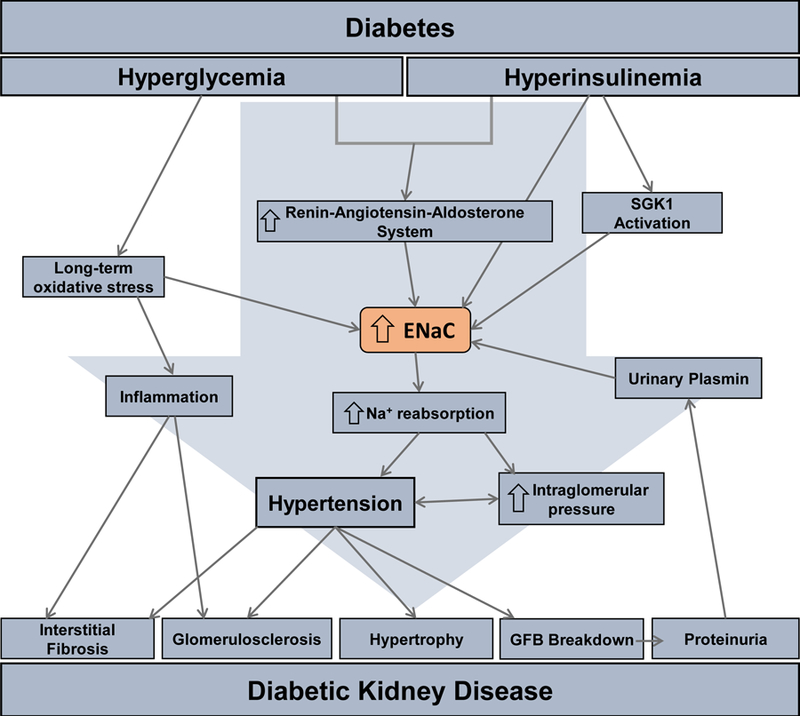
Schematic for epithelial Na+ channel (ENaC) induced tubular renal injury in DKD. Hyperglycemia and hyperinsulinemia induced via diabetes cause over-activation of the renin-angiotensin-aldosterone system (RAAS), long-term oxidative stress, and serum and glucocorticoid-regulated kinase (SGK) 1 activation that all directly cause the increase in the ENaC activation and/or expression. This increase in ENaC leads to various factors shown in the above pathway, such as hypertension. The culmination of these factors result in the development of the major characteristics of DKD.
4.2. Sodium hydrogen exchanger (NHE)
Sodium-hydrogen exchangers (NHE) directly and indirectly contribute to the maintenance of blood volume and whole body acid-base homeostasis. The inward movement of Na+ down its electrochemical gradient supplies the energy for the active transport of H+ against its gradient. In the human genome there are 9 isoforms of NHE belonging to one of 3 subfamilies: cation-proton antiporters (CPA1 and CPA2) and Na-transporting carboxylic acid decarboxylase (NaT-DC). NHE1–4 and 6–9 are members of the CPA1 family and are found in various parts of the kidney (Bobulescu, Di Sole, & Moe, 2005; Bobulescu & Moe, 2009; Brett, Donowitz, & Rao, 2005; A. B. Chang, Lin, Keith Studley, Tran, & Saier, 2004; Orlowski & Grinstein, 2004). NHE isoforms 1–4 and 8 are located on either the apical or basolateral membranes of renal epithelial cells, whereas NHE6, 7, and 9 are found only on membranes of organelles. NHE3 and NHE8 are located on the apical membrane of the PT; NHE2 and NHE3 are on the apical membrane of the thick ascending limb (TAL) of the loop of Henle; NHE 2 is on the apical membrane of the distal convoluted tubule (DCT) and connecting tubule; NHE4 is on the basolateral membrane of the entire nephron; and NHE1 is located on the basolateral membrane everywhere except the macula densa and intercalated cells (Chambrey et al., 2001; Chambrey et al., 1998; Goyal, Mentone, & Aronson, 2005; Nakamura, Tanaka, Teko, Mitsui, & Kanazawa, 2005; Orlowski & Grinstein, 2004; Xu, Chen, & Ghishan, 2005).
Given the location and the general function of NHE exchangers, it is expected that certain NHE isoforms are involved in the progression of DKD. STZ-induced T1DM in a mouse model with a loss of function mutation in NHE1 (swe/swe mouse created on the C57BL/6 mouse background) resulted in the development of DKD characteristics. Swe/swe mice without STZ-induced T1DM exhibited renal tubular epithelial cell atrophy, and STZ treatment resulted in the additional development of albuminuria and increased tubulointerstital pathology (Khan, Wu, Sedor, Abu Jawdeh, & Schelling, 2006; Wu et al., 2003). Numerous studies have found that NHE3 in particular is implicated in DKD development. A link between SGLT2 and NHE3 has been proposed to contribute to the renoprotective effects of SGLT2 inhibitors and to potentially contribute to the reduction in sodium reabsorption during treatment with SGLT2 inhibitors. However, contradictory explanations have been reported, suggesting that SGLT2 inhibitors increase rather than reduce sodium absorption and that the nephro-protective effects of these inhibitors is not dependent on the TGF system (Wakisaka, 2016; Wakisaka, Nagao, & Yoshinari, 2016; Wright, Loo, & Hirayama, 2011; Zeni, Norden, Cancarini, & Unwin, 2017). The connection between NHE3 and SGLT2 has even been hypothesized to have benefits in treating some of the characteristics of DKD, including the alteration of renal hemodynamics (Pessoa, Campos, Carraro-Lacroix, Girardi, & Malnic, 2014; Tonneijck et al., 2017). The “tubular theory” for hyperfiltration (a hallmark of DKD) suggests that the relationship between the glomerulus and the tubule is the key to explaining diabetes-induced renal dysfunction and abnormalities, and proposes that hyperfiltration is caused by increased sodium reabsorption combined with tubular hypertrophy and up-regulation of SGLTs and NHE3. The theory suggests that the combination of these factors inhibit TGF (Tonneijck et al., 2017; Tuttle, 2017; Zeni et al., 2017). It has also been discovered that the GLP-1 receptor agonist, liraglutide, reduces GFR and albumin excretion in patients with T2DM by GLP-1 mediated inhibition of NHE3 and DPP-4 assembly in the PT brush border. Inhibiting this relationship causes a reduction in sodium reabsorption and GFR by activating the TGF system (Muskiet, Smits, Morsink, & Diamant, 2014; Tonneijck et al., 2017).
4.3. NKCC2 and NCC
The furosemide-sensitive cotransporter NKCC2 and the thiazide-sensitive cotransporter NCC play important roles in renal salt handling and extracellular volume regulation in the TAL and DCT, respectively. Similar to other sodium transporters, expression of both total and active forms of NKCC2 and NCC is increased under diabetic conditions, which has been reported in several rodent models (Cipriani et al., 2012; Eriguchi et al., 2018; Riazi, Khan, Tiwari, Hu, & Ecelbarger, 2006; Riazi, Maric, & Ecelbarger, 2006). Metformin, an antidiabetic drug that is widely used to treat patients with diabetes mellitus, was shown to increase urinary sodium excretion by reducing phosphorylation of NCC. Interestingly, the activity of other renal sodium transporters, such as NKCC2, ENaC, and NHE3 did not show significant changes during metformin treatment (Hashimoto et al., 2018). Similar to this finding, our data also revealed that metformin-treated Dahl SS rats fed a high salt diet had no difference in the activity of ENaC (Pavlov et al., 2017).
Hyperinsulinism is associated with increased expression of NCC along with Na+/K+ ATPase and ENaC (Bickel, Verbalis, Knepper, & Ecelbarger, 2001). In vitro studies of insulin effects revealed that insulin induces activation and phosphorylation of NCC, which could contribute to sodium balance and the progression of DKD in hyperinsulinemic states (Chavez-Canales et al., 2013). Therefore, there is some evidence demonstrating the potential contribution of sodium cotransporters in the TAL and DCT (especially for thiazide-sensitive transporter NCC). However, additional studies are warranted.
5. POTASSIUM CHANNELS
Although renal potassium channels have not been definitively identified as causal in the development of DKD, it is likely that some of these channels are involved in the disease progression. The kidney is responsible for maintaining whole-body potassium homeostasis, which is essential for the proper control of blood glucose, as insulin is secreted from pancreatic beta cells in response to a potassium induced depolarization (Ekmekcioglu, Elmadfa, Meyer, & Moeslinger, 2016). Various clinical studies indicate that insufficient serum potassium or dietary potassium intake is associated with the onset of T2DM. Low potassium diets and hypokalemia contribute to impaired insulin secretion and glucose intolerance (Rowe, Tobin, Rosa, & Andres, 1980; Sagild, Andersen, & Andreasen, 1961). It has also been shown that the treatment of hypertension with thiazide diuretics, which commonly cause potassium depletion as a side-effect, has been associated with increased risk of new-onset diabetes (Zillich, Garg, Basu, Bakris, & Carter, 2006). In addition to diabetes onset, potassium may also play a role in the progression of diabetes to more severe cardiovascular and renal impairment. High potassium diets have been established to reduce the risk of development of cardiovascular disease in healthy patients. However, it has been less clear whether increasing dietary potassium intake in patients with diabetes would have similar protective results. A study by Smyth et al. examined how potassium intake may correlate with renal outcomes in nearly 30,000 patients with established diabetes or other vascular disease. This study found that higher potassium was associated with decreased risk for all renal outcomes in these patients. Interestingly, they found that only potassium, not sodium, was predictive for renal outcome (Smyth et al., 2014). A similar study involving over 600 Japanese patients with T2DM also showed that high urinary excretion of K+ (indicative of higher potassium intake), but not sodium, was associated with better cardiorenal outcomes (Araki et al., 2015). These studies together indicate that the improper maintenance of potassium homeostasis, and possible dysfunction of renal K+ channels, may be involved in the development of renal impairments in diabetes. For example, the voltage-gated potassium channel gene subfamily, KCNQ1, which localizes to the brush border of the PT, has been proposed as a marker for DKD. Genetic variants in this gene have significant association with susceptibility for DKD and microalbuminuria in multiple studies involving East Asian and European populations (Lim et al., 2012; Ohshige et al., 2010; Unoki et al., 2008).
The Ca2+ - activated K+ channel 3.1 (KCa3.1) has been identified as a potential target in DKD. This voltage-independent potassium channel is expressed in multiple cell types implicated in tubulointerstitial fibrosis including renal PT cells, fibroblasts, inflammatory cells, and endothelial cells (Huang, Pollock, & Chen, 2014b). In vitro, as well as in vivo studies in diabetic mouse models have shown that KCa3.1 is activated by high glucose and produces a proinflammatory response that contributes to renal damage in DKD. Moreover, blocking KCa3.1 suppresses the proinflammatory cytokine chemokine ligand 20 (CCL20), which prevents macrophage accumulation and improves renal fibrosis in diabetic mice, making it a potential therapeutic target for the treatment of DKD (Huang, Pollock, & Chen, 2014a; Huang et al., 2013). One study found that renal injury in DKD may be exacerbated by insufficient autophagy in proximal tubular cells, and that blocking KCa3.1 restores normal autophagy, which may prevent some degree of renal injury in diabetic kidney disease (Huang et al., 2016). KCa3.1 blockers have been studied in clinical trials to treat sickle cell disease and although results showed it may be ineffective for this purpose, these drugs were safe and well-tolerated by patients in the trial (Wulff & Castle, 2010). This supports the claim that KCa3.1 blockers may be beneficial in the treatment of DKD.
As previously discussed, the malfunction of podocytes and loss of nephrin in the GFB is a hallmark of DKD. Studies show that both insulin and exposure to high glucose affect the activity and expression of the large-conductance Ca2+-activated K+ (BK) channel present in podocytes. Treatment of mouse podocytes with high glucose caused a marked reduction of BK channel current and a decrease in surface expression of BK channels, as well as nephrin, which likely interacts with the channel. However, insulin acts to stimulate BK channel activity and expression, which is blocked by the presence of high glucose. BK channels have been shown to interact with transient receptor potential canonical (TRPC) channels and their large conductance may provide the driving force facilitating movement of Ca2+ into the podocyte through TRPC channels. BK channels in the podocytes are responsive to insulin and glucose and interact with TRPC channels and nephrin, both of which are implicated in DKD. Thus it is feasible that the malfunction of these channels is involved in the progression of DKD (Kim & Dryer, 2011).
In addition to tubular potassium channels, there are renal vascular K+ channels which also contribute to DKD (Salomonsson, Brasen, & Sorensen, 2017). As an example, the involvement of ATP-sensitive K+ channels (KATP channels) in renal afferent arteriolar dilation was reported during STZ-induced T1DM (Ikenaga, Bast, Fallet, & Carmines, 2000). Similarly, we have shown recently that Dahl salt-sensitive rats with STZ-induced diabetes had an increased vasodilator response to the KATP channel activator, pinacidil (Miller et al., 2018). In addition to increased KATP channel activity in diabetes, it was also shown that other renal vascular K+ channels, including BK channels and members of the inward rectifier (Kir) family, contribute to afferent arteriolar dilation in diabetic animal models (Brindeiro, Fallet, Lane, & Carmines, 2008; Carmines & Fujiwara, 2002).
6. CALCIUM AND MAGNESIUM CHANNELS
6.1. Transient receptor potential (TRP) channels
The TRP superfamily of cation channels vary in permeability, selectivity, and mode of activation. TRP channels play a pivotal role in the influx of calcium, magnesium, and other ions across the plasma membrane and contribute to a diversity of functions, including their physiological and pathophysiological roles in the kidney (Abramowitz & Birnbaumer, 2009; Marko, Mannaa, Haschler, Kramer, & Gollasch, 2017; Tomilin, Mamenko, Zaika, & Pochynyuk, 2016). The TRP family is sub-divided into the following groups based on function and sequence: melastatin-related TRP (TRPM), ankryin transmembrane TRP (TRPA), vanilloid-receptor-related TRP (TRPV), mucolipin TRP (TRPML), polycystin TRP (TRPP), and canonical TRP (TRPC). Each group has a varied number of members and are expressed in all cell membranes, excluding mitochondrial membranes, throughout tissue types including the brain, lungs, smooth muscle, and kidneys. Abnormal activity and mutations in these channels have been linked to an assortment of kidney disorders such as nephrotic syndromes, glomerular diseases, polycystic kidney disease (PKD), and DKD (Abramowitz & Birnbaumer, 2009; Harris & Torres, 2009; Nilius & Owsianik, 2011; Tomilin et al., 2016; Woudenberg-Vrenken, Bindels, & Hoenderop, 2009). More recently the TRPC subfamily has become a target for research into possible therapeutic treatments for DKD.
The TRPC subfamily is composed of seven channels that are non-selectively permeable to calcium and sodium (Abramowitz & Birnbaumer, 2009; Vazquez, Wedel, Aziz, Trebak, & Putney, 2004; Woudenberg-Vrenken et al., 2009). TRPC 1–7 are all activated through phospholipase C (PLC) coupled receptors; however, they differ in mode of operation. Some are store-operated, whereas others operate via receptors (Dietrich, Mederos y Schnitzler, Kalwa, Storch, & Gudermann, 2005). Store-operated calcium entry (SOC) occurs when inositol 1,4,5-trisphosphate (IP3) or another signal causes the release of intracellular calcium stores from the endoplasmic reticulum (ER), reducing the calcium concentration in the ER. This Ca2+ decrease leads to the activation of SOC channels (Dietrich et al., 2005). Receptor-operated calcium entry (ROC) occurs when an agonist binds to and activates the PLC coupled receptor, which is located on the cell membrane separate from the actual channel (Dietrich et al., 2005). Activation of both SOC and ROC TRPC channels lead to increased intracellular calcium levels (Abramowitz & Birnbaumer, 2009). In the kidney, TRPC channels are present in renal tubules and the glomerulus where their malfunction, overexpression, or mutation is linked to certain renal diseases. Of these channels, only TRPC6 has been genetically linked to a renal disease (Reiser et al., 2005; Winn et al., 2005).
6.2. TRPC6 and DKD
Numerous gain-of-function mutations in Trpc6 gene have been identified to ultimately lead to the development of Focal Segmental Glomerulosclerosis (FSGS) (Heeringa et al., 2009; Reiser et al., 2005; Winn et al., 2005). A recent analysis of human disease-causing Trpc6 mutations also revealed a loss-of-function mutation in TRPC6 as an additional cause of hereditary FSGS (Riehle et al., 2016), which demonstrates that not only activation, but also inhibition of TRPC6 activity might lead to FSGS. With its location on the membrane of the podocyte, TRPC6 participates in unison with other integral players of the slit diaphragm such as podocin and nephrin (Dryer & Reiser, 2010; Ilatovskaya & Staruschenko, 2015; Reiser et al., 2005). TRPC6 normally remains dormant in the cell membrane until activated by a stimulus (Fig. 5) (Ilatovskaya & Staruschenko, 2015). Ang II is increased during the progression of DKD and has been found to activate TRPC6 channels (Anderson, Roshanravan, Khine, & Dryer, 2014; Ilatovskaya et al., 2018; Ilatovskaya, Levchenko, Lowing, et al., 2015; Ilatovskaya et al., 2014; Ilatovskaya, Palygin, Levchenko, Endres, & Staruschenko, 2017; Nijenhuis et al., 2011; Sonneveld et al., 2014). In addition, hyperglycemia together with Ang II leads to overexpression of the TRPC6 channel in the podocyte and the subsequent drastic increase in intracellular calcium flowing through TRPC6. Hyperglycemia alone is insufficient to cause this same response (Sonneveld et al., 2014). Ang II influences TRPC6 mRNA and protein levels by increasing the expression of the channel in the podocyte. The Ang II-mediated activation of the TRPC6 channel leads to engorgement of the podocyte with calcium, which causes podocyte cell death and breakdown of the GFB. This inevitably leads to albuminuria which is a hallmark of DKD (Fig 5) (Adebiyi, Soni, John, & Yang, 2014; Eckel et al., 2011; Evans, Lee, & Ragolia, 2009; Ilatovskaya, Levchenko, Lowing, et al., 2015; Ilatovskaya et al., 2014; Ilatovskaya et al., 2017; Nijenhuis et al., 2011; Qin Zou, 2015; Reiser et al., 2005; Sonneveld et al., 2014; Zhang, Ding, Fan, & Liu, 2009). Our recent studies using a TRPC6 knockout on the Dahl SS rat background (SSTrpc6−/−) indicate that TRPC6 channel inhibition may have at least partial renoprotective effects in the context of type 1 DN (Spires et al., 2018). Further studies revealed the contribution of Nox4-mediated oxidative stress in the regulation of TRPC6 in DKD (Ilatovskaya et al., 2018). In agreement with our studies, Kim et al. reported that the genetic inactivation of TRPC6 in Sprague-Dawley rats confers this protection in a model of severe nephrosis (Kim, Yazdizadeh Shotorbani, & Dryer, 2018). Interestingly, Wang et al. reported that TRPC6 KO Akita mice exhibit prominent mesangial expansion in the diabetic group, which suggests enhanced susceptibility of glomerular cell types to the adverse effects of the TRPC6 KO with regards to hyperglycemia (Staruschenko, 2019; L. Wang, Chang, Buckley, & Spurney, 2019).
Figure 5.
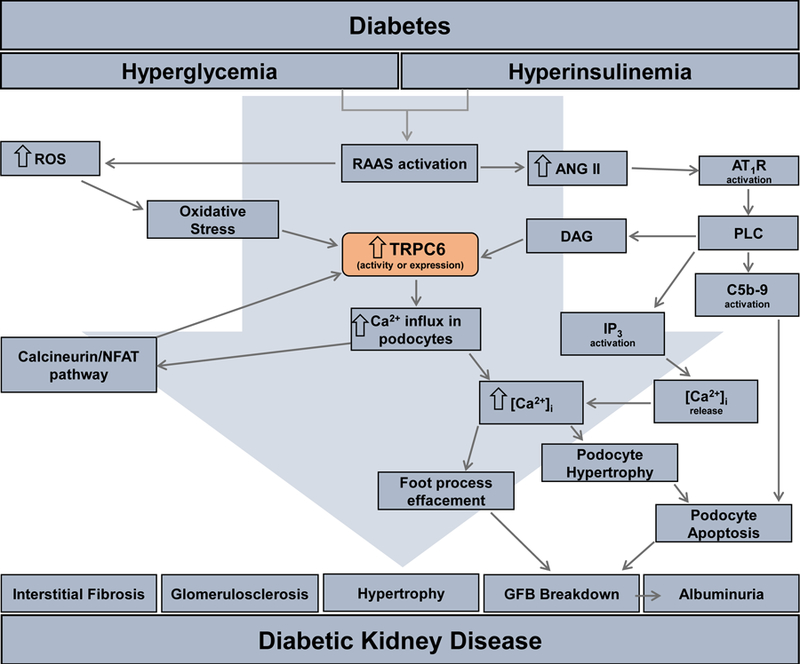
Schematic for TRPC6-induced glomerular renal injury in the progression of DKD. Activation of the renin-angiotensin-aldosterone system (RAAS) cause an increase in angiotensin II (ANG II) that acts through the angiotensin II receptor type 1 (AT1R). This receptor activates phospholipase C (PLC) on the podocyte cell membrane. PLC activates 3 additional targets/pathways in the podocyte: diacylglycerol (DAG) that cause calcium influx through the transient receptor potential canonical 6 (TRPC6) channel which in turn increases intracellular calcium [Ca2+]i; inositol triphosphate (IP3) activates calcium release from the intracellular stores; the membrane attack complex of complement C (C5b-9), a transmembrane channel involved in some immune responses, induces podocyte apoptosis. Increases in RAAS also leads to an increase in reactive oxygen species (ROS) that begets oxidative stress activating the calcineurin/NFAT pathway that increases the transcription of Trpc6. The culmination of the increase in [Ca2+]I from over-activation of TRPC6 leads to podocyte hypertrophy/apoptosis and foot process effacement ending in breakdown of the glomerular filtration barrier (GFB). The ultimate result of this pathway is the development of the DKD characteristic albuminuria.
6.4. Voltage-gated calcium channels
Although less thoroughly studied than TRPC channels, voltage-gated calcium channels have also been shown to play a role in DKD. In the clinic, calcium channel blockers are commonly prescribed in conjunction with RAAS inhibitors to achieve better control over blood pressure, which is required for hypertensive patients with renal complications. These drugs are promising candidates in the treatment of DKD as they have been shown to be effective antihypertensive medications that are safe and well-tolerated with few reported side effects. The antihypertensive effect of calcium channel blockers is mainly attributed to the blockade of L-type Ca2+ channels, but N- and T-type channels may play a role in renal protective action due to their effects on glomerular capillary pressure, renal fibrotic process, sympathetic nerve activity and aldosterone synthesis (Sugano, Hayashi, Hosoya, & Yokoo, 2013). Blockers of both the N- and L-type calcium channels have been tested in patients with diabetes to determine whether they impacted renal function. Multiple clinical studies have found that N- and L- type calcium channel blockers are renoprotective and improve proteinuria/microalbuminuria in diabetic patients. However, there is debate as to whether a specific L-type blocker, amlodipine, or a general N-/L-type blocker, cilnidipine, is more protective in the progression of DKD. Ando et al. compared the results of administering the antagonist of both the N- and L-type channels, cilnidipine, and the specific L-type calcium channel blocker, amlodipine, to patients with hypertension and T2DM with microalbuminuria over a 12-month duration and did not find a substantial difference in renal outcomes (Ando et al., 2013). In contrast, a different study in hypertensive diabetic patients found significant differences in patient outcomes between the two drugs. Masuda et al. found that cilnidipine treatment resulted in significantly lower insulin resistance, higher estimated GFR, lower urinary albumin, and lower urinary creatinine. These parameters indicate better renal function and less renal damage, suggesting that cilnidipine rather than amlodipine, better preserves kidney function and is a more appropriate candidate for slowing the progression of DKD (Masuda et al., 2011). Both calcium channel blockers are already safely being prescribed to diabetic patients with hypertension who are at the highest risk for developing DKD; however, more clinical research is needed to determine which drug is most renoprotective. Basic science research on these channels indicates that inhibiting the N-type channel is especially important for the protection against kidney injury and the preservation of function. Thus, it has been predicted that antagonizing the N-type channel would produce a more balanced dilation of the afferent and efferent arterioles, more effectively reducing glomerular pressure. The N-type voltage-gated calcium channel, Cav2.2, is expressed in podocytes in the glomerulus as well as in the DCT. A global deletion of this channel in the db/db diabetic mouse ameliorates many of the renal manifestations of diabetes that are known to contribute to DKD. This knockout mouse had a significant reduction in hyperfiltration, renal injury, blood pressure, and proteinuria. Similarly, applying pharmaceutical antagonists to Cav2.2 resulted in renoprotection. Blocking this channel in cultured podocytes caused a reduction in transforming growth factor beta (TGF-β) mediated nephrin loss. These studies suggest that treatment with specific Cav2.2 channel blockers may slow the progression of DKD by protecting podocytes and reducing glomerular injury (Ohno et al., 2016).
6.5. Magnesium homeostasis and TRPM6 and TRPM7 channels
Renal magnesium handling is not classically associated with DKD, as magnesium has long been considered to be independent of endocrine control, often termed an “orphan ion” (Yee, 2018). However, recent studies suggest that magnesium should not be overlooked in the context of DKD. Next to potassium, magnesium is the second most abundant intracellular cation and the kidney is responsible for maintaining its homeostasis. Like potassium, magnesium is associated with increased longevity and cardiovascular health, and is also an important anti-inflammatory molecule. Several studies have found an association between serum magnesium and diabetes. Earlier studies have suggested a link between hypomagnesemia and hyperglycemia, as well as an association between hypomagnesemia and the complications of diabetes (White & Campbell, 1993). A meta-analysis including over 500,000 patients found an inverse correlation between serum magnesium levels and risk for cardiovascular disease (Qu et al., 2013). Furthermore, this protection has been shown to extend to patients with CKD, who are at heightened risk for cardiovascular events (Kanbay, Goldsmith, Uyar, Turgut, & Covic, 2010), and hypertension where magnesium supplementation has been shown to be effective in reducing blood pressure. Additionally, patients with diabetes have lower serum magnesium, on average, than healthy controls. Diabetic patients with microalbuminuria, indicative of progression towards DKD, have significantly lower magnesium than those without (Corsonello et al., 2000). Poor glycemic control in these patients likely causes enhanced urinary magnesium loss, which is associated with a more rapid progression from diabetes to DKD.
There is considerable evidence suggesting that Mg2+ deficiency is a significant risk factor for the development of insulin resistance and T2DM (Barbagallo & Dominguez, 2007). A recent population-based cohort study by Kieboom et al. revealed that low serum Mg2+ levels are associated with an increased risk of prediabetes. Furthermore, common variants in magnesium-regulating genes, including the magnesium transporters, SLC41A2 (Solute Carrier Family 41 Member 2) and TRPM6, modify diabetes risk through altering serum Mg2+ levels (Kieboom et al., 2017). Transient receptor potential melastatin 6 and 7 (TRPM6 and TRPM7) channels play a central role in magnesium homeostasis, which is critical for maintaining glucose and insulin metabolism. Several loss-of-function mutations in Trpm6 have been identified among patients with an autosomal-recessive form of hypomagnesemia with secondary hypocalcemia (Lainez et al., 2014; Schlingmann et al., 2002; Walder et al., 2002). Various factors and hormones, including epidermal growth factor, pH, and insulin, contribute to the expression and function of this important channel (de Baaij, Hoenderop, & Bindels, 2015). There is also genetic evidence establishing the potential contribution of TRPM6 channels to the development of diabetes. For instance, Song et al., reported that two common non-synonymous SNPs in Trpm6 might confer susceptibility to T2DM in women with low magnesium intake (Y. Song et al., 2009). Another study revealed that SNPs in Trpm6 have been associated with gestational diabetes. Loss of insulin-induced activation of TRPM6 channels results in impaired glucose tolerance during pregnancy (Nair et al., 2012). Insulin binding causes the activation of two cascades resulting in more TRPM6 channels as well as NCC transporters to be inserted in the apical membrane. Hyperinsulinemia and insulin resistance may cause an uncoupling of these cascades, in which NCC increases concomitantly with insulin activity while TRPM6 becomes unresponsive to insulin activity, both effectively contributing to hypertension. A recent genome-wide meta-analysis of Mg2+ homeostasis and metabolic phenotypes identified two loci associated with urinary magnesium near Trpm6 (Corre et al., 2018). TRPM6 activity may also be inhibited by oxidative stress, further reducing Mg2+ uptake in diabetic patients. Diabetic rat models have also shown altered expression levels of TRPM6. Hyperfiltration and increased urinary flow rates in DKD patients are inversely correlated with Mg2+ reabsorption in the TAL and DCT and may also affect osmotic diuresis and passive reabsorption in the PT (Gommers, Hoenderop, Bindels, & de Baaij, 2016). In addition to TRPM6 and TRPM7 channels, there are other magnesium carriers in the kidney which might contribute to Mg2+ handling under normal conditions as well as during DKD. Therefore, current studies provide evidence supporting the critical contribution of magnesium channels in diabetes and the potential beneficial role of Mg2+ supplementation in diabetic patients. However, additional clinical and fundamental research is needed to identify specific mechanisms contributing to magnesium deficiency in diabetes. Despite gaps in research on the complicated role of renal magnesium handling in DKD, it is certain that magnesium imbalance contributes to or is affected by multiple pathways integral to the progression of DKD including blood pressure control, oxidative stress, inflammation and hyperfiltration.
7. CONCLUSIONS AND FUTURE PERSPECTIVES
DKD as a result of diabetes mellitus is a medical pandemic affecting many patients today. Due to the critical role of renal ion transport in kidney damage caused by DKD, these transporters and their pathways represent promising targets for therapeutics. Understanding the roles of the numerous transporters in the disease progression of DKD requires knowledge of both their individual functions as well as their collective interactions, cascades, and mechanisms of regulation. This review provides only a brief description of the involvement of some of the most influential transporters in DKD. More studies are certainly necessary to further validate and evaluate the roles of these and other ion transporters in the pathogenesis of DKD.
ACKNOWLEDGEMENTS
Work in the authors’ laboratories is supported by NHLBI grants R35 HL135749, P01 HL116264, and T32 HL007852, American Heart Association grants 16EIA26720006 and 18PRE34030127, and Department of Veteran Affairs I01 BX004024. We would also like to thank Nnamdi Uche for his literary proofing assistance.
ABBREVIATIONS
- AGEs
Advanced glycation end-products
- BK
Large-conductance Ca2+-activated K+ channel
- CD
collecting duct
- CKD
Chronic kidney disease
- DCT
Distal convoluted tubule
- DKD
Diabetic kidney disease
- DN
Diabetic nephropathy
- DPP-4
Dipeptidyl peptidase-4
- ENaC
Epithelial Na+ channel
- ESRD
End stage renal disease
- ET-1
Endothelin 1
- FSGS
Focal Segmental Glomerulosclerosis
- GFB
Glomerular filtration barrier
- GFR
Glomerular filtration rate
- GLP-1
Glucagon-like peptide-1
- GLUT
Glucose transporter
- KATP
ATP-sensitive K+ channels
- mTOR
Mechanistic target of rapamycin
- Na+/K+
ATPase Sodium potassium ATPase
- NCC
Na+−2Cl− cotransporter
- NHE
Na+/H+ exchanger
- NKCC
Na+-K+−2Cl− cotransporter
- NO
nitric oxide
- PT
Proximal tubule
- RAAS
Renin-angiotensin-aldosterone system
- ROS
Reactive oxygen species
- SGK1
Serum and glucocorticoid-regulated kinase
- SGLT1
Sodium-glucose transporter 1
- SGLT2
Sodium-glucose transporter 2
- SLC41A2
Solute carrier family 41 member 2
- STZ
streptozotocin
- T1DM
Type 1 diabetes mellitus
- T2DM
Type 2 diabetes mellitus
- TAL
Thick ascending limb
- TGF
Tubuloglomerular feedback
- TGF-β
Transforming growth factor beta
- TRP
Transient receptor potential channel
- TRPC
Transient receptor potential canonical channel
- TRPM
Transient receptor potential melastatin channel
REFERENCES
- Abdul-Ghani MA, Norton L, & DeFronzo RA (2015). Renal sodium-glucose cotransporter inhibition in the management of type 2 diabetes mellitus. Am J Physiol Renal Physiol, 309(11), F889–900. 10.1152/ajprenal.00267.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramowitz J, & Birnbaumer L (2009). Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J, 23(2), 297–328. 10.1096/fj.08-119495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adebiyi A, Soni H, John TA, & Yang F (2014). Lipid rafts are required for signal transduction by angiotensin II receptor type 1 in neonatal glomerular mesangial cells. Exp Cell Res, 324(1), 92–104. 10.1016/j.yexcr.2014.03.011 [DOI] [PubMed] [Google Scholar]
- Anders H-J, Huber TB, Isermann B, & Schiffer M (2018). CKD in diabetes: diabetic kidney disease versus nondiabetic kidney disease. Nature Reviews Nephrology, 14(6), 361–377. 10.1038/s41581-018-0001-y [DOI] [PubMed] [Google Scholar]
- Andersen H, Friis UG, Hansen PB, Svenningsen P, Henriksen JE, & Jensen BL (2015). Diabetic nephropathy is associated with increased urine excretion of proteases plasmin, prostasin and urokinase and activation of amiloride-sensitive current in collecting duct cells. Nephrol Dial Transplant, 30(5), 781–789. 10.1093/ndt/gfu402 [DOI] [PubMed] [Google Scholar]
- Anderson M, Roshanravan H, Khine J, & Dryer SE (2014). Angiotensin II activation of TRPC6 channels in rat podocytes requires generation of reactive oxygen species. J Cell Physiol, 229(4), 434–442. 10.1002/jcp.24461 [DOI] [PubMed] [Google Scholar]
- Ando K, Ueshima K, Tanaka S, Kosugi S, Sato T, Matsuoka H, … Fujita T (2013). Comparison of the antialbuminuric effects of L-/N-type and L-type calcium channel blockers in hypertensive patients with diabetes and microalbuminuria: the study of assessment for kidney function by urinary microalbumin in randomized (SAKURA) trial. Int J Med Sci, 10(9), 1209–1216. 10.7150/ijms.5508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki S, Haneda M, Koya D, Kondo K, Tanaka S, Arima H, … Maegawa H (2015). Urinary Potassium Excretion and Renal and Cardiovascular Complications in Patients with Type 2 Diabetes and Normal Renal Function. Clin J Am Soc Nephrol, 10(12), 2152–2158. 10.2215/cjn.00980115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora MK, & Singh UK (2013). Molecular mechanisms in the pathogenesis of diabetic nephropathy: an update. Vascul Pharmacol, 58(4), 259–271. 10.1016/j.vph.2013.01.001 [DOI] [PubMed] [Google Scholar]
- Barbagallo M, & Dominguez LJ (2007). Magnesium metabolism in type 2 diabetes mellitus, metabolic syndrome and insulin resistance. Arch Biochem Biophys, 458(1), 40–47. 10.1016/j.abb.2006.05.007 [DOI] [PubMed] [Google Scholar]
- Bautista R, Manning R, Martinez F, Avila-Casado Mdel C, Soto V, Medina A, & Escalante B (2004). Angiotensin II-dependent increased expression of Na+-glucose cotransporter in hypertension. Am J Physiol Renal Physiol, 286(1), F127–133. 10.1152/ajprenal.00113.2003 [DOI] [PubMed] [Google Scholar]
- Bickel CA, Verbalis JG, Knepper MA, & Ecelbarger CA (2001). Increased renal Na-K-ATPase, NCC, and beta-ENaC abundance in obese Zucker rats. Am J Physiol Renal Physiol, 281(4), F639–648. 10.1152/ajprenal.2001.281.4.F639 [DOI] [PubMed] [Google Scholar]
- Bobulescu IA, Di Sole F, & Moe OW (2005). Na+/H+ exchangers: physiology and link to hypertension and organ ischemia. Curr Opin Nephrol Hypertens, 14(5), 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobulescu IA, & Moe OW (2009). Luminal Na(+)/H (+) exchange in the proximal tubule. Pflugers Arch, 458(1), 5–21. 10.1007/s00424-008-0595-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner BM (1983). Hemodynamically mediated glomerular injury and the progressive nature of kidney disease. Kidney Int, 23(4), 647–655. [DOI] [PubMed] [Google Scholar]
- Brett CL, Donowitz M, & Rao R (2005). Evolutionary origins of eukaryotic sodium/proton exchangers. Am J Physiol Cell Physiol, 288(2), C223–239. 10.1152/ajpcell.00360.2004 [DOI] [PubMed] [Google Scholar]
- Brindeiro CMT, Fallet RW, Lane PH, & Carmines PK (2008). Potassium channel contributions to afferent arteriolar tone in normal and diabetic rat kidney. American Journal of Physiology-Renal Physiology, 295(1), F171–F178. 10.1152/ajprenal.00563.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosius FC 3rd, Alpers CE, Bottinger EP, Breyer MD, Coffman TM, Gurley SB, … Takahashi T (2009). Mouse models of diabetic nephropathy. J Am Soc Nephrol, 20(12), 2503–2512. 10.1681/asn.2009070721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosius FC 3rd, Briggs JP, Marcus RG, Barac-Nieto M, & Charron MJ (1992). Insulin-responsive glucose transporter expression in renal microvessels and glomeruli. Kidney Int, 42(5), 1086–1092. [DOI] [PubMed] [Google Scholar]
- Brosius FC, & Heilig CW (2005). Glucose transporters in diabetic nephropathy. Pediatr Nephrol, 20(4), 447–451. 10.1007/s00467-004-1748-x [DOI] [PubMed] [Google Scholar]
- Buhl KB, Oxlund CS, Friis UG, Svenningsen P, Bistrup C, Jacobsen IA, & Jensen BL (2014). Plasmin in urine from patients with type 2 diabetes and treatment-resistant hypertension activates ENaC in vitro. J Hypertens, 32(8), 1672–1677; 10.1097/HJH.0000000000000216 [DOI] [PubMed] [Google Scholar]
- Carmines PK, & Fujiwara K (2002). Altered electromechanical coupling in the renal microvasculature during the early stage of diabetes mellitus. Clin Exp Pharmacol Physiol, 29(1–2), 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambrey R, St John PL, Eladari D, Quentin F, Warnock DG, Abrahamson DR, … Paillard M (2001). Localization and functional characterization of Na+/H+ exchanger isoform NHE4 in rat thick ascending limbs. Am J Physiol Renal Physiol, 281(4), F707–717. 10.1152/ajprenal.2001.281.4.F707 [DOI] [PubMed] [Google Scholar]
- Chambrey R, Warnock DG, Podevin RA, Bruneval P, Mandet C, Belair MF, … Paillard M (1998). Immunolocalization of the Na+/H+ exchanger isoform NHE2 in rat kidney. Am J Physiol, 275(3 Pt 2), F379–386. [DOI] [PubMed] [Google Scholar]
- Chang AB, Lin R, Keith Studley W, Tran CV, & Saier MH Jr. (2004). Phylogeny as a guide to structure and function of membrane transport proteins. Mol Membr Biol, 21(3), 171–181. 10.1080/09687680410001720830 [DOI] [PubMed] [Google Scholar]
- Chang CT, Wu MS, Tian YC, Chen KH, Yu CC, Liao CH, … Yang CW (2007). Enhancement of epithelial sodium channel expression in renal cortical collecting ducts cells by advanced glycation end products. Nephrol Dial Transplant, 22(3), 722–731. 10.1093/ndt/gfl668 [DOI] [PubMed] [Google Scholar]
- Chavez-Canales M, Arroyo JP, Ko B, Vazquez N, Bautista R, Castaneda-Bueno M, … Gamba G (2013). Insulin increases the functional activity of the renal NaCl cotransporter. J Hypertens, 31(2), 303–311. 10.1097/HJH.0b013e32835bbb83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SL, Heilig KO, Brosius FC, & Heilig CW (2003). Diabetes increases glomerular GLUT1, and antisense-GLUT1 protects against diabetic glomerulosclerosis.
- Cherney DZ, Perkins BA, Soleymanlou N, Maione M, Lai V, Lee A, … von Eynatten M (2014). Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation, 129(5), 587–597. 10.1161/circulationaha.113.005081 [DOI] [PubMed] [Google Scholar]
- Chin E, Zamah AM, Landau D, Gronbcek H, Flyvbjerg A, LeRoith D, & Bondy CA (1997). Changes in facilitative glucose transporter messenger ribonucleic acid levels in the diabetic rat kidney. Endocrinology, 138(3), 1267–1275. 10.1210/endo.138.3.5015 [DOI] [PubMed] [Google Scholar]
- Cipriani P, Kim SL, Klein JD, Sim JH, von Bergen TN, & Blount MA (2012). The role of nitric oxide in the dysregulation of the urine concentration mechanism in diabetes mellitus. Front Physiol, 3, 176 10.3389/fphys.2012.00176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corre T, Arjona FJ, Hayward C, Youhanna S, de Baaij JHF, Belge H, … Devuyst O (2018). Genome-Wide Meta-Analysis Unravels Interactions between Magnesium Homeostasis and Metabolic Phenotypes. Journal of the American Society of Nephrology, 29(1), 335–348. 10.1681/asn.2017030267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsonello A, Ientile R, Buemi M, Cucinotta D, Mauro VN, Macaione S, & Corica F (2000). Serum ionized magnesium levels in type 2 diabetic patients with microalbuminuria or clinical proteinuria. Am J Nephrol, 20(3), 187–192. 10.1159/000013582 [DOI] [PubMed] [Google Scholar]
- Coward RJ, Welsh GI, Yang J, Tasman C, Lennon R, Koziell A, … Saleem MA (2005). The human glomerular podocyte is a novel target for insulin action. Diabetes, 54(11), 3095–3102. [DOI] [PubMed] [Google Scholar]
- D’Agord Schaan B, Lacchini S, Bertoluci MC, Irigoyen MC, Machado UF, & Schmid H (2001). Increased renal GLUT1 abundance and urinary TGF-beta 1 in streptozotocin-induced diabetic rats: implications for the development of nephropathy complicating diabetes. Horm Metab Res, 33(11), 664–669. 10.1055/s-2001-18683 [DOI] [PubMed] [Google Scholar]
- de Baaij JHF, Hoenderop JGJ, & Bindels RJM (2015). Magnesium in Man: Implications for Health and Disease (Vol. 95). [DOI] [PubMed] [Google Scholar]
- Defronzo RA (2009). Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes, 58(4), 773–795. 10.2337/db09-9028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo RA, Hompesch M, Kasichayanula S, Liu X, Hong Y, Pfister M, … Griffen SC (2013). Characterization of renal glucose reabsorption in response to dapagliflozin in healthy subjects and subjects with type 2 diabetes. Diabetes Care, 36(10), 3169–3176. 10.2337/dc13-0387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo RA, Lewin A, Patel S, Liu D, Kaste R, Woerle HJ, & Broedl UC (2015). Combination of empagliflozin and linagliptin as second-line therapy in subjects with type 2 diabetes inadequately controlled on metformin. Diabetes Care, 38(3), 384–393. 10.2337/dc14-2364 [DOI] [PubMed] [Google Scholar]
- Dekkers CCJ, Gansevoort RT, & Heerspink HJL (2018). New Diabetes Therapies and Diabetic Kidney Disease Progression: the Role of SGLT-2 Inhibitors. Curr Diab Rep, 18(5), 27 10.1007/s11892-018-0992-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng D, & Yan N (2016). GLUT, SGLT, and SWEET: Structural and mechanistic investigations of the glucose transporters. Protein Sci, 25(3), 546–558. 10.1002/pro.2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich A, Mederos y Schnitzler M, Kalwa H, Storch U, & Gudermann T (2005). Functional characterization and physiological relevance of the TRPC3/6/7 subfamily of cation channels. Naunyn Schmiedebergs Arch Pharmacol, 371(4), 257–265. 10.1007/s00210-005-1052-8 [DOI] [PubMed] [Google Scholar]
- Dryer SE, & Reiser J (2010). TRPC6 channels and their binding partners in podocytes: role in glomerular filtration and pathophysiology. American Journal of Physiology Renal Physiology, 299(4), F689–F701. 10.1152/ajprenal.00298.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckel J, Lavin PJ, Finch EA, Mukerji N, Burch J, Gbadegesin R, … Winn MP (2011). TRPC6 enhances angiotensin II-induced albuminuria. J Am Soc Nephrol, 22(3), 526–535. 10.1681/asn.2010050522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekmekcioglu C, Elmadfa I, Meyer AL, & Moeslinger T (2016). The role of dietary potassium in hypertension and diabetes. J Physiol Biochem, 72(1), 93–106. 10.1007/s13105-015-0449-1 [DOI] [PubMed] [Google Scholar]
- Eriguchi M, Bernstein EA, Veiras LC, Khan Z, Cao DY, Fuchs S, … Giani JF (2018). The Absence of the ACE N-Domain Decreases Renal Inflammation and Facilitates Sodium Excretion during Diabetic Kidney Disease. J Am Soc Nephrol, 29(10), 2546–2561. 10.1681/ASN.2018030323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JF, Lee JH, & Ragolia L (2009). Ang-II-induced Ca(2+) influx is mediated by the 1/4/5 subgroup of the transient receptor potential proteins in cultured aortic smooth muscle cells from diabetic Goto-Kakizaki rats. Mol Cell Endocrinol, 302(1), 49–57. 10.1016/j.mce.2008.12.004 [DOI] [PubMed] [Google Scholar]
- Farber SJ, Berger EY, & Earle DP (1951). Effect of diabetes and insulin of the maximum capacity of the renal tubules to reabsorb glucose. J Clin Invest, 30(2), 125–129. 10.1172/jci102424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrannini E, Muscelli E, Frascerra S, Baldi S, Mari A, Heise T, … Woerle HJ (2014). Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest, 124(2), 499–508. 10.1172/jci72227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrannini E, & Solini A (2012). SGLT2 inhibition in diabetes mellitus: rationale and clinical prospects. Nat Rev Endocrinol, 8(8), 495–502. 10.1038/nrendo.2011.243 [DOI] [PubMed] [Google Scholar]
- Ferrannini G, & Ryden L (2018). Sodium-glucose transporter 2 inhibition and cardiovascular events in patients with diabetes: information from clinical trials and observational real-world data. Clin Sci (Lond), 132(18), 2003–2012. 10.1042/CS20171374 [DOI] [PubMed] [Google Scholar]
- Ficociello LH, Rosolowsky ET, Niewczas MA, Maselli NJ, Weinberg JM, Aschengrau A, … Krolewski AS (2010). High-normal serum uric acid increases risk of early progressive renal function loss in type 1 diabetes: results of a 6-year follow-up. Diabetes Care, 33(6), 1337–1343. 10.2337/dc10-0227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo LA, Wright EM, & Vallon V (2015). Probing SGLT2 as a therapeutic target for diabetes: basic physiology and consequences. Diab Vasc Dis Res, 12(2), 78–89. 10.1177/1479164114561992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg SK, Henry RR, Banks P, Buse JB, Davies MJ, Fulcher GR, … Strumph P (2017). Effects of Sotagliflozin Added to Insulin in Patients with Type 1 Diabetes. N Engl J Med, 377(24), 2337–2348. 10.1056/NEJMoa1708337 [DOI] [PubMed] [Google Scholar]
- Gembardt F, Bartaun C, Jarzebska N, Mayoux E, Todorov VT, Hohenstein B, & Hugo C (2014). The SGLT2 inhibitor empagliflozin ameliorates early features of diabetic nephropathy in BTBR ob/ob type 2 diabetic mice with and without hypertension. Am J Physiol Renal Physiol, 307(3), F317–325. 10.1152/ajprenal.00145.2014 [DOI] [PubMed] [Google Scholar]
- Gheith O, Farouk N, Nampoory N, Halim MA, & Al-Otaibi T (2016). Diabetic kidney disease: world wide difference of prevalence and risk factors. J Nephropharmacol, 5(1), 49–56. [PMC free article] [PubMed] [Google Scholar]
- Ghezzi C, Loo DDF, & Wright EM (2018). Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia, 61(10), 2087–2097. 10.1007/s00125-018-4656-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gommers LM, Hoenderop JG, Bindels RJ, & de Baaij JH (2016). Hypomagnesemia in Type 2 Diabetes: A Vicious Circle? Diabetes, 65(1), 3–13. 10.2337/db15-1028 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Rodriguez E, Gaeggeler HP, & Rossier BC (2007). IGF-1 vs insulin: respective roles in modulating sodium transport via the PI-3 kinase/Sgk1 pathway in a cortical collecting duct cell line. Kidney International, 71(2), 116–125. [DOI] [PubMed] [Google Scholar]
- Gorboulev V, Schurmann A, Vallon V, Kipp H, Jaschke A, Klessen D, … Koepsell H (2012). Na(+)-D-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes, 61(1), 187–196. 10.2337/db11-1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal S, Mentone S, & Aronson PS (2005). Immunolocalization of NHE8 in rat kidney. Am J Physiol Renal Physiol, 288(3), F530–538. 10.1152/ajprenal.00229.2004 [DOI] [PubMed] [Google Scholar]
- Grzeszczak W, Moczulski DK, Zychma M, Zukowska-Szczechowska E, Trautsolt W, & Szydlowska I (2001). Role of GLUT1 gene in susceptibility to diabetic nephropathy in type 2 diabetes. Kidney Int, 59(2), 631–636. 10.1046/j.1523-1755.2001.059002631.x [DOI] [PubMed] [Google Scholar]
- Guzman J, Jauregui AN, Merscher-Gomez S, Maiguel D, Muresan C, Mitrofanova A, … Fornoni A (2014). Podocyte-specific GLUT4-deficient mice have fewer and larger podocytes and are protected from diabetic nephropathy. Diabetes, 63(2), 701–714. 10.2337/db13-0752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagg S, Thorn LM, Putaala J, Liebkind R, Harjutsalo V, Forsblom CM, … FinnDiane Study, G. (2013). Incidence of stroke according to presence of diabetic nephropathy and severe diabetic retinopathy in patients with type 1 diabetes. Diabetes Care, 36(12), 4140–4146. 10.2337/dc13-0669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanukoglu I, & Hanukoglu A (2016). Epithelial sodium channel (ENaC) family: Phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene, 579(2), 95–132. 10.1016/j.gene.2015.12.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada N, & Inagaki N (2012). Role of sodium-glucose transporters in glucose uptake of the intestine and kidney. J Diabetes Investig, 3(4), 352–353. 10.1111/j.2040-1124.2012.00227.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haring HU, Merker L, Seewaldt-Becker E, Weimer M, Meinicke T, Broedl UC, & Woerle HJ (2014). Empagliflozin as add-on to metformin in patients with type 2 diabetes: a 24-week, randomized, double-blind, placebo-controlled trial. Diabetes Care, 37(6), 1650–1659. 10.2337/dc13-2105 [DOI] [PubMed] [Google Scholar]
- Harris PC, & Torres VE (2009). Polycystic kidney disease. Annu Rev Med, 60, 321–337. 10.1146/annurev.med.60.101707.125712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H, Nomura N, Shoda W, Isobe K, Kikuchi H, Yamamoto K, … Sohara E (2018). Metformin increases urinary sodium excretion by reducing phosphorylation of the sodium-chloride cotransporter. Metabolism, 85, 23–31. 10.1016/j.metabol.2018.02.009 [DOI] [PubMed] [Google Scholar]
- Hediger MA, & Rhoads DB (1994). Molecular physiology of sodium-glucose cotransporters. Physiol Rev, 74(4), 993–1026. 10.1152/physrev.1994.74.4.993 [DOI] [PubMed] [Google Scholar]
- Heeringa SF, Moller CC, Du J, Yue L, Hinkes B, Chernin G, … Hildebrandt F (2009). A novel TRPC6 mutation that causes childhood FSGS. PLOS ONE, 4(11), e7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig C, Zaloga C, Lee M, Zhao X, Riser B, Brosius F, & Cortes P (1995). Immunogold localization of high-affinity glucose transporter isoforms in normal rat kidney. Lab Invest, 73(5), 674–684. [PubMed] [Google Scholar]
- Heilig CW, Brosius FC 3rd, & Cunningham C (2006). Role for GLUT1 in diabetic glomerulosclerosis. Expert Rev Mol Med, 8(4), 1–18. 10.1017/s1462399406010490 [DOI] [PubMed] [Google Scholar]
- Heilig CW, Brosius FC 3rd, & Henry DN (1997). Glucose transporters of the glomerulus and the implications for diabetic nephropathy. Kidney Int Suppl, 60, S91–99. [PubMed] [Google Scholar]
- Hershon KS (2016). Options for empagliflozin in combination therapy in type 2 diabetes mellitus. Int J Gen Med, 9, 155–172. 10.2147/ijgm.s100288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hills CE, Bland R, Bennett J, Ronco PM, & Squires PE (2006). High glucose up-regulates ENaC and SGK1 expression in HCD-cells. Cellular Physiology and Biochemistry, 18(6), 337–346. [DOI] [PubMed] [Google Scholar]
- Hodgkinson AD, Millward BA, & Demaine AG (2001). Polymorphisms of the glucose transporter (GLUT1) gene are associated with diabetic nephropathy. Kidney Int, 59(3), 985–989. 10.1046/j.1523-1755.2001.059003985.x [DOI] [PubMed] [Google Scholar]
- Hsu CC, Kao WL, Steffes MW, Gambir T, Brancati FL, Heilig CW, … Coresh J (2011). Genetic variation of glucose transporter-1 (GLUT1) and albuminuria in 10,278 European Americans and African Americans: a case-control study in the Atherosclerosis Risk in Communities (ARIC) study. BMC Med Genet, 12, 16 10.1186/1471-2350-12-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Lin MZ, Cheng D, Braet F, Pollock CA, & Chen XM (2016). KCa3.1 mediates dysfunction of tubular autophagy in diabetic kidneys via PI3k/Akt/mTOR signaling pathways. Sci Rep, 6, 23884 10.1038/srep23884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Pollock CA, & Chen XM (2014a). High glucose induces CCL20 in proximal tubular cells via activation of the KCa3.1 channel. PLoS One, 9(4), e95173 10.1371/journal.pone.0095173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Pollock CA, & Chen XM (2014b). Role of the potassium channel KCa3.1 in diabetic nephropathy. Clin Sci (Lond), 127(7), 423–433. 10.1042/cs20140075 [DOI] [PubMed] [Google Scholar]
- Huang C, Shen S, Ma Q, Chen J, Gill A, Pollock CA, & Chen XM (2013). Blockade of KCa3.1 ameliorates renal fibrosis through the TGF-beta1/Smad pathway in diabetic mice. Diabetes, 62(8), 2923–2934. 10.2337/db13-0135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel CS, Lu C, Loo DD, Hirayama BA, Voss AA, & Wright EM (2011). Glucose transport by human renal Na+/D-glucose cotransporters SGLT1 and SGLT2. Am J Physiol Cell Physiol, 300(1), C14–21. 10.1152/ajpcell.00388.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenaga H, Bast JP, Fallet RW, & Carmines PK (2000). Exaggerated impact of ATP-sensitive K(+) channels on afferent arteriolar diameter in diabetes mellitus. J Am Soc Nephrol, 11(7), 1199–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilatovskaya DV, Blass G, Palygin O, Levchenko V, Pavlov TS, Grzybowski MN, … Staruschenko A (2018). A NOX4/TRPC6 Pathway in Podocyte Calcium Regulation and Renal Damage in Diabetic Kidney Disease. J Am Soc Nephrol, 29(7), 1917–1927. 10.1681/ASN.2018030280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilatovskaya DV, Levchenko V, Brands MW, Pavlov TS, & Staruschenko A (2015). Cross-talk between insulin and IGF-1 receptors in the cortical collecting duct principal cells: implication for ENaC-mediated Na+ reabsorption. Am J Physiol Renal Physiol, 308(7), F713–719. 10.1152/ajprenal.00081.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilatovskaya DV, Levchenko V, Lowing A, Shuyskiy LS, Palygin O, & Staruschenko A (2015). Podocyte injury in diabetic nephropathy: implications of angiotensin II-dependent activation of TRPC channels. Sci Rep, 5, 17637 10.1038/srep17637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilatovskaya DV, Palygin O, Chubinskiy-Nadezhdin V, Negulyaev YA, Ma R, Birnbaumer L, & Staruschenko A (2014). Angiotensin II has acute effects on TRPC6 channels in podocytes of freshly isolated glomeruli. Kidney Int, 86(3), 506–514. 10.1038/ki.2014.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilatovskaya DV, Palygin O, Levchenko V, Endres BT, & Staruschenko A (2017). The Role of Angiotensin II in Glomerular Volume Dynamics and Podocyte Calcium Handling. Sci Rep, 7(1), 299 10.1038/s41598-017-00406-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilatovskaya DV, & Staruschenko A (2015). TRPC6 channel as an emerging determinant of the podocyte injury susceptibility in kidney diseases. Am J Physiol Renal Physiol, 309(5), F393–397. 10.1152/ajprenal.00186.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irsik DL, Blazer-Yost BL, Staruschenko A, & Brands MW (2017). The normal increase in insulin after a meal may be required to prevent postprandial renal sodium and volume losses. Am J Physiol Regul Integr Comp Physiol, 312(6), R965–R972. 10.1152/ajpregu.00354.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irsik DL, & Brands MW (2018). Physiologic Hyperinsulinemia Caused by Acute Hyperglycemia Minimizes Renal Sodium Loss by Direct Action on the Kidney. Am J Physiol Regul Integr Comp Physiol. 10.1152/ajpregu.00016.2018 [DOI] [PMC free article] [PubMed]
- Jabbour SA, Hardy E, Sugg J, & Parikh S (2014). Dapagliflozin is effective as add-on therapy to sitagliptin with or without metformin: a 24-week, multicenter, randomized, double-blind, placebo-controlled study. Diabetes Care, 37(3), 740–750. 10.2337/dc13-0467 [DOI] [PubMed] [Google Scholar]
- Jefferson JA, Shankland SJ, & Pichler RH (2008). Proteinuria in diabetic kidney disease: a mechanistic viewpoint. Kidney Int, 74(1), 22–36. 10.1038/ki.2008.128 [DOI] [PubMed] [Google Scholar]
- Kamran M, Peterson RG, & Dominguez JH (1997). Overexpression of GLUT2 gene in renal proximal tubules of diabetic Zucker rats. J Am Soc Nephrol, 8(6), 943–948. [DOI] [PubMed] [Google Scholar]
- Kanbay M, Goldsmith D, Uyar ME, Turgut F, & Covic A (2010). Magnesium in chronic kidney disease: challenges and opportunities. Blood Purif, 29(3), 280–292. 10.1159/000276665 [DOI] [PubMed] [Google Scholar]
- Khan S, Wu KL, Sedor JR, Abu Jawdeh BG, & Schelling JR (2006). The NHE1 Na+/H+ exchanger regulates cell survival by activating and targeting ezrin to specific plasma membrane domains. Cell Mol Biol (Noisy-le-grand), 52(8), 115–121. [PubMed] [Google Scholar]
- Kieboom BCT, Ligthart S, Dehghan A, Kurstjens S, de Baaij JHF, Franco OH, … Hoorn EJ (2017). Serum magnesium and the risk of prediabetes: a population-based cohort study. Diabetologia, 60(5), 843–853. 10.1007/s00125-017-4224-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, & Dryer SE (2011). Effects of insulin and high glucose on mobilization of slo1 BKCa channels in podocytes. J Cell Physiol, 226(9), 2307–2315. 10.1002/jcp.22567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Yazdizadeh Shotorbani P, & Dryer SE (2018). Trpc6 inactivation confers protection in a model of severe nephrosis in rats. J Mol Med (Berl), 96(7), 631–644. 10.1007/s00109-018-1648-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleyman TR, Kashlan OB, & Hughey RP (2018). Epithelial Na(+) Channel Regulation by Extracellular and Intracellular Factors. Annu Rev Physiol, 80, 263–281. 10.1146/annurev-physiol-021317-121143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepsell H (2017). The Na(+)-D-glucose cotransporters SGLT1 and SGLT2 are targets for the treatment of diabetes and cancer. Pharmacol Ther, 170, 148–165. 10.1016/j.pharmthera.2016.10.017 [DOI] [PubMed] [Google Scholar]
- Kohan DE, Fioretto P, Tang W, & List JF (2014). Long-term study of patients with type 2 diabetes and moderate renal impairment shows that dapagliflozin reduces weight and blood pressure but does not improve glycemic control. Kidney Int, 85(4), 962–971. 10.1038/ki.2013.356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan DE, Rossi NF, Inscho EW, & Pollock DM (2011). Regulation of blood pressure and salt homeostasis by endothelin. Physiological Reviews, 91(1), 1–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima N, Williams JM, Slaughter TN, Kato S, Takahashi T, Miyata N, & Roman RJ (2015). Renoprotective effects of combined SGLT2 and ACE inhibitor therapy in diabetic Dahl S rats. Physiol Rep, 3(7). 10.14814/phy2.12436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima N, Williams JM, Takahashi T, Miyata N, & Roman RJ (2013). Effects of a new SGLT2 inhibitor, luseogliflozin, on diabetic nephropathy in T2DN rats. J Pharmacol Exp Ther, 345(3), 464–472. 10.1124/jpet.113.203869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs CS, Seshiah V, Swallow R, Jones R, Rattunde H, Woerle HJ, & Broedl UC (2014). Empagliflozin improves glycaemic and weight control as add-on therapy to pioglitazone or pioglitazone plus metformin in patients with type 2 diabetes: a 24-week, randomized, placebo-controlled trial. Diabetes Obes Metab, 16(2), 147–158. 10.1111/dom.12188 [DOI] [PubMed] [Google Scholar]
- Lainez S, Schlingmann KP, van der Wijst J, Dworniczak B, van Zeeland F, Konrad M, … Hoenderop JG (2014). New TRPM6 missense mutations linked to hypomagnesemia with secondary hypocalcemia. Eur J Hum Genet, 22(4), 497–504. 10.1038/ejhg.2013.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leehey DJ, Singh AK, Alavi N, & Singh R (2000). Role of angiotensin II in diabetic nephropathy. Kidney Int Suppl, 77, S93–98. [DOI] [PubMed] [Google Scholar]
- Lim XL, Nurbaya S, Salim A, Tai ES, Maeda S, Nakamura Y, & Ng DP (2012). KCNQ1 SNPS and susceptibility to diabetic nephropathy in East Asians with type 2 diabetes. Diabetologia, 55(9), 2402–2406. 10.1007/s00125-012-2602-5 [DOI] [PubMed] [Google Scholar]
- Linden KC, DeHaan CL, Zhang Y, Glowacka S, Cox AJ, Kelly DJ, & Rogers S (2006). Renal expression and localization of the facilitative glucose transporters GLUT1 and GLUT12 in animal models of hypertension and diabetic nephropathy. Am J Physiol Renal Physiol, 290(1), F205–213. 10.1152/ajprenal.00237.2004 [DOI] [PubMed] [Google Scholar]
- Liu JJ, Lee T, & DeFronzo RA (2012). Why Do SGLT2 inhibitors inhibit only 30–50% of renal glucose reabsorption in humans? Diabetes, 61(9), 2199–2204. 10.2337/db12-0052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZH, Guan TJ, Chen ZH, & Li LS (1999). Glucose transporter (GLUT1) allele (XbaI-) associated with nephropathy in non-insulin-dependent diabetes mellitus. Kidney Int, 55(5), 1843–1848. 10.1046/j.1523-1755.1999.00449.x [DOI] [PubMed] [Google Scholar]
- Lupsa BC, & Inzucchi SE (2018). Use of SGLT2 inhibitors in type 2 diabetes: weighing the risks and benefits. Diabetologia, 61(10), 2118–2125. 10.1007/s00125-018-4663-6 [DOI] [PubMed] [Google Scholar]
- Mansley MK, Watt GB, Francis SL, Walker DJ, Land SC, Bailey MA, & Wilson SM (2016). Dexamethasone and insulin activate serum and glucocorticoid-inducible kinase 1 (SGK1) via different molecular mechanisms in cortical collecting duct cells. Physiol Rep, 4(10). 10.14814/phy2.12792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maqbool M, Cooper ME, & Jandeleit-Dahm KAM (2018). Cardiovascular Disease and Diabetic Kidney Disease. Semin Nephrol, 38(3), 217–232. 10.1016/j.semnephrol.2018.02.003 [DOI] [PubMed] [Google Scholar]
- Marcus RG, England R, Nguyen K, Charron MJ, Briggs JP, & Brosius FC 3rd. (1994). Altered renal expression of the insulin-responsive glucose transporter GLUT4 in experimental diabetes mellitus. Am J Physiol, 267(5 Pt 2), F816–824. 10.1152/ajprenal.1994.267.5.F816 [DOI] [PubMed] [Google Scholar]
- Marko L, Mannaa M, Haschler TN, Kramer S, & Gollasch M (2017). Renoprotection: focus on TRPV1, TRPV4, TRPC6 and TRPM2. Acta Physiol (Oxf), 219(3), 589–612. 10.1111/apha.12828 [DOI] [PubMed] [Google Scholar]
- Marks J, Carvou NJ, Debnam ES, Srai SK, & Unwin RJ (2003). Diabetes increases facilitative glucose uptake and GLUT2 expression at the rat proximal tubule brush border membrane. J Physiol, 553(Pt 1), 137–145. 10.1113/jphysiol.2003.046268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T, Ogura MN, Moriya T, Takahira N, Matsumoto T, Kutsuna T, … Izumi T (2011). Beneficial effects of L- and N-type calcium channel blocker on glucose and lipid metabolism and renal function in patients with hypertension and type II diabetes mellitus. Cardiovasc Ther, 29(1), 46–53. 10.1111/j.1755-5922.2009.00126.x [DOI] [PubMed] [Google Scholar]
- Mather A, & Pollock C (2011). Glucose handling by the kidney. Kidney Int Suppl(120), S1–6. 10.1038/ki.2010.509 [DOI] [PubMed] [Google Scholar]
- McCormick JA, Bhalla V, Pao AC, & Pearce D (2005). SGK1: a rapid aldosterone-induced regulator of renal sodium reabsorption. Physiology, 20, 134–139. [DOI] [PubMed] [Google Scholar]
- Miller BS, Blumenthal SR, Shalygin A, Wright KD, Staruschenko A, Imig JD, & Sorokin A (2018). Inactivation of p66Shc Decreases Afferent Arteriolar KATP Channel Activity and Decreases Renal Damage in Diabetic Dahl SS Rats. Diabetes, 67(11), 2206–2212. 10.2337/db18-0308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muskiet MH, Smits MM, Morsink LM, & Diamant M (2014). The gut-renal axis: do incretin-based agents confer renoprotection in diabetes? Nat Rev Nephrol, 10(2), 88–103. 10.1038/nrneph.2013.272 [DOI] [PubMed] [Google Scholar]
- Nair AV, Hocher B, Verkaart S, van Zeeland F, Pfab T, Slowinski T, … Hoenderop JG (2012). Loss of insulin-induced activation of TRPM6 magnesium channels results in impaired glucose tolerance during pregnancy. Proceedings of the National Academy of Sciences, 109(28), 11324–11329. 10.1073/pnas.1113811109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Tanaka S, Teko Y, Mitsui K, & Kanazawa H (2005). Four Na+/H+ exchanger isoforms are distributed to Golgi and post-Golgi compartments and are involved in organelle pH regulation. J Biol Chem, 280(2), 1561–1572. 10.1074/jbc.M410041200 [DOI] [PubMed] [Google Scholar]
- Ng DP, Canani L, Araki S, Smiles A, Moczulski D, Warram JH, & Krolewski AS (2002). Minor effect of GLUT1 polymorphisms on susceptibility to diabetic nephropathy in type 1 diabetes. Diabetes, 51(7), 2264–2269. [DOI] [PubMed] [Google Scholar]
- Nijenhuis T, Sloan AJ, Hoenderop JG, Flesche J, van Goor H, Kistler AD, … van der Vlag J (2011). Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am J Pathol, 179(4), 1719–1732. 10.1016/j.ajpath.2011.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, & Owsianik G (2011). The transient receptor potential family of ion channels. Genome Biol, 12(3), 218 10.1186/gb-2011-12-3-218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizar JM, Dong W, McClellan RB, Labarca M, Zhou Y, Wong J, … Bhalla V (2016). Na+-sensitive elevation in blood pressure is ENaC independent in diet-induced obesity and insulin resistance. Am J Physiol Renal Physiol, 310(9), F812–820. 10.1152/ajprenal.00265.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizar JM, Shepard BD, Vo VT, & Bhalla V (2018). Renal tubule insulin receptor modestly promotes elevated blood pressure and markedly stimulates glucose reabsorption. JCI Insight, 3(16). 10.1172/jci.insight.95107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S, Yokoi H, Mori K, Kasahara M, Kuwahara K, Fujikura J, … Mukoyama M (2016). Ablation of the N-type calcium channel ameliorates diabetic nephropathy with improved glycemic control and reduced blood pressure. Sci Rep, 6, 27192 10.1038/srep27192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshige T, Tanaka Y, Araki S, Babazono T, Toyoda M, Umezono T, … Maeda S (2010). A single nucleotide polymorphism in KCNQ1 is associated with susceptibility to diabetic nephropathy in japanese subjects with type 2 diabetes. Diabetes Care, 33(4), 842–846. 10.2337/dc09-1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlowski J, & Grinstein S (2004). Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflugers Arch, 447(5), 549–565. 10.1007/s00424-003-1110-3 [DOI] [PubMed] [Google Scholar]
- Pao AC (2016). There and back again: insulin, ENaC, and the cortical collecting duct. Physiol Rep, 4(10). 10.14814/phy2.12809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao SS, Paulsen IT, & Saier MH Jr. (1998). Major facilitator superfamily. Microbiol Mol Biol Rev, 62(1), 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patney V, Chaudhary K, & Whaley-Connell A (2018). Treatment of Diabetic Kidney Disease With Hypertension Control and Renin Angiotensin System Inhibition. Adv Chronic Kidney Dis, 25(2), 158–165. 10.1053/j.ackd.2017.11.002 [DOI] [PubMed] [Google Scholar]
- Patney V, Whaley-Connell A, & Bakris G (2015). Hypertension Management in Diabetic Kidney Disease. Diabetes Spectr, 28(3), 175–180. 10.2337/diaspect.28.3.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavkov ME, Collins AJ, Coresh J, & Nelson RG (2018). Kidney Disease in Diabetes (Cowie C, Casagrande S, Menke A, Cissell M, Eberhardt M, Meigs J, Gregg E, Knowler W, Barrett-Connor E, Becker D, Brancati F, Boyko E, Herman W, Howard B, Narayan K, Rewers M, & Fradkin EJE Eds. Vol. Chapter 22): National Institutes of Health. [Google Scholar]
- Pavlov TS, Ilatovskaya DV, Levchenko V, Li L, Ecelbarger CM, & Staruschenko A (2013). Regulation of ENaC in mice lacking renal insulin receptors in the collecting duct. FASEB J, 27(7), 2723–2732. 10.1096/fj.12-223792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov TS, Levchenko V, Ilatovskaya DV, Li H, Palygin O, Pastor-Soler NM, … Staruschenko A (2017). Lack of Effects of Metformin and AICAR Chronic Infusion on the Development of Hypertension in Dahl Salt-Sensitive Rats. Front Physiol, 8(227), 227 10.3389/fphys.2017.00227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov TS, & Staruschenko A (2017). Involvement of ENaC in the development of salt-sensitive hypertension. Am J Physiol Renal Physiol, 313(2), F135–F140. 10.1152/ajprenal.00427.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PMT, & Kohan DE (2015). Collecting Duct Principal Cell Transport Processes and Their Regulation. Clinical J Am Soc Nephrol, 10(1), 135–146. 10.2215/cjn.05760513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessoa TD, Campos LC, Carraro-Lacroix L, Girardi AC, & Malnic G (2014). Functional role of glucose metabolism, osmotic stress, and sodium-glucose cotransporter isoform-mediated transport on Na+/H+ exchanger isoform 3 activity in the renal proximal tubule. J Am Soc Nephrol, 25(9), 2028–2039. 10.1681/asn.2013060588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen SB, Fenton RA, & Rieg T (2015). Sodium-glucose cotransport. Curr Opin Nephrol Hypertens, 24(5), 463–469. 10.1097/mnh.0000000000000152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell DR, DaCosta CM, Gay J, Ding ZM, Smith M, Greer J, … Zambrowicz B (2013). Improved glycemic control in mice lacking Sglt1 and Sglt2. Am J Physiol Endocrinol Metab, 304(2), E117–130. 10.1152/ajpendo.00439.2012 [DOI] [PubMed] [Google Scholar]
- Powell DR, Smith M, Greer J, Harris A, Zhao S, DaCosta C, … Ding ZM (2013). LX4211 increases serum glucagon-like peptide 1 and peptide YY levels by reducing sodium/glucose cotransporter 1 (SGLT1)-mediated absorption of intestinal glucose. J Pharmacol Exp Ther, 345(2), 250–259. 10.1124/jpet.113.203364 [DOI] [PubMed] [Google Scholar]
- Qin Zou CZ, Yuqing Guo. (2015). TRPC3 and TRPC6 Contribute to the Pathogenesis of Hypertension. Am J Mol Biol, 5(4), 124–133. 10.4236/ajmb.2015.54011 [DOI] [Google Scholar]
- Qu X, Jin F, Hao Y, Li H, Tang T, Wang H, … Dai K (2013). Magnesium and the Risk of Cardiovascular Events: A Meta-Analysis of Prospective Cohort Studies. PLOS ONE, 8(3), e57720 10.1371/journal.pone.0057720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray EC, Miller RG, Demko JE, Costacou T, Kinlough CL, Demko CL, … Kleyman TR (2018). Urinary Plasmin(ogen) as a Prognostic Factor for Hypertension. Kidney Int Rep, 3(6), 1434–1442. 10.1016/j.ekir.2018.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, Wei C, … Pollak MR (2005). TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet, 37(7), 739–744. 10.1038/ng1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazi S, Khan O, Tiwari S, Hu X, & Ecelbarger CA (2006). Rosiglitazone regulates ENaC and Na-K-2Cl cotransporter (NKCC2) abundance in the obese Zucker rat. Am J Nephrol, 26(3), 245–257. 10.1159/000093783 [DOI] [PubMed] [Google Scholar]
- Riazi S, Maric C, & Ecelbarger CA (2006). 17-beta Estradiol attenuates streptozotocin-induced diabetes and regulates the expression of renal sodium transporters. Kidney Int, 69(3), 471–480. 10.1038/sj.ki.5000140 [DOI] [PubMed] [Google Scholar]
- Rieg T, & Vallon V (2018). Development of SGLT1 and SGLT2 inhibitors. Diabetologia, 61(10), 2079–2086. 10.1007/s00125-018-4654-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riehle M, Buscher AK, Gohlke BO, Kassmann M, Kolatsi-Joannou M, Brasen JH, … Harteneck C (2016). TRPC6 G757D Loss-of-Function Mutation Associates with FSGS. J Am Soc Nephrol, 27(9), 2771–2783. 10.1681/ASN.2015030318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstock J, Jelaska A, Frappin G, Salsali A, Kim G, Woerle HJ, & Broedl UC (2014). Improved glucose control with weight loss, lower insulin doses, and no increased hypoglycemia with empagliflozin added to titrated multiple daily injections of insulin in obese inadequately controlled type 2 diabetes. Diabetes Care, 37(7), 1815–1823. 10.2337/dc13-3055 [DOI] [PubMed] [Google Scholar]
- Rowe JW, Tobin JD, Rosa RM, & Andres R (1980). Effect of experimental potassium deficiency on glucose and insulin metabolism. Metabolism, 29(6), 498–502. [DOI] [PubMed] [Google Scholar]
- Ruggenenti P, Porrini EL, Gaspari F, Motterlini N, Cannata A, Carrara F, … Remuzzi G (2012). Glomerular hyperfiltration and renal disease progression in type 2 diabetes. Diabetes Care, 35(10), 2061–2068. 10.2337/dc11-2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagild U, Andersen V, & Andreasen PB (1961). Glucose tolerance and insulin responsiveness in experimental potassium depletion. Acta Med Scand, 169, 243–251. [DOI] [PubMed] [Google Scholar]
- Salomonsson M, Brasen JC, & Sorensen CM (2017). Role of renal vascular potassium channels in physiology and pathophysiology. Acta Physiol, 221(1), 14–31. 10.1111/apha.12882 [DOI] [PubMed] [Google Scholar]
- Sano M, Takei M, Shiraishi Y, & Suzuki Y (2016). Increased Hematocrit During Sodium-Glucose Cotransporter 2 Inhibitor Therapy Indicates Recovery of Tubulointerstitial Function in Diabetic Kidneys. J Clin Med Res, 8(12), 844–847. 10.14740/jocmr2760w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheen AJ, & Delanaye P (2018). Renal outcomes with dipeptidyl peptidase-4 inhibitors. Diabetes Metab, 44(2), 101–111. 10.1016/j.diabet.2017.07.011 [DOI] [PubMed] [Google Scholar]
- Schena FP, & Gesualdo L (2005). Pathogenetic mechanisms of diabetic nephropathy. J Am Soc Nephrol, 16 Suppl 1, S30–33. [DOI] [PubMed] [Google Scholar]
- Schiffer M, Susztak K, Ranalletta M, Raff AC, Bottinger EP, & Charron MJ (2005). Localization of the GLUT8 glucose transporter in murine kidney and regulation in vivo in nondiabetic and diabetic conditions. Am J Physiol Renal Physiol, 289(1), F186–193. 10.1152/ajprenal.00234.2004 [DOI] [PubMed] [Google Scholar]
- Schleicher ED, Wagner E, & Nerlich AG (1997). Increased accumulation of the glycoxidation product N(epsilon)-(carboxymethyl)lysine in human tissues in diabetes and aging. J Clin Invest, 99(3), 457–468. 10.1172/JCI119180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlingmann KP, Weber S, Peters M, Niemann NL, Vitzthum H, Klingel K, … Konrad M (2002). Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nature Genetics, 31(2), 166–170. [DOI] [PubMed] [Google Scholar]
- Secrest MH, Udell JA, & Filion KB (2017). The cardiovascular safety trials of DPP-4 inhibitors, GLP-1 agonists, and SGLT2 inhibitors. Trends Cardiovasc Med, 27(3), 194–202. 10.1016/j.tcm.2017.01.009 [DOI] [PubMed] [Google Scholar]
- Singh VP, Bali A, Singh N, & Jaggi AS (2014). Advanced glycation end products and diabetic complications. Korean J Physiol Pharmacol, 18(1), 1–14. 10.4196/kjpp.2014.18.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth A, Dunkler D, Gao P, Teo KK, Yusuf S, O’Donnell MJ, … Clase CM (2014). The relationship between estimated sodium and potassium excretion and subsequent renal outcomes. Kidney Int, 86(6), 1205–1212. 10.1038/ki.2014.214 [DOI] [PubMed] [Google Scholar]
- Song J, Knepper MA, Verbalis JG, & Ecelbarger CA (2003). Increased renal ENaC subunit and sodium transporter abundances in streptozotocin-induced type 1 diabetes. Am J Physiol Renal Physiol, 285(6), F1125–1137. 10.1152/ajprenal.00143.2003 [DOI] [PubMed] [Google Scholar]
- Song P, Onishi A, Koepsell H, & Vallon V (2016). Sodium glucose cotransporter SGLT1 as a therapeutic target in diabetes mellitus. Expert Opin Ther Targets, 20(9), 1109–1125. 10.1517/14728222.2016.1168808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Hsu Y-H, Niu T, Manson JE, Buring JE, & Liu S (2009). Common genetic variants of the ion channel transient receptor potential membrane melastatin 6 and 7 (TRPM6 and TRPM7), magnesium intake, and risk of type 2 diabetes in women. BMC Medical Genetics, 10(1), 4 10.1186/1471-2350-10-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonneveld R, van der Vlag J, Baltissen MP, Verkaart SA, Wetzels JF, Berden JH, … Nijenhuis T (2014). Glucose specifically regulates TRPC6 expression in the podocyte in an AngII-dependent manner. Am J Pathol, 184(6), 1715–1726. 10.1016/j.ajpath.2014.02.008 [DOI] [PubMed] [Google Scholar]
- Spatola L, Finazzi S, Angelini C, Dauriz M, & Badalamenti S (2018). SGLT1 and SGLT1 Inhibitors: A Role to Be Assessed in the Current Clinical Practice. Diabetes Ther, 9(1), 427–430. 10.1007/s13300-017-0342-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires D, Ilatovskaya DV, Levchenko V, North PE, Geurts AM, Palygin O, & Staruschenko A (2018). Protective role of Trpc6 knockout in the progression of diabetic kidney disease. Am J Physiol Renal Physiol, 315(4), F1091–F1097. 10.1152/ajprenal.00155.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staruschenko A (2012). Regulation of transport in the connecting tubule and cortical collecting duct. Compreh Physiol, 2(2), 1541–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staruschenko A (2017). Hypertension and Diabetes Mellitus: The Chicken and Egg Problem. Hypertension, 69(5), 787–788. 10.1161/HYPERTENSIONAHA.117.08671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staruschenko A (2019). TRPC6 in diabetic kidney disease: Good guy or bad guy? Kidney Int, 10.1016/j.kint.2018.10.027 [DOI] [PMC free article] [PubMed]
- Sugano N, Hayashi K, Hosoya T, & Yokoo T (2013). Mechanistic view of renal protective action of calcium channel blockade. Curr Hypertens Rev, 9(3), 187–192. [DOI] [PubMed] [Google Scholar]
- Svenningsen P, Skott O, & Jensen BL (2012). Proteinuric diseases with sodium retention: Is plasmin the link? Clin Exp Pharmacol Physiol, 39(1), 117–124. 10.1111/j.1440-1681.2011.05524.x [DOI] [PubMed] [Google Scholar]
- Szablewski L (2017). Distribution of glucose transporters in renal diseases. J Biomed Sci, 24(1), 64 10.1186/s12929-017-0371-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahrani AA, Barnett AH, & Bailey CJ (2013). SGLT inhibitors in management of diabetes. Lancet Diabetes Endocrinol, 1(2), 140–151. 10.1016/s2213-8587(13)70050-0 [DOI] [PubMed] [Google Scholar]
- Tanaka S, Sugiura Y, Saito H, Sugahara M, Higashijima Y, Yamaguchi J, … Tanaka T (2018). Sodium-glucose cotransporter 2 inhibition normalizes glucose metabolism and suppresses oxidative stress in the kidneys of diabetic mice. Kidney Int, 94(5), 912–925. 10.1016/j.kint.2018.04.025 [DOI] [PubMed] [Google Scholar]
- Tarnow L, Grarup N, Hansen T, Parving HH, & Pedersen O (2001). Diabetic microvascular complications are not associated with two polymorphisms in the GLUT-1 and PC-1 genes regulating glucose metabolism in Caucasian type 1 diabetic patients. Nephrol Dial Transplant, 16(8), 1653–1656. [DOI] [PubMed] [Google Scholar]
- Thorens B, & Mueckler M (2010). Glucose transporters in the 21st Century. Am J Physiol Endocrinol Metab, 298(2), E141–145. 10.1152/ajpendo.00712.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari S, Nordquist L, Halagappa VK, & Ecelbarger CA (2007). Trafficking of ENaC subunits in response to acute insulin in mouse kidney. American Journal of Physiology. Renal Physiology, 293(1), F178–F185. [DOI] [PubMed] [Google Scholar]
- Tomilin V, Mamenko M, Zaika O, & Pochynyuk O (2016). Role of renal TRP channels in physiology and pathology. Semin Immunopathol, 38(3), 371–383. 10.1007/s00281-015-0527-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonneijck L, Muskiet MH, Smits MM, van Bommel EJ, Heerspink HJ, van Raalte DH, & Joles JA (2017). Glomerular Hyperfiltration in Diabetes: Mechanisms, Clinical Significance, and Treatment. J Am Soc Nephrol, 28(4), 1023–1039. 10.1681/asn.2016060666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle KR (2017). Back to the Future: Glomerular Hyperfiltration and the Diabetic Kidney. Diabetes, 66(1), 14–16. 10.2337/dbi16-0056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umanath K, & Lewis JB (2018). Update on Diabetic Nephropathy: Core Curriculum 2018. Am J Kidney Dis, 71(6), 884–895. 10.1053/j.ajkd.2017.10.026 [DOI] [PubMed] [Google Scholar]
- Unoki H, Takahashi A, Kawaguchi T, Hara K, Horikoshi M, Andersen G, … Maeda S (2008). SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet, 40(9), 1098–1102. 10.1038/ng.208 [DOI] [PubMed] [Google Scholar]
- Unruh ML, Pankratz VS, Demko JE, Ray EC, Hughey RP, & Kleyman TR (2017). Trial of Amiloride in Type 2 Diabetes with Proteinuria. Kidney Int Rep, 2(5), 893–904. 10.1016/j.ekir.2017.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallon V, Blantz RC, & Thomson S (2003). Glomerular hyperfiltration and the salt paradox in early [corrected] type 1 diabetes mellitus: a tubulo-centric view. J Am Soc Nephrol, 14(2), 530–537. [DOI] [PubMed] [Google Scholar]
- Vallon V, & Komers R (2011). Pathophysiology of the diabetic kidney. Compr Physiol, 1(3), 1175–1232. 10.1002/cphy.c100049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallon V, Platt KA, Cunard R, Schroth J, Whaley J, Thomson SC, … Rieg T (2011). SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol, 22(1), 104–112. 10.1681/asn.2010030246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaulont S, & Kahn A (1994). Transcriptional control of metabolic regulation genes by carbohydrates. Faseb j, 8(1), 28–35. [DOI] [PubMed] [Google Scholar]
- Vazquez G, Wedel BJ, Aziz O, Trebak M, & Putney JW Jr. (2004). The mammalian TRPC cation channels. Biochim Biophys Acta, 1742(1–3), 21–36. 10.1016/j.bbamcr.2004.08.015 [DOI] [PubMed] [Google Scholar]
- Wakisaka M (2016). Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N Engl J Med, 375(18), 1799–1800. 10.1056/NEJMc1611290 [DOI] [PubMed] [Google Scholar]
- Wakisaka M, Nagao T, & Yoshinari M (2016). Sodium Glucose Cotransporter 2 (SGLT2) Plays as a Physiological Glucose Sensor and Regulates Cellular Contractility in Rat Mesangial Cells. PLoS One, 11(3), e0151585 10.1371/journal.pone.0151585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walder RY, Landau D, Meyer P, Shalev H, Tsolia M, Borochowitz Z, … Sheffield VC (2002). Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nature Genetics, 31(2), 171–174. [DOI] [PubMed] [Google Scholar]
- Wang L, Chang J-H, Buckley AF, & Spurney RF (2019). Knockout of TRPC6 promotes insulin resistance and exacerbates glomerular injury in Akita mice. Kidney International. [DOI] [PMC free article] [PubMed]
- Wang Q, Song B, Jiang S, Liang C, Chen X, Shi J, … Ma HP (2015). Hydrogen Sulfide Prevents Advanced Glycation End-Products Induced Activation of the Epithelial Sodium Channel. Oxid Med Cell Longev, 2015, 976848 10.1155/2015/976848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XX, Levi J, Luo Y, Myakala K, Herman-Edelstein M, Qiu L, … Levi M (2017). SGLT2 Protein Expression Is Increased in Human Diabetic Nephropathy: SGLT2 PROTEIN INHIBITION DECREASES RENAL LIPID ACCUMULATION, INFLAMMATION, AND THE DEVELOPMENT OF NEPHROPATHY IN DIABETIC MICE. J Biol Chem, 292(13), 5335–5348. 10.1074/jbc.M117.779520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Heilig K, Saunders T, Minto A, Deb DK, Chang A, … Heilig CW (2010). Transgenic overexpression of GLUT1 in mouse glomeruli produces renal disease resembling diabetic glomerulosclerosis. Am J Physiol Renal Physiol, 299(1), F99–f111. 10.1152/ajprenal.00466.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, do Carmo JM, Aberdein N, Zhou X, Williams JM, da Silva AA, & Hall JE (2017). Synergistic Interaction of Hypertension and Diabetes in Promoting Kidney Injury and the Role of Endoplasmic Reticulum Stress. Hypertension, 69(5), 879–891. 10.1161/HYPERTENSIONAHA.116.08560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasik AA, & Lehtonen S (2018). Glucose Transporters in Diabetic Kidney Disease-Friends or Foes? Front Endocrinol (Lausanne), 9, 155 10.3389/fendo.2018.00155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JR Jr., & Campbell RK (1993). Magnesium and diabetes: a review. Ann Pharmacother, 27(6), 775–780. 10.1177/106002809302700619 [DOI] [PubMed] [Google Scholar]
- Wilding JP, Woo V, Soler NG, Pahor A, Sugg J, Rohwedder K, & Parikh S (2012). Long-term efficacy of dapagliflozin in patients with type 2 diabetes mellitus receiving high doses of insulin: a randomized trial. Ann Intern Med, 156(6), 405–415. 10.7326/0003-4819-156-6-201203200-00003 [DOI] [PubMed] [Google Scholar]
- Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, … Rosenberg PB (2005). A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science, 308(5729), 1801–1804. [DOI] [PubMed] [Google Scholar]
- Woudenberg-Vrenken TE, Bindels RJ, & Hoenderop JG (2009). The role of transient receptor potential channels in kidney disease. Nat Rev Nephrol, 5(8), 441–449. 10.1038/nrneph.2009.100 [DOI] [PubMed] [Google Scholar]
- Wright EM, Loo DD, & Hirayama BA (2011). Biology of human sodium glucose transporters. Physiol Rev, 91(2), 733–794. 10.1152/physrev.00055.2009 [DOI] [PubMed] [Google Scholar]
- Wu KL, Khan S, Lakhe-Reddy S, Wang L, Jarad G, Miller RT, … Schelling JR (2003). Renal tubular epithelial cell apoptosis is associated with caspase cleavage of the NHE1 Na+/H+ exchanger. Am J Physiol Renal Physiol, 284(4), F829–839. 10.1152/ajprenal.00314.2002 [DOI] [PubMed] [Google Scholar]
- Wulff H, & Castle NA (2010). Therapeutic potential of KCa3.1 blockers: recent advances and promising trends. Expert Rev Clin Pharmacol, 3(3), 385–396. 10.1586/ecp.10.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Chen R, & Ghishan FK (2005). Subcloning, localization, and expression of the rat intestinal sodium-hydrogen exchanger isoform 8. Am J Physiol Gastrointest Liver Physiol, 289(1), G36–41. 10.1152/ajpgi.00552.2004 [DOI] [PubMed] [Google Scholar]
- Yale JF, Bakris G, Cariou B, Yue D, David-Neto E, Xi L, … Meininger G (2013). Efficacy and safety of canagliflozin in subjects with type 2 diabetes and chronic kidney disease. Diabetes Obes Metab, 15(5), 463–473. 10.1111/dom.12090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N (2015). Structural Biology of the Major Facilitator Superfamily Transporters. Annu Rev Biophys, 44, 257–283. 10.1146/annurev-biophys-060414-033901 [DOI] [PubMed] [Google Scholar]
- Yee J (2018). Magnesium: An Important Orphan. Adv Chronic Kidney Dis, 25(3), 217–221. 10.1053/j.ackd.2018.04.001 [DOI] [PubMed] [Google Scholar]
- Zambrowicz B, Freiman J, Brown PM, Frazier KS, Turnage A, Bronner J, … Powell DR (2012). LX4211, a dual SGLT1/SGLT2 inhibitor, improved glycemic control in patients with type 2 diabetes in a randomized, placebo-controlled trial. Clin Pharmacol Ther, 92(2), 158–169. 10.1038/clpt.2012.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeni L, Norden AGW, Cancarini G, & Unwin RJ (2017). A more tubulocentric view of diabetic kidney disease. J Nephrol, 30(6), 701–717. 10.1007/s40620-017-0423-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ding J, Fan Q, & Liu S (2009). TRPC6 up-regulation in Ang II-induced podocyte apoptosis might result from ERK activation and NF-kappaB translocation. Exp Biol Med (Maywood), 234(9), 1029–1036. 10.3181/0901-rm-11 [DOI] [PubMed] [Google Scholar]
- Zhang H, Schin M, Saha J, Burke K, Holzman LB, Filipiak W, … Brosius FC 3rd. (2010). Podocyte-specific overexpression of GLUT1 surprisingly reduces mesangial matrix expansion in diabetic nephropathy in mice. Am J Physiol Renal Physiol, 299(1), F91–98. 10.1152/ajprenal.00021.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zillich AJ, Garg J, Basu S, Bakris GL, & Carter BL (2006). Thiazide diuretics, potassium, and the development of diabetes: a quantitative review. Hypertension, 48(2), 219–224. 10.1161/01.HYP.0000231552.10054.aa [DOI] [PubMed] [Google Scholar]
- Zou H, Zhou B, & Xu G (2017). SGLT2 inhibitors: a novel choice for the combination therapy in diabetic kidney disease. Cardiovasc Diabetol, 16(1), 65 10.1186/s12933-017-0547-1 [DOI] [PMC free article] [PubMed] [Google Scholar]